

Abstract
The 5th edition of the World Health Organization (WHO) Classification of Head and Neck Tumours (2022) comes out only five years after the previous edition, however it presents important updates that run in parallel with the rapid progression involving the increasingly sophisticated molecular investigation and its interpretation, some of which already have therapy-related impact. This manuscript provides an overview of the leading changes introduced in the classification of Odontogenic and Maxillofacial Bone Tumours that encompasses cysts of the jaws, odontogenic tumours, giant cell lesions and bone cysts, and bone and cartilage tumours. This is the first edition that Essential and Desirable Diagnostic Features were added for each entity, so that the most important clinical, microscopic and/or radiologic features were encapsulated and briefly highlighted. Surgical ciliated cyst was added to the group of odontogenic cysts, adenoid ameloblastoma was a newly recognized benign epithelial odontogenic tumour, and segmental odontomaxillary dysplasia was introduced in the group of fibro-osseous tumours and dysplasia. In addition, rhabdomyosarcoma with TFCP2 rearrangement, was introduced into the group of malignant jawbone tumours. The unique genetic aberrations distinguish it from other types of rhabdomyosarcomas. On the other hand, melanotic neuroectodermal tumour of infancy and osteoid osteoma were deleted from the benign bone and cartilageneous tumours, as was the hematolymphoid tumour of solitary plasmacytoma of bone. We systematically reviewed each entity in this chapter and provided important updated findings for selected topics that can further aid in the diagnostic process for challenging cases, broaden insights on the logic of the present classification, and finally, emphasize the potential that some of the molecular results may have in the near future to set new treatment approaches.
Keywords: Update, WHO Classification, Odontogenic cysts, Odontogenic tumours, Bone and cartilage tumours, Surgical ciliated cyst, Adenoid ameloblastoma, Segmental odontomaxillary dysplasia, Rhabdomyosarcoma with TFCP2 rearrangement
Introduction
The 2022 World Health Organization (WHO) Classification of Odontogenic and Maxillofacial Bone Tumours (5th edition) [1] comes out only five years after its predecessor (4th edition, 2017) [2], while it took over a decade to update the edition published at the beginning of the twenty-first century (3rd edition, 2005) [3]. It is the fast pace of advanced and progressively changing molecular technology and its potential clinical relevance that was a major impetus for the WHO to reduce the time interval between new editions. Some of the ensuing novel molecular findings may have clinical application and can set the stage to the beginning of a new era of treatment approaches, different from the hitherto commonly accepted modalities.
There is little conceptually different between the new and latest editions but the new edition contains significant reorganization. There seems to be an effort to provide consensus definitions and more clearly articulated diagnostic features. In addition to the standard description of microscopic findings, every lesion contains Essential as well as Desirable Diagnostic Features. Despite the plethora of new molecular findings, only one new entity is defined in the 2022 edition by its molecular findings, namely a new rhabdomyosarcoma of bone with TFCP2 rearrangement and a predilection for the jaws [1]. While the molecular findings in odontogenic cysts and tumours play a significant role in pathogenesis, at least for now, none are defining characteristics. Other notable changes included the addition of surgical ciliated odontogenic cyst, introducing a newly recognized benign epithelial odontogenic tumour – adenoid ameloblastoma, and adding segmental odontomaxillary dysplasia to the fibro-osseous tumours and dysplasia group of lesions. On the other hand, melanotic neurectodermal tumour of infancy and osteoid osteoma were deleted from the benign bone and cartilaginous tumours as was also the hematolymphoid tumour of solitary plasmacytoma of bone.
The following paragraphs highlight the main changes introduced in selected entities in the 2022 WHO classification of Odontogenic and Maxillofacial Bone Tumours. Table 1 presents odontogenic and maxillofacial bone tumours 2022 edition versus 2017.
Table 1.
WHO Classification of Odontogenic and maxillofacial bone tumours, 2022 versus 2017. Main headings are arranged in the order of the 2022 classification; within sub-headings, lesions are presented in the original order of each classification
| 2022 Classification | 2017 Classification* | |
|---|---|---|
| Cysts of the jaws | Odontogenic cysts of inflammatory origin | Odontogenic & non-odontogenic developmental cysts |
| Radicular cyst | Radicular cyst | Dentigerous cyst |
| Inflammatory collateral cysts | Inflammatory collateral cysts | Odontogenic keratocyst |
| Post-surgical ciliated cyst | Lat. periodontal cyst and botryoid cyst | |
| Nasopalatine duct cyst | Gingival cyst | |
| Gingival cyst | Glandular odontogenic cyst | |
| Dentigerous cyst | Calcifying odontogenic cyst | |
| Orthokeratinized odontogenic cyst | Orthokeratinized odontogenic cyst | |
| Lat. periodontal cyst and botryoid cyst | Nasopalatine duct cyst | |
| Calcifying odontogenic cyst | ||
| Glandular odontogenic cyst | ||
| Odontogenic keratocyst | ||
| Odontogenic Tumours | Odontogenic Tumours | |
| Benign epithelial odontogenic tumours | Benign epithelial odontogenic tumours | |
| Adenomatoid odontogenic tumour | Ameloblastoma | |
| Squamous odontogenic tumour | -Ameloblastoma, unicystic type | |
| Calcifying epithelial odontogenic tumour | -Ameloblastoma, extraosseous/peripheral type | |
| Ameloblastoma, extraosseous/peripheral | -Metastasizing ameloblastoma | |
| Ameloblastoma, unicystic | Squamous odontogenic tumour | |
| Ameloblastoma, conventional | Calcifying epithelial odontogenic tumour | |
| Adenoid ameloblastoma | Adenomatoid odontogenic tumour | |
| Metastasizing ameloblastoma | ||
| Benign mixed epithelial and mesenchymal odontogenic tumours | Benign mixed epithelial and mesenchymal odontogenic tumours | |
| Odontoma | Ameloblastic fibroma | |
| Primordial odontogenic tumour | Primordial odontogenic tumour | |
| Ameloblastic fibroma | Odontoma | |
| Dentinogenic ghost tumour | -Odontoma, compound type | |
| -Odontoma, complex type | ||
| Dentinogenic ghost tumour | ||
| Benign mesenchymal odontogenic tumours | Benign mesenchymal odontogenic tumours | |
| Odontogenic fibroma | Odontogenic fibroma | |
| Cementoblastoma | Odontogenic myxoma/myxofibroma | |
| Cemento-ossifying fibroma | Cementoblastoma | |
| Odontogenic myxoma | Cemento-ossifying fibroma (discussed under the heading of Fibro-osseous and osteochondromatous lesions) | |
| Malignant odontogenic tumours | Malignant odontogenic tumours | |
| Sclerosing odontogenic carcinoma | Odontogenic carcinomas | |
| Ameloblastic carcinoma | - Ameloblastic carcinoma | |
| Clear cell odontogenic carcinoma | - Primary intraosseous carcinoma, NOS | |
| Ghost cell odontogenic carcinoma | - Sclerosing odontogenic carcinoma | |
| Primary intraosseous carcinoma, NOS | -Clear cell odontogenic carcinoma | |
