

Abstract
Background:
Mounting evidence has revealed an inverse association between cigarette smoking and the risk of Parkinson’s disease (PD). Meanwhile, cigarette smoking has been found to be associated with cognitive impairment in PD patients. However, the neural mechanisms of the association between cigarette smoking and PD are not fully understood.
Objective:
The aim of this study is to explore the neural mechanisms of the association between cigarette smoking and PD.
Methods:
A total of 129 PD patients and 69 controls were recruited from the Parkinson’s Progression Markers Initiative (PPMI) cohort, including 39 PD patients with regular smoking history (PD-S), 90 PD patients without regular smoking history (PD-NS), 26 healthy controls with regular smoking history (HC-S), and 43 healthy controls without regular smoking history (HC-NS). Striatal dopamine transporter (DAT) binding and gray matter (GM) volume of the whole brain were compared among the four groups.
Results:
PD patients showed significantly reduced striatal DAT binding compared with healthy controls, and HC-S showed significantly reduced striatal DAT binding compared with HC-NS. Moreover, smoking and PD showed a significant interaction effect in the left medial prefrontal cortex (mPFC). PD-S showed reduced GM volume in the left mPFC compared with PD-NS.
Conclusion:
The degeneration of dopaminergic neurons in PD results in a substantial reduction of the DAT and dopamine levels. Nicotine may act as a stimulant to inhibit the action of striatal DAT, increasing dopamine levels in the synaptic gap. The inverse alteration of dopamine levels between PD and nicotine addiction may be the reason for the inverse association between smoking and the risk of PD. In addition, the mPFC atrophy in PD-S may be associated with cognitive impairment.
Keywords: cigarette smoking, dopamine transporter imaging, medial prefrontal cortex, Parkinson’s disease, striatum
Introduction
Cigarette smoking is a leading cause of premature deaths, accounting for about 100 million deaths in the 20th century and an estimated 1 billion deaths in the 21st century. 1 The association between cigarette smoking and a significantly increased risk of lung, heart, and vascular disease, as well as many cancers, is largely indisputable. 2 Parkinson’s disease (PD) is the second most common neurodegenerative disorder that affects 2–3% of elderly people >65 years old worldwide, 3 which is characterized by bradykinesia, resting tremor, and muscular rigidity and other non-motor symptoms. A meta-analysis of observational studies revealed the inverse association between cigarette smoking and the risk of PD. 4 Recently, a prospective study further demonstrated a causally protective effect of smoking on the risk of PD in a 65-year follow-up of 30,000 male British doctors. 5 However, the mechanism of the inverse association between smoking and PD is not fully illustrated.
PD is a progressive neurodegenerative disorder mainly caused by the degeneration of the dopaminergic neurons within the nigrostriatal pathway. 6 As a vital part of the nigrostriatal pathway, the striatum includes the caudate and putamen. 7 Reduced dopamine transporter (DAT) binding is the result of a reduction in the number of neurons projecting from the substantia nigra to the striatum in PD.8,9 The loss of dopamine neurons results in a substantial reduction of the DAT and dopamine levels. 10 Chronic cigarette smoking results in a dysregulated reward processing, which is mediated by neuroadaptations in the mesolimbic dopamine system, particularly the striatum. 11 Nicotine is the primary addictive ingredient within tobacco, which acts as a presynaptic nicotinic acetylcholine receptors (nAChR) agonist, facilitates dopamine release, and makes cigarettes highly addictive. 12 Nicotine can boost extracellular dopamine levels by binding to nAChR in the striatum, and repeated nicotine stimulation may ‘hijack’ natural reward circuits by increasing the drive to get nicotine. 13 Accumulating neuroimaging evidences have consistently shown that cigarette smokers exhibited abnormal function in the striatum.14–17 Boosted dopamine levels by nicotine effects of chronic smoking in healthy smokers may delay or even prevent the onset of PD. Here, we speculate that elevated dopamine levels in healthy smokers and reduced dopamine levels in PD may be the reason for the inverse association between cigarette smoking and the risk of PD. DAT is responsible for the reuptake of free dopamine from the synaptic cleft back into the axonal button. 18 Nicotine stimulates dopamine release, 19 which may act as the ‘DAT-blockers’ to increase dopamine levels and inhibit the action of DAT. Therefore, in this study, we hypothesize that smokers may show decreased striatal DAT binding compared with non-smokers. DAT imaging is an established diagnostic tool for degenerative parkinsonism. To prove this hypothesis, striatal DAT binding was compared using DAT imaging among PD patients and healthy controls with or without regular smoking history from the Parkinson’s Progress Markers Initiative (PPMI) cohort.
Meanwhile, several studies reported the harmful impact of smoking on PD. For example, smoking was associated with global cognitive impairment in PD patients, even those who had quit smoking. 20 Besides, smoking history showed an independent and dose-dependent association with impulse control disorders in PD patients. 21 Mounting evidence has demonstrated that cigarette smoking is linked with neurobiological and neurocognitive abnormalities. 22 Most neuroimaging studies examining the neurobiological consequences of smoking have focused on the volume of cortical and subcortical gray matter (GM).23–25 The most consistently reported brain areas of reduced GM volume include the prefrontal cortex (PFC), insula, thalamus, and cerebellum.24,26,27 PFC is critical for cognitive control, which is heavily influenced by dopamine levels. 28 Here, we hypothesize that PD patients with regular smoking history show reduced GM volume in the PFC, which may result in the cognitive impairment.
In this study, therefore, to explore the neural mechanism of the association between cigarette smoking and PD, we compared striatal DAT binding and GM volume of the whole brain among the PD patients with regular smoking history (PD-S), PD patients without regular smoking history (PD-NS), healthy controls with regular smoking history (HC-S), and healthy controls without regular smoking history (HC-NS) using the PPMI cohort. Clarifying this issue might improve the understanding of the neurobiological substrates of the association between cigarette smoking and PD.
