

Abstract
Although prostate cancer is a major cause of cancer-related mortality worldwide, most patients will have a relatively indolent clinical course. Contrary to most other types of cancer, even the diagnosis of locally advanced or metastatic disease is not always lethal. The present review aimed to summarize what is known regarding the underlying mechanisms related to the indolent course of subsets of prostate cancer, at various stages. The data suggested that no specific gene alteration by itself was responsible for carcinogenesis or disease aggressiveness. However, pathway analysis identified genetic aberrations in multiple critical pathways that tend to accumulate over the course of the disease. The progression from indolence into aggressive disease is associated with a complex interplay in which genetic and epigenetic factors are involved. The effect of the immune tumor microenvironment is also very important. Emerging evidence has suggested that the upregulation of pathways related to cellular aging and senescence can identify patients with indolent disease. In addition, a number of tumors enter a long-lasting quiescent state. Further research will determine whether halting tumor evolution is a feasible option, and whether the life of patients can be markedly prolonged by inducing tumor senescence or long-term dormancy.
Keywords: indolence, epigenetic, genetic, prostate, cancer
1. Introduction
The diagnosis of prostate cancer (PCa) is usually not a death sentence. Overall, the relative survival rate in five years is almost 100 percent (1). Even when the disease progresses, it usually has a relatively slow clinical course. Prostate cancer is usually diagnosed in elderly men who often have several comorbidities and reduced expected lifespan. This suggests that if PCa patients were able to survive 15 or 20 years with the disease, prostate cancer-specific mortality would greatly decrease.
Androgen deprivation therapy (ADT) is the mainstay of therapy in early disseminated prostate cancer. Despite the initial response to treatment, the disease eventually relapses into an androgen independent state. Several attempts have been made to therapeutically target the mechanisms of resistance to ADT (2-4). However, the responses to these novel treatments are generally short lived (5). New candidate treatment regimens or combinations that counter the effects of resistance mechanisms are not always successful (6,7). Similar to all biological systems, cancer cells have molecular redundancies and it is rare that a viable combination of therapeutic compounds can kill every single cancer cell while sparing normal cells. In addition, tumors within individual patients are highly heterogeneous, hence there is a marked probability that at least a few tumor clones will survive a certain treatment (8). Most research in prostate cancer is following ‘down the rabbithole’ of discovering and subsequently targeting the emerging resistance mechanisms in order to eliminate every single cancer cell (3-7). Not much research has been directed towards discovering novel ways to slow the progression rate of the disease.
2. Indolence in localized disease
Early stage localized PCa can be successfully treated with radical prostatectomy (RP) and/or radiation therapy (9). The 5-year biochemical disease-free survival in patients with Grade Group 1 through 5 disease after RP is 96, 88, 63, 48 and 26% respectively (10). Among patients with T0-2 clinical stage disease, only 11% eventually die from prostate cancer (11). However, the effects of RP and radiotherapy are not so dramatic. These high survival rates mostly result from the indolent clinical course of the tumors. In early localized disease, excellent survival rates can be achieved even without intervention. In a 2015 meta-analysis of active surveillance studies, only 8 out of 7,627 patients eventually died from prostate cancer, with a median follow up of 3.5 years (range of 1.5-7.5 years) (12). In a prospective randomized trial, radical prostatectomy did not increase survival compared to watchful waiting in patients above 65 years old with early-stage prostate cancer (13). Longer follow up confirms the relatively indolent course of most localized prostate cancers. The 15-year metastasis-free survival in patients with Gleason 6 or less and PSA between 10-20 ng/ml is 94%. In Gleason 3+4 and PSA <20 ng/ml is 84%, while in Gleason 4+3 and PSA <20 ng/ml is 63% (13).