| Odontogenic carcinosarcoma | -Ghost cell odontogenic carcinoma | |
| Odontogenic sarcomas | Odontogenic carcinosarcoma | |
| Odontogenic sarcomas | ||
| Giant cell lesions and bone cysts | Giant cell lesions and bone cysts | |
| Central giant cell granuloma | Central giant cell granuloma | |
| Peripheral giant cell granuloma | Peripheral giant cell granuloma | |
| Cherubism | Cherubism | |
| Aneurysmal bone cyst | Aneurysmal bone cyst | |
| Simple bone cyst | Simple bone cyst | |
| Bone and cartilage tumours | Fibro-osseous and osteochondromatous lesions | |
| Fibro-osseous tumours and dysplasias | ||
| Cemento-ossifying dysplasia | Ossifying fibroma | |
| Segmental odontomaxillary dysplasia | Familial gigantiform cementoma | |
| Fibrous dysplasia | Fibrous dysplasia | |
| Juvenile trabecular ossifying fibroma | Cemento-ossifying dysplasia | |
| Psammomatoid ossifying fibroma | Osteochondroma | |
| Familial gigantiform cementoma | ||
| Benign maxillofacial bone and cartilage tumours | Benign maxillofacial bone and cartilage tumours | |
| Osteoma | Chondroma | |
| Osteochondroma | Osteoma | |
| Osteoblastoma | Melanotic neuroectodermal tumour of infancy | |
| Chondroblastoma | Chondroblastoma | |
| Chondromyxoid fibroma | Chondromyxoid fibroma | |
| Desmoplastic fibroma of bone | Osteoid osteoma | |
| Osteoblastoma | ||
| Desmoplastic fibroma | ||
| Malignant maxillofacial bone and cartilage tumours |
Malignant maxillofacial bone and cartilage tumours |
|
| Osteosarcoma of the jaw | Chondrosarcoma | |
| Chondrosarcoma family | -Chondrosarcoma, grade 1 | |
| Mesenchymal chondrosarcoma | -Chondrosarcoma, grade 2/3 | |
| Rhabdomyosarcoma with TFCP2 rearrangement | Mesenchymal chondrosarcoma | |
| Osteosarcoma, NOS | ||
| -Low-grade central osteosarcoma | ||
| -Chondroblastic osteosarcoma | ||
| -Parosteal osteosarcoma | ||
| -Periosteal osteosarcoma | ||
| Haematolymphoid tumours | ||
| Solitary plasmacytoma of bone | ||
*Original order of the main classes of lesions: Odontogenic carcinomas; Odontogenic carcinosarcoma; Odontogenic sarcomas; Benign epithelial odontogenic tumours; Benign mixed epithelial and mesenchymal odontogenic tumours; Benign mesenchymal odontogenic tumours; Odontogenic cysts of inflammatory origin; Odontogenic and non-odontogenic developmental cysts; Malignant maxillofacial bone and cartilage tumours; Benign maxillofacial bone and cartilage tumours; Fibro-osseous and osteochondromatous lesions; Giant cell lesions and bone cysts; Haematolymphoid tumours
Cysts of the Jaws
The post-surgical ciliated cyst, while not new, is new to the classification [1]. It is a rare cyst caused by the traumatic implantation of respiratory epithelium into the gnathic bones, most commonly diagnosed in the 5th-6th decades. It is entrapment of maxillary sinus or nasal mucosae that serves as the origin for the cysts and constitute the microscopic hallmark finding. The relatively few mandibular cases are assumedly caused during autologous nasal osteocartilagenous grafts for genioplasty or examples of other simultaneous maxillary and mandibular orthognathic surgical procedures [4] (Fig. 1). There is usually a long interval of up to 20 years between the causal surgery until diagnosis, although a considerable shorter interval has been reported in association with sinus floor augmentation prior to dental implant placement [5]. Cysts are usually asymptomatic; radiographically, they usually present as well-defined unilocular radiolucencies. Treatment consists of enucleation with no expected recurrence.
Fig. 1.

Surgical ciliated cyst of the edentulous R maxilla (arrow) following sinus surgery, showing a unilocular, radiolucent, well-demarcated and corticated lesion located on the anterior floor of the maxillary sinus. Inset shows cyst lining entirely composed of upper respiratory epithelium. Courtesy of Dr. Maria A. Copete, MSc, DDS, Professor, College of Dentistry, University of Saskatchewan, Canada
Definition of calcifying odontogenic cyst (COC) has been changed so that it currently refers only to the presence of characteristic ghost cells that may undergo calcification, while the ameloblastoma-like lining epithelium is a desirable diagnostic feature. In addition, those odontoma-associated COCs are no longer separated from the rest of COCs, despite the fact that they are separated by others, as 24% of COCs occur with odontomas and 3.5% with other odontogenic tumours [6]. The pathogenesis of COC has been linked to the identification of mutations in CTNNB1 gene (Wnt molecular pathway) that encodes the beta-catenin protein product [7]. The CTNNB1/Wnt aberrations are shared with other head and neck, ghost cell-containing tumours, both non-odontogenic (i.e., adamantinomatous craniopharyngioma and pilomatrixoma) and odontogenic (dentinogenic ghost cell tumour, ghost cell odontogenic carcinoma and odontogenic carcinoma with dentinoid, the last not being included in the 2022 classification) [7, 8].
Glandular odontogenic cyst (GOC) is defined by epithelial lining that resembles glandular tissue [1]. In the 2017 edition, ten specific histopathological criteria were listed, with the concept that the fulfillment of seven of them could strongly support a diagnosis of GOC [2]. Two of these criteria were then considered to be present in all lesions, namely the thickness of the epithelium and the luminal layer of hob-nail cells, present at least focally. In the 2022 classification, all histological features are characteristic but none are essential. Hob-nail cells are the only parameter considered to be the most characteristic finding of all GOCs, others being present with less consistency, therefore there is no reference to a specific number of criteria to support a diagnosis of GOC [1]. There is a certain extent of microscopic similarity and overlap between GOC and some central mucoepidermoid carcinomas (CMEC). MAML2 gene rearrangements, once thought to be exclusively present in CMEC, have recently been reported also in one aggressive lesion that fulfilled the diagnosis of GOC [9]. As the knowledge is very limited so far, we only can hypothetically raise the possibility of a transition occurring in aggressive GOC to CMEC if MAML2 rearrangements are identified. Clinical considerations and treatment decisions based on MAML2 should be taken with caution.
Odontogenic keratocyst (OKC) maintains its status as a cyst in both 2017 and 2022 classifications [1, 2]. The most frequent genetic modification associated with OKC pathogenesis occurs in the PTCH1 gene (Sonic Hedgehog (SHH) signaling pathway) and this has been identified in up to 93% of sporadic cases [10]. Interestingly, activating mutation in the BRAF p.V600E gene, mainly related to ameloblastoma, but not expression of its mutated protein product, has been reported in OKC [11, 12]. The molecular findings may open non-surgical, pharmaceutical options for treatment of OKCs, mainly large, destructive cysts, both sporadic and syndromic. Research is now focusing on small molecule selective inhibitors for SHH-related targets [13]. In addition, involvement of components from the connective tissue, i.e., fibroblasts [14], are also under investigation for the future development of possible therapeutic targets.
Odontogenic Tumours – Benign (Fig. 2)
Fig. 2.