Methods
Participants
The data used in this study were all obtained from the PPMI database (www.ppmi-info.org). 29 The study was approved by the institutional review board of all PPMI sites involved, and signed informed consent was obtained from all participants recruited. Data were downloaded on May 1, 2021. In all, 132 de novo PD patients and 70 control subjects with smoking history questionnaires were enrolled in this study. One PD patient with poor quality of T1-weighted images, one PD patient with missing DAT scan, and one control subject with image format conversion error were excluded. The smoking history questionnaires included dozens of smoking questions ranging from ‘sqm1’ to ‘smq9’ (Supplementary Table S1). In this study, the subjects with ‘sqm2 = yes’ were regarded as smoked subjects, while the subjects with ‘sqm2 = no’ were regarded as non-smoked subjects. ‘sqm2’ referred to the question ‘In your lifetime, have you ever regularly smoked cigarettes, that is, at least one cigarette per day for 6 months or longer?’ One PD patient who didn’t know/prefer not to answer the ‘smq2’ was excluded. Finally, 129 PD patients (39 PD-S and 90 PD-NS patients) and 69 controls (26 HC-S and 43 HC-NS subjects) were included in this study (Figure 1).
Figure 1.

Flow diagram of the study population.
In addition to the demographic variables (age, sex, and education), the clinical variables were collected, including the Hoehn and Yahr (H-Y) stages, the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), Montreal Cognitive Assessment (MOCA), the Geriatric Depression Scale (GDS), the Scale for Outcomes for Parkinson’s Disease–autonomic function (SCOPA-AUT), State and Trait Anxiety Scale (STAI), the Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease (QUIP), the University of Pennsylvania Smell Identification Test (UPSIT), and the Epworth Sleepiness Scale (ESS). Moreover, cardiovascular risk factors (CVRF) were further collected, including the hypertension, diabetes, hypercholesterolemia, and hyperlipidemia.
DAT data processing
According to the PPMI imaging protocol, the DAT scan was acquired using single-photon emission computed tomography (SPECT) imaging. DAT imaging using [123I] FP-CIT SPECT was performed at PPMI imaging centers. Then, DAT images were sent to the Institute for Neurodegenerative Disorders for processing and calculation of striatal binding ratios (SBRs). For iterative reconstruction, SPECT raw projection data were imported to a HERMES (Hermes Medical Solutions, Skeppsbron 44, 111 30 Stockholm, Sweden) system. Second, the reconstructed files were transferred to the PMOD (PMOD Technologies, Zurich, Switzerland) for subsequent processing. Third, attenuation correction and standard Gaussian 3D 6.0 mm filter was applied. Fourth, these files were normalized to Montreal Neurologic Institute (MNI) space. Fifth, the highest striatal uptake of the trans-axial slice was identified, and the eight hottest striatal slices around it were averaged to generate a single slice image. Sixth, striatal regions of interest (ROIs) were then placed on the target regions (caudate and putamen, Figure 2(a)) and reference region (occipital cortex). Finally, count densities for each ROI were extracted and used to calculate SBRs for each of the four striatal ROIs. SBRs were calculated as follows: SBRs = (target region / reference region) – 1. Detailed DAT processing procedures can be found online (https://www.ppmi-info.org/access-data-specimens/download-data/) and in published documents.30,31
Figure 2.

(a) Illustrating exact position of the striatal regions of interest, including left caudate (pink), right caudate (red), left putamen (sky blue), and right putamen (yellow). PD patients (PD-S and PD-NS) showed lower SBRs than healthy controls (HC-S and HC-NS), and HC-S showed lower SBRs than HC-NS in each of the four striatal nuclei, including the left caudate (b), right caudate (c), left putamen (d), and right putamen (e). SBRs, striatal binding ratios.
MRI data acquisition and preprocessing
All baseline 3D T1-weighted imaging (T1WI) data were obtained from the PPMI database, which were acquired using Siemens 3.0T scanners according to a standardized protocol. The scanning parameters were as follows: repetition time (TR) = 2300 ms, echo time (TE) = 2.98 ms, inversion time = 900 ms, slice thickness = 1 mm, field of view = 256 mm, and matrix size = 240 × 256.
Before preprocessing, the magnetic resonance imaging (MRI) images of raw DICOM format were reviewed and converted into the Neuroimaging Informatics Technology Initiative (NII) format using MRICRON software. All NII images were preprocessed and analyzed using the CAT12 toolbox (Computational Anatomy Toolbox; http://dbm.neuro.uni-jena.de/cat/) implemented in SPM12 (http://www.fil.ion.ucl.ac.uk/spm/software/spm12/). CAT12 served as the platform for preprocessing the structural MRI data and offered a processing pipeline for voxel-based morphometry (VBM). For processing and analysis steps, pre-set parameters in accordance with standard protocol (http://www.neuro.uni-jena.de/cat12/CAT12-Manual.pdf) were used, applying default settings unless indicated otherwise. The procedure of data analysis was as follows:
T1 images are normalized to a template space and segmented into GM, white matter (WM), and cerebrospinal fluid (CSF).
Display slices for each subject to check the quality of spatial registration, including if the native volume had artifacts or if the native volume had a wrong orientation. No subject had to be excluded because of poor quality.
Total intracranial volume (TIV) was estimated.
Mean correlation, weighted overall image quality, and Mahalanobis distance algorithms were used to quantify image quality after segmentation (‘VBM data homogeneity’ function). No subject had to be excluded because of poor quality.
Last, segmented GM images were smoothed with an 8 mm full-width-half-maximum (FWHM) isotropic Gaussian kernel.
For excluding artifacts on the GM/WM border (i.e. incorrect voxel classification), an absolute GM threshold of 0.1 was applied to the VBM data.
In addition, the volume of each striatal nucleus (caudate and putamen) was estimated according to the Neuromorphometrics atlas and normalized by TIV for each subject (structure/TIV × 103).
