

Abstract
Objective:
The estrogen metabolite 2-methoxyestradiol has shown antitumorigenic action in some epithelial tumors. In the present work we investigate its effects in ovarian cancer used alone or in combination with other apoptotic-inducing reagents such as tumor necrosis factor-related apoptosis-inducing ligand.
Methods:
To assess the effect of 2-methoxyestradiol, dose response and time courses in ovarian cancer and normal cells were conducted. Apoptosis was confirmed through DNA laddering, by flow cytometry, and Western blotting of proteins involved in the apoptotic cascade.
Results:
2-Methoxyestradiol induced apoptosis in ovarian cancer cells but not in normal counterparts. 2-Methoxyestradiol activates both the intrinsic and extrinsic apoptotic pathways. 2-Methoxyestradiol–mediated apoptosis involves reactive oxygen species generation and caspase-dependent and caspase-independent mechanisms. We also demonstrate that 2-methoxyestradiol selectively induces an additive/synergistic apoptotic response in ovarian cancer cells when used in combination with tumor necrosis factor-related apoptosis-inducing ligand.
Conclusions:
2-Methoxyestradiol, alone or in combination with tumor necrosis factor-related apoptosis-inducing ligand, should be considered as a potential treatment for ovarian cancer.
Keywords: AIF, 2ME, TRAIL, death receptors, chemotherapy, ovarian cancer
BACKGROUND
Epithelial ovarian cancer constitutes the most deadly gynecologic malignancy.1 Worldwide, approximately 200,000 women are newly diagnosed and almost 125,000 women annually die from ovarian cancer.2 Currently, despite complete clinical response to extensive surgery followed by adjuvant chemotherapy, the 5-year overall survival remains less than 40%.1,3
The high lethality of ovarian cancer is partially explained by primary or secondary resistance to chemotherapy.4 The mechanisms of resistance are not completely understood, however, epidemiological studies on early events during ovarian carcinogenesis seem to assign a central role to the hormonal environment.5–7 Estrogens exert a promoting effect leading to cell proliferation, cell survival, metastasis, and drug resistance.8,9 Estrogens mitogenic effects are mediated through activation of the IL6/STAT-3 signaling pathway10; this pathway is highly expressed in ovarian cancer cells. Furthermore, ovarian cancer cells secrete 17-beta-estradiol exerting an autocrine antiapoptotic effect that leads to tumor progression.10 In contrast, progesterone commands inhibitory signaling pathways, reducing cell growth, invasiveness, and promoting cell death.8
Some estrogens metabolites, such as 2-methoxyestradiol (2ME), may have antitumorigenic activity11; in fact, 2ME exhibits potent apoptotic activity in a wide variety of tumor cells. Also, antiangiogenic activity has been demonstrated through inhibition of HIF1α, reduction of vascular epidermal growth factor (VEGF), and induction of apoptosis in endothelial cells.12 Other molecular mechanisms of 2ME have been reported, including microtubule destabilization by inhibition of colchicine-binding site. Although the exact mechanisms of 2ME action are still unclear, its potential effectiveness in preventing tumor growth has been suggested for a number of tumor types. A phase II randomized, double-blind trial showed that 2ME stabilized or reduced prostate specific antigen levels in hormone-refractory prostate cancer.13
Compared to estradiol, 2ME possesses a 500 times lower binding affinity for estrogen receptor (ER) α and 3200 times lower affinity for estrogen receptor β.14,15 Thus, antiproliferative and proapoptotic activities of 2ME are most probably mediated by estrogen receptor (ER) independent mechanisms. 2-Methoxyestradiol ER-independent cell cycle G2/M arrest was reported in a variety of cancer cells, probably through microtubules toxicity16 and cell cycle inhibition.17 In prostate cancer cells, 2ME inhibits cell growth via the beta-catenin pathway18 and modulates p44/42 mitogen-activated protein kinase19; also, 2ME attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated metastasis of gastric cancer.20 It has been shown that 2-methoxyestradiol ER-independent apoptosis was mediated by activation of both the intrinsic and extrinsic apoptotic pathways.21–25
A novel strategy against ovarian carcinoma has been to overcome resistance by reestablishing sensitivity to apoptotic agents via the activation of alternative pathways. Several publications have shown that a member of the tumor necrosis factor (TNF) family, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), may induce apoptosis in a wide variety of epithelial cancers.26–29 TRAIL induces apoptosis in cancerous but not in corresponding normal cells both in vitro and in vivo30–32 thus we suggested that TRAIL may be a good candidate for the treatment of gynecological malignancies.31,33–35 TRAIL mediates cell death by binding to death receptors, TRAIL-R1 (also called DR4) and TRAIL-R2 (also called DR5), and induction of caspase-mediated apoptosis. Two types of cell responses to TRAIL mediate apoptosis: one requires the activation of the intrinsic cascade (type II cells) and the other is independent of that activation (type I cells). Unfortunately, TRAIL itself is barely effective in inducing death of ovarian cancers.34–38
The mechanisms of TRAIL resistance are not completely understood. Preliminary studies have shown that resistance does not correlate with the expression of inhibitory proteins involved in death receptor mediated apoptosis (FLIP, IAPs, survivin, decoy receptors, etc).39,40 We and others suggested that survival signals mediated by growth factors (ie, EGF stimulation, erb-B2 overexpression) and the subsequent activation of downstream kinases (ie Akt) affect TRAIL mediated apoptosis by inhibiting both the mitochondrial pathway (also referred as the intrinsic pathway) and the extrinsic pathway through reduction of FLIP levels.41 The majority of the ovarian cancer cells should be considered type II cells, because mitochondrial activation is required for apoptosis. In fact complete inhibition of the intrinsic pathway by overexpression of Bcl-2 in ovarian cancer cells produce resistance to chemotherapeutic agents.42
Herein, we investigate if pretreatment of ovarian cancer cells with 2ME overcomes resistance to TRAIL or produces an additive effect on cancer cell death. We found that 2ME induce apoptosis and enhance TRAIL-mediated apoptosis in ovarian cancer. The mechanisms behind the effect of 2ME are studied.
MATERIALS AND METHODS
Primary tissue of the human female reproductive tract was obtained with patient consent from the Gynecology Department of the Hospital of the Pontificia Universidad Católica de Chile in Santiago, Chile. Ethical approval for the study was obtained from both the ethical committees of the Faculty of Medicine and the Faculty of Biological Sciences at the Pontificia Universidad Católica de Chile and the South Eastern Health Service of Santiago, Chile.
Primary Culture and Cell Lines
Primary cultures of normal and cancerous human ovarian epithelium were derived from tissue obtained from premenopausal and postmenopausal women undergoing either ovarian cystectomy or unilateral salpingo-oophorectomy hysterectomy with bilateral salpingo-oophorectomy and from benign or malignant tumors or from ascitic fluid using minor modifications to our previously published protocols.31 Briefly, ovarian tumor samples obtained from both the primary and peritoneal metastatic sites were removed under sterile conditions and collected into warm saline serum. A section was sent to pathology to independently verify and classify the cancer type based on routinely used markers and histology. The stage of the menstrual cycle was determined from patient menstrual history. The tissue was washed twice in cold phosphate-buffered saline (PBS), minced and incubated with collagenase type IV (2mg/mL, Sigma Aldrich Co, USA) in antibiotic-antimycotic (Invitrogen, NY,) containing calcium and magnesium-free Hanks’ buffered salt solution (HBSS; Invitrogen, NY) for 30 minutes at 37°C with agitation. After digestion the majority of the stromal and immune cells are visualized as individual cells, while cells of ovarian epithelial origin are present as aggregates. This digestion medium is filtered through a 70-μ nylon mesh (PGC Scientific, Md) and the remaining clumps (containing epithelial cells) washed in HBSS. The solution was centrifuged at 3000 rpm for 3 minutes. The cells were resuspended in DMEM/F12 (Invitrogen, NY, USA) with 10% fetal bovine serum (FBS; Invitrogen, NY) containing 100-U/mL penicillin G, and 100-mg/mL streptomycin sulfate (Invitrogen, NY). Cells from ovarian cancer ascitic fluid were extracted by syringe. Approximately 400 mL was transferred to a conical tube and centrifuged at 3000 rpm for 5 minutes. 70% of the supernatant was replaced with MCBD/M199 supplemented with 10% FBS and used to seed 6 cm2 Petri dishes. The cells were incubated at 37°C at 5% CO2 for 3 days to allow adhesion. The medium was subsequently removed and the cells washed before further experimentation. For the primary culture of human epithelial ovarian normal cells, ovarian tissue was obtained from women undergoing oophorectomy for nonmalignant gynecological disorders. The tissue was washed in calcium, magnesium, and phenol red–free Hanks solution (Invitrogen, NY) containing penicillin and streptomycin. The ovarian tissue was incubated in Dispase II (Sigma-Aldrich Co) in HBBS for 30 minutes at 37°C, with agitation. The epithelial layer was then scraped off using a tissue culture scrapper. The cells were suspended in MCDB 105/M199 medium and centrifuged at 3000 rpm for 5 minutes. The cell pellet was resuspended in the same medium complemented with 10% FBS, 100-U/mL penicillin G, and 100-mg/mL streptomycin sulfate. The cells were plated in 6 cm2 Petri dishes (Falcon, Becton-Dickinson, NJ) at 37° C at 5% CO2 for 3 days to allow adhesion before confirmation of epithelial origin by cytokeratin and vimentin. The ovarian cancer cell lines A2780, AD10, OVCAR-3, and UCI 101 were obtained from Dr. G Scott Rose (University of California, Calif). The normal ovarian cell line, HOSE, was kindly donated from Dr Carmen Romero (University of Chile). Cell lines were grown in DMEM/F12 medium supplemented with 10% FBS, 100-U/mL penicillin G, and 100-mg/mL streptomycin sulfate. For protein and RNA experiments cells were plated at 50% confluence in 10 cm2 Petri dishes (Falcon, Becton-Dickinson). The medium was changed to charcoal-treated medium containing 5% FBS for 24 hours before the addition of 2ME or TRAIL. All tissue culture reagents were obtained from Invitrogen Life Technologies (Carlsbad, Calif).
Reagents
All estrogen metabolites were purchased from Steroloids, Inc (Wilton, NH). Gluthation (GST)–TRAIL fusion protein was produced as previously described34 and stored in aliquots at −20°C. An equal volume of vehicle (DMEM/F12 medium) was added as a control in each experiment. The tetrapeptide caspase inhibitor ZVAD-fmk, the caspase-8 inhibitor Z-IETD-fmk and the caspase-9 inhibitor Z-LEHD-fmk (Enzyme Systems Products, Livermore, Calif) were resuspended in DMSO (Sigma-Aldrich Co) at a concentration of 10 mM and added to cells at a final concentration of 50 μM. N-acetylcystein (NAC) and ICI-182780 (ICI) were purchased from Sigma-Aldrich Group (Germany). The chemotherapeutics drugs (doxorubicin, paclitaxel, and cisplatin) were a kind gift of the Cancer Center of the Pontificia Universidad Católica de Chile, and used at a final concentration of 5 μM.
Measurement of Cell Viability
To assess estrogen, estrogen metabolites [(4-hydroxyestradiol (40H), 4-methoxyestradiol (4ME), 2-methoxyestradiol (2ME), 2-hydroxyestradiol (20H) and 17-β estradiol (E2)] and TRAIL-mediated cytotoxicity primary cultured cells or cancer cell lines were plated at 1–5 × 104 cells/well in 96-well microtiter plates and allowed to adhere to the plates 24 hours in DMEM/F12 with 10% FBS. Cells were incubated in 5% charcoal-stripped FBS containing DMEM/F12 medium for 24 hours before addition of estrogen metabolites, at concentrations stated in figure legends, from 0 to 72 hours. GST-TRAIL fusion protein was added under the same conditions and incubated for 18 hours. In experiments where cells were co-treated with 2ME and GST-TRAIL, the procedure was identical for that of above mentioned estrogen metabolites, with GST-TRAIL being added to the medium during the last 18 hours of the treatment period. Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) dye reduction assay was used following manufacturer recommendations (Cell Titer 96 Aqueous One Solution Cell proliferation Assay, Promega, Madison, Wis). All MTS data points were repeated simultaneously five times and each experiment was carried out at least three times. Results of representative experiments are given as the mean ± the standard deviation (SD) and of multiple experiments as the mean ± the standard error of the mean (SE).
DNA Fragmentation Assay
Cells were plated (2 × 106) onto 100-mm tissue culture dishes, allowed to adhere overnight and then treated using same conditions described in MTS assay. The cells were trypsinized, washed in cold PBS, and harvested into cold lysis buffer containing 10 mM Tris-HCl, pH 8.0, 150 nM NaCl, 2 mM MgCl2, 1 mM dithiothreitol (DTT), and 0.5% NP-40 on ice for 40 minutes. Lysates were centrifuged at 13,000 rpm and the pellets resuspended in cold buffer containing 10 mM Tris-HCl, pH 8.0, 350 mM NaCl, 1 mM MgCl2, and 1 mM DTT before incubation on ice for 20 min. Lysates were then extracted once with phenol/chloroform and the DNA precipitated with 10 mM MgCl2 and 2.5 volumes of 100% ethanol overnight at −20°C. DNA was collected by centrifugation at 14,000g for 20 minutes, resuspended in TE Buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) plus 0.1 mg/mL RNase A, and incubated at 37°C for 1 hour. Proteinase K (1 mg/mL) was added and the mixture was further incubated at 37°C for 1 hour. Fragmented DNA was separated by electrophoresis in a 1.5% agarose gel and visualized with ethidium bromide (0.5 μg/mL).
Flow Cytometry Analysis
To assess apoptosis, cells were plated at 5 × 105 in 60-mm tissue culture dishes, allowed to adhere overnight and then treated under same conditions described previously.31 The cells were trypsinized, harvested, centrifuged, washed in cold PBS, and resuspended in a cold solution of 1 mL 1× PBS and 4 mL 70% ethanol. The cells were incubated overnight at 4°C, washed in 1 × PBS and resuspended in a solution of 250 μL of propidium iodide (50 μg/mL) in 1× PBS and 1 μL RNase (20 μg/mL).43 Cells were incubated (protected from light) for 15 minutes at room temperature before flow cytometric analysis. Cells undergoing apoptosis were determined as a percent of cells with sub-G0/G1 DNA content in the DNA histogram compared to the total number of cells present using a FACSort system (Becton Dickinson, Mansfield, Mass) and the Cell Quest software (Becton Dickinson, San Jose, Calif).
