

Abstract
Recent advances in pharmacological immune modulation against tumor cells has dramatically changed the paradigm of cancer treatment. Checkpoint inhibitor therapy is a form of cancer immunotherapy already in clinical setting but also under active basic and clinical investigation. Nevertheless, some patients are primary unresponsive or develop ulterior resistance to these family of drugs. This review aims to update the basic molecular mechanism of resistance as well as the current strategies for checkpoint inhibitor selection in order to propose new approaches to individualize the use of these novel therapies.
Keywords: Immunotherapy, checkpoint inhibitors, cytotoxic T-lymphocyte antigen 4, programmed death receptor-1, programmed death ligand-1
Introduction
New therapies based on modulating immune response, also known as cancer immunotherapy, has emerged as a new and promising option for tumor treatment in recent years. Relationship between immune system and tumor evolution have been described since the latest 18th century. William Coley described a group of patients suffering from locally advanced soft tissue sarcoma that presented a clinical regression after tumor infection caused by Streptococcus pyogenes (also known as Erysipellas)[1]. Ulterior efforts were focused on discover the relationship between immune system and tumor cells. In the 20th century, Paul Ehrlich proposed the concept of cancer immunosurveillance: emergence of cancer cells seems to be a frequent event, however, host natural immunity keeps suppressed ulterior development of tumor tissues; cancer occurs when this immune response is weakened. Linsley et al.[2] enriched this concept focusing on T cells and its key role on immune reaction. In the last decades, molecular mechanisms of immune response were progressively clarified: T cell activation implies an antigen presentation by the major histocompatibility complex (MHC) located on the surface of antigen presenting cells (APC) to the corresponding T cell receptor (TCR) on T lymphocytes[3]. Costimulatory molecules CD28 and B7 interaction is required for full activation, which is regulated by inhibitor checkpoints tov avoid autoimmunity phenomenon. Agata et al.[4] describes the cytotoxic T-lymphocyte antigen-4 (CTLA-4) receptor on activated effector T cells (Teff) and regulatory T cells (Treg). Sharma et al.[5] demonstrate that CTLA-4 inhibits both proliferation and IL-2 secretion by T cells completing the function of CD28/B7 signal. Finally, Sanchez-Vega et al.[6] described and cloned the programmed death 1 checkpoint receptor (PD-1), while Liu et al.[7] and Peng et al.[8] discovered its ligand PD-L1.
Ulterior clinical testing resulted in the approval of Anti-CTLA-4 first, and Anti-PD1 and Anti-PD-L1 monoclonal antibodies later; the last two presenting a long lasting clinical responses, with impressive plateau in survival curves in a broad spectrum of tumors, which initiate an immunotherapy era in solid and hematological tumors treatment. Despite of the initial enthusiasm, many patients do not respond to checkpoint blockade or progressed after an initial response. Molecular resistance mechanisms to checkpoint inhibitors are currently under a marked active research, however, the emerging knowledge of primary or secondary tumor resistance mechanisms does not translate into rational use of checkpoint inhibitors in clinical settings. Translation between basic research in resistance mechanisms of immunotherapy and clinical practice is key to optimize treatments, reduce cost and prolong survival in cancer patients. This review aims to update the basic molecular mechanism of resistance as well as the current strategies for checkpoint inhibitor selection in order to propose new approaches to individualize the use of these novel therapies.
Immune response to tumor cells and the immune synapse
Immune system responds to cancer cells emergence through a coordinated response of distinct interacting cells that detect and eliminate tumor cells. The Figure 1 described the relationship among immune cells, dentritic cells and tumor cells[1] (the “immune synapse”). An APC process and displays tumor antigen complexed with MHCs on their surfaces. T-cell recognize these complexes using the TCR. CTLA-4 expression and function is linked with T cell activation, and is upregulated following TCR engagement[2]. CTLA-4 dampens TCR signaling through competition with CD28, a costimulatory receptor for the B7 ligands B7-1 (CD80) and B7-2 (CD86)[3]. B7-1 and B7-2 receptors provide positive costimulatory signals through CD28, resulting in a competitive inhibition of both molecules by CTLA-4, this blockade is necessary to attenuate T cell activation.
Figure 1.
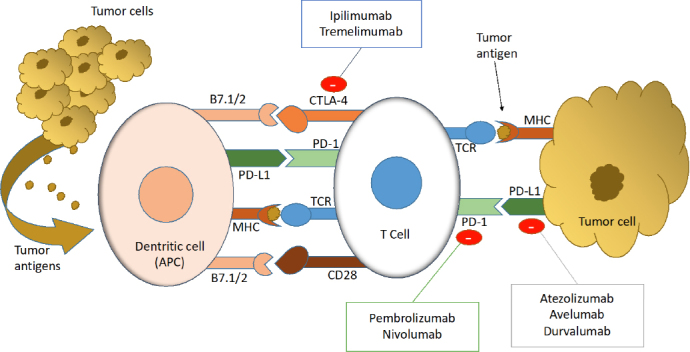
The immune synapse and response to tumor cells. APC: antigen presenting-cell; PD-L1: programmed death ligand-1; PD-1: programmed death-1; CTLA-4: cytotoxic T-lymphocyte antigen-4; TCR: T cell receptor; MHC: major histocompatibily complex; B7.1/2: ligand B7-1 and 2; CD28: cluster of differentiation-28
Additionally, PD-1 is expressed upon activation of T and B lymphocytes[4], and regulates immune response maintaining T cell responses within a desirable physiological range. Its ligands PD-L1 and PD-L2 are widely expressed in nonlymphoid tissues and also in tumor cells. The PD-1/PD-L1 regulatory system is induced by immune responses, forming a negative feedback loop to attenuate local T cell responses and minimizing tissue damage. PD-1 regulates T cell activation through interaction with PD-L1 and PD-L2. Inflammatory cytokines such as (interferon-gamma) INF-γ induce PD-L1 expression, and it is a key mechanism to maintain the immune response under control.
