

Abstract
Benign peripheral nerve tumors and malignant peripheral nerve tumors are rarely found in patients with upper limb tumors. A four-year retrospective study was conducted on patients with tumors in the upper limb area admitted to the Emergency Clinical Hospital, Bucharest, Romania. Seventeen patients were admitted within this time range, 15 of which were benign and two malignant. All patients required surgical intervention after thorough clinical and imaging evaluation. Benign masses were removed, follow-up examination revealing no local recurrent, as well as good function recovery. On the other hand, malignant tumors due to their highly aggressive features, both determined local recurrence, one requiring upper limb amputation, the other presenting metastases.
Keywords: tumor, nerve, upper limb, histopathological findings, surgical treatment
⧉ Introduction
Nerve tumors represent uncommon upper extremity tumors in adults and include benign peripheral nerve sheath tumors (BPNSTs) and malignant peripheral nerve sheath tumors (MPNSTs) [1,2]. Benign nerve tumors present slow growing evolution and are minimally symptomatic, whereas malignant masses should be suspected when a more aggressive growth pattern is encountered [1, 3].
Schwannoma (neurilemmoma) is the most common tumor type of the peripheral nerve, presenting as slow growing, encapsulated lesions, occasionally of considerable size (as presented in Figure 1A,1B,1C) [1, 4]. Schwannomas encountered in the limbs have different clinical presentations, from asymptomatic features to pain of various intensities, or paresthesia resulting from peripheral nerve compression [4].
Figure 1.

(A–C) Large median nerve schwannoma of the proximal forearm (personal archive)
Neurofibromas occur in two disease patterns: sporadic or associated with neurofibromatosis [1, 5]. Malignant transformation is rare in isolated neurofibromas. Literature reports reveal an occurrence of malignancy in up to 13% cases of von Recklinghausen disease [5,6].
Clinical symptoms are usually indistinguishable from schwannomas although surgical techniques differ. Due to their encapsulated structure, schwannomas are more amenable to intraneural dissection, whereas neurofibromas involve a more challenging excision that can cause structural nerve damage and require nerve reconstruction procedures [2, 7]. Figure 2 (A and B) presents an ulnar nerve neurofibroma in the elbow region.
Figure 2.

(A and B) Ulnar nerve neurofibroma at the elbow level in a 54-year-old female patient: clinical and intraoperative aspect (personal archive).
MPNSTs constitute approximately 2% of sarcoma tumors. They arise de novo or from malignant degeneration of a benign tumor, commonly from plexiform neurofibromas. Half of the MPNSTs is associated with neurofibromatosis type 1 (NF1). Early diagnosis and treatment can offer the highest chance of survival, but, if left untreated, the natural history is metastasis and eventual death [8].
Peripheral nerve tumors are evaluated and located anatomically using imaging tests, such as echography, computed tomography, or magnetic resonance imaging (MRI). The latter is the most accurate in establishing tumor relative location to the nerve and renders precise adjacent tissue involvement [1, 9,10,11]. Such examinations do not differentiate peripheral nerve tumors from other types, but may aid in revealing malignant features, such as necrosis, adjacent invasion, and increased dimensions. The definitive diagnosis, regardless of its benign or malignant nature is done by histopathological (HP) examination and immunohistochemistry [1, 9].
Surgical treatment of malignant tumors involves radical resection or limb amputation in severe cases [12]. Radiotherapy (RT) may provide local control and chemotherapy has been proven to be ineffective [13]. Traumatic neuroma may occasionally need to be differentiated from a benign or malignant peripheral nerve tumor and consists of a non-neoplastic proliferation of connective tissue and neural fibers. Although a variety of symptoms occurs, it can cause persistent symptoms and debilitating sequelae [14].
Table 1 reviews peripheral nerve tumors, describing their clinical and HP features [15,16,17,18].
Table 1.
Features of peripheral nerve tumors
|
Tumor |
Cell origin |
Gross pathology |
Microscopic pathology |
IHC marker |
|
Schwannoma |
Schwann |
Well-circumscribed, encapsulated (epineurium), ovoid, firm, light tan-yellow, may have degenerative changes (cystic, hemorrhagic) |
Schwann cells; biphasic architecture (Antoni A/Antoni B); nuclear palisading (Verocay bodies); may have degenerative changes (cystic, hemorrhagic) |
S100 protein +++; SOX10 +++; type IV collagen +; CD34 + (subcapsular areas); H3K27me3 intact; NFP + (entrapped axons); GFAP, CK – (peripheral sites); EMA – (+ in capsule); claudin-1, GLUT1 – |
|
Neurofibroma |
Schwann, fibroblasts, perineurial cells |
Well-circumscribed, unencapsulated, ovoid, grayish-tan, gelatinous to firm |
Loose spindle cell proliferation with haphazard arrangement; small hyperchromatic wavy nuclei; myxoid or collagenous (shredded carrot appearance) matrix; mast cells are common multinodular growth pattern (plexiform type); pseudo-Meissner bodies (diffuse and plexiform type); lack of degenerative changes (cystic, hemorrhagic) |
S100 protein and SOX10 +++ (in Schwann cells); type IV collagen +; EMA, GLUT1 +++ (perineurial cells); CD34 +++ (fibroblasts); NFP + (entrapped axons) |
|
Granular cell tumor |
Schwann |
Uninodular unencapsulated subcutaneous/submucosal firm masses; often with overlying epithelial hyperplasia (verrucous appearance); pale yellow-cream cut surface, with finely granular appearance |
Irregular borders; monotonous epithelioid/polygonal cells organized as nests, trabeculae, sheets; indistinct cell borders, small, round, central nuclei; abundant, finely granular eosinophilic cytoplasm; perineural growth |
S100 protein, SOX10, nestin, inhibin, calretinin +++; CD68, NSE +++; MITF, TFE3 ++; type IV collagen +; SMA, desmin, myogenin, GFAP, HMB45, Melan-A, NFP – |
|
Cellular neurothekeoma |
Unclear |
Ill-defined borders, multinodular |
Spindle and epithelioid cells; micronodular or lobulated architecture; areas of myxoid matrix, not abundant; whorled pattern; multinucleated giant cells (osteoclastic or Touton) may be present; slightly increased mitotic activity |
S100 protein, SOX10 –; CD63, NSE, MITF +; HMB45, desmin, keratin –; SMA, p63 +/– |
|
Nerve sheath myxoma |
Schwann |
Well circumscribed, nodular/multi-nodular; rubbery-firm consistency; white-translucent appearance |
Lobular architecture; dense fibrous septa and abundant myxoid matrix; spindle and epithelioid cells, which form cords, networks, syncytial nests, and ring-like structures |
S100 protein +++; GFAP, CD57 ++/+++; type IV collagen +; SMA, desmin, CD68, synaptophysin, chromogranin A, HMB45 –; CD34 + in rare fibroblasts; NFP + (entrapped axons); EMA – |
|
Perineurioma |
Perineurial |
Well circumscribed, nodular, unencapsulated; firm to rubbery; yellow-tan or white cut surface |
Growth patterns: fascicular, storiform, whorled, lamellar; spindle cells with bipolar cytoplasmic processes; thin, wavy or tapering/round and pale nuclei; collagenous stroma, focally myxoid |
EMA +/+++; claudin-1, GLUT1 +++; CD34 +/–; S100 protein, SOX10, GFAP, SMA, desmin – |
|
MPNST |
Schwann |
Usually deep, large (>5 cm), fusiform tumors, tan-white, gelatinous/fleshy cut surface; areas of necrosis and hemorrhage |
Monomorphic to highly pleomorphic spindle cells, high and conspicuous mitotic activity, geographic necrosis; growth pattern: fascicular, hemangiopericytoma-like, hypercellular and hypocellular areas; myxoid to collagenous stroma; may present heterologous differentiation and/or preexisting benign nerve sheath tumor |
S100 – or focally +; SOX10 + patchy/–; type IV collagen +/++; GFAP –/+; EMA – (except MPNST with perineurial differentiation); CD34 +/++; NFP +/+++; loss of nuclear H3K27me3 expression; HMB45, Melan-A –; markers for heterologous elements |
CD: Cluster of differentiation; CK: Cytokeratin; EMA: Epithelial membrane antigen; GFAP: Glial fibrillary acidic protein; GLUT1: Glucose transporter 1; H3K27me3: Trimethylation of histone H3 on lysine 27; HMB45: Human melanoma black 45; MITF: Microphthalmia-associated transcription factor; MPNST: Malignant peripheral nerve sheath tumor; NFP: Neurofilament protein; NSE: Neuron-specific enolase; SMA: Smooth muscle actin; SOX10: SRY-box transcription factor 10; TFE3: Transcription factor E3
⧉ Patients, Materials and Methods
This paper represents a retrospective cross-sectional study of peripheral nerve tumors in the upper limbs. The patients were admitted to Emergency Clinical Hospital, Bucharest, Romania, between 2016 and 2019. Inclusion criteria were upper limb nerve tumors. Exclusion criteria were non peripheral nerve originating upper limb tumors. All the patients gave formal consent. Data were gathered from the patients’ admission charts, Hospital physical and digital archives, as well as from the Department of Pathology archives. In addition, the results were compared to present literature, as drawn from PubMed® database.