Western Blotting
Cells were harvested in cold PBS and the pellet resuspended in lysis buffer (10 mM TRIS-HCl pH 7.4, 150 mM NaCl, 0.5% Triton X-100) for 20 minutes on ice. After this incubation, the lysate was sonicated and centrifuged at 14,000g for 20 minutes at 40°C and pellet was discarded. To isolate cytosol, mitochondrial and nuclear fractions, cells were suspended in 200 μL of suspension buffer (20 mM HEPES (pH 7.5), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 150 mM sucrose) disrupted in a glass homogenizer. The homogenate was centrifuged at 750g for 10 minutes to isolate nuclei and discard unbroken cells. The mitochondrial pellet was obtained by centrifugation at 10,000g for 15 minutes and resuspended in 40 μL of lysis buffer. The supernatant obtained by centrifugation at 100,000g for 60 minutes at 4°C was the cytosolic fraction. Protein concentration was determined by Bradford assay. About 100 μg of crude membrane extract was loaded in each lane, separated by 10% to 15% polyacrylamide gel electrophoresis in the presence of sodium dodecylsulfate, transferred to nitrocellulose membranes, and incubated overnight with primary antibodies (1:1000): Bcl-2, phosphoBcl-2 (DAKO, Calif), cytochrome c (BD Biosciences, USA), poly(ADP-ribose) polymerase (PARP), apoptosis-inducing factor (AIF), Bcl-xL, BAX, b-actin (Santa Cruz, Calif), Flip, Bid/tBid (Calbiochem Calif), SMAC/Diablo (BD Transduction Laboratories, Calif). Secondary antibody, goat anti-mouse/rabbit immunoglobulin G (IgG) secondary antibody coupled to hydrogen peroxidase (1:5000, Bio-Rad Labs, Calif), was applied for 1 hour at room temperature. The reaction was developed with chemiluminescence using ECL Western blot analysis system (NEN, Western lightning, Perkin-Elmer).
Measurement of Caspase-3, 8, and 9 Activities
Cultured cells were harvested and washed once in cold PBS. After a brief centrifugation (3000 rpm, 5 minutes) cells were incubated in lysis buffer containing 20 mM Hepes (pH 7.4), 100 mM NaCl, 0.5% (v/v) NP-40, and 10 mM DTT on ice for 30 minutes. Following centrifugation at 13,000 rpm for 10 minutes at 4°C, supernatants were collected, transferred to a 96-well plate and corresponding substrate to caspase-3, 8, and 9 (Calbiochem-Novabiochem Corp, Calif) added. The samples were incubated for 24 hours at room temperature. Optical density at 405 nm was measured using an ELISA plate reader (EL310 Boots-Celtech). Casapase activity was expressed as percentage in respect to control that was set at 100%.
Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was isolated using the Chomczynski method.44 cDNA was generated using reverse transcriptase (Superscript II, Invitrogen). TRAIL-R1 or DR4 sense: 5′-CCGCGGCCACACCCAGAAAGT-3,’ TRAIL-R1 or DR4 antisense: 5′-GTACATGGGAGG-CAAGCAAACAAA-3′ (generates a fragment of 415 bp); TRAIL-R2 or DR5 sense: 5′-GGGAGCCGCTCAT-GAGGAAGTTGG-3,′ TRAIL-R2 or DR5 antisense: 5′-GGCAAGTCTCTCTCCCAGCGTCTC-3′ (generates a fragment of 181 bp); DcR1 sense: 5′-GCCGGAAGTGTAGCAGGTG-3′ DcR1 antisense: 5′-GAGATGTGTGGACCCCCA-3′ (generates a fragment of 672 bp); DcR2 sense: 5′-CCCCCGGCAGGACGAAGTT-3 DcR2 antisense: 5′-CTCCTCCGCTGCTGGGGTTTT-3′ (generates a fragment of 418 bp); superoxide dismutase (SOD) type I sense: 5′-CTAGCGAGTTATGGCGAC-3,’ SOD type I antisense: 5′-GAATGTTTATTGGGCGATC-3′ (generates a fragment of 483 bp; BiosChile, Santiago, Chile). Semi-quantitative polymerase chain reaction (PCR) reactions were performed from cDNA generated from cell lines and primary cultured cells maintained under maintenance conditions, using Taq polymerase (Invitrogen). As an internal control, primers amplifying a region of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used to test the integrity of the starting cDNA as previously Publisher.31
Immunocytochemistry
Ovarian cancer cells were resuspended in DMEM/F12 with 10% FBS and plated in 6 cm2 Petri dishes (tissue culture grade, Falcon BD Labware, NJ) containing glass cover slides. Once plated, the cell culture medium was changed to DMEM/F12 with 5% charcoal-treated FBS for 24 hours. Cells were washed twice in PBS and fixed in 1% paraformaldehyde. Cells were washed again three times in PBS and pseudoperoxidase activity blocked by 10 minutes incubation with 10% hydrogen peroxidase (Labvision, UK). Cells were washed as before and blocked in serum-free protein block solution (DAKO, Calif) for 30 minutes. TRAIL-R2/DR5 (Imgenex, Calif), antibodies were added at a dilution of 1:250 for 18 hours. The slides were washed three times and incubated with secondary anti-rabbit IgG peroxidase labelled antibody (Kirkegaard & Perry Laboratorios, Md) for 1 hour at room temperature. Covers were washed three times and DAB Plus substrate (Labvision UK) added until a colour change was detected (approximately 5–10 minutes). The reaction was stopped by washing in distilled water, and cells were examined by light microscopy.
Statistical Analysis
Statistical Analysis is performed using the Mann-Whitney test, with P < .05 regarded as statistically significant (GraphPad Instat 3). Data are represented as a percentage in respect to vehicle control. Standard deviation of the means is given from a minimum of 3 individual experiments performed, each consisting of 5 replicates. Bars represent ± SD (standard deviation) of the mean.
RESULTS
2-Methoxyestradiol Induces Apoptosis in Ovarian Cancer Cells
To determine if estrogen and estrogen metabolites altered cellular viability in ovarian cancer cells, we incubated the A2780 ovarian cancer cell line for 48 hours with 2 μM concentration of the different metabolites or ethanol vehicle. Concentrations and times of exposure were based on previous work done in endometrial cancer cells.35 As shown in Figure 1, analysis of cell cytotoxicity, as measured by the MTS assay, demonstrated that treatment with 2 μM E2, 40H or 4ME, for 48 hours, had no significant effect on cell cytotoxicity. In contrast, 2ME and its precursor 20H significantly increased cell cytotoxicity. To further assess the effects of 2ME in ovarian cancer cells, concentration and time curves were built. Incubation for 48 hours using different concentrations of 2ME resulted in a dose-dependent increase in cell cytotoxicity starting at concentrations as low as 0.5 μM and increasing out to 5 μM (Figure 2A). To confirm that the cell cytotoxicity induced by 2ME was due to apoptosis, cell lysates were analyzed for cleaved PARP expression by Western blotting (W-B). Figure 2B shows that PARP cleavage is detectable in wide range of concentrations matching the pattern of cell cytotoxicity observed by MTS. As shown in Figure 2C, incubation with 2ME at 5 μM for 48 hours resulted in an increase in DNA laddering. As observed in Figure 3, incubation with 2ME at 5 μM resulted in a progressive increase in cell cytotoxicity starting at 24 hours and achieving a plateau around 72 hours (longer incubation times are not shown). Poly(ADP-ribose) polymerase cleavage is detectable from 12 hours after 2ME incubation, achieving a peak at 24 hours, before progressively declining, yet remaining detectable, out to 72 hours (Figure 3B).
Figure 1.
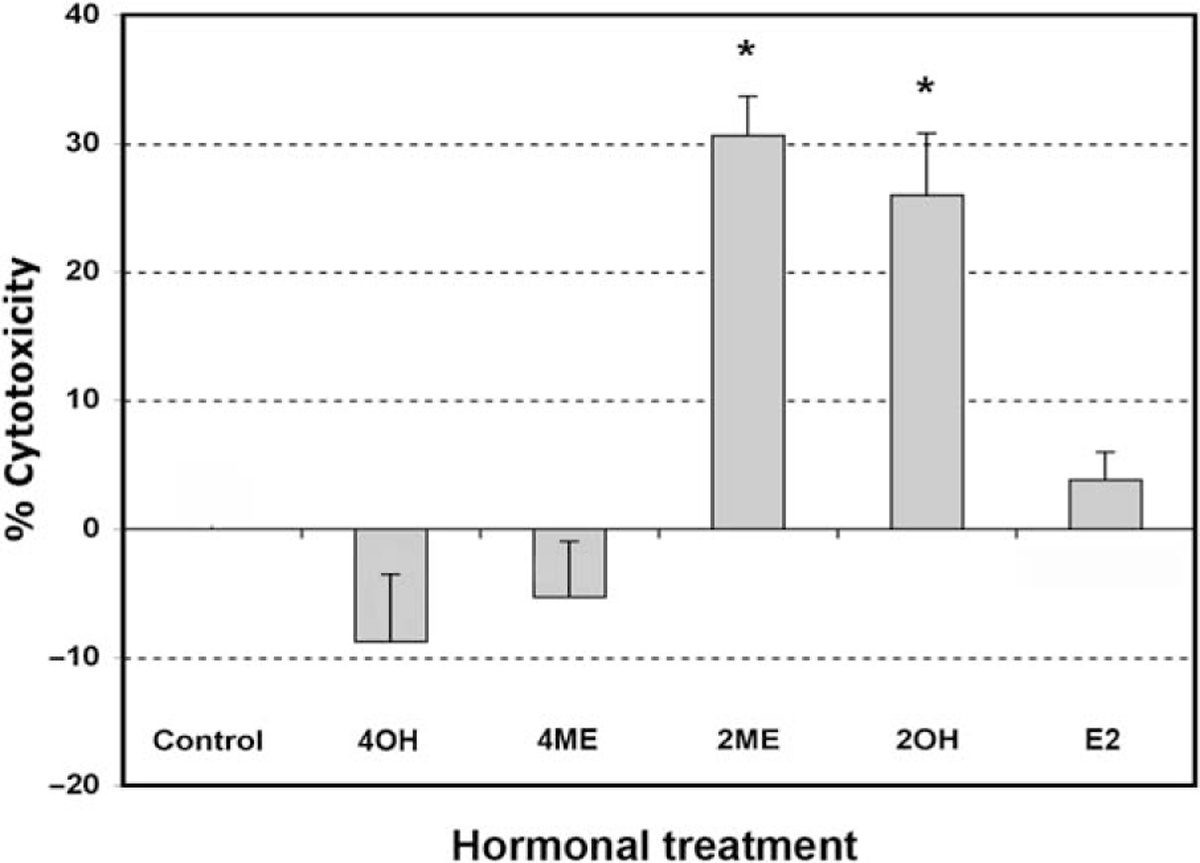
2-Methoxyestradiol (2ME) and 2-hydroxyestradiol, but not other estrogen metabolites, increase cytotoxicity in the ovarian cancer cell line A2780. Cell cytotoxicity was measured by the MTS assay, and the data represented as a percentage in respect to control (ethanol, C) in the presence or absence of 4-hydroxyestradiol (40H), 4-methoxyestradiol (4ME), 2-methoxyestradiol (2ME), 2-hydroxyestradiol (20H), and 17-beta estradiol (E2). All concentrations were 2 μM in ethanol vehicle. Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Mann-Whitney test statistical significance (★) was set at P < .05 with respect to control (vehicle).
Figure 2.
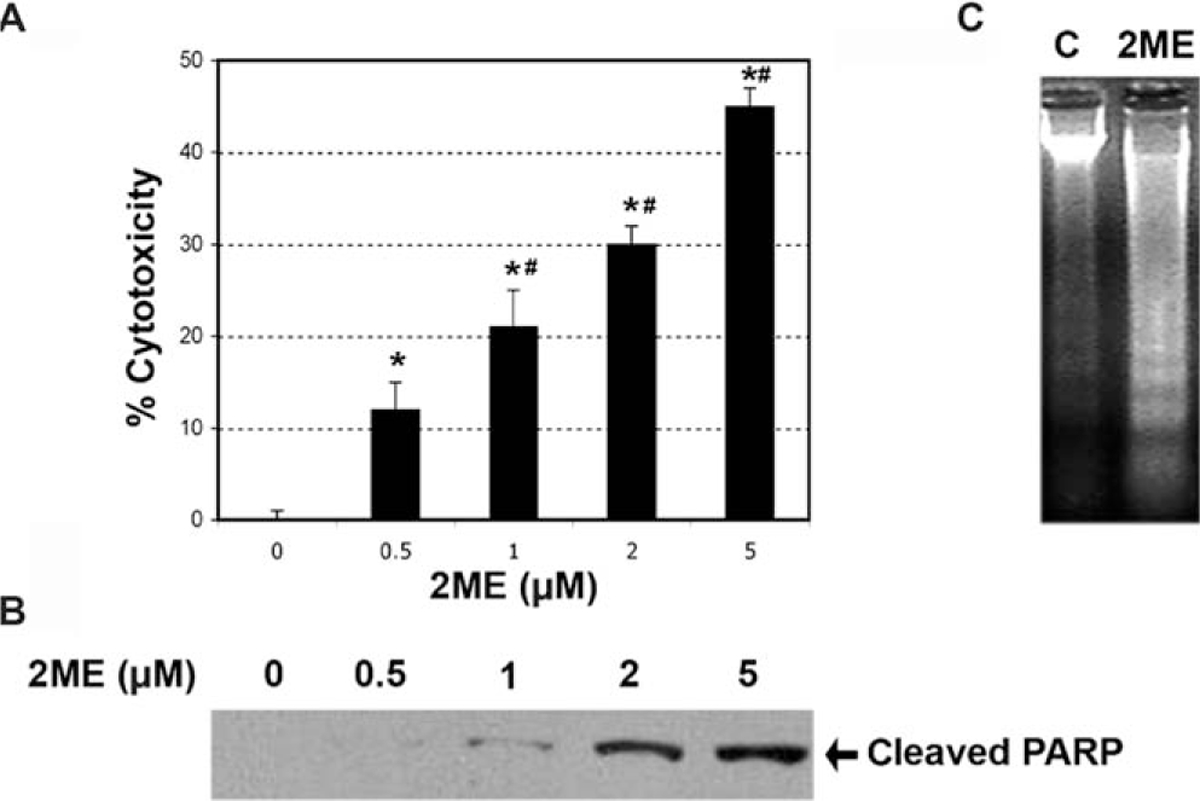
2-Methoxyestradiol (2ME) induces apoptosis in a concentration-dependent manner. A, Concentration-response curve in the presence of 2-methoxyestradiol (2ME, μM) for 48 hours in A2780 cells. Cell cytotoxicity was measured by the MTS assay, and the data represented as a percentage in respect to control. Standard deviation of the means is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Mann-Whitney test ★ = P < .05 with respect to control (vehicle), # = P < .05 in respect to previous concentration. B, Western blot analysis in A2780 cells demonstrating the presence of cleaved PARP after the addition of varying concentrations of 2ME for 48 hours. C, DNA fragmentation analysis of apoptosis induced by 2ME for 48 hours in comparison to vehicle treated controls (C).
Figure 3.