Insights into the normal immune processes and the regulation of costimulatory molecules inform our understanding of cancer immunosurveillance, mechanism of action of checkpoint inhibitors and its resistance mechanisms. Increasing the knowledge of these mechanisms is critical for the development of two future therapeutic goals: the optimization of available drugs administration in daily clinical practice and the emergence of new strategies against drug resistance.
Mechanisms of resistance to checkpoint blockade
In primary resistance, patients do not respond to the initial treatment to checkpoint inhibitors, while in secondary resistance, patients respond initially to checkpoint inhibitors and then progressed. The underlying molecular mechanisms of primary resistance resides in the lack of response to immunotherapy, which includes adaptive immune resistance[5]. Adaptive resistance is a mechanism which a tumor tissue is recognized by the immune system but it protects itself by adapting to the immune response, and this effect may have a clinical translation in the form of primary resistance, mixed responses or acquired resistance. In contrast, the acquired resistance, initiates with an early response of tumor to immunotherapy that after a period of time, re-growth, relapses and progresses. Therefore, the resistant mechanisms can be divided into two major responses based on both intrinsic tumor factors or microenvironment features (called tumor cell-intrinsic and extrinsic factors). Table 1 describes the main molecular pathways implicated in mechanisms of resistance to checkpoint inhibitors[6].
Table 1.
Checkpoint inhibitors, current indications and associated biomarker
| MoAb | Target | FDA/EMEA Approval | Biomarker of response/resistance |
|---|---|---|---|
| Ipilimumab | CTLA-4 | Melanoma | No |
| RCC (*) | No | ||
| Tremelimumab | CTLA-4 | No | No |
| Pembrolizumab | PD-1 | NSCLC 1st line | TPS PD-L1 expression ≥ 50%[62] |
| NSCLC after 1st line | TPS PD-L1 expression > 1%[62] | ||
| H&NSCC | TPS PD-L1 expression ≥ 50%[62] | ||
| UC | CPS ≥ 10[62] | ||
| cHL | Not required | ||
| Nivolumab | PD-1 | Melanoma (&) | No |
| NSCLC, H&NSCC, UC, cHL | Not required | ||
| Atezolizumab | PD-L1 | UC 1st line ($) | ≥ 5% PD-L1 expression[63] |
| UC after 1st line, NSCLC | Not required | ||
| Avelumab | PD-L1 | MCC | Not required |
| Durvalumab | PD-L1 | NSCLC (ç) | ≥ 1% PD-L1 expression |
MoAB: monoclonal antibody; FDA: food and drug administration; EMEA: European Medicines Agency; CTLA-4: cytotoxic T-lymphocyte antigen-4; PD-1: programmed dead cell receptor-1; PD-L1: programmed dead ligand-1; RCC: renal cell carcinoma; NSCLC: non small-cell lung cancer; H&NSCC: head and neck squamous cell carcinoma; UC: urothelial carcinoma; cHL: classic hodgkin lymphoma; MCC: merkel cell carcinoma; TPS: tumor proportion score; CPS: combined proportion score; *: In combination with Nivolumab; &: In combination with Ipilimumab; $: If no progression disease after chemoradiotherapy in locally-advanced tumors
Primary resistance mechanism
Tumor-cell mechanism
Primary Tumor-cell resistance mechanisms suppose a wide variety of intrinsic molecular cancer-cell alterations that primary block the immune system action in response to checkpoint inhibitors.
RTK/RAS pathway
The mitogen-activated protein kinase (MAPK) pathway activation presents inhibitory effects on T cell recruitment and function. In this sense, BRAF inhibition in combination with adoptive T cell transfer is more effective at inducing long-term clinical regression of BRAF-mutant tumor in BRAF-mutated human melanoma tumor-cell models, providing a strong rationale for the potential application of combining BRAF inhibitors and immunotherapy[7].
PI3K/PTEN/mTOR
The importance of PTEN status in tumor cells was explored in preclinical models of melanoma cells[8], also confirming a strong correlation between tumor PTEN loss and T cell infiltration at tumor sites and supporting the rationale for further evaluate combinatorial strategies targeting PI3K-AKT to increase immune response to checkpoint inhibitors.
