

Abstract
Negative symptoms and cognitive deficits contribute strongly to disability in schizophrenia, and are resistant to existing medications. Recent drug development has targeted enhanced NMDA function by increasing mGluR2/3 signaling. However, the clinical utility of such agents remains uncertain, and markers of brain circuit function are critical for clarifying mechanisms and understanding individual differences in efficacy. We conducted a double-blind, placebo-controlled, randomized cross-over (14 day washout) pilot study evaluating adjunctive use of the mGluR2 positive allosteric modulator AZD8529 (80mg daily for 3 days), in chronic stable patients with schizophrenia (n=26 analyzed). We focused on 3T fMRI response in frontostriatal regions during an n-back working memory task, testing the hypothesis that AZD8529 produces fMRI changes that correlate with improvement in negative symptoms and cognition. We found that AZD8529 did not produce significant group-average effects on symptoms or cognitive accuracy. However, AZD8529 did increase n-back fMRI activation in striatum (p<0.0001) and anterior cingulate/paracingulate (p=0.002). Greater drug-versus-placebo effects on caudate activation significantly correlated with greater reductions in PANSS negative symptom scores (r=−0.42, p=0.031), and exploratory correlations suggested broader effects across multiple symptom domains and regions of interest. These findings demonstrate that fMRI responses to mGluR2 positive modulation relate to individual differences in symptom reduction, and could be pursued for future biomarker development. Negative clinical results at the group level should not lead to premature termination of investigation of this mechanism which may benefit an important subset of individuals with schizophrenia. Imaging biomarkers may reveal therapeutic mechanisms, and help guide treatment towards specific populations.
Introduction
Negative symptoms of schizophrenia, such as amotivation and flat affect, are prevalent and contribute to disability and poor outcomes [1]. Negative symptoms are also resistant to existing treatments, creating a critical need for novel therapeutic targets and agents [2]. Cognitive deficits in schizophrenia are also prominent, disabling, and treatment resistant [3], and there are important interactions between motivational and cognitive deficits at both psychological and neurobiological levels [4–7]. In particular, both cognitive and motivational impairments are linked to dysfunction in frontostriatal circuitry. This circuitry includes dorsolateral and anterior cingulate regions in the prefrontal cortex, which are critical for executive functions such as working memory, and ventral and dorsal striatal regions linked to motivation and reinforcement learning [5, 7]. In fMRI reward tasks, hypofunction in striatum correlates with global negative symptom severity, and amotivation specifically [8–11]. Hypofunction in ventral and dorsal striatum and connected prefrontal regions is also associated with schizophrenia and negative symptoms during working memory and other cognitive fMRI paradigms [12–16].
Blockade of dopamine receptors is the primary mechanism of all existing antipsychotics, and this blockade may have competing positive and negative effects on both negative symptoms and cognition [17, 18]. Inspired by the glutamate hypothesis of schizophrenia pathophysiology, more recent drug development efforts have focused on ameliorating putative deficits in NMDA signaling [19, 20]. Interest has been fueled by multiple studies in rodents showing positive effects of agonism at type II metabotropic glutamate receptors (mGluR2/3) and mGluR2 positive allosteric modulators (PAMs) in NMDA-antagonist models of psychosis, including improvements in cognition and motivation [21–25]. In a rodent functional imaging study, an mGluR2 PAM opposed the effects of NMDA-antagonists in specific brain regions including prefrontal cortex, dorsal and ventral striatum, and mediodorsal thalamus [26], and an mGluR2-selective agonist reduced ketamine-induced fMRI abnormalities in healthy humans [27]. The mGluR2 receptor is thus a promising target for modulating frontostriatal circuitry and improving negative symptoms and cognitive deficits in schizophrenia.
The extent to which mGluR2 signaling impacts cognitive or reward function in humans remains largely unknown. On the strength of preclinical studies, mGluR2/3 agonists and more recently mGluR2 PAMs have been brought into clinical trials in schizophrenia [22, 28] and other psychiatric disorders [29, 30]. These trials have demonstrated an excellent safety profile of mGluR 2/3 agonists and modulators, but their efficacy in reducing symptoms has been inconsistent. Encouraging initial efforts, the mGluR2/3 agonist pomaglumetad improved both negative and positive symptoms in the first large-scale trial [31]. However, later clinical trials with pomeglumetad did not show consistent benefits [32, 33]. Two different mGluR 2/3 agonists reduced ketamine-induced psychotic symptoms in healthy volunteers [34]. An mGluR2 PAM (ADX/JNJ) also reduced ketamine-induced negative symptoms in healthy volunteers [35], but only trend improvement was found in a preliminary study of schizophrenia associated with prominent residual negative symptoms [36]. The single published clinical trial in schizophrenia with the mGluR2 PAM studied here (AZD8529) did not improve symptoms or cognition [37].
It remains unclear why the promise of these agents has so far not been borne out in clinical trials, but the field is at risk of prematurely discarding an intervention that could still benefit an important subset of patients. Heterogeneity of individual response is likely to be a major contributor to negative clinical trials [22, 38]. Indeed, re-analyses of the pomeglumetad schizophrenia trials suggested efficacy in specific subgroups, including those in early stages of illness (< 3 years) and those whose antipsychotic treatment had included dopamine D2 blockade but not serotonin 5HT2A blockade [39]. There is some evidence for inverted U-shaped dose response curves with mGluR2 PAMs [40], and individual differences in drug metabolism or sensitivity could impede identification of effects averaged across a sample.
Further progress in understanding this heterogeneity, evaluating early signals of efficacy, and predicting clinical effects will require greater understanding of drug effects on specific neural circuits, and development of neurophysiological biomarkers such as pharmacological fMRI (phMRI) [41, 42]. Even when effects are not robust on average, incorporation of neurophysiological biomarkers can reveal brain-behavioral correlations that give insight into drug mechanisms and individual differences in drug response, consistent with the precision medicine and experimental therapeutic approaches advocated by NIMH and others [43, 44].
To date, no imaging studies have been published using mGluR2/3 agonists or modulators in schizophrenia (or any other psychiatric populations). To address this gap and gain insight into neural circuit effects and potential response heterogeneity, we performed this phMRI study evaluating adjunctive treatment with the mGluR2 PAM AZD8529. We hypothesized the drug would increase fMRI activation in prefrontal and striatal circuitry, and that this change in activation would correlate with improvement in negative symptoms and cognitive performance.