Statistical analysis
Clinical and imaging characteristics analyses were performed using IBM SPSS Statistics 23 software (IBM Corporation, New York). The distribution of continuous variables for normality was tested using one-sample Kolmogorov–Smirnov test. Then, parametric one-way analysis of variance (ANOVA) was used to assess differences for the data of normal distribution and homogeneity of variance between four groups. Non-parametric Kruskal–Wallis H test was used to assess differences for the data of non-normal distribution and heterogeneity of variance between four groups. And non-parametric Mann–Whitney U test was used to assess differences for the data of non-normal distribution and heterogeneity of variance between two groups (such as H-Y stages). Besides, Chi-square tests were used to evaluate categorical variables. To account for multiple comparisons reported, a family-wise error rate was applied to each set of analyses. A Bonferroni correction was made to adjust for the number of comparisons of clinical and imaging characteristics, respectively. The corrected significance level is 0.05 divided by the total number of comparisons provided for that table, that is, 0.05/15 = 0.0033 for clinical characteristics comparisons in Table 1 and 0.05/15 = 0.0033 for imaging characteristics comparisons in Table 2.
Table 1.
Clinical characteristics.
| PD-S | PD-NS | HC-S | HC-NS | p values | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (n = 39) | (n = 90) | (n = 26) | (n = 43) | 4 groups | PD-S versus PD-NS | PD-S versus HC-S | PD-S versus HC-NS | PD-NS versus HC-S | PD-NS versus HC-NS | HC-S versus HC-NS | |
| Age (years) | 61.5 ± 10.1 | 59.0 ± 8.7 | 59.1 ± 10.5 | 60.5 ± 11.2 | 0.545 a | – | – | – | – | – | – |
| Sex (male/female) | 28/11 | 56/34 | 18/8 | 28/15 | 0.735 b | – | – | – | – | – | – |
| Education (years) | 16.2 ± 2.1 | 16.8 ± 2.7 | 16.5 ± 2.6 | 17.4 ± 2.4 | 0.149 c | – | – | – | – | – | – |
| Regularly current smokers (n) | 3 | 0 | 1 | 0 | n/a | n/a | n/a | n/a | n/a | n/a | n/a |
| H-Y stages | 1.4 ± 0.6 | 1.6 ± 0.5 | n/a | n/a | n/a | 0.175 d | n/a | n/a | n/a | n/a | n/a |
| MDS-UPDRS Part I | 4.7 ± 3.3 | 4.8 ± 3.3 | 2.9 ± 2.6 | 3.1 ± 2.5 | 0.003 c * | 0.882 | 0.020 | 0.023 | 0.006 | 0.004 | 0.721 |
| MDS-UPDRS Part II | 5.8 ± 3.9 | 5.6 ± 4.2 | 0.3 ± 0.9 | 0.3 ± 0.7 | <0.001 c * | 0.707 | <0.001 | <0.001 | <0.001 | <0.001 | 0.957 |
| MDS-UPDRS Part III | 19.0 ± 7.3 | 20.0 ± 7.8 | 0.7 ± 1.0 | 0.9 ± 1.4 | <0.001 c * | 0.572 | <0.001 | <0.001 | <0.001 | <0.001 | 0.893 |
| MDS-UPDRS Total Score | 29.4 ± 11.4 | 30.4 ± 12.1 | 3.9 ± 3.5 | 4.2 ± 3.3 | <0.001 c * | 0.788 | <0.001 | <0.001 | <0.001 | <0.001 | 0.799 |
| MOCA | 27.5 ± 2.4 | 27.5 ± 1.8 | 28.1 ± 1.1 | 28.1 ± 1.1 | 0.31 c | – | – | – | – | – | – |
| GDS | 1.9 ± 1.2 | 1.9 ± 2.2 | 1.1 ± 1.4 | 1.1 ± 1.8 | 0.001 c * | 0.137 | 0.007 | <0.001 | 0.075 | 0.007 | 0.679 |
| SCOPA-AUT | 9.0 ± 5.4 | 7.7 ± 4.8 | 4.9 ± 3.2 | 5.6 ± 2.8 | <0.001 c * | 0.224 | <0.001 | 0.002 | 0.003 | 0.015 | 0.404 |
| STAI | 58.2 ± 15.0 | 63.0 ± 17.5 | 54.5 ± 12.1 | 53.3 ± 12.1 | 0.004 c | – | – | – | – | – | – |
| QUIP | 0.4 ± 0.6 | 0.2 ± 0.60 | 0.6 ± 1.1 | 0.1 ± 0.3 | 0.004 c | – | – | – | – | – | – |
| UPSIT | 23.3 ± 8.4 | 23.3 ± 7.6 | 33.4 ± 5.9 | 35.1 ± 4.2 | <0.001 c * | 0.904 | <0.001 | <0.001 | <0.001 | <0.001 | 0.373 |
| ESS | 5.3 ± 2.7 | 5.7 ± 3.6 | 5.7 ± 2.9 | 5.5 ± 3.6 | 0.954 c | – | – | – | – | – | – |
ESS, Epworth Sleepiness Scale Score; GDS, Geriatric Depression Scale; HC-NS, healthy controls without regular smoking history; HC-S, healthy controls with regular smoking history; H-Y stages, Hoehn and Yahr stages; MDS-UPDRS, Movement Disorders Society Unified Parkinson’s Disease Rating Scale; MOCA, Montreal Cognitive Assessment; PD-NS, PD patients without regular smoking history; PD-S, PD patients with regular smoking history; QUIP, Questionnaire for Impulsive-Compulsive Disorders; SCOPA-AUT, Scale for Outcomes for Parkinson’s Disease–autonomic function; STAI, State trait anxiety score; UPSIT, University of Pennsylvania Smell Identification Test. Fewer than five participants in each group missed any one assessment.
