

Abstract
The initiative of the 5th edition of the WHO classification of the Head and Neck Tumours establishing a new section dedicated to familial/heritable tumor syndromes with tumors and lesions in the head and neck region was much needed to better understand the tumours, diseases, and associated syndromes, as well as establish recommendations for monitoring and treating these patients. (WHO Classification of Tumours Editorial Board. Head and Neck tumours. Lyon (France): International Agency for Research on Cancer; 2022. https://publications.iarc.fr/). Within the newly established chapter on genetic tumor syndromes, we have described the main manifestations on the head and neck region in 15 syndromes. This review highlights the important findings within these syndromes, especially on the update on syndromes with tumors involving the head and neck region, as Gorlin syndrome/nevoid basal cell carcinoma syndrome associated with odontogenic keratocysts; Brooke-Spiegler syndrome/familial cylindromatosis and the associated membranous-type salivary gland basal cell adenoma, PTEN hamartoma tumor syndrome/Cowden syndrome with associated facial skin and mucosal lesions and characteristic multinodular thyroid lesions, Von Hippel Lindau syndrome and the associated middle ear endolymphatic sac tumor, as well as the fascinating genetic aspects of the diverse Head and Neck Paragangliomas. We will also discuss hyperparathyroidism-jaw tumor syndrome is characterized by parathyroid tumors in association with fibro-osseous jaw tumors, as well as head and neck desmoid tumors associated with familial adenomatous polyposis with Gardner syndrome variant familial, multicentric head and neck squamous cell carcinoma, tuberous sclerosis and neurofibromatosis type 1-associated head and neck lesions.
Keywords: Familial cancer syndromes, Familial tumor syndromes, Familial genetic syndromes, Head and neck tumors, Thyroid, Salivary gland
Introduction
It has been known that some hereditary aspects can contribute to the development of tumors involving the head and neck for more than 120 years. As an example, nevoid basal cell carcinoma syndrome, Gorlin syndrome, or basal cell nevus syndrome (NBCCS) is believed to have been initially described in the literature in 1894. However, it was not until 1959 when NBCCS was recognized as a distinct entity [1, 2].
Cancer caused more than 600,000 deaths in the United States in 2000 (Cancer Statistics, 2020). This estimate includes patients who have a genetic predisposition to neoplastic disease, including head and neck neoplasms.
A significant number of patients with inherited head and neck tumors manifest disease that appears to be sporadic disease even in some syndromic cases. A careful clinical documentation of family history or familial syndromes that can be associated with head and neck disease can help identify germline susceptibility-driven neoplasia (Table 1). Clinically recognizable tumour-predisposing syndromes have been more widely recognized during the last decades due to the rapidly increasing use of sequencing of DNA in both diagnostic pathology and clinical genetics. What appears to be a sporadic tumour is becoming less often so with more intense investigation and new syndromes and tumours associated with these syndromes are being reported.
Table 1.
Familial cancer syndromes involving the head and neck: genetic
| Disease/phenotype | Inheritance | Locus | Gene(s) | Encoded protein(s) | Normal protein function |
|---|---|---|---|---|---|
| Naevoid basal cell carcinoma syndrome: | |||||
| Naevoid basal cell carcinoma syndrome 1 | AD | 9q22.3 | PTCH1 | PTCH1 | Plays a role in embryonic development and regulates hedgehog signalling |
| Naevoid basal cell carcinoma syndrome 2 | AD | 10q24.3 | SUFU | SUFU | Inhibits hedgehog signalling |
| Neurofibromatosis 1 (NF1) | AD | 17q11 | NF1 | Neurofibromin (NF1) | Regulation of RAS signalling |
| Familial adenomatous polyposis with the Gardner syndrome variant | AD | 5q22.2 | APC | APC | Negative regulation of β-catenin and WNT signalling |
| Brooke-Spiegler syndrome | AD | 16q12.1 | CYLD | CYLD | Negatively regulates NF-κB activation |
| PTEN hamartoma tumor syndrome/Cowden syndrome: | |||||
| Cowden syndrome 1 | AD | 10q23.31 | PTEN | PTEN | A phosphatase; regulation of cell division (tumour suppressor) |
| Cowden syndrome 4 | AD | 10q23.31 | KLLN | Killin | Triggers apoptosis |
| Cowden syndrome 5 | AD | 3q26.32 | PIK3CA | PIK3CA (p110) | A catalytic subunit of PI3K |
| Cowden syndrome 6 | AD | 14q32.33 | AKT1 | AKT1 | Modulation of the AKT/mTOR signalling pathway |
| Cowden syndrome 7 | AD | 20p11.23 | SEC23B | SEC23B | ER-associated protein secretion |
| Hereditary paraganglioma syndromes: | |||||
| -SDH-deficient: | Dysfunction of the SDH complex enzyme involved in a broad spectrum of tumorigenic pathways including hypoxic regulation | ||||
| PGL1 | AD; PT | 11q23.1 | SDHD | SDHD | |
| PGL2 | AD; PT | 11q12.2 | SDHAF2 | SDHAF2 | |
| PGL3 | AD | 1q23.3 | SDHC | SDHC | |
| PGL4 | AD | 1p36.1-p35 | SDHB | SDHB | |
| PGL5 | AD | 5p15 | SDHA | SDHA | |
| -MEN2 | AD | 10q11.21 | RET | RET | Normal development of nerve cells |
| -VHL | AD | 3p25.3 | VHL | VHL | Ubiquitination and degradation of HIF (tumour suppressor) |
| -NF1 | AD | 17q11 | NF1 | Neurofibromin (NF1) | Regulation of RAS signalling |
| -Other genes: | |||||
| MAX; TMEM127; FH; KIF1B; EGLN1,2,3; EPAS1; MEN1 | |||||
| Multiple endocrine neoplasia type 2B | AD | 10q11.21 | RET | RET | Normal development of nerve cells |
| Hyperparathyroidism-Jaw tumour syndrome | AD | 1q25-q31 | CDC73 | Parafibromin | Parafibromin is a critical component of the PAF1C that binds to RNA polymerase II and controls transcription |
| Li–Fraumeni syndrome, TP53-associated | AD | 17p13.1 | TP53 | p53 | Regulation of cell division (tumour suppressor) |
| Li–Fraumeni syndrome, CHEK2-associated | AD | 22q12.1 | CHEK2 | CHEK2 | Induction of cell-cycle arrest and apoptosis after DNA damage |
| Fanconi anemia | Most AR FANC-Bis: X-linked and FANC-R: AD | 16q24.3 | FANCA (FA1) | FANCA | Some are involved in the repair of DNA damage |
| Dyskeratosis congenita | Most X-linked, AD, AR | 3q26.2 | DKCA | DKCA | Defective telomere maintenance caused by germline mutations in genes important in telomere biology |
| Ataxia telangiectasia syndrome | AR | 11q22.3 | ATM | ATM | Absence of ATM protein affects genomic instability or DNA damage response, leading to the development of tumours |
| Bloom syndrome | AR | 15q26.1 | BLM | BLM | BLM protein normally suppresses hyperrecombination |
| VonHippel Lindau syndrome | AD | 3p25.3 | VHL | VHL | Ubiquitination and degradation of HIF (tumour suppressor) |
| Tuberous sclerosis syndrome | |||||
| Tuberous sclerosis type 1 | AD | 9q34 | TSC1 | Hamartin | Interacts with tuberin; inhibits growth |
| Tuberous sclerosis type 2 | AD | 16p13.3 | TSC2 | Tuberin | Interacts with hamartin; inhibits growth |
Familial tumor syndromes are important to be identify clinically because family members require high degrees of monitoring and genetic counseling. Study of these individuals and families has led to the discovery of genes that are an intrinsic aspect of cell regulation and will continue to be relevant in defining mechanisms of neoplastic development in any tissue.