WNT pathway
Some studies support the stabilization of b-catenin resulting in constitutive WNT signaling activation as a key mechanism in melanoma models, resulting in primary resistance of checkpoint inhibitors[9]. WNT signaling is frequently dysregulated in melanoma. b-catenin levels control tumor differentiation and regulate both MAPK and PI3K signaling. A correlation between activation of the WNT/b-catenin signaling pathway and the absence of a T cell gene expression signature would result in poor clinical responses. However, relationship between immune expression and WNT signaling alterations still remains unclear.
INF-γ pathway
INF-γ pathway plays a pivotal role in molecular resistance mechanisms to immune checkpoint inhibitors. Anti-CTLA blockade could be overcome in patients treated with melanoma in base to primary or secondary alterations in INF-γ pathway genes[10], at least in mouse models. In fact, mice bearing melanoma tumors with knockdown of INF-γ receptor 1 have impaired tumor rejection upon anti-CTLA-4 treatment, illustrating that loss of the INF-γ signaling pathway is associated with primary resistance to checkpoint blockade. In this context, recent studies involving the INF-g-JAK1/JAK2-STAT1/STAT2/STAT3-IRF1 axis describes an inducible expression of PD-L1 (with IRF1 binding to its promoter) and PD-L1 (with STAT3 bind to its promoter) confirming the primary role in immune synapse regulation.
Cyclin and cell cycle pathway
Cyclin-dependent kinase 5 (Cdk5) function[11] allows some tumor models as medulloblastoma (MB) to evade immune elimination via INF-g-induced PD-L1 up-regulation. Thus, MB PD-L1 expression results in potent CD4(+) T cell-mediated tumor rejection. In this tumor model, loss of Cdk5 results in persistent expression of IRF2 and IRF2BP2, which likely leads to reduced PD-L1 expression on tumors thereby promoting anti-tumor immunity.
Epigenetic control: DNA methylation
Epigenetic modification of DNA may play an important role in primary resistance to immunotherapy. In preclinical models, deacetylase inhibitors increase MHC and tumor-associated antigen expression, resulting in a decrease of competing endogenous lymphocytes and a proliferative advantage for the adoptively transferred cells[12]. Epigenetic modulation can induce the expression of a major T cell co-stimulatory molecule on cancer cells, which in turn overcome immune tolerance, and induce an efficient anti-tumor CTL response.
PD-L1 expression and molecular control
A key mechanism in tumor resistance to immunotherapy implies a constitutively expression of cell surface ligands that may inhibit antitumor T cell responses. This mechanism involves, but not only, proteins related to immune synapse discussed above. Specifically, increased PD-L1 expression may inhibit antitumor T cell responses, and it is a critical aspect that explains the main mechanism of checkpoint inhibitors. An increase of PD-1 constitutive expression is described on classical Hodgkin lymphoma (cHL) and mediastinal large B-cell lymphoma cell lines[13], identifying PD-L1 and PD-L2 as key targets at the 9p24.1 amplification region, that includes the Janus kinase 2 (JAK2) locus. 9p24.1 amplification is a disease-specific structural alteration that increases both the gene dosage of PD-1 ligands and their induction by JAK2, defining the PD-1 pathway and JAK2 as complementary therapeutic targets. In fact, in non-small cell lung cancer cell lines[14], oncogenic activation of the AKT-mTOR pathway promotes immune escape by driving expression of PD-L1. Additionally, EGFR-driven lung tumors inhibit antitumor immunity by activating PD-1/PD-L1 pathway to suppress T cell function and increase levels of proinflammatory cytokines[15].
MYC pathway also regulates the expression of PD-L1 and CD47[16], an innate immune regulator. Thus, MYC inactivation in mouse tumors down-regulated CD-47 and PD-L1 expression and enhanced the antitumor immune response; in contrast, when MYC was inactivated in tumors with enforced expression of CD47 or PD-L1, the immune response was suppressed, and tumors continued to grow.
Microenvironment mechanisms
Other components besides tumor cells contribute to primary or secondary resistance to checkpoint inhibitors, which include Tregs, myeloid derived suppressor cells (MDSCs), M2 macrophages, fibroblast, and other stromal cells.
Regulatory T cells are critical lymphocytes maintaining self-tolerance. Tregs can be identified by expression of FoxP3 transcription factor[17], and are known to suppress Teff responses by secretion of inhibitory cytokines such as IL-10 and TGF-beta or direct cell contact[18]. Many human tumors are infiltrated by Tregs[19], and its depletion from the tumor microenvironment can enhance or restore anti-tumor immunity. Clinical responses to checkpoint inhibitors have been associated with augmentation of preexisting immune responses, however, many other tumors have a non-inflammed microenvironment.
Preliminary studies highlight the MDSCs as major regulators of immune responses in cancer. MDSCs were characterized by the expression of CD11b and Gr-1 markers[20] and they are implicated in promoting tumor cell invasion and metastasis[21]. By other hand, the presence of MDSCs correlates with reduced survival in colorectal and breast cancer patients[22]. The presence of MDSCs in the tumor microenvironment also correlates with decreasing efficacy of immunotherapies, including checkpoint inhibitors[23].