3. Indolence in high-risk disease and biochemical recurrence
Contrary to most other cancers, in prostate cancer, even the diagnosis of locally advanced or metastatic disease is not always a death sentence. A study conducted in 1997 found that patients with locally advanced disease had a corrected 15-year survival rate of 57% (11). Even 6% of patients discovered with initial metastatic disease did not die from prostate cancer after 15 years (11). Several attempts have been made to develop a genetic signature to inform us regarding which patients will have a reduced survival. This has led to the development of several experimental molecular assays, such as Decipher, and others (14-16). After RP, patients with low, intermediate or high risk Decipher scores have 10-year cumulative metastasis rates of 5.5, 15 and 26.7% respectively (17). This means that patients with the most aggressive genetic signatures will potentially be 73.3% metastasis-free at 10 years based on this method (17). Patients with biochemical-only recurrence also have a mostly indolent disease course. Post-RP biochemical relapse has a 37% likelihood of metastatic disease in 5 years (18). Median time to clinical metastases after PSA elevation is approximately 8 years (18). Even for patients who develop biochemical relapse <1.2 years after RP, the ten-year cancer-specific mortality rate is about 10 percent (19). Similarly, biochemical relapse after prostate radiotherapy yields high survival rates (20). Post-RP salvage radiotherapy results in 10-year prostate cancer specific survival rates of 86 percent (21).
4. Indolence in advanced disease
Even in the recurrent or metastatic setting, a subset of patients can achieve remarkable and durable responses to ADT and novel antiandrogen therapies (22). Among men with biochemical recurrence who are placed on immediate ADT, the 5-year overall survival rate is 91.2% (22). Among patients who develop distant metastatic disease, the 5-year prostate cancer-specific mortality is 57% (23). In the phase 3 AFFIRM study, there was a group of long-term responders to enzalutamide after treatment with docetaxel, who had a median survival of 7.9 years (24). In the STAMPEDE trial, almost half of the patients with metastatic disease who received ADT plus abiraterone acetate in the hormone-naive setting, were free from disease progression after 4.5 years (25). While subsets of prostate cancers can have a remarkably indolent course and show good response to therapy, other subsets can be particularly aggressive and refractory to treatments (26,27). Hence, the fundamental question that arises is what is the underlying cause of these differences? Is it an inherent property of the tumors, dictated by their genetic and epigenetic signature? Is it a matter of the tumor microenvironment, including the immune microenvironment? In this review, we will attempt to summarize what is already known regarding the underlying molecular mechanisms related to the indolent course of subsets of prostate cancers. Our hope is to provide evidence that the mechanisms that drive the aggressive variants are reversible.
5. Genetic determinants of indolence and aggressiveness in prostate cancer
Several distinct genomic subsets of PCa exist. Unsupervised clustering of molecular profiling (which include gene mutations, fusions, copy number alterations, gene expression levels and DNA methylation) indicate that 74% of all tumors can be assigned in one out of seven classes based on oncogenic drivers: fusions that involve 1) ERG, 2) ETV1, 3) ETV4, 4) FLI1, or mutations in 5) SPOP, 6) FOXA1, 7) IDH1(28). The relative distribution of these subgroups is similar in tumors derived from both primary and metastatic sites. This molecular taxonomy cannot accurately predict whether the tumors will be aggressive or indolent. The tumor mutational burden in prostate cancer is relatively low (28). Overall, increased number of copy number alterations (CNA) is associated with worse prognosis (29,30). One of the most frequent events in the prostate cancer genome with prognostic significance is a loss in the short arm of chromosome 8 (31,32). Loci frequently lost (>40%) include 8p21.2 and 8p23.2(31). The latter is associated with advanced disease and is most commonly found in progressors vs. non progressors (50 vs. 31%) (31). The most frequent gains (>50%) include 11p15.4, 2p25.1, 13q34, and 11q13.1. The latter is associated with biochemical recurrence independent of tumor stage and grade. Genes that overlap with this region include MEN1, MAP4K2, SF1, PPP2R5B and others (31). Fusions of androgen-regulated promoters with members of the ETS family of transcription factors are also very common. About 53% of prostate cancers have ETS-family fusions (33-36). TMPRSS2-ERG fusion analysis for CNAs reveals three important regions of copy-number loss: Two regions spanning PTEN and TP53 and the third spanning the region at 3p14, which likely contains FOXP1, RYBP and SHQ1 genes (30).
