

Abstract
PROteolysis TArgeting Chimeras (PROTACs) are hetero-bifunctional small molecules that can simultaneously recruit target proteins and E3 ligases to form a ternary complex, promoting target protein ubiquitination and degradation via the Ubiquitin-Proteasome System (UPS). PROTACs have gained increasing attention in recent years due to certain advantages over traditional therapeutic modalities and enabling targeting of previously “undruggable” proteins. To better understand the mechanism of PROTAC-induced Target Protein Degradation (TPD), several computational approaches have recently been developed to study and predict ternary complex formation. However, mounting evidence suggests that ubiquitination can also be a rate-limiting step in PROTAC-induced TPD. Here, we propose a structure-based computational approach to predict target protein ubiquitination induced by cereblon (CRBN)-based PROTACs by leveraging available structural information of the CRL4A ligase complex (CRBN/DDB1/CUL4A/Rbx1/NEDD8/E2/Ub). We generated ternary complex ensembles with Rosetta, modeled multiple CRL4A ligase complex conformations, and predicted ubiquitination efficiency by separating the ternary ensemble into productive and unproductive complexes based on the proximity of the ubiquitin to accessible lysines on the target protein. We validated our CRL4A ligase complex models with published ternary complex structures and additionally employed our modeling workflow to predict ubiquitination efficiencies and sites of a series of cyclin-dependent kinases (CDKs) after treatment with TL12–186, a pan-kinase PROTAC. Our predictions are consistent with CDK ubiquitination and site-directed mutagenesis of specific CDK lysine residues as measured using a NanoBRET ubiquitination assay in HEK293 cells. This work structurally links PROTAC-induced ternary formation and ubiquitination, representing an important step toward prediction of target “degradability.”
Keywords: PROTAC, degrader, CRL4A ligase complex, computational drug design, ubiquitination
Abbreviations: CDK, cyclin-dependent kinase; CRBN, Cereblon; CRL, Cullin-ring ubiquitin ligase; CTD, C-terminal domain; DCAF, DDB1-CUL4 associated factor; DDB1, UV-damaged DNA binding; NTD, N-terminal domain; PDB, protein data bank; PPI, protein-protein interaction; PROTAC, proteolysis targeting chimeras; Rbx1, RING protein; RING, really interesting new gene; RMSD, root-mean-square deviation; SR, substrate receptor; TPD, target protein degradation; Ub, ubiquitin; UPS, ubiquitin-proteasome system
PROteolysis TArgeting Chimeras (PROTACs) are a promising therapeutic modality that utilizes the Ubiquitin-Proteasome System (UPS) to selectively induce target protein degradation (TPD) (1). PROTAC molecules have three components: a warhead ligand that binds the target protein, an E3 ligand that binds the E3 ligase, and a linker connecting these two ligands. PROTACs can interact with both the target protein and the E3 ligase simultaneously. This induced molecular proximity triggers ubiquitination of the target protein, leading to proteasomal degradation. The induced TPD mechanism confers PROTACs with several advantages over traditional small molecules, including the ability to target proteins previously considered “undruggable” due to lack of a functional binding pocket, event-driven rather than occupancy-driven pharmacology leading to prolonged duration of effect, and enhanced specificity (2, 3).
In recent years, interest has rapidly accelerated in understanding the mechanism of PROTAC-induced TPD from a therapeutic perspective, with specific focus on identifying the rate-limiting event/s of in vitro and in vivo drug actions. Known steps include binary complex formation between the PROTAC and the target protein, and the PROTAC and the E3 ligase (PROTAC/target and PROTAC/E3); ternary complex formation between the target protein, the PROTAC, and the E3 ligase (target/PROTAC/E3); initial ubiquitination of the target; ubiquitin chain elongation; and proteasomal degradation. Various techniques have been applied to characterize the kinetics of binary and ternary interactions including surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), and NanoBRET (4), based on the premise that ternary complex formation is a key driver of TPD (5, 6). In addition, many ternary complex structures have been solved for different target/E3 systems, including: Brd4/VHL (5, 7, 8), Brd4/CRBN (9), SMARC2/VHL (10), and Bcl2-L-1/VHL (11). These kinetics studies and reported structures have demonstrated that the type and length of linkers affect PROTAC activity (9, 12, 13) and that protein–protein interactions (PPIs) play a role in the ternary complex formation process (5). However, the extent to which PPI-mediated cooperativity contributes to ternary complex formation remains an open question. Although cooperativity has been identified in most reported cases (5, 7, 10, 14), noncooperative (13) and even anticooperative (9, 13) examples have also been identified.
To help better understand the structural underpinnings of PROTAC-induced ternary complex formation, many computational approaches to model this phenomenon have been developed (15, 16, 17, 18). Most modeling workflows share two main steps: (1) protein–protein docking to study the PPI between target and E3; and (2) linker conformation sampling to screen different PROTAC conformations, since most reported PROTACs have long and flexible linkers. Both Drummond et al. and Zaidman et al. used published ternary complex crystal structures to validate their pipelines and found a good match between their models and the PDB structures (15, 16, 17). Taking into consideration the dynamics of the ternary system, Bai et al. (18) (our previous study) evaluated a modeling approach using the number of ternary complex models generated as a proxy for the conformational flexibility of a given complex and reported a positive correlation with cellular degradation. This idea about conformational flexibility of playing a significant role in ternary complexes has been further validated by Eron et al. (19) using hydrogen-deuterium exchange–mass spectrometry (HDX-MS). Furthermore, recent studies have reported that ternary complex binding affinities do not always correlate with target protein cellular degradation (20, 21), indicating that although TPD always requires ternary complex formation, ternary complex formation does not always lead to TPD, suggesting that at least in some cases, additional factors can drive degradation.
A current key challenge in the field is to address the knowledge gap relating to the events that occur after ternary complex formation and before target degradation. Target protein ubiquitination is an essential step following ternary complex formation during PROTAC-induced TPD (Figure 1). Target ubiquitination is sequentially catalyzed by three enzymes: ubiquitin (Ub)-activating enzyme (E1), Ub-conjugating enzyme (E2), and ligase enzyme (E3) (22). PROTACs are involved in the E2/E3 Ub conjugation cascade where E2/Ub binds E3 resulting in the transfer of Ub to a lysine (Lys) residue on the surface of the target protein (23, 24, 25). Although intuitively, ternary complex formation should correlate with ubiquitination, Vieux et al. (26) demonstrated that for a Brd4BD1/CRBN targeted series of PROTACs, there was no correlation between ternary complex formation and target ubiquitination rate. In another recent paper, an interesting concept was mentioned that may help to explain this observation. The authors posit that not every ternary complex conformation can be considered a “productive” conformation that can induce ubiquitination, meaning that at least in theory, there may be physical limitations to ubiquitination depending on the orientation or distance of accessible lysines on the target to the Ub on E2 (27).
Figure 1.

PROTAC-induced target protein degradation (TPD). PROTAC binds either target protein or E3 enzyme (CRBN) to form a binary complex. Then a ternary complex (target protein – PROTAC – CRBN) forms, followed by ubiquitination, involving the CRL4A complex. Target protein is shown in slate; substrate receptor protein or DCAF (CRBN) is shown in forest; adaptor protein (DDB1) is shown in pale green; Cul4A is shown in light blue; Rbx1 is shown in pink; NEDD8 is shown in wheat; E2 (UBE2D1) is shown in pale yellow, ubiquitin is shown in sky blue, and proteasome is shown in light purple.
To better understand the relationship between ternary complex formation and ubiquitination, and to further study how “productive versus unproductive ternary complex conformations” may affect ubiquitination, here we propose a structure-based computational approach to predict PROTAC-induced target protein ubiquitination by integrating E3 ligase complex structural information with ternary complex ensemble modeling (Fig. 1). The primary goal of establishing this workflow is to advance in silico capabilities for predicting target degradability. In this work, we use the CRBN-based Cullin-Ring ubiquitin Ligase 4A (CRL4A) complex as our E3 model system because CRBN-based PROTACs are among the most frequently studied PROTACs (28). CRL4A belongs to the Really Interesting New Gene (RING) E3 family, one major E3 family (29, 30). In the CRL4A ligase complex, CUL4A operates as a scaffold protein that recruits UV-Damaged DNA Binding (DDB1) protein as the adaptor protein at the tip of the N-terminal domain (NTD), and RING protein (Rbx1) at the C-terminal end (CTD) (31, 32). DDB1 has a large rotatable range (33, 34) and can interact with many different DDB1-CUL4 Associated Factors (DCAFs—also known as substrate receptors, SR), including CRBN (35). DCAFs can connect the CRL4A complex with various target proteins. At the C-terminal end of CUL4A, the ubiquitin-like protein NEDD8 covalently attaches to CUL4A at the C-terminal “winged-helix B” (WHB) domain, inducing a conformational change in the Rbx1/CUL4A CTD, enabling the engagement of the E2/Ub complex with the Rbx1/CUL4/NEDD8 complex (36, 37, 38). The aggregate of these PPIs in the CRL4A complex results in the target protein being presented to the E2/Ub for ubiquitination (Fig. 1).