Tumor-associated macrophages (TAMs), including M1 and M2 macrophages are another subgroup of white cells that seem to influence responses to immune therapies. M1 macrophages are involved in promoting anti-tumor immunity, while M2 macrophages possess pro-tumorigenic properties[24,25]. Some reports suggest that macrophages can suppress T cell responses through PD-L1[26].
How the genomic landscape of a tumor is shaped by anti-tumor immunity has been explored using large-scale genomic data sets of tumor biopsies across different tumor types[27], defining some recurrently mutated genes that showed positive association with antitumor activity, including beta-2-microglobulin (β2M), HLA-A, -B and -C and Caspase 8. A resulting T cell gene expression profile can predict response to checkpoint inhibitors in a broad spectrum of tumor types.
Secondary resistance mechanisms
The potential mechanism of resistance after primary response to checkpoint blockade include development of escape mutations variants in cancer cells, lack of T cell recognition by downregulation of tumor antigen presentation and loss of T cell function[5]. Mutations, genetic deletions or epigenetic changes could lead to loss of expression of mutational neoantigens presented by MHC molecules, resulting in acquired resistance to checkpoint inhibitors.
Inhibitory immune checkpoints that are often expressed in the tumor microenvironment include Lag-3, Tim-3 and TIGIT: these receptors, although they belong to the same class or receptor as PD-1 and CTLA-4 exhibit unique functions, especially at tissue sites where they regulate distinct aspects of immunity and are under active investigation[28].
In a recent study, acquired resistance to PD-1 blockade immunotherapy in patients with melanoma was associated with defects in the pathway involved in interferon-receptor signaling JAK1or JAK2, concurrent with deletion of the wild-type allele[29]. A truncating mutation in the gene encoding the β2M was also identified. This mutation led to loss of surface expression of major MHC class I interfering the antigen presentation.
Another evidence of loss of antigen presenting machinery leading to acquired resistance to cancer immunotherapy is provided identifying a polyclonal CD8+ T cell response against KRAS G12D in tumor-infiltrating lymphocytes obtained from a patient with metastatic colorectal cancer[30]. Objective regression was observed of lung metastasis after the infusion of HLA-C*8:02-restricted tumor infiltrating lymphocytes that were composed of four different T cell clonotypes that specifically targeted KRAS G12D. However, one lung lesion progressed on evaluation 9 months after therapy: the lesion was resected and found to have lost the chromosome 6 haplotype encoding the HLA-C*8:02 class I MHC molecule. The loss of expression of this molecule provided new evidence of secondary immune evasion.
Checkpoint inhibitors and current predictive biomarkers
Checkpoint inhibitors in clinical practice
Clinical development of checkpoint inhibitors has been complemented in some cases with the inclusion in clinical practice of predictive biomarkers. Table 1 correlates drug, tumor indications and approved biomarker in every histological type.
Ipilimumab
This is a fully human IgG1 monoclonal antibody that blocks CTLA-4 increasing the number of reactive T-effector cells which mobilize a direct T-cell immune response against tumor cells[31,32]. Reduction of Treg function mediated by CTLA-4 blockade and Treg depletion (increasing the intratumoral Teff/Treg ratio) can also reduce Treg function contributing to an antitumor effect[33]. As monotherapy, Ipilimumab is indicated for the treatment of advanced melanoma[34] and, in combination with Nivolumab, it is indicated for the treatment of advanced melanoma and renal cell carcinoma (RCC)[35].
Ipilimumab was the first checkpoint inhibitor that showed activity in solid tumors, demonstrating a clear benefit in overall survival (OS) in two phase 3 studies involving patients with metastatic melanoma[36,37]. It is also the first monoclonal antibody that showed increased activity in combination with other checkpoint inhibitor, Nivolumab, a monoclonal antibody which binds to the PD-1 receptor[38].
There is no required biomarker needed in the treatment of melanoma in monotherapy. Relative to Nivolumab combination in the treatment of RCC, low tumor PD-L1 expression predicts progression-free survival (PFS) and OS, but it is not a condition to administrate with Ipilimumab[35].
Tremelimumab
This is a fully human anti-CTLA4 monoclonal antibody under current investigation. The antitumor activity in melanoma patients showed contradictory results[39,40]. Ongoing studies in pancreatic cancer[41], hepatocellular carcinoma[42], NCSCL[43] or malignant mesothelioma[44,45] will define its role in the standard of care in the future clinical practice. Currently, the combination of Tremelimumab and Durvalumab, an antibody against the PD-L1 receptor (see above) seems to be an active combination in head and neck carcinomas and NSCLC (see above).
Pembrolizumab
Pembrolizumab is a humanized monoclonal antibody (IgG4/kappa isotype) which binds to de PD-1 receptor and blocks its interactions with ligands PD-L1 and PD-L2, potentiating anti-tumor responses. As monotherapy, Pembrolizumab is indicated for the adjuvant treatment of stage III melanoma after complete resection and, in the metastatic setting, for advanced melanoma, non-small cell lung carcinoma (NSCLC), relapse or refractory cHL, platinum-resistant urothelial carcinoma and head and neck squamous cell carcinoma (HNSCC)[46].