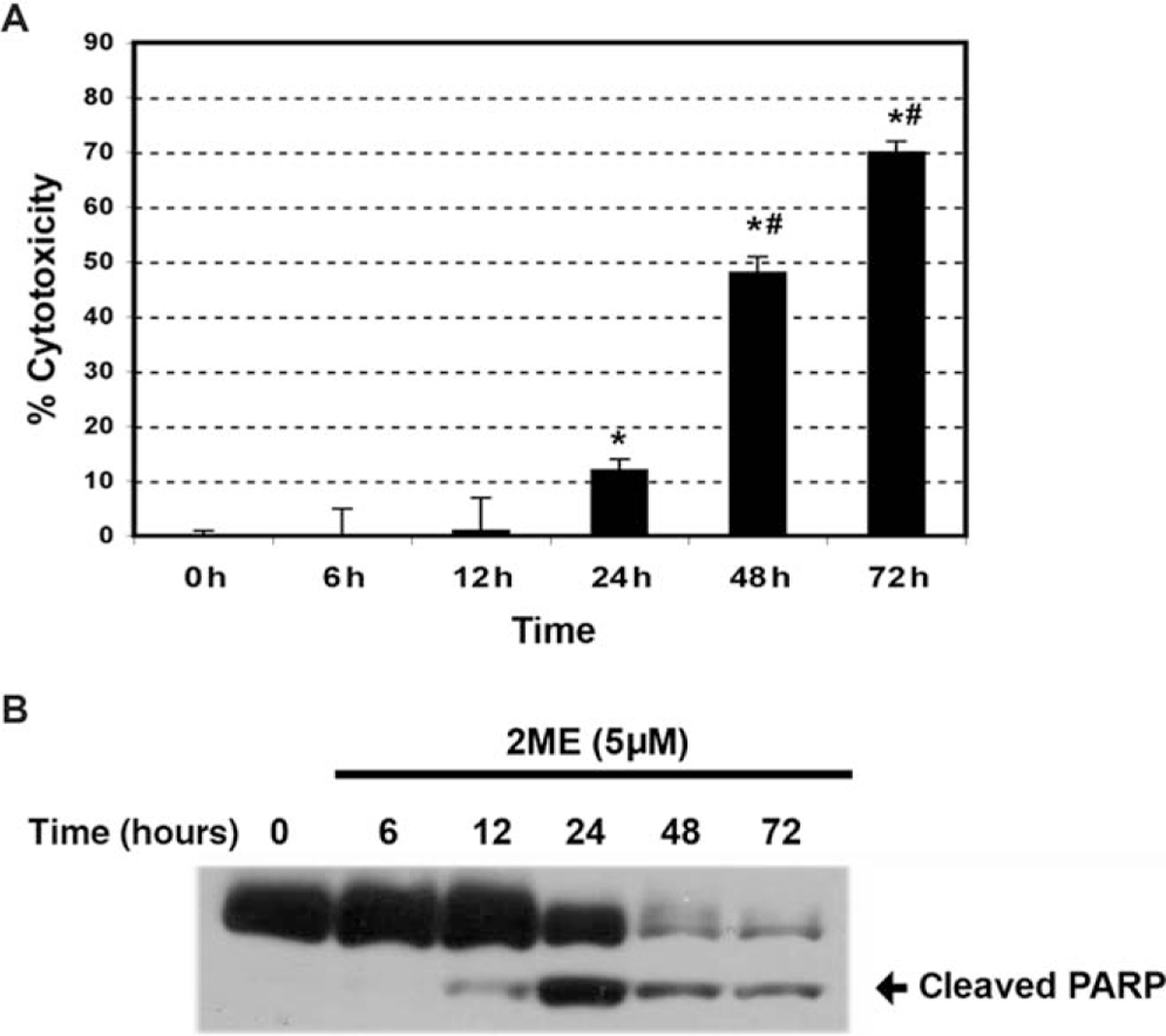
2-Methoxyestradiol (2ME) increases A2780 cells cytotoxicity and induces apoptosis in a time-dependent manner. A, Time-course in response to 2-methoxyestradiol (2ME, 5 μM). Cell cytotoxicity was measured by the MTS assay, and the data represented as a percentage in respect to control (vehicle). Standard deviation of the means is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Mann-Whitney test set statistical significance at ★= P < .05 with respect to control and previous time points, # = P < .05 with respect to 24 hours. B, Western blot analysis of cleaved PARP after varying incubation times (hours) of 2ME (5 μM).
2-Methoxyestradiol Induced Apoptosis is not ER-dependent
2-Methoxyestradiol has been previously shown to be a weak agonist of estrogen receptors, ERα and ERβ, with a much lower affinity compared to that of 17β-estradiol.14,15 Previously, we demonstrated in endometrial and cervical cancer cells that 2ME induces apoptosis independently of ER expression.35 To assess whether 2ME induces apoptosis in ER-independent manner, we first determined the ER levels in the A2780 ovarian cancer cells. As reported by others, this cell line did not express detectable levels of either ERα or ERβ by reverse transcriptase-polymerase chain reaction (RT-PCR) or W-B (data not shown). To further confirm this hypothesis, we preincubated A2780 cells with increasing concentrations (0.5 to 2 μM) of the pure estrogen receptor antagonist ICI 182780 (ICI) before treating with 2ME (5 μM). As observed in Figure 4, ICI did not reduce 2ME-induced cytotoxicity, confirming an ER-independent mechanism. .ICI by itself did not induce any cytotoxic effect in this cell line
Figure 4.
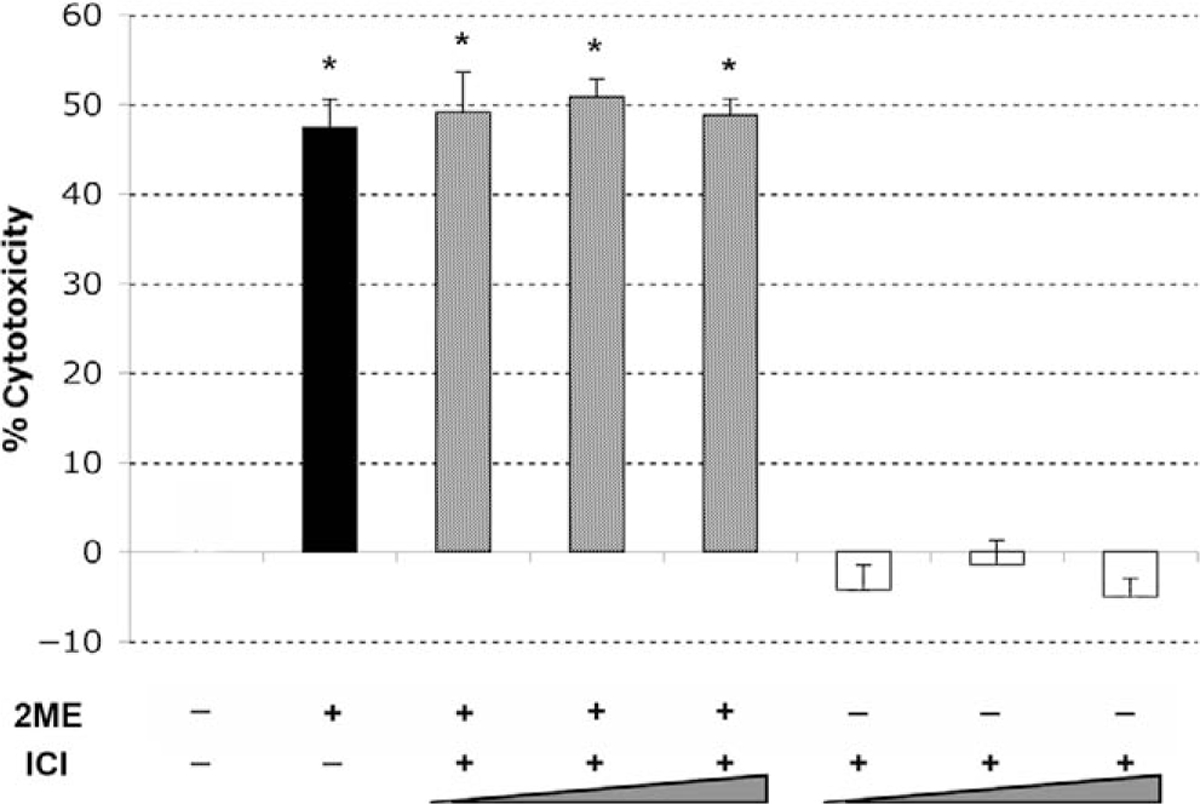
2-Methoxyestradiol (2ME) mediates apoptosis via an estrogen receptor independent pathway. Cell cytotoxicity was measured by the MTS assay. The competitive estrogen receptor antagonist ICI 182780 (ICI, 0.5, 1, and 2 μM) was added for 30 minutes prior to 2-methoxyestradiol (2ME, 5 μM) addition for 48 hours. Data represented as a percentage in respect to control. Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Mann-Whitney test set statistical significance at ★ = P < .05 with respect to control.
2-Methoxyestradiol Induces Cell Cytotoxicity in Primary Cultured Cancerous but not in Normal Ovarian Cells
Having characterized the effects of 2ME in the A2780 cell line, we determined if this action was characteristic to other ovarian cancer cell lines and to primary tissue cultures established from normal ovary, benign tumor, and malignant lesions. As shown in Figure 5, 2ME (5 μM) induces similar cell cytotoxicity in all cancer cell lines studied. In addition, 2ME induced cell cytotoxicity in primary cultures established from a benign epithelial tumor (ie serous cystoadenoma) and malignant lesions, which included a case of serous low malignant potential tumor, (indicated as cancer 1), and cases of advanced serous-papilar ovarian carcinomas, (indicated as ascitis-1 and −2). Interestingly, 2ME did not demonstrate any significant cytotoxic effect in primary tissue cultures derived from normal ovary. To further test this observation we used the HOSE cell line, which was obtained by the immortalization of human ovarian surface epithelial cells using human papilloma viral oncogenes.45 This cell maintains the epithelial phenotype while processing a higher proliferation rate than primary cultured cells (Figure 5).
Figure 5.
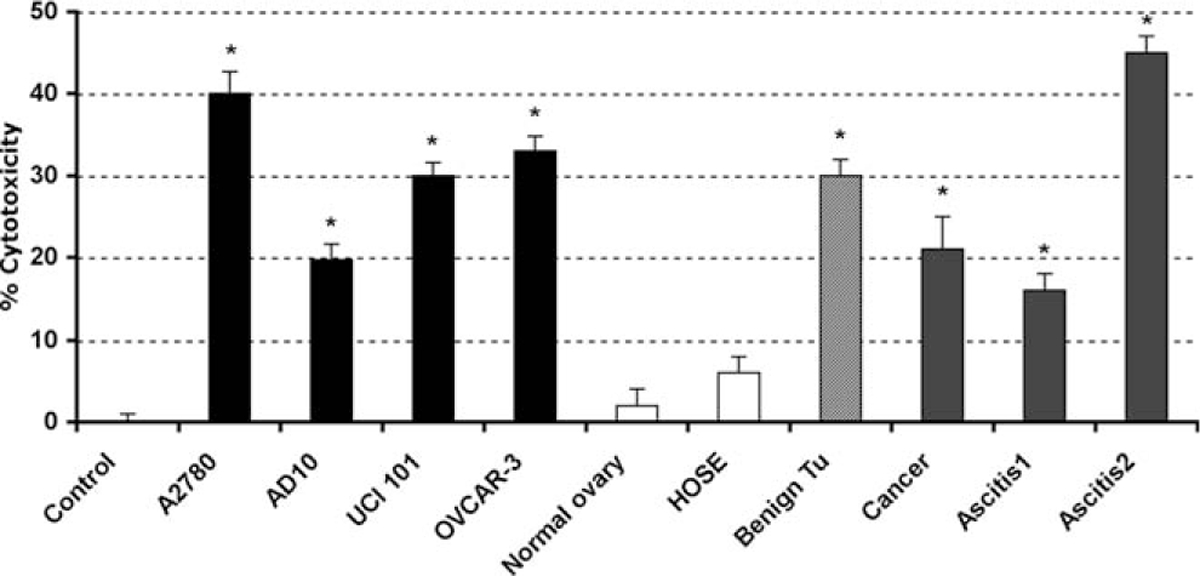
2-Methoxyestradiol (2ME) selectively induces cytotoxicity in cancerous but not normal ovarian cells. Cell cytotoxicity was measured by the MTS assay. 2-methoxyestradiol (2ME, 5 μM for 48 hours) was added to the cancer cells lines A2780, AD10, UCI 101, and OVCAR-3 and to the HOSE cell line, normal, benign tumor, and cancer primary tissue cultures. Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Mann-Whitney test set statistical significance at ★ = P < .05 with respect to control.
2-Methoxyestradiol Induces Apoptosis Through Both Intrinsic and Extrinsic Pathways in Ovarian Cancer Cells
To address whether the mechanism of 2ME-mediated apoptosis involved caspase activation, in vitro caspase activity assays were performed. Caspases involving both the extrinsic and intrinsic pathways were analyzed (initiator caspases-8 and 9, and an executor caspase, caspase-3). After 24 hours of 2ME exposure we observed activation of caspase-3, a caspase used at the end of both the extrinsic and intrinsic pathway. In addition, we observed activation of caspase-8 and 9, which respectively support the participation of both pathways (Figure 6A). Comparing the 2 latter caspases, we observed a notable increase in caspase-9 activity, which suggested a major role for the intrinsic pathway in 2ME mediated apoptosis. Caspase activation was further confirmed by immunoblotting (data not shown). To further characterize the participation of each pathway we used specific caspase inhibitors. As observed in Figure 6B, the addition of specific inhibitors for caspase-8 and 9 resulted in partial reversion of cell cytotoxicity induced by 2ME. In accordance with in vitro caspase assays, the caspase-9 inhibition resulted in a higher reversion of cell viability compared with the caspase-8 inhibition, further supporting a more predominant role for the intrinsic pathway. Interestingly, the combined inhibition of both pathways, using caspase-8 and 9 inhibitors together, did not result in complete reversion of the cytotoxic effect of 2ME. To assess if other caspases could be involved, a nonselective caspase inhibitor, ZVAD-fmk, was used. As observed by the MTS assay in Figure 6b and by flow cytometry in Figure 6C, the complete inhibition of caspases did not completely abrogate the 2ME effect. The remaining cytotoxicity was similar to that observed when inhibitors of caspase-8 and 9 were added together. This result shows that the intrinsic and extrinsic pathways are not capable of completely blocking 2ME-mediated apoptosis and thus suggests the presence of a caspase-independent pathway.
Figure 6.