Taken together, the diversity of DCAFs and the mobility of DDB1 lead the CRL4A ligase complex to have a large ubiquitination zone (340 Å × 110 Å × 30 Å), useful for degrading various target proteins (33, 34). Two recent papers report a positive correlation between the number of accessible lysines in the ubiquitination zone and target degradability or degradation efficiency, respectively (39, 40). Zhang et al. (40) investigated kinase degradability in a CRL4 ligase system (without neddylation) using a machine learning approach, and Dixon et al. (39) explored degradation efficiency using a neddylated CRL2 ligase system combining biophysical data, molecular dynamics simulations, and modeling of the CRL2 ensemble. In this work, we propose a pipeline combining different conformations of the CRL4A ligase complex ensemble (CRBN/DDB1/CUL4A/Rbx1/NEDD8/E2/Ub) with the ternary complex ensemble to predict whether a given ternary complex conformation is productive or unproductive based on the proximity of the target surface exposed Lys residues to the C-terminal of Ub. Furthermore, we attempt to predict target ubiquitination efficiency, potential ubiquitination sites, and validate our predictions with a NanoBRET assay to measure ubiquitination of the wild-type CDK protein and the CDK protein carrying a mutation at specific lysine residues. Although many other factors can affect PROTAC-induced ubiquitination/degradation, our structure-based modeling approach specifically addresses the relationship between ternary complex formation and lysine proximity for ubiquitination with CRBN-based PROTAC systems in this study.
Results
Computational approach development
Our proposed approach includes three parts. Part One is to generate the ternary complex (target protein/PROTAC/CRBN) ensemble that encompasses the diverse interacting modes among these three components. Part Two involves modeling different CRL4A ligase complex conformations due to the mobility of DDB1. Part Three consists of aligning each conformation from the ternary complex ensemble with each conformation from the CRL4A ligase complex ensemble through CRBN, which is common to both ensembles. The resulting ensemble of target/PROTAC/CRBN/DDB1/CUL4A/Rbx1/NEDD8/E2/Ub complexes allows for classification of “productive” or “unproductive” ternary complexes based on the proximity of Ub to exposed lysine/s on the target. Unproductive ternary complex conformations are excluded, and productive ternary complex conformations are used to predict target ubiquitination efficiency and the specific lysines on the target that are involved in degradation. Details regarding specific parameters applied in different software suites and others are described in Experimental procedures.
Part One: Generating the ternary complex ensemble
The ternary complex ensemble is generated using an approach reported in our previous paper (18). Briefly, target protein/E3 docking is performed using the Rosetta software suite (41, 42), PROTAC linker conformers are created using RDKit (43), and ternary complex is generated by aligning linker conformers to the docking decoy ensemble. To simplify downstream processing of the alignment between the sizeable ternary complex ensemble and the CRL4A ligase complex ensemble, the ternary complex ensemble is clustered using the Multiscale Modeling Tools for Structural Biology (MMTSB) clustering script (44) with an RMSD threshold of 3 Å. Models in the same cluster are considered as the same conformation and are ranked based on the ternary complex interface energy calculated by Rosetta. The top models of each cluster/conformation are selected to advance in the workflow.
Part Two: Modeling the CRL4A ligase complex ensemble
One key challenge of modeling the CRL4A ligase complex is the significant mobility of DDB1, the adaptor protein. DDB1 has three WD40 domains (BPA, BPB, and BPC), and the BPB domain rotates along its interface with BPA/BPC (33), leading to diverse CRL4A ligase complex conformations. To capture this conformational flexibility, all 36 available full-length DDB1 structures from PDB were collected and clustered using the RMSD cutoff of 2 Å (Table S1). After clustering, 12 different DDB1 conformations were identified (three depicted in Fig. 2A) and aligned to DDB1/CUL4A (PDBID: 2HYE) through the BPB domain of DDB1, leading to 12 DDB1/CUL4A complex conformations. CRBN was added using a solved CRBN/DDB1 structure as the template (PDBID: 4TZ4), aligning CRBN/DDB1 to DDB1/CUL4A through the BPA/BPC domain of DDB1. To engage E2/Ub at the CTD of CUL4A, neddylation is required and the CRL1 complex structure (PDBID: 6TTU) was utilized as the template (45), since CRL family members, including CUL1 and CUL4, share sequence (34∼48%) and structure homology (Table S2 and Fig. S1A) (38). After modeling neddylation, the CRL4A complex (CRBN/DDB1/CUL4A/Rbx1/NEDD8/E2/Ub) was created with Rbx1/NEDD8/E2/Ub derived from 6TTU. UBE2D1 is utilized as the ubiquitination priming E2 in CRL4A complex models. Finally, Rosetta was used to optimize these CRL4A ligase complex models.
Figure 2.

CRL4 ligase complex models with different DDB1 conformations.A, different DDB1 conformations. B, CRL4A ligase complex conformation with reasonable distance (≤4 Å distance) between CRBN and E2 enzyme. C, CRL4A ligase complex conformation with a big gap (>4 Å distance) between CRBN and E2 enzyme. D, CRL4A ligase complex conformation with clashing between CRBN and E2 enzyme. CRBN is shown in forest; DDB1 is shown in pale green; Cul4A is shown in light blue; Rbx1 is shown in pink; NEDD8 is shown in wheat; E2 (UBE2D1) is shown in pale yellow; and ubiquitin is shown in sky blue.
The 12 CRL4A ligase complex conformations (Table S3) can be further classified into three groups based on the relative positions of CRBN and E2: (1) “ring-forming CRL4A ligase complexes,” with a potential interface (≤4 Å distance) between CRBN and E2 (conformations 1, 2, 4, 5, and 9; Fig. 2B); (2) “open CRL4A ligase complexes,” with a greater distance (>4 Å) between CRBN and E2 (conformations 3, 10, 11, and 12; Fig. 2C); and (3) “clashing CRL4A ligase complexes,” with a clash between CRBN/DDB1 and E2/NEDD8/Ub (conformations 6, 7, and 8; Fig. 2D). The CRL1 complex cryo-EM structure indicates an interface between the E2 enzyme (UBE2D2) and the substrate receptor protein (β-TRCP; one of many CRL1 ligase substrate receptors) (45). Baek et al. also reported that a mutation of UBE2D (H32A) at this interface causes a 200-fold reduction in substrate (target protein) ubiquitination priming when combined with another mutation on NEDD8 (I44A), while single mutant NEDD8 (I44A) only reduces substrate priming by tenfold. We hypothesized that this interface between E2 and the substrate receptor protein is conserved between CRL1 and CRL4, helping to stabilize the whole CRL complex by closing the “ring” of this system, providing a scaffolding to increase the probability of interaction between Ub and target protein, thereby resulting in improved ubiquitination efficiency. We revisit this assumption in the Discussion. Consequently, only the “ring-forming CRL4A ligase complex” group (conformations 1, 2, 4, 5, and 9) was selected for advancement in our workflow. One possible alternative scenario is that an interface could form between the target protein and E2/NEDD8. This hypothetical CRL4A complex would also serve to stabilize the complex for ubiquitin transfer. This situation could occur for the second conformation group (conformations 3, 10, 11, and 12) where the gap between CRBN and E2 would be filled by the target protein. For the current study, this alternative scenario is not considered but is elaborated on in the Discussion section and may be revisited in the future.
Part Three: Combining the ternary complex and CRL4A ligase complex ensembles
The ternary complex conformations from the ensemble are aligned with all five CRL4A ligase complex conformations via the common constant, CRBN, to generate the complete “ternary + CRL4A ligase” complex (target/PROTAC/CRBN/DDB1/CUL4A/Rbx1/NEDD8/E2/Ub) required for transfer of the ubiquitin to the target (Fig. 3). The resulting complex ensemble can be categorized into three different scenarios. In the first scenario, the target protein is in proximity to the E2/Ub (Fig. 3, B and C). One or several surface-exposed lysine residues on the target are sufficiently close (≤16 Å) to the C-terminal of ubiquitin to serve as potential ubiquitination site/s (Fig. 3D). These ternary complex conformations are classified as “productive ternary complex conformations.” In contrast, if the target protein is far away (>16 Å) from E2/Ub (Fig. 3, E and F) or clashes with E2/Ub (Fig. 3, G and H), no target ubiquitination can occur. These ternary complex conformations are considered as “unproductive ternary complex conformations” and all the models belonging to these conformations, based on the clustering step in Part One, are excluded. The cutoff distance (16 Å) chosen here originated from a benchmarking study using ternary complexes with known degradation outcomes (shown in Results), where solved ternary complex structures were aligned to the CRL4A ligase ensemble and distances between Lys residues and Ub were calculated. This cutoff distance could be adjusted/optimized in future studies. Analysis of interaction energies indicated no significant difference between productive and unproductive ternary complexes (Fig. S2), suggesting that ternary interaction energy is not a critical parameter using this modeling approach. In summary, we hypothesize that for a given ternary complex ensemble, the larger the fraction of productive ternary complexes, the higher the ubiquitination efficiency.
Figure 3.