Clinical suspicion coupled with molecular tumor analysis and testing for germline mutations can help differentiate the clinical mimicry within tumors and facilitate diagnosis and management.
The initiative of the 5th edition of the WHO classification of the Head and Neck Tumours establishing a new section dedicated to familial/heritable tumor syndromes with tumors and lesions in the head and neck region was much needed to better understand the tumours, diseases, and associated syndromes, as well as establish recommendations for monitoring and treating these patients. (WHO Classification of Tumours Editorial Board. Head and neck tumours. Lyon (France): International Agency for Research on Cancer; 2022. (WHO classification of tumours series, 5th ed.; vol. 9). https://publications.iarc.fr/).
Within the newly established chapter on genetic tumor syndromes, we have described the main manifestations on the head and neck region in 15 syndromes.
We will review and update the main syndromes involving the head and neck region: Nevoid basal cell carcinoma syndrome, Brooke-Spiegler syndrome or familial cylindromatosis, phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome/Cowden syndrome (CS), Von Hippel Lindau syndrome, Head and Neck Paragangliomas, Familial multicentric head and neck squamous cell carcinoma (HNSCC) within others (Table 2).
Table 2.
Familial cancer syndromes involving the head and neck: disease phenotype
| Disease phenotype (gene) | Main H&N presentation | H&N lesion | H&N lesion | H&N lesion | Associated lesions |
|---|---|---|---|---|---|
| 1 | 2 | 3 | |||
| Nevoid basal cell carcinoma syndrome: | Multiple basal cell carcinomas; | Cleft lip/palate | Ocular anomalies | Macrocephaly | Calcification of the falx cerebri; |
| Nevoid basal cell carcinoma syndrome 1 (PTCH1) | Odontogenic keratocyst (OKC) of the jaw | Palmar and plantar pits; Bifid or fused ribs; Medulloblastoma | |||
| Nevoid basal cell carcinoma syndrome 2 (SUFU) | |||||
| Neurofibromatosis 1 (NF1) | Multiple café-au-lait macules; Cutaneous neurofibromas; Orbitofacial neurofibromatosis | Optic pathway glioma | Iris hamartomas (Lisch nodules) | Orbital dysplasia | Neurofibroma and MPNST; Skinfold freckling in the axillary, inguinal, and submammary regions; Duodenal neuroendocrine tumor |
| Pheochromocytoma and paragangliomas | |||||
| Malignant peripheral nerve sheath tumour (MPNST); Rhabdomyosarcoma, Glomus tumours of the digits | |||||
| Familial adenomatous polyposis with Gardner syndrome variant (APC) | Desmoid fibromatosis: maxillary sinus, nasopharynx, and oral cavity | Bone osteomas | Dental structures: supernumerary teeth | Compound odontomas located in the anterior maxilla, or complex odontomas in the posterior mandible or anterior maxilla | > 100 adenomatous colorectal polyps; Gastric and duodenal polyps; |
| Cribriform morular thyroid carcinoma; Hepatobiliary tree and pancreas carcinoma; | |||||
| Childhood hepatoblastoma (1%); Adrenocortical adenomas /adenocarcinomas; medulloblastoma (< 1%); Adamantinomatous craniopharyngioma | |||||
| Brooke-Spiegler syndrome (CYLD) | Salivary gland basal cell adenoma, membranous-type, multicentric and bilateral | Spiradenoma, cylindroma and spiradenocylindroma mainly involve the skin of the head and neck | Trichoepitheliomas predominantly affect the nasolabial folds | ||
| PTEN hamartoma tumor syndrome/Cowden syndrome: | Papillomas on the lips, tongue, buccal mucosa; | Mucocutaneous neuromas | Adult Lhermitte-Duclos disease (cerebellar tumors); | ||
| Cowden syndrome 1 (PTEN) | Facial trichilemmomas | Breast cancer; | |||
| Cowden syndrome 4 (KLLN) | Non-medullary thyroid cancer; | ||||
| Cowden syndrome 5 (PIK3CA) | Megalocephaly; Endometrial carcinoma; | ||||
| Cowden syndrome 6 (AKT1) | |||||
| Cowden syndrome 7 (SEC23B) | Multiple GI hamartomas or ganglioneuromas | ||||
| Hereditary paraganglioma syndromes: | |||||
| SDH-deficient syndromes: | |||||
| PGL1 (SDHD) | H&N Paraganglioma (most common head and neck PGL: ~ 50%) | Pheochromocytoma; Renal cell carcinoma; | |||
| Gastrointestinal stromal tumor; | |||||
| Pituitary tumor | |||||
| PGL2 (SDHAF2) | H&N Paraganglioma | ||||
| PGL3 (SDHC) | H&N Paraganglioma | Renal cell carcinoma; | |||
| Gastrointestinal stromal tumor | |||||
| PGL4 (SDHB) | H&N Paraganglioma | Renal cell carcinoma; | |||
| Gastrointestinal stromal tumor; | |||||
| Pituitary tumor | |||||
| Abdomen and thorax paraganglioma | |||||
| PGL5 (SDHA) | H&N Paraganglioma | Pheochromocytoma; Renal cell carcinoma; | |||
| Gastrointestinal stromal tumor; | |||||
| Pituitary tumor | |||||
| Multiple endocrine neoplasia type 2/MEN2 (RET) | H&N Paraganglioma rare | Medullary thyroid carcinoma; | |||
| Parathyroid proliferations; | |||||
| Oral ganglioneuromas; Mucosal neuromas oropharynx, lips, and eyelids | Pheochromocytoma | ||||
| VonHippel-Lindau (VHL) | H&N Paraganglioma rare | Central nervous system and retinal hemangioblastoma | Clear cell renal cell carcinoma; | ||
| Neuroendocrine tumors; | |||||
| Endolymphatic sac tumor of middle ear | Pancreatic serous cystadenomas | ||||
| Neurofibromatosis type 1 (NF1) | H&N Paraganglioma rare | Neurofibroma and MPNST; Ocular manifestations; Duodenal neuroendocrine tumor | |||
| Others syndromes/genes: MAX; TMEM127; FH; KIF1B; EGLN1,2,3; EPAS1; MEN1 | H&N Paraganglioma rare | ||||
| Multiple endocrine neoplasia type 2B/MEN2B (RET) | Oral ganglioneuromas; Mucosal neuromas in MEN 2B can involve the oropharynx, lips, and eyelids | Thickened corneal nerves | Medullary thyroid carcinoma; Pheochromocytoma; Parathyroid proliferations; | ||