⧉ Results
During the study period, 17 patients with upper limb nerve tumors were treated in the Clinic of Plastic Surgery, Emergency Clinical Hospital, Bucharest, of which 15 were benign mases and two were malignancies. Table 2 presents the characteristics of this group of patients and Figure 3 illustrate the topography of upper limb peripheral nerve tumors.
Table 2.
Upper limb tumor patients
|
Patient No. |
Gender |
Age [years] |
Location |
Nerve involved |
Type |
|
1. |
F |
69 |
Second digit, distal phalanx (L) |
Dorsal digital nerve |
Neuroma |
|
2. |
M |
34 |
Medio-palmar (R) |
Median nerve |
Schwannoma |
|
3. |
M |
48 |
Distal forearm (R) |
Ulnar nerve |
Schwannoma |
|
4. |
M |
43 |
Second digit, distal phalanx (L) |
Radial collateral digital nerve branch |
Schwannoma |
|
5. |
M |
62 |
Third digit, proximal phalanx (L) |
Radial collateral digital nerve |
Schwannoma |
|
6. |
M |
37 |
Hypothenar eminence (R) |
Cutaneous branch |
Schwannoma |
|
7. |
M |
38 |
Distal forearm (L) |
Median nerve |
Schwannoma |
|
8. |
M |
36 |
Proximal forearm (L) |
Median nerve |
Schwannoma |
|
9. |
F |
62 |
Fifth digit, distal phalanx (R) |
Radial collateral digital nerve |
Neuroma |
|
10. |
M |
52 |
Distal dorsal forearm (R) |
Cutaneous branch |
Neurofibroma |
|
11. |
F |
56 |
Distal dorsal forearm (R) |
Superficial radial nerve |
Neurofibroma |
|
12. |
M |
57 |
Third digit, distal phalanx (R) |
Dorsal digital nerve |
Neurofibroma |
|
13. |
M |
58 |
Second digit, middle phalanx (R) |
Collateral digital nerve |
Neurofibroma |
|
14. |
F |
68 |
Fifth finger, middle phalanx (L) |
Collateral radial digital nerve |
Neurofibroma |
|
15. |
F |
67 |
Medio-palmar (L) |
Median nerve |
Neurofibroma |
|
16. |
M |
65 |
Arm (L) |
Median nerve |
MPNST |
|
17. |
M |
43 |
Arm (R) |
Musculocutaneous nerve |
MPNST |
L: Left; F: Female; M: Male; MPNST: Malignant peripheral nerve sheath tumor; R: Right
Figure 3.
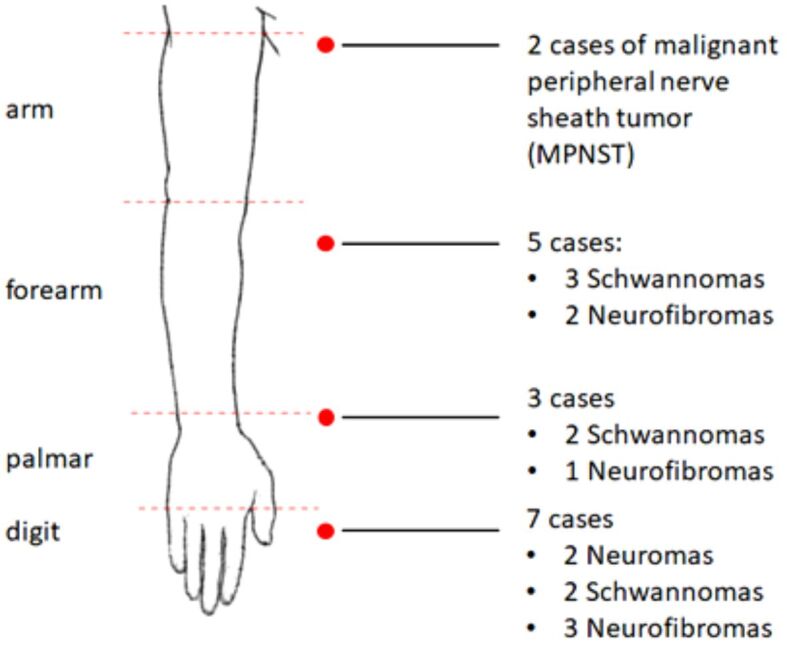
Topography of upper limb peripheral nerve tumors
Our most frequent findings were schwannomas, which occurred only in male patients, with an age range between 34 and 62 years, two cases occurring in the digits, two in the palmar area and three in the forearm. Secondly, neurofibromas occurred equally in females and males, with an age range between 52 and 68 years, three cases affecting the digits, one the palmar area and two the forearm. Figures 4,5,6,7,8,9,10 show the intraoperative and HP features found in patients with schwannomas in our group.
Figure 4.

(A and B) Intraoperative aspect of median nerve schwannoma of the forearm
Figure 5.
