

Abstract
In the 5th edition of the World Health Organization (WHO) Classification of Head and Neck Tumours, the discussion of hematolymphoid proliferations is substantially reorganized and expanded in comparison to the prior edition. The 5th edition includes, in addition to hematolymphoid neoplasms, reactive lymphoid proliferations. Much more information on hematolymphoid proliferations that commonly affect cervical lymph nodes, in addition to those affecting extranodal sites in the head and neck, is included. For the first time, there are dedicated sections on multiple entities, including recently described lymphoproliferative disorders such as EBV+ mucocutaneous ulcer and pediatric-type follicular lymphoma, and several types of histiocytic neoplasms. Tremendous advances have been made in understanding the genetic features that underlie the pathogenesis of hematolymphoid neoplasms, and these have been incorporated into the WHO Classification.
Keywords: Lymphoma, Lymphoid hyperplasia, Lymphadenitis, Genetics
Introduction
Five years have passed since the publication of the 4th edition of the World Health Organization Classification of Head and Neck Tumours. In the 5th edition [1], there is a chapter dedicated to hematolymphoid proliferations and neoplasia. Previously, individual types of hematolymphoid neoplasms were discussed among other types of neoplasms in chapters that were dedicated to neoplasms of a specific anatomic site. The most important changes and updates are described below.
Reactive Lymphoid Hyperplasia
A section on reactive lymphoid hyperplasia is included in the WHO Classification of Head and Neck Tumors for the first time. Cervical lymph nodes and extranodal lymphoid tissue in the head and neck can be affected by a wide variety of reactive lymphoid hyperplasia and lymphadenitis. They include viral infections (infectious mononucleosis, cytomegaloviral lymphadenitis (Fig. 1), HIV-associated lymphoid hyperplasia and others), bacterial, mycobacterial and protozoal infections, lymphoid hyperplasias related to autoimmune diseases, and lymphoid proliferations of unknown etiology. Certain disorders addressed in this section, including infectious mononucleosis [2], Kikuchi disease [3–5] and atypical marginal zone hyperplasia [6–8], are associated with histologic changes that can mimic lymphoma, underscoring the importance of familiarity with these disorders.
Fig. 1.

Cytomegaloviral lymphadenitis. In this case, there was a proliferation of monocytoid B cells that was so prominent that it raised the question of nodal marginal zone lymphoma (A). Flow cytometry showed polyclonal B cells. Also seen were areas with small lymphocytes and small clusters of epithelioid histiocytes. The circle highlights a CMV-infected cell (B). High power shows a CMV-infected cell, with a large nuclear inclusion and multiple small granular cytoplasmic inclusions (C). An immunostain for CMV confirms the presence of CMV inclusions (D, immunoperoxidase technique on a paraffin section)
EBV+ Mucocutaneous Ulcer
EBV+ mucocutaneous ulcer (MCU) is a localized, typically self-limited lymphoproliferative disorder with a polymorphous composition, including large, atypical EBV+ B cells with features of Reed–Sternberg cells and variants, involving skin, oral cavity, tonsils and other extranodal sites. Patients often have an underlying immunodeficiency, likely facilitating development of this EBV-related lymphoproliferation [9, 10]. The large atypical cells are typically CD30+, CD15−/+, Pax5+, MUM1+, CD10−, BCL6−, OCT2+. EBER is positive by in situ hybridization, with variable expression of CD20, from weak to strong. Admixed smaller lymphocytes are a mix of B cells and T cells. The differential diagnosis includes EBV+ diffuse large B-cell lymphoma and classic Hodgkin lymphoma. The well circumscribed nature of EBV+ MCU helps to exclude lymphoma; disease in more than one anatomic site excludes EBV+ MCU. Recognition of EBV+ MCU is critical, as this disease does not require treatment with chemotherapy. EBV+ MCU may resolve spontaneously, or respond to decreased immunosuppression (in cases with iatrogenic immunosuppression) or local therapy [9, 10].
IgG4-Related Disease
IgG4-related disease (IgG4RD) is a fibrosing, immune-mediated, inflammatory mass-forming disorder that commonly involves salivary glands and orbit [11–13]. Other head and neck sites and sites away from the head and neck (pancreas and many others) may be involved [14, 15]. Patients are mostly middle-aged and older adults, with a male preponderance which is less pronounced in cases with head and neck involvement than for IgG4-RD in other sites [12, 13]. They present with symptoms related to a mass lesion. Evaluation may reveal localized or multifocal disease in extranodal sites, with or without lymph nodal involvement.
Microscopic examination reveals fibrosis (typically storiform fibrosis) and a lymphoplasmacytic infiltrate rich in IgG4+ plasma cells, with an elevated IgG4+:IgG+ ratio (> 40%), sometimes with obliterative phlebitis, but typically without necrosis, necrotizing vasculitis or a component of neutrophils [14, 15]. Reactive lymphoid follicles are prominent in some cases [11, 12, 13]. Diagnosis rests on identifying the characteristic histologic features and excluding the many neoplastic and inflammatory disorders that can mimic IgG4RD [12]. In the head and neck, important entities to exclude are MALT lymphoma, which in contrast to IgG4RD harbors clonal B cells, and sometimes clonal plasma cells, and ANCA-associated vasculitis [12].