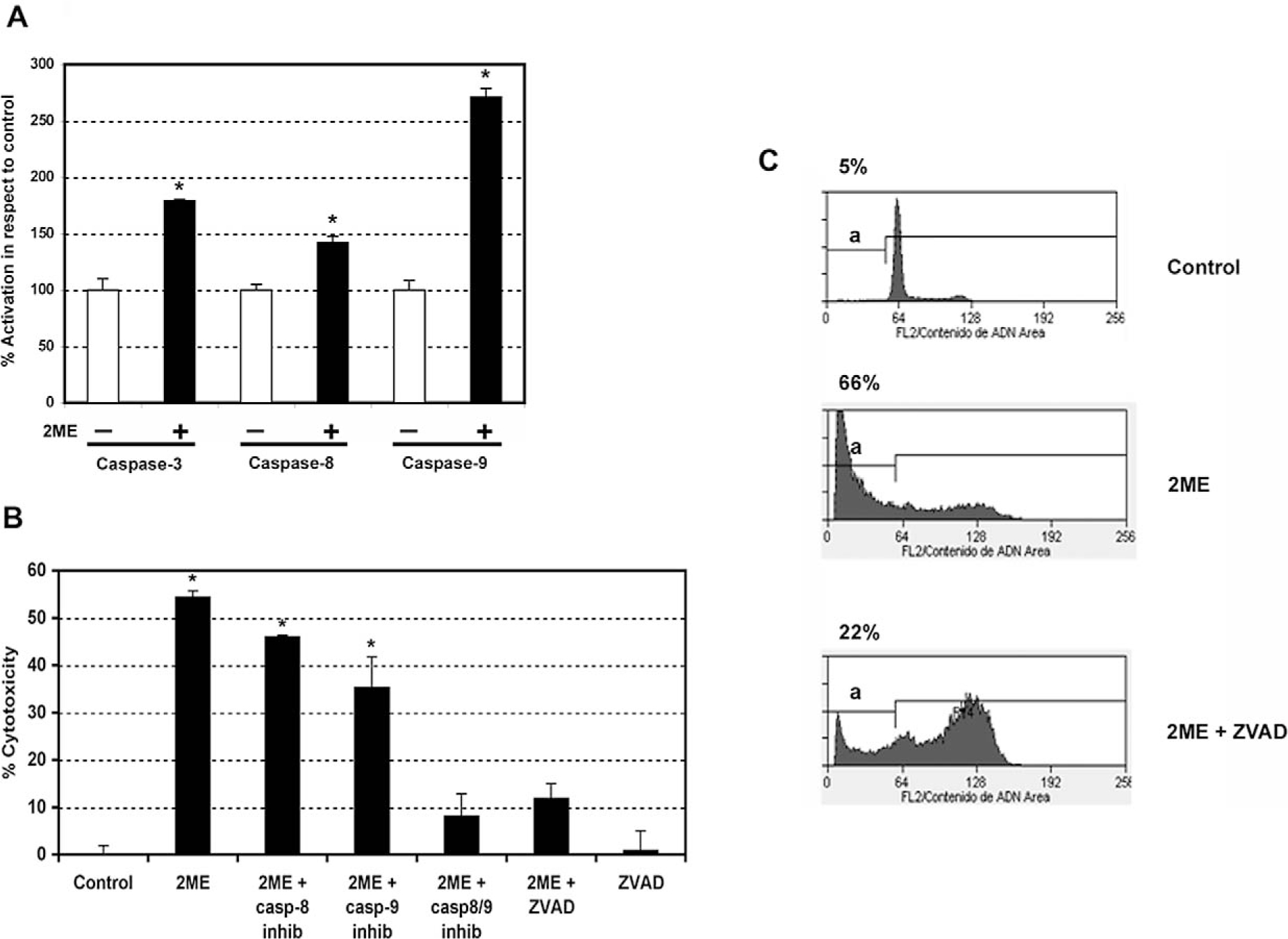
2-Methoxyestradiol (2ME) induces apoptosis via caspases-8 and 9 in A2780 cells. A, Caspase activity assays (caspase-3, 8, and 9) in A2780 cells after the addition of 2ME 5 μM for 24 hours. Cells in the absence of 2ME are set at 100%. Mann-Whitney test ★ = P < .05 with respect to control. B, Cell cytotoxicity was measured by the MTS assay, and the data represented as a percentage in respect to control (vehicle, C). Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. The caspase inhibitors-8, 9, and ZVAD (all 50 μM) were added for 30 minutes previous to and for 24 hours after 2-methoxyestradiol (2ME, 5 μM) addition for 48 hours. Using the Mann-Whitney test, significance was set at P < .05, with ★ = significant in respect to vehicle. C, Representative flow cytometry analysis of A2780 demonstrating each phase of the cell cycle and an apoptotic sub-GO/G1 peak (a), after treatment with ethanol vehicle (control), 2ME 5 μM for 48 hours, and 2ME (5 μM) for 48 hours in the presence of ZVAD (50 μM). The percentage of apoptotic cells is indicated on top of each figure.
2-Methoxyestradiol Induces Mitochondrial Release of Cytochrome C, SMAC/Diablo, AIF and Reactive Oxygen Species Generation Leading to Apoptosis Through Caspase-dependent and Caspase-independent Pathways
Previously, we demonstrated that the intrinsic pathway plays a major role in determining sensitivity of ovarian cancer to proapoptotic reagents such as chemotherapy and TRAIL.34 Recently, we have also reported that one of the main mechanisms driving the apoptosis induced by 2ME involves the activation of the mitochondrial pathway.35 In Figure 7A we characterize several members of the Bcl-2 family known for triggering or inhibiting the activation of mitochondrial pathway. As observed in Figure 7A, 2ME exposure resulted in the progressive loss of the cytosolic form of BAD and its translocation to the mitochondria (reaching a maximum at 24 hours). In addition, the proapoptotic member, Bax, a component of the voltage-dependent anion channels (VDACs), was translocated into the mitochondria. These changes coincided with cytochrome c and SMAC/Diablo release to the cytosol. Coincidentally, we observed phosphorylation and downregulation of the antiapoptotic Bcl-2 protein. Bcl-xL was also downregulated at similar time points. We also tested the FLIP protein, a member interacting at the level of the DISC complex, known to possess antiapoptotic properties by inhibiting caspase-8 activation upon binding of death receptor ligands. 2-Methoxyestradiol also caused a transient upregulation of Fas-Associated Death Domain-Like IL-1-Converting Enzyme– inhibitory protein (FLIP), followed by a downregulation that is maximal at 24 hours. The protein AIF is released from the mitochondria and is known to trigger caspase-independent apoptosis. As shown in Figure 7B, at 6 hours after 2ME exposure, AIF began to be released from mitochondria to the cytosol and was later translocated to the nucleus.
Figure 7.
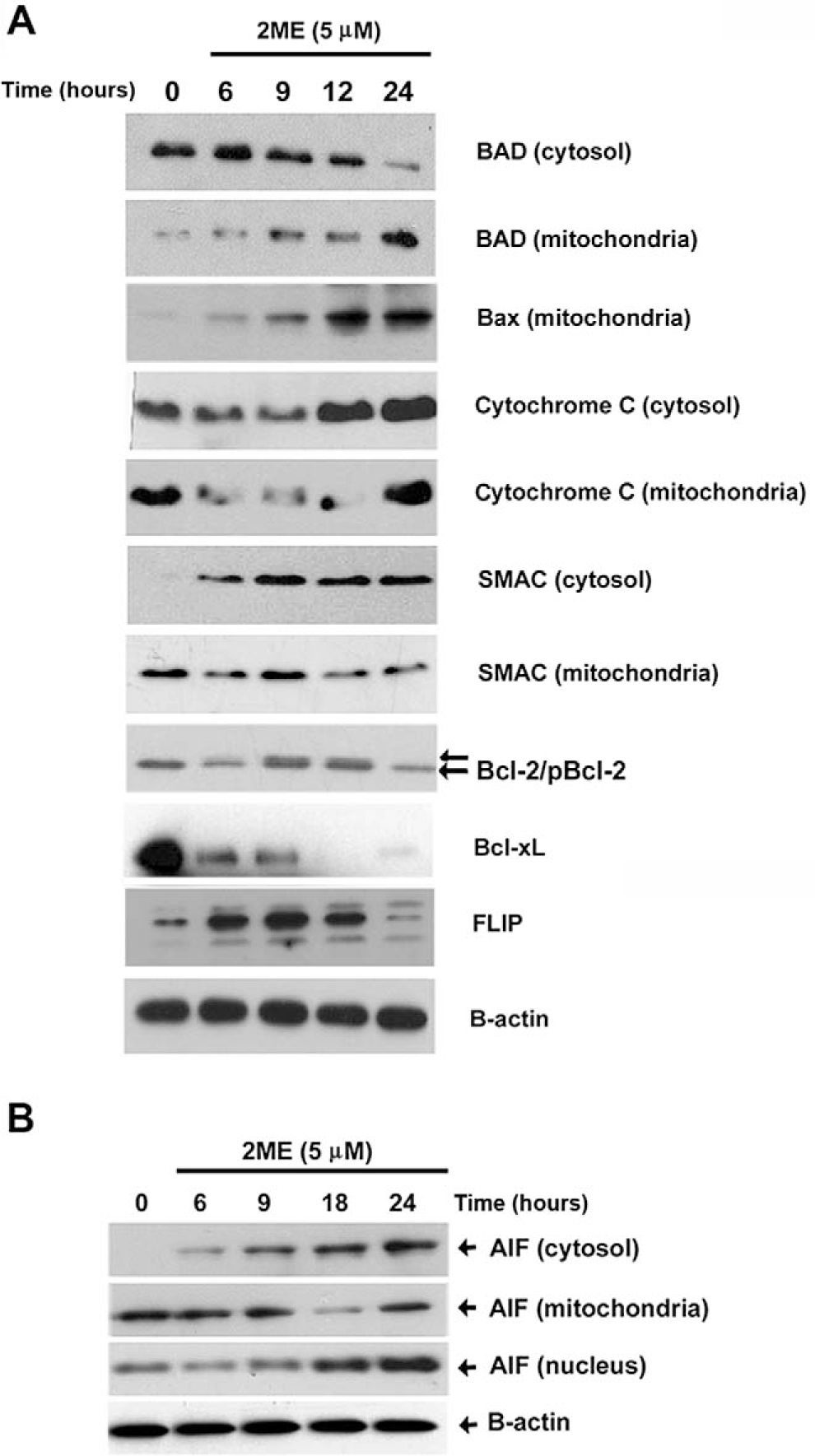
2-Methoxyestradiol (2ME) mediates activation of the intrinsic and extrinsic apoptotic pathways in A2780 cells. Western blot analysis of intrinsic and extrinsic apoptotic pathway proteins at varying time points after 2ME (5 μM) addition (hours). Western blot analysis of cytosolic and membrane factions was performed to examine the translocation of (A) BAD, BAX Cytochrome c, SMAC/Diablo, Bcl-2, Bcl-xL, and FLIP and (B) apoptosis inducing factor (AIF) apoptotic protein between the cytoplasm (cytosol), mitochondrial membrane (mitochondria), and nucleus.
It has been shown that 2ME affects the reactive oxygen species (ROS) generation in certain epithelial cancers.35 Therefore, we studied the participation of ROS generation to explore a mechanism by which 2ME could activate the intrinsic pathway. Figure 8A shows a concentration dependent reduction in mRNA levels of SOD type I upon 2ME treatment. As shown in Figure 8B, the coincubation with increasing concentrations of the antioxidant NAC progressively and completely abrogated (at 10 nM) the cytotoxicity induced by 2ME. Together, these results support the role of ROS generation and the mitochondrial apoptotic pathway in 2ME mediated apoptosis.
Figure 8.
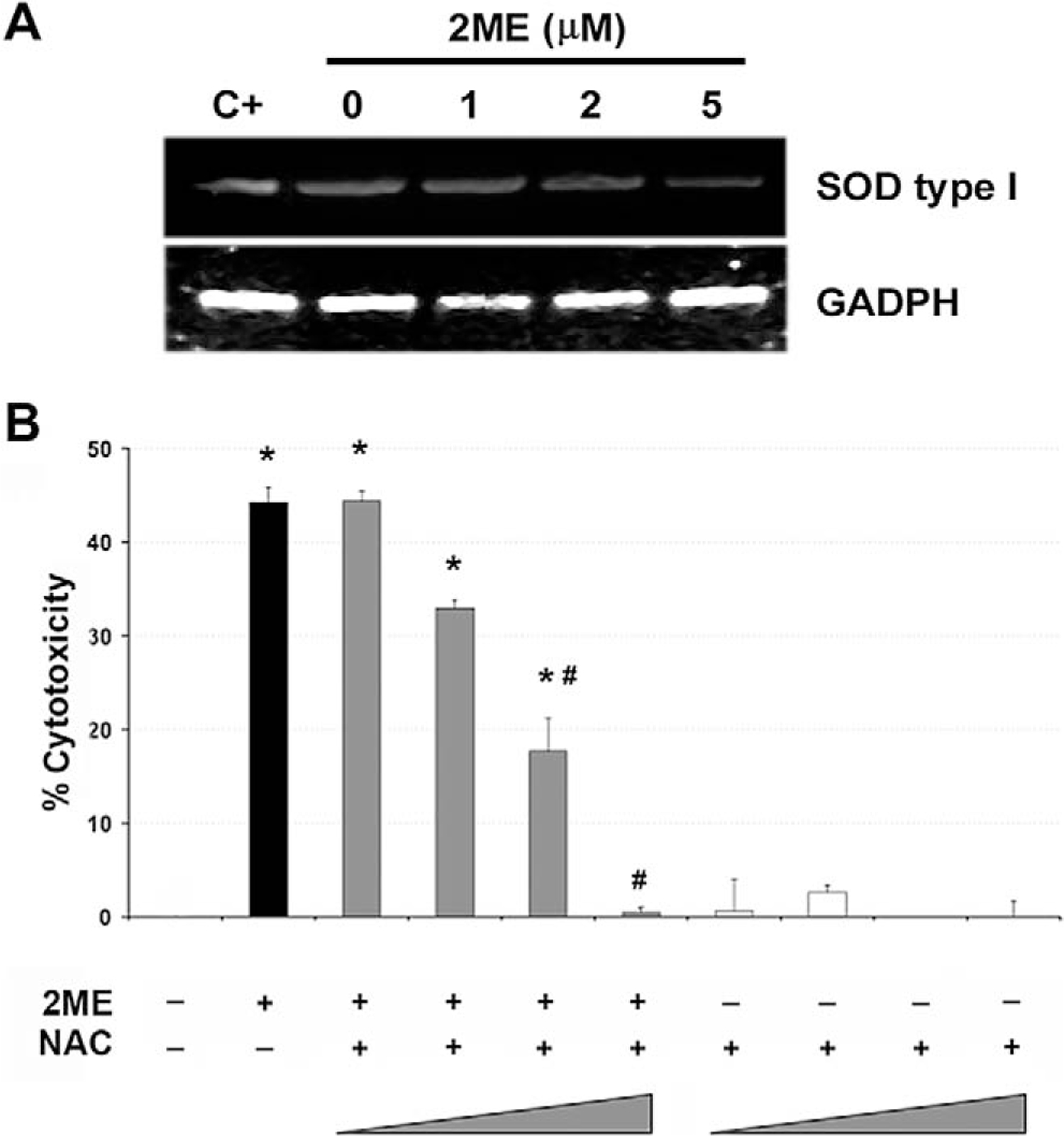
Downregulation of superoxide dismutase type 1 by 2-methoxyestradiol (2ME). A, RT-PCR analysis of superoxide dismutase (SOD) type I mRNA in A2780 cells in response to varying concentrations of 2ME for 9 hours. GAPDH is shown as a loading control. B, Cell viability was measured by the MTS assay, and the data represented as a percentage in respect to control (vehicle, C). Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. The antioxidant N-acetylcystein (NAC) was coincubated at 0.1, 1, 5, and 10 mM with 2ME (5 μM) for 48 hours. Using the Mann-Whitney test, significance was set at P < .05, with ★ = significant in respect to vehicle, # = significant in respect to 2ME (5 μM).