One-way ANOVA
Chi-square test
Kruskal–Wallis H test
Mann–Whitney U test
– indicates post hoc test was not further performed if there was no statistical difference among the four groups in one-way ANOVA or Kruskal–Wallis H test.
Significance level for comparisons is p < 0.0033 (after Bonferroni correction).
Table 2.
Imaging characteristics.
| PD-S | PD-NS | HC-S | HC-NS | p values | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (n = 39) | (n = 90) | (n = 26) | (n = 43) | 4 groups | PD-S versus PD-NS | PD-S versus HC-S | PD-S versus HC-NS | PD-NS versus HC-S | PD-NS versus HC-NS | HC-S versus HC-NS | |
| Caudate_L SBRs | 2.08 ± 0.55 | 2.07 ± 0.57 | 2.93 ± 0.49 | 3.26 ± 0.77 | <0.001 a * | 0.956 | <0.001 | <0.001 | <0.001 | <0.001 | 0.031 |
| Caudate_R SBRs | 2.10 ± 0.63 | 2.03 ± 0.54 | 2.92 ± 0.49 | 3.24 ± 0.75 | <0.001 a * | 0.539 | <0.001 | <0.001 | <0.001 | <0.001 | 0.036 |
| Putamen_L SBRs | 0.76 ± 0.29 | 0.83 ± 0.35 | 2.09 ± 0.51 | 2.40 ± 0.65 | <0.001 a * | 0.419 | <0.001 | <0.001 | <0.001 | <0.001 | 0.006 |
| Putamen_R SBRs | 0.87 ± 0.38 | 0.83 ± 0.31 | 2.08 ± 0.48 | 2.36 ± 0.69 | <0.001 a * | 0.659 | <0.001 | <0.001 | <0.001 | <0.001 | 0.013 |
| TIV | 1525.33 ± 148.30 | 1535.29 ± 167.98 | 1505.83 ± 149.68 | 1507.83 ± 151.58 | 0.742 a | – | – | – | – | – | – |
| mPFC volume | 0.356 ± 0.061 | 0.382 ± 0.050 | 0.401 ± 0.050 | 0.360 ± 0.055 | 0.001 a * | 0.012 | 0.001 | 0.748 | 0.116 | 0.026 | 0.002 |
| Normalized mPFC volume | 0.234 ± 0.035 | 0.250 ± 0.025 | 0.270 ± 0.025 | 0.240 ± 0.033 | <0.001 a * | 0.005 | <0.001 | 0.382 | 0.008 | 0.064 | <0.001 |
| Caudate_L volume | 2.51 ± 0.41 | 2.57 ± 0.37 | 2.53 ± 0.29 | 2.44 ± 0.36 | 0.278 a | – | – | – | – | – | – |
| Caudate_R volume | 2.58 ± 0.44 | 2.67 ± 0.45 | 2.64 ± 0.32 | 2.52 ± 0.43 | 0.248 a | – | – | – | – | – | – |
| Putamen_L volume | 3.15 ± 0.49 | 3.26 ± 0.52 | 3.16 ± 0.48 | 3.21 ± 0.55 | 0.683 a | – | – | – | – | – | – |
| Putamen_R volume | 3.00 ± 0.38 | 3.13 ± 0.45 | 3.07 ± 0.43 | 3.10 ± 0.49 | 0.547 a | – | – | – | – | – | – |
| Normalized Caudate_L volume | 1.64 ± 0.23 | 1.69 ± 0.28 | 1.69 ± 0.19 | 1.63 ± 0.24 | 0.799 b | – | – | – | – | – | – |
| Normalized Caudate_R volume | 1.70 ± 0.25 | 1.76 ± 0.32 | 1.76 ± 0.21 | 1.68 ± 0.29 | 0.640 b | – | – | – | – | – | – |
| Normalized Putamen_L volume | 2.07 ± 0.31 | 2.14 ± 0.36 | 2.12 ± 0.37 | 2.14 ± 0.38 | 0.795 a | – | – | – | – | – | – |
| Normalized Putamen_R volume | 1.98 ± 0.24 | 2.06 ± 0.34 | 2.06 ± 0.36 | 2.07 ± 0.34 | 0.536 a | – | – | – | – | – | – |
HC-NS, healthy controls without regular smoking history; HC-S, healthy controls with regular smoking history; L, left; mPFC, medial prefrontal cortex; PD-NS, PD patients without regular smoking history; PD-S, PD patients with regular smoking history; R, right; SBRs, striatal binding ratios; TIV, Total intracranial volume.
One-way ANOVA
Kruskal–Wallis H test
Mann–Whitney U test
– indicates post hoc test was not further performed if there was no statistical difference among the four groups in the one-way ANOVA or Kruskal–Wallis H test.
Significance level for comparisons is p < 0.0033 (after Bonferroni correction).
Statistical analyses of imaging data were performed using the CAT12/SPM12 statistical module. The interaction effects of GM volume alteration between cigarette smoking and PD were determined using a full-factorial model with 2 × 2 ANOVA. TIV was included as a covariate to remove variance related to this global parameter of brain morphometry. To correct for multiple comparisons, we employed cluster-level family-wise error (FWE) correction at voxel level with p < 0.001 and then corrected at the cluster level with non-stationary cluster extent correction. The significantly different brain region in the ANOVA was saved as a mask using xjview (https://www.alivelearn.net/xjview/). Then, this mask was applied to extract the GM volume of every subject using the ROI signal extractor tool of Data Processing & Analysis for Brain Imaging (DPABI) 32 for post hoc tests.
Results
In this study, a total of 129 PD patients (39 PD-S and 90 PD-NS) and 69 controls (26 HC-S and 43 HC-NS) were included. Baseline demographics and clinical variables were summarized in Table 1. There were no significant differences among PD-S, PD-NS, HC-S, and HC-NS in age, sex, and education. No difference was observed in the H-Y stages between the PD-S and PD-NS. Furthermore, MDS-UPDRS (specifically the part I, part II, part III, and total score), MOCA, GDS, SCOPA-AUT, STAI, QUIP, UPSIT, and ESS were also compared. Apart from MOCA, STAI, QUIP, and ESS, the clinical variables of MDS-UPDRS (part I, part II, part III, and total score), GDS, SCOPA-AUT, and UPSIT were different in PD subgroups compared with controls subgroups. In addition, there were no significant differences of CVRF among the four groups (Supplementary Table S2).