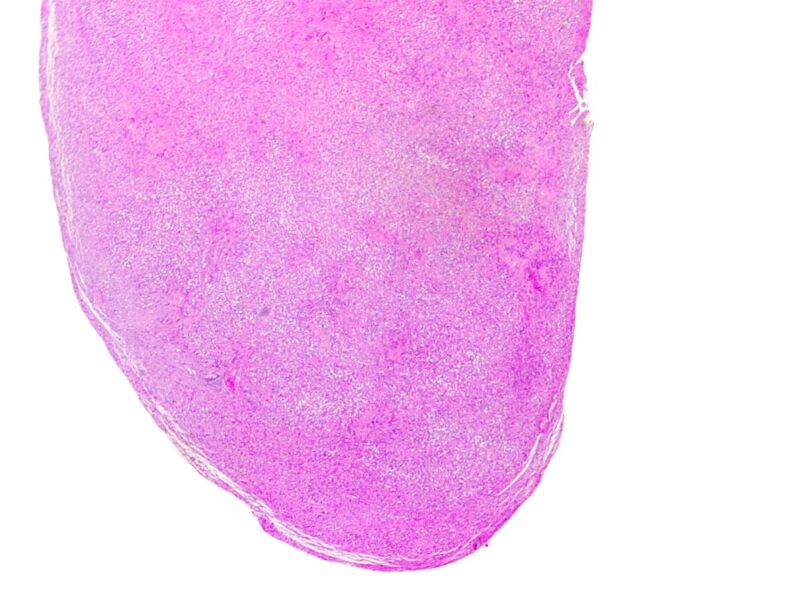
Schwannoma. Encapsulated proliferation, showing admixed hypercellular Antoni A areas with focal nuclear palisading, and loose, less cellular Antoni B areas (HE staining, ×25)
Figure 6.
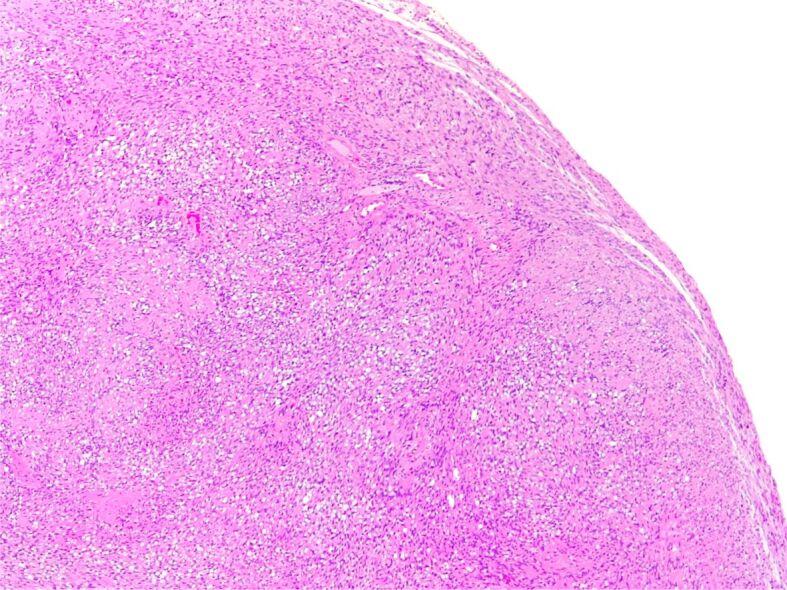
Schwannoma. Hypercellular Antoni A areas, with focal nuclear palisading (HE staining, ×50)
Figure 7.
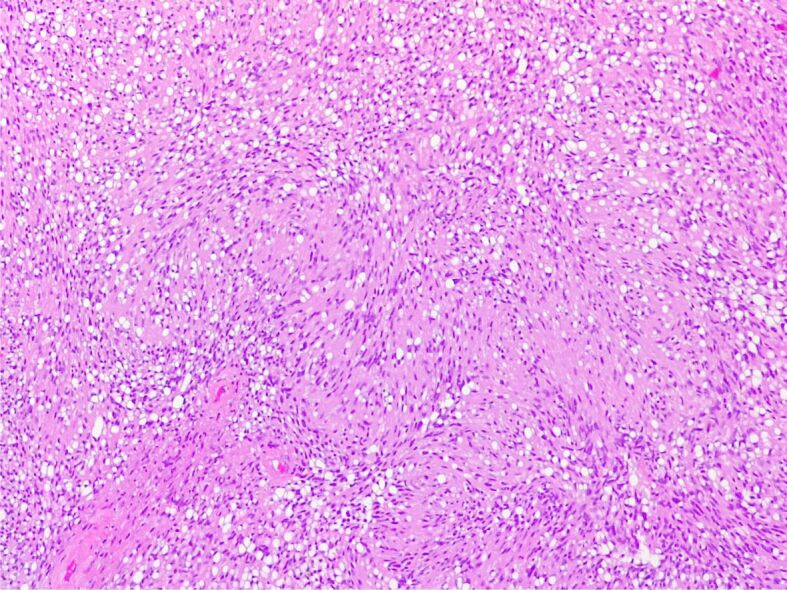
Schwannoma. Hypercellular Antoni A areas with nuclear palisading and forming Verocay bodies (HE staining, ×100)
Figure 8.
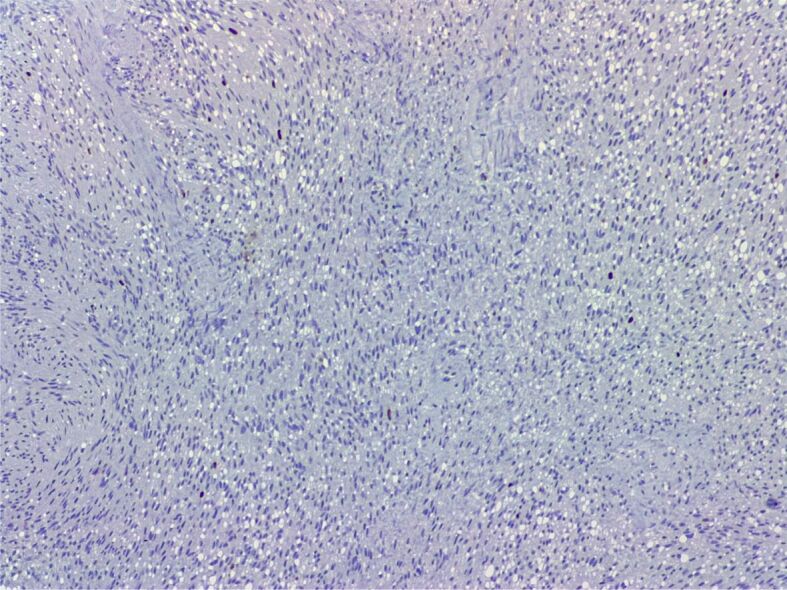
Schwannoma. Spindle cell tumor with low Ki67 proliferation index (Anti-Ki67 antibody immunomarking, ×100)
Figure 9.
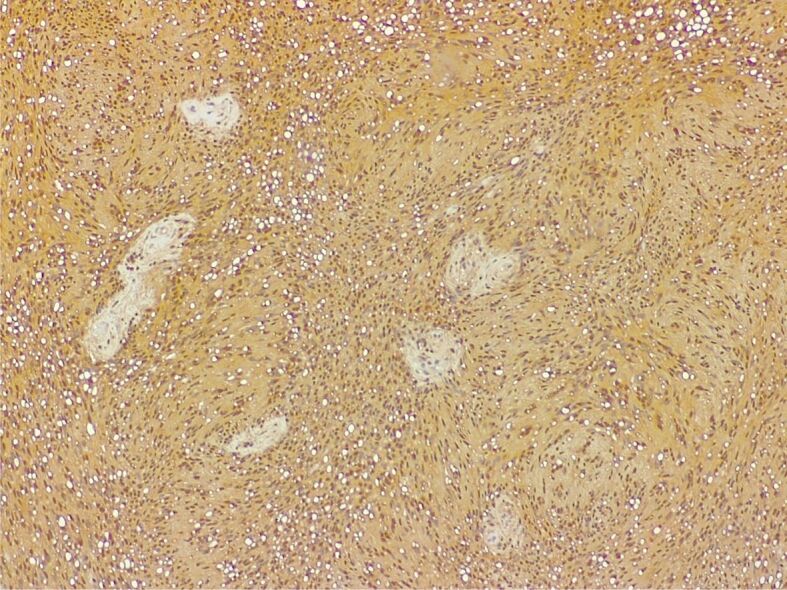
Schwannoma. S100 protein immunostaining showing diffuse and intense positivity of the proliferation, both nuclear and cytoplasmic (Anti-S100 antibody immunomarking, ×100)
Figure 10.