Materials and methods
Participants
45 adult individuals were randomized into the study. After exclusions (See Supplemental Methods for inclusion/exclusion criteria and excluded participants), 26 participants (10 female) had complete fMRI data analyzed and reported here. Participants were stable outpatients with a DSM-IV diagnosis of schizophrenia (n=25) or schizoaffective disorder depressed type (n=1), on a stable antipsychotic regimen which was continued throughout the study. Most (n=22) were on 2nd/3rd-generation antipsychotics, two were on 1st-generation antipsychotics, and two were on both; mean daily dosage was 510±283 mg chlorpromazine equivalents (CPZ). Additional psychotropic medications taken on a routine basis included anticholinergics (n=8), anxiolytics (n=9), antidepressants (n=10), anticonvulsants/lithium (n=4/1), thyroid replacement hormone (n=1). Demographic and clinical characterization of the analyzed sample is included in Table 1. Participants underwent standard medical, neurological, psychiatric, and neurocognitive evaluations (detailed in Supplementary Methods). Participants were provided with a complete description of the study and written informed consent was obtained. All study procedures were approved by the University of Pennsylvania IRB and conducted in accordance with the Declaration of Helsinki.
Table 1.
Demographic and Clinical Variables.
| Variable | PLACEBO | DRUG | p-value |
|---|---|---|---|
| Age (years) | 42.5 (9.6) a | - | - |
| Gender (% female) | 38% | - | - |
| Smoke (% yes) | 77% | - | - |
| Education (years) | 12.7 (1.9) | - | - |
| bParental Education (years) | 12.7 (2.3) | - | - |
| PANSS Negative | 13.7 (3.9) | 13.5 (4.2) | 0.77c |
| PANSS Positive | 13.7 (4.5) | 13.9 (4.8) | 0.77 |
| PANSS General | 24.2 (6.0) | 24.3 (4.6) | 0.90 |
| PANSS Total | 51.6 (10.7) | 51.7 (9.7) | 0.96 |
| dAZD8529 Blood Level (ng/mL) | - | 112 (50) | - |
Mean (SD) reported for dimensional variables
Parental Education data from n=25 of total 26.
2-tailed p-values from paired t-test; mixed model results are reported in main text
AZD8529 PK blood levels on day of scan, from n=22 of total 26.
Study drug and design
The adjunctive pharmacological agent used here, AZD8529, is an mGluR2-selective positive allosteric modulator (PAM). Like all mGluR2 PAMS, AZD8529 acts on an allosteric site to potentiate mGluR2 activation only in the presence of endogenous glutamate [21]. The use of direct agonists at mGluR2/3 carries significant limitations for both mechanistic investigation and therapeutic applications, which can be surmounted by the use of PAMs [45]. Targeting the allosteric site allows selective activation of mGluR2 and not mGluR3, and maintains physiologic activity-dependent receptor activation, which may reduce non-physiological and adverse effects, as well as habituation [21, 22]. In addition, PAMs including AZD8529 exhibit greater oral bioavailability and CNS penetration than agonists, which enhances their potential clinical utility. The dosing of AZD8529 was 80mg daily for three days, a regimen designed to achieve after 3 days the steady-state level approximating the level from the chronic 40 mg every-other-day dosing used in Litman et al. [37]. This approximation was based on prior pharmacokinetic studies using various dosing regimens (AZD8529 Multiple Ascending Dose Study D1960C00002, data on file at AstraZeneca); as can be seen in Table 1 we achieved average blood levels (112 ng/mL) that were somewhat higher than those reported in Litman et al. (Cmax 73ng/mL, Cmin 38ng/mL) while still well within the range previously determined to be safe and tolerable.
This was a Phase 1 pilot study, designed and registered in 2009 as NCT00986531. The overall design was a double blind, placebo-controlled, counterbalanced within-subject crossover, with a 14-day washout between drug and placebo phases. Primary outcome measures included fMRI activation and cognitive battery performance; secondary outcomes included EEG, negative and other symptoms from the Positive and Negative Syndrome Scale (PANSS), and safety assessments including vital signs, electrocardiogram (ECG), suicidality and other adverse events. Key outcome measures were obtained once following 3 days of treatment with oral AZD8529 (80 mg daily), and once following 3 days of treatment with an identical-appearing placebo (see Supplementary Methods for details of visit schedule, and Figure S1 for study design schematic).
Imaging task
During both MRI sessions, subjects completed a previously described fractal n-back task (0, 1, 2, and 3-back block design; Figure 1) [46]. To minimize practice effects, two equivalent forms of the tasks with unique stimuli were administered in a counterbalanced order. Our primary behavioral measure in the scanner n-back task was d′, which summarizes overall task performance while accounting for the number of correct responses and the number of false positives [47]. Due to technical problems, n-back in-scanner behavioral data was incomplete and excluded for two participants. See Supplement for further fMRI task details.
Figure 1. N-Back fMRI Paradigm.
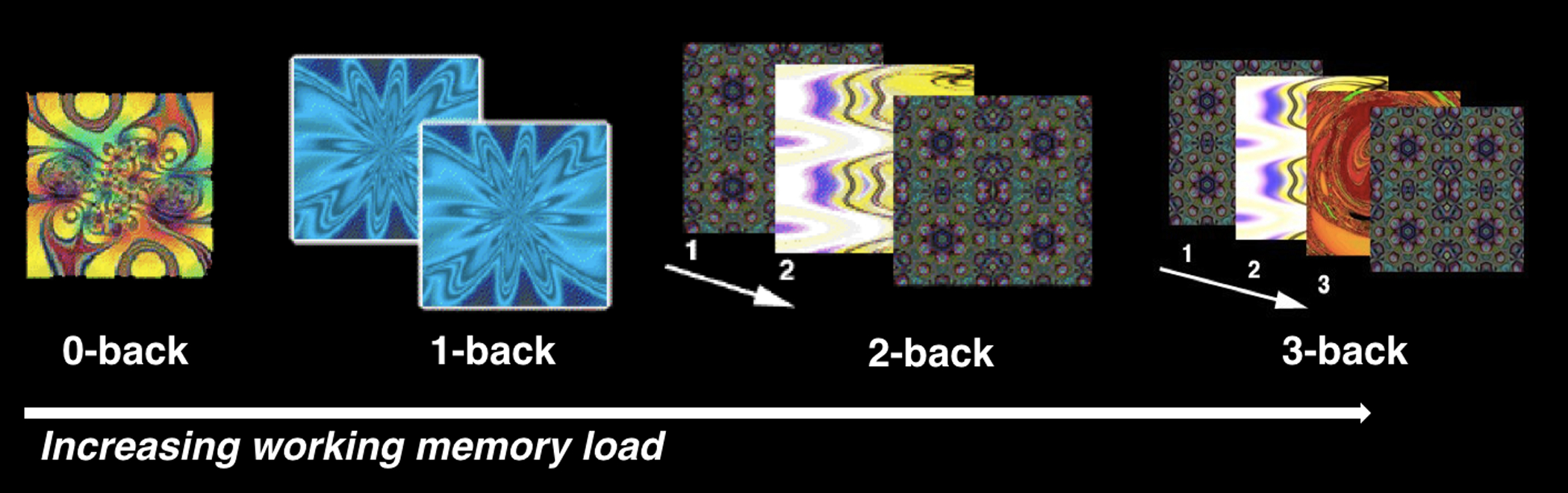
A. Working memory was tested with complex geometric figures (fractals), parametrically varying levels of memory load (0-back, 1-back, 2-back, 3-back) across blocks. In the 0-back condition, participants responded with a button press to a specified target fractal. For the 1-back condition, participants responded if the current fractal was identical to the previous one; in the 2-back and 3-back conditions, participants responded if the current fractal was identical to the item presented two or three trials previously, respectively. Each condition consisted of a 20-trial block (60 s); each level was repeated over three blocks. The target-foil ratio was 1:3 in all blocks. Each fractal was presented for 500ms, followed by an inter-stimulus interval of 2500ms. Total n-back task duration (including fixation and instruction periods) was 900 seconds.