The relationship between IgG4RD and eosinophilic angiocentric fibrosis (EAF) is somewhat controversial. Cases of EAF have histologic features consistent with IgG4RD, usually accompanied by increased numbers of IgG4+ plasma cells; many authorities consider EAF to be part of the spectrum of IgG4RD [16–18]. In contrast to IgG4RD, however, obliterative phlebitis is typically absent [17]. some cases do not have elevated IgG4+ plasma cells (sometimes attributed to a late stage of disease) [16], and there is lack of consistent response to steroids. Evaluation of larger numbers of cases of EAF is likely required to resolve the uncertainties surrounding this rare entity.
Follicular Lymphoma
Follicular lymphoma is a common B-cell lymphoma, usually low-grade, usually harboring a BCL2 rearrangement that contributes to the longevity of the neoplastic cells. The main advances related to typical follicular lymphoma include better characterization of the many genetic abnormalities that contribute to lymphomagenesis and disease progression [19].
An uncommon type of follicular lymphoma, pediatric-type follicular lymphoma (PTFL), is discussed in a separate section in the 5th edition for the first time. PTFL mainly affects children and young adults, with a male preponderance. Patients have localized peripheral lymphadenopathy, which is most often cervical [20–22]. Lymph nodes are replaced by a proliferation of large, round and irregular follicles occupied by medium-sized blastoid cells, and sometimes, centroblasts. Interfollicular involvement is uncommon. Foci of diffuse large B-cell lymphoma exclude the diagnosis. Flow cytometry shows clonal B cells. The usual immunophenotype, by flow cytometry and/or immunohistochemistry, is CD19+, CD20+, CD10+, BCL6+, BCL2− (occasionally weakly positive), CD5− [20, 22]. In contrast to typical, “adult-type” follicular lymphoma, PTFL lacks rearrangements of BCL2, and infrequently shows mutations of chromatin modifier genes such as KMT2D, CREBBP and EZH2 [21, 23]. It also lacks rearrangements of BCL6, MYC and IRF4 [21, 23]. Instead, PTFL is characterized by frequent mutations of MAPK pathway genes (~ 50% of cases) [21, 23], of TNFRSF14 (~ 50% of cases) and of IRF8 (~ 15% of cases) [23]. Distinguishing PTFL from adult-type follicular lymphoma is of paramount importance, as PTFL patients are usually effectively treated by excision alone and follow-up is typically uneventful.
Large B-Cell Lymphoma with IRF4 Rearrangement
Large B-cell lymphoma with IRF4 rearrangement is a recently recognized, rare type of large B-cell lymphoma characterized by a follicular, follicular and diffuse, or entirely diffuse growth pattern, strong expression of IRF4/MUM1 and rearranged IRF4, usually with an immunoglobulin heavy chain, and less often a light chain gene partner. This type of lymphoma affects patients over a broad age range, although the majority are younger than 18; males and females are affected equally. This lymphoma most often involves head and neck sites, e.g., Waldeyer’s ring and cervical lymph nodes. Neoplastic cells are usually centroblasts and less often medium-sized blastoid cells [24]. The usual immunophenotype is CD20+, CD3−, CD5−/+, CD10+/−, BCL6+, BCL2+/−, IRF4+ (strong), BLIMP1/PRDM1− [25]. Rearrangements of BCL2 or MYC exclude the diagnosis. Disease is usually localized and the prognosis is favorable [25, 26].
Other Lymphomas
A variety of other lymphomas, including extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) (Fig. 2), mantle cell lymphoma and many others, are also discussed in detail in the 5th edition, but space constraints limit detailed discussion of these entities. A major advance in the understanding of MALT lymphoma is the more refined genetic characterization of this type of lymphoma in different anatomic sites [27–30 ]. Similarly, the genetic changes underlying the pathogenesis and progression of mantle cell lymphoma [31–34], including cyclin D1-negative mantle cell lymphoma [35, 36], Burkitt lymphoma [37, 38, 39], and T lymphoblastic lymphoma [40–45] are now better defined. The pathogenesis of plasmablastic lymphoma, including genetic differences between EBV+ and EBV− plasmablastic lymphoma [46, 47], and the possible role of EBV miRNAs in lymphomagenesis [48], have been studied in detail.
Fig. 2.