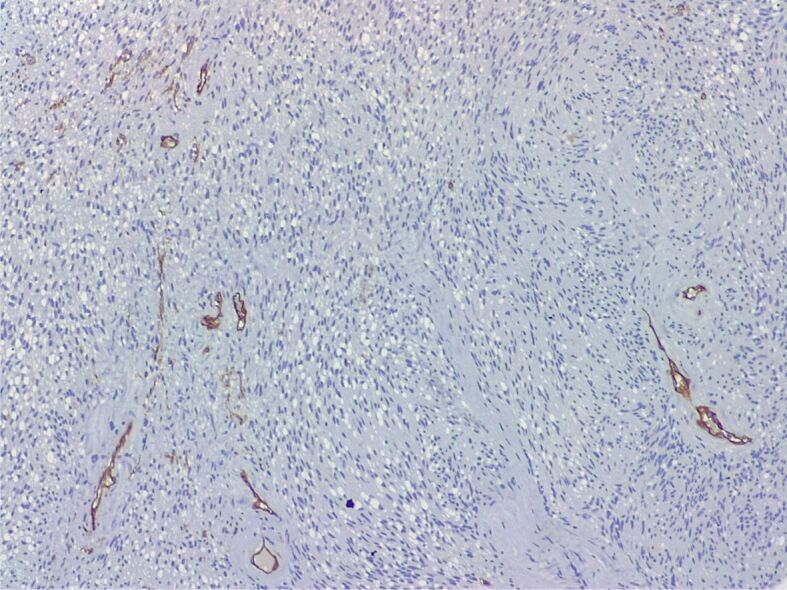
Schwannoma. CD34 immunostaining, negative in the spindle cell proliferation, with positive internal control in small vascular structures (Anti-CD34 antibody immunomarking, ×100)
Two cases of neuromas were found, both occurring in the digits in a 62-year-old female patient and, respectively in a 69-year-old female patient. All 15 cases of peripheral benign upper limb nerve tumors were clinically evaluated and performed imaging examinations. All tumors of the aforementioned cases were resected, with no recurrence and good functional recovery during follow-up examinations.
Two male patients with MPNSTs were also included in the study. A 65-year-old male patient known with cardiovascular history, presented initially with a 6-month-old enlarged mass in the left arm region, associated with intense local pain. After imaging evaluation, the patient underwent surgical intervention, with intraoperative findings of a large tumor adherent to the median nerve. Complete resection of the tumor was performed. Definitive HP examination revealed MPNST (Figures 11 and 12). Microscopic examination revealed sarcomatous tumor proliferation, with fusiform cells arranged in overlapped, short fascicles. Considerate cytological and nuclear pleomorphism were noted. Mitotic activity was enhanced with frequent atypical mitoses. Cells with epithelioid aspect were noted, with ample eosinophilic cytoplasm, as well as giant, odd multi-nucleated cells. Hyalinization areas were noted, which include tumoral cells. Immunohistochemical (IHC) evaluation confirmed the diagnosis: S100, cluster of differentiation 34 (CD34) positive within tumoral cells; negative desmin and smooth muscle actin (SMA) within tumoral cells, but positive within vascular walls; negative SRY-box transcription factor 10 (SOX10), p16, glial fibrillary acidic protein (GFAP) within tumoral cells, 30% Ki67 index. Early recurrence was observed one month after surgery. Postoperative therapy with three cycles of Epirubicin was ineffective, with rapid tumor growth. After a thorough clinical and imagistic evaluation for secondary determinations, no distant lesions were found. Upper limb necessity amputation was decided at the level of the upper third of the left arm.
Figure 11.
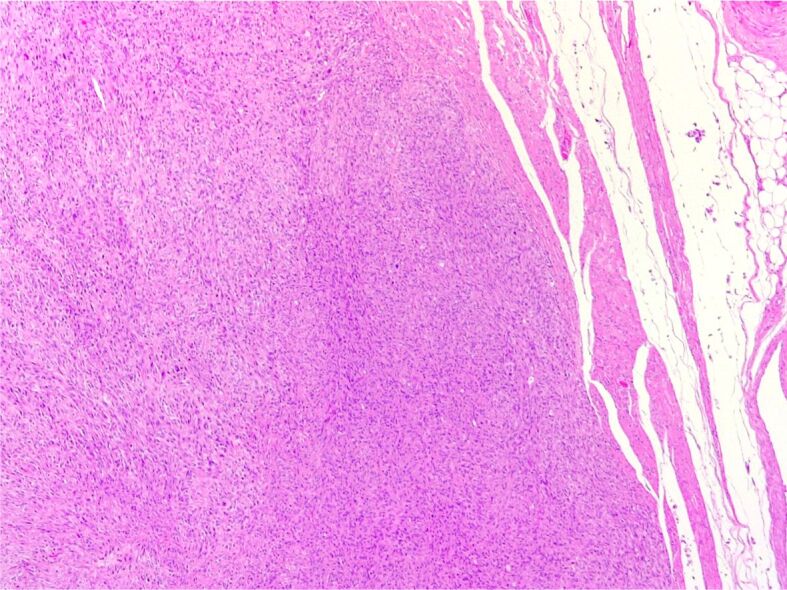
MPNST. Unencapsulated, infiltrating tumor, composed of variably pleomorphic spindle cells, with a vaguely fascicular and whorled growth pattern (HE staining, ×50). HE: Hematoxylin–Eosin; MPNST: Malignant peripheral nerve sheath tumor
Figure 12.
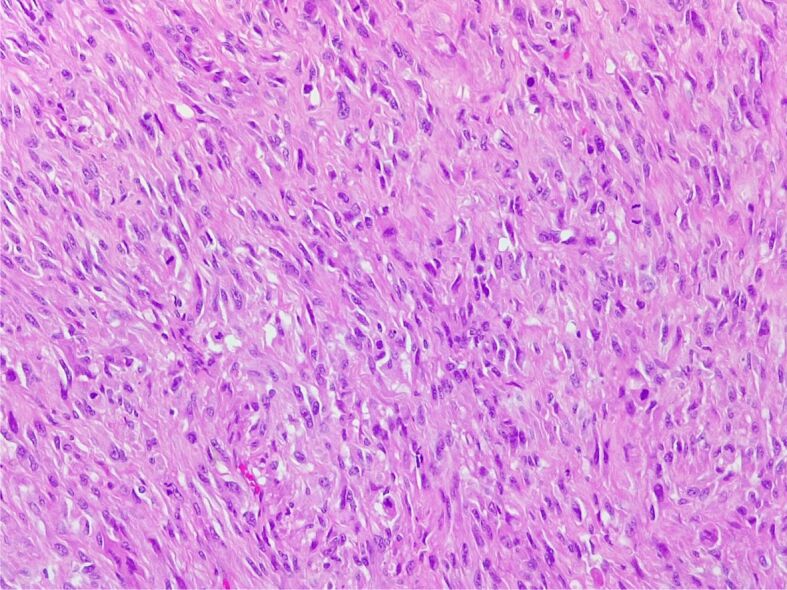
MPNST. Hypercellular area composed of moderately pleomorphic spindle cells, with collagenous stroma, with high mitotic count (HE staining, ×200)
The second case was of a 43-year-old male that presented a soft tissue tumor in the proximal aspect of the arm. It should be noted that, five years anterior to this tumor, patient had a surgical intervention for a nodular malignant melanoma localized on the right anterior hemithorax, Breslow 2.13 mm, Clark IV, requiring chemotherapy and RT after excision, as well as axillary lymphadenectomy. Surgical intervention was performed, with tumor excision, HP findings being of MPNST. Figure 13A,13B,13C presents imagistic and intraoperative findings in this patient. In evolution, the patient developed two local recurrences and distal pulmonary and digestive metastases. He required sustained oncological treatment and multiple surgical interventions addressing excision of each localized tumoral mass.