Image acquisition, processing and timeseries analysis
Images were acquired with a Siemens Trio 3T (Erlangen, Germany) system, in the following order: structural, resting perfusion, emotion identification BOLD, continuous performance BOLD, n-back BOLD, B0 map. BOLD fMRI data were preprocessed and analyzed using FEAT (FMRI Expert Analysis Tool) within FSL (FMRIB’s Software Library, www.fmrib.ox.ac.uk/fsl). Individual-level timeseries analysis was carried out using a general linear model in FSL. A canonical hemodynamic response function was used to model 4 conditions (0-back, 1-back, 2-back, 3-back) with separate regressors; six motion correction parameters were included as nuisance regressors, and rest condition was designated the unmodeled baseline. See Supplementary Methods for further acquisition and processing details.
Group-level region of interest image analysis
For our primary analysis of group-level effects, a region of interest (ROI) approach was used. Based on the expected intersection of working memory effects and mGluR2 effects within frontostriatal circuitry as described above, we examined three bilateral ROIs: DLPFC, ACC, and striatum (STR). To obtain unbiased functional ROIs we generated 10mm spheres around areas of peak activation from the Neurosynth meta-anlysis of working memory fMRI (see Supplementary Methods and Figure S2 for detailed ROI definitions). For each ROI, individual subject activation measures (contrast parameter estimates, arbitrary units) were extracted for statistical analysis.
Group-level exploratory voxelwise analyses
In addition to analysis in these three ROIs, we conducted exploratory whole-brain voxelwise analyses to test for effects outside predicted regions. Individual-level statistical maps were transformed into MNI space and resampled to 2mm isotropic voxels. Drug-placebo fixed effects were calculated within each subject and these contrasts were then subjected to group-level analyses in FSL using FLAME1. All voxelwise analyses were FWE-corrected using 5000 permutations in FSL’s randomise, with threshold-free cluster enhancement (TFCE)[48] to ensure rigorous control of multiple comparisons.
Mixed model analysis and other statistical tests
Drug effects were examined using a linear mixed effects model (lmer procedure) implemented in R version 3.2.5, with participants as a random factor, drug (AZD8529 vs. placebo) as a fixed effect; some analyses included working memory load level as an additional fixed factor. There was no effect of the two n-back task forms on any outcome and so this variable was not included. Descriptive statistics and uncorrected post-hoc t-tests were used to explicate significant results from the mixed models; normality and variance were examined for key variables. Pearsons correlations were used to relate drug-induced change in ROI activation to drug-induced change in symptoms. For all statistical tests, significance was determined using an alpha criterion of p<0.05, two-tailed.
Results
Safety, Clinical Symptoms, Cognitive Performance
Overall, the drug was well-tolerated. There were two serious adverse events, both involving hyperglycemia in participants with pre-existing type 2 diabetes (one on study drug, one on placebo); neither was considered study-related, they were withdrawn from the study. There were no clinically significant ECG abnormalities reported.
No significant effects of drug were found on PANSS symptoms, out-of scanner cognitive battery overall accuracy (p=0.16), or the d’ measure of in-scanner n-back performance (p=0.46). The d’ measure did show an expected effect of worse performance at higher n-back levels (F(3,165)=46.47, p <.0001), with no drug x level interaction (p=0.78).
Effects on n-back fMRI activation
BOLD activation showed expected modulation by working memory load in ACC (F(3,178)=5.25, p=0.002) and DLPFC (F(3,178) = 3.14, p=0.027) although load effects were not significant in striatum (F(3,178)=0.92, p=0.43). This pattern reflected increasing activation at higher load levels, although with some reduction at the highest 3-back level (Figure 2). Relative to placebo, drug increased activation across load levels in ACC (F(1, 178) = 10.04, p=0.002) and more dramatically in striatum (F(1,178)=25.4,p<0.0001); no significant effects were seen in DLPFC (F(1, 178)=2.10, p=0.15). No regions showed significant interaction effects of drug with working memory load level (p’s >0.3). Exploratory voxelwise analysis did not identify other regions showing significant drug effects. Participants who showed greater striatal activation by the drug also showed greater reductions in PANSS negative symptom scores (correlation of drug-placebo difference scores, r=−0.42, p=0.031; Figure 3). This relationship with the primary PANSS negative symptom score was not significant in other ROIs and was not accounted for by other PANSS domains; however, exploratory correlations suggested broader drug effects across multiple symptom domains and regions of interest (see Supplementary Results and Tables S1 and S2). fMRI findings were not explained by demographic variables, head motion, anxiety, extrapyramidal symptoms, or other potential confounds (see Supplementary Results).
Figure 2. Drug Effects on N-back fMRI Activation.