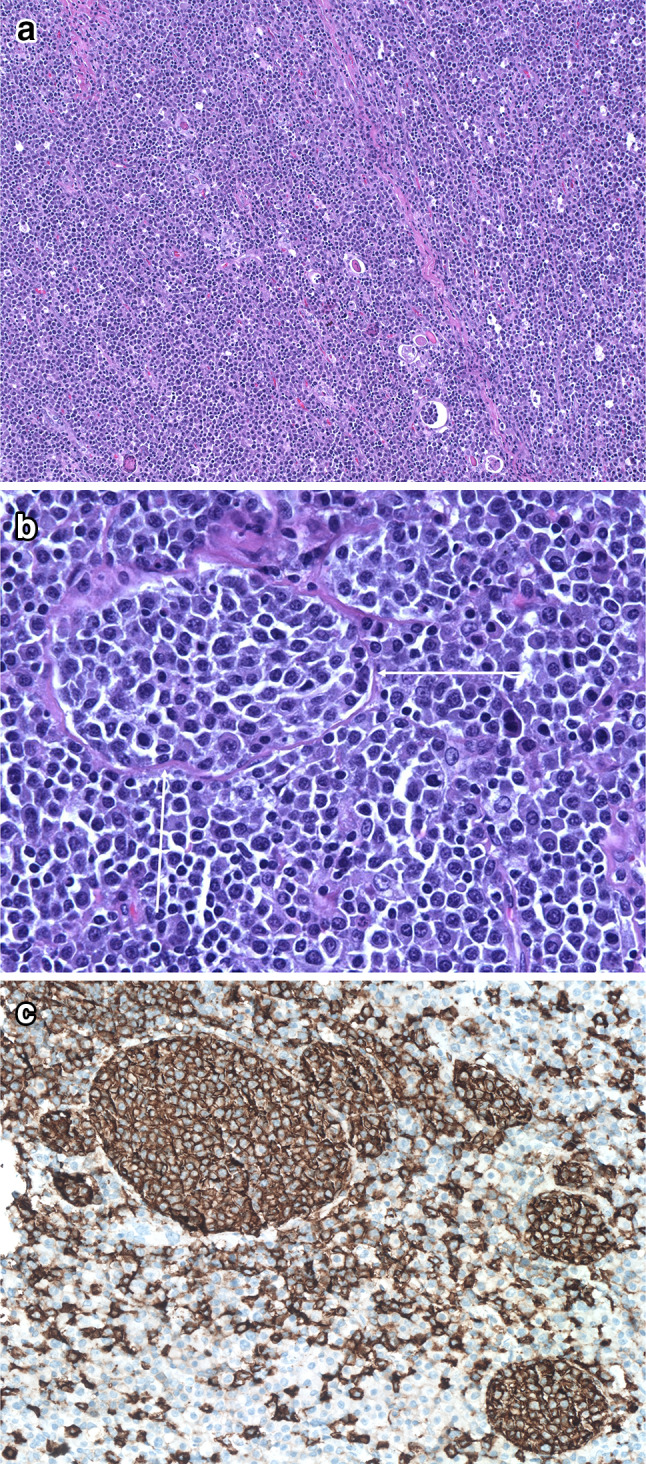
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) of the thyroid. Low power shows a dense, diffuse lymphoid infiltrate obliterating most of the tissue. Only rare recognizable thyroid follicles remain (A). High power shows that the infiltrate is composed of marginal zone B cells and many plasmacytoid forms. Note the discrete nodule, indicated with arrows, which represents a lymphoepithelial lesion of the distinctive “MALT-ball type”, often found in MALT lymphomas of the thyroid (B). An immunostain for CD20 highlights several MALT-ball lymphoepithelial lesions (C, immunoperoxidase technique on a paraffin section)
An interesting advance in extranodal NK/T-cell lymphoma (Fig. 3) is its proposed division into three molecular subtypes: (1) TSIM subtype (presumably, TP53, STAT, IMmune), characterized by mutated TP53 and JAK/STAT pathway genes, 9p24.1 amplification (JAK2, PD-L1/2), del(6q21), NK-cell origin and PD-L1 overexpression; (2) MB subtype (MGA, BRDT), with mutated MGA, LOH at 1p22.1/BRDT and MYC overexpression; and (3) HEA subtype (HDAC9, EP300, ARID1A), with mutated HDAC9, EP300 and ARID1A; T-cell origin; NFkB activation; and histone chaperone DAXX expression [49]. With asparaginase-based therapy, the MB subtype had an inferior prognosis compared to other subtypes. Knowledge of these molecular subtypes opens the door to targeted therapy. Extranodal NK/T-cell lymphoma of TSIM subtype could be susceptible to checkpoint inhibitor therapy, MB subtype to MYC inhibitors, and HEA subtype to HDAC inhibitors [49].
Fig. 3.