Distribution of benign odontogenic tumours according to tissue of origin (epithelial, mixed epithelial and mesenchymal, mesenchymal), radiological appearance, peak age decade/s and frequent location. AdAM: adenomatoid ameloblastoma; AF: ameloblastic fibroma; AM: ameloblastoma; AOT: adenomatoid odontogenic tumour; CEOT: calcifying epithelial odontogenic tumour; COsF: cemento-ossifying fibroma; DGCT: dentinogenic ghost cell tumour; MetAM: metastasizing ameloblastoma; OdF: odontogenic fibroma; OdM: odontogenic myxoma; POT: primordial odontogenic tumour; SOT: squamous odontogenic tumour; UAM: unicystic ameloblastoma; mand-post: mandible posterior; max-ant: maxilla anterior; cx: complex odontoma; cd: compound odontoma
Most (~ 70%) of sporadic adenomatoid odontogenic tumours (AOT) have been identified to carry mutations in KRAS gene (mitogen activating protein kinase, MAPK, pathway; p.G12V and p.G12R loci), but they have not been connected to their clinico-pathological features [15]. Multiple AOTs may be encountered in patients with neurocutaneous Schimmelpenning syndrome, which is caused by postzygotic mutations in the RAS gene (MAPK pathway) and is characterized by presence of nevus sebaceus, ophthalmic, neurologic, skeletal, urologic, and cardiovascular alterations. In addition to AOTs, other oral manifestations include dental defects, papillary lesions in the oral mucosa and giant cell lesions of the jaws [16].
Calcifying epithelial odontogenic tumour (CEOT) is now recognized to have three histopathological subtypes: clear cell, cystic/microcystic and non-calcified/Langerhans cell rich [1]. The latter is still disputed for the possibility of its being better classified as the amyloid sub-type of odontogenic fibroma, given that it shares microscopic and clinical properties more in common with odontogenic fibroma than with CEOT [17]. Areas with CEOT-like features may be seen in AOT and the diagnosis of these cases should be AOT. Mutations in tumour suppressor genes (PTEN, CDKN2A, PTCH1), oncogenes (JAK3, MET) have been identified in CEOT, however so far, these do not contribute to clinical properties or treatment decisions.
After being omitted in the 2017 edition as a descriptive term of ameloblastoma (AM) [2], the term "conventional" was re-introduced in the upcoming edition [1]. In the 2017 classification, the possibility of moving unicystic ameloblastoma (UAM) mural sub-type to conventional AM was raised, based on the need for aggressive surgical treatment for both tumours [2]. In the 2022 classification, UAM mural sub-type has been retained within UAMs [1]. As both conventional AM and UAM have been found to harbor BRAFp.V600E mutations [18], aggressive and destructive tumours could be candidates for BRAF-targeted therapy that has the potential to reduce tumour size and ultimately enable a conservative surgical procedure [19]. Preliminary data of biological treatment show effectiveness in selected cases [20].
Adenoid ameloblastoma (AdAM) is a newly recognized entity separate from the AM group of tumours, defined as an epithelial neoplasm characterized by cribriform architecture and duct-like structures, with dentinoid being often present [1]. There are about 40 published cases, with a peak incidence in the 4th decade (age range 25-52y), a slight female predilection and demographically similar to conventional AM [21, 22]. It has a propensity for the mandible (64.7%) and usually manifests clinically as a painless swelling, occasionally with pain and paresthesia [21]. Radiologically, most (~ 82%) tumours present as radiolucencies with occasional radio-opaque foci with ill-defined margins and cortical perforation at time of diagnosis. The essential histopathological features of AdAM consist of an ameloblatoma-like component, duct-like structures, whorled cellular condensations reminiscent of morules and cribriform architecture (Fig. 3). About two-thirds of tumours contain varying amounts of dentinoid. There are overlapping microscopic features with AOT and dentinogenic ghost cell tumour (DGCT), but the combination of the essential features of AdAM are expected to distinguish it from these other entities. There is also considerable overlap between AdAM and odontogenic carcinoma with dentinoid, with limited current criteria to separate them [22]. Positively stained nuclear beta catenin colocalizes with the epithelial morules. Ki-67 proliferation marker is usually high. AdAM is characterized by an aggressive biological behavior with local infiltration and a recurrence rate that ranges between 45.5% [21] and 70% [22]. BRAFp.V600E mutations, usually identified in AM/UAM, are absent in AdAM. Whether AdAM is a unique standalone tumour or a histologic variant of AM will require further investigation.
Fig. 3.
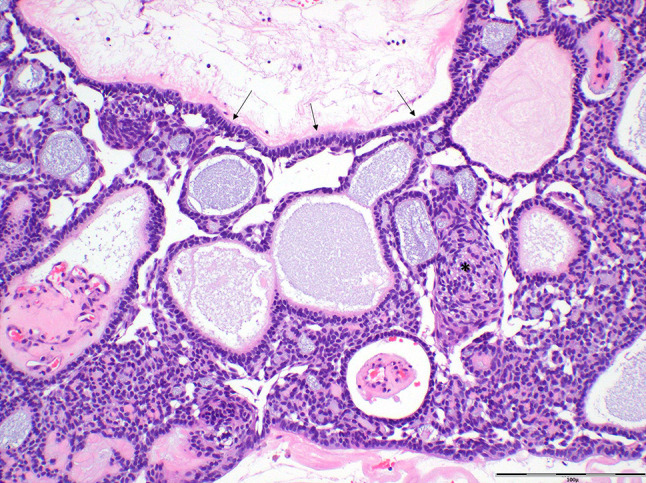
Photomicrograph of a case of adenoid ameloblastoma highlighting the major histopathological features: cribriform architecture, ameloblastoma-like component (including basal palisading and reverse polarity) (arrows), duct-like structures and whorled cellular condensations reminiscent of morules (asterisk) (hematoxylin and eosin; scale bar 100µ)
Metastasizing AM is being currently considered in the AM group of benign epithelial tumours [1], similar to the 2017 classification [2]. In contrary, in the 2005 classification, it was regarded as a malignant odontogenic tumour [3]. Moving a neoplasm that metastasizes and has a 30% mortality rate into the benign category was and continues to be controversial [23, 24].
Odontomas are now considered as hamartomas and are currently the second most commonly accessioned odontogenic lesion after ameloblastoma, although their real frequency is probably higher as many of them are unreported [25]. WNT/beta-catenin pathway activation in embryonic SOX-2 positive dental stem cells can drive odontoma formation [26]. Developing odontomas may be comprised of soft tissue closely resembling dental papilla with prominent epithelial strands and limited or no evidence of dental hard tissue induction. These features overlap with ameloblastic fibroma (AF), sometimes causing a problem differentiating between them. Lesions previously diagnosed as ameloblastic fibro-dentinoma (AFD) and ameloblastic fibro-odontoma (AFO) have a soft tissue component that resembles AF, and a component of dental hard tissue matrix, which resembles but is less prominent and less well organized than odontoma. The status of AFD and AFO has been debated as they appear to be intermediate between AF and odontoma. Currently, AFD and AFO are classified as developing odontomas although presence of BRAF p.V600E mutations in AFD and AFO is similar to AF, but differs from odontoma, which lacks BRAF p.V600E mutations [27]. In addition, several AFO/AFD cases with locally aggressive biological behavior, large size and recurrence may better fit a neoplastic type of lesion rather than an odontoma/hamartoma, but these represent a minority overall of lesions diagnosed as AFD or AFO. Further molecular study is expected to clarify whether AFD and AFO are separate entities, intermediate lesions with a spectrum of behavior that ultimately result in formation of odontoma, or are a mixture of developing odontoma and AF.