2-Methoxyestradiol Enhances TRAIL Mediated Apoptosis in Ovarian Cancer Cells
Having demonstrated that 2ME induces apoptosis through activation of the mitochondrial pathway we decided to assess whether 2ME could affect TRAIL sensitivity in ovarian cancer cells. To test this, the A2780 and UCI 101 ovarian cancer cells were incubated with either TRAIL alone for 18 hours, 2ME alone for 48 hours, or the combination (2ME for 48 hours with addition of TRAIL for the last 18 hours of the treatment period). As shown in Figure 9A, TRAIL alone brought about slight cytotoxicity in both cell lines (18% in A2780 and less than 5% in UCI 101 cancer cells). 2-Methoxyestradiol also mediated moderate cell cytotoxicity in both cell lines. In contrast, the combination of TRAIL and 2ME significantly enhances cell cytotoxicity in both cell lines (an additive effect in A2780 and synergism in UCI 101). This additive to synergistic effect observed by MTS was confirmed at the protein level with the detection of complete cleavage of PARP (loss of uncleaved form) in presence of the combined treatment (Figure 9B). We further confirmed these results in primary tissue cultures of ovarian cancers (Figure 10). In benign epithelial (ie serous cystoadenoma) and malignant ovarian tumors an additive effect of TRAIL and 2ME in cytotoxicity was observed (Figure 10). Favorably, for potential future cancer treatment, the combination of 2ME plus TRAIL did not lower the viability of cells of normal ovarian origin. Next, we determined if 2ME pretreatment could lower the concentrations of TRAIL required to bring about the additive effect on apoptosis. To this end, A2780 cells were treated with 2ME (5 μM) for 48 hours with varying doses of TRAIL, ranging from 50 to 500 ng/mL during the final 18 hours (Figure 11). As anticipated, TRAIL alone generated low levels of cytotoxity. However, pretreatment with 2ME permitted the observation of synergism when lower concentrations (50 to 100 ng/mL) of TRAIL were evaluated. Increasing the concentration of TRAIL, beyond 50 ng/mL, did not further enhance loss of cell viability or maintain the observation of synergism.
Figure 9.
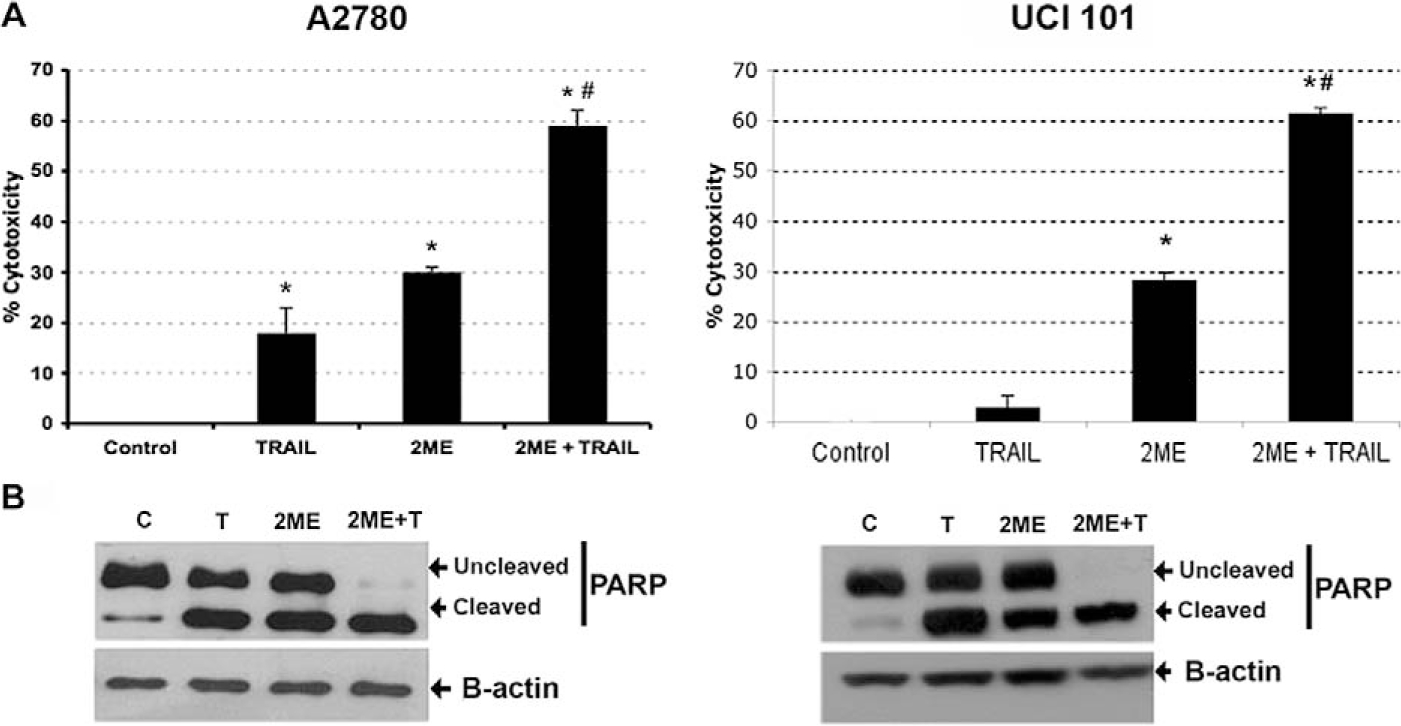
2-Methoxyestradiol (2ME) enhances TRAIL-mediated apoptosis in the ovarian cancer cell lines A2780 and UCI 101. Cytotoxicity was measured by the MTS assay in A2780 cells and UCI 101 cell line (Panel A). Cells were treated with ethanol vehicle (Control), TRAIL 200 ng/mL for 18 hours, 2ME for 48 hours or 2ME for 48 hours with TRAIL addition occurring 18 hours before the end of the experiment (2ME + TRAIL). Using the Mann-Whitney test, statistical significance in change in cytotoxicity in respect to vehicle treated control (★) and in respect to 2ME administration (#) for each drug was set at P < .05. Panel B, western blot analysis of PARP in the presence of above-stated treatments, b-actin is used as a loading control.
Figure 10.
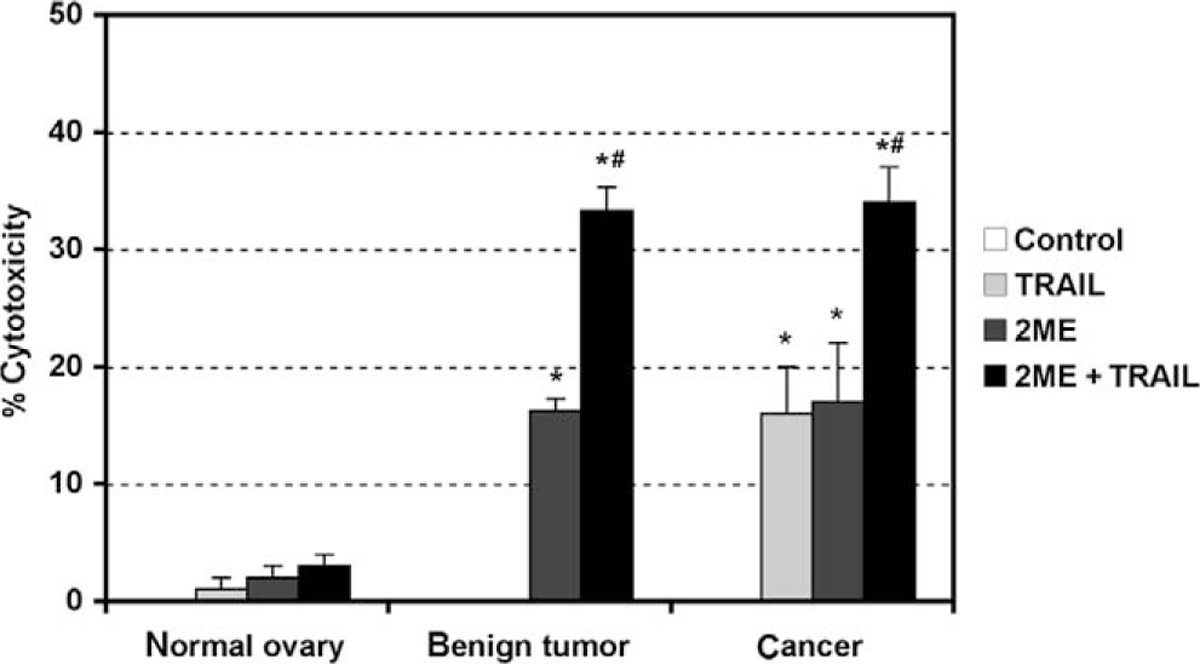
2-Methoxyestradiol (2ME) in combination with TRAIL induces apoptosis in cancerous and benign tumor cells but not in normal cultured human cells of ovarian origin. MTS cell cytotoxicity assays after 2ME (5 μM) addition for 48 hours in cultured normal ovary, benign tumor, and ovarian cancer cells. Data represent percentage in respect to control. Standard derivation of the means is shown, each consisting of 5 replicates. Statistical significance in respect to vehicle treated control (★) and in respect to 2ME or TRAIL treatment (#) for each cell type was set at P < .05 using the Mann-Whitney test.
Figure 11.
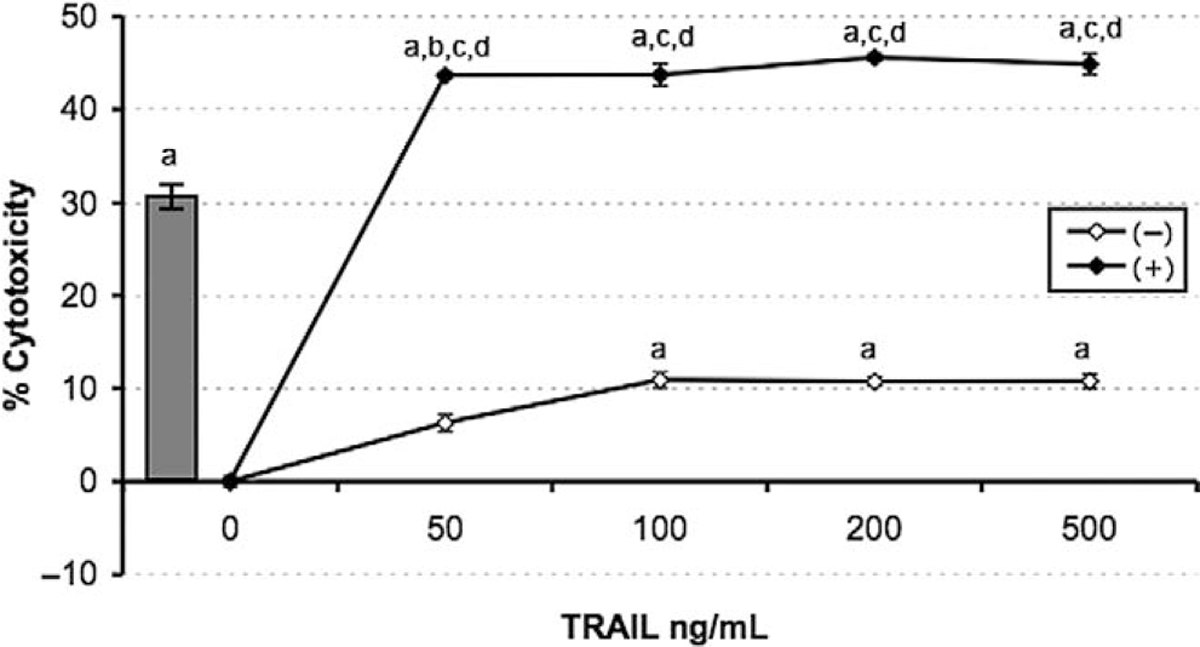
2-methoxyestradiol sensitizes A2780 ovarian cancer cells to TRAIL-mediated apoptosis. Cell viability was measured by the MTS assay in response to the apoptotic drugs 2ME and TRAIL. TRAIL concentration was varied between 50 and 500 ng/mL for 18 hours after the presence (black dot) or absence (white dot) of 2ME fixed at 5 uM added 24 hours previously. The grey bar represents 2ME (5 uM) alone for 48 hours. Data is represented as a percentage in respect to control and standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. Using the Mann-Whitney test, significance was set at P < .05, with a = significant in respect to vehicle, b = significant in respect to previous lower concentration, c = significant in respect to the matched concentrations of the same drug alone, d = significant in respect to fixed-drug concentration (grey bar).
Mechanisms by Which 2ME Enhances TRAIL Mediated Apoptosis
We next evaluated the mechanisms by which 2ME could enhance the response of A2780 cells to the apoptotic actions of TRAIL. It has been demonstrated in epithelial cancer cells that 2ME can upregulate TRAIL-R2 expression.24 As shown in Figure 12A, in A2780 cells mRNA levels of TRAIL-R1 (also named DR4) were not detected in the presence or absence of 2ME. Low levels of TRAIL-R2 mRNA (DR5) were detected under basal conditions. 2-Methoxyestradiol significantly increased TRAIL-R2 after 9 hours of incubation. No change was observed in mRNA levels of decoy receptors TRAIL-R3 (also named DcR1) and TRAIL-R4 (also named DcR2) under any condition. Changes in TRAIL receptors were confirmed by densitometry as shown in Figure 12B. The upregulation of TRAIL-R2 was further confirmed at the protein level by immunocytochemistry (Figure 12C).
Figure 12.
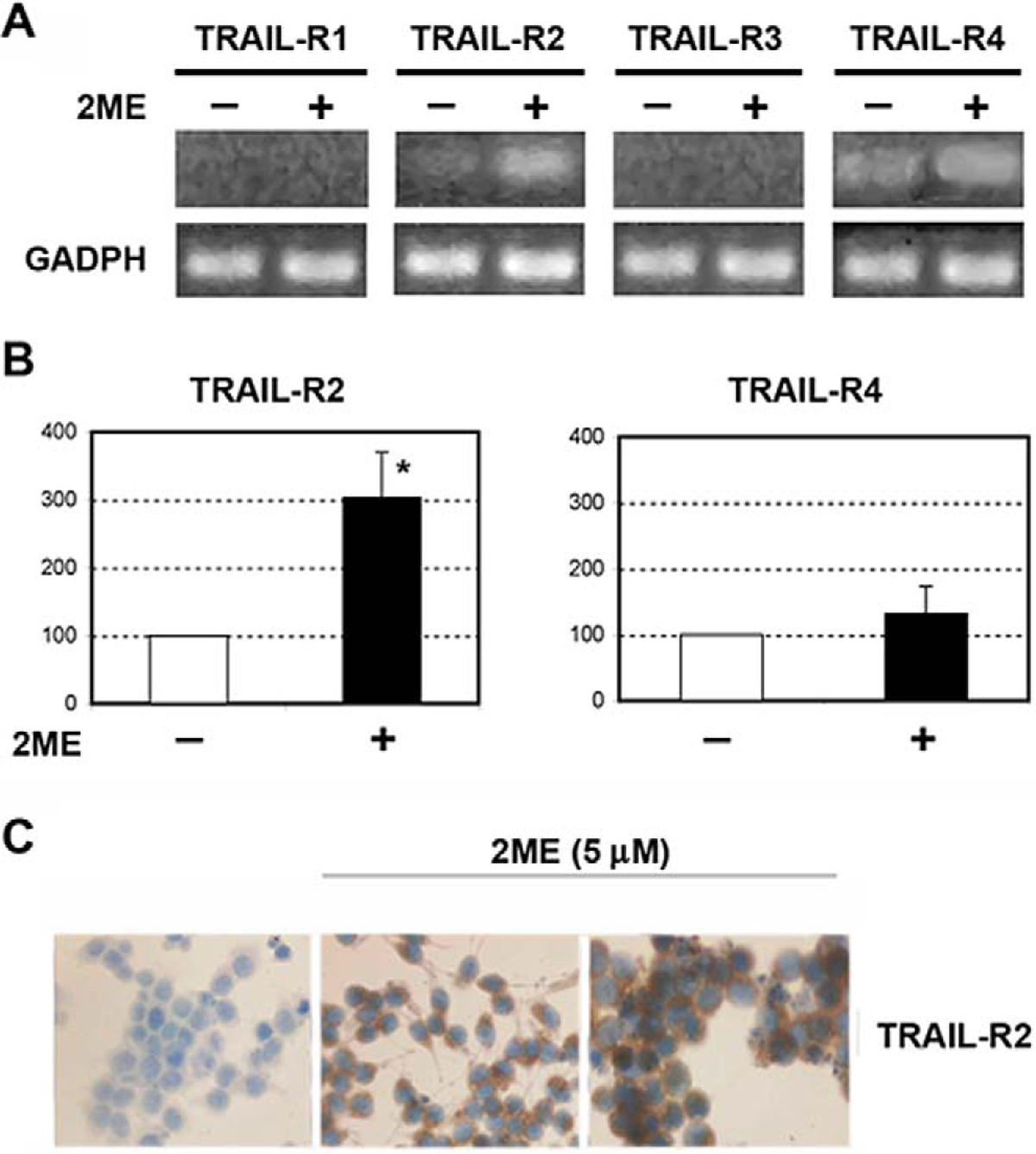
2-Methoxyestradiol (2ME) upregulates TRAIL-R2 in ovarian cancer cell line A2780. A, RT-PCR of TRAIL-R1, TRAIL-R2, TRAIL-R3, and TRAIL-R4 mRNA with 2ME 5 μM for 9 hours. B, Densitometric analysis of the TRAIL-R2 and TRAIL R4 RT-PCR. Statistical significance (★) in respect to vehicle treated control for each cell type was set at P < .05 using the Mann-Whitney test. C, Immunocytochemistry demonstrating that the increases in mRNA levels are mirrored at the protein level after 24 hours of 2ME treatment.