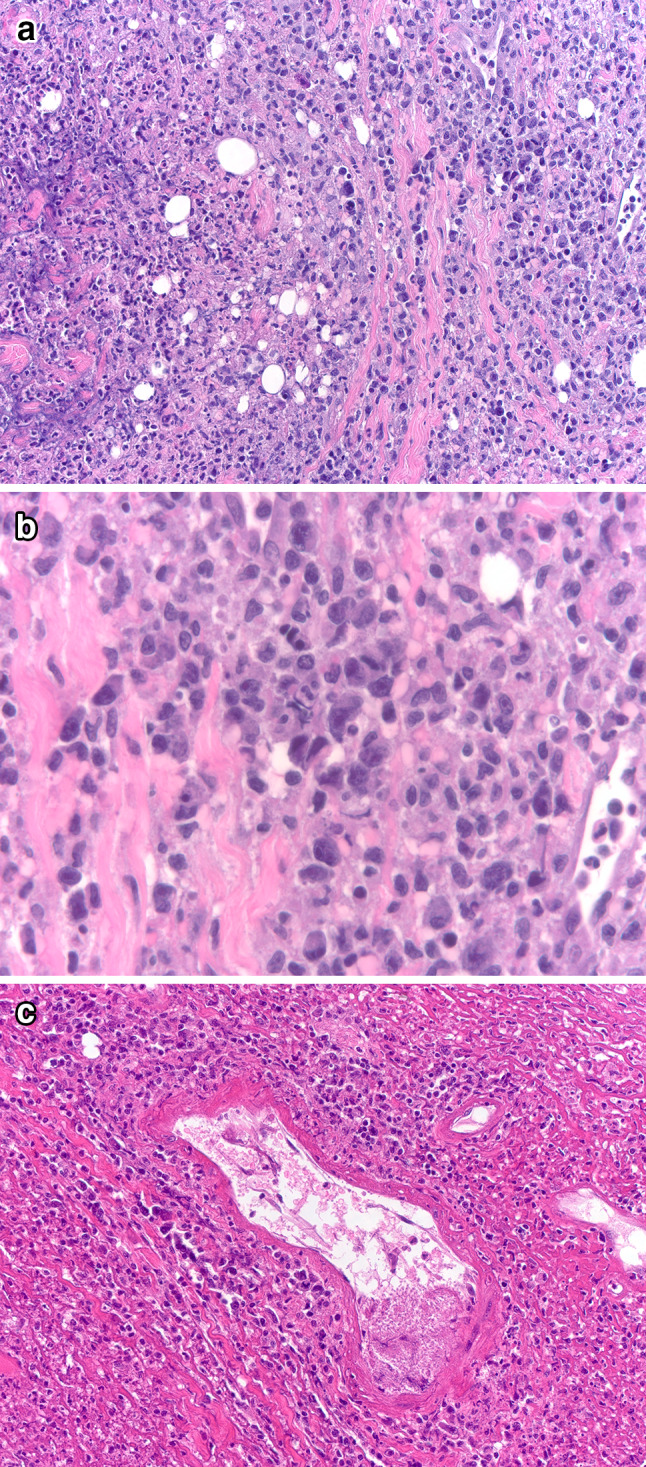
Extranodal NK/T-cell lymphoma, nasal-type, involving oral cavity. Low power shows a diffuse atypical lymphoid infiltrate with areas of necrosis (left of field) (A). Higher power shows pleomorphic lymphoid cells of a range of sizes (B). In areas the infiltrate has a perivascular localization, in a background of necrotic debris; the blood vessel shown has mural necrosis (C). Neoplastic cells expressed CD3, CD56 (partial) and cytotoxic granule proteins; in situ hybridization for EBER was positive (not shown)
Histiocytic Proliferations
Previously, Langerhans cell histiocytosis was the only histiocytic neoplasm included in the WHO Classification of Head and Neck Tumours. In this 5th edition, histiocytic-dendritic cell neoplasms that are included are juvenile xanthogranuloma, Erdheim−Chester disease, Rosai−Dorfman disease and follicular dendritic cell sarcoma [50], in addition to Langerhans cell histiocytosis (Table 1). The nature of some of these diseases, juvenile xanthogranuloma [51, 52], Erdheim−Chester disease [53–55] and Rosai−Dorfman disease [56–59], has been uncertain, but in recent years, a substantial subset of each of these diseases has been shown to carry pathogenetic mutations, most often activating mutations of the MAPK or MAPK-ERK (mitogen-activated protein kinase/extracellular signal-related kinase) pathway and other abnormalities, so that these diseases, or at least those cases associated with pathogenetic mutations, should be considered neoplastic (see Table 1). An intriguing feature is the infrequent finding of more than one type of histiocytic proliferation in a single patient, sometimes as a composite tumor (Fig. 4), possibly representing divergent differentiation of a common precursor with mutations of MAPK pathway genes.
Table 1.