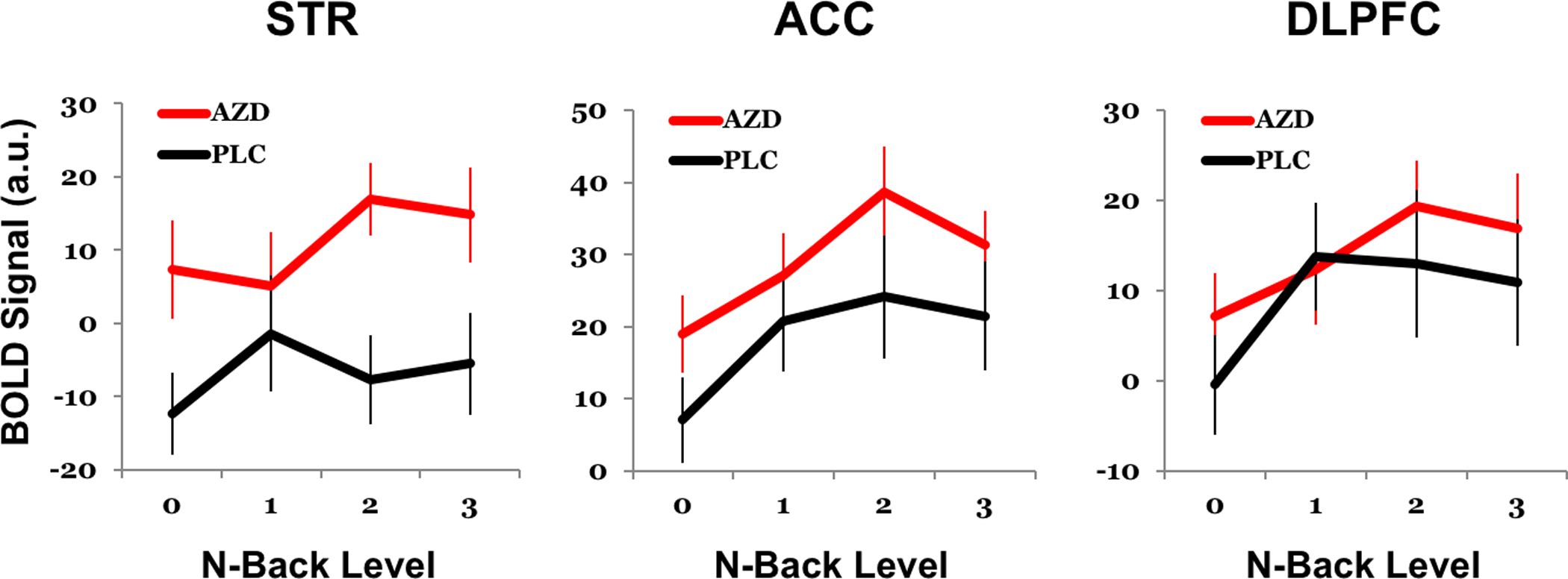
Drug-induced increases in activation during the n-back task were most robust in striatum (STR, p<0.0001 across levels), then Anterior cingulate cortex (ACC, p=0.002), but was not significant in dorsolateral prefrontal cortex (DLPFC, p=0.15). Activation in all regions tended to be higher on levels requiring working memory, but effects of n-back level were only significant in ACC and DLPFC. Drug effects appeared strongest at the 2-back level, which was also the level associated with greatest activation across sessions, but drug x level interactions were not statistically significant. AZD=AZD8529, PLC=Placebo. Error bars show SEM.
Figure 3. fMRI-Symptom Relationship.
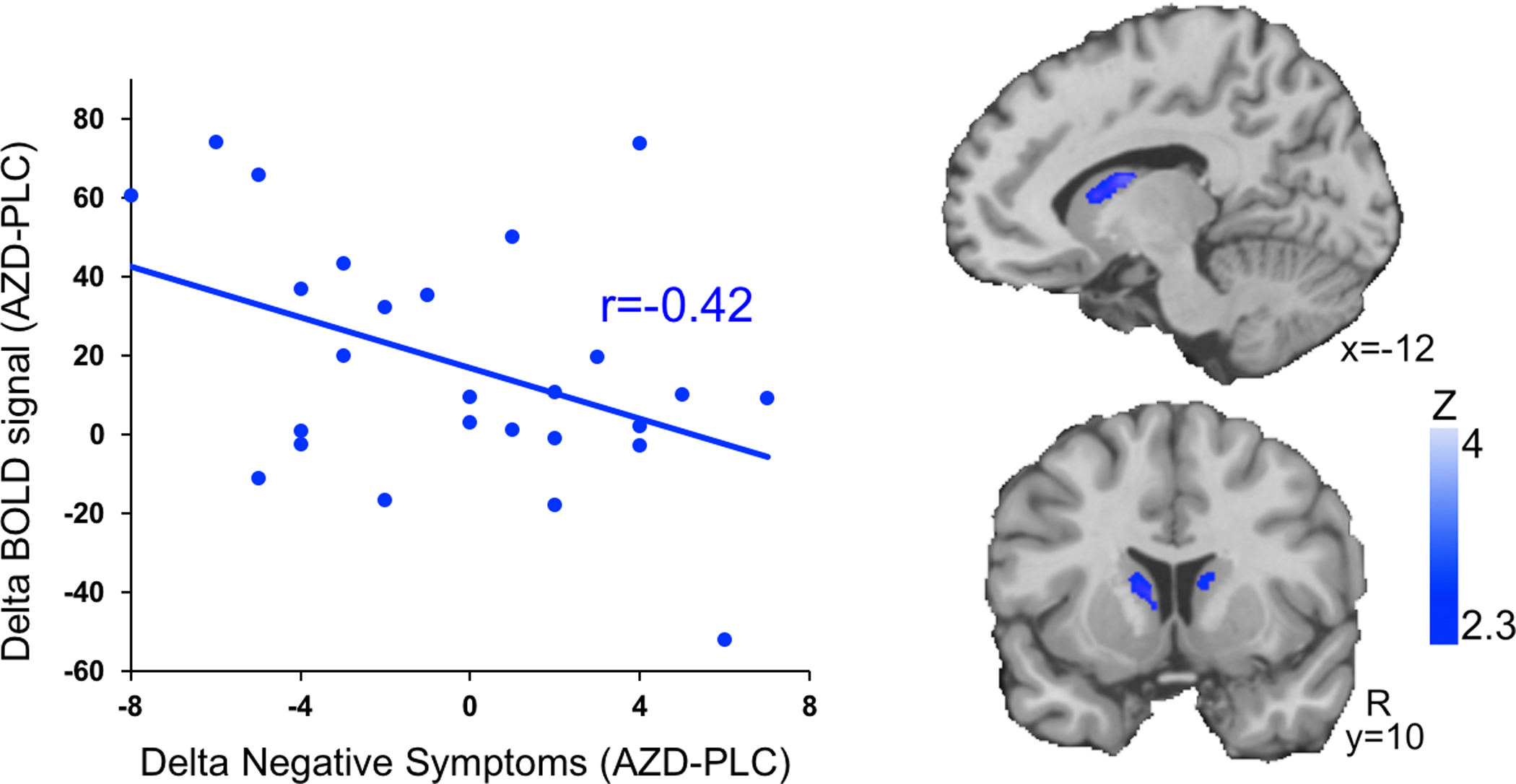
Scatterplot (left) shows that drug-induced increases in activation in a working-memory responsive region of caudate (y-axis, independent neurosynth ROI) correlates (r=−0.42, p=0.03) with drug-induced improvement in negative symptoms (x-axis, PANSS negative scores). Voxelwise images (right) demonstrate the region of caudate showing this effect (image thresholded at z>2.33 and masked by anatomical striatum for display). AZD=AZD8529, PLC=Placebo.
Discussion
Our study is the first to examine the neural circuit effects of enhancing mGluR2 functioning in schizophrenia. Applying the mGluR2 PAM AZD8529 during working memory fMRI, we find that AZD8529 increases activation in task-activated frontostriatal regions (significantly in STR and ACC with a non-significant trend in DLPFC), and also observe that the magnitude of increased activation in striatum correlates with reduction in negative symptoms.