Productive ternary complex conformation versus unproductive ternary complex conformation.A, different target protein/PROTAC/CRBN ternary conformations. B and C, ternary complex conformation 1 is combined with the CRL4A ligase complex; accessible Lys residues are shown on the target protein surface. D, interface of the target protein and E2-Ub, showing distance between target protein surface Lys residues and C-terminus of Ub. E and F, ternary complex conformation 2 is combined with the CRL4A ligase complex and target protein is far away from Ub. G and H, ternary complex conformation 3 combined with CRL4A ligase complex with a clash between target protein and E2. Target protein is shown in slate (conformation 1), blue (conformation 2), and purple (conformation 3); Lys residues are shown in stick, orange; CRBN is shown in forest; DDB1 is shown in pale green; Cul4A is shown in light blue; Rbx1 is shown in pink; NEDD8 is shown in wheat; E2 (UBE2D1) is shown in pale yellow; ubiquitin is shown in sky blue; and PROTACs are shown in sphere, gray.
Validation of CRL4A ligase complex ensemble using reported ternary complex crystal structures
To validate our computational approach, three published CRBN-based ternary complex crystal structures with different target proteins and degraders were utilized: 6BOY (Brd4/dBET6/CRBN) (9), 5HXB (GSTP1/CC-885/CRBN) (46), and 5FQD (CK1α/lenalidomide/CRBN) (34). Due to the limited number of available CRBN-based ternary complex structures, both PROTAC-containing (6BOY) and Molecular Glue (MG; 5HXB and 5FQD)-containing structures were included. We aligned CRBN from each solved ternary complex structure with our final CRL4A ligase complex ensemble (c1, c2, c4, c5, and c9). In the Brd4/dBET6/CRBN case, when combined with three CRL4A ligase complex conformations (c1, c2, and c4), Ub accessible Lys residues on the surface of Brd4 were identified (Table S4). For example, in Brd4/dBET6/CRBN/CRL4A ligase conformation 2 complex (Fig. 4, A and D), Lys141 of Brd4 is around 12 Å away from Ub and may serve as a potential ubiquitination site. In Brd4/dBET6/CRBN/CRL4A ligase conformation 1, Lys155 is close to Ub (∼15 Å); and in Brd4/dBET6/CRBN/CRL4A ligase conformation 4, Lys141 is close to Ub (∼16 Å). Similar results were observed for the MG cases (Table S4): (1) GSTP1/CC-885/CRBN showed accessible Lys residues close to Ub when aligned with CRL4A ligase conformations 1 (Lys622, ∼12 Å), 2 (Lys541, ∼14 Å), and 4 (Lys541, ∼12 Å and Lys622, ∼14 Å, Fig. 4, B and E); (2) CK1α/lenalidomide/CRBN showed accessible lysine residues close to Ub when aligned with CRL4A ligase conformation 2 (Lys51, ∼14 Å, Fig. 4, C and F). Interestingly, Lys51 observed in CK1α/lenalidomide/CRBN/CRL4A ligase conformation 2 does not match the ubiquitinated target lysines detected in previous in vitro (Lys8, Lys62, and Lys179) (34) and in vivo (Lys65 and Lys225) (47) ubiquitination assays, but Lys62 was close to Ub when we aligned 5FQD with CRL4A ligase conformation 12 (Fig. S3). In the Computational approach section, we mentioned this CRL4A ligase conformation category (c3, c10, c11, and c12) may be available for ubiquitination if the target protein can fill the relatively large gap between CRBN and the E2/NEDD8 region of CRL4A. In the CK1α/lenalidomide/CRBN/CRL4A ligase conformation 12 case, CK1α bridged CRBN and E2 in support of this idea.
Figure 4.

Published ternary complex structures align with CRL4A ligase complex models.A and D, Brd4 – dBET6 (PROTAC) – CRBN (PDBID: 6BOY) aligned with CRL4A ligase complex conformation 2. Residue Lys141 of Brd4 is ∼11.8 Å to C-terminal of Ub. B and E, GSTP1 – CC-885 (molecular glue) – CRBN (PDBID: 5HXB) aligned with CRL4A ligase complex conformation 4. Residue Lys541 and Lys622 of GSTP1 are 11.8 Å and 14.3 Å to C-terminal of Ub, respectively. C and F, CK1α – lenalidomide (molecular glue) – CRBN (PDBID: 5FQD) aligned with CRL4A ligase complex conformation 2. Residue Lys51 of CK1α is 13.5 Å to C-terminal of Ub. Target proteins (Brd4, GSTP1, and CK1α) are shown in slate; Lys residues are shown in stick, orange; CRBN is shown in forest; DDB1 is shown in pale green; Cul4A is shown in light blue; Rbx1 is shown in pink; NEDD8 is shown in wheat; E2 (UBE2D1) is shown in pale yellow; ubiquitin is shown in sky blue and PROTAC and MGs are shown in sphere, gray.
To conclude, when integrating our CRL4A ligase ensemble (five conformations: c1, c2, c4, c5, and c9) with three different reported ternary complex structures, at least one lysine residue on the target protein surface was observed in close proximity to the Ub (∼11–16 Å). These model-identified lysines can be considered as potential ubiquitination sites that may facilitate or contribute to the target protein degradation observed for these degraders. Furthermore, we observed that different potential Ub sites were identified when aligning the ternary complex structure with different CRL4A ligase complex conformations. This observation may help explain why multiple lysine residues were detected in ubiquitination assays (in vitro and in vivo), such as in the CK1α/lenalidomide/CRBN case.
Productive and unproductive models help explain different degradation results of CDK family members induced by TL12–186
TL12–186 is a CRBN-based pan-kinase PROTAC designed with a low selectivity kinase inhibitor (TL13–87) warhead, a flexible linker (PEG), and pomalidomide as the CRBN ligand. TL12–186 induces degradation of multiple kinases, including many Cyclin-Dependent Kinase (CDK) family members, with different efficiencies (Dmax50) (21). Therefore, TL12–186 is a good tool molecule to help understand the PROTAC-induced TPD mechanism. Riching et al. (4) monitored TL12–186-induced degradation of 16 CDK family members in vitro. Degradation profiles generated across the family showed striking diversity that was not correlated with basal expression level, and cell-cycle-dependent degradation was further analyzed for CDK2. The authors hypothesized that the remaining cell-cycle-associated CDK proteins, CDK1, CDK4, and CDK6 may also exhibit cell cycle dependence given the similarities in degradation profiles within this subclass. For the remaining 12 CDKs, it is not understood why different degradation efficiencies were observed across proteins with such high structural homology of their kinase domains (Fig. 5A).
Figure 5.

Kinase domain alignment of different CDKs; lysine distribution of CDK7, CDK8, and CDK12; and ubiquitination of CDK7, CDK8, CDK9, CDK12, and CDK13.A, CDK5 (PDBID: 4AU8), CDK7 (PDBID: 1UA2), CDK8 (PDBID: 5HBE), CDK9 (PDBID: 4EC8), CDK12 (PDBID: 4NST) and CDK13 (PDBID: 5EFQ) are aligned based on their kinase domain. B–D, Lys residue distribution of CDK7, CDK8, and CDK12, kinase domain. CDK5 is shown in lemon, CDK7 is shown in slate, CDK8 is shown in marine, CDK9 is shown in cyan, CDK12 is shown in green cyan and CDK13 is shown in blue. Lys residues are shown in orange. E, ubiquitination of endogenous CDK7, CDK8, CDK9, CDK12, and CDK13 proteins was measured by NanoBRET ubiquitination assay following treatment with 1 μM serial dilution of TL12–186 PROTAC. Data are presented as mean ± SD of n = 4 technical replicates.
Here, we apply our computational workflow to generate CRBN/TL12–186/CDKx ternary complexes, classify them as productive or unproductive, and identify potential ubiquitination sites for all the CDKs investigated in the Riching et al. paper with available PDB structures (CDK1, CDK2, CDK4, CDK6, CDK5, CDK7, CDK8, CDK9, CDK12, and CDK13). Because degradation of CDK1, 2, 4, and 6 is potentially affected by the cell cycle, we focus on the other six CDKs (CDK5, 7, 8, 9, 12, and 13). Among these six CDKs, CDK8 has the lowest degradation efficiency (Table 1). One potential explanation for CDK8 low degradation efficiency is the reported low binding affinity (KINOMEscan score is 96) to TL12–186 (21). However, CDK9 also has low binding affinity (KINOMEscan score is 47) to TL12–186, but it shows high degradation efficiency; furthermore, CDK7 has higher binding affinity (KINOMEscan score is 0.1) than CDK9, but it shows lower degradation efficiency than CDK9 (21). This implies that binary formation may not be the rate-limiting step for degradation efficiency in this case study. We also studied the numbers and positions of lysine residues on the CDK kinase domain surfaces (Table 1, Fig. 5, B and C, Table S5, and Fig. S4, A–D). Interestingly, CDK8 has 33 lysines, the largest number of surface lysine residues. On the contrary, CDK7 only has 14, half the surface lysines as CDK8 but shows twofold higher degradation (Fig. 5, B and C). Therefore, it appears there is no direct correlation between the number of surface lysine residue and PROTAC-induced TPD in this case study.
Table 1.