Figure 13.
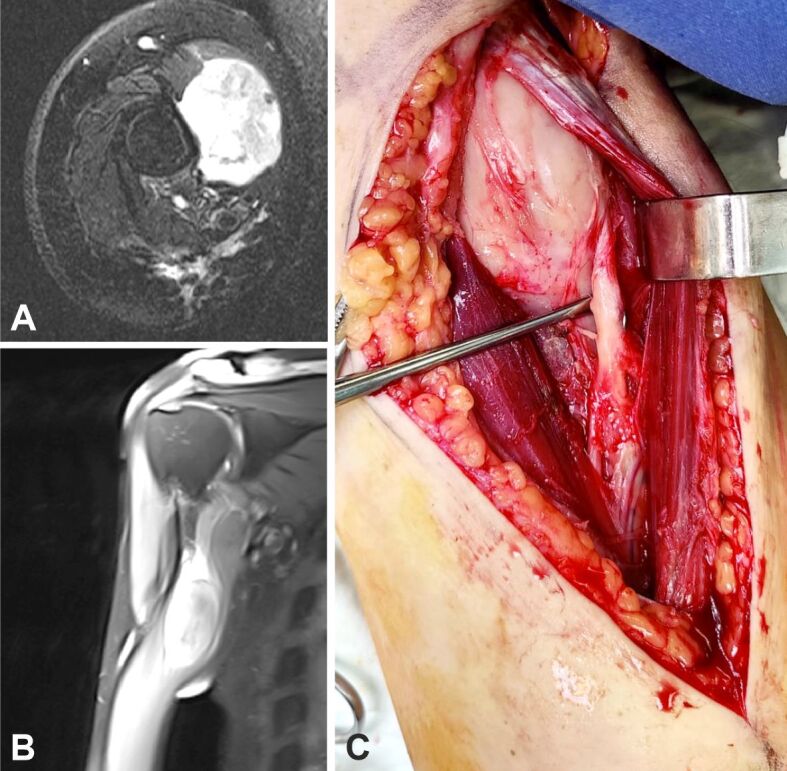
(A–C) Imagistic and intraoperative findings in a patient with MPNST of the right arm. MPNST: Malignant peripheral nerve sheath tumor
⧉ Discussions
Soft tissue limb tumors are frequently encountered, currently, due to the technological advancements, ease of access to efficient imaging methods, therefore a faster diagnosis and incipient therapeutic approach is possible for this pathology. Limb tumors present a widespread histological variability, which makes the anatomopathological diagnosis an essential asset in the management of such cases [19].
Benign tumors are a frequent finding at the level of the limbs, with good prognosis, but are accompanied by challenges such as: nervous compressions, invasive tumors, erroneous diagnosis, malignant transformation and lastly loss of function. Patients’ adherence to rehabilitation protocols and periodic follow-ups should not be neglected [7, 20].
In most situations, limb malignant tumors require mutilating surgical interventions, with secondary severe functional deficits, having psychic and socio-professional repercussions. Whenever possible, limb salvage is desired, but the amputation represents a valid approach, being the only therapeutic option in cases where reconstructive procedures are not indicated, the vital prognosis having the utmost importance [21,22]. For the lower limb, in case of amputations, prostheses represent a good result with rapid social and professional reintegration.
A particular type of soft tissues upper limb tumors are represented by nerve tumors, posing diagnostic and therapeutic challenges.
Peripheral nerve sheath tumors represent a broad spectrum of tumors with neuroectodermal origin derived from the neural sheath, or from non-neural sheath origin. More than 90–95% are benign and include schwannoma, perineurioma or neurofibroma as the most common subtypes. Examples of hybrid BPNSTs have been also described, such as schwannoma/neurofibroma or neurofibroma/perineurioma [23,24]. Surgical management is not required in all patients with BPNSTs. The size of a tumor is not usually used as a criterion for surgical intervention. We must acknowledge that conservative approach carries the risk of neurological deterioration as well as the likelihood of malignant change in patients with neurofibromas. Pain, paresthesia, and neurological impairments are all potentially debilitating conditions that warrant surgical intervention. BPNSTs can be removed in a subtotal manner (50% or more) or gross total excision that may or may not leave behind the tumor capsule attached to the nerve fascicles. The method to be used is determined by the macroscopic nature of the tumor intraoperatively [25,26].
In our study, the 15 cases of benign tumoral masses elicited various symptoms, such as pain, paresthesia, and moderate motor deficits. There were 13 benign tumors and two neuromas among these cases. All the aforementioned benign tumors were totally resected, preserving the affected nerve, which ensured an optimal functional result in all cases, with remittance of presurgical symptomatology.
Neuromas impose clinical differential diagnosis with benign tumors, usually neurofibromas [18]. Usually, a traumatic episode followed by nerve contusion or transection is recalled by the patient.
Neurofibromas are benign tumors, with very rare local recurrence after entire resection of the tumor. Malignant transformation risk is very small. Neurofibromas are determined by a mutation in the NF1 gene and there are three types of tumors: localized, diffuse, and plexiform. The localized form is the most prevalent. Even though most neurofibromas have a very low risk of transforming into a malignant tumor, the plexiform neurofibroma is pathognomonic for the NF1 and caries a higher risk of malignancy transformation [27,28].
When neurofibroma is suspected, schwannoma should be ruled out first, due to its higher incidence. Both lesions appear on MRI as well-defined fusiform masses with a diameter of less than 5 cm [29]. Neurofibromas, on the other hand, can be recognized from schwannomas by the location of the tumors. Neurofibromas are concentric, but schwannomas are eccentric in relation to the nerve. Pathological examination is the “gold standard” for diagnosing neurofibromas in general. Elongated Schwann cells, fibroblasts, and perineurial-like cells are intermixed with undulating bundles of nerve fibers, collagen, and mucus on a histological section. To achieve a definitive diagnosis, IHC stainings are also used. S100 positivity is less evident in neurofibromas, except myxoid-types, than in schwannomas. The neurofilament can be highlighted by immunostaining the perineurium around the tumor with epithelial membrane antigen (EMA) [1, 18, 30]. Isolated neurofibromas are treated with excision when symptoms justify it.
However, Healey & McCormack indicate that when the diagnosis is clear and nerve fiber excision is required, a small amount of tumor may be left behind due to the low risk of malignant transformation and recurrence. The repercussions of a neurofibroma’s malignant degeneration must be weighed against the risk of nerve injury after removal [29, 31].
Our study contained six neurofibromas, all represented by localized subtypes. The cases in this study presented a distal topography, two being in the distal dorsal forearm, one in the palmar area, and three affecting the digits.