In advanced NSCLC, Pembrolizumab is indicated in the first-line treatment if tumor expression of PD-L1 presents ≥ 50% of tumor proportion score (TPS) and, after chemotherapy (at least one prior chemotherapy regimen), if tumor expression of PD-L1 presents ≥ 1% of TPS. In HNSCC, it is indicated only in patients whose tumors progressing on or after platinum containing chemotherapy and express PD-L1 with ≥ 50% TPS. In advanced urothelial carcinoma in patients not eligible for cisplatin-containing chemotherapy, Pembrolizumab is indicated if tumor express PD-L1 with a combined positive score (CPS) ≥ 10. No biomarkers of response are needed in melanoma or Hodgkin lymphoma.
Nivolumab
Nivolumab is a human immunoglobulin G4 monoclonal antibody which binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2. Combined Nivolumab and Ipilimumab (anti-CTLA-4) mediated inhibition results in improved anti-tumor responses in metastatic melanoma. Thus, in monotherapy it is indicated in melanoma patients both in the adjuvant and in the metastatic setting. It is also approved in combination with Ipilimumab in advanced melanoma. In NSCLC, it is used in locally advanced or metastatic patients after prior chemotherapy. In RCC as monotherapy or in combination with Ipilimumab, it is indicated for the first-line treatment of adults with intermediate/poor-risk tumors. Finally, Nivolumab is an active treatment and it is indicated in relapsed or refractory cHL, recurrent or metastatic platinum-resistant SCCHN and after platinum-based therapy of urothelial carcinoma[35].
Relative to Nivolumab monotherapy in melanoma patients, an increase in PFS and OS for the combination of Nivolumab and Ipilimumab is established only in patients with low tumor PD-L1 expression.
Atezolizumab
Atezolizumab is a monoclonal antibody that directly binds to PD-L1 and provides a dual blockade of the PD-L1 and B7.1 receptors, releasing PD-L1/PD-1 mediated inhibition of the immune response, including reactivating the antitumor immune response. Atezolizumab spares the PD-L2/PD-1 interaction allowing PD-L2/PD-1 mediated inhibitory signals to persist. Atezolizumab is indicated for the treatment of urothelial carcinoma after a platinum-containing regimen, and in NSCLC after prior chemotherapy[47].
Patients with previously untreated urothelial carcinoma should be selected for treatment based on the tumor expression of PD-L1: if patients are considered ineligible for platinum-containing regimen, PD-L1 expression 5% permit Atezolizumab administration. After first line chemotherapy regimen, urothelial carcinoma and NSCLC patients can receive Atezolizumab irrespectively of PD-L1 expression.
Avelumab
Avelumab is a human monoclonal IgG1 antibody directed against the PD-L1 receptor. It is indicated as monotherapy for the treatment of adult patients with metastatic Merkel cell carcinoma (MCC)[48], a rare, aggressive skin cancer. In the pivotal phase 2 trial[49] patient selection was not based on PD-L1 expression. A recent phase III trial[50] demonstrated a PFS combining Avelumab plus Axitinib among patients with PD-L1 positive advanced renal-cell carcinoma compared with the first line standard-of-care Sunitinib. Avelumab is under current investigation in urothelial carcinoma[51], breast cancer[52], NSCLC and other solid tumors.
There is no described biomarker of response or primary resistance in MCC patients currently treated with Avelumab.
Durvalumab
Durvalumab is a human monoclonal IgG1 antibody directed against the PD-L1 receptor. It is indicated for the treatment of locally advanced, unresectable NSCLC whose disease has not progressed following platinum-based chemoradiation therapy[53,54]. In clinical trials, Durvalumab presented antitumor activity against NSCLC alone or in combination with Tremelimumab[55]. This combination seems to be active against recurrent or metastatic HNSCC in low or no PD-L1 tumor cell expression, suggesting an increasing in tumor or microenvironment immunogenicity due to combination treatment[56]. Durvalumab is also active against urothelial carcinoma[57,58]. The combination with a poly (ADP-Ribose) polymerase inhibitor Olaparib and a vascular endothelial growth factor receptor 1-3 inhibitor Cediranib presented antitumoral activity in preliminary studies[59].
In NSCLC patients, Durvalumab is indicated in patients whose tumors express PD-L1 on ³ 1% of total tumor cells.
Predictive biomarkers in current clinical practice
Table 2 shows the response checkpoint inhibitor biomarkers approved in daily clinical practice using for solid tumor treatment.
Table 2.
Biomarkers for checkpoint inhibitors
| Biomarker | Tumor Type | Definition |
|---|---|---|
| Tumor Proportion Score | NSCLC | Percentage of tumor cells expressing the protein PD-L1 relative to the total number of tumor cells |
| Combined Proportion Score | UC H&NSCC | Percentage of PD-L1 expression in tumor and infiltrating immune cells relative to the total number of tumor cells |
| MSI-H | Tumor Agnostic | Microsatellite Instability |
PD-L1: programmed dead ligand-1; NSCLC: non small-cell lung cancer; H&NSCC: head and neck squamous cell carcinoma; UC: urothelial carcinoma
PD-L1 expression and predictive scores
Knowledge of the role played by each receptor modulating the immune synapse has allowed the development of some useful response biomarkers. The most important biomarker in routine clinical practice is determination of the expression of PD-L1 on tumor cells surface. Thus, the first clinical studies with Nivolumab in a wide spectrum of tumors[60] found that response to checkpoint inhibitors were restricted to patients who expressed PD-L1, while patients without PD-L1 tumor expression were frequently unresponsive.