Different CDK family members with multikinase degrader
| Target protein | PROTAC (TL12–186) | Dmax50 (nM)a | # Surface lysine | # Ternary complex models | % Productive | Ubiquitination EC50 (nM) |
|---|---|---|---|---|---|---|
| CDK5 | 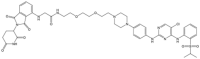 |
62 | 22 | 4157 | 57 | Not tested |
| CDK7 | 233 | 14 | 14,452 | 24 | 428 | |
| CDK8 | >500 | 33 | 29,920 | 14 | 888 | |
| CDK9 | 58 | 26 | 9802 | 33 | 297 | |
| CDK12 | 7 | 31 | 4359 | 77 | 16 | |
| CDK13 | 7 | 29 | 12,018 | 62 | 7 |
Dmax50 values are from Riching et al. (48).
Ternary complex ensembles were generated for all CDKs. No correlation was observed between ternary complex interaction energy (calculated by Rosetta) and degradation efficiency. This was not surprising since anticooperative interactions between the target protein and E3 are known to result in efficient target degradation (9, 13). In our previous study (18), a positive correlation was observed between the number of ternary complex models generated by this workflow and degradation efficiency. However, here, CDK8 ternary complex modeling resulted in the highest number of models, yet it had the lowest degradation efficiency (Table 1). This may be because of the disconnect between ternary formation and degradation, as discussed previously (18). In our new approach, we incorporated a CRL4A ligase complex ensemble modeling step to filter out unproductive ternary complex models (Table 1). Interestingly, using this approach, only a small percentage (<20%) of the CDK8 ternary complex ensemble was classified as productive. In contrast, a majority of the CDK12 and CDK13 ternary complex ensembles, which have the highest degradation, were classified as productive (>60%). Consistent with our hypothesis, rank ordering of the six CDKs investigated supports the idea that the percentage of the ternary complex ensemble identified as productive may result in increased ubiquitination efficiency, with an increased likelihood of degradation.
Application of models to predict ubiquitination efficiency of CDK proteins: Experimental corroboration using NanoBRET ubiquitination assay
As ubiquitination has been shown to be correlated with degradation rate (4), we sought to measure ubiquitination of CDK proteins to better understand and validate our model predictions of productive ternary complexes. To study ubiquitination of CDK proteins in live cells, NanoBRET assays were performed with HEK293 CRISPR-edited cell lines containing HiBiT fused to specific endogenous CDK proteins and ectopic expression of LgBiT and HaloTag-Ub. Complementation of HiBiT and LgBiT produces a bright luminescent signal, which serves as the energy donor for NanoBRET, with HaloTag-Ub as the energy acceptor. When CDK proteins are ubiquitinated, a NanoBRET signal is produced, which increases in correlation with extent of ubiquitination. Treatment with a serial dilution of 1 μM TL12–186 for 3 h resulted in different ubiquitination potencies of CDK proteins, with EC50 ranking in strong agreement with the model predictions of productive ternary complexes (Table 1), as well as matching the previously published Dmax50 ranking (48). Ubiquitination was most potent on CDK12 and CDK13 (Fig. 5E and Table 1), with these proteins having the highest percentage of productive ternary conformations (77% and 62%, respectively) as predicted by the model. CDK9 exhibited a reduced ubiquitination potency compared with CDK12 and CDK13 (Table 1) consistent with a lower percent productive ternary conformations (∼33%) and CDK7, with ∼24% productive ternary complex models, exhibited even weaker ubiquitination. Finally, CDK8 with the lowest percent productive ternary complex models (∼14%) had very little ubiquitination above baseline levels across the concentration series, suggesting very low ubiquitination efficiency. Taken together, our ubiquitination results show strong alignment with our computational modeling predictions for five of the six CDK proteins, consistent with our hypothesis that degradation efficiency is strongly dependent on ubiquitination efficiency.
Application of computational workflow to predict CDK lysine ubiquitination sites: Experimental corroboration using site-directed mutagenesis
To further pressure-test our approach, we used the ternary-CRL4A ligase models that derived from productive ternary complexes to identify CDK lysine residues that could serve as potential ubiquitination sites (Fig. S5). For example, due to the proximity of CDK5 Lys56 to Ub (Fig. 6, A and B), Lys 56 was identified as a likely ubiquitination site. Since our approach involved integration of two ensembles, multiple lysines were identified as potential ubiquitination sites for each CDK protein. To investigate the validity of our lysine ubiquitination site identification approach, we ran a Ubifast assay, which involved screening the whole HEK293 proteome for ubiquitinated lysines after treatment with 1 μM TL12–186 (Fig. S6). Surprisingly, few CDK lysines were identified as significantly ubiquitinated post TL12–186 treatment by Ubifast, and fewer still matched our model predictions. However, upon further examination of the data, we also observed that many of the Ubifast-identified ubiquitination sites were not predicted to be solvent exposed, indicating that at 3 h, we may be missing the kinetic window for direct observation of ubiquitinated target. Consequently, we elected to select ubiquitinated CDK lysines for site-directed mutagenesis studies purely based on the overlap between our model prediction and a Ubifast-identified ubiquitination site. Based on this approach, we identified CDK5 Lys56, CDK9 Lys88, and CDK2 Lys6 for further study (Fig. 6C).
Figure 6.

Point mutant generation with predicted ubiquitin accessible lysine residues of CDK5, CDK2, and CDK9.A and B, CDK5 – CRL4A ligase complex conformation 2 model with Lys56 as the predicted ubiquitin accessible lysine residue. CDK5 is shown in lemon; Lys residues are shown in stick, orange; CRBN is shown in forest; DDB1 is shown in pale green; Cul4A is shown in light blue; Rbx1 is shown in pink; NEDD8 is shown in wheat; E2 (UBE2D1) is shown in pale yellow; ubiquitin is shown in sky blue; and TL12–186 is shown in sphere, gray. C, predicted ubiquitin accessible lysine residues ubiquitination level measured by UbiFast assay. Data are presented as mean ± SD of n = 2 technical replicates. D, CDK5, (E) CDK2, and (F) CDK9, both WT and mutant, kinetic ubiquitination measured by NanoBRET ubiquitination assay. Data are presented as mean ± SD of n = 4 technical replicates.
With experimental validation in hand, single lysine-to-arginine point mutations in these three CDKs were incorporated into plasmids that were transiently transfected into HEK293 cells (which can lead to higher protein expression levels). After treatment with 1 and 10 μM TL12–186, ubiquitination was significantly reduced to baseline levels in the CDK5 K56R mutant in a dose-dependent manner (Fig. 6D), indicating that Lys56 is a critical ubiquitination site of TL12–186 induced CDK5 ubiquitination, as our model predicted. In the CDK2 K6R case, reduced ubiquitination was observed with 10 μM treatment, but no change was captured with 1 μM treatment (Fig. 6E). In the CDK9 K88R case, ubiquitination did not decrease, in fact, an even higher ubiquitination extent was observed with 1 μM treatment (Fig. 6F). Interestingly, these results also align with the Ubifast data (Fig. 6C). To explain the difference in results for these three mutants, we first excluded the possibility of ternary complex destabilization, at least theoretically, as these mutated resides are not at the interface of target protein and CRBN; therefore, they are not expected to lead to conformational change. One potential explanation is the presence of additional Ub accessible lysine residues identified in proximity of the Ub for a given ternary-CRL4A ligase model. For example, in both the CDK9 (Fig. S7, A and B) and CDK2 models (Fig. S7, C and D), there were other lysine residues close to Ub (within 16 Å), such as CDK9 Lys18 and CDK2 Lys24. The increase in ubiquitination efficiency of CDK9 K88R may be due to an increased affinity for an alternative site following mutation. Furthermore, this suggests that a given lysine residue on a given target using a given PROTAC may be “totally responsible” (CDK5), “partially responsible” (CDK2 and CDK9), or “not responsible” for ubiquitination, and our approach may help to classify these three scenarios.
Discussion
To better understand the mechanism of PROTAC-induced TPD and the relationship between ternary complex formation and target protein ubiquitination, we propose a computational workflow that integrates both ternary complex and CRL4A ligase complex structural information to aid in the identification of “productive” ternary complexes within the ensemble; where productive ternary complexes are classified based on the proximity of at least one surface lysine on the target protein to the C-terminus of the ubiquitin on the E2. The workflow enables prediction of target protein ubiquitination efficiency based on the percentage of the productive ternary complexes relative to the total number of models in the ensemble. Furthermore, we utilize the workflow to predict, at least in some cases, the specific lysine residues on the target that may serve as ubiquitination sites. Finally, we validate our predictions using mass-spectrometry-based ubiquitination analysis and NanoBRET ubiquitination assay with wild-type and lysine mutant CDK proteins.
Our approach demonstrates potential utility in identifying ternary complex conformations that will lead to efficient target ubiquitination and identifying specific lysines as potential ubiquitination sites; however, several challenges remain. First, the accuracy of our prediction could be affected by the available protein structures. If full-length target protein is not utilized, we may miss potential lysine residues as ubiquitination sites. Furthermore, missing domains may cause a clash with the CRL4A ligase complex and lead to inaccurate predictions. Full-length target protein is not only an important consideration for this structure-based computational modeling approach, it is also likely a key factor for wet-lab assays to assess in vitro or in vivo ubiquitination. Truncated protein constructs may lead to less reliable results. In this study, we used full-length structure for the majority of the CDK proteins for modeling (except CDK12 and CDK13), while all CDK proteins are full-length in ubiquitination assays.