Cemento-ossifying fibroma (COsF), which has already been defined as a benign mesenchymal odontogenic tumour in the 2017 classification, but was then detailed under the heading of Fibro-osseous and osteochondromatous lesions [2], has become an integral part of the benign mesenchymal odontogenic tumours in the 2022 classification [1] and is completely separated from the non-odontogenic juvenile trabecular and psammomatoid types. Pathogenesis of COsF in a minority of tumours is linked to inactivating mutations in the tumour suppressor gene CDC73 (HRPT2), usually in those cases that are part of hyperparathyroidism-jaw tumour syndrome [28]. COsF can also be part of gnathodiaphyseal dysplasia, which is characterized by GDD1 gene mutations [27]. The microscopic features of sporadic and syndrome-related COsF are essentially the same, with a certain range of diversity between the fibrous and calcified components [29]. It should be emphasized that the peripheral ossifying fibroma should not be regarded as the peripheral counterpart of COsF, but rather a reactive gingival hyperplasia with mineralization [30].
Odontogenic myxoma with a greater amount of collagen was termed myxofibroma in the 2017 classification [2], while in the 2022 edition it is termed fibromyxoma [1]. Activating mutations in the MAPK/ERK signaling pathway have been identified in this tumour and as such, may serve as targets for pharmacologic therapy [31]. The microscopic differential diagnosis for odontogenic myxoma includes normal dental papilla, hyperplastic dental follicle, myxoid neurofibroma, chondromyxoid fibroma, odontogenic fibroma and low-grade fibromyxoid sarcoma, especially in the context of fibromyxoma [32].
Odontogenic Tumours—Malignant
Ameloblastic carcinoma (AMCa) is defined in the 2022 WHO as a primary odontogenic carcinoma histologically resembling AM [1] and not as the malignant counterpart of AM, as it has been in the 2017 classification [2]. AMCa, although rare, constitutes 30% of the malignant odontogenic tumours [1]. Most of them occur de novo, but some might arise in pre-existing longstanding, untreated or recurrent AM. Microscopically, AMCa essentially resembles AM with variable features of malignancy, however the threshold for diagnosis is still poorly defined. AMCa should show at least moderate cellular or nuclear atypia, nuclear hyperchromatism, increased mitotic activity, crowding of basal cells with their expansion into the other epithelial layers; central necrosis, if present, can support the diagnosis. The 5-year survival after complete surgical removal is ~ 70%, irrespective of the microscopic findings or presence of a pre-existing AM; local recurrence is ~ 40%, distant metastases (mainly lungs) have been found in ~ 33% of cases, far higher than in regional cervical lymph nodes (~ 13%), with novel treatment modalities being pursued [33]. The differential diagnosis includes entities in keeping with the predominant microscopic findings in AMCa, so that tumours with predominant presence of basaloid cells should be differentiated from tumours such as basal cell AM, basaloid squamous cell carcinoma and adamantinoma-like Ewing tumour [34]; true spindle cell AMCa should be distinguished from spindle cell/sarcomatoid squamous cell carcinoma, and tumours with clear cells should be differentiated from clear cell odontogenic carcinoma (COdC).
COdC is characterized by EWSR1 gene rearrangement in about 80% of cases [reviewed in 35]. Recurrence rate is as high as 40%, regional lymph node metastases are more common than distant ones and death rate is about 11% [35]. Differential diagnosis includes jawbone clear cell-containing tumours, such as CEOT, amyloid-rich odontogenic fibroma, odontogenic carcinoma with dentinoid, primary or metastatic tumours of salivary glands (e.g., mucoepidermoid carcinoma, clear cell carcinoma, epithelial myoepithelial carcinoma), and metastatic tumours (i.e., renal cell carcinoma, melanoma).
Giant Cell Lesions and Bone Cysts (Fig. 4)
Fig. 4.

Giant cell lesions, Fibro-osseous lesions and bone and cartilage benign and malignant maxillofacial lesions with emphasis on peak age decade/s (each scale bar = 1 decade; thick scale/s = peak frequency) and leading genetic aberrations. ABC: aneurysmal bone cyst; ABC*: secondary aneurysmal bone cyst; CGCG: central giant cell granuloma; JTOF: juvenile trabecular ossifying fibroma; FoCD: focal cemento-ossifying dysplasia; FamFLCD: familial florid cemento-osseous dysplasia; FLCD: florid cemento-osseous dysplasia; PSOF: psammomatoid ossifying fibroma; PCD: periapical cemento-ossifying dysplasia; SBC*: secondary simple bone cyst; SOD: segmental odontomaxillary dysplasia
Central giant cell granuloma (CGCG) is now defined as a lesion comprised of osteoclasts [1], while in the 2017 classification these were termed osteoclast-type giant cells [2]. It is now believed that the mononuclear stroma harbors osteoclast precursors that mature and differentiate into osteoclasts following different molecular inductions. In about 70% of sporadic CGCGs, mutually exclusive somatic mutations in KRAS, FGFR1 and TRPV4 genes were identified that all lead to the activation of the MAPK signaling pathway [36]. Interestingly, the TRPV4 gene encodes for a calcium channel that was found to be mutated in hereditary channelopathies, which are characterized by peripheral nervous system and skeletal changes. Recently, CGCG-like lesions have been described as part of a syndrome caused by germline TRPV4 mutation [37], further enhancing the apparent TRPV4 gene mutations-calcium channels-CGCG interconnections. H3F3A mutation characteristic of giant cell tumours of long bones, has not been identified in CGCG [36]. Multiple giant cell lesions that microscopically are indistinguishable from CGCG, occur in several syndromes, most of which are known to be caused by mutations in the MAPK pathway [38]. Peripheral giant cell granuloma (PGCG), a reactive gingival/alveolar lesion, is now also defined as osteoclast-containing [1], and not as osteoclast-type giant cell-containing, as was in the 2017 classification [2]. Like in CGCG, about 70% of cases of PGCG harbor mutations in the KRAS gene, including those lesions associated with dental implants [36].
In spite of the ongoing use of the term aneurysmal bone cyst (ABC), it should be emphasized that this lesion has been recognized as a neoplasm already in the 2017 classification [2]. The leading genetic aberration identified in about 70% of ABCs is the USP6—CDH11 fusion [39]. ABC-like cystic haemorrhagic areas may be part of the microscopic features in various lesions but these do not show the genetic mutations characteristic of true ABCs (Fig. 4).
Bone and Cartilage Tumours—Fibro-Osseous Tumours and Dysplasias (Fig. 4)
Cemento-osseous dysplasia, the most common benign fibro-osseous lesion of the jawbones, has three well established sub-types, defined according to anatomical location and extent of jawbone involvement: periapical, focal and florid; the familial florid cemento-osseous dysplasia (FFCOD) is the 4th sub-type introduced in the 2022 classification [1]. FFCOD onset is earlier than florid, often affects tooth eruption and is usually prone to cause considerable jawbone expansion. Genetic analysis revealed only one family with FFCOD mutations in the ANO5 gene. FFCOD raises a challenging differential diagnosis with familial gigantiform cementoma and syndromes characterized by benign fibro-osseous lesions, such as hyperparathyroidism jaw tumour syndrome and gnathodiaphyseal dysplasia [40]. Currently, we still rely on clinical and radiological features to distinguish familial gigantiform cementoma, which presents with diffuse expansion in multiple quadrants early in the disease process resulting in marked facial disfigurement, from FFCOD, which presents with typical florid cemento-osseous dysplasia lesions that may exhibit localized areas of expansion [40].