On the other hand, a preliminary study of Atezolizumab followed an adaptive design to allow some PD-L1-positive enriched cohorts[61]. Herbst et al.[61] establish IHQ expression levels in relation to the percentage of tumor cells expressing the protein, as 0 (< 1%), 1 (1%-5%), 2 (> 5%-10%) or 3 (> 10%). Again, a positive correlation was found between patients with tumors with positive PD-L1 expression and the inhibitory checkpoint response, in this case against the direct blockade of PD-L1. The key concept of a positive, preferred association between PD-L1 expression and tumor response, independent of PD-L1 expression has emerged as a key concept in oncology.
Thus, in advanced melanoma, there is a correlation between presence of intratumoral CD8+ T cells and response to Pembrolizumab[62], confirming the need for a preliminary presence of peritumoral CD8+ T-cells together with the inhibitory response of the PD-1/PD-L1 axis as requirements for a potent antitumor response. Likewise, the presence of CD8- T cells in subsequent biopsies after progression in these patients tends to decrease, reflecting an important mechanism of secondary resistance.
In fact, response to checkpoint inhibitors based on expression of PD-L1 presents a continuous gradation: there is a clear positive correlation between protein expression and tumor response, and this correlation is especially intense in patients with high PD-L1 expression of PD-L1. This phenomenon was described in studies that led to the approval of Pembrolizumab in NSCLC. In the KEYNOTE-001 study, Garon et al.[63] assessed PD-L1 expression in solid tumor samples in training group and a validation group, defining the percentage of membranous tumor PD-L1 expression as a proportion score. Thus, patients who presented a PD-L1 of at least 50% presented higher response rates and longer OS. These responses remain prolonged over time[64]. Currently, the NCCN[65] NSCLC Panel recommends IHC testing for PD-L1 expression before first-line treatment in patients with metastatic NSCLC, however, there are no unified criteria for the administration of the entire set of all the different checkpoint inhibitors in relation to expression levels of PD-L1. In spite of this, assessing PD-L1 expression in locally-advanced disease after chemo-radiotherapy and in first line after progression to a systemic treatment seems to be a reasonable strategy.
Solid bladder urothelial carcinoma is another type of tumor in which good responses have been verified with checkpoint inhibitors. The active drugs administer are Prembrolizumab[66], Atezolizumab[67], Nivolumab[68], Avelumab[69] or Durvalumab[58]. However, the value of PD-L1 expression as a predictive response factor showed disparate results. A specific response score (PD-L1 or CPS) was determined in urothelial carcinoma patients treated with Pembrolizumab: his was defined as the percentage of PD-L1 expression in tumor and infiltrating immune cells relative to the total number of tumor cells, with positive markers scoring higher than 10%. Unexpectedly, the benefit of the monoclonal antibody appeared to be independent of PD-L1 expression on tumor and infiltrating cells[66].
Finally, checkpoint inhibitors are active drugs against HNSCC. Both Nivolumab[70] and Pembrolizumab[71,72] showed antitumor activity in recent clinical trials. In the same way, the latest studies with Pembrolizumab include determination of PD-L1[73]. In a second interim analysis of this Phase III trial comparing single-agent Pembrolizumab versus platinum-based chemotherapy or the combination, patients with CPS > 20 showed a better OS.
MSI-H status
Microsatellite Instability is a biological condition that results from inactivation of the DNA repair system known as Mismatch Repair (deficient Mismatch Repair or dMMR). It is characterized by a large increase in point mutations, which are detected by a marked increase in sequence instability in chromosomal regions called microsatellites (High Microsatellite Instability or MSI-H). Inactivation of MLH1, MSH2, MSH6 or PMS2 Mismatch Repair protein can occur sporadically (usually by inactivated expression caused by promoter methylation) or less frequently as a germline mutation (Lynch Syndrome)[74,75].
MSI-H condition presents a high predictive value in terms of response to checkpoint inhibitor therapies[76]. MSI-H tumors shows a marked clinical response in comparison with MMR proficient tumors. Currently, the US Food and Drug Administration has approved Pembrolizumab for the treatment of unresectable or advanced solid tumors after standard treatment (especially in colorectal cancer after oxaliplatin, irinotecan and fluoropyrimidine progression, or in first line advanced NSCLC). It is important to note that antitumor activity is not restricted to a specific tumor type (“site agnostic”). Therefore, now in 2019, the possibility of using Nivolumab and the combination of Nivolumab-Ipilimumab has been opened. In fact, MSI-H is predictive of Lynch Syndrome across a much broader tumor spectrum than currently appreciated, such urothelial, melanoma, gastric or gem cell tumors. However, use of checkpoint inhibitors in this scenario is currently under active investigation.