Another concern of this approach is the assumption about the interface between CRBN and NEDD8/E2-Ub, which we make based on the Cryo-EM structure of CRL1 ligase complex (45). Although we have been unable to find additional direct evidence supporting this idea, a similar hypothesis has been discussed in previous neddylation studies (36, 49). Both Duda et al. and Saha et al. mentioned that the big gap (∼50 Å) between the target protein and E2-Ub needs to be bridged and stabilized for effective transfer of Ub to target protein. In their studies, they demonstrated that neddylation induces a conformational change in CRL that increases the proximity of the target relative to the Ub. Furthermore, this “ring-forming” CRL model is also briefly mentioned in Duda et al. paper. Therefore, in this work, we keep this “ring-forming” assumption, and we are interested to further validate it in our future study.
As outlined in the “Computational approach” section, we classify 12 distinct CRL4A ligase complex conformations into three categories: Category 1) “ring-forming CRL4A ligase complexes” with a potential interface between CRBN and NEDD8/E2/Ub (conformation c1, c2, c4, c5, and c9); Category 2) “open CRL4A ligase complexes,” where CRBN is far away from NEDD8/E2/Ub (conformation c3, c10, c11, and c12); and Category 3) “clashing CRL4A ligase complexes,” with a clash between CRBN/DDB1 and NEDD8/E2/Ub (conformation c6, c7, and c8). In our current approach, only Category 1 is included in the workflow. However, we recognize that Category 2 may also happen in real case when the target protein fills the gap between CRBN/DDB1 and NEDD8/E2/Ub forming an alternative stabilized complex to promote ubiquitination. Several studies partially support the Category 2 hypothesis. One example is the CK1α/lenalidomide/CRBN case (PDBID: 5FQD) (34), described in the Results session. When PDB structure 5FQD was aligned with our five CRL4A ligase conformations from Category 1, a reasonable CK1α/lenalidomide/CRL4A ligase complex model with accessible lysine residues close to the Ub was obtained (Fig. 4, C and F), yet these lysine residues did not match published ubiquitination data (34, 47). However, when the PDB structure was aligned to CRL4A conformation 12 from Category 2, Lys62 was in proximity of the Ub (Fig. S3), in alignment with a CK1α ubiquitination site from a published study (34). In this case, a potential interface forms not between CRBN and NEDD8/E2/Ub, but between the target protein and NEDD8/E2/Ub. Although we did not include Category 2 conformations in our current approach due to a lack of direct structural evidence and an increase in modeling complexity, there are testable hypotheses generated from this work, and they could be interesting to follow-up on.
An additional Category 2 scenario is where a ring-between-RING (RBR) E3 ligase protein bridges the gap between the two ends of the target protein/degrader/ligase complex and acts to transfer the ubiquitin between the E2 and the target protein. For example, in a series of recently solved PDB structures (PDBID: 7B5S, 7B5R, 7B5M, 7B5L, and 7B5N), Horn-Ghetko et al. (50) reported that Ub was first transferred from E2 to ARIH1, an RBR-type E3 ligase, then to the target protein (Cyclin E) in the CRL1 ligase system. A similar scenario has also been described in the CRL5 ligase system with ARIH2, a different RBR-type E3 ligase (51). To date, ARIH1 has been shown to interact with multiple CRLs, while ARIH2 is specific for CRL5 (29). To our knowledge, it is still unclear that if there is an RBR-type E3 ligase involved in CRL4A system. Therefore, we did not include it in our current approach.
Additional future computational workflow improvements include adding additional DDB1 conformations as part of the CRL4A ligase ensemble. In the current workflow, only 12 CRL4A ligase complex conformations were generated based on the number of unique DDB1 structures available. Given the well-established mobility of DDB1, the current ensemble likely does not adequately capture the conformational flexibility of this protein. Adding additional conformations may help improve the accuracy of “productive versus unproductive” ternary complex prediction. Furthermore, several other steps could be integrated in the future pipeline, including a Ub chain extension step. The current implementation of the workflow focuses solely on the priming step of ubiquitination without considering downstream polyubiquitination steps. One recent study observed that TRIP12 (Thyroid Hormone Receptor–Interacting Protein 12, a HECT-type E3 ligase) could generate K29/K48-brached Ub chains and enhance PROTAC-induced target protein degradation (52). Inclusion of such downstream target protein degradation enhancers into future iterations of the workflow may improve its predictive power and/or make it more generalizable.
Despite these challenges, our model demonstrates remarkable predictive power in identifying productive complexes that align with measured ubiquitination potency ranking, ultimately matching the previously observed degradation ranking of CDK proteins. Furthermore, our model demonstrates the capacity to predict specific target lysine residues for ubiquitination based upon their proximity to the Ub. For proteins in which more than one lysine residue was identified in productive complexes, mutation of one to arginine resulted in either reduced ubiquitination (CDK2) or a potential shift in lysine preference (CDK9), suggesting that our model may ultimately enable “tuning” of ubiquitination efficacy upon optimization of ternary complex conformations with available lysines. Such tuning may be further aided by analyzing ternary complex models with other PROTAC linkers containing different geometry or flexibility. Moreover, combining this approach with lead compound optimization could have broad utility in design and mechanistic characterization of efficacious PROTACs, as ubiquitination thus far remains the best predictor of successful degradation. The ability of our model to predict successful ubiquitination represents a significant advance forward in rational design of PROTAC molecules. A future extension of our structural modeling strategy may also aid understanding of polyubiquitination events leading to proteasomal degradation.
To summarize, in this work we present a structure-based computational approach to predict CRBN-based PROTAC-induced target protein ubiquitination by integrating ternary complex and CRL4A ligase complex structural information. Our method represents a small step toward: (1) linking ternary complex formation and target ubiquitination information to advance understanding of PROTAC-induced TPD; (2) arming researchers with a new tool to evaluate PROTAC molecules and their targets; and (3) providing a novel idea to better understand/explore the E3 ligase toolbox from a structural perspective.
Experimental procedures
Ternary complex ensemble modeling
Ternary complex ensembles were generated with the protocol as previously described (18), with slight modification. Linker conformers were sampled with RDKit with 0.5 Å RMSD as cutoff: a new conformer would be kept only with larger than 0.5 Å RMSD, compared with retained conformers. In the protein–protein docking step, instead of using global docking to generate 50,000 decoys without any constraints, a three-step docking method, with constraints, was applied. The constraints parameter was decided based on the median of the linker conformer length distribution. In the first step of docking, starting ternary complex structure was generated with PyMOL (version 2.4.0), and 20 decoys were generated with Rosetta (version 3.12) global docking. Then, top ten decoys were used as the start structures for the next round of global docking. Total 10 × 1000 decoys were generated, and top 5000 decoys were clustered by MMTSB clustering script (http://blue11.bch.msu.edu/mmtsb/Cluster.pl) with an RMSD threshold of 3 Å. The top one decoy of each cluster was ranked based on docking scores, and top 200 decoys (from 200 clusters) were passed for the next step. In the last docking step, 200 × 1000 decoys were generated with Rosetta local docking, still with “-spin” flag. Top 20,000 decoys were considered for the ternary complex generation. Ternary complex models were generated with ternary_model_prediction.py (18). Ternary complex models were minimized by Rosetta minimize_ppi and clustered as previously described.
CRL4A ligase complex ensemble modeling
All 36 full-length DDB1 PDB structures were downloaded and clustered with 2 Å RMSD as cutoff: 3I8E (conformation/cluster 1, c1), 3E0C (c2), 3EI4 (c3), 3E54 (c4), 4TZ4 (c5), 6FCV (c6), 4A08 (c7), 4A0B (c8), 4A0L (c9), 3EI3 (c10), 6PAI (c11), and 2B5L (c12) were used for CRL4A ligase complex model generation. To generate CRL4A ligase complex model, CUL4A (PDBID: 2HYE, with DDB1) was aligned to CUL1 (PDBID: 6TTU) based on partial CTD region (residues 416–672). Because of neddylation induced conformation change, residues 688 to 759 were extracted from CUL4A and aligned to 6TTU separately. UBE2D1 (PDB ID: 5FER) was aligned to 6TTU and DDB1 (from 2HYX)/CUL4A (from 2HYE)/NEDD8 (from 6TTU)/Rbx1 (from 6TTU)/UBE2D1 (from 5FER)/Ub (from 6TTU) was extracted. Then, each DDB1 conformation was aligned to this complex based on BPB domain of DDB1. Finally, CRBN was built in based on the CRBN/DDB1 complex (PDBID: 4TZ4). All 12 CRL4A ligase complex models (CRBN/DDB1/CUL4A/NEDD8/Rbx1/E2/Ub) were optimized with Rosetta relax.
Excluding unproductive ternary complexes
Top one ternary complex model of each cluster was aligned to each CRL4A ligase complex model based on CRBN, which was the shared part in both ternary complex model and CRL4A ligase complex model. Distances between each lysine residue on the surface of target protein and C-terminal of ubiquitin were calculated with PyMOL. Ternary complex model was considered as productive only when there was/were lysine residue(s) close to ubiquitin without any clashing.