There has been a single published schizophrenia clinical trial with AZD8529, which did not show benefits of chronic dosing (28 days) on positive or negative symptoms or cognition [37]. Here we also find no average effects of briefer treatment (3 days) with AZD8529 on these domains. However, our inclusion of fMRI measures allowed us to identify drug effects at the brain level and understand potential heterogeneity in drug response. This outcome provides support for the experimental medicine approach recently implemented by NIMH [43, 44], demonstrating that studies that would be “negative” if only standard clinical endpoints were included can be informative when brain-based biomarkers are incorporated.
The location of our findings is consistent with the known distribution and function of mGluR2 receptors in animal and human studies. mGluR2 expression is high in prefrontal regions including dorsolateral and anterior cingulate and in subcortical regions, particularly striatum, and metabotropic glutamate receptors including mGluR2 modulate glutamatergic function in these regions [26, 27, 34, 49–52].
Prior fMRI studies of working memory in schizophrenia typically report reduced task activation, although this effect depends in part on task difficulty [53]. Here we did not have a healthy control group; however, consistent with most prior literature, our n-back task has previously shown fMRI hypoactivation associated with psychotic symptoms and global psychopathology [54, 55]. Thus, the increased activation seen here is consistent with a normalization of neural circuit function. However, AZD8529 did not improve working memory performance itself, seemingly reflecting greater sensitivity of fMRI compared to behavior [56]. It could also indicate that there are other important contributors to impaired behavioral performance that are not impacted by AZD8529, or that higher doses or longer duration of treatment are necessary for clear cognitive benefits. We observed robust drug-induced increases in activation in ACC but not DLPFC; the reason for this difference is unclear, but a stronger increase in DLPFC activity might be needed to achieve cognitive improvement.
While working memory is most closely associated with prefrontal cortex, striatal regions including the dorsal caudate are also activated during working memory fMRI [57, 58]. The striatal region showing drug effects correlating with negative symptom change in this study lies within that dorsal caudate area (Supplementary Figure S3). The same dorsal caudate region exhibits functional connectivity with dorsolateral prefrontal cortex, which is reduced in association with duration of untreated psychosis [59]. Some prior studies have also related impaired dorsal striatal activation to schizophrenia and negative symptoms during working memory fMRI. In particular, Ehrlich et al. [14] found that higher negative symptom severity in schizophrenia correlated with lower working memory fMRI activation in dorsal striatum (but not significantly in DLPFC). Koch et al. [15] found reduced dorsal caudate activation during working memory in schizophrenia, but did not examine negative symptoms specifically. More broadly, our results are consistent with the reward imaging literature showing that hypofunction in striatum correlates with negative symptom severity. While most prior studies have focused on the ventral striatum [8], human and animal studies also link dorsal striatum hypofunction to negative symptoms and amotivation; dorsal striatum may be particular important for the formation of action-reward associations [9, 60].
Metabotropic glutamate signaling – imaging and schizophrenia
Although our study does not address the question of baseline abnormalities in mGluR2 signaling in schizophrenia, it suggests that enhancing this signaling in striatum improves an mGluR2 signaling deficit either directly, or by compensating for other abnormalities. Existing literature regarding mGluR2 expression abnormalities in schizophrenia is mixed, and expression abnormalities may be limited to early stages of illness [61], although alterations in function may occur independently of expression levels. Genome-wide association studies to date have identified risk variants in mGluR3, not mGluR2 [62], but there are poorly-understood interactions between mGluR2 and mGluR3 pathways [63]. A strength of AZD8529 and other mGluR2-selective PAMS is that unlike direct agonists, effects can be clearly attributed to mGluR2 and not mGluR3, aiding biological interpretability as well as therapeutic specificity. In animal studies, mGluR2 has been shown to interact with other (non-glutamatergic) neurotransmitter systems associated with schizophrenia pathophysiology and/or antipsychotic response including dopamine [52], serotonin [64], and GABA [65]. The mGluR2 effects are thought to be mediated mainly by presynaptic autoreceptors that reduce neurotransmitter release but there is also some post-synaptic expression of mGluR2, perhaps contributing to complex (inverted-U) dose reponses [22, 40]. Regardless of whether AZD8529 effects are normalizing a pathophysiological hypofunction of mGluR2 signaling or increasing it to supra-normal levels, our results suggest that this effect may be beneficial for ameliorating negative symptoms.
Imaging of glutamatergic effects
There is limited work in humans examining effects of glutamatergic signaling on neural circuitry. Most studies have employed the NMDA antagonist ketamine, demonstrating primarily increases in regional cerebral blood flow, blood-oxygen-level-dependent (BOLD) signal, resting-state connectivity, and glutamate/glutamine spectroscopic levels [41, 66–68]. Studies of ketamine effects on fMRI task-activation have shown variable increases or decreases, including a decrease in striatal reward responses [69] and dorsolateral prefrontal memory responses [41, 70]. Task-fMRI ketamine effects also vary according to individual differences such as depression status [71, 72]. Two studies demonstrated that mGluR2/3 agonists as well as a selective mGluR2 agonist reduced ketamine-induced BOLD [27, 34]. To our knowledge, no published studies have examined effects of AZD8529 or other mGluR2 PAMs using neuroimaging. One study using EEG found that another mGluR2 PAM altered sleep EEG structure [73]. Although much more work is needed in this area, overall results suggest that AZD8529 and other agents that increase mGluR2 signaling have effects opposite to those of ketamine.
Limitations
There are several limitations in the current study. The sample size is relatively small, and replication in a larger sample will be important. Our study lacked a healthy control group, so we cannot judge to what extent observed drug effects are dependent on baseline brain pathophysiology of schizophrenia. We studied a chronic medicated sample, and all patients were on a medication with potential serotonergic effects, thus we could not examine putatively responsive subgroups of early-stage psychosis and D2-exclusive medication exposure identified by Kinon et al. [39]. Early-stage schizophrenia may exhibit distinct glutamatergic abnormalities [74] and more robust clinical response to mGluR agonism [39], and antipsychotic treatment in our patients may have limited the efficacy of AZD8529, perhaps through downregulating mGluR2 [39] or inducing D2 supersensitivity [75]. Future work could examine mGluR2 PAMS in early-stage psychosis and at-risk populations including medication-naive individuals. Patients were also symptomatically stable with relatively low symptom severity; this increased the safety of the study, but may have reduced sensitivity for detecting drug-related improvements. In addition, most of the participants were smokers, so the extent to which findings generalize to nonsmokers is unclear. AZD8529 is also being studied in nicotine and other addictions, and future studies could examine the relationship of the fMRI effects we see to addiction phenotypes. Another limitation is that our design did not include long-term treatment with AZD8529 as would be needed for actual clinical use. Although we selected a dose designed to approximate over 3 days the steady-state level expected from chronic dosing, medications may have long-term or indirect effects that take weeks to emerge even at steady-state. We only used a single dose, and mGluR2 PAMs may show complex/inverted-U dose responses. This would be expected to obscure average drug effects at a given dose given inter-individual variability in pharmacokinetics and pharmacodynamics. While we show that some clinical response heterogeneity is related to heterogeneity in brain responses, this leaves open the question of what factors drive these differences in brain response, pointing to the need for future work examining different stages of illness and different medication exposures. Aspects of our study design that substantially limit our ability to address questions of specificity regarding negative symptoms and the role of motivation circuitry include: use of the PANSS for our primary negative symptom measure since current cutting edge measures such as CAINS and BNSS were not fully developed at time of study design; the lack of a measure of primary/deficit negative symptoms or a specific depression measure; and lack of an fMRI task directly manipulating reward. Lastly, the test-retest reliability of fMRI and hence its sensitivity to drug effects is generally higher in task-activated regions [76], so we cannot rule out drug effects in other regions that would be better detected with other imaging tasks or task-free fMRI approaches.