| Oral and intestinal ganglioneuromas; Marfanoid body habitus | |||||
| Hyperparathyroidism-Jaw tumour syndrome (CDC73) | Cemento-ossifying fibroma | Parathyroid adenoma and carcinoma | Fibro-osseous jaw lesions | Renal and uterine tumours | |
| Li-Fraumeni syndrome: | H&N and laryngeal carcinomas | Squamous cell carcinomas larynx, pharynx and oral cavity | Breast, soft tissue, bone, brain, adrenal glands, and female genital tract carcinomas | ||
| Li–Fraumeni syndrome (TP53-associated) | |||||
| Li–Fraumeni syndrome (CHEK2-associated) | |||||
| Fanconi anemia (FANCA) | Head and neck squamous cell carcinoma (FA-HNSCC) frequently present as poorly differentiated | Oral potentially malignant disorders (OPMD) | Increased risk for hematologic and solid malignancies | ||
| Dyskeratosis congenita (DKCA) | Oral leukoplakia | Oral leukoplakia seen on the tongue | Triad of oral leukoplakia, nail dystrophy, and reticulate hyperpigmentation; | ||
| Bone marrow failure; Increased malignancy; Lung and liver diseases | |||||
| Ataxia telangiectasia syndrome (ATM) | Head and neck squamous cell carcinoma (HNSCC) | Lymphoid neoplasms of the B and T-cell lineage in the head and neck, including involvement of the Waldeyer ring | Telangiectases in sun exposed areas as bulbar conjunctiva and bridge of nose | Neurologic manifestations and conjunctival telangiectasias | |
| Bloom syndrome (BLM) | Head and neck squamous cell carcinoma (HNSCC) | Rash on cheeks and nose | Small size and lack of subcutaneous tissue | ||
| Men and infertile and women have early menopause | |||||
| Colorectal and breast cancer | |||||
| Von-Hippel Lindau syndrome (VHL) | Endolymphatic sac tumor, bilateral | Hemangioblastomas cerebellum, spinal cord, brain, and retina | Kidney cysts | ||
| and renal cell carcinomas; | |||||
| Adrenal pheochromocytomas and extraadrenal paragangliomas; | |||||
| Pancreatic neuroendocrine tumors; Cysts and serous cystadenomas | |||||
| Tuberous sclerosis syndrome: | Predominantly cutaneous and oral manifestations; | Fibrous cephalic plaques in forehead and scalp | Dental enamel loss on the incisal border and tooth | Cutaneous angiofibromas; Shagreen patches; Subungual fibromas; Cardiac rhabdomyomas; Pulmonary lymphangioleiomyomatosis; | |
| Tuberous sclerosis type 1 (TSC1) | Facial angiofibromas; | ||||
| Tuberous sclerosis type 2 (TSC2) | Shagreen patches and poliosis in scalp | Renal angiomyolipomas |
Gorlin Syndrome/Nevoid Basal Cell Carcinoma Syndrome
Gorlin syndrome/Naevoid basal cell carcinoma syndrome (NBCCS) is a rare autosomal dominantly inherited disorder that is characterized by cancer and neoplastic predisposition including basal cell carcinomas (BCC), odontogenic keratocysts (OKC) and medulloblastomas, as well as dyskeratotic palmar and plantar pitting, range of skeletal and other developmental abnormalities. Gorlin syndrome is associated with germline mutations in components of the Sonic Hedgehog pathway, including Patched1 (PTCH1) and Suppressor of fused (SUFU) SUFU mutation carriers [3–5].
Basal cell carcinomas most commonly develop on the face, but also appear on the trunk and limbs. Additional well-recognized clinical features are often present, occurring in 70–80% of patients, and include OKC, dyskeratotic palmar and plantar pitting, rib and spine abnormalities, and early calcification of the falx cerebri. A small proportion of patients may have frontal bossing, hypertelorism, macrocephaly, and cleft lip and/or palate and desmoplastic medulloblastoma during childhood, and ovarian neoplasms and cardiac fibromas, mesenteric keratocysts, rhabdomyosarcomas, and meningiomas.
Odontogenic keratocysts typically occur in the posterior mandible and show an area of lucency by image. Greater than 80% are unilocular [4]. There may be soft keratinaceous debris within the cystic space. Histologically, lesions exhibit a uniformly thick squamous epithelial lining, typically 5–10 cells in thickness, with parakeratosis and basal/parabasal cell palisading, in contrast to other odontogenic cysts, which are orthokeratinized, do not display parakeratosis and basal/parabasal cell palisading (Fig. 1). Satellite tumors are associated with higher risk of recurrence, estimated to be 10–40%. Basal and parabasal cell hyperplasia is also often present, and often exhibit a high Ki67 proliferation index, and with p53 immunopositivity of the basal and suprabasal cell layers, and display loss of heterozygosity at several loci. Due to these reasons this entity has been re-classified as a neoplasm rather than a developmental phenomenon. Odontogenic keratocyst screening should begin at age 2 with annual orthopantogram beginning around age 8 for PTCH1 PV carriers only [6, 7]. The most notable molecular finding in OKCs is frequent mutations of the tumour suppressor gene PTCH1(9q22.3-q31), in syndromic and up to 80% of sporadic OKCs [8, 9].
Fig. 1.
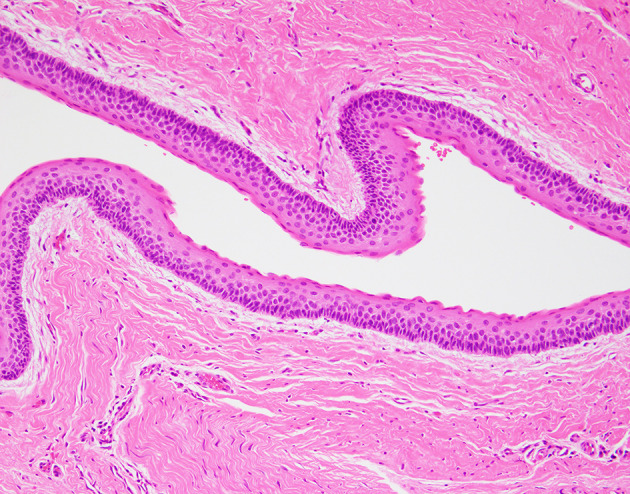
Odontogenic keratocyst in Nevoid BCC syndrome: Gorlin syndrome/Naevoid basal cell carcinoma syndrome is characterized by cancer predisposition syndrome associated with basal cell carcinomas, odontogenic keratocysts and medulloblastomas. The epithelial cyst lining of an odontogenic keratocyst displays cuboidal to columnar basal cells with hyperchromatic basal cell nuclei and focal reverse nuclear polarity and characteristic surface parakeratosis with corrugated surface parakeratin
As with BCCs, OKCs occurring in patients with NBCCS are histologically indistinguishable from those occurring sporadically. The diagnostic criteria for nevoid basal cell carcinoma syndrome are described in Table 3.
Table 3.