Striatal imaging variables were summarized in Table 2. One-way ANOVA showed significant differences of SBRs in both left and right striatal nuclei (caudate and putamen) among PD-S, PD-NS, HC-S, and HC-NS (p < 0.001). Specifically, post hoc tests showed that PD patients (PD-S and PD-NS) had lower SBRs than controls (HC-S and HC-NS). Moreover, HC-S showed lower SBRs than HC-NS in these four striatal nuclei (Figure 2). In addition, the volume of each striatal nucleus (caudate and putamen) was estimated. However, none of the striatal GM volume was different in cohorts of the four groups.
In addition, the interaction effects of GM volume alteration between cigarette smoking and PD were determined with 2 × 2 ANOVA. A significant interaction effect was detected in the left medial prefrontal cortex (mPFC) (Figure 3). Then, post hoc tests revealed that PD-S showed decreased GM volume in the left mPFC compared with PD-NS and HC-S.
Figure 3.
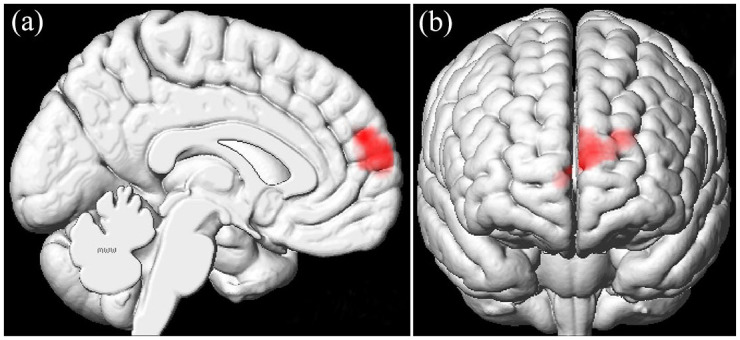
Using 2 × 2 analysis of variance (ANOVA), significant interaction effect was detected in the left mPFC (BA10) (medial frontal gyrus) (peak MNI coordinate: –22.5, 57, 24; F values: 21.0359; cluster size: 1599). mPFC, medial prefrontal cortex.
Discussion
To our knowledge, this study is the first to explore the neural substrates for the association between cigarette smoking and PD by analyzing both DAT and GM volume. The DAT results showed that PD patients had lower striatal DAT binding than controls, and HC-S had lower striatal DAT binding than HC-NS in both left and right striatal nuclei (caudate and putamen). In addition, a significant interaction effect between smoking and PD was detected in the left mPFC. Compared with PD-NS and HC-S, PD-S showed decreased GM volume in the left mPFC.
In this study, PD patients showed significantly decreased striatal DAT binding compared with healthy controls, which is in line with the previous studies. 33 However, the influence of smoking on striatal DAT binding of PD patients (PD-S versus PD-NS) was not detected. One explanation may be that there is a statistical ‘floor’ effect 34 that the DAT damage is severe enough in PD regardless of smoking history. Furthermore, we found that HC-S had significantly decreased striatal DAT binding in both left and right striatal nuclei (caudate and putamen) relative to HC-NS. A recent meta-analysis included seven previous small-sample studies of DAT imaging in smokers. It demonstrated a significant reduction in DAT availability in the current smokers compared with non-smokers. 35 Furthermore, this study demonstrated a significant decrease in DAT availability in healthy individuals with regular smoking history (25 ex-smokers and only 1 current smoker, Table 1) relative to 43 healthy non-smokers. DAT mediates the reuptake of free dopamine from the synaptic cleft back into the axonal button. 18 Nicotine stimulates dopamine release, which has been demonstrated in non-human primates 36 and human. 19 The stimulants may act as the ‘DAT-blockers’ to increase dopamine concentration in the synaptic gap and inhibit the action of DAT. This is supported by a DAT imaging study that nicotine may act as a stimulant on striatal DAT to reduce primarily elevated DAT density in adults with attention deficit hyperactivity disorder (ADHD). 37 Furthermore, cigarette smoking has been demonstrated to inhibit monoamine oxidase (MAO) activity.38,39 MAO is a metabolic enzyme to breakdown dopamine. Thus, inhibited MAO is likely to result in greater dopamine concentration. Therefore, we believe that cigarettes may act as a stimulant to impact striatal DAT, increasing dopamine concentration in the synaptic gap and inhibiting the action of DAT. This study demonstrated that chronic smoking decreased DAT availability in healthy smokers using [123I] FP-CIT SPECT. [123I] FP-CIT uptake may compete with intrinsic boosted dopamine levels by nicotine effects of chronic smoking and result in decreased DAT availability in healthy smokers. Therefore, we do not suggest that decreased DAT availability is caused by nigrostriatal degeneration in healthy smokers. We suggest that boosted dopamine levels by nicotine effects in healthy smokers may delay or even prevent the onset of PD. The reduction of striatal DAT binding was detected in both PD and healthy smokers in this study. However, the different neurobiological mechanisms between PD and nicotine addiction result in inverse dopamine levels in PD and healthy smokers. This inverse alteration of dopamine levels in PD and healthy smokers may provide the reason for the inverse association between cigarette smoking and the risk of PD.4,5 In the future, more studies are needed to confirm this hypothesis that chronic smoking affects striatal dopamine, DAT, and MAO.