The 2022 classification has added Segmental odontomaxillary dysplasia (SOD) within the group of fibro-osseous lesions [1]. This is defined as a non-hereditary, unilateral developmental disorder characterized by segmental maxillary and soft tissue enlargement with dento-osseous abnormalities and occasional homolateral, usually subtle, cutaneous manifestations [1]. This rare condition is slightly more frequent in males, with no precise etiologic factor and suspected mutations in PIK3CA or ACTB genes [41]. SOD is asymptomatic with onset in 1st-2nd decades that usually halts around puberty. Imaging highlights the coarse bony trabeculation and dentition-related changes. Surgical treatment is considered for esthetic or functional purposes.
Unlike the 2017 classification, juvenile trabecular ossifying fibroma (JTOF) and psammomatoid ossifying fibroma (PsOF), are currently separated from the odontogenic COsF and are individually discussed as benign fibro-osseous lesions [1]. Of interest, the term juvenile was included in the name of both variants in the 2017 edition [2]. Neither variant exclusively affects juveniles, but both do have a strong predilection for the 1st-2nd decades, although the trabecular variant more so than the psammomatoid variant. Accordingly, juvenile was dropped from the psammomatoid terminology in 2022 [1]. JTOF essential features consist of onset in childhood (mean age 11.3 years), rapid expansion, well-demarcation on imaging and hypercellular stroma with prominent anastomosing osteoid trabeculae [reviewed in 42]. The pathogenesis is assumed to be associated with MDM2 and RASAL1 gene amplifications [43]. JTOF consists of unique aggregates of curvilinear strands of edema, haemorrhage, osteoclasts and pseudocystic degeneration, which are also seen in macroscopic specimens [42]. A recurrence rate of ~ 20% was reported [42]; due to the young age of patients, disfiguring surgical procedures should be avoided. PsOF is defined as a benign fibro-osseous neoplasm of the craniofacial skeleton characterized by spherical ossicles histologically with a peak incidence in the 2nd – 4th decades [42]. Molecular studies are similar to JTOF [43]. Histopathologically, the tumours show hypercellular spindle cell stroma in which multiple, spherical, relatively uniform ossicles are generated. These are called "psammoma" bodies, better termed as ossicles, differ from classical psammoma bodies as they are considerable larger, not sharply defined and lack lamellar pattern. Recurrence rate is 30%-56% following surgical excision [44].
Bone and Cartilage Tumours—Benign Maxillofacial Bone and Cartilage Tumours (Fig. 4)
Osteoblastoma, either intra-osseous or periosteal, occurs most commonly in the 2nd-3rd decades and have a slight female preponderance [45]. Gene rearrangement in FOS or less frequent in FOSB genes, are encountered and are also shared by osteoid osteoma (deleted from the 2022 classification) and cementoblastoma, possibly inferring a common pathogenesis in these entities.
Chondroblastoma in the head and neck region is located around the temporomandibular joint and squamous part of the temporal bone [1]. Those tumours that harbor p.Lys36Met mutations occurring in either the H3-3A or H3-3B genes, are expected to be immunohistochemically positive for the histone mutant-specific antibody K36M [46].
Chondromyxoid fibroma (CMF), a rare benign chondroid neoplasm, with a zonal architecture composed of chondroid, myxoid and myofibroblastic areas, can develop either within bones or on bone surface [1]. In about 90% of tumours, the genetic driver event involves mutation in the glutamate receptor gene, GRM1, which seems to be unique for CMF, while it is rare-to-absent in other cartilaginous tumours [47].
Desmoplastic fibroma of bone (DFB) is a locally aggressive fibroblastic/myofibroblastic tumour composed of benign spindle cells embedded in a collagenous background, mimicking desmoid-type fibromatosis [1]. In the jaws, 82% affect the mandible, almost 70% are diagnosed before the age of 30 years [reviewed in 48]. Clinically, DFB is asymptomatic, however swelling, facial asymmetry, pain, trismus may be present. Radiologically, DFB manifests as a well-defined radiolucency, uni- or multi-locular. Phenotypically, spindle cells are primarily positive for vimentin and smooth muscle actin; Ki67 proliferative marker is low. Lack of nuclear beta-catenin expression is in conformity with the absence of CTNNB1 mutations. This panel should be used to differentiate DFB from other intraosseous spindle cell lesions: desmoid tumour (nuclear expression of beta catenin, CTNN1 or APC mutations), fibrous dysplasia (GNAS mutation), low-grade fibrosarcoma, low-grade central osteosarcoma (positive for SATB2; MDM2, CDK4 gene aberrations), low-grade myofibroblastic sarcoma (positive for smooth muscle actin, desmin, nuclear beta catenin), synovial sarcoma (positive for TLE1; SS18 gene rearrangement), spindle cell rhabdomyosarcoma of the jaw (TFCP2 gene rearrangement) and myoepithelial tumours (positive for cytokeratin, S100; EWSR1 gene rearrangement). Recurrence rate after curettage is ~ 31%, enucleation – 25%, and resection only 10%.
Bone and Cartilage Tumours—Malignant Maxillofacial Bone and Cartilage Tumours
Osteosarcoma is a rare bone malignancy, with only 6% of all tumours involving the jawbones [1]. The pathogenesis and precise cell of origin are still unknown. Mutations in the TP53 and RB1 genes were found to be frequent, chromosomal instability was identified in MDM2, CDK4 and RAS genes, although the driver mutation remains unknown. Jaw osteosarcomas are staged using the 8th edition of the AJCC/UICC staging of bone and soft tissue sarcomas [49]. Histological grade of the tumour and stage of disease are the most important predictive factors. Surgical resection with clear margins is still the accepted standard of care, although more recent data showed a survival advantage for chemotherapy, especially in patients with positive margins, high-grade tumours and recurrent disease [50].
The chondrosarcoma family in the 2022 classification, [1] is replacing the chondrosarcoma in the 2017 classification [2]. This family of tumours is defined as malignant bone neoplasms arising in the medullary cavity that produces cartilaginous matrix. There is a periosteal variant. Dedifferentiated tumours show abrupt transition into a high-grade, non-cartilaginous sarcoma. Clear cell chondrosarcoma is a low-grade malignancy of lobules of cartilage with abundant clear cells. Pathogenesis shows that conventional, periosteal and dedifferentiated chondrosarcoma, but not clear cell chondrosarcoma, harbor somatic mutations in the IDH1 and IDH2 genes, however their rate of detection in the facial bone is quite low [51]. Staging is done according to the 8th edition of the AJCC/UICC staging of bone and soft tissue sarcomas [49].
Almost all mesenchymal chondrosarcomas harbor a specific HEY1/NCOA2 fusion gene. Areas with small cells may also show loss of Tp53, Rb expression and homozygous loss of CDKN2A/p16 [52].