Diagnostic criteria for nevoid basal cell carcinoma syndrome
| Major criteria |
| Basal cell carcinoma before 20 years of age or excessive numbers of basal cell carcinomas out of proportion to prior sun exposure and skin type |
| Odontogenic keratocyst before 20 years of age |
| Palmar or plantar pitting |
| Lamellar calcification of the falx cerebri |
| Medulloblastoma, typically desmoplastic |
| First degree relative with nevoid basal cell carcinoma syndrome |
| Minor criteria |
| Rib abnormalities |
| Other specific skeletal malformations and radiologic changes (i.e., vertebral anomalies, kyphoscoliosis, short fourth metacarpals, postaxial polydactyly) |
| Macrocephaly |
| Cleft lip or palate |
| Ovarian or cardiac fibroma |
| Lymphomesenteric cysts |
| Ocular abnormalities (i.e., strabismus, hypertelorism, congenital cataracts, glaucoma, coloboma) |
Adapted from Bree and Shah [48]
Requirement for diagnosis Two major diagnostic criteria and one minor diagnostic criterion, or One major and three minor diagnostic criteria, or Identification of a heterozygous germline PTCH1 or SUFU pathogenic variant on molecular genetic testing
Screening for PTCH1 Mutation Carriers
Basal cell carcinoma screening annually by age 10, with increased frequency after first basal cell carcinoma observed
Baseline echocardiogram in infancy, dental exams with jaw X-ray every 12 to 18 months beginning at age 8, and an ovarian ultrasound by age 18
Low risk of medulloblastoma: no radiographic screening unless concerning neurologic exam, head circumference change, or other unusual signs or symptoms
If medulloblastoma: radiation-sparing treatment given risk of radiation-induced skin cancers
Screening for SUFU Mutation Carriers
Same as PTCH1 mutation carriers, except for no jaw X-rays, as keratocysts have not been described
Additional medulloblastoma screening: consider every-4-month brain MRI through age 3 and then every-6-month brain MRI until the age of 5a
Radiation-sparing treatments are again recommended if a brain tumor should occur
Brooke-Spiegler Syndrome or Familial Cylindromatosis
Brooke-Spiegler syndrome or familial cylindromatosis or CYLD cutaneous syndrome is an inherited autosomal dominant disease characterized by multiple spiradenomas, cylindromas, spiradenocylindromas and trichoepitheliomas mainly involving the skin of the head and neck as well as a distinctive salivary gland tumour, basal cell adenoma (BCA), membranous-type.
Salivary gland involvement is usually seen after the age of 40 years and about 2% of patients with BSS develop salivary gland tumors. Salivary gland involvement by BCA, membranous-type which is a type characteristic of this syndrome [10]. Some BCACs (~ 15%) occur in the setting of this familial/multiple cylindromatosis syndrome.
Basal cell adenoma and basal cell adenocarcinoma (BCAC) are uncommon biphasic salivary gland tumors having morphologic similarities to other biphasic salivary gland neoplasms with differentiation toward the intercalated ducts of the salivary gland. This tumour involves primarily the parotid gland. These tumors are usually bilateral/multicentric and well-circumscribed.
Tumor nests characteristically show biphasic basaloid morphology with peripheral palisading of dark cells with more centrally located paler cells and ducts (Fig. 2) [10]. Epithelial and myoepithelial markers highlight the dual cell composition. CYLD1 alterations are also common, more so with membranous BCA [10, 11]. The prognosis shows recurrence rates to be low (< 2%); except for the membranous subtype (~ 25%), which occurs in this syndrome. Malignant transformation can occur, more frequently with the membranous subtype [12]. BCA are differentiated from BCAC by the absence of invasion, absence of necrosis and lower mitotic index: BCAC is characterized by infiltration, necrosis and mitoses (especially when > 4 per 2 mm2). BCAs are differentiated from cellular pleomorphic adenoma by the basaloid appearance, peripheral palisading and lack of blending with associated myoepithelial type stroma.
Fig. 2.
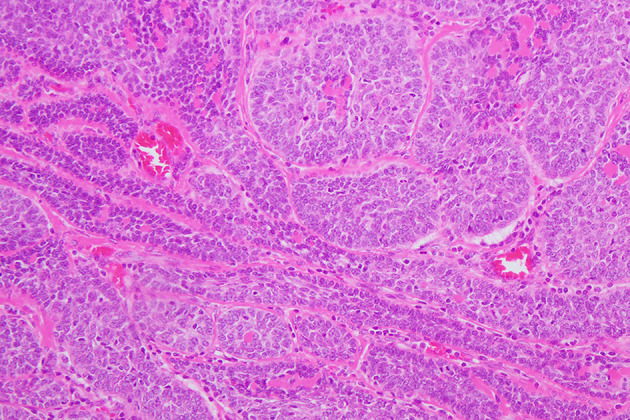
Basal cell adenoma, membranous pattern in Brooke Spiegler syndrome: Brooke-Spiegler syndrome or familial cylindromatosis is characterized by multiple spiradenomas, cylindromas, spiradenocylindromas and trichoepitheliomas mainly involving the skin of the head and neck as well as a distinctive salivary gland tumour, basal cell adenoma, membranous-type. Sharply delimited islands of basaloid cells are separated by basement membrane material, also seen as small droplets intermixed within tumour islands
Genodermatosis: PTEN Hamartoma Tumor Syndrome (PHTS)
Genodermatosis affecting skin and mucosa of the head and neck have been well characterized [13]. PTEN hamartoma tumor syndrome refers to a spectrum of disorders caused by mutations in the PTEN gene. CS is part of the PHTS, along with Bannayan–Riley–Ruvalcaba syndrome (BRRS), Proteus syndrome (PS), and Proteus-like syndrome.
Cowden syndrome is the paradigmatic PTEN-related disorder, and a genodermatosis characterized by multiple hamartomas, neoplasms of ectodermal, mesodermal and endodermal origin, as well as mucosal papillomatosis, and skin lesions, trichilemmomas and with a high risk for benign and malignant tumors of the thyroid, breast, and endometrium. Trichilemmomas and mucocutaneous papillomatous papules are one of the first signs of the disease [13]. A trichilemmoma is rarely a sporadic finding, and the literature regularly reports numerous lesions at presentation in individuals with a clinical and/or genetic diagnosis. Consequently, trichilemmoma is a clinically significant sign of CS when at least ≥ 3 are present. The most distinctive and peculiar facial features of CS consist of multiple small and keratotic papules concentrated around the skin-mucosa interface of the mouth, nose and eye.
PTEN immunohistochemistry (IHC) in trichilemmomas may be helpful in screening these tumors for association with CS. The loss of PTEN within the lesional cells and the maintenance of PTEN immunostaining by the endothelial cells is characteristic of this syndrome.
Early recognition of these skin lesions may help with diagnosing an underlying malignancy and early cancer screening [13]. Cutaneous findings that appear in childhood may be the first sign of a hereditary tumor syndrome. Early detection of genodermatoses allows the patient and at-risk family members to be screened for associated malignancies.
BRRS is a characterized by macrocephaly, intestinal hamartomatous polyposis, multiple thyroid nodules and tumors, lipomas, and pigmented macules of the glans penis.
Thyroid pathologic findings in patients with PHTS include multinodular goiter, multiple adenomatous nodules, follicular adenoma, follicular carcinoma, and less frequently papillary carcinoma. According to the diagnostic criteria for CS, follicular carcinoma is a major criterion, and multinodular goiter, adenomatous nodules, and follicular adenomas are minor criteria, with a frequency of 50–67% [14].