Due to the localized subtype and distal topography, complete resection of the neurofibromas was possible without any significant functional impact. Particularly, one of the neurofibromas was described as myxoid-type, presenting an uncommon feature, S100-positive immunomarker, a more frequent finding in schwannoma.
Schwannomas are solitary, slow-growing, encapsulated neoplasms that are entirely composed of Schwann cells from myelin sheaths. The painless lump will persist for a long time, with insidious onset of symptoms. Clinical differential diagnosis includes neurofibroma or ganglion cyst. Pain, paresthesia, and other symptoms may occur when the tumor becomes larger and gradually compresses the affected nerve [32].
MRI and ultrasonography can be used in the clinic to diagnose and localize these tumors. Schwannomas have low signal intensity on T1-weighted (T1W) MRI and high signal intensity on T2-weighted (T2W) MRI. Schwannomas can be distinguished from neurofibromas by their concomitant enhancement and eccentric location of the nerve. Biphasic structures, such as Antoni A (dense) and Antoni B (loose), as well as nuclear palisading and high S100 protein positivity, are hallmark pathological features of schwannoma [32,33,34].
In our patients, the histological diagnosis revealed classical aforementioned patterns, the immunohistochemistry tests further characterizing the cytological aspects of the tumor.
Takase et al. believe that one of the main reasons of postoperative neurological impairments is damage to the fascicles around the tumor during extracapsular excision. To reduce the chances of nerve damage, intracapsular enucleation was recommended [35,36,37,38]. It is widely assumed that this type of tumor can be easily removed from the nerve trunk without causing further neurological damage. Date et al. used the intracapsular enucleation method. The results of extracapsular and intracapsular enucleation of schwannoma were compared, and the latter procedure was determined to be preferable due to a lower risk of complications. When adhesions between the epineurium and the capsule were present, Date et al. performed microenucleation of the tumor (“tumor was excised piece-by-piece”) [39,40].
Section of fascicles entering the tumor, according to Park et al., may result in postoperative neurological impairments [41]. On this subject, Donner et al. held a different viewpoint. Based on intraoperative stimulation, the authors found that fascicles entering the tumor are frequently nonfunctional and that their transection does not result in additional neurological impairments [42]. The incidence of neurological impairments after schwannoma excision ranges from 1.5% to 80% [32, 35, 41,42]. Short-term observations account for a disproportionately high percentage of complications. In the early postoperative phase, Adani et al. reported aggravation of paresthesias in 23 of 24 patients [32]. Within 12 months of the procedure, they spontaneously resolved in all the patients. Siqueira et al. found a 15.2% postoperative complication rate [38]. Numerous studies have found that postoperative complications are more common in patients with larger tumors, tumors with a proximal localization, and tumors arising from the ulnar nerve [35, 37, 39].
Our operative technique in peripheral nerve tumoral cases consists of microsurgical dissection of nerve under optic magnification, aided by loupes or surgical microscope, using microsurgical instruments. In our patient group with schwannomas, extracapsular resection was performed. However, in two cases, an interfascicular microdissection was required to separate tumoral mass from the main fascicles of the nerves: one with median nerve schwannoma in the proximal forearm at the level of the anterior interosseous nerve and one with a schwannoma affecting the median nerve fascicles within the distal third of the forearm. Electric selective stimulation of nerve fascicles was useful, guiding intraoperative dissection, rendering salvage of a maximum number of fascicles. A transitory flexor pollicis longus motor deficit was observed in the patient with a schwannoma affecting the median nerve in its proximal forearm, with function recovery three months postoperatively.
MPNST is a rare tumor, with a 0.001% incidence rate, that exhibits extraordinary developmental plasticity and can arise after irradiation [24]. MPNST has three documented etiologies: tumors can arise in combination with NF1, because of prior RT (RT-induced) or occur sporadically. The autosomal dominant disorder NF1, which is the most common human cancer genetic predisposition syndrome, is linked to half of MPNSTs. MPNST arising in the setting of NF1 presented worse outcomes compared to sporadic disease, with inferior responses to cytotoxic chemotherapy [8, 43]. RT-induced soft tissue sarcomas are extremely rare, with only around 5% of them being MPNST. The risk of MPNST after RT is difficult to assess, however it has been recorded as 0.06%, most typically in the context of breast cancer or lymphoma [44].
Our 43-year-old patient with MPNST required RT for a melanoma five years prior to presentation, which was localized on right hemithorax, in the vicinity of current tumor. In this young patient, aggressive tumoral evolution was noted, with rapid metastasis and sequential local recurrences.
Most MPNSTs show as potentially painful, rapidly expanding lumps close to major nerves, with the tumors spreading along the distal and proximal perineural and epineural tissue. Fluorodeoxyglucose–positron emission tomography (FDG–PET) has been proposed as the key clinical task of differentiating benign neurofibromas from MPNST in patients with NF1 [45].
MRI helped in tumor identification and localization in our patients with good identification of tumoral extension in affected compartments, allowing accurate description of relationship to the large structures, vessels, and the affected nerve, ensuring an adequate preoperative planning, diminishing therefore intraoperative incidents. Both patients with MPNST presented involvement of a major nerve, the musculocutaneous nerve and median nerve in the arm region. Tumor dissection was performed under loupes magnification due to the intimate relationship to major vasculo-nervous structures. Surgical interventions were thoroughly planned due to the general altered status of oncological patients, having a higher anesthetic risk and increased incidence of intra- and postoperative complications.
It might be difficult to identify MPNST from other sarcomas due to the absence of morphological criteria or IHC/molecular assays. The diagnosis is obvious when the gross specimen emerges from a nerve. If this is not the case, a range of IHC stainings may be needed to differentiate between muscle, vascular, and other non-neural tissue sarcomas. MPNSTs are staged and treated like malignant soft tissue sarcomas in general. Surgical resection with a broad margin alone is frequently insufficient for primary cancers of the limbs, whereas amputation of the extremity proximal to the tumor may be required [46]. In certain circumstances, neoadjuvant RT can save a limb, and postoperative adjuvant RT can reduce the incidence of local recurrence in individuals who are unable to achieve broad excision margins with surgery alone. The following factors have a negative prognostic: proximal primary lesion, large dimensions of tumor and neurofibromatosis. Despite current multimodality therapy, the 5-year survival ranges from 35% to 50% [47,48].
The anatomical location of these tumors has a substantial influence on resectability and prognosis. Resectability and outcomes are worsened when proximal structures such as the roots or plexus are involved rather than distant structures such as nerves or terminal branches. A broad local resection is performed for MPNSTs of the distal limb, depending on the amount of metastatic disease. This technique involves removing the nerve, any nearby soft tissues (adherent or not), and a several-centimeter margin of the entering and exiting nerve, all of which were found to be tumor-free on frozen and permanent tissue sections. When it comes to sarcomas of the extremities, limb-sparing surgery has become more popular in recent years, but amputation or forequarter amputation is still performed on imposed indication [49].
MPNST is a very aggressive subtype of sarcoma, as confirmed in both cases belonging to this study. The 65-year-old patient present a rapid evolution both preoperatively until tumor resection, as well as postoperative with early recurrence and chemotherapy resistant features. The most favorable therapeutic approach that controlled the tumoral process and improved vital prognosis was necessity amputation, the patient presenting no secondary distant metastases.