In addition, we found smoking and PD showed a significant interaction effect in the left mPFC. PD-S showed decreased GM volume of mPFC relative to PD-NS and HC-S. In addition to motor dysfunction, dopaminergic degeneration is also associated with cognitive deficits in PD. 40 Both PD patients and mice with ventral tegmental area (VTA) dopamine depletion can lead to attenuated delta activity (1–4 Hz) in the mPFC during interval timing. 41 Interval timing is a task of estimating an interval of several seconds as guided by a cue and requires executive resources such as working memory and attention to time. 41 Furthermore, optogenetic stimulation of the mPFC neurons expressing D1 dopamine receptors at delta frequencies can compensate for the impaired temporal control of action caused by VTA dopamine depletion. 41 The rodents with 6-hydroxydopamine (6-OHDA)-induced lesion in the medial forebrain successfully recapitulates PD motor impairment and several non-motor symptoms. 42 Nicotine, the addictive component of cigarettes, activates nAChR in the VTA, resulting in dopamine release in the frontal cortex, mesolimbic area, and corpus striatum. Dopamine release results in a pleasurable experience, which is critical for the reinforcing effects (effects that promote self-administration) of nicotine. 43 The mPFC plays a crucial role in addictive behavior. A subset of the mPFC neurons forms neuronal ensembles to encode the coupling between the reward of drug use and the associated contexts. And reactivation of the neuronal ensembles caused by drug-associated contexts during abstinence can provoke drug relapse. 44 The mPFC contributes to goal-directed behavior in response to motivational salience and reward expectation. 45 Functional MRI studies have shown that decreased reward-related mPFC activity is related to impaired motivation and poor self-control in individuals with addictions. 46 Total smoking pack-years have been found to be associated with global cognitive impairment in PD patients with smoking history even in patients who have quit smoking. 20 In this study, PD-S showed reduced GM volume of mPFC compared with PD-NS and HC-S. Taken together, these findings suggest that reduced GM volume of mPFC may be associated with cognitive impairment in smoking PD patients.
In this study, we found a laterality observation that the GM alteration of mPFC volume was on the left side. Such a hemispheric laterality difference maybe because of the lateralization of dopaminergic systems. It is well known that PD is characterized as dopaminergic dysfunction. The dopaminergic neurons of the substantia nigra are more vulnerable in the left hemisphere in right-handed patients with PD, 47 and a stronger left-sided nigrostriatal FC was associated with a lower risk for PD. 48 This was further supported by the fact that nicotine intake can be affected in humans via dopaminergic agonists and antagonists. 49 Many reward-related functional MRI studies revealed an obvious left-sided bias to the activation of cortical and subcortical regions involved in reward processing.50–52 In addition, structural MRI studies demonstrated a strong left-sided bias to the abnormality of cortical and subcortical regions in smokers, such as left PFC and insula, 53 left anterior cingulate cortex, 54 left thalamus, and amygdala. 55
This study has several limitations. First, in this retrospective analysis, more detailed smoking behavior characteristics on smoking history were not collected, such as years smoked, cigarettes smoked per day, smoking initiation age, and nicotine dependence levels. Thus, further association analysis cannot be performed between altered imaging biomarkers and smoking behavior characteristics. Second, this retrospective study design does not allow us to determine causality; we cannot determine whether the alterations of DAT binding and GM anatomy predispose to smoking initiation or whether chronic smoking influences DAT binding and GM anatomy. This question could be answered by prospective longitudinal studies. Future longitudinal evaluations are needed to explore the alterations of DAT binding and GM anatomy before and after the initiation of smoking.
Conclusion
In this study, we reported baseline clinical and imaging characteristics of PD patients and healthy controls with or without regular smoking history. PD patients showed significantly reduced striatal DAT binding compared with healthy controls. PD is characterized by selective degeneration of dopaminergic neurons, and it results in a substantial reduction of the DAT and dopamine levels. Interestingly, HC-S showed significantly reduced striatal DAT binding compared with HC-NS. Nicotine may act as a stimulant to inhibit the action of striatal DAT, increasing dopamine levels in the synaptic gap. Despite the reduction of striatal DAT binding in both PD and healthy smokers, the inverse alteration of dopamine levels because of different neurobiological mechanisms between PD and nicotine addiction may be the reason for the inverse association between smoking and the risk of PD. In addition, smoking and PD showed a significant interaction effect in the left mPFC. The mPFC atrophy may be associated with cognitive impairment in PD-S.
Supplemental Material
Supplemental material, sj-docx-1-tan-10.1177_17562864221092566 for Association between cigarette smoking and Parkinson’s disease: a neuroimaging study by Chao Wang, Cheng Zhou, Tao Guo, Peiyu Huang, Xiaojun Xu and Minming Zhang in Therapeutic Advances in Neurological Disorders
Supplemental material, sj-docx-2-tan-10.1177_17562864221092566 for Association between cigarette smoking and Parkinson’s disease: a neuroimaging study by Chao Wang, Cheng Zhou, Tao Guo, Peiyu Huang, Xiaojun Xu and Minming Zhang in Therapeutic Advances in Neurological Disorders
Acknowledgments
Data used in the preparation of this article were obtained from the PPMI database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI, a public private partnership, is funded by The Michael J. Fox Foundation for Parkinson’s Research and funding partners, including AbbVie, Allergan, Avid, Biogen, Bristol-Myers Squibb, Covance, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, SANOFI GENZYME, Servier, Takeda, Teva, UCB, and GolubCapital. In addition, the authors would like to thank the efforts of the statistical expert (Zexin Chen) for providing the statistical advice.
Footnotes
Author contributions: Chao Wang: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Project administration; Resources; Software; Writing – original draft; Writing – review & editing.
Cheng Zhou: Formal analysis; Methodology; Software; Writing – original draft.
Tao Guo: Methodology; Software; Validation.
Peiyu Huang: Methodology; Software; Supervision; Validation.
Xiaojun Xu: Project administration; Supervision.
Minming Zhang: Funding acquisition; Project administration; Writing – review & editing.