CDK protein structures preparation
Available CDK protein structures were downloaded from PDB: CDK1 (6GU3), CDK2 (2G9X), CDK4 (2W96), CDK5 (4AU8), CDK6 (5L2T), CDK7 (1UA2), CDK8 (5HBE), CDK9 (4EC8), CDK12 (4NST), and CDK13 (5EFQ). The structure chosen for each CDK is either because it is the unique available structure for one CDK or because it contains the ligand that is highly similar to warhead (TL13–87). Binary complex of warhead and each CDK protein was generated with Schrodinger (Ligand Docking) (53). Binary complexes were minimized with Rosetta and passed to next the step, ternary complex ensemble modeling, which was described above.
Cell samples preparation for Ubifast assay
HEK293 cells (ATCC# CRL-1573) were cultured in MEM (ATCC) supplemented with 10% FBS (Gibco). Cells were treated for 3 h with 1 μM TL12–186 (Tocris) with the presence of 5 μM MG-132 (Sigma-Aldrich) or treated with only 5 μM MG-132 for 3 h; n = 2 was chosen for each treatment, allowing the investigation of a number of alternative treatment conditions within the same multiplexed experimental (selected results shown). By the end of treatment, HEK293 cells were harvested by Trypsin-0.25% EDTA (Gibco) and washed with cold DPBS. Cell pellets were snap frozen in liquid nitrogen and then stored at −80 °C.
Sample preparation for MS, cell lysis, and trypsin digestion
Protein digestion was carried out using modified S-Trap midi protocol (54) (Protifi). Briefly, cell pellets were lysed in 5% sodium dodecyl sulfate (SDS)(Sigma), 100 mM Triethylammonium bicarbonate (TEAB)(Sigma), pH 7.5, and sonicated with three 10 s intervals. Samples were clarified by centrifugation for 10 min at 16,000g. 5% SDS, 100 mM Tris(2-carboxyethyl)phosphine (Fisher), 400 mM 2-chloroacetamide (Sigma) was added at 1:10 ratio to the samples. Samples were heated for 10 min at 80 °C and then cooled down to RT. Samples were acidified with 12% phosphoric acid (Fisher) at 1:10 ratio. S-Trap buffer was added to the acidified sample at 6:1 ratio and mixed. Samples were loaded unto the S-Trap midi spin column by centrifugation at 4000g for 30 s. Four consecutive washes were carried out with 3 ml of S-Trap buffer by centrifugation at 4000g for 30 s. Samples were digested with MS-grade trypsin (Worthington Biochemical) at 1:20 wt:wt in 50 mM TEAB for 15 h at 37 °C. Digested peptides were eluted by centrifugation at 4000g followed by two consecutive elutions starting with 0.2% formic acid (FA)(Fisher), followed by 50% MeCN (Fisher) containing 0.2% FA. This eluted samples were dried completely with a centrifugal evaporator.
Ubiquitinated peptide enrichment and TMT labeling
Samples were prepared using modified Ubifast approach (55). Briefly, S-trap digested samples were resuspended in 1 ml of 1× Immunoaffinity Purification (IAP) bind buffer following the protocol outlined in the PTMScan HS Ubiquitin/SUMO remnant kit (#59322, Cell Signaling Technologies). Samples were sonicated at RT for 5 min, followed by centrifugation for 5 min at 10,000g at 4 °C to clarify the samples of insoluble particulates. Peptide concentrations were determined by colorimetric assay (Thermo Fisher Scientific). Samples were mass normalized to 1.5 mg of tryptic peptides and spiked with internal standard of custom ubiquitin-remnant peptides (i.e., peptides contain one internal lysine each, where the epsilon amine is modified with diglycine) in 1.5 ml final volume IAP bind buffer. Twenty microliters of PTMScan HS antibody-bead slurry in ice-cold phosphate buffered saline (PBS) was added to each clarified sample. Samples were incubated on an end-over-end rotator in a cold room at 4 °C for 2 h. Samples were subsequently washed with 1.5 ml of ice-cold IAP buffer followed by 1.5 ml ice-cold PBS and finally with 1.5 ml 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 8.5. The beads were resuspended in 200 μl of 100 mM HEPES, pH 8.5 for on-bead TMTpro (Thermo Fisher Scientific) labeling. TMTpro reagents (Lot #VI306841) were reconstituted in 20 μl anhydrous MeCN (Sigma). TMTpro labeling was carried out on-bead by adding 10 μl of reconstituted TMTpro reagent to respective samples in parallel. TMTpro labels for the MG-132-only treatment were 126 & 127N, and for the MG-132 plus TL12–186 treatment were 130C & 131N. The samples were incubated at RT for 10 min. The TMT labeling reaction was quenched by addition of 8 μl of 5% hydroxylamine followed by 5 min incubation at RT. The beads were washed with 1.5 ml ice-cold IAP wash buffer twice. TMTpro labeled samples on-bead were serially reconstituted in 1.5 ml IAP wash buffer and combined. The combined beads were washed with 1.5 ml ice-cold PBS. TMTpro labeled K-ε-GG peptides were eluted from beads with 70 μl of 0.15% TFA incubated for 5 min at RT twice sequentially. The eluted peptides were loaded on a C18 SPE spin tip (Pierce) and the flow-through was processed by pipettor-based C18 Ziptip (Millipore) following the protocols provided by the manufacturer. In summary, C18 tips were activated by 70 μl of MeCN and equilibrated with 70 μl 0.1% FA before loading of the 140 μl 0.15% TFA sample. The C18 tips were washed with 70 μl of 0.1% FA followed by elution with 70 μl of 70% MeCN 0.1% FA. Eluted peptides were pooled and dried down using speed vac. Peptides were reconstituted in 20 μl of 5% MeCN and 0.1% FA for LC/MS analysis.
LC-MS analysis
TMTpro labeled K-ε-GG enriched samples were analyzed on Orbitrap Exploris 480 mass spectrometer with FAIMS (Thermo Fisher Scientific) coupled to an UltiMate 3000 RSLCnano high-performance liquid chromatograph (Thermo Fisher Scientific). Two injections each of 6 μl were made for the LC-MS analysis. Samples were directly injected onto a μPAC C18 200 cm Analytical column (PharmaFluidics) heated to a temperature of 40 °C. Samples were analyzed using a 300 min LC -MS method at a flow rate of 300 nl/min. Mobile phase consisted of Solvent A (0.1% FA) and Solvent B (80% MeCN 0.1% FA). The LC method consisted of following gradient ramps: (min:%B) 1:3; 10:10; 245:45; 265:60; 270:98; 282:98; 283:1;289:1), followed by a 10 min equilibration run at 900 nl/min. MS1 was acquired at 120K resolution with AGC target of 3e5, mass range of 350 to 1550 m/z. MS2 was acquired at resolution of 60,000, AGC target of 2e5 with max fill time set at 200 ms, stepped HCD of 28, 33, and 55, isolation width set at 0.7 m/z. The first sample injection was analyzed with three different FAIMS CV of −30, −54, and −78, with a replicate injection analyzed with FAIMS CV of −42, −66, and −90; each CV was set at a cycle time of 2.5 s for both analyses. The data were acquired with APD on and precursor filter set at minimum 70% match.
Data analysis
Raw Orbitrap MS file data analysis was carried out in Proteome Discoverer 2.4 (Thermo Fisher Scientific). Spectral recalibration and main search functions utilized fully specific trypsin cleavage, with dynamic GG modification on lysine, static peptide N-terminal modification of TMTpro on any N-terminus, and static carbamidomethylation on cysteine. Both spectral recalibration and main searches utilized a Uniprot.fasta database of reviewed human proteins (SwissProt, downloaded March 8, 2018 with 25,889 entries) using the Sequest HT node with the following settings: fully specific trypsin, max missed cleavage sites of 2, minimum peptide length of 6, MS1 mass tolerance of 10 ppm, MS2 mass tolerance of 0.02 Da, static modifications of peptide N-terminus for TMTpro and cysteine carbamidomethyl, dynamic modification of peptide for methionine oxidation, lysine diglycine (i.e., ubiquitin remnant) and lysine TMTpro; dynamic modification of protein N-terminus for acetylation, loss of methionine, and loss of methionine coupled with acetylation. The peptide spectral matches were filtered using the Percolator (56) node for target FDR of 1% at the peptide level, with Target/Decoy Selection set to Concatenated with Validation based on q-value. The Reporter Ions Quantifier node used an Integration Tolerance of 20 ppm (Most Confident Centroid). For the consensus workflow on the Reporter Ions Quantifier node, Apply Quan Value Correction was set to True, Average Reporter S/N was set to 10 and Co-Isolation Threshold was set to 50 used for peptide quantification; a custom Quantification Method for TMTpro lot # VI306841 was created with only selected TMT Reporter Ion Isotope Distributions active to allow for utilization of the Total Peptide Amount setting under the Normalization and Scaling menu. ANOVA significance calculations based on individual peptides were carried out to determine the significance of the change.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (57) partner repository with the dataset identifier PXD030213 and 10.6019/PXD030213. Detailed MS information for all ubiquitinated (diGly) peptide sequences assigned is also summarized in Table S6.