Immunohistochemical staining for the assessment of PTEN expression levels supply additional support to the diagnosis of CS (Fig. 3). Loss of PTEN expression in thyroid nodules by IHC, whether in all nodules or in a subset of nodules, is both sensitive and specific for CS/PHTS. Loss of PTEN IHC staining occurs in isolated thyroid nodules, either some of the adenomatous nodules, follicular adenomas, nodular hyperplasia and/or follicular carcinoma, related to PTEN gene mutations/deletions. Background thyroid follicles as well as the endothelial cells retain PTEN. PTEN IHC serves as a screening molecular correlate to predict for germline PTEN mutation. Recognition of this syndrome is important so that cancer screening and genetic counseling can be initiated.
Fig. 3.
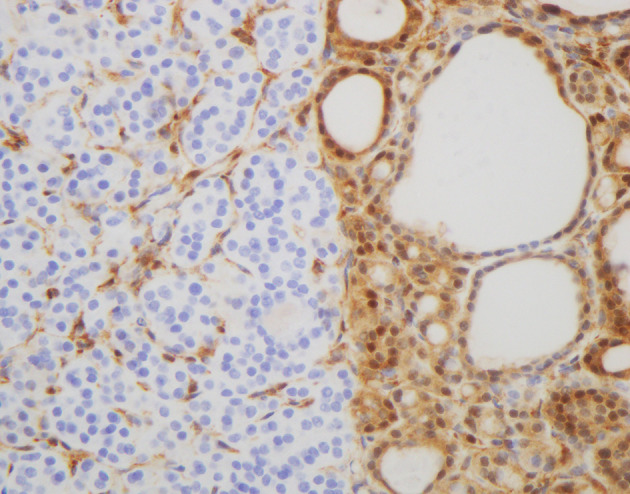
Adenomatous nodule in Cowden syndrome (PHTS): Cowden syndrome is the principal PTEN-related disorder, which are genodermatosis characterized by multiple mucosal papillomatosis, and facial trichilemmomas and with a high risk for benign and malignant tumors of the thyroid, breast, and endometrium. PTEN immunostaining showing complete loss of immunoreactivity for PTEN of the follicular cells in one nodule. The endothelial cells and the adjacent thyroid follicular cells are positive
Immunohistochemistry for PTEN should be used in context with other defining clinical criteria, and also including family history, genetic counseling, and possibly germline PTEN genotyping to confirm a diagnosis of CS.
The revised diagnostic criteria for PTEN hamartoma tumor syndrome/CS are described in Table 4.
Table 4.
Revised diagnostic criteria for PTEN hamartoma tumor syndrome/Cowden syndrome
| Major criteria |
| Breast cancer |
| Endometrial cancer (epithelial) |
| Thyroid cancer (follicular) |
| Gastrointestinal hamartomas (including ganglioneuromas, but excluding hyperplastic polyps; > 3) Lhermitte–Duclos disease (adult) |
| Macrocephaly (> 97 percentile: 58 cm for females, 60 cm for males) |
| Macular pigmentation of the glans penis |
| Multiple mucocutaneous lesions (any of the following) |
| Multiple trichilemmomas (> 3, at least one biopsy proven) |
| Acral keratoses (> 3 palmoplantar keratotic pits and/or acral hyperkeratotic papules) |
| Mucocutaneous neuromas (> 3) |
| Oral papillomas (particularly on tongue and gingiva), multiple (> 3) OR biopsy proven OR dermatologist diagnosed |
| Minor criteria |
| Autism spectrum disorder |
| Colon cancer |
| Esophageal glycogenic acanthosis (> 3) |
| Lipomas (> 3) |
| Mental retardation (ie, IQ < 75) |
| Renal cell carcinoma |
| Testicular lipomatosis |
| Thyroid cancer (papillary or follicular variant of papillary) |
| Thyroid structural lesions (eg, adenoma, multinodular goiter) |
| Vascular anomalies (including multiple intracranial developmental venous anomalies) |
Adapted from Pilarski et al. [49]
Operational diagnosis in an individual (either of the following) Three or more major criteria, but one must include macrocephaly, Lhermitte–Duclos disease, or gastrointestinal hamartomas; Two major and three minor criteria
Operational diagnosis in a family where one individual meets revised PTEN hamartoma tumor syndrome clinical diagnostic criteria or has a PTEN mutation Any two major criteria with or without minor criteria; One major and two minor criteria; Three minor criteria
Von Hippel Lindau Syndrome
Vin-Hippel Lindau syndrome (VHLS) is an autosomal dominant tumor syndrome caused by inactivating mutations in the VHL gene that inactivate the VHL protein.
The most important head and neck tumor is the endolymphatic sac tumours (ELST). The middle ear ELST is a well-recognized tumour associated with von Hippel Lindau syndrome in over a third of the cases. Data from the “International ELST Registry” [15] show a prevalence of ELST of 3.6% in VHL. VHL germline mutations were present in almost 40% of ELSTs, with an apparent sporadic presentation. These findings support genetic testing in all patients with ELSTs [15–18] and all VHLS patients should be radiographically screened for ELSTs.
Central nervous system hemangioblastomas occurs in about 80% of patients [19, 20]. Ocular manifestations are expected in roughly half of VHL patients. Retinal hemangioblastomas and retinal capillary hemangioblastomas are often the initial manifestation of the disease [21, 22], are often multiple and are bilateral in about 50% [23].
Other manifestations include pheochromocytomas, paragangliomas, kidney cysts and renal cell carcinomas, papillary cystadenoma of epididymis, and neuroendocrine tumors mostly in the pancreatoduodenal area.
The ELST are papillary and/or cystic with a single layer of low cuboidal to columnar cells are arranged in simple, interdigitating papillary projections with fibrovascular cores (Fig. 4). The acinar spaces filled with inspissated thyroid-colloid-like material. The cytoplasm is ample, clear, vacuolated with indistinct cell membranes. The nuclei are consistently small, round, and hyperchromatic with coarse nuclear chromatin and small nucleoli. [24–30]. Pleomorphism, increased mitotic figures, and necrosis are inconspicuous.
Fig. 4.
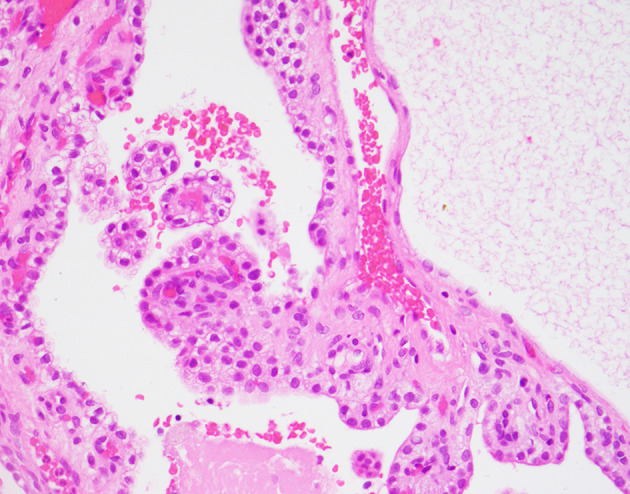
Ear Endolymphatic sac tumour in VHL syndrome: The middle ear endolymphatic sac tumour is a well-recognized tumour associated with von Hippel Lindau syndrome in over a third of the cases and it is the initial manifestation of VHL in > 30% of individuals with these lesions. The neoplastic cells show a low cuboidal to columnar cell appearance, lining papillary projections and dilated cystic spaces. Secretory colloid-like material is frequently seen in cystic spaces
Immunohistochemistry shows that the lesional cells are positive for cytokeratin 7, HIF-alpha, EMA, GLUT1, PAX8 and CAIX.