New approaches to optimize the activity of checkpoint inhibitors: rational development of clinical trials
The most important strategies to optimize the antitumoral activity of checkpoint inhibitors in clinical research are described in Figure 2. Notably, a first strategy focuses on combining anti PD-1 or PD-L1 monoclonal antibodies with a second drug that blocks other molecular signaling pathways, especially those related to mechanisms of resistance to checkpoint inhibitors. An alternative approach involves the design of monoclonal antibodies with dual activity, which can modulate the action of PD-1 or PD-L1 blockade. Bispecific antibodies can act in base to a synergistic effect by simultaneously modulating the activity of two immunoregulating targets. Finally, development of new response biomarkers, such as the infiltration of TAMs in tumor specimens, can optimize the rational use of checkpoint inhibitors in clinical setting. The following table summarizes several approaches in current clinical and translational research to optimize the use of checkpoint inhibitors.
Figure 2.
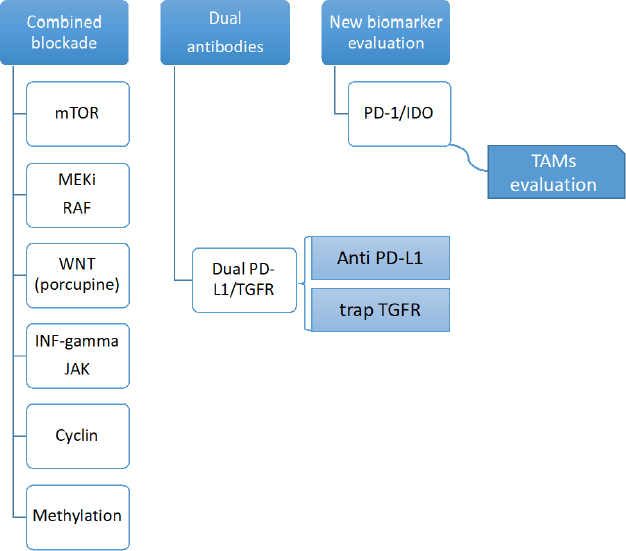
Strategies to optimize the antitumor activity of checkpoint inhibitors
The following table lists some ongoing clinical studies that show different strategies for the improvement of immunotherapy based on checkpoint inhibitors.
Conclusion
Development of checkpoint blockade-based therapy is an important medical issue in the treatment of some aggressive tumors, such as melanoma, NSCLC or urothelial carcinoma. Knowledge of the mechanisms of tumor response and resistance are currently under active investigation. However, this is not reflected in our clinical practice: the percentage of PD-L1 expression, TPS or the CPS are only partially informative biomarkers to evaluate primary resistance.
By other hand, other studies focused on clinical factors, such as laboratory data, specific site of metastasis or clinical comorbidities could be added to a comprehensive evaluation of resistance of checkpoint inhibitors[77,78].
To date, there are no validated studies in clinical practice to test for secondary resistance to checkpoint inhibitors, and in primary resistance (based on the existence of MSI or based on the PD-L1 expression) these appear to be insufficient. A concerted effort is, therefore, required to understand all these biological mechanisms and to apply them in routine clinical practice.
Declarations
Authors’ contributions
Contributed equally to this work: Rodriguez-Pascual J, Ayuso-Sacido A, Belda-Iniesta C
Availability of data and materials
Not applicable.
Financial support and sponsorship
We are grateful for the financial support from the “Fondo de Investigaciones Sanitarias” (PI17/01489), the Miguel Servet Program (CPII16/00056) del Instituto de Salud Carlos III, and the Ministerio de Economiía y Competitividad - FEDERER (RTC-2016-4990-1, RTC-2015-3846-1).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
References
- 1.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8:1069–86. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 2.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–9. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, et al. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 4.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 5.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321–37.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu C, Peng W, Xu C, Lou Y, Zhang M, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. doi: 10.1158/1078-0432.CCR-12-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peng W, Chen JQ, Liu C, Malu S, Creasy C, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–16. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–5. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404.e9. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353:399–403. doi: 10.1126/science.aae0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009;69:8693–99. doi: 10.1158/0008-5472.CAN-09-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lastwika KJ, Wilson W, Li QK, Norris J, Xu H, et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016;76:227–38. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 15.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casey SC, Tong L, Li Y, Do R, Walz S, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–66. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–18. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–52. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 22.Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118:2254–65. doi: 10.1182/blood-2010-12-325753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63:247–57. doi: 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 25.Hu W, Li X, Zhang C, Yang Y, Jiang J, et al. Tumor-associated macrophages in cancers. Clin Transl Oncol. 2016;18:251–8. doi: 10.1007/s12094-015-1373-0. [DOI] [PubMed] [Google Scholar]
- 26.Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–62. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 32.O’Day SJ, Hamid O, Urba WJ. Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4): a novel strategy for the treatment of melanoma and other malignancies. Cancer. 2007;110:2614–27. doi: 10.1002/cncr.23086. [DOI] [PubMed] [Google Scholar]
- 33.Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26:5275–83. doi: 10.1200/JCO.2008.17.8954. [DOI] [PubMed] [Google Scholar]
- 34. Annex I. Summary Of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/yervoy-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 35. Annex I. Summary Of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/opdivo-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 36.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 38.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camacho LH, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol. 2009;27:1075–81. doi: 10.1200/JCO.2008.19.2435. [DOI] [PubMed] [Google Scholar]