Rhabdomyosarcoma with TFCP2 (TFCP2-RMS) rearrangement is a new tumour that has been introduced in the 2022 classification [1]. It is defined as a high-grade RMS characterized by fusion of TFCP2 gene to EWSR1 or FUS gene. This specific genetic aberration distinguishes TFCP2-RMS from other types of RMS and diagnosis can be confirmed by FISH study with break-apart probe for TFCP2 or by sequencing. The tumour affects young adults, with about a third being less than 18-year-old [53, 54]. TFCP2-RMS has predilection for the craniofacial bones, especially the mandible and is characterized by frequent bone perforation and infiltration into the adjacent soft tissues. Histopathologically, TFCP2-RMS are usually bi-phasic with spindle and epithelioid areas in solid sheets or fascicles with scant stroma, brisk mitotic activity, conspicuous nucleoli and frequent necrosis. Immunohistochemical stains are positive for pan-cytokeratin and desmin/myogenin/MyoD1, reflecting the bi-phasic morphological appearance (Fig. 5). Additional positive immunostains include p63, CK7, SATB2, S100, CD30, CD4, caldesmon and others. Therefore, an array of malignancies can be listed in the differential diagnosis, such as metastatic carcinoma, triton tumour and anaplastic lymphoma. TFCP2-RMS is associated with poor patient prognosis, with advanced clinical stage and distant metastases already present at time of diagnosis [53]. Despite aggressive multi-modal therapy, disease recurrence is frequent and there is only a 14-month median survival time. As less than 30 cases have been reported so far [54], data on prognosis is rather preliminary and more targeted treatment approaches likely to emerge.
Fig. 5.

A 48-year-old male with a lesion of the anterior maxilla. Microscopically, the tumour cells showed a bi-phasic morphology with both spindled (A) and epithelioid (B) cells (A, B—hematoxylin and eosin; scale bar 50µ). Cytogenetic study was positive for TFCP2 gene aberration and confirmed the diagnosis of TFCP2-RMS. Photomics are courtesy of Robert D. Foss, DDS, MS, The Joint Pathology Center, Head & Neck Pathology, Silver Spring, MD., USA
Authors' Contribution
Both authors contributed to the writing and editing of the manuscript.
Funding
(Information that explains whether and by whom the research was supported) – N/A.
Data Availability
(data transparency) – N/A.
Code Availability
(software application or custom code) – N/A.
Declarations
Conflict of interest
(Include appropriate disclosures) – Authors have no conflicts of interests to disclose.
Ethical Approval
This manuscript does not contain studies with human or animals.
Consent to Participate
N/A
Consent for Publication
N/A
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.WHO Classification of Tumours Editorial Board. Head and neck tumours. Lyon (France): International Agency for Research on Cancer; 2022. (WHO classification of tumours series, 5th ed.; vol. 9). https://publications.iarc.fr/
- 2.El-Naggar AK, John KC, Grandis JR, Takata T, Slootweg PJ. WHO Classification of Head and Neck Tumours. 4th ed. Lyon: IARC; 2017
- 3.Barnes L, Eveson JW, Reichart P, Sidransky D. WHO Classification of Head and Neck Tumours. 3rd ed. Lyon: IARC; 2005
- 4.Bourgeois SL, Jr, Nelson BL. Surgical ciliated cyst of the mandible secondary to simultaneous Le Fort I osteotomy and genioplasty: report of case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:36–39. doi: 10.1016/j.tripleo.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 5.Kahn A, Matalon S, Bassam Salem R, Kats L, Chaushu L, Vered M, Rosen E. Sinus floor augmentation—associated surgical ciliated cysts: case series and a systematic review of the literature. Appl Sci. 2021;11:1903. [Google Scholar]
- 6.Ledesma-Montes C, Gorlin RJ, Shear M, Praetorius F, Mosqueda-Taylor A, Altini M, Unni K, de Almeida OP, Carlos-Bregni R, de León ER, Phillips V, Delgado-Azañero W, Meneses-García A. International collaborative study on ghost cell odontogenic tumours: calcifying cystic odontogenic tumour, dentinogenic ghost cell tumour and ghost cell odontogenic carcinoma. J Oral Pathol Med. 2008;37:302–08. doi: 10.1111/j.1600-0714.2007.00623.x. [DOI] [PubMed] [Google Scholar]
- 7.Yukimori A, Oikawa Y, Morita KI, Nguyen CT, Harada H, Yamaguchi S, Kayamori K, Yamaguchi A, Ikeda T, Sakamoto K. Genetic basis of calcifying cystic odontogenic tumors. PLoS One. 2017;12:e0180224. doi: 10.1371/journal.pone.0180224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gondak RO, Mariano FV, de Sousa SF, de Siqueira EC, Díaz KP, Martins LA, Altemani A, Mosqueda-Taylor A, Gomez RS, Gomes CC. CTNNB1 and APC mutations in odontogenic carcinoma with dentinoid. Oral Surg Oral Med Oral Pathol Oral Radiol. 2020;129:e249–56. doi: 10.1016/j.oooo.2019.08.017. [DOI] [PubMed] [Google Scholar]
- 9.Greer RO, Eskendri J, Freedman P, Ahmadian M, Murakami-Walter A, Varella-Garcia M. Assessment of biologically aggressive, recurrent glandular odontogenic cysts for mastermind-like 2 (MAML2) rearrangements: histopathologic and fluorescent in situ hybridization (FISH) findings in 11 cases. J Oral Pathol Med. 2018;47:192–97. doi: 10.1111/jop.12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stojanov IJ, Schaefer IM, Menon RS, Wasman J, Gokozan HN, Garcia EP, Baur DA, Woo SBG, Sholl LM. Biallelic PTCH1 inactivation is a dominant genomic change in sporadic keratocystic odontogenic tumors. Am J Surg Pathol. 2020;44:553–60. doi: 10.1097/PAS.0000000000001407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain KS, Bodhankar K, Desai RS, Bansal S, Shirsat P, Prasad P, Shah A. Absence of BRAFV600E immunohistochemical expression in sporadic odontogenic keratocyst, syndromic odontogenic keratocyst and orthokeratinized odontogenic cyst. J Oral Pathol Med. 2020;49:1061–67. doi: 10.1111/jop.13081. [DOI] [PubMed] [Google Scholar]
- 12.Kurppa KJ, Catón J, Morgan PR, Ristimäki A, Ruhin B, Kellokoski J, Elenius K, Heikinheimo K. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 2014;232:492–98. doi: 10.1002/path.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhai J, Zhang H, Zhang J, Zhang R, Hong Y, Qu J, Chen F, Li T. Effect of the sonic hedgehog inhibitor GDC-0449 on an in vitro isogenic cellular model simulating odontogenic keratocysts. Oral Dis. 2019;25:1600–07. doi: 10.1038/s41368-018-0034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang HC, Jiang WP, Sima ZH, Li TJ. Fibroblasts isolated from a keratocystic odontogenic tumor promote osteoclastogenesis in vitro via interaction with epithelial cells. Oral Dis. 2015;21:170–77. doi: 10.1111/odi.12231. [DOI] [PubMed] [Google Scholar]