As we demonstrated that ROS generation was involved in 2ME-mediated apoptosis (Figure 8b), we studied its role in 2ME-enhanced TRAIL-mediated apoptosis. The presence of the antioxidant NAC did not revert TRAIL-mediated cytotoxicity demonstrating that 2ME and TRAIL use different apoptotic mechanisms in the A2780 cell line. Interestingly, the cytotoxicity observed with combined 2ME and TRAIL treatment was only partially reversed upon NAC addition (Figure 13).
Figure 13.
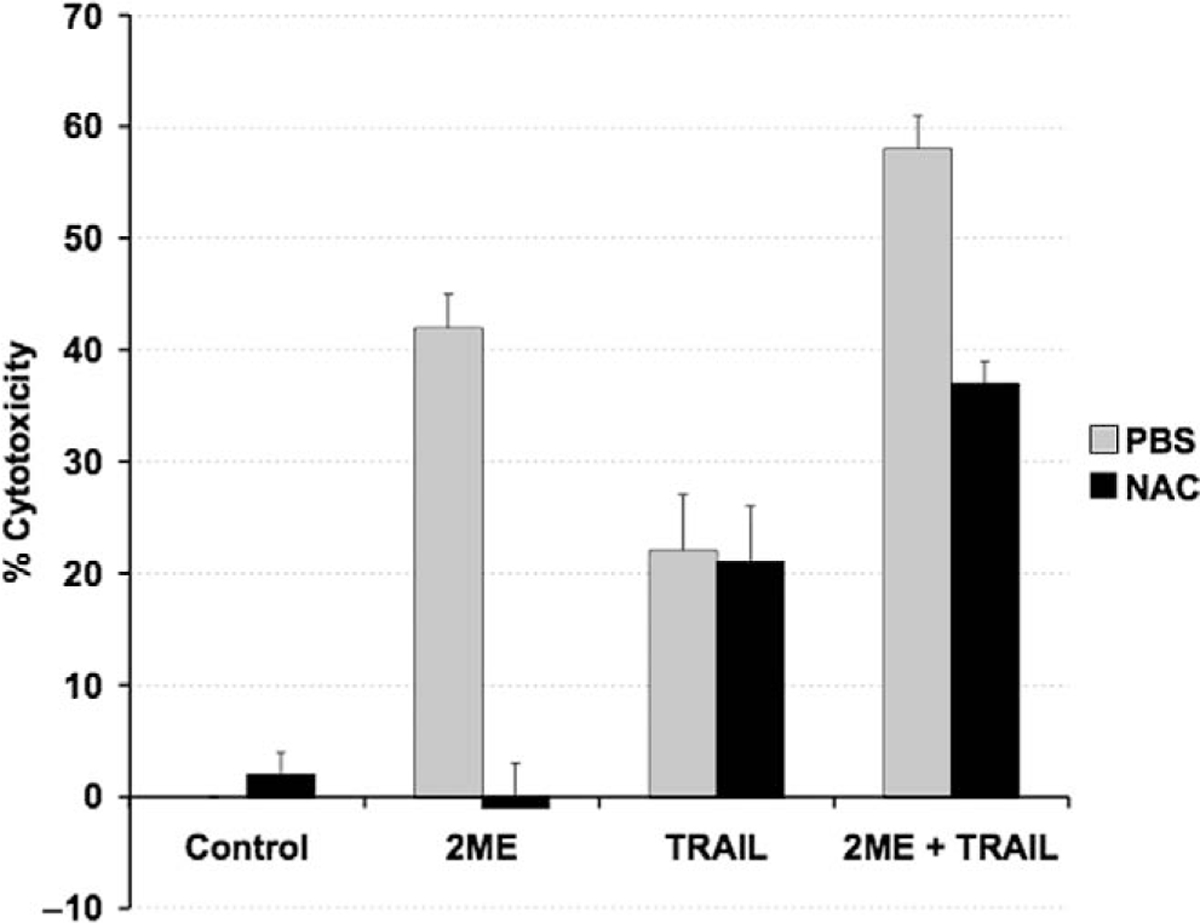
N-acetylcystein (NAC) prevents only 2-methoxyestradiol (2ME)–induced apoptosis but not TRAIL-mediated apoptosis. Cell viability was measured by the MTS assay, and the data represented as a percentage in respect to control (vehicle, C). Standard deviation of the mean is shown from a minimum of 3 experiments performed, each consisting of 5 replicates. The antioxidant NAC was coincubated at 10 μM with 2ME (5 μM) for 48 hours and TRAIL (200 ng/mL) for 18 hours. Using the Mann-Whitney test, significance was set at P < .05, with a = significant in respect to vehicle, b = significant in respect to 2ME (5 uM) in A2780 cells.
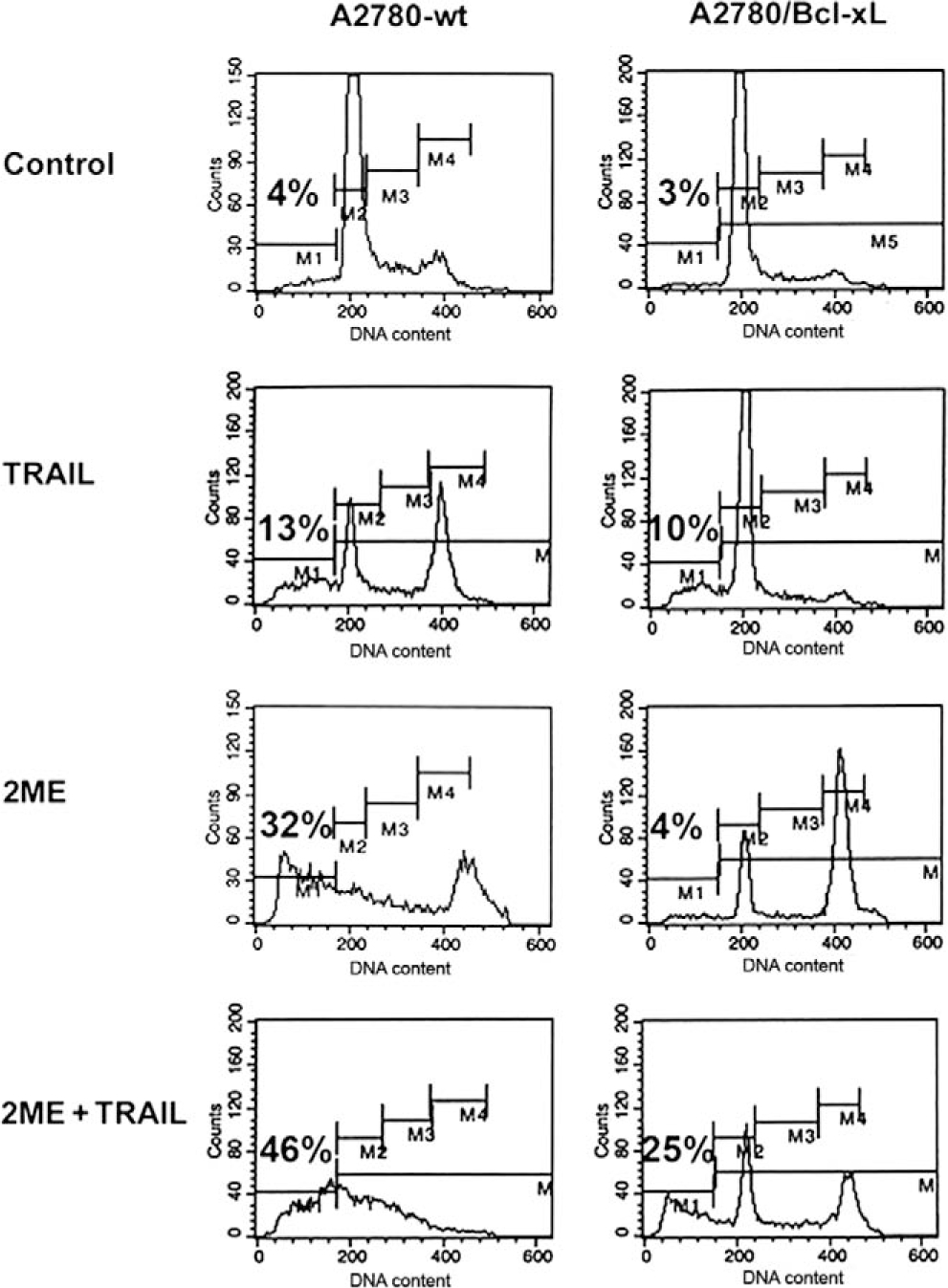
Having demonstrated the effects of 2ME on the mitochondrial pathway in ovarian cancer cells, we decided to explore the effects of inhibiting this pathway in the interaction between 2ME and TRAIL. To this end, in A2780 cells we overexpressed Bcl-xL, an antiapoptotic member of Bcl-2 family which acts at the mitochondrial membrane inhibiting VDAC channels formation. Figure 14 shows the assessment of apoptosis through measurement of sub-G0/G1 DNA content in the DNA histogram generated by flow cytometry. As indicated as M1 in the panels on Figure 14, we observed that upon 24 hours of 2ME incubation there is an increase in the subG0/G1 DNA content compared with the vehicle control. We also observed an arrest in G2/M (indicated as M4 in the figure). This arrest was also observed at early time points, before apoptosis was induced (data not shown). TRAIL alone also increased the subG0/G1 content and a coincidental arrest in G2/M. The combination resulted in a complete loss of the normal curve of DNA content histogram with a significant proportion of cells in the subG0/G1 fraction. The panels on the right side of Figure 14, demonstrate that overexpression of Bcl-xL significantly reduced the proportion of cells in the sub G0/G1 fraction when 2ME was present. Interestingly, the inhibition of apoptosis was accompanied by an increase in G2/M fraction. These results suggest that 2ME induces an arrest in G2/M independently of the activation of the apoptotic mitochondrial machinery. The overexpression of Bcl-xL almost completely reverted 2ME mediated apoptosis. Bcl-xL overexpression notably reduced the subG0/G1 fraction and increased the G2/M fraction observed with the combination of TRAIL and 2ME. To further assess these inhibitory effects of Bcl-xL overexpression on the mitochondrial pathway we studied its actions on cytochrome c and AIF release upon treatment with each reagent used alone or in the combination. As shown in Figure 15A, Bcl-xL overexpression is demonstrated by W-B (upper panel). Analysis of Bid, a protein downstream of the death-inducing signaling complex (DISC) complex that acts upstream of the Bcl-2 members, demonstrated a loss of cytosolic expression and the appearance of the cleaved form (tBid) at 24 hours after 2ME incubation (as shown in lower panel). Incubation with TRAIL also mediated the appearance of tBid. The combination of 2ME plus TRAIL increased tBid expression compared with each reagent individually in both cell lines (empty vector v/s Bcl-xL overexpression). These results support the relevance of the mitochondrial pathway in the combined effect of 2ME plus TRAIL in the A2780 ovarian cancer cell line. The overexpression of Bcl-xL almost completely abrogated the cytochrome c release induced by the combination of 2ME plus TRAIL (Figure 15B). A slight and transient cytochrome c release to cytosol was induced between 3 and 6 hours of incubation but afterwards a complete inhibition was achieved. In addition, Bcl-xL overexpression resulted in complete inhibition of AIF release to the cytosol. Together, these results support the pleiotropic effects of 2ME on enhancing TRAIL sensitivity.
Figure 14.
Flow cytometry analysis of 2-methoxyestradiol (2ME) and TRAIL-mediated apoptosis in A2780-wt (vector) and A2780/Bcl-XL cells. Representative flow cytometry analysis of A2780-wt (left side) and A2780/Bcl-XL cells (right side), demonstrating each phase of the cell cycle (GO/G1 = M2, S = M3, G2/M = M4) and an apoptotic sub-GO/G1 peak (M1), after treatment with ethanol vehicle (Control), TRAIL 200 ng/mL for 18 hours, 2ME (5 μM) for 48 hours or 2ME for 48 hours with TRAIL addition occurring 18 hours before the end of the experiment (2ME + TRAIL).
Figure 15.