- 40.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH Jr, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014;25:1750–5. doi: 10.1093/annonc/mdu205. [DOI] [PubMed] [Google Scholar]
- 42.Sangro B, Gomez-Martin C, de la Mata M, Iñarrairaegui M, Garralda E, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59:81–8. doi: 10.1016/j.jhep.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 43.Planchard D, Yokoi T, McCleod MJ, Fischer JR, Kim YC, et al. A Phase III Study of Durvalumab (MEDI4736) With or Without Tremelimumab for Previously Treated Patients With Advanced NSCLC: Rationale and Protocol Design of the ARCTIC Study. Clin Lung Cancer. 2016;17:232–6.e1. doi: 10.1016/j.cllc.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Calabrò L, Morra A, Fonsatti E, Cutaia O, Amato G, et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2013;14:1104–11. doi: 10.1016/S1470-2045(13)70381-4. [DOI] [PubMed] [Google Scholar]
- 45.Maio M, Scherpereel A, Calabrò L, Aerts J, Perez SC, et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017;18:1261–73. doi: 10.1016/S1470-2045(17)30446-1. [DOI] [PubMed] [Google Scholar]
- 46. Annex I. Summary Of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/keytruda-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 47. Annex I. Summary Of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/tecentriq-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 48. bavencio-epar-product-information_en.pdf. Available from: https://www.ema.europa.eu/en/documents/product-information/bavencio-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 49.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374–85. doi: 10.1016/S1470-2045(16)30364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019;380:1103–15. doi: 10.1056/NEJMoa1816047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. 2018;19:51–64. doi: 10.1016/S1470-2045(17)30900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat. 2018;167:671–86. doi: 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med. 2017;377:1919–29. doi: 10.1056/NEJMoa1709937. [DOI] [PubMed] [Google Scholar]
- 54. Annex I. Summary Of product characteristics. Available from: https://www.ema.europa.eu/en/documents/product-information/imfizi-epar-product-information_en.pdf. [Last accessed on 27 Sep 2019].
- 55.Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siu LL, Even C, Mesía R, Remenar E, Daste A, et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1-Low/Negative Recurrent or Metastatic HNSCC: The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol. 2019;5:195–203. doi: 10.1001/jamaoncol.2018.4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma: Updated Results From a Phase 1/2 Open-label Study. JAMA Oncol. 2017;3:e172411. doi: 10.1001/jamaoncol.2017.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol. 2016;34:3119–25. doi: 10.1200/JCO.2016.67.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JM, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination With Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J Clin Oncol. 2017;35:2193–202. doi: 10.1200/JCO.2016.72.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;27;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Natur. 2014;515:568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 64.Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, et al. Five-Year Overall Survival for Patients With Advanced Non–Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J Clin Oncol. 2019;37:2518–27. doi: 10.1200/JCO.19.00934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gubens MA, Davies M. NCCN Guidelines Updates: New Immunotherapy Strategies for Improving Outcomes in Non-Small Cell Lung Cancer. J Natl Compr Canc Netw. 2019;17:574–8. doi: 10.6004/jnccn.2019.5005. [DOI] [PubMed] [Google Scholar]
- 66.Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. 2017;376:1015–26. doi: 10.1056/NEJMoa1613683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389:67–76. doi: 10.1016/S0140-6736(16)32455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18:312–22. doi: 10.1016/S1470-2045(17)30065-7. [DOI] [PubMed] [Google Scholar]
- 69.Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, et al. Avelumab, an Anti-Programmed Death-Ligand 1 Antibody, In Patients With Refractory Metastatic Urothelial Carcinoma: Results From a Multicenter, Phase Ib Study. J Clin Oncol. 2017;35:2117–24. doi: 10.1200/JCO.2016.71.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N Engl J Med. 2016;375:1856–67. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17:956–65. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 72.Cohen EEW, Soulières D, Le Tourneau C, Dinis J, Licitra L, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393:156–67. doi: 10.1016/S0140-6736(18)31999-8. [DOI] [PubMed] [Google Scholar]
- 73.Wiegand S, Wichmann G, Dietz A. Perspectives of Induction With Chemo and/or Immune Check Point Inhibition in Head and Neck Organ Preservation Treatment. Front Oncol. 2019;9:191. doi: 10.3389/fonc.2019.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 75.Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87.e3. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–56. doi: 10.1002/cam4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berardi R, Rinaldi S. The role of an immune checkpoint score in resected non-small cell lung cancer patients’ prognosis. J Thorac Dis. 2017;9:3480–2. doi: 10.21037/jtd.2017.09.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cortellini A, Buti S, Santini D, Perrone F, Giusti R, et al. Clinical Outcomes of Patients with Advanced Cancer and Pre-Existing Autoimmune Diseases Treated with Anti-Programmed Death-1 Immunotherapy: A Real-World Transverse Study. Oncologist. 2019;24:e327–37. doi: 10.1634/theoncologist.2018-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.