Hemangioblastomas are circumscribed and may be associated with a pseudocyst. These are composed of proliferations of abundant mature vasculature thought to be reactive and the neoplastic stromal cell component of hemangioblast progenitor cells. IHC of hemangioblastoma is particular and thus reviewed here. The neoplastic cells are positive for pancytokeratin, CK7, HIF-1α, EMA, GLUT1, CAIX (membranous), and PAX-8 (nuclear), with limited S100 protein, GFAP, and vimentin, and negative for TTF-1, thyroglobulin, PSA, CD10, P504S, p63, synaptophysin, GATA3, and RCC [26, 27, 30].
Head and Neck Paragangliomas
Paragangliomas have a hereditary predisposition in about 40% of affected patients. Parasympathetic-derived paragangliomas usually arise in the head-and-neck, as carotid body and along cervical branches of the glossopharyngeal and vagus nerves. Most familial paragangliomas are multifocal.
Succinate dehydrogenase (SDH)-deficient tumor syndromes are the most common paragangliomas in the head and neck region. SDH-deficient tumour syndromes are a group of syndromes characterized by SDH deficient neoplasia, usually associated with pathogenic germline variants in one of the SDH genes. The paragangliomas within these syndromes are divided in: PGL1 (germline SDHD mutation), PGL2 (germline SDHAF2 mutation), PGL3 (germline SDHC mutation), PGL4 (germline SDHB mutation), PGL5 (germline SDHA mutation), and Carney triad (restricted to syndromic, but non-hereditary disease). The SDH genes encode the subunits of succinate dehydrogenase (SDHA, SDHB, SDHC, SDHD, SDHAF2) or with epimutation (promoter hypermethylation) of the SDHC gene.
The most common germline mutation in patients with head and neck paragangliomas is in SDHD (~ 50%), followed by SDHB (~ 30%), and SDHC (~ 16%) [31]. SDHD mutation is associated predominantly with single or multifocal tumours in the head and neck, whereas SDHB mutation is more often associated with thoracoabdominal tumors. Paragangliomas in the head and neck are rarely associated with mutations of VHL, RET, or NF1[17].
Some morphological features may be suggestive of genetic predisposition, although the morphology of most familial paragangliomas overlaps that of sporadic paragangliomas. SDHx-related paragangliomas from the head and neck usually have small cells with clear cytoplasm. Unlike sporadic paragangliomas, SDHx-associated tumours are rarely associated with a spindled morphology or densely granular cytoplasm. VHL-associated tumors may have clear cells with vacuolated cytoplasm and may show edema in the stroma. They express markers of neuroendocrine differentiation including insulinoma associated protein1 (INSM1), synaptophysin, and chromogranin A, as well as nuclear GATA3 and cytoplasmic catecholamine-synthesizing enzymes, tyrosine hydroxylase and dopamine β-hydroxylase [32, 33] (Fig. 5).
Fig. 5.

Middle ear paraganglioma: SDHx-related paragangliomas from the head and neck usually have small cells with clear cytoplasm (a). The tumor cells express markers of neuroendocrine differentiation, including insulinoma associated protein1/INSM1 (b), synaptophysin, and chromogranin A, as well as nuclear GATA3 and cytoplasmic catecholamine-synthesizing enzymes, tyrosine hydroxylase and dopamine β-hydroxylase. SDHB-immunohistochemistry is negative in tumour cells of SDHx-related tumors (c), and only capillary endothelial cells are positive as internal controls. On the other hand, tumour cells in paragangliomas with non-SDH-mutation demonstrate numerous immunoreactive granules in the cytoplasm
Alpha-inhibin is expressed in pseudohypoxic tumours (including SDHx-related and VHL-related types) and CAIX is expressed strongly in VHL-associated tumours [34, 35]. Paragangliomas express somatostatin receptors, and SDH-deficient paragangliomas have been reported to demonstrate stronger staining for SSTR2A and SSTR3 [36].
The new WHO 5th edition book recommends that, in view of the strongly syndromic nature of SDH-deficient neoplasia, surveillance and follow up for other SDH-deficient neoplasms is indicated even in cases where no germline mutation has been identified [37]. The main putative risk factor for metastasis is hereditary mutation of SDHB [38]. Familial cases require lifelong surveillance to evaluate for multicentric disease.
Familial Multicentric HNSCC
Li-Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS) is an autosomal dominant cancer predisposition syndrome caused by germline, broad spectrum of mutations, involving the coding regions of the TP53 gene.
Li-Fraumeni Syndrome is mostly associated with cancers of the breast, adrenocortical carcinoma, soft tissue, bone, brain, and female genital tract. Although cancers of the head and neck region are uncommon, the diagnosis of multicentric HNSCC particularly, occurring in the larynx, the pharynx and the oral cavity can be seen in LFS [39, 40]. Various sarcomas are present as head and neck malignancies as rhabdomyosarcoma and mandibular osteosarcoma, synovial cell sarcoma and pleomorphic myxoid liposarcoma seen in LFS pediatric patients.
Li-Fraumeni Syndrome is characterized by the early onset of a broad spectrum of cancers as breast and adrenocortical carcinomas, brain tumors, leukemia, and soft tissue and bone sarcomas and have an overall high lifetime cancer risk, while this syndrome has many tumor associations, this chapter is focused on head and neck tumours.
Fanconi Anemia
Fanconi anemia (FA) is the most common hereditary cancer syndrome, and is a heterogeneous genetic disorder resulting from mutations of one of the 22 genes involved in DNA repair and maintenance of genomic stability.
The most common solid cancer in FA patients is also HNSCC. Compared to the general population, risk of HNSCC in FA is increased by 500- to 800-fold and the diagnosis of HNSCC often precedes the diagnosis of FA [41]. FA-HNSCC frequently occur as multifocal disease, preceded by oral potentially malignant disorders presenting as lichenoid keratosis or leukoplakia.
Other Syndromes Involving the Head and Neck Region
Familial Adenomatous Polyposis with Gardner Syndrome Variant
Patients with a familial adenomatous polyposis due to germline APC mutations, which is characterized by over 100 adenomatous colorectal polyps, may present also multiple extracolonic manifestations, as Gardner syndrome variant. Desmoid tumors may arise in soft tissue of the head and neck, but may also originate in the maxillary sinus, nasopharynx, and oral cavity [42]. Other pathologies involving the head and neck are osteomas. Multiple osteomas occur in 50–90% patients. They are benign neoplasms mostly present in the craniofacial bones, most commonly the mandible [43, 44]. Supernumerary teeth and multifocal odontomas are also found in this syndrome. The thyroid carcinomas are usually multifocal and bilateral in this syndrome and are of a specific type: cribriform morular thyroid carcinoma [45], which is specific for this syndrome.
Hyperparathyroidism-Jaw Tumor Syndrome
Hyperparathyroidism-jaw tumor (HPT-JT) syndrome is characterized by parathyroid tumors in association with fibro-osseous jaw tumors and uterine and renal lesions. HPT-JT syndrome is autosomal dominant disorder caused by germline mutations of the cell division cycle 73 (CDC73) gene that encodes the parafibromin. Primary hyperparathyroidism is the main finding of HPT-JT syndrome, usually caused by a single-gland parathyroid involvement, different from other variants of hereditary hyperparathyroidism, in which a multiglandular involvement is more frequent and parathyroid carcinoma is more frequently seen in this syndrome. Clues to the diagnosis include younger age of onset, large tumours with associated markedly elevated serum PTH levels and severe hypercalcemia.