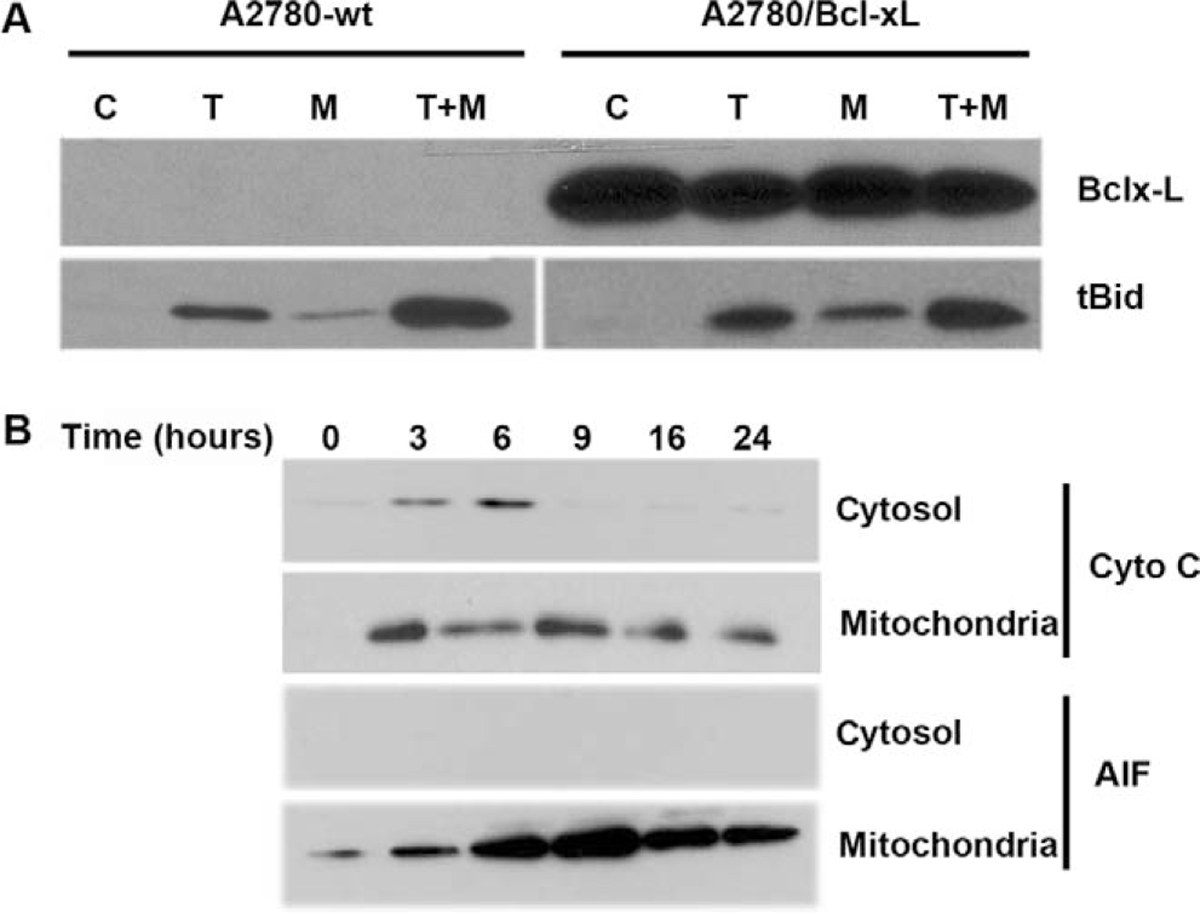
Bcl-xL overexpression does not affect Bid cleavage (tBid) upon 2-methoxyestradiol (2ME) or 2ME plus TRAIL treatment and abrogates cytochrome c and apoptosis inducing factor (AIF) release induced by 2ME. A, Western blot analysis of Bcl-xL and tBid after treatment with ethanol vehicle (Control), TRAIL 200 ng/mL for 18 hours, 2ME for 48 hours or 2ME for 48 hours with TRAIL addition occurring 18 hours before the end of the experiment (2ME + TRAIL) in the A2780/wt (vector) and A2780/Bcl-xL cell lines. B, Western blot analysis of intrinsic apoptotic proteins, cytochrome c and AIF between the cytoplasm (cytosol) and the mitochondrial membrane (mitochondria) at varying time points after 2ME (5 μM) addition (hours) in the A2780/Bcl-xL cell line.
Comparison of the Specificity and Cytotoxicity of 2ME with Current Chemotherapeutic Drugs
To evaluate the reported adverse side effects of current chemotherapeutics on normal tissue we treated the normal human ovarian surface epithelial (HOSE) cell line with cisplatin, doxorubicin, and paclitaxel. As demonstrated in previous figures, 2ME did not significantly reduce viability, in contrary to the anticipated effect observed with each of the chemotherapies tested (Figure 16). To further test the benefits of 2ME in the treatment of ovarian cancer we compared the efficacy of the TRAIL and 2ME alone and in combination with currently used chemotherapeutic reagents in ovarian cancer cells. Figure 16 demonstrates the levels of cytotoxicity observed in comparison with cisplatin, doxorubicin, and paclitaxel in the UCI 101 cell line. The combination of 2ME plus TRAIL produced higher cell cytotoxicity than any of the chemotherapeutic reagents used alone or different combinations (data not shown). The A2780 cells and the known doxorubicin resistant cell line AD10, both showed additive effects in the presence of TRAIL and 2ME, however the level of cytotoxicity did not exceed that of the chemotherapies (results not shown). As previous reports have shown a potential synergistic effect ofthe combination between 2ME and chemotherapeutic reagents, a combination of 2ME plus the chemotherapy was compared. Results demonstrate additive effects of 2ME with each of the 3 chemotherapies, and that these levels of cytotoxicity were not higher than the combinations with TRAIL.
Figure 16.
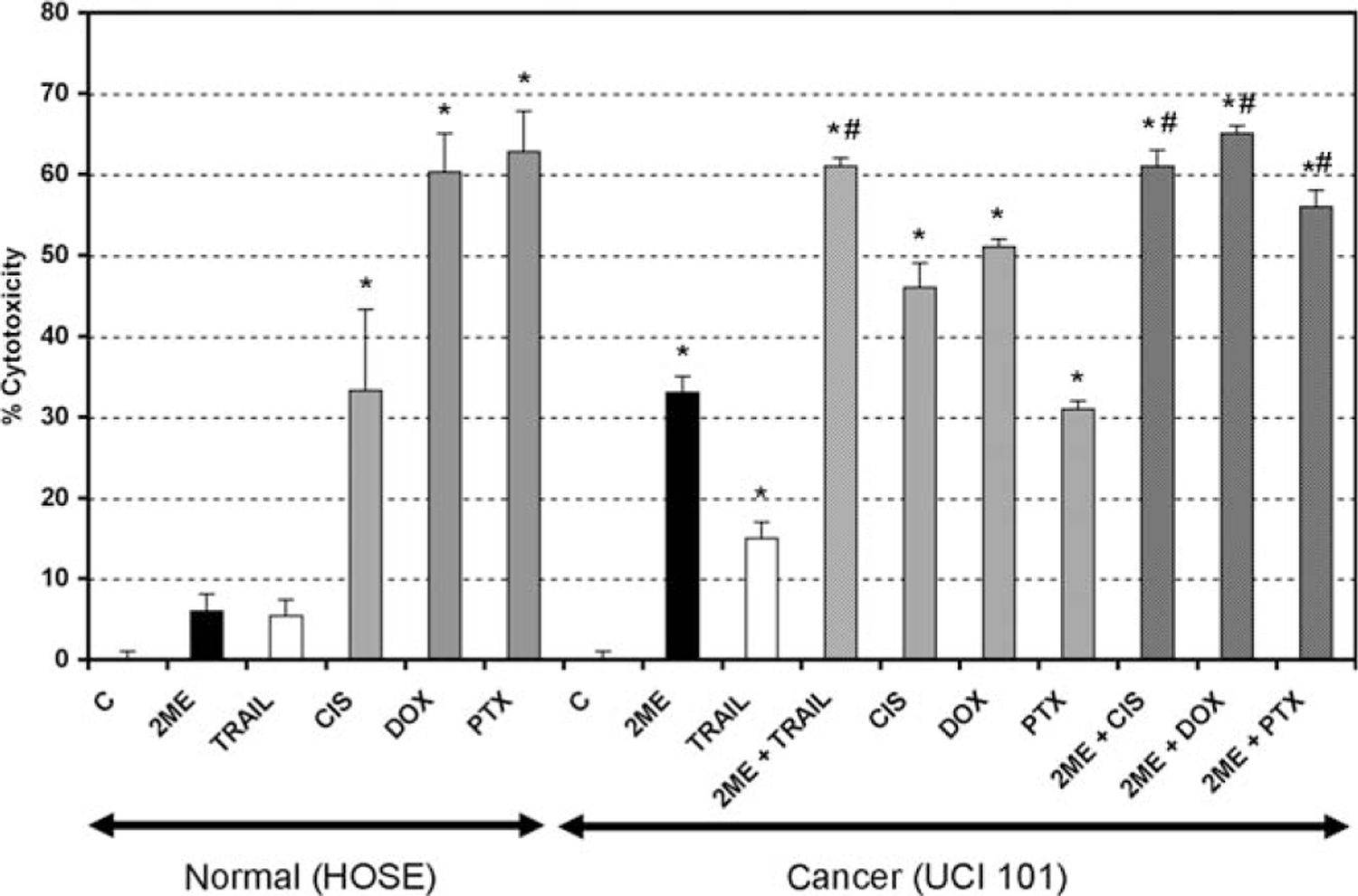
The combination of 2-methoxyestradiol (2ME) plus TRAIL produced similar cytotoxicity to that observed with current chemotherapeutic reagents. Cell cytotoxicity was measured by the MTS assay in a human normal ovarian surface epithelium cell line (HOSE) and in the UCI 101 ovarian cancer cell line. Cells were treated with ethanol vehicle (C), TRAIL 200 ng/mL for 18 hours, 2ME and the chemotherapeutics doxorubicin (DOX, 5 μM), paclitaxel (PTX, 5 μM), and cisplatin (CIS, 5 μM), were administered for 48 hours. Furthermore, 2ME for 48 hours with TRAIL or chemotherapeutic addition occurring 18 hours before the end of the experiment were tested. Using the Mann-Whitney test, statistical significance in change in cell cytotoxicity in respect to vehicle treated control (★) and in respect to individual administration (#) for each drug was set at P < .05.
DISCUSSION
During recent years growing evidence has supported the hormonal participation, especially of the estrogens, in the carcinogenic process affecting the ovarian epithelium. However, how the estrogens play a role in the genesis of ovarian cancer is not fully understood. It appears not to be a simple over-exposure to estrogens that leads to cancer and thus the imbalance in the level of varying estrogen metabolites may be a plausible theory. Such an imbalance of metabolites may predispose or protect cells of developing cancer. Herein we demonstrate that 2ME and its precursor form, 20H, induce cell cytotoxicity as a result of apoptosis in a concentration and time-dependent manner in ovarian cancer cells.
As described in other epithelial tumors, such as breast or endometrial carcinomas, the estrogen metabolites alone, at higher than physiological concentrations, may display unique, estrogenic or antiestrogenic behavior.46,47 In our hands, physiological (picomolar concentrations) or low pharmacological concentrations (lower than 0.5 μM) were not capable of triggering apoptosis in the ovarian cancer cells. This may suggest that endogenous 2ME is not mediating an inhibitory or proapoptotic stimulus under physiological conditions.
2-Methoxyestradiol is derived by sequential hepatic hydroxylation and methylation from its parent compounds, estradiol or estrone.48 Here we demonstrate that 2ME induces cell death through an ER-independent pathway in ovarian cancer cells. Our first evidence of ER-independence comes from the observation that 2ME mediates cell death in an ER-negative ovarian cancer cell line (A2780). Furthermore, a pure estrogen receptor antagonist (ICI) did not inhibit 2ME-mediated cytotoxicity in the same cells. These findings are consistent with our previous work in other ER-negative gynecologic cancer cell lines, which also experienced cell death upon incubation with 2ME.35
2-Methoxyestradiol mediates apoptosis through triggering both the intrinsic and the extrinsic apoptotic pathways. Our results suggest that 2ME preferentially uses the intrinsic pathway in mediating cell death. This assumption is based on a higher activity of caspase-9 and a near total reversion of apoptosis upon exogenous expression of Bcl-xL. 2-Methoxyestradiol activation of the extrinsic pathway includes upregulation of death receptors, specifically TRAIL-R2, downregulation of FLIP, and cleavage of caspase-8 and Bid. The cleaved form of Bid, also called tBid, may explain the observed participation of both the extrinsic and intrinsic pathways. A principal mechanism driving the activation of the mitochondrial pathway appears to be the generation of reactive oxygen species. Here we demonstrated that 2ME downregulates the antioxidant enzyme SOD type I which would allow the accumulation of ROS. The participation of the intrinsic pathway is further corroborated by the 2ME-downregulation of Bcl-2 and Bcl-xL expression which leads to changes in the mitochondrial transmembrane potential (through VDAC channel formation), resulting in cytochrome c and SMAC/Diablo release.
How 2ME mediates its apoptotic effects in an ER-independent manner remains unclear. The functional downregulation of proteins interacting with 2ME, such as SOD type I, could be one explanation. Binding to unknown intranuclear factor(s) leading to the unique transcriptional regulation of the downstream genes (ie bcl-2 family members) could constitute an alternative mechanism. However, further studies are required to better understand how 2ME exerts its effects independently of the binding to the ER receptor.
2-Methoxyestradiol mediated apoptosis is accompanied by an increase in the number of cells in the S or G2/M phase of the cell cycle as has been previously reported in other cancer cell types.23,49 We recently published that 2ME induced an arrest in G2/M phase and that apoptosis is associated with downregulation of antiapoptotic members of Bcl-2 family (Bcl-xL and Bcl-2) in endometrial cancer cells.35 Herein, we demonstrated that the 2ME induced G2/M arrest occurs independently of activation of the mitochondrial pathway in ovarian cancer cells, as the overexpression of Bcl-xL did not affect the G2/M arrest while inhibiting the cytochrome c release. Furthermore, 2ME appears to have independent and separate effects upon both pathways. The Bcl-xL overexpression efficiently inhibits the 2ME effect on the intrinsic pathway but does not affect the activation of the extrinsic pathway. In fact, upon combination treatment the enhancement in caspase-8 activation and Bid cleavage remains similar in Bcl-xL over-expressing cells compared to the wild type.
Previously we demonstrated that 2ME induces apoptosis in epithelial cancers through activation of caspases. Herein, we present the first evidence that 2ME can induce cell death through a caspase-independent pathway, as demonstrated by AIF release and lack of total apoptotic inhibition by casapase inhibitors. Upon release, AIF translocates to the nucleus where it induces chromatin condensation, DNA degradation and finally cell death. These observations are interesting in light of our previous studies in endometrial cancer cells, where 2ME mediates cell death without AIF release and caspase inhibitors can totally reverse the 2ME mediated apoptosis. This further demonstrates the heterogeneity between cancers and supports the dogma that all cancers differ in their genesis and cannot be treated in the same therapeutic regimen.
Important for any potential oncology therapy is that our results demonstrate that 2ME selectively induces cell death in ovarian cancer without affecting corresponding normal tissues. This finding is consistent with our recent report on the effects of 2ME in other reproductive tissues (endometrium, cervix, and fallopian tube) in which 2ME only exhibits cytotoxicity on cells of malignant origin.35 In addition to this evidence, we demonstrate herein that 2ME also affects benign and lower grade malignant lesions arising from the ovarian epithelium (Figure 10). Equally important is the observation in normal ovarian epithelial cells that current chemotherapy regimes bring about a substantial decrease in cell viability while 2ME has no significant effect (Figure 16). These findings have prompted our interest in using pharmacological concentrations of 2ME in the treatment of other benign conditions affecting the genital tract, such as fibroids or endometriosis.