Summary & Conclusions
Notwithstanding these limitations, our findings from the first fMRI study of mGluR2 modulation in schizophrenia support further investigation of this pharmacological mechanism. In particular, our findings suggest the potential of increased mGluR2 signaling to improve disabling symptoms of schizophrenia by activating fronto-striatal brain regions. Our results also support the future development of fMRI and other imaging biomarkers to reveal therapeutic mechanisms and tailor drug development and treatment towards specific neural circuits and patient populations. Ultimately, this effort will move us towards the ultimate goal of personalized medicine with optimal outcomes for every patient.
Supplementary Material
Acknowledgments
This study was supported by AstraZeneca Pharmaceuticals LP. DHW was also supported by NIMH grant K23MH085096 and R01MH101111. TDS was supported by NIMH grant R01MH112847, R01MH113550 and ACTTION. The work was also supported by NIMH grants R01MH060722, P50MH064045, and T32MH019112. The authors thank Elizabeth Hanson, Raphael Gerraty, and Janina Seubert for assistance with data acquisition; Jeffrey Valdez for assistance with neuroimaging analysis; Warren Bilker for assistance with statistical analysis; and Monica Calkins for assistance with symptom assessment.
Footnotes
Conflict of Interest
Drs. Smith, Zukin, and Cross are former employees of AstraZeneca Pharmaceuticals LP, the study sponsor. The other authors declare no conflict of interest.
Supplementary information is available at Molecular Psychiatry’s website (http://www.nature.com/mp)
References
- 1.Foussias G, Agid O, Fervaha G, Remington G. Negative symptoms of schizophrenia: Clinical features, relevance to real world functioning and specificity versus other CNS disorders. European Neuropsychopharmacology 2014; 24: 693–709. [DOI] [PubMed] [Google Scholar]
- 2.Fusar-Poli P, Papanastasiou E, Stahl D, Rocchetti M, Carpenter W, Shergill S et al. Treatments of negative symptoms in schizophrenia: Meta-analysis of 168 randomized placebo-controlled trials. Schizophrenia Bulletin 2015; 41: 892–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacology, Biochemistry, and Behavior 2011; 99: 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gard DE, Fisher M, Garrett C, Genevsky A, Vinogradov S. Motivation and its relationship to neurocognition, social cognition, and functional outcome in schizophrenia. Schizophrenia Research 2009; 115: 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anticevic A, Schleifer C, Youngsun TC. Emotional and cognitive dysregulation in schizophrenia and depression: Understanding common and distinct behavioral and neural mechanisms. Dialogues in Clinical Neuroscience 2015; 17: 421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foussias G, Siddiqui I, Fervaha G, Mann S, McDonald K, Agid O et al. Motivated to do well: An examination of the relationships between motivation, effort, and cognitive performance in schizophrenia. Schizophrenia Research 2015; 166: 276–282. [DOI] [PubMed] [Google Scholar]
- 7.Barch DM, Ceaser A. Cognition in schizophrenia: Core psychological and neural mechanisms. Trends in Cognitive Sciences 2012; 16: 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radua J, Schmidt A, Borgwardt S, Heinz A, Schlagenhauf F, McGuire P et al. Ventral striatal activation during reward processing in psychosis: A neurofunctional meta-analysis. JAMA Psychiatry 2015; 72: 1243–1251. [DOI] [PubMed] [Google Scholar]
- 9.Mucci A, Merlotti E, Ucok A, Aleman A, Galderisi S. Primary and persistent negative symptoms: Concepts, assessments and neurobiological bases. Schizophrenia Research 2017; 186: 19–28. [DOI] [PubMed] [Google Scholar]
- 10.Wolf DH, Satterthwaite TD, Kantrowitz JJ, Katchmar N, Vandekar L, Elliott MA et al. Amotivation in schizophrenia: Integrated assessment with behavioral, clinical, and imaging measures. Schizophrenia Bulletin 2014; 40: 1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stepien M, Manoliu A, Kubli R, Schneider K, Tobler PN, Seifritz E et al. Investigating the association of ventral and dorsal striatal dysfunction during reward anticipation with negative symptoms in patients with schizophrenia and healthy individuals. PloS One 2018; 13: e0198215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf DH, Gerraty R, Satterthwaite TD, Loughead J, Campellone T, Elliott MA et al. Striatal intrinsic reinforcement signals during recognition memory: Relationship to response bias and dysregulation in schizophrenia. Front Behav Neurosci 2011; 5: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf DH, Turetsky BI, Loughead J, Elliott MA, Pratiwadi R, Gur RE et al. Auditory oddball fmri in schizophrenia: Association of negative symptoms with regional hypoactivation to novel distractors. Brain Imaging and Behavior 2008; 2: 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrlich S, Yendiki A, Greve DN, Manoach DS, Ho BC, White T et al. Striatal function in relation to negative symptoms in schizophrenia. Psychological Medicine 2012; 42: 267–282. [DOI] [PubMed] [Google Scholar]
- 15.Koch K, Wagner G, Nenadic I, Schachtzabel C, Schultz C, Roebel M et al. Fronto-striatal hypoactivation during correct information retrieval in patients with schizophrenia: An fMRI study. Neuroscience 2008; 153: 54–62. [DOI] [PubMed] [Google Scholar]
- 16.Vink M, Ramsey NF, Raemaekers M, Kahn RS. Striatal dysfunction in schizophrenia and unaffected relatives. Biological Psychiatry 2006; 60: 32–39. [DOI] [PubMed] [Google Scholar]
- 17.Krause M, Zhu Y, Huhn M, Schneider-Thoma J, Bighelli I, Nikolakopoulou A et al. Antipsychotic drugs for patients with schizophrenia and predominant or prominent negative symptoms: A systematic review and meta-analysis. European Archives of Psychiatry and Clinical Neuroscience 2018; 268: 625–639. [DOI] [PubMed] [Google Scholar]
- 18.Omachi Y, Sumiyoshi T. Dose reduction/discontinuation of antipsychotic drugs in psychosis; effect on cognition and functional outcomes. Front Psychiatry 2018; 9: 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang YS, Marder SR, Green MF. Repurposing drugs for cognition in schizophrenia. Clinical Pharmacology and Therapeutics 2017; 101: 191–193. [DOI] [PubMed] [Google Scholar]
- 20.Moghaddam B, Javitt D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 2012; 37: 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman EJ, Bubser M, Conn PJ, Jones CK. Metabotropic glutamate receptors for new treatments in schizophrenia. Handbook of Experimental Pharmacology 2012: 297–365. [DOI] [PubMed] [Google Scholar]