Conflict of interest statement: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Zhejiang Provincial Natural Science Foundation of China under Grant No. LY21H180003 and the 13th Five-Year Plan for National Key Research and Development Program of China under Grant No. 2016YFC1306600. We have posted this paper in the preprints server (https://www.researchsquare.com/article/rs-1029131/v1).
ORCID iD: Chao Wang  https://orcid.org/0000-0002-5268-9570
https://orcid.org/0000-0002-5268-9570
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Chao Wang, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, No.88 Jiefang Road, Hangzhou 310009, China.
Cheng Zhou, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Tao Guo, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Peiyu Huang, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Xiaojun Xu, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
Minming Zhang, Department of Radiology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
References
- 1. Pirie K, Peto R, Reeves GK, et al. The 21st century hazards of smoking and benefits of stopping: a prospective study of one million women in the UK. Lancet 2013; 381: 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doll R, Peto R, Boreham J, et al. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ 2004; 328: 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers 2017; 3: 17013. [DOI] [PubMed] [Google Scholar]
- 4. Li X, Li W, Liu G, et al. Association between cigarette smoking and Parkinson’s disease: a meta-analysis. Arch Gerontol Geriatr 2015; 61: 510–516. [DOI] [PubMed] [Google Scholar]
- 5. Mappin-Kasirer B, Pan H, Lewington S, et al. Tobacco smoking and the risk of Parkinson disease: a 65-year follow-up of 30,000 male British doctors. Neurology 2020; 94: e2132–e2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimohama S, Sawada H, Kitamura Y, et al. Disease model: Parkinson’s disease. Trends Mol Med 2003; 9: 360–365. [DOI] [PubMed] [Google Scholar]
- 7. Whetsell WO, Jr. The mammalian striatum and neurotoxic injury. Brain Pathol 2002; 12: 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and Clinical Implications. N Engl J Med 1988; 318: 876–880. [DOI] [PubMed] [Google Scholar]
- 9. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia Nigra regional selectivity. Brain 1991; 114(Pt 5): 2283–2301. [DOI] [PubMed] [Google Scholar]
- 10. Uhl GR. Dopamine transporter: basic science and human variation of a key molecule for dopaminergic function, locomotion, and parkinsonism. Mov Disord 2003; 18(Suppl. 7): S71–S80. [DOI] [PubMed] [Google Scholar]
- 11. Wise RA. Roles for nigrostriatal–not just mesocorticolimbic–dopamine in reward and addiction. Trends Neurosci 2009; 32: 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci 1997; 20: 92–98. [DOI] [PubMed] [Google Scholar]
- 13. Volkow ND, Fowler JS, Wang GJ, et al. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 2009; 56(Suppl. 1): 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bühler M, Vollstädt-Klein S, Kobiella A, et al. Nicotine dependence is characterized by disordered reward processing in a network driving motivation. Biol Psychiatry 2010; 67: 745–752. [DOI] [PubMed] [Google Scholar]
- 15. David SP, Munafò MR, Johansen-Berg H, et al. Ventral striatum/nucleus accumbens activation to smoking-related pictorial cues in smokers and nonsmokers: a functional magnetic resonance imaging study. Biol Psychiatry 2005; 58: 488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sweitzer MM, Geier CF, Joel DL, et al. Dissociated effects of anticipating smoking versus monetary reward in the caudate as a function of smoking abstinence. Biol Psychiatry 2014; 76: 681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C, Huang P, Shen Z, et al. Increased striatal functional connectivity is associated with improved smoking cessation outcomes: a preliminary study. Addict Biol 2021; 26: e12919. [DOI] [PubMed] [Google Scholar]
- 18. Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci 1993; 14: 43–49. [DOI] [PubMed] [Google Scholar]
- 19. Brody AL, Mandelkern MA, Olmstead RE, et al. Ventral striatal dopamine release in response to smoking a regular vs a denicotinized cigarette. Neuropsychopharmacology 2009; 34: 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doiron M, Dupré N, Langlois M, et al. Smoking history is associated to cognitive impairment in Parkinson’s disease. Aging Ment Health 2017; 21: 322–326. [DOI] [PubMed] [Google Scholar]
- 21. Valença GT, Glass PG, Negreiros NN, et al. Past smoking and current dopamine agonist use show an independent and dose-dependent association with impulse control disorders in Parkinson’s disease. Parkinsonism Relat Disord 2013; 19: 698–700. [DOI] [PubMed] [Google Scholar]
- 22. Durazzo TC, Meyerhoff DJ, Nixon SJ. Chronic cigarette smoking: implications for neurocognition and brain neurobiology. Int J Environ Res Public Health 2010; 7: 3760–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang Z, Zhang Y, Cheng J, et al. Meta-analysis of brain gray matter changes in chronic smokers. Eur J Radiol 2020; 132: 109300. [DOI] [PubMed] [Google Scholar]
- 24. Sutherland MT, Riedel MC, Flannery JS, et al. Chronic cigarette smoking is linked with structural alterations in brain regions showing acute nicotinic drug-induced functional modulations. Behav Brain Funct 2016; 12: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fritz HC, Wittfeld K, Schmidt CO, et al. Current smoking and reduced gray matter volume-a voxel-based morphometry study. Neuropsychopharmacology 2014; 39: 2594–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang C, Xu X, Qian W, et al. Altered human brain anatomy in chronic smokers: a review of magnetic resonance imaging studies. Neurol Sci 2015; 36: 497–504. [DOI] [PubMed] [Google Scholar]