TRAIL has become a promising alternative for the treatment of a wide variety of epithelial tumors.50 Unfortunately, the majority of the ovarian cancer cells are resistant to TRAIL-mediated apoptosis and the mechanisms explaining its resistance are not well understood. Having demonstrated that 2ME acts by inducing changes in both the extrinsic and intrinsic pathway and that previous reports from our laboratory have demonstrated that 2ME can sensitize endometrial cancer cells to other cancer therapies, we tested the effects of 2ME in combination with TRAIL in ovarian cancer cells. Using the A2780 and UCI 101 cell lines we demonstrate that 2ME and TRAIL can synergize to enhance cell death depending on the concentrations of each compound used. In the case of A2780 the preincubation with 2ME reduces the concentration of TRAIL required to achieve maximum cell death, an observation that is favorable for clinical practice. This synergy may be explained by the upregulation of death receptors by 2ME and thus allowing an enhanced apoptotic signal upon TRAIL binding. Moreover, 2ME also facilitates the activation of the DISC complex through the downregulation of the inhibitory protein FLIP thus favoring the extrinsic pathway. The differential use of the extrinsic and intrinsic pathways by 2ME and TRAIL is highlighted by our experiment using the antioxidant, NAC. Here we demonstrated that NAC can completely reverse the action of 2ME, without altering TRAIL mediated apoptosis. Surprisingly, in the presence of both reagents there was only a partial reversion in cytotoxicity in the presence of NAC, suggesting that mechanism of action in the presence of TRAIL and 2ME is different when each reagent is added alone. Previously, our group and other researchers have postulated the existence of a reinforcement loop as a mechanism by which reagents, acting through the mitochondria (ie 4HPR and chemotherapy), are able to enhance TRAIL-mediated apoptosis.33,51,52 On combined treatment with any those reagents with TRAIL, a reciprocal activation of the intrinsic and extrinsic cascades takes place creating the reinforcement loop. In 2ME plus TRAIL combination, a similar crosstalk has been demonstrated in pancreatic cancer cells.53
Having demonstrated the beneficial effect of 2ME and the combination with TRAIL in ovarian cancer cell lines, primary tissue cultures of ovarian normal cells, benign lesions and ovarian cancers, we compared this regime to chemotherapies currently employed in ovarian cancer. Our results in 3 distinct ovarian cell lines demonstrate that either 2ME alone or the combination of 2ME and TRAIL is as effective or is more effective that either of the conventional chemotherapies. However, as with any in vitro study, the next step has to be the testing of these observation in mouse xenograft models of human cancer to ascertain if the stromal environment of an in vivo tumour may protect the cancer cell from 2ME-mediated apoptosis and to evaluate the possibility that ovarian tumours may highly express enzymes which rapidly metabolize 2ME and thus limit this drugs potency.
As it is well established that chemotherapy reduces normal cell viability and we have demonstrated that 2ME and TRAIL do not bring about cytotoxicity in primary cultured normal ovarian cells, we postulate that 2ME and TRAIL should be considered as potential new cancer therapeutics in ovarian cancer treatment and suggest the need for clinical trials.
ACKNOWLEDGMENTS
We thank Drs Horacio Croxatto and Pedro Orihuela for their advice on the action of 2ME, Dr David Mayerson for recruiting patients and collecting samples, and Dr Carmen Romero of the University of Chile for the use of the normal human ovarian cell line. This work was supported by FONDECYT 1060495 (GO) and 1050744 (MC).
Contributor Information
Sumie Kato, Department of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile.
Anil Sadarangani, Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile.
Soledad Lange, Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile.
Ana M. Delpiano, Department of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile
Macarena Vargas, Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile.
Jorge Brañes, Department of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile.
Jorge Carvajal, Department of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile.
Stanley Lipkowitz, Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Institute of Cancer, National Institutes of Health, Bethesda, Maryland.
Gareth I. Owen, Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile.
Mauricio A. Cuello, Department of Obstetrics and Gynecology, Faculty of Medicine, Pontificia Universidad Católica de Chile, Santiago, Chile.
REFERENCES
- 1.Bhoola S, Hoskins WJ. Diagnosis and management of epithelial ovarian cancer. Obstet Gynecol. 2006;107:1399–1410. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. [DOI] [PubMed] [Google Scholar]
- 3.Aletti GD, Gallenberg MM, Cliby WA, Jatoi A, Hartmann LC. Current management strategies for ovarian cancer. Mayo Clin Proc. 2007;82:751–770. [DOI] [PubMed] [Google Scholar]
- 4.Lage H, Denkert C. Resistance to chemotherapy in ovarian carcinoma. Recent Results Cancer Res. 2007;176:51–60. [DOI] [PubMed] [Google Scholar]
- 5.Beral V, Bull D, Green J, Reeves G. Ovarian cancer and hormone replacement therapy in the Million Women Study. Lancet. 2007;369:1703–1710. [DOI] [PubMed] [Google Scholar]
- 6.Glud E, Kjaer SK, Thomsen BL, et al. Hormone therapy and the impact of estrogen intake on the risk of ovarian cancer. Arch Intern Med. 2004;164:2253–2259. [DOI] [PubMed] [Google Scholar]
- 7.Whittemore AS, Harris R, Itnyre J. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. IV. The pathogenesis of epithelial ovarian cancer. Collaborative Ovarian Cancer Group. Am J Epidemiol. 15 1992;136:1212–1220. [DOI] [PubMed] [Google Scholar]
- 8.Ho SM. Estrogen, progesterone and epithelial ovarian cancer. Reprod Biol Endocrinol. 2003;1:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syed V, Mukherjee K, Godoy-Tundidor S, Ho SM. Progesterone induces Apoptosis in TRAIL-resistant ovarian cancer cells by circumventing c-FLIP(L) overexpression. J Cell Biochem. 2007;102:442–52. [DOI] [PubMed] [Google Scholar]
- 10.Syed V, Ulinski G, Mok SC, Ho SM. Reproductive hormone-induced, STAT3-mediated interleukin 6 action in normal and malignant human ovarian surface epithelial cells. J Natl Cancer Inst. 2002;94:617–629. [DOI] [PubMed] [Google Scholar]
- 11.Utsumi T, Leese MP, Chander SK, et al. The effects of 2-methoxyoestrogen sulphamates on the in vitro and in vivo proliferation of breast cancer cells. J Steroid Biochem Mol Biol. 2005;94:219–227. [DOI] [PubMed] [Google Scholar]
- 12.Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, Van Waes C. 2-methoxyestradiol inhibits hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin Cancer Res. 2004;10:8665–8673. [DOI] [PubMed] [Google Scholar]
- 13.Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13:739–749. [DOI] [PubMed] [Google Scholar]
- 14.Merriam GR, MacLusky NJ, Picard MK, Naftolin F. Comparative properties of the catechol estrogens, I: methylation by catechol-O-methyltransferase and binding to cytosol estrogen receptors. Steroids. 1980;36:1–11. [DOI] [PubMed] [Google Scholar]
- 15.Van Aswegen CH, Purdy RH, Wittliff JL. Binding of 2-hydroxyestradiol and 4-hydroxyestradiol to estrogen receptors from human breast cancers. J Steroid Biochem. 1989;32:485–492. [DOI] [PubMed] [Google Scholar]
- 16.Sidor C, D’Amato R, Miller KD. The potential and suitability of 2-methoxyestradiol in cancer therapy. Clin Cancer Res. 2005;11:6094–6095; author reply 6095–6096. [DOI] [PubMed] [Google Scholar]
- 17.Qadan LR, Perez-Stable CM, Anderson C, et al. 2-Methoxyestradiol induces G2/M arrest and apoptosis in prostate cancer. Biochem Biophys Res Commun. 2001;285:1259–1266. [DOI] [PubMed] [Google Scholar]
- 18.Van Veldhuizen PJ, Ray G, Banerjee S, et al. 2-Methoxyestradiol modulates beta-catenin in prostate cancer cells: a possible mediator of 2-methoxyestradiol-induced inhibition of cell growth. Int J Cancer. 2008;122:567–571. [DOI] [PubMed] [Google Scholar]
- 19.Casarez EV, Dunlap-Brown ME, Conaway MR, Amorino GP. Radiosensitization and modulation of p44/42 mitogen-activated protein kinase by 2-Methoxyestradiol in prostate cancer models. Cancer Res. 2007;67:8316–8324. [DOI] [PubMed] [Google Scholar]
- 20.Lin HL, Yang MH, Wu CW, et al. 2-Methoxyestradiol attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated metastasis of gastric cancer. Int J Cancer. 2007;121: 2547–2555. [DOI] [PubMed] [Google Scholar]
- 21.Thews O, Lambert C, Kelleher DK, Biesalski HK, Vaupel P, Frank J. Possible protective effects of alpha-tocopherol on enhanced induction of reactive oxygen species by 2-methoxyestradiol in tumors. Adv Exp Med Biol. 2005;566:349–355. [DOI] [PubMed] [Google Scholar]
- 22.Joubert A, Maritz C, Joubert F. Bax/Bcl-2 expression levels of 2-methoxyestradiol-exposed esophageal cancer cells. Biomed Res. 2005;26:131–134. [DOI] [PubMed] [Google Scholar]
- 23.Zhou NN, Zhu XF, Zhou JM, et al. 2-Methoxyestradiol induces cell cycle arrest and apoptosis of nasopharyngeal carcinoma cells. Acta Pharmacol Sin. 2004;25:1515–1520. [PubMed] [Google Scholar]
- 24.LaVallee TM, Zhan XH, Johnson MS, et al. 2-methoxyestradiol up-regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res. 2003;63:468–475. [PubMed] [Google Scholar]
- 25.Bu S, Blaukat A, Fu X, Heldin NE, Landstrom M. Mechanisms for 2-methoxyestradiol-induced apoptosis of prostate cancer cells. FEBS Lett. 2002;531:141–151. [DOI] [PubMed] [Google Scholar]
- 26.Gibson SB. Epidermal growth factor and trail interactions in epithelial-derived cells. Vitam Horm. 2004;67:207–227. [DOI] [PubMed] [Google Scholar]
- 27.Kurbanov BM, Geilen CC, Fecker LF, Orfanos CE, Eberle J. Efficient TRAIL-R1/DR4-mediated apoptosis in melanoma cells by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Invest Dermatol. 2005;125:1010–1019. [DOI] [PubMed] [Google Scholar]
- 28.Ray S, Bucur O, Almasan A. Sensitization of prostate carcinoma cells to Ap02L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis. 2005;10:1411–1418. [DOI] [PubMed] [Google Scholar]
- 29.Shi J, Zheng D, Liu Y, et al. Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res. 2005;65:1687–1692. [DOI] [PubMed] [Google Scholar]
- 30.Meurette O, Fontaine A, Rebillard A, et al. Cytotoxicity of TRAIL/anticancer drug combinations in human normal cells. Ann N Y Acad Sci. 2006;1090:209–216. [DOI] [PubMed] [Google Scholar]
- 31.Sadarangani A, Kato S, Espinoza N, et al. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis. 2007;12:73–85. [DOI] [PubMed] [Google Scholar]
- 32.Shi J, Zheng D, Man K, Fan ST, Xu R. TRAIL: a potential agent for cancer therapy. Curr Mol Med. 2003;3:727–736. [DOI] [PubMed] [Google Scholar]
- 33.Cuello M, Coats AO, Darko I, et al. N-(4-hydroxyphenyl) retinamide (4HPR) enhances TRAIL-mediated apoptosis through enhancement of a mitochondrial-dependent amplification loop in ovarian cancer cell lines. Cell Death Differ. 2004;11:527–541. [DOI] [PubMed] [Google Scholar]
- 34.Cuello M, Ettenberg SA, Nau MM, Lipkowitz S. Synergistic induction of apoptosis by the combination of trail and chemotherapy in chemoresistant ovarian cancer cells. Gynecol Oncol. 2001;81:380–390. [DOI] [PubMed] [Google Scholar]
- 35.Kato S, Sadarangani A, Lange S, et al. The oestrogen metabolite 2-methoxyoestradiol alone or in combination with tumour necrosis factor-related apoptosis-inducing ligand mediates apoptosis in cancerous but not healthy cells of the human endometrium. Endocr Relat Cancer. 2007;14:351–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane D, Cartier A, L’Esperance S, Cote M, Rancourt C, Piche A. Differential induction of apoptosis by tumor necrosis factor-related apoptosis-inducing ligand in human ovarian carcinoma cells. Gynecol Oncol. 2004;93:594–604. [DOI] [PubMed] [Google Scholar]
- 37.Tomek S, Horak P, Pribill I, et al. Resistance to TRAIL-induced apoptosis in ovarian cancer cell lines is overcome by co-treatment with cytotoxic drugs. Gynecol Oncol. 2004;94:107–114. [DOI] [PubMed] [Google Scholar]
- 38.Catley L, Tai YT, Chauhan D, Anderson KC. Perspectives for combination therapy to overcome drug-resistant multiple myeloma. Drug Resist Updat. 2005;8:205–218. [DOI] [PubMed] [Google Scholar]
- 39.Leverkus M, Neumann M, Mengling T, et al. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 2000;60:553–559. [PubMed] [Google Scholar]
- 40.Vignati S, Codegoni A, Polato F, Broggini M. Trail activity in human ovarian cancer cells: potentiation of the action of cytotoxic drugs. Eur J Cancer. 2002;38:177–183. [DOI] [PubMed] [Google Scholar]
- 41.Cuello M, Ettenberg SA, Clark AS, et al. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001;61:4892–4900. [PubMed] [Google Scholar]
- 42.Beale PJ, Rogers P, Boxall F, Sharp SY, Kelland LR. BCL-2 family protein expression and platinum drug resistance in ovarian carcinoma. Br J Cancer. 2000;82:436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gibson EM, Henson ES, Haney N, Villanueva J, Gibson SB. Epidermal growth factor protects epithelial-derived cells from tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by inhibiting cytochrome c release. Cancer Res. 2002;62:488–496. [PubMed] [Google Scholar]
- 44.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. [DOI] [PubMed] [Google Scholar]
- 45.Tsao SW, Mok SC, Fey EG, et al. Characterization of human ovarian surface epithelial cells immortalized by human papilloma viral oncogenes (HPV-E6E7 ORFs). Exp Cell Res. 1995;218:499–507. [DOI] [PubMed] [Google Scholar]
- 46.Yager JD. Endogenous estrogens as carcinogens through metabolic activation. J Natl Cancer Inst Monogr. 2000;(27):67–73. [DOI] [PubMed] [Google Scholar]
- 47.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu Rev Pharmacol Toxicol. 1996;36:203–232. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe K, Takanashi K, Imaoka S, et al. Comparison of cytochrome P-450 species which catalyze the hydroxylations of the aromatic ring of estradiol and estradiol 17-sulfate. J Steroid Biochem Mol Biol. 1991;38:737–743. [DOI] [PubMed] [Google Scholar]
- 49.Li L, Bu S, Backstrom T, Landstrom M, Ulmsten U, Fu X. Induction of apoptosis and G2/M arrest by 2-methoxyestradiol in human cervical cancer HeLaS3 cells. Anticancer Res. 2004;24:873–880. [PubMed] [Google Scholar]
- 50.Song JJ, An JY, Kwon YT, Lee YJ. Evidence for two modes of development of acquired tumor necrosis factor-related apoptosis-inducing ligand resistance. Involvement of Bcl-xL. J Biol Chem. 2007;282:319–328. [DOI] [PubMed] [Google Scholar]
- 51.Kondo K, Yamasaki S, Inoue N, et al. Prospective antitumor effects of the combination of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and cisplatin against esophageal squamous cell carcinoma. Surg Today. 2006;36:966–974. [DOI] [PubMed] [Google Scholar]
- 52.Singh TR, Shankar S, Chen X, Asim M, Srivastava RK. Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 2003;63:5390–5400. [PubMed] [Google Scholar]
- 53.Basu A, Castle VP, Bouziane M, Bhalla K, Haldar S. Crosstalk between extrinsic and intrinsic cell death pathways in pancreatic cancer: synergistic action of estrogen metabolite and ligands of death receptor family. Cancer Res. 2006;66:4309–4318. [DOI] [PubMed] [Google Scholar]