Histiocytic and dendritic cell neoplasms
| Disease | Clinical | Histology | IHC | Genetic features | Outcome |
|---|---|---|---|---|---|
| Juvenile xanthogranuloma (JXG) |
Children > adults; One or more skin lesions most common presentation; isolated or disseminated extracutaneous JXG is uncommon; Pink macules or yellow-tan papules |
Xanthomatous histiocytes and Touton giant cells with admixed lymphocytes, eosinophils, neutrophils, plasma cells, mast cells; early lesions may have small oval to spindled histiocytes with eosinophilic cytoplasm |
Positive: CD68, CD163, factor XIIIa, CD4, CD14, fascin Negative: CD1a, langerin, S-100 |
Mutated NRAS, KRAS, ARAF, MAP2K1 and rare RNF11-BRAF fusion in systemic JXG; Mutated CSF-1R, NTRK1 fusions; less commonly, mutated CSF-3R, KIT, ALK, MET, JAK3, RAF1, as well as RAF fusions and others |
Good overall; inferior outcome for rare cases with disseminated JXG |
| Erdheim−Chester disease (ECD) |
Most patients are middle-aged and older adults, M>F, nearly all with bilateral symmetric osteosclerosis of the lower extremity long bones, and often with extraosseous disease; almost any site can be involved; Increased incidence of clonal hematopoiesis and myeloid neoplasms |
Histiocytes with foamy and/or eosinophilic cytoplasm +/− chronic inflammatory cells, Touton giant cells, fibrosis; Some patients also have RDD or LCH |
Positive: CD68, CD163, factor XIIIa, CD4, CD14 Negative: CD1a, langerin Variable: S100 Cases with BRAF p.Val600Glu mutation may be BRAF+ by IHC |
Activating mutations of (MAPK) pathway and/or cell-proliferation related pathways in nearly all cases BRAF Val600Glu is most common (50–60%); MAP2K1, KRAS, or NRAS mutations can be found in BRAF-wt cases |
Nearly all patients are symptomatic and require therapy; CNS, cardiac, multisystem involvement: worse prognosis. Prognosis may be improving with availability of targeted therapies |
| Rosai−Dorfman disease (RDD) |
Wide age range affected; median age third decade; All races affected but more common among Blacks; Lymphadenopathy, especially cervical, most common. Other lymph nodes and/or many different extranodal sites can be involved |
Histiocytes with enlarged, vesicular nuclei, distinct nucleoli, and abundant pale cytoplasm, sometimes with emperipolesis; admixed lymphocytes, plasma cells, +/− lymphoid follicles |
Histiocytes: S100+, OCT2+, cyclin D1+; weak CD4, CD68 and CD163; negative langerin, CD1a Lymphocytes: mixed polytypic B cells and T cells Plasma cells: polytypic; may show increased IgG4+ plasma cells |
Mutations of MAPK/ERK pathway genes in about 50%, including KRAS, NRAS, MAP2K1, ARAF, CSF1R and rarely BRAF V600E; A few patients have ALPS with mutated TNFRSF6 or H syndrome with mutated SLC29A3 |
Prognosis is good in most; spontaneous regression is common; occasional multifocal disease or involvement of critical site may be associated with significant morbidity |
| Langerhans cell histiocytosis (LCH) |
Multisystem disease most common in infants; multifocal disease in young children and unifocal disease in adults; Bone is most common site; skin, lymph nodes, others can be involved; Liver, spleen and bone marrow can be involved in multisystem disease |
Large cells with oval or irregular nuclei, some with prominent longitudinal folds, inconspicuous nucleoli and scant to moderate pale cytoplasm without dendritic processes; admixed eosinophils, lymphocytes, +/− neutrophils, plasma cells |
Positive: S100, CD1a, langerin; In LCH with BRAF V600E, mutation-specific BRAF is positive |
Mutations of MAPK pathway genes in 85%, BRAF V600E (most common), MAP2K1, ARAF, others Subset: clonal IGH, IGK or TCR gene rearrangements, c/w transdifferentiation of B- or T-cell neoplasm |
Cases with high stage disease or underlying B- or T-cell neoplasm have worse prognosis; unifocal disease has excellent prognosis; late-onset neurodegenerative CNS LCH has a poor prognosis |
| Follicular dendritic cell sarcoma (FDCS) | Most cases affect adults who have unifocal lymphadenopathy or extranodal mass lesion | Fascicular or whorled proliferation of spindle cells with admixed lymphocytes; minority arise from hyaline-vascular Castleman disease |
Positive (one or more may be lost): CD21, CD23, CD35, D2-40 Negative: CD45 |
MAPK pathway mutations are rare. NF-κB pathway mutations are common |
Behaves as a low-grade malignancy; better prognosis with small lesions (< 5 cm), bland histology, few mitoses, absent necrosis, localized disease |
Fig. 4.

Nasal Rosai−Dorfman disease (RDD), with relapse showing RDD and Langerhans cell histiocytosis (LCH). A patient with a history of allergic rhinitis had a lesion that consists of a loose proliferation of histiocytes with pale, sometimes vacuolated cytoplasm, with interspersed small lymphoid aggregates, beneath intact respiratory-type epithelium (A). Higher power shows histiocytes with large oval nuclei, distinct nucleoli and abundant pale cytoplasm with emperipolesis, and many admixed small lymphocytes and plasma cells (B); the distinctive histiocytes were S100+ (not shown). Two years later, disease recurred. In addition to areas of RDD, there were areas rich in eosinophils and with histiocytes with less abundant cytoplasm than that of the RDD histiocytes shown above (C). In this area, histiocytes have complex nuclear contours and prominent nuclear folds, smooth chromatin, absent nucleoli and pale pink cytoplasm, with admixed eosinophils (D, oil immersion). The Langerhans cells are langerin+ (E, immunoperoxidase technique on a paraffin section). They were also S100+ and CD1a+ (not shown)
In conclusion, the 5th edition of the World Health Organization (WHO) Classification of Head and Neck Tumours provides an authoritative, up-to-date reference for a wide variety of reactive and neoplastic hematologic neoplasms that can involve the head and neck. Compared to the prior edition, the space dedicated to these lesions is expanded, and a larger number of entities is discussed. The detailed descriptions of histologic, immunophenotypic and genetic features are very helpful in formulating a differential diagnosis and establishing a diagnosis.