The jaw tumours arise more often in the mandible than the maxilla with about 15% being multiple cemento-ossifying fibroma. These tumors may present only in a minority of cases [46]. Clinically important disease outside the head and neck is infrequent.
Tuberous Sclerosis
Manifestations of tuberous sclerosis in the head and neck are predominantly cutaneous and oral. Angiofibroma occurs in the face characteristically arising in the centrofacial cheek areas, nasolabial folds and chin. Oral manifestations are frequent and specifically include angiofibromas in the anterior gingiva, lips, tongue, and palate and dental enamel pits loss on the incisal border and tooth wear are also observed [47].
Neurofibromatosis Type 1
Neurofibromatosis type 1 is recognized in patients with orbitofacial neurofibromas and numerous neurofibromas at other sites. Multiple café-au-lait macules are frequently identified at birth, plexiform neurofibromas are considered as congenital, and cutaneous neurofibromas are identified at puberty. Orbitofacial NFs is often characterized by progressive, large disfiguring tumors involving orbit and periorbital tissues and associated with remodeling of adjacent skull bones. Severe scoliosis, sphenoid wing dysplasia, non-ossifying fibromas, and congenital tibial bowing are known bone findings in this syndrome. Some neoplasms are more frequently found in NF1 as optic pathway glioma/pilocytic astrocytoma and other gliomas, rhabdomyosarcoma, glomus tumours, gastrointestinal stromal tumours, pheochromocytoma, breast cancer in females, and duodenal somatostatinomas. This syndrome is described in the head and neck book focusing on the head and neck pathology findings.
Other important syndromes with involvement of the head and neck, as with oral mucosa leukoplakia (dyskeratosis congenita), and oropharyngeal carcinoma (Bloom syndrome) are described in detail in the WHO 5th edition Head and Neck.
Conclusions
Pathologists have a very important role in recognizing tumours of the head and neck tumours associated with the heritable syndromes addressed in this section. Pathologists must be aware of the morphologic features associated with distinct syndromes that should prompt consideration of germline screening. The routine use of NGS will undoubtedly continue to widen the spectrum of known predisposing genetic variants among patients with tumours of the head and neck. The identification of syndromes has made possible identification of at-risk individuals in affected families, who can then be offered genetic counseling. Information on underlying genetic variants can also have an important impact on the choice of therapy and surveillance guidelines. Consequently, the recognition of specific germline susceptibility syndromes can assist in providing information to facilitate early detection to prevent aggressive disease.
Virtually all of these disorders have now been characterized at the molecular level; patients with these disorders may also require distinct forms of therapy and personalized clinical follow-up.
Funding
No external funding was obtained for this study.
Declarations
Conflict of interest
All authors have contributed to this work and both authors declare they have no conflict of interest as it relates to this study.
Ethical Approval
All evaluations performed in this analysis do not involve any individual patient’s data, but was still performed in accordance with the ethical standards of the institutional review board. The opinions or assertions contained herein are the private views of the authors.
Consent Statement
No personally identifiable information is included and thus informed consent is not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Howell JB, Caro MR. The basal-cell nevus: its relationship to multiple cutaneous cancers and associated anomalies of development. AMA Arch Dermatol. 1959;79(1):67–77. doi: 10.1001/archderm.1959.01560130069008. [DOI] [PubMed] [Google Scholar]
- 2.Gorlin RJ, Goltz RW. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med. 1960;262:908–912. doi: 10.1056/NEJM196005052621803. [DOI] [PubMed] [Google Scholar]
- 3.Evans DG, et al. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30(6):460–464. doi: 10.1136/jmg.30.6.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bresler SC, Padwa BL, Granter SR. Nevoid basal cell carcinoma syndrome (Gorlin Syndrome) Head Neck Pathol. 2016;10(2):119–124. doi: 10.1007/s12105-016-0706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stojanov IJ, et al. Biallelic PTCH1 inactivation is a dominant genomic change in sporadic keratocystic odontogenic tumors. Am J Surg Pathol. 2020;44(4):553–560. doi: 10.1097/PAS.0000000000001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foulkes WD, et al. Cancer surveillance in Gorlin syndrome and Rhabdoid tumor predisposition syndrome. Clin Cancer Res. 2017;23(12):e62–e67. doi: 10.1158/1078-0432.CCR-17-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karhade DS, Afshar S, Padwa BL. What is the prevalence of undiagnosed nevoid basal cell carcinoma syndrome in children with an odontogenic keratocyst? J Oral Maxillofac Surg. 2019;77(7):1389–1391. doi: 10.1016/j.joms.2019.01.045. [DOI] [PubMed] [Google Scholar]
- 8.Li TJ. The odontogenic keratocyst: a cyst, or a cystic neoplasm? J Dent Res. 2011;90(2):133–142. doi: 10.1177/0022034510379016. [DOI] [PubMed] [Google Scholar]
- 9.Qu J, et al. PTCH1 alterations are frequent but other genetic alterations are rare in sporadic odontogenic keratocysts. Oral Dis. 2019;25(6):1600–1607. doi: 10.1111/odi.13135. [DOI] [PubMed] [Google Scholar]
- 10.Robinson RA. Basal cell adenoma and basal cell adenocarcinoma. Surg Pathol Clin. 2021;14(1):25–42. doi: 10.1016/j.path.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Rito M, et al. Frequent and differential mutations of the CYLD gene in basal cell salivary neoplasms: linkage to tumor development and progression. Mod Pathol. 2018;31(7):1064–1072. doi: 10.1038/s41379-018-0018-6. [DOI] [PubMed] [Google Scholar]
- 12.Nagao T, et al. Carcinoma in basal cell adenoma of the parotid gland. Pathol Res Pract. 1997;193(3):171–178. doi: 10.1016/S0344-0338(97)80074-X. [DOI] [PubMed] [Google Scholar]
- 13.Nose V. Genodermatosis affecting the skin and mucosa of the head and neck: clinicopathologic, genetic, and molecular aspect–PTEN-hamartoma tumor syndrome/cowden syndrome. Head Neck Pathol. 2016;10(2):131–138. doi: 10.1007/s12105-016-0708-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laury AR, et al. Thyroid pathology in PTEN-hamartoma tumor syndrome: characteristic findings of a distinct entity. Thyroid. 2011;21(2):135–144. doi: 10.1089/thy.2010.0226. [DOI] [PubMed] [Google Scholar]
- 15.Bausch B, et al. Characterization of endolymphatic sac tumors and von Hippel-Lindau disease in the International Endolymphatic Sac Tumor Registry. Head Neck. 2016;38(Suppl 1):E673–E679. doi: 10.1002/hed.24067. [DOI] [PubMed] [Google Scholar]
- 16.Codreanu CM, et al. Endolymphatic sac tumors in von Hippel-Lindau disease: report of three cases. Otol Neurotol. 2010;31(4):660–664. doi: 10.1097/MAO.0b013e3181d8d863. [DOI] [PubMed] [Google Scholar]