- 22.Ellaithy A, Younkin J, Gonzalez-Maeso J, Logothetis DE. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci 2015; 38: 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawaura K, Karasawa J, Hikichi H. Stimulation of the metabotropic glutamate (mglu) 2 receptor attenuates the MK-801-induced increase in the immobility time in the forced swimming test in rats. Pharmacological Reports 2016; 68: 80–84. [DOI] [PubMed] [Google Scholar]
- 24.Griebel G, Pichat P, Boulay D, Naimoli V, Potestio L, Featherstone R et al. The mglur2 positive allosteric modulator, sar218645, improves memory and attention deficits in translational models of cognitive symptoms associated with schizophrenia. Scientific Reports 2016; 6: 35320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavreysen H, Langlois X, Ahnaou A, Drinkenburg W, te Riele P, Biesmans I et al. Pharmacological characterization of JNJ-40068782, a new potent, selective, and systemically active positive allosteric modulator of the mglu2 receptor and its radioligand [3H]JNJ-40068782. The Journal of Pharmacology and Experimental Therapeutics 2013; 346: 514–527. [DOI] [PubMed] [Google Scholar]
- 26.Hackler EA, Byun NE, Jones CK, Williams JM, Baheza R, Sengupta S et al. Selective potentiation of the metabotropic glutamate receptor subtype 2 blocks phencyclidine-induced hyperlocomotion and brain activation. Neuroscience 2010; 168: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta MA, Schmechtig A, Kotoula V, McColm J, Jackson K, Brittain C et al. Group ii metabotropic glutamate receptor agonist prodrugs LY2979165 and LY2140023 attenuate the functional imaging response to ketamine in healthy subjects. Psychopharmacology 2018; 235: 1875–1886. [DOI] [PubMed] [Google Scholar]
- 28.Gray LJ, Hannan AJ, Zhang X. Metabotropic glutamate receptors as targets for novel antipsychotic treatments. Current Pharmaceutical Biotechnology 2012; 13: 1522–1534. [DOI] [PubMed] [Google Scholar]
- 29.Acri JB, Cross AJ, Skolnick P. From bench to bedside: Mglur2 positive allosteric modulators as medications to treat substance use disorders. Psychopharmacology 2017; 234: 1347–1355. [DOI] [PubMed] [Google Scholar]
- 30.Kent JM, Daly E, Kezic I, Lane R, Lim P, De Smedt H et al. Efficacy and safety of an adjunctive mglu2 receptor positive allosteric modulator to a SSRI/SNRI in anxious depression. Progress in Neuropsychopharmacology & Biological Psychiatry 2016; 67: 66–73. [DOI] [PubMed] [Google Scholar]
- 31.Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV et al. Activation of mglu2/3 receptors as a new approach to treat schizophrenia: A randomized phase 2 clinical trial. Nature Medicine 2007; 13: 1102–1107. [DOI] [PubMed] [Google Scholar]
- 32.Stauffer VL, Millen BA, Andersen S, Kinon BJ, Lagrandeur L, Lindenmayer JP et al. Pomaglumetad methionil: No significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophrenia Research 2013; 150: 434–441. [DOI] [PubMed] [Google Scholar]
- 33.Kinon BJ, Zhang L, Millen BA, Osuntokun OO, Williams JE, Kollack-Walker S et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY140023 monohydrate in patients with dsm-iv schizophrenia. Journal of Clinical Psychopharmacology 2011; 31: 349–355. [DOI] [PubMed] [Google Scholar]
- 34.Kantrowitz JT, Grinband J, Goff DC, Lahti AC, Marder SR, Kegeles LS et al. Proof of mechanism and target engagement of glutamatergic drugs for the treatment of schizophrenia: Rcts of pomaglumetad and ts-134 on ketamine-induced psychotic symptoms and pharmacobold in healthy volunteers. Neuropsychopharmacology 2020; 45: 1842–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salih H, Anghelescu I, Kezic I, Sinha V, Hoeben E, Van Nueten L et al. Pharmacokinetic and pharmacodynamic characterisation of jnj-40411813, a positive allosteric modulator of mglur2, in two randomised, double-blind phase-i studies. Journal of Psychopharmacology 2015; 29: 414–425. [DOI] [PubMed] [Google Scholar]
- 36.De Boer P, Sinha V, Hoeben E, Ion-George A, Kezic I, Daly E et al. Characterization of the clinical effect of a positive allosteric modulator of the metabotropic glutamate receptor-2. Poster presented at 68th Annual Scientific Convention of Society of Biological Psychiatry: San Francisco, 2013. [Google Scholar]
- 37.Litman RE, Smith MA, Doherty JJ, Cross A, Raines S, Gertsik L et al. Azd8529, a positive allosteric modulator at the mglur2 receptor, does not improve symptoms in schizophrenia: A proof of principle study. Schizophrenia Research 2016; 172: 152–157. [DOI] [PubMed] [Google Scholar]
- 38.Marek GJ. When is a proof-of-concept (poc) not a poc? Pomaglumetad (ly2140023) as a case study for antipsychotic efficacy. Current Pharmaceutical Design 2015; 21: 3788–3796. [DOI] [PubMed] [Google Scholar]
- 39.Kinon BJ, Millen BA, Zhang L, McKinzie DL. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biological Psychiatry 2015; 78: 754–762. [DOI] [PubMed] [Google Scholar]
- 40.Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AFT et al. Mglur2 versus mglur3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: Postsynaptic mglur3 strengthen working memory networks. Cerebral Cortex 2018; 28: 974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: A randomized clinical trial. JAMA Psychiatry 2018; 75: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wandschneider B, Koepp MJ. Pharmaco fMRI: Determining the functional anatomy of the effects of medication. Neuroimage Clin 2016; 12: 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Insel TR. The NIM experimental medicine initiative. World Psychiatry 2015; 14: 151–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhakta SG, Chou HH, Rana B, Talledo JA, Balvaneda B, Gaddis L et al. Effects of acute memantine administration on matrics consensus cognitive battery performance in psychosis: Testing an experimental medicine strategy. Psychopharmacology 2016; 233: 2399–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sheffler DJ, Gregory KJ, Rook JM, Conn PJ. Allosteric modulation of metabotropic glutamate receptors. Advances in Pharmacology 2011; 62: 37–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loughead J, Ray R, Wileyto EP, Ruparel K, Sanborn P, Siegel S et al. Effects of the alpha4beta2 partial agonist varenicline on brain activity and working memory in abstinent smokers. Biological Psychiatry 2010; 67: 715–721. [DOI] [PubMed] [Google Scholar]
- 47.Snodgrass JG, Corwin J. Pragmatics of measuring recognition memory: Applications to dementia and amnesia. J Exp Psychol Gen 1988; 117: 34–50. [DOI] [PubMed] [Google Scholar]
- 48.Smith SM, Nichols TE. Threshold-free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. NeuroImage 2009; 44: 83–98. [DOI] [PubMed] [Google Scholar]