Author Contributions
Judith Ferry wrote this review. Additional declarations for articles in life science journals that report the results of studies involving humans and/or animals: Not applicable for this update on the WHO Classification.
Funding
No external funding.
Data Availability
Not applicable.
Code Availability
Not applicable.
Declarations
Conflicts of interest
The author declares that they have no conflict of interest.
Ethical Approval
Not applicable for this review.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO Classification of Tumours Editorial Board . Head and neck tumours. Lyon: International Agency for Research on Cancer; 2022. [Google Scholar]
- 2.Louissaint A, Jr, Ferry JA, Soupir CP, Hasserjian RP, Harris NL, Zukerberg LR. Infectious mononucleosis mimicking lymphoma: distinguishing morphological and immunophenotypic features. Mod Pathol. 2012;25:1149–59. doi: 10.1038/modpathol.2012.70. [DOI] [PubMed] [Google Scholar]
- 3.Lin H, Su C, Huang C, Hwang C, Chien C. Kikuchi’s disease: a review and analysis of 61 cases. Otolaryngol Head Neck Surg. 2003;128:650–3. doi: 10.1016/S0194-59980223291-X. [DOI] [PubMed] [Google Scholar]
- 4.Kuo T. Kikuchi’s disease (histiocytic necrotizing lymphadenitis). A clinicopathologic study of 79 cases with an analysis of histologic subtypes, immunohistology and DNA ploidy. Am J Surg Pathol. 1995;19:798–809. doi: 10.1097/00000478-199507000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Tsang WYW, Chan JKC, Ng CS. Kikuchi’s lymphadenitis. A morphologic analysis of 75 cases with special reference to unusual features. Am J Surg Pathol. 1994;18:219–31. [PubMed] [Google Scholar]
- 6.Attygalle AD, Liu H, Shirali S, et al. Atypical marginal zone hyperplasia of mucosa-associated lymphoid tissue: a reactive condition of childhood showing immunoglobulin lambda light-chain restriction. Blood. 2004;104:3343–8. doi: 10.1182/blood-2004-01-0385. [DOI] [PubMed] [Google Scholar]
- 7.Elicora SS, Guven M, Varli AF, Yilmaz MS, Alponat S. Rare posterior pharyngeal mass: atypical marginal zone hyperplasia. J Pediatr Hematol Oncol. 2016;38:152–4. doi: 10.1097/MPH.0000000000000494. [DOI] [PubMed] [Google Scholar]
- 8.Kaur P, Levy NB. Atypical marginal zone hyperplasia of tonsil with immunoglobulin light chain restriction. Am J Hematol. 2012;87:424–5. doi: 10.1002/ajh.22132. [DOI] [PubMed] [Google Scholar]
- 9.Dojcinov SD, Venkataraman G, Raffeld M, Pittaluga S, Jaffe ES. EBV positive mucocutaneous ulcer--a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405–17. doi: 10.1097/PAS.0b013e3181cf8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart M, Thakral B, Yohe S, et al. EBV-positive mucocutaneous ulcer in organ transplant recipients: a localized indolent posttransplant lymphoproliferative disorder. Am J Surg Pathol. 2014;38:1522–9. doi: 10.1097/PAS.0000000000000282. [DOI] [PubMed] [Google Scholar]
- 11.Geyer JT, Ferry JA, Harris NL, et al. Chronic sclerosing sialadenitis (Kuttner tumor) is an IgG4-associated disease. Am J Surg Pathol. 2010;34:202–10. doi: 10.1097/PAS.0b013e3181c811ad. [DOI] [PubMed] [Google Scholar]
- 12.Ferry JA, Klepeis V, Sohani AR, et al. IgG4-related orbital disease and its mimics in a western population. Am J Surg Pathol. 2015;39:1688–700. doi: 10.1097/PAS.0000000000000497. [DOI] [PubMed] [Google Scholar]
- 13.Ferry JA, Deshpande V. IgG4-related disease in the head and neck. Semin Diagn Pathol. 2012;29:235–44. doi: 10.1053/j.semdp.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181–92. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 15.Deshpande V. The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol. 2012;29:191–6. doi: 10.1053/j.semdp.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Deshpande V, Khosroshahi A, Nielsen GP, Hamilos DL, Stone JH. Eosinophilic angiocentric fibrosis is a form of IgG4-related systemic disease. Am J Surg Pathol. 2011;35:701–6. doi: 10.1097/PAS.0b013e318213889e. [DOI] [PubMed] [Google Scholar]
- 17.Ahn J, Flanagan M. Eosinophilic angiocentric fibrosis: a review and update of its association with immunoglobulin G4-related disease. Arch Pathol Lab Med. 2018;142:1560–3. doi: 10.5858/arpa.2017-0223-RS. [DOI] [PubMed] [Google Scholar]
- 18.Thompson LDR. Algorithmic approach to fibroinflammatory sinonasal tract lesions. Head Neck Pathol. 2021;15:120–9. doi: 10.1007/s12105-020-01272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horn H, Schmelter C, Leich E, et al. Follicular lymphoma grade 3B is a distinct neoplasm according to cytogenetic and immunohistochemical profiles. Haematologica. 2011;96:1327–34. doi: 10.3324/haematol.2011.042531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louissaint A, Jr, Ackerman AM, Dias-Santagata D, et al. Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood. 2012;120:2395–404. doi: 10.1182/blood-2012-05-429514. [DOI] [PubMed] [Google Scholar]