Whole exome sequencing reveals that there is only a small number of recurrent genes with alterations among various subtypes (28). In primary tumors, the most frequently altered gene is PTEN (17%), followed by TP53 (8%) (28). This suggests that no specific gene alteration by itself is solely responsible for carcinogenesis or disease aggressiveness. However, when distinct pathways as a whole are analyzed, the hypothesis changes. Taylor et al reported androgen receptor (AR) pathway gene aberrations in 56 percent of primaries and 100% of metastases (30). While AR gene amplifications and mutations were almost exclusively found in metastatic disease, it appeared that aberrations in NCOA2 and NCOR2 genes were important in primary tumors (30). PI3K signaling pathway was affected in 42% of primary tumors and 100% of metastases. The RB signaling pathway was affected in 34% of primary sites and 74% of metastases. The RAS/RAF signaling pathway was affected in 43% of primary sites and 90% of metastases (30). These findings support the notion that genetic factors are associated with the development and progression of prostate cancer. It is unlikely that clinically significant prostate cancer is a result of a single altered gene. The frequencies of altered critical pathways in primary tumors (many of which are indolent) suggest that clinically significant prostate cancers are unlikely to also result from a single altered molecular pathway. On the contrary, the evidence points towards combinations of genetic aberrations, which result in several altered molecular pathways. Accumulation of critical genomic aberrations over time and divergent clonal evolution are also hallmarks of the disease progression towards a more aggressive state (37) (Table I). Identifying and targeting key aberrant genes or pathways simultaneously may theoretically ‘force’ the disease to regress into a more indolent state. However, it is unknown whether the aggressive state is actually reversible once it occurs.
Table I.
Factors that maintain tumor indolence and mechanisms mediating a switch into aggressive disease.
| Indolence factor | First author, year | Escape mechanism/aggressiveness induction | (Refs.) |
|---|---|---|---|
| Low mutation rate | Taylor et al, 2010 | Additional genetic aberrations | (30) |
| Bonollo et al, 2020 | CAF effects | (88) | |
| Aggarwal et al, 2018 | Epigenetic modifications | (50) | |
| Slow proliferation | Taylor et al, 2010 | Additional genetic aberrations | (30) |
| Bonollo et al, 2020 | CAF effects | (88) | |
| Sugiura et al, 2021 | Cell cycle gene hypermethylation | (43) | |
| Sejda et al, 2020 | Neurotrophic signaling | (98) | |
| Androgen dependence | Taylor et al, 2010; | Additional genetic aberrations | (30,37) |
| Beltran et al, 2016 | |||
| Blom et al, 2019 | TME factors | (89) | |
| Ngollo et al, 2014; | Epigenetic adaptation | (40,59) | |
| Fu et al, 2006 | |||
| Nutrient scarcity/hypoxia | West et al, 2001 | VEGF upregulation | (129) |
| Ngollo et al, 2014; | Epigenetic adaptation | (40,48) | |
| Ge et al, 2020 | |||
| Taylor et al, 2010; | Additional genetic aberrations | (30,37) | |
| Beltran et al, 2016 | |||
| Bonollo et al, 2020 | Crosstalk with CAFs | (88) | |
| Immunosurveillance | Ness et al, 2014 | Dysfunctional TILs | (71) |
| Nardone et al, 2016 | High regulatory Foxp3+ | (75) | |
| Mariathasan et al, 2018 | High M2 macrophages | (77) | |
| Heninger et al, 2016 | MHC Class 1 silencing | (78) | |
| Fibroblast/stromal-induced inhibition of tumor growth | Blom et al, 2019 | CAFs activity/epigenetic changes in CAFs | (89) |
| Bonollo et al, 2020 | Increased stromal stiffness | (88) | |
| Sejda et al, 2020 | Perineural invasion | (98) | |
| March et al, 2021 | Neurotrophic growth factors | (99) | |
| Senescence phenotype | Ewald et al, 2010 | Treatment resistance/therapy escape | (110) |
| Wang et al, 2020 | Secretome sends tumorigenic signals to neighboring cells | (108) | |
| Low visceral tropism | Beltran et al, 2016 | Additional mutations/CNA in critical genes | (37) |
| Davies et al, 2020 | Neuroendocrine differentiation | (51) | |
| Yegnasubramanian et al, 2008 | Epigenetic adaptation | (49) | |
| Dormancy induction | Recasens et al, 2019 | Additional genetic aberrations | (148) |
| Decker et al, 2017 | Beta-adrenergic signaling | (149) | |
| Cackowski et al, 2017 | TYRO3, MERTK activity | (145) |
There are several factors that contribute to an indolent phenotype in subsets of prostate cancers. They include inherent properties of a tumor (such as slow proliferation rate, low visceral tropism), effects of treatment, immunosurveillance, TME-related effects, induction of dormancy/quiescence/senescence phenotype. However, as genetic and epigenetic alterations continue to accumulate, combined with the TME-endothelial compartment crosstalk, many tumors eventually escape dormancy and switch to aggressive disease.