- 49.Ohishi H, Neki A, Mizuno N. Distribution of a metabotropic glutamate receptor, mglur2, in the central nervous system of the rat and mouse: An immunohistochemical study with a monoclonal antibody. Neuroscience Research 1998; 30: 65–82. [DOI] [PubMed] [Google Scholar]
- 50.Phillips T, Rees S, Augood S, Waldvogel H, Faull R, Svendsen C et al. Localization of metabotropic glutamate receptor type 2 in the human brain. Neuroscience 2000; 95: 1139–1156. [DOI] [PubMed] [Google Scholar]
- 51.Ghose S, Gleason KA, Potts BW, Lewis-Amezcua K, Tamminga CA. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: A mechanism for antipsychotic drug action? American Journal of Psychiatry 2009; 166: 812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson KA, Mateo Y, Lovinger DM. Metabotropic glutamate receptor 2 inhibits thalamically-driven glutamate and dopamine release in the dorsal striatum. Neuropharmacology 2017; 117: 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karlsgodt KH, Sanz J, van Erp TG, Bearden CE, Nuechterlein KH, Cannon TD. Re-evaluating dorsolateral prefrontal cortex activation during working memory in schizophrenia. Schizophrenia Research 2009; 108: 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolf DH, Satterthwaite TD, Calkins ME, Ruparel K, Elliott MA, Hopson RD et al. Functional neuroimaging abnormalities in youth with psychosis spectrum symptoms. JAMAPpsychiatry 2015; 72: 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shanmugan S, Wolf DH, Calkins ME, Moore TM, Ruparel K, Hopson RD et al. Common and dissociable mechanisms of executive system dysfunction across psychiatric disorders in youth. American Journal ofPpsychiatry 2016; 173: 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rasch B, Papassotiropoulos A, de Quervain DF. Imaging genetics of cognitive functions: Focus on episodic memory. NeuroImage 2010; 53: 870–877. [DOI] [PubMed] [Google Scholar]
- 57.Niendam TA, Laird AR, Ray KL, Dean YM, Glahn DC, Carter CS. Meta-analytic evidence for a superordinate cognitive control network subserving diverse executive functions. Cognitive, Affective & Behavioral Neuroscience 2012; 12: 241–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Backman L, Nyberg L. Dopamine and training-related working-memory improvement. Neuroscience and Biobehavioral Reviews 2013; 37: 2209–2219. [DOI] [PubMed] [Google Scholar]
- 59.Manivannan A, Foran W, Jalbrzikowski M, Murty VP, Haas GL, Tarcijonas G et al. Association between duration of untreated psychosis and frontostriatal connectivity during maintenance of visuospatial working memory. Biol Psychiatry Cogn Neurosci Neuroimaging 2019; 4: 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Balleine BW, O’Doherty JP. Human and rodent homologies in action control: Corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology 2010; 35: 48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li ML, Hu XQ, Li F, Gao WJ. Perspectives on the mglur2/3 agonists as a therapeutic target for schizophrenia: Still promising or a dead end? Progress in Neuropsychopharmacology & Biological Psychiatry 2015; 60: 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lane TA, Boerner T, Bannerman DM, Kew JN, Tunbridge EM, Sharp T et al. Decreased striatal dopamine in group ii metabotropic glutamate receptor (mglu2/mglu3) double knockout mice. BMC Neurosci 2013; 14: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shah UH, Gonzalez-Maeso J. Serotonin and glutamate interactions in preclinical schizophrenia models. ACS Chemical Neuroscience 2019; 10: 3068–3077. [DOI] [PubMed] [Google Scholar]
- 65.Johnson KA, Niswender CM, Conn PJ, Xiang Z. Activation of group ii metabotropic glutamate receptors induces long-term depression of excitatory synaptic transmission in the substantia nigra pars reticulata. Neuroscience Letters 2011; 504: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maltbie EA, Kaundinya GS, Howell LL. Ketamine and pharmacological imaging: Use of functional magnetic resonance imaging to evaluate mechanisms of action. Behav Pharmacol 2017; 28: 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bryant JE, Frolich M, Tran S, Reid MA, Lahti AC, Kraguljac NV. Ketamine induced changes in regional cerebral blood flow, interregional connectivity patterns, and glutamate metabolism. J Psychiatr Res 2019; 117: 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bojesen KB, Andersen KA, Rasmussen SN, Baandrup L, Madsen LM, Glenthoj BY et al. Glutamate levels and resting cerebral blood flow in anterior cingulate cortex are associated at rest and immediately following infusion of s-ketamine in healthy volunteers. Front Psychiatry 2018; 9: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Francois J, Grimm O, Schwarz AJ, Schweiger J, Haller L, Risterucci C et al. Ketamine suppresses the ventral striatal response to reward anticipation: A cross-species translational neuroimaging study. Neuropsychopharmacology 2016; 41: 1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D’Souza DC et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Molecular Psychiatry 2013; 18: 1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reed JL, Nugent AC, Furey ML, Szczepanik JE, Evans JW, Zarate CA Jr., Effects of ketamine on brain activity during emotional processing: Differential findings in depressed versus healthy control participants. Biol Psychiatry Cogn Neurosci Neuroimaging 2019; 4: 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sterpenich V, Vidal S, Hofmeister J, Michalopoulos G, Bancila V, Warrot D et al. Increased reactivity of the mesolimbic reward system after ketamine injection in patients with treatment-resistant major depressive disorder. Anesthesiology 2019; 130: 923–935. [DOI] [PubMed] [Google Scholar]
- 73.Ahnaou A, de Boer P, Lavreysen H, Huysmans H, Sinha V, Raeymaekers L et al. Translational neurophysiological markers for activity of the metabotropic glutamate receptor (mglur2) modulator jnj-40411813: Sleep EEG correlates in rodents and healthy men. Neuropharmacology 2016; 103: 290–305. [DOI] [PubMed] [Google Scholar]
- 74.Krystal JH, Anticevic A. Toward illness phase-specific pharmacotherapy for schizophrenia. Biological Psychiatry 2015; 78: 738–740. [DOI] [PubMed] [Google Scholar]
- 75.Gill KM, Cook JM, Poe MM, Grace AA. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophrenia Bulletin 2014; 40: 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Plichta MM, Schwarz AJ, Grimm O, Morgen K, Mier D, Haddad L et al. Test-retest reliability of evoked bold signals from a cognitive-emotive fmri test battery. NeuroImage 2012; 60: 1746–1758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.