- 15.Coura BP, Bernardes VF, de Sousa SF, França JA, Pereira NB, Pontes HAR, Batista AC, da Cruz Perez DE, Albuquerque Junior RLC, de Souza LB, Martins MD, Diniz MG, Gomez RS, Gomes CC. KRAS mutations drive adenomatoid odontogenic tumor and are independent of clinicopathological features. Mod Pathol. 2019;32:799–806. doi: 10.1038/s41379-018-0194-4. [DOI] [PubMed] [Google Scholar]
- 16.Chaves RRM, Júnior AACP, Gomes CC, de Castro WH, Gomez RS. Multiple adenomatoid odontogenic tumors in a patient with Schimmelpenning syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol. 2020;129:e12–17. doi: 10.1016/j.oooo.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Roza ALOC, Sousa EM, Leite AA, Amaral-Silva GK, Morais TML, Wagner VP, Schuch LF, Vasconcelos ACU, de Arruda JAA, Mesquita RA, Fonseca FP, Abrahão AC, Agostini M, de Andrade BAB, da Silveira EJD, Martínez-Flores R, Rondanelli BM, Alberdi-Navarro J, Robinson L, Marin C, Assunção Júnior JNR, Valiati R, Fregnani ER, Santos-Silva AR, Lopes MA, Hunter KD, Khurram SA, Speight PM, Mosqueda-Taylor A, van Heerden WFP, Carlos R, Wright JM, de Almeida OP, Romañach MJ, Vargas PA. Central odontogenic fibroma: an international multicentric study of 62 cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2021;131:549–57. doi: 10.1016/j.oooo.2020.08.022. [DOI] [PubMed] [Google Scholar]
- 18.González-González R, López-Verdín S, Lavalle-Carrasco J, Molina-Frechero N, Isiordia-Espinoza M, Carreón-Burciaga RG, Bologna-Molina R. Current concepts in ameloblastoma-targeted therapies in B-raf proto-oncogene serine/threonine kinase V600E mutation: Systematic review. World J Clin Oncol. 2020;11:31–42. doi: 10.5306/wjco.v11.i1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zlotogorski-Hurvitz A, Soluk Tekkeşin M, Passador-Santos F, Martins Montalli VA, Salo T, Mauramo M, Kats L, Buchner A, Vered M. Conceptual changes in ameloblastoma: suggested re-classification of a "veteran" tumor. Oral Dis. 2021 doi: 10.1111/odi.13770. [DOI] [PubMed] [Google Scholar]
- 20.Hirschhorn A, Campino GA, Vered M, Greenberg G, Yacobi R, Yahalom R, Barshack I, Toren A, Amariglio N, Rechavi G. Upfront rational therapy in BRAF V600E mutated pediatric ameloblastoma promotes ad integrum mandibular regeneration. J Tissue Eng Regen Med. 2021 doi: 10.1002/term.3254. [DOI] [PubMed] [Google Scholar]
- 21.Jayasooriya PR, Abeyasinghe WAMUL, Liyanage RLPR, Uthpali GN, Tilakaratne WM. diagnostic enigma of adenoid ameloblastoma: literature review based evidence to consider it as a new sub type of ameloblastoma. Head Neck Pathol. 2021 doi: 10.1007/s12105-021-01358-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loyola AM, Cardoso SV, de Faria PR, Servato JP, Eisenberg AL, Dias FL, Thavaraj S, Gomes CC, Gomez RS. Adenoid ameloblastoma: clinicopathologic description of five cases and systematic review of the current knowledge. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:368–77. doi: 10.1016/j.oooo.2015.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Reichart P, Sciubba JJ, Philipsen HP. Splitters or lumpers: the 2017 WHO classification of head and neck tumours. J Am Dent Assoc. 2018;149:567–71. doi: 10.1016/j.adaj.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 24.Soluk-Tekkeşin M, Wright JM. The world health organization classification of odontogenic lesions: a summary of the changes of the 2017 (4th) Edition. Turk Patoloji Derg. 2018;34:1–18. doi: 10.5146/tjpath.2017.01410. [DOI] [PubMed] [Google Scholar]
- 25.Soluk-Tekkesin M, Cakarer S, Aksakalli N, Alatli C, Olgac V. New world health organization classification of odontogenic tumours: impact on the prevalence of odontogenic tumours and analysis of 1231 cases from Turkey. Br J Oral Maxillofac Surg. 2020;58:1017–22. doi: 10.1016/j.bjoms.2020.06.033. [DOI] [PubMed] [Google Scholar]
- 26.Fujii S, Nagata K, Matsumoto S, Kohashi KI, Kikuchi A, Oda Y, Kiyoshima T, Wada N. Wnt/β-catenin signaling, which is activated in odontomas, reduces sema3A expression to regulate odontogenic epithelial cell proliferation and tooth germ development. Sci Rep. 2019;9:4257. doi: 10.1038/s41598-019-39686-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coura BP, Bernardes VF, de Sousa SF, Diniz MG, Moreira RG, de Andrade BAB, Romañach MJ, Pontes HAR, Gomez RS, Odell EW, Gomes CC. Targeted next-generation sequencing and allele-specific quantitative pcr of laser capture microdissected samples uncover molecular differences in mixed odontogenic tumors. J Mol Diagn. 2020;22:1393–99. doi: 10.1016/j.jmoldx.2020.08.005. [DOI] [PubMed] [Google Scholar]
- 28.de Mesquita Netto AC, Gomez RS, Diniz MG, Fonseca-Silva T, Campos K, De Marco L, Carlos R, Gomes CC. Assessing the contribution of HRPT2 to the pathogenesis of jaw fibrous dysplasia, ossifying fibroma, and osteosarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:359–67. doi: 10.1016/j.oooo.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Riminucci M, Collins MT, Corsi A, Boyde A, Murphey MD, Wientroub S, Kuznetsov SA, Cherman N, Robey PG, Bianco P. Gnathodiaphyseal dysplasia: a syndrome of fibro-osseous lesions of jawbones, bone fragility, and long bone bowing. J Bone Miner Res. 2001;16:1710–18. doi: 10.1359/jbmr.2001.16.9.1710. [DOI] [PubMed] [Google Scholar]
- 30.Brierley DJ, Crane H, Hunter KD. Lumps and bumps of the gingiva: a pathological miscellany. Head Neck Pathol. 2019;13:103–13. doi: 10.1007/s12105-019-01000-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pereira NB, Bastos VC, de Souza JC, Diniz MG, Vitório JG, Kitten GT, de Oliveira AL, de Avelar GF, Castro WH, Bernardes VF, Dias AAM, Gomez RS, Gomes CC. First insights for targeted therapies in odontogenic myxoma. Clin Oral Investig. 2020;24:2451–58. doi: 10.1007/s00784-019-03107-4. [DOI] [PubMed] [Google Scholar]
- 32.Alhousami T, Sabharwal A, Gupta S, Aguirre A, Park E, Kramer JM. Fibromyxoma of the jaw: case report and review of the literature. Head Neck Pathol. 2018;12:44–51. doi: 10.1007/s12105-017-0823-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giridhar P, Mallick S, Upadhyay AD, Rath GK. Pattern of care and impact of prognostic factors in the outcome of ameloblastic carcinoma: a systematic review and individual patient data analysis of 199 cases. Eur Arch Otorhinolaryngol. 2017;274:3803–10. doi: 10.1007/s00405-017-4631-7. [DOI] [PubMed] [Google Scholar]