The most important objectives in limb salvage surgery are complete excision of the tumor, uphold hand function without any risk of local recurrence and allow for a good survival prognostic. Extremity salvage surgery should allow for a similar survival rate as the amputation option [21, 44]. Combining the surgical and oncological standards and adhering to a systematic therapeutical plan for the limb salvage surgery, the plastic surgeon can strive to achieve full patient satisfaction, limb function and survival. Functional reconstruction in sarcomas of the extremities is a relatively novel approach, with limited experience in the literature. Advanced microsurgical procedures, such as nerve transfer and free functional muscle transfers, allow reconstructive surgeons to save limbs while also restoring function following the loss of important motor and sensory structures during soft tissue tumor excision in the extremities [50,51].
Careful follow-up is mandatory for patients with upper limb nerve tumors. Adequate functional recovery is ensured through sustained rehabilitation program. Advanced multi-modal oncological treatment is essential for patients with peripheral nerve malignancies and should be conducted according to a detailed evaluation of clinicopathological tumor features and also specific molecular markers. Accurate staging systems of these highly aggressive and metastasizing tumors are mandatory in prognostic assessment and therapeutical management [52].
⧉ Conclusions
The upper limb presents a wide variety of tumors, with rare findings of nerve sheath tumors. Benign tumors are a more frequent finding than malignant tumors, the latter being highly aggressive, life-threatening, with destructive functional consequences. Tumor relation to the nerve, such as invasion, compression, and peripheral adjacent tissues infiltration may determine clinical signs and symptoms. Imaging techniques are highly valuable, aiding in presurgical evaluation and adequate surgical planning therefore avoiding needless damage to important nerve fascicles and helping in acquire optimal nerve recovery. HP evaluation represents the definitive diagnosis. The most suitable surgical intervention for BPNSTs is represented by complete removal, with maximum preservation of residual neurological function, whereas BPNSTs require a wide margin resection, or even amputation to obtain the most favorable prognosis.
Conflict of interest
The authors declare that they have no conflict of interests.
References
- 1.Zhou HY, Jiang S, Ma FX, Lu H. Peripheral nerve tumors of the hand: clinical features, diagnosis, and treatment. World J Clin Cases. 2020;8(21):5086–5098. doi: 10.12998/wjcc.v8.i21.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strike SA, Puhaindran ME. Nerve tumors of the upper extremity. Clin Plast Surg. 2019;46(3):347–350. doi: 10.1016/j.cps.2019.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Hsu CS, Hentz VR, Yao J. Tumours of the hand. Lancet Oncol. 2007;8(2):157–166. doi: 10.1016/S1470-2045(07)70035-9. [DOI] [PubMed] [Google Scholar]
- 4.Sheikh MM, De Jesus O. In: StatPearls, editor. Treasure Island FL USA: StatPearls Publishing; Schwannoma Updated 2021 Aug 30.https://www.ncbi.nlm.nih.gov/books/NBK562312/ [Google Scholar]
- 5.Antônio JR, Goloni-Bertollo EM, Trídico LA. Neurofibromatosis: chronological history and current issues. An Bras Dermatol. 2013;88(3):329–343. doi: 10.1590/abd1806-4841.20132125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tucker T, Wolkenstein P, Revuz J, Zeller J, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65(2):205–211. doi: 10.1212/01.wnl.0000168830.79997.13. [DOI] [PubMed] [Google Scholar]
- 7.Payne WT, Merrell G. Benign bony and soft tissue tumors of the hand. J Hand Surg Am. 2010;35(11):1901–1910. doi: 10.1016/j.jhsa.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, Maki RG. Malignant peripheral nerve sheath tumors. Oncologist. 2014;19(2):193–201. doi: 10.1634/theoncologist.2013-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilavaki M, Chourmouzi D, Kiziridou A, Skordalaki A, Zarampoukas T, Drevelengas A. Imaging of peripheral nerve sheath tumors with pathologic correlation: pictorial review. Eur J Radiol. 2004;52(3):229–239. doi: 10.1016/j.ejrad.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Sperandio M, Di Poce I, Ricci A, Di Trapano R, Costanzo E, Di Cello P, Pelle F, Izzo L, Simonetti G. Malignant peripheral nerve sheath tumour: CT and MRI findings. Case Rep Radiol. 2013;2013:517879–517879. doi: 10.1155/2013/517879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilsson J, Sandberg K, Søe Nielsen N, Dahlin LB. Magnetic resonance imaging of peripheral nerve tumours in the upper extremity. Scand J Plast Reconstr Surg Hand Surg. 2009;43(3):153–159. doi: 10.1080/02844310902734572. [DOI] [PubMed] [Google Scholar]
- 12.Kim DH, Murovic JA, Tiel RL, Moes G, Kline DG. A series of 397 peripheral neural sheath tumors: 30-year experience at Louisiana State University Health Sciences Center. J Neurosurg. 2005;102(2):246–255. doi: 10.3171/jns.2005.102.2.0246. [DOI] [PubMed] [Google Scholar]
- 13.Forthman CL, Blazar PE. Nerve tumors of the hand and upper extremity. Hand Clin. 2004;20(3):233–242 v. doi: 10.1016/j.hcl.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Watson J, Gonzalez M, Romero A, Kerns J. Neuromas of the hand and upper extremity. J Hand Surg Am. 2010;35(3):499–510. doi: 10.1016/j.jhsa.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 15.Choi JH, Ro JY. The 2020 WHO Classification of tumors of soft tissue: selected changes and new entities. Adv Anat Pathol. 2021;28(1):44–58. doi: 10.1097/PAP.0000000000000284. [DOI] [PubMed] [Google Scholar]
- 16.Fisher C. Immunohistochemistry in diagnosis of soft tissue tumours. Histopathology. 2011;58(7):1001–1012. doi: 10.1111/j.1365-2559.2010.03707.x. [DOI] [PubMed] [Google Scholar]
- 17.Lindberg MR. Diagnostic pathology: soft tissue tumors. 3. Elsevier; 2019. pp. 506–571.https://www.elsevier.com/books/diagnostic-pathology-soft-tissue-tumors/lindberg/978-0-323-66110-2 [Google Scholar]
- 18.Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123(3):295–319. doi: 10.1007/s00401-012-0954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sluijmer HCE, Becker SJE, Ring DC. Benign upper extremity tumors: factors associated with operative treatment. Hand (N Y) 2013;8(3):274–281. doi: 10.1007/s11552-013-9518-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plate AM, Lee SJ, Steiner G, Posner MA. Tumorlike lesions and benign tumors of the hand and wrist. J Am Acad Orthop Surg. 2003;11(2):129–141. doi: 10.5435/00124635-200303000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Jubbal KT, D’Souza G, Abrams RA, Kulidjian AA. Management of soft tissue tumors of the upper extremity: a review. SICOT J. 2017;3:47–47. doi: 10.1051/sicotj/2017001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duran-Moreno J, Kontogeorgakos V, Koumarianou A. Soft tissue sarcomas of the upper extremities: maximizing treatment opportunities and outcomes. Oncol Lett. 2019;18(3):2179–2191. doi: 10.3892/ol.2019.10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shanouda S, Kaya G. Benign cutaneous peripheral nerve sheath tumor with hybrid features: report of two cases with schwannoma/perineurioma and schwannoma/neurofibroma components. Dermatopathology (Basel) 2017;4(1-4):1–6. doi: 10.1159/000478854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chikkannaiah P, Boovalli MM, Nathiyal V, Venkataramappa S. Morphological spectrum of peripheral nerve sheath tumors: an insight into World Health Organization 2013 Classification. J Neurosci Rural Pract. 2016;7(3):346–354. doi: 10.4103/0976-3147.182768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wasa J, Nishida Y, Tsukushi S, Shido Y, Sugiura H, Nakashima H, Ishiguro N. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010;194(6):1568–1574. doi: 10.2214/AJR.09.2724. [DOI] [PubMed] [Google Scholar]