- 27. Shen Z, Huang P, Wang C, et al. Cerebellar gray matter reductions associate with decreased functional connectivity in nicotine-dependent individuals. Nicotine Tob Res 2018; 20: 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ott T, Nieder A. Dopamine and cognitive control in prefrontal cortex. Trends Cogn Sci 2019; 23: 213–234. [DOI] [PubMed] [Google Scholar]
- 29. The Parkinson progression marker initiative (PPMI). Prog Neurobiol 2011; 95: 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tinaz S, Chow C, Kuo PH, et al. Semiquantitative analysis of dopamine transporter scans in patients with Parkinson disease. Clin Nucl Med 2018; 43: e1–e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tagare HD, DeLorenzo C, Chelikani S, et al. Voxel-based logistic analysis of PPMI control and Parkinson’s disease DaTscans. NeuroImage 2017; 152: 299–311. [DOI] [PubMed] [Google Scholar]
- 32. Yan CG, Wang XD, Zuo XN, et al. DPABI: data processing & analysis for (resting-state) brain imaging. Neuroinformatics 2016; 14: 339–351. [DOI] [PubMed] [Google Scholar]
- 33. Palermo G, Ceravolo R. Molecular imaging of the dopamine transporter. Cells 2019; 8: 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bohnen NI, Kuwabara H, Constantine GM, et al. Grooved pegboard test as a biomarker of nigrostriatal denervation in Parkinson’s disease. Neurosci Lett 2007; 424: 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ashok AH, Mizuno Y, Howes OD. Tobacco smoking and dopaminergic function in humans: a meta-analysis of molecular imaging studies. Psychopharmacology (Berl) 2019; 236: 1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cumming P, Rosa-Neto P, Watanabe H, et al. Effects of acute nicotine on hemodynamics and binding of [11C]raclopride to dopamine D2,3 receptors in pig brain. NeuroImage 2003; 19: 1127–1136. [DOI] [PubMed] [Google Scholar]
- 37. Krause KH, Dresel SH, Krause J, et al. Stimulant-like action of nicotine on striatal dopamine transporter in the brain of adults with attention deficit hyperactivity disorder. Int J Neuropsychopharmacol 2002; 5: 111–113. [DOI] [PubMed] [Google Scholar]
- 38. Lewis A, Miller JH, Lea RA. Monoamine oxidase and tobacco dependence. Neurotoxicology 2007; 28: 182–195. [DOI] [PubMed] [Google Scholar]
- 39. Berlin I, Anthenelli RM. Monoamine oxidases and tobacco smoking. Int J Neuropsychopharmacol 2001; 4: 33–42. [DOI] [PubMed] [Google Scholar]
- 40. Kaasinen V, Nurmi E, Brück A, et al. Increased frontal [(18)F]fluorodopa uptake in early Parkinson’s disease: sex differences in the prefrontal cortex. Brain 2001; 124: 1125–1130. [DOI] [PubMed] [Google Scholar]
- 41. Kim YC, Han SW, Alberico SL, et al. Optogenetic stimulation of frontal D1 neurons compensates for impaired temporal control of action in dopamine-depleted mice. Curr Biol 2017; 27: 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marshall CA, King KM, Kortagere S. Limitations of the rat medial forebrain lesion model to study prefrontal cortex mediated cognitive tasks in Parkinson’s disease. Brain Res 2019; 1702: 105–113. [DOI] [PubMed] [Google Scholar]
- 43. Benowitz NL. Nicotine addiction. N Engl J Med 2010; 362: 2295–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bossert JM, Stern AL, Theberge FR, et al. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci 2011; 14: 420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 2002; 159: 1642–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goldstein RZ, Alia-Klein N, Tomasi D, et al. Is decreased prefrontal cortical sensitivity to monetary reward associated with impaired motivation and self-control in cocaine addiction? Am J Psychiatry 2007; 164: 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scherfler C, Seppi K, Mair KJ, et al. Left hemispheric predominance of nigrostriatal dysfunction in Parkinson’s disease. Brain 2012; 135: 3348–3354. [DOI] [PubMed] [Google Scholar]
- 48. Ellmore TM, Castriotta RJ, Hendley KL, et al. Altered nigrostriatal and nigrocortical functional connectivity in rapid eye movement sleep behavior disorder. Sleep 2013; 36: 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Caskey NH, Jarvik ME, Wirshing WC, et al. Modulating tobacco smoking rates by dopaminergic stimulation and blockade. Nicotine Tob Res 2002; 4: 259–266. [DOI] [PubMed] [Google Scholar]
- 50. Delgado MR, Nystrom LE, Fissell C, et al. Tracking the hemodynamic responses to reward and punishment in the striatum. J Neurophysiol 2000; 84: 3072–3077. [DOI] [PubMed] [Google Scholar]
- 51. Koepp MJ, Gunn RN, Lawrence AD, et al. Evidence for striatal dopamine release during a video game. Nature 1998; 393: 266–268. [DOI] [PubMed] [Google Scholar]
- 52. Thut G, Schultz W, Roelcke U, et al. Activation of the human brain by monetary reward. Neuroreport 1997; 8: 1225–1228. [DOI] [PubMed] [Google Scholar]
- 53. Zhang X, Salmeron BJ, Ross TJ, et al. Factors underlying prefrontal and insula structural alterations in smokers. NeuroImage 2011; 54: 422–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liao Y, Tang J, Liu T, et al. Differences between smokers and non-smokers in regional gray matter volumes: a voxel-based morphometry study. Addiction Biol 2012; 17: 977–980. [DOI] [PubMed] [Google Scholar]
- 55. Hanlon CA, Owens MM, Joseph JE, et al. Lower subcortical gray matter volume in both younger smokers and established smokers relative to non-smokers. Addiction Biol 2016; 21: 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-tan-10.1177_17562864221092566 for Association between cigarette smoking and Parkinson’s disease: a neuroimaging study by Chao Wang, Cheng Zhou, Tao Guo, Peiyu Huang, Xiaojun Xu and Minming Zhang in Therapeutic Advances in Neurological Disorders
Supplemental material, sj-docx-2-tan-10.1177_17562864221092566 for Association between cigarette smoking and Parkinson’s disease: a neuroimaging study by Chao Wang, Cheng Zhou, Tao Guo, Peiyu Huang, Xiaojun Xu and Minming Zhang in Therapeutic Advances in Neurological Disorders