Expression plasmids preparation for ubiquitination assays
CDK2 (NM_001798), CDK5 (NM_004935), and CDK9 (NM_001261) expression plasmids were obtained from Kazusa DNA Research Institute as pFN21A HaloTag CMV Flexi vectors (Promega). CDK2 and CDK5 were cloned into pFC32K vector (Promega) to generate a C-terminal NanoLuc fusion and CDK9 was cloned into pFN31K vector (Promega) to generate an N-terminal NanoLuc fusion, corresponding to the same termini tagging used previously for endogenous CDK proteins (48). Site-directed mutagenesis was performed on each vector to generate the lysine to arginine point mutants, CDK2 (K6R), CDK5 (K56R), and CDK9 (K88R), while maintaining the same NanoLuc tag placement as the corresponding wild-type CDK protein. The N-terminal HaloTag-Ubiquitin vector (Promega) consists of the first ubiquitin repeat, amino acids 1 to 76, of the human polyubiquitin-B precursor (NM_018955).
NanoBRET CDK ubiquitination assays
For ubiquitination experiments with HiBiT fusions to endogenous CDK proteins, 8 × 105 HEK293 cells clonally edited via CRISPR/Cas9 to express HiBiT on the N-terminus of CDK7, CDK8, and CDK9 or the C-terminus of CDK12 and CDK13 as previously described (48) were transfected with FuGENE HD (Promega) and 2 μg of HaloTag-Ubiquitin (Promega) in 6-well plates, and incubated overnight at 37 °C, 5% CO2. The following day, 2 × 104 transfected cells were replated into white 96-well tissue culture plates (Corning) containing 5% v/v LgBiT BacMam particles and HaloTag NanoBRET 618 Ligand (Promega). Background control wells lacking HaloTag NanoBRET 618 Ligand were also included. Cells were allowed to adhere overnight by incubating at 37 °C, 5% CO2 before treatment with a serial dilution of 1 μM TL12–186 (Tocris) for 3 h. NanoBRET NanoGlo (Promega) was prepared as a 5× concentration in Opti-MEM (Gibco) and added to the plate before collecting NanoBRET measurements on a CLARIOstar Plus microplate reader (BMG Labtech). For ubiquitination experiments with transient expression of either wild-type CDK protein or a single lysine-to-arginine point mutant, 8 × 105 HEK293 cells were transfected using FuGENE HD (Promega) with 2 μg of HaloTag-Ubiquitin and 0.02 μg NanoLuc CDK fusion in 6-well plates. The following day, 2 × 104 transfected cells were replated into white 96-well tissue culture plates in the presence or absence of HaloTag NanoBRET 618 Ligand, and the plate was incubated overnight at 37 °C, 5% CO2. The following day, medium was replaced with CO2-Independent medium (Gibco) containing 1× Vivazine (Promega), and plates were incubated at 37 °C, 5% CO2, for 1 h to allow luminescence to equilibrate prior to addition of DMSO or the indicated concentrations of TL12–186 (Tocris). Plates were then read every 5 min for a period of 5 h on a CLARIOstar Plus microplate reader (BMG Labtech) set to 37 °C. Dual-filtered luminescence was collected with a 460/80 nm bandpass filter (donor, NanoBiT or NanoLuc CDK protein) and a 610 nm long-pass filter (acceptor, HaloTag NanoBRET ligand) using an integration time of 0.5 s. Background subtracted NanoBRET ratios expressed in milliBRET units were calculated from the equation:
Fold increase in BRET was calculated by normalizing mBRET ratios to the average mBRET ratios for DMSO controls.
Data availability
Data are contained within the article and its supporting information. The entire CRL4A ligase and ternary complex ensembles can be obtained from corresponding author (Nan Bai) upon reasonable request.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare the following competing financial interest(s): N. B., A. M., H. W., T. M. A., S.-C. O., Y. Z., X. S., D. N. B., H. R., B. W. G., B. M. R., and S. C. H. are employees and stockholders of Amgen. K. M. R., D. L. D., and M. U. are employees and stockholders of Promega Corporation.
Acknowledgments
Special thanks are given to Ryan Potts, Amit Vaish, and Avijit Ghosh for thoughtful suggestions and continued support (48).
This study was funded jointly by Amgen and Promega where relevant, according to the author guidelines.
Author contributions
N. B. and S. C. H. conceptualization; N. B., K. M. R., A. M., T. M. A., D. N. B. and X. S. formal analysis; N. B., K. M. R., A. M. and H. W. methodology; T. M. A., S.-C. O., Y. Z., H. R., B. W. G., D. L. D., M. U., and B. M. R. validation; N. B., K. M. R., A. M., H. W., and S. C. H. writing—original draft; N. B., K. M. R., A. M., D. N. B., T. M. A., S.-C. O., and H. R., and S. C. H. writing—review and editing.
Edited by George DeMartino
Contributor Information
Nan Bai, Email: nbai@amgen.com.
Kristin M. Riching, Email: Kristin.Riching@promega.com.
Sara C. Humphreys, Email: shumph01@amgen.com.
Supporting information
References
- 1.Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. Protacs: Chimeric molecules that target proteins to the Skp1–cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neklesa T.K., Winkler J.D., Crews C.M. Targeted protein degradation by PROTACs. Pharmacol. Ther. 2017;174:138–144. doi: 10.1016/j.pharmthera.2017.02.027. [DOI] [PubMed] [Google Scholar]
- 3.Pettersson M., Crews C.M. PROteolysis TArgeting Chimeras (PROTACs)—past, present and future. Drug Discov. Today Technol. 2019;31:15–27. doi: 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riching K.M., Mahan S., Corona C.R., McDougall M., Vasta J.D., Robers M.B., Urh M., Daniels D.L. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem. Biol. 2018;13:2758–2770. doi: 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- 5.Gadd M.S., Testa A., Lucas X., Chan K.-H., Chen W., Lamont D.J., Zengerle M., Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 2017;13:514. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes S.J., Ciulli A. Molecular recognition of ternary complexes: A new dimension in the structure-guided design of chemical degraders. Essays Biochem. 2017;61:505–516. doi: 10.1042/EBC20170041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Testa A., Hughes S.J., Lucas X., Wright J.E., Ciulli A. Structure-based design of a macrocyclic PROTAC. Angew. Chem. Int. Ed. Engl. 2020;59:1727–1734. doi: 10.1002/anie.201914396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dragovich P.S., Pillow T.H., Blake R.A., Sadowsky J.D., Adaligil E., Adhikari P., Chen J., Corr N., dela Cruz-Chuh J., Del Rosario G. Antibody-mediated delivery of chimeric BRD4 degraders. Part 2: Improvement of in vitro antiproliferation activity and in vivo antitumor efficacy. J. Med. Chem. 2021;64:2576–2607. doi: 10.1021/acs.jmedchem.0c01846. [DOI] [PubMed] [Google Scholar]
- 9.Nowak R.P., DeAngelo S.L., Buckley D., He Z., Donovan K.A., An J., Safaee N., Jedrychowski M.P., Ponthier C.M., Ishoey M. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018;14:706. doi: 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farnaby W., Koegl M., Roy M.J., Whitworth C., Diers E., Trainor N., Zollman D., Steurer S., Karolyi-Oezguer J., Riedmueller C. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019;15:672–680. doi: 10.1038/s41589-019-0294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung C.-w., Dai H., Fernandez E., Tinworth C.P., Churcher I., Cryan J., Denyer J., Harling J.D., Konopacka A., Queisser M.A. Structural insights into PROTAC-mediated degradation of Bcl-xL. ACS Chem. Biol. 2020;15:2316–2323. doi: 10.1021/acschembio.0c00266. [DOI] [PubMed] [Google Scholar]
- 12.Lebraud H., Wright D.J., Johnson C.N., Heightman T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent. Sci. 2016;2:927–934. doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zorba A., Nguyen C., Xu Y., Starr J., Borzilleri K., Smith J., Zhu H., Farley K.A., Ding W., Schiemer J. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E7285–E7292. doi: 10.1073/pnas.1803662115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roy M.J., Winkler S., Hughes S.J., Whitworth C., Galant M., Farnaby W., Rumpel K., Ciulli A. SPR-measured dissociation kinetics of PROTAC ternary complexes influence target degradation rate. ACS Chem. Biol. 2019;14:361–368. doi: 10.1021/acschembio.9b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond M.L., Williams C.I. In silico modeling of PROTAC-mediated ternary complexes: Validation and application. J. Chem. Inf. Model. 2019;59:1634–1644. doi: 10.1021/acs.jcim.8b00872. [DOI] [PubMed] [Google Scholar]
- 16.Drummond M.L., Henry A., Li H., Williams C.I.J.B. Improved accuracy for modeling PROTAC-mediated ternary complex formation and targeted protein degradation via new in silico methodologies. J. Chem. Inf. Model. 2020;60:5234–5254. doi: 10.1021/acs.jcim.0c00897. [DOI] [PubMed] [Google Scholar]