- 26.Guha D, Davidson B, Nadi M, Alotaibi NM, Fehlings MG, Gentili F, Valiante TA, Tator CH, Tymianski M, Guha A, Zadeh G. Management of peripheral nerve sheath tumors: 17 years of experience at Toronto Western Hospital. J Neurosurg. 2018;128(4):1226–1234. doi: 10.3171/2017.1.JNS162292. [DOI] [PubMed] [Google Scholar]
- 27.Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC, Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci U S A. 2009;106(46):19479–19484. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spurlock G, Knight SJL, Thomas N, Kiehl TR, Guha A, Upadhyaya M. Molecular evolution of a neurofibroma to malignant peripheral nerve sheath tumor (MPNST) in an NF1 patient: correlation between histopathological, clinical and molecular findings. J Cancer Res Clin Oncol. 2010;136(12):1869–1880. doi: 10.1007/s00432-010-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Healey JH, McCormack RR. In: Tumors of the hand and upper limb. Bogumill GP, Fleegler EJ, editors. New York NY USA: Churchill Livingstone; 1993. Nerve tumors; pp. 205–222.https://books.google.ro/books/about/Tumors_of_the_Hand_and_Upper_Limb.html?id=zKZrAAAAMAAJ&redir_esc=y [Google Scholar]
- 30.Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88(3):443–456. doi: 10.1093/neuros/nyab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skovronsky DM, Oberholtzer JC. Pathologic classification of peripheral nerve tumors. Neurosurg Clin N Am. 2004;15(2):157–166. doi: 10.1016/j.nec.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Adani R, Baccarani A, Guidi E, Tarallo L. Schwannomas of the upper extremity: diagnosis and treatment. Chir Organi Mov. 2008;92(2):85–88. doi: 10.1007/s12306-008-0049-0. [DOI] [PubMed] [Google Scholar]
- 33.Lai CS, Chen IC, Lan HC, Lu CT, Yen JH, Song DY, Tang YW. Management of extremity neurilemmomas: clinical series and literature review. Ann Plast Surg. 2013;71(Suppl 1):S37–S42. doi: 10.1097/SAP.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 34.Wippold FJ, Lubner M, Perrin RJ, Lämmle M, Perry A. Neuropathology for the neuroradiologist: Antoni A and Antoni B tissue patterns. AJNR Am J Neuroradiol. 2007;28(9):1633–1638. doi: 10.3174/ajnr.A0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takase K, Yamamoto K, Imakiire A. Clinical pathology and therapeutic results of neurilemmoma in the upper extremity. J Orthop Surg (Hong Kong) 2004;12(2):222–225. doi: 10.1177/230949900401200216. [DOI] [PubMed] [Google Scholar]
- 36.Jiang S, Shen H, Lu H. Multiple schwannomas of the digital nerves and common palmar digital nerves: an unusual case report of multiple schwannomas in one hand. Medicine (Baltimore) 2019;98(10):e14605–e14605. doi: 10.1097/MD.0000000000014605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SM, Seo SW, Lee JY, Sung KS. Surgical outcome of schwannomas arising from major peripheral nerves in the lower limb. Int Orthop. 2012;36(8):1721–1725. doi: 10.1007/s00264-012-1560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siqueira MG, Socolovsky M, Martins RS, Robla-Costales J, Di Masi G, Heise CO, Cosamalón JG. Surgical treatment of typical peripheral schwannomas: the risk of new postoperative deficits. Acta Neurochir (Wien) 2013;155(9):1745–1749. doi: 10.1007/s00701-013-1818-6. [DOI] [PubMed] [Google Scholar]
- 39.Date R, Muramatsu K, Ihara K, Taguchi T. Advantages of intra-capsular micro-enucleation of schwannoma arising from extremities. Acta Neurochir (Wien) 2012;154(1):173–178; discussion 178. doi: 10.1007/s00701-011-1213-0. [DOI] [PubMed] [Google Scholar]
- 40.Kang HJ, Shin SJ, Kang ES. Schwannomas of the upper extremity. J Hand Surg Br. 2000;25(6):604–607. doi: 10.1054/jhsb.2000.0472. [DOI] [PubMed] [Google Scholar]
- 41.Park MJ, Seo KN, Kang HJ. Neurological deficit after surgical enucleation of schwannomas of the upper limb. J Bone Joint Surg Br. 2009;91(11):1482–1486. doi: 10.1302/0301-620X.91B11.22519. [DOI] [PubMed] [Google Scholar]
- 42.Donner TR, Voorhies RM, Kline DG. Neural sheath tumors of major nerves. J Neurosurg. 1994;81(3):362–373. doi: 10.3171/jns.1994.81.3.0362. [DOI] [PubMed] [Google Scholar]
- 43.Gabhane SK, Kotwal MN, Bobhate SK. Morphological spectrum of peripheral nerve sheath tumors: a series of 126 cases. Indian J Pathol Microbiol. 2009;52(1):29–33. doi: 10.4103/0377-4929.44958. [DOI] [PubMed] [Google Scholar]
- 44.LaFemina J, Qin LX, Moraco NH, Antonescu CR, Fields RC, Crago AM, Brennan MF, Singer S. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol. 2013;20(1):66–72. doi: 10.1245/s10434-012-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stucky CCH, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, Wasif N. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol. 2012;19(3):878–885. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 46.Kar M, Deo SVS, Shukla NK, Malik A, Datta Gupta S, Mohanti BK, Thulkar S. Malignant peripheral nerve sheath tumors (MPNST) – clinicopathological study and treatment outcome of twenty-four cases. World J Surg Oncol. 2006;4:55–55. doi: 10.1186/1477-7819-4-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, Lozza L, Collini P, Olmi P, Casali PG, Pilotti S, Gronchi A. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107(5):1065–1074. doi: 10.1002/cncr.22098. [DOI] [PubMed] [Google Scholar]
- 48.Ferner RE, Golding JF, Smith M, Calonje E, Jan W, Sanjayanathan V, O’Doherty M. [18F]2-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) as a diagnostic tool for neurofibromatosis 1 (NF1) associated malignant peripheral nerve sheath tumours (MPNSTs): a long-term clinical study. Ann Oncol. 2008;19(2):390–394. doi: 10.1093/annonc/mdm450. [DOI] [PubMed] [Google Scholar]
- 49.Riad S, Biau D, Holt GE, Werier J, Turcotte RE, Ferguson PC, Griffin AM, Dickie CI, Chung PW, Catton CN, O’Sullivan B, Wunder JS. The clinical and functional outcome for patients with radiation-induced soft tissue sarcoma. Cancer. 2012;118(10):2682–2692. doi: 10.1002/cncr.26543. [DOI] [PubMed] [Google Scholar]
- 50.Dobke M, Mackert GA. Upper extremity sarcoma: impact of current practice guidelines and controversies on reconstructive approaches. SICOT J. 2017;3:15–15. doi: 10.1051/sicotj/2017003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krauss EM, Tung TH, Moore AM. Free functional muscle transfers to restore upper extremity function. Hand Clin. 2016;32(2):243–256. doi: 10.1016/j.hcl.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 52.Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, Zhang W, McCutcheon IE, Slopis JM, Lazar AJ, Pollock RE, Lev D. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249(6):1014–1022. doi: 10.1097/SLA.0b013e3181a77e9a. [DOI] [PubMed] [Google Scholar]