- 17.Codreanu C, Tran Ba Huy P. Isolate vertigo crisis revealing an endolymphatic sac tumor. Rom J Morphol Embryol. 2010;51(2):387–389. [PubMed] [Google Scholar]
- 18.Wick CC, et al. Endolymphatic sac tumors. Otolaryngol Clin North Am. 2015;48(2):317–330. doi: 10.1016/j.otc.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Vortmeyer AO, et al. Nervous system involvement in von Hippel-Lindau disease: pathology and mechanisms. Acta Neuropathol. 2013;125(3):333–350. doi: 10.1007/s00401-013-1091-z. [DOI] [PubMed] [Google Scholar]
- 20.Wanebo JE, et al. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):82–94. doi: 10.3171/jns.2003.98.1.0082. [DOI] [PubMed] [Google Scholar]
- 21.Binderup MLM, et al. Retinal hemangioblastoma: prevalence, incidence and frequency of underlying von Hippel-Lindau disease. Br J Ophthalmol. 2018;102(7):942–947. doi: 10.1136/bjophthalmol-2017-310884. [DOI] [PubMed] [Google Scholar]
- 22.Ruppert MD, et al. Ocular manifestations of von Hippel-Lindau disease. Cureus. 2019;11(8):e5319. doi: 10.7759/cureus.5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reich M, et al. Genotype-phenotype correlation in von Hippel-Lindau disease. Acta Ophthalmol. 2021 doi: 10.1111/aos.14843. [DOI] [PubMed] [Google Scholar]
- 24.Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases. Cancer. 1989;64(11):2292–2302. doi: 10.1002/1097-0142(19891201)64:11<2292::aid-cncr2820641119>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 25.el-Naggar AK, et al. Tumors of the middle ear and endolymphatic sac. Pathol Annu. 1994;29(Pt 2):199–231. [PubMed] [Google Scholar]
- 26.Bisceglia M, D'Angelo VA, Wenig BM. Endolymphatic sac papillary tumor (Heffner tumor) Adv Anat Pathol. 2006;13(3):131–138. doi: 10.1097/00125480-200605000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Skalova A, et al. Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): report of two cases with analysis of the VHL gene. Pathol Res Pract. 2008;204(8):599–606. doi: 10.1016/j.prp.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 28.Michaels L. Origin of endolymphatic sac tumor. Head Neck Pathol. 2007;1(2):104–111. doi: 10.1007/s12105-007-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell D, et al. Endolymphatic sac tumor (aggressive papillary tumor of middle ear and temporal bone): sine qua non radiology-pathology and the University of Texas MD Anderson Cancer Center experience. Ann Diagn Pathol. 2011;15(2):117–123. doi: 10.1016/j.anndiagpath.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Thompson LDR, et al. CAIX and pax-8 commonly immunoreactive in endolymphatic sac tumors: a clinicopathologic study of 26 cases with differential considerations for metastatic renal cell carcinoma in von hippel-lindau patients. Head Neck Pathol. 2019;13(3):355–363. doi: 10.1007/s12105-018-0973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith JD, et al. Head and neck paragangliomas: a two-decade institutional experience and algorithm for management. Laryngoscope Investig Otolaryngol. 2017;2(6):380–389. doi: 10.1002/lio2.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kriegsmann K, et al. Insulinoma-associated protein 1 (INSM1) in thoracic tumors is less sensitive but more specific compared with synaptophysin, chromogranin A, and CD56. Appl Immunohistochem Mol Morphol. 2020;28(3):237–242. doi: 10.1097/PAI.0000000000000715. [DOI] [PubMed] [Google Scholar]
- 33.Juhlin CC, Zedenius J, Hoog A. Clinical routine application of the second-generation neuroendocrine markers ISL1, INSM1, and secretagogin in neuroendocrine neoplasia: staining outcomes and potential clues for determining tumor origin. Endocr Pathol. 2020;31(4):401–410. doi: 10.1007/s12022-020-09645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinato DJ, et al. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br J Cancer. 2013;108(2):429–437. doi: 10.1038/bjc.2012.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Favier J, et al. Carbonic anhydrase 9 immunohistochemistry as a tool to predict or validate germline and somatic VHL mutations in pheochromocytoma and paraganglioma-a retrospective and prospective study. Mod Pathol. 2020;33(1):57–64. doi: 10.1038/s41379-019-0343-4. [DOI] [PubMed] [Google Scholar]
- 36.Kimura N, et al. Familial cervical paragangliomas with lymph node metastasis expressing somatostatin receptor type 2A. Endocr Pathol. 2010;21(2):139–143. doi: 10.1007/s12022-009-9098-7. [DOI] [PubMed] [Google Scholar]
- 37.Amar L, et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol. 2021;17(7):435–444. doi: 10.1038/s41574-021-00492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCrary HC, et al. Characterization of malignant head and neck paragangliomas at a single institution across multiple decades. JAMA Otolaryngol Head Neck Surg. 2019;145(7):641–646. doi: 10.1001/jamaoto.2019.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bouaoun L, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat. 2016;37(9):865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- 40.Prime SS, et al. A review of inherited cancer syndromes and their relevance to oral squamous cell carcinoma. Oral Oncol. 2001;37(1):1–16. doi: 10.1016/s1368-8375(00)00055-5. [DOI] [PubMed] [Google Scholar]
- 41.Scheckenbach K, et al. Squamous cell carcinomas of the head and neck in Fanconi anemia: risk, prevention, therapy, and the need for guidelines. Klin Padiatr. 2012;224(3):132–138. doi: 10.1055/s-0032-1308989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saito Y, et al. Risk factors for the development of desmoid tumor after colectomy in patients with familial adenomatous polyposis: multicenter retrospective cohort study in Japan. Ann Surg Oncol. 2016;23(Suppl 4):559–565. doi: 10.1245/s10434-016-5380-3. [DOI] [PubMed] [Google Scholar]
- 43.Lee BD, et al. A case report of Gardner syndrome with hereditary widespread osteomatous jaw lesions. Oral Surg Oral Med Oral Pathol Oral Radiol Endodontol. 2009;107(3):e68–72. doi: 10.1016/j.tripleo.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 44.Nah KS. Osteomas of the craniofacial region. Imaging Sci Dent. 2011;41(3):107–113. doi: 10.5624/isd.2011.41.3.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyraz B, et al. Cribriform-morular thyroid carcinoma is a distinct thyroid malignancy of uncertain cytogenesis. Endocr Pathol. 2021;32(3):327–335. doi: 10.1007/s12022-021-09683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carpenter TO, et al. Case 32–2021: a 14-year-old girl with swelling of the jaw and hypercalcemia. N Engl J Med. 2021;385(17):1604–1613. doi: 10.1056/NEJMcpc2107351. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen QD, DarConte MD, Hebert AA. The cutaneous manifestations of tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):321–325. doi: 10.1002/ajmg.c.31649. [DOI] [PubMed] [Google Scholar]
- 48.Bree AF, Shah MR, for the BCNS Colloquium Group Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS) Am J Med Genet Part A. 2011;155:2091–2097. doi: 10.1002/ajmg.a.34128. [DOI] [PubMed] [Google Scholar]
- 49.Pilarski R, Burt R, Kohlman W, et al. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–1616. doi: 10.1093/jnci/djt277. [DOI] [PubMed] [Google Scholar]