- 21.Louissaint A, Jr, Schafernak KT, Geyer JT, et al. Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood. 2016;128:1093–100. doi: 10.1182/blood-2015-12-682591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agostinelli C, Akarca AU, Ramsay A, et al. Novel markers in pediatric-type follicular lymphoma. Virchows Arch. 2019;475:771–9. doi: 10.1007/s00428-019-02681-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt J, Ramis-Zaldivar JE, Nadeu F, et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood. 2017;130:323–7. doi: 10.1182/blood-2017-03-776278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Attarbaschi A, Abla O, Arias Padilla L, et al. Rare non-Hodgkin lymphoma of childhood and adolescence: a consensus diagnostic and therapeutic approach to pediatric-type follicular lymphoma, marginal zone lymphoma, and nonanaplastic peripheral T-cell lymphoma. Pediatr Blood Cancer. 2020;67:e28416. doi: 10.1002/pbc.28416. [DOI] [PubMed] [Google Scholar]
- 25.Salaverria I, Philipp C, Oschlies I, et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood. 2011;118:139–47. doi: 10.1182/blood-2011-01-330795. [DOI] [PubMed] [Google Scholar]
- 26.Au-Yeung RKH, Arias Padilla L, Zimmermann M, et al. Experience with provisional WHO-entities large B-cell lymphoma with IRF4-rearrangement and Burkitt-like lymphoma with 11q aberration in paediatric patients of the NHL-BFM group. Br J Haematol. 2020;190:753–63. doi: 10.1111/bjh.16578. [DOI] [PubMed] [Google Scholar]
- 27.Moody S, Thompson JS, Chuang SS, et al. Novel GPR34 and CCR6 mutation and distinct genetic profiles in MALT lymphomas of different sites. Haematologica. 2018;103:1329–36. doi: 10.3324/haematol.2018.191601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chanudet E, Ye H, Ferry J, et al. A20 deletion is associated with copy number gain at the TNFA/B/C locus and occurs preferentially in translocation-negative MALT lymphoma of the ocular adnexa and salivary glands. J Pathol. 2009;217:420–30. doi: 10.1002/path.2466. [DOI] [PubMed] [Google Scholar]
- 29.Chanudet E, Huang Y, Ichimura K, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010;24:483–7. doi: 10.1038/leu.2009.234. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Climent JA. G-protein coupled receptor (GPCR) mutations in lymphoid malignancies: linking immune signaling activation and genetic abnormalities. Haematologica. 2018;103:1252–5. doi: 10.3324/haematol.2018.196998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aukema SM, Croci GA, Bens S, et al. Mantle cell lymphomas with concomitant MYC and CCND1 breakpoints are recurrently TdT positive and frequently show high-grade pathological and genetic features. Virchows Arch. 2021;479:133–45. doi: 10.1007/s00428-021-03022-8. [DOI] [PubMed] [Google Scholar]
- 32.Silkenstedt E, Linton K, Dreyling M. Mantle cell lymphoma—advances in molecular biology, prognostication and treatment approaches. Br J Haematol. 2021;195:162–73. doi: 10.1111/bjh.17419. [DOI] [PubMed] [Google Scholar]
- 33.Hill HA, Qi X, Jain P, et al. Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020;4:2927–38. doi: 10.1182/bloodadvances.2019001350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Tang G, Medeiros LJ, et al. MYC rearrangement but not extra MYC copies is an independent prognostic factor in patients with mantle cell lymphoma. Haematologica. 2021;106:1381–9. doi: 10.3324/haematol.2019.243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salaverria I, Royo C, Carvajal-Cuenca A, et al. CCND2 rearrangements are the most frequent genetic events in cyclin D1(-) mantle cell lymphoma. Blood. 2013;121:1394–402. doi: 10.1182/blood-2012-08-452284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Garcia D, Navarro A, Valdes-Mas R, et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1(–) mantle cell lymphoma. Blood. 2019;133:940–51. doi: 10.1182/blood-2018-07-862151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wagener R, Bens S, Toprak UH, et al. Cryptic insertion of MYC exons 2 and 3 into the immunoglobulin heavy chain locus detected by whole genome sequencing in a case of “MYC-negative” Burkitt lymphoma. Haematologica. 2020;105:e202-e5. doi: 10.3324/haematol.2018.208140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med 2014;4. [DOI] [PMC free article] [PubMed]