- 17.Zaidman D., Prilusky J., London N. PRosettaC: Rosetta based modeling of PROTAC mediated ternary complexes. J. Chem. Inf. Model. 2020;60:4894–4903. doi: 10.1021/acs.jcim.0c00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai N., Miller S.A., Andrianov G.V., Yates M., Kirubakaran P., Karanicolas J. Rationalizing PROTAC-mediated ternary complex formation using Rosetta. J. Chem. Inf. Model. 2021;61:1368–1382. doi: 10.1021/acs.jcim.0c01451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eron S.J., Huang H., Agafonov R.V., Fitzgerald M.E., Patel J., Michael R.E., Lee T.D., Hart A.A., Shaulsky J., Nasveschuk C.G., Phillips A.J., Fisher S.L., Good A. Structural characterization of degrader-induced ternary complexes using hydrogen–deuterium exchange mass spectrometry and computational modeling: Implications for structure-based design. ACS Chem. Biol. 2021;16:2228–2243. doi: 10.1021/acschembio.1c00376. [DOI] [PubMed] [Google Scholar]
- 20.Bondeson D.P., Smith B.E., Burslem G.M., Buhimschi A.D., Hines J., Jaime-Figueroa S., Wang J., Hamman B.D., Ishchenko A., Crews C.M. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 2018;25:78–87.e75. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H.-T., Dobrovolsky D., Paulk J., Yang G., Weisberg E.L., Doctor Z.M., Buckley D.L., Cho J.-H., Ko E., Jang J. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 2018;25:88–99.e86. doi: 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickart C.M. Back to the future with ubiquitin. Cell. 2004;116:181–190. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 23.Deshaies R.J., Joazeiro C.A.P. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 24.Hurley J.H., Lee S., Prag G. Ubiquitin-binding domains. Biochem. J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henneberg L.T., Schulman B.A. Decoding the messaging of the ubiquitin system using chemical and protein probes. Cell Chem. Biol. 2021;28:889–902. doi: 10.1016/j.chembiol.2021.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vieux E.F., Agafonov R.V., Emerson L., Isasa M., Deibler R.W., Simard J.R., Cocozziello D., Ladd B., Lee L., Li H. A method for determining the kinetics of small-molecule-induced ubiquitination. SLAS Discov. 2021;26:547–559. doi: 10.1177/24725552211000673. [DOI] [PubMed] [Google Scholar]
- 27.Han B. A suite of mathematical solutions to describe ternary complex formation and their application to targeted protein degradation by heterobifunctional ligands. J. Biol. Chem. 2020;295:15280–15291. doi: 10.1074/jbc.RA120.014715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schapira M., Calabrese M.F., Bullock A.N., Crews C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019;18:949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- 29.Harper J.W., Schulman B.A. Cullin-RING ubiquitin ligase regulatory circuits: A quarter century beyond the F-box hypothesis. Annu. Rev. Biochem. 2021;90:403–429. doi: 10.1146/annurev-biochem-090120-013613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng N., Shabek N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017;86:129–157. doi: 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]
- 31.Zheng N., Schulman B.A., Song L., Miller J.J., Jeffrey P.D., Wang P., Chu C., Koepp D.M., Elledge S.J., Pagano M. Structure of the Cul1–Rbx1–Skp1–F box Skp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 32.Hannah J., Zhou P. Distinct and overlapping functions of the cullin E3 ligase scaffolding proteins CUL4A and CUL4B. Gene. 2015;573:33–45. doi: 10.1016/j.gene.2015.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer E.S., Scrima A., Böhm K., Matsumoto S., Lingaraju G.M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 34.Petzold G., Fischer E.S., Thomä N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4 CRBN ubiquitin ligase. Nature. 2016;532:127–130. doi: 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- 35.Angers S., Li T., Yi X., MacCoss M.J., Moon R.T., Zheng N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 36.Duda D.M., Borg L.A., Scott D.C., Hunt H.W., Hammel M., Schulman B.A. Structural insights into NEDD8 activation of cullin-RING ligases: Conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichermeier K.M. Doctoral dissertation. California Institute of Technology; 2020. Quantitative Characterization of Composition and Regulation of Cullin-RING Ubiquitin Ligases. [Google Scholar]
- 38.Baek K., Scott D.C., Schulman B.A. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr. Opin. Struct. Biol. 2021;67:101–109. doi: 10.1016/j.sbi.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon T., MacPherson D., Mostofian B., Dauzhenka T., Lotz S., McGee D., Shechter S., Shrestha U.R., Wiewiora R., McDargh Z.A., Pei F., Pal R., Ribeiro J.V., Wilkerson T., Sachdeva V., et al. Atomic-resolution prediction of degrader-mediated ternary complex structures by combining molecular simulations with hydrogen deuterium exchange. bioRxiv. 2021 doi: 10.1101/461830. [preprint] [DOI] [Google Scholar]
- 40.Zhang W., Burman S.S.R., Chen J., Donovan K.A., Cao Y., Zhang B., Zeng Z., Zhang Y., Li D., Fischer E.S., Tokheim C., Liu X.S. Machine learning modeling of protein-intrinsic features predicts tractability of targeted protein degradation. bioRxiv. 2021 doi: 10.1101/462040. [preprint] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lyskov S., Gray J.J. The RosettaDock server for local protein–protein docking. Nucleic Acids Res. 2008;36:W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leaver-Fay A., Tyka M., Lewis S., Lange O., Thompson J., Jacak R., Kaufman K., Renfrew P., Smith C., Sheffler W. ROSETTA3: This article is licensed under a Creative Commons Attribution 3.0 Unported Licence. An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landrum G. 2006. RDKit: Open-Source Cheminformatics. [Google Scholar]
- 44.Feig M., Karanicolas J., Brooks C.L., III MMTSB tool set: Enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Baek K., Krist D.T., Prabu J.R., Hill S., Klügel M., Neumaier L.-M., von Gronau S., Kleiger G., Schulman B.A. NEDD8 nucleates a multivalent cullin–RING–UBE2D ubiquitin ligation assembly. Nature. 2020;578:461–466. doi: 10.1038/s41586-020-2000-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matyskiela M.E., Lu G., Ito T., Pagarigan B., Lu C.-C., Miller K., Fang W., Wang N.-Y., Nguyen D., Houston J. A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature. 2016;535:252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 47.Krönke J., Fink E.C., Hollenbach P.W., MacBeth K.J., Hurst S.N., Udeshi N.D., Chamberlain P.P., Mani D.R., Man H.W., Gandhi A.K. Lenalidomide induces ubiquitination and degradation of CK1α in del (5q) MDS. Nature. 2015;523:183–188. doi: 10.1038/nature14610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riching K.M., Schwinn M.K., Vasta J.D., Robers M.B., Machleidt T., Urh M., Daniels D.L. CDK family PROTAC profiling reveals distinct kinetic responses and cell cycle–dependent degradation of CDK2. SLAS Discov. 2021;26:560–569. doi: 10.1177/2472555220973602. [DOI] [PubMed] [Google Scholar]
- 49.Saha A., Deshaies R.J. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell. 2008;32:21–31. doi: 10.1016/j.molcel.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horn-Ghetko D., Krist D.T., Prabu J.R., Baek K., Mulder M.P.C., Klügel M., Scott D.C., Ovaa H., Kleiger G., Schulman B.A. Ubiquitin ligation to F-box protein targets by SCF–RBR E3–E3 super-assembly. Nature. 2021;590:671–676. doi: 10.1038/s41586-021-03197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lumpkin R.J., Ahmad A.S., Blake R., Condon C.J., Komives E.A. The mechanism of NEDD8 activation of CUL5 ubiquitin E3 ligases. Mol. Cell. Proteomics. 2021;20:100019. doi: 10.1074/mcp.RA120.002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaiho-Soma A., Akizuki Y., Igarashi K., Endo A., Shoda T., Kawase Y., Demizu Y., Naito M., Saeki Y., Tanaka K. TRIP12 promotes small-molecule-induced degradation through K29/K48-branched ubiquitin chains. Mol. Cell. 2021;81:1411–1424.e7. doi: 10.1016/j.molcel.2021.01.023. [DOI] [PubMed] [Google Scholar]
- 53.New York, NY . Schrödinger, LLC; 2021. Maestro. [Google Scholar]
- 54.HaileMariam M., Eguez R.V., Singh H., Bekele S., Ameni G., Pieper R., Yu Y. S-Trap, an ultrafast sample-preparation approach for shotgun proteomics. J. Proteome Res. 2018;17:2917–2924. doi: 10.1021/acs.jproteome.8b00505. [DOI] [PubMed] [Google Scholar]
- 55.Udeshi N.D., Mani D.C., Satpathy S., Fereshetian S., Gasser J.A., Svinkina T., Olive M.E., Ebert B.L., Mertins P., Carr S.A. Rapid and deep-scale ubiquitylation profiling for biology and translational research. Nat. Commun. 2020;11:359. doi: 10.1038/s41467-019-14175-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.The M., MacCoss M.J., Noble W.S., Käll L. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0. J. Am. Soc. Mass Spectrom. 2016;27:1719–1727. doi: 10.1007/s13361-016-1460-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are contained within the article and its supporting information. The entire CRL4A ligase and ternary complex ensembles can be obtained from corresponding author (Nan Bai) upon reasonable request.