- 34.Ko YCK, Varma S, Zhu CF, Zhu SX, Vennam S, Poh CF, Jordan RC, Kong C, Pollack JR, West RB. Gene expression profiling of head and neck tumors identifies foxp1 and sox10 expression as useful for distinguishing ameloblastoma from basaloid salivary gland tumors. Am J Surg Pathol. 2020;44:665–72. doi: 10.1097/PAS.0000000000001421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guastaldi FPS, Faquin WC, Gootkind F, Hashemi S, August M, Iafrate AJ, Rivera MN, Kaban LB, Jaquinet A, Troulis MJ. Clear cell odontogenic carcinoma: a rare jaw tumor. a summary of 107 reported cases. Int J Oral Maxillofac Surg. 2019;48:1405–10. doi: 10.1016/j.ijom.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes CC, Gayden T, Bajic A, Harraz OF, Pratt J, Nikbakht H, Bareke E, Diniz MG, Castro WH, St-Onge P, Sinnett D, Han H, Rivera B, Mikael LG, De Jay N, Kleinman CL, Valera ET, Bassenden AV, Berghuis AM, Majewski J, Nelson MT, Gomez RS, Jabado N. TRPV4 and KRAS and FGFR1 gain-of-function mutations drive giant cell lesions of the jaw. Nat Commun. 2018;9:4572. doi: 10.1038/s41467-018-06690-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ragamin A, Gomes CC, Bindels-de Heus K, Sandoval R, Bassenden AV, Dib L, Kok F, Alves J, Mathijssen I, Medici-Van den Herik E, Eveleigh R, Gayden T, Pullens B, Berghuis A, van Slegtenhorst M, Wilke M, Jabado N, Mancini GMS, Gomez RS. De novo TRPV4 Leu619Pro variant causes a new channelopathy characterised by giant cell lesions of the jaws and skull, skeletal abnormalities and polyneuropathy. J Med Genet:jmedgenet. 2020 doi: 10.1136/jmedgenet-2020-107427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomes CC, Diniz MG, Bastos VC, Bernardes VF, Gomez RS. Making sense of giant cell lesions of the jaws (GCLJ): lessons learned from next-generation sequencing. J Pathol. 2020;250:126–33. doi: 10.1002/path.5365. [DOI] [PubMed] [Google Scholar]
- 39.Oliveira AM, Perez-Atayde AR, Inwards CY, Medeiros F, Derr V, Hsi BL, Gebhardt MC, Rosenberg AE, Fletcher JA. SP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol. 2004;165:1773–80. doi: 10.1016/S0002-9440(10)63432-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nel C, Yakoob Z, Schouwstra CM, van Heerden WF. Familial florid cemento-osseous dysplasia: a report of three cases and review of the literature Dentomaxillofac Radiol. 2021;50:20190486. doi: 10.1259/dmfr.20190486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson TM, Rafferty K, Ryan E, Ganguly A, Koutlas IG. Segmental ipsilateral odontognathic dysplasia (mandibular involvement in segmental odontomaxillary dysplasia?) and identification of PIK3CA somatic variant in lesional mandibular gingival tissue. Head Neck Pathol. 2021;15:368–73. doi: 10.1007/s12105-020-01185-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chrcanovic BR, Gomez RS. Juvenile ossifying fibroma of the jaws and paranasal sinuses: a systematic review of the cases reported in the literature. Int J Oral Maxillofac Surg. 2020;49:28–37. doi: 10.1016/j.ijom.2019.06.029. [DOI] [PubMed] [Google Scholar]
- 43.Tabareau-Delalande F, Collin C, Gomez-Brouchet A, Bouvier C, Decouvelaere AV, de Muret A, Pagès JC, de Pinieux G. Chromosome 12 long arm rearrangement covering MDM2 and RASAL1 is associated with aggressive craniofacial juvenile ossifying fibroma and extracranial psammomatoid fibro-osseous lesions. Mod Pathol. 2015;28:48–56. doi: 10.1038/modpathol.2014.80. [DOI] [PubMed] [Google Scholar]
- 44.Sarode SC, Sarode GS, Waknis P, Patil A, Jashika M. Juvenile psammomatoid ossifying fibroma: a review. Oral Oncol. 2011;47:1110–16. doi: 10.1016/j.oraloncology.2011.06.513. [DOI] [PubMed] [Google Scholar]
- 45.Jones AC, Prihoda TJ, Kacher JE, Odingo NA, Freedman PD. Osteoblastoma of the maxilla and mandible: a report of 24 cases, review of the literature, and discussion of its relationship to osteoid osteoma of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;102:639–50. doi: 10.1016/j.tripleo.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Amary MF, Berisha F, Mozela R, Gibbons R, Guttridge A, O'Donnell P, Baumhoer D, Tirabosco R, Flanagan AM. The H3F3 K36M mutant antibody is a sensitive and specific marker for the diagnosis of chondroblastoma. Histopathology. 2016;69:121–27. doi: 10.1111/his.12945. [DOI] [PubMed] [Google Scholar]
- 47.Nord KH, Lilljebjörn H, Vezzi F, Nilsson J, Magnusson L, Tayebwa J, de Jong D, Bovée JV, Hogendoorn PC, Szuhai K. GRM1 is upregulated through gene fusion and promoter swapping in chondromyxoid fibroma. Nat Genet. 2014;46:474–77. doi: 10.1038/ng.2927. [DOI] [PubMed] [Google Scholar]
- 48.Karimi A, Derakhshan S, Moradzadeh Khiavi M, Mosavat F, Mirjalili F. Desmoplastic fibroma of the jaws: a case series and review of literature. Iran J Pathol. 2020;15:134–43. doi: 10.30699/ijp.2020.103833.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR, Winchester DP. 2017 The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more "personalized" approach to cancer staging. CA Cancer J Clin 67: 93–9 [DOI] [PubMed]
- 50.Liang L, Zhang T, You Y, He Q, Fan Y, Liao G. An individual patient data meta-analysis on the effect of chemotherapy on survival in patients with craniofacial osteosarcoma. Head Neck. 2019;41:2016–23. doi: 10.1002/hed.25668. [DOI] [PubMed] [Google Scholar]
- 51.Tallegas M, Miquelestorena-Standley É, Labit-Bouvier C, Badoual C, Francois A, Gomez-Brouchet A, Aubert S, Collin C, Tallet A, de Pinieux G. IDH mutation status in a series of 88 head and neck chondrosarcomas: different profile between tumors of the skull base and tumors involving the facial skeleton and the laryngotracheal tract. Hum Pathol. 2019;84:183–91. doi: 10.1016/j.humpath.2018.09.015. [DOI] [PubMed] [Google Scholar]
- 52.Meijer D, de Jong D, Pansuriya TC, van den Akker BE, Picci P, Szuhai K, Bovée JV. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes Chromosomes Cancer. 2012;51:899–909. doi: 10.1002/gcc.21974. [DOI] [PubMed] [Google Scholar]
- 53.Le Loarer F, Cleven AHG, Bouvier C, Castex MP, Romagosa C, Moreau A, Salas S, Bonhomme B, Gomez-Brouchet A, Laurent C, Le Guellec S, Audard V, Giraud A, Ramos-Oliver I, Cleton-Jansen AM, Savci-Heijink DC, Kroon HM, Baud J, Pissaloux D, Pierron G, Sherwood A, Coindre JM, Bovée JVMG, Larousserie F, Tirode F. A subset of epithelioid and spindle cell rhabdomyosarcomas is associated with TFCP2 fusions and common ALK upregulation. Mod Pathol. 2020;33:404–19. doi: 10.1038/s41379-019-0323-8. [DOI] [PubMed] [Google Scholar]
- 54.Xu B, Suurmeijer AJH, Agaram NP, Zhang L, Antonescu CR. Head and neck rhabdomyosarcoma with TFCP2 fusions and ALK overexpression: a clinicopathological and molecular analysis of 11 cases. Histopathology. 2021;79:347–57. doi: 10.1111/his.14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
(data transparency) – N/A.
(software application or custom code) – N/A.