- 39.Abate F, Ambrosio MR, Mundo L, et al. Distinct viral and mutational spectrum of endemic burkitt lymphoma. PLoS Pathog. 2015;11:e1005158. doi: 10.1371/journal.ppat.1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2010;23:307–18. doi: 10.1016/j.beha.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Van Vlierberghe P, Pieters R, Beverloo HB, Meijerink JP. Molecular-genetic insights in paediatric T-cell acute lymphoblastic leukaemia. Br J Haematol. 2008;143:153–68. doi: 10.1111/j.1365-2141.2008.07314.x. [DOI] [PubMed] [Google Scholar]
- 42.Balbach ST, Makarova O, Bonn BR, et al. Proposal of a genetic classifier for risk group stratification in pediatric T-cell lymphoblastic lymphoma reveals differences from adult T-cell lymphoblastic leukemia. Leukemia. 2016;30:970–3. doi: 10.1038/leu.2015.203. [DOI] [PubMed] [Google Scholar]
- 43.Bongiovanni D, Saccomani V, Piovan E. Aberrant signaling pathways in T-cell acute lymphoblastic leukemia. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed]
- 44.Anand P, Guillaumet-Adkins A, Dimitrova V, et al. Single-cell RNA-seq reveals developmental plasticity with coexisting oncogenic states and immune evasion programs in ETP-ALL. Blood. 2021;137:2463–80. doi: 10.1182/blood.2019004547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khanam T, Sandmann S, Seggewiss J, et al. Integrative genomic analysis of pediatric T-cell lymphoblastic lymphoma reveals candidates of clinical significance. Blood. 2021;137:2347–59. doi: 10.1182/blood.2020005381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y, Xu Z, Lin W, et al. Comprehensive genomic profiling of EBV-positive diffuse large B-cell lymphoma and the expression and clinicopathological correlations of some related genes. Front Oncol. 2019;9:683. doi: 10.3389/fonc.2019.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Menter T, Juskevicius D, Alikian M, et al. Mutational landscape of B-cell post-transplant lymphoproliferative disorders. Br J Haematol. 2017;178:48–56. doi: 10.1111/bjh.14633. [DOI] [PubMed] [Google Scholar]
- 48.Ambrosio MR, Mundo L, Gazaneo S, et al. MicroRNAs sequencing unveils distinct molecular subgroups of plasmablastic lymphoma. Oncotarget. 2017;8:107356–73. doi: 10.18632/oncotarget.22219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong J, Cui BW, Wang N, et al. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell. 2020;37:403–19. doi: 10.1016/j.ccell.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Massoth LR, Hung YP, Ferry JA, et al. Histiocytic and dendritic cell sarcomas of hematopoietic origin share targetable genomic alterations distinct from follicular dendritic cell sarcoma. Oncologist. 2021;26:e1263-e72. doi: 10.1002/onco.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839–42. doi: 10.1038/s41591-019-0653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paxton CN, O’Malley DP, Bellizzi AM, et al. Genetic evaluation of juvenile xanthogranuloma: genomic abnormalities are uncommon in solitary lesions, advanced cases may show more complexity. Mod Pathol. 2017;30:1234–40. doi: 10.1038/modpathol.2017.50. [DOI] [PubMed] [Google Scholar]
- 53.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120:2700–3. doi: 10.1182/blood-2012-05-430140. [DOI] [PubMed] [Google Scholar]
- 54.Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6:154–65. doi: 10.1158/2159-8290.CD-15-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ozkaya N, Rosenblum MK, Durham BH, et al. The histopathology of Erdheim–Chester disease: a comprehensive review of a molecularly characterized cohort. Mod Pathol. 2018;31:581–97. doi: 10.1038/modpathol.2017.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garces S, Medeiros LJ, Patel KP, et al. Mutually exclusive recurrent KRAS and MAP2K1 mutations in Rosai–Dorfman disease. Mod Pathol. 2017;30:1367–77. doi: 10.1038/modpathol.2017.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baraban E, Sadigh S, Rosenbaum J, et al. Cyclin D1 expression and novel mutational findings in Rosai–Dorfman disease. Br J Haematol. 2019;186:837–44. doi: 10.1111/bjh.16006. [DOI] [PubMed] [Google Scholar]
- 58.Chakraborty R, Abdel-Wahab O, Durham BH. MAP-kinase-driven hematopoietic neoplasms: a decade of progress in the molecular Age. Cold Spring Harb Perspect Med. 2021 doi: 10.1101/cshperspect.a034892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai–Dorfman–Destombes disease. Blood. 2018;131:2877–90. doi: 10.1182/blood-2018-03-839753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.
Not applicable.