6. Epigenetic determinants of prognosis in prostate cancer
The low mutation rate in PCa suggests that other factors might also determine the clinical course of the disease. It is now well established that epigenetic modifications play an important role in prostate cancer (38-41) (Table I). Epigenetics is the study of heritable changes in gene expression, without the presence of changes in the DNA sequence itself (42). While cells can alter their epigenome as a response to various conditions, it is known that epigenetic abnormalities frequently accumulate in cancer (43,44). Epigenetic changes can predispose genes to mutations, while genes that modify the epigenome are frequently mutated (45-47). DNA methylation has been implicated in the lineage plasticity of PCa (48). Several hypermethylated cell cycle genes and growth suppressor genes have been linked to worse prognosis (43). Aberrant DNA hypomethylation has also been observed more frequently in late stages of PCa (49). Epigenetic reprogramming is associated with loss of luminal epithelial identity and the transition from a typical prostate adenocarcinoma towards an aggressive neuroendocrine PCa (NEPC) (37,50,51). Neuroendocrine PCa cell lines possess a unique chromatin accessibility profile, distinct from prostate adenocarcinoma (52). Inactivation of TP53 and/or RB1 leads to upregulation of DNA methyltransferase family member 1 (DNTM1) (53,54). DNA methylation is linked to the activity of EZH2, which serves as a recruitment platform for DNMTs (55). EZH2 is a central regulator of neuroendocrine differentiation and the transition from an androgen receptor (AR)-dependent disease towards an aggressive state that is independent of AR signaling and indifferent to the effects of antiandrogens (51,56-58). The activity of AR can also be directly regulated by epigenetic modifiers, such as histone deacetylases (HDAC) (59). Post-transcriptional mechanisms, such as mRNA splicing or regulation by miRNA also play a role in the progression of prostate cancer (60-62). EZH2 can act both as a transcriptional activator or repressor, depending on post-translational modifications of EZH2 (63-65).
7. The role of tumor microenvironment
Prostate tumors with ‘bad’ morphologic or genetic features can still run an indolent clinical course. This suggests that the cellular and secreted factors in the tumor immune microenvironment (TIME) might play a role in the balance between tumor clearance and tumor progression, as well as the response to treatment. However, PCa in general has an immunologically ‘cold’ TIME (65). Overall mutation rates, as well as DDR gene defects in PCa are low, especially in the early disease setting (66). Hence, neoantigen expression is diminished compared to many other cancers. This results in a non-inflamed TIME, where tumor cells proliferate freely and evade immune-mediated elimination. Although the presence of cytotoxic and helper T-lymphocytes within tumor margins have been associated with favorable prognosis across several cancer types, PCa exhibits unique features (66-69). Studies suggest that high density of stromal CD8+ tumor infiltrating lymphocytes (TILs), and high PD-L1 expression are not associated with better outcomes in PCa (66,70-72). Some studies indicate that they might even be detrimental (73). Prostate cancer-infiltrating TILs are frequently dysfunctional (71,74) (Table I). However, high proportions of Foxp3+ regulatory TILs are associated with worse progression-free and overall survival in prostate cancer (75). In addition, high levels of M2 macrophages are pro-tumorigenic, suggesting that TGFβ might play a role in immune exclusion in prostate cancer (76,77). Epigenetic silencing of MHC Class I expression is common in advanced prostate tumors (78,79). Several signaling pathways, including the INF axis can also affect the expression of MHC Class I and the subsequent activation and expansion of CD8+ TILs within the invasive margins of a tumor (80). PTEN loss and other DDR defects also impact the TIME by modulating the activation of cellular INF pathways (66,81-83). It is known that the PTEN axis can confer sensitivity to T-cell-based immunotherapies (84). The development of bone metastases promotes the interaction between the tumor cells and the bone microenvironment. This further decreases the immunogenicity of the lesions (85). However, a few prostate cancers are immunologically ‘hot’ tumors and show durable responses when treated with immunotherapeutic agents (85-87). This suggests that the TIME has the potential to affect the clinical course of a patient who develops PCa, given the right circumstances.
Cancer associated fibroblasts (CAFs) constitute the most abundant cell population in the TME. They have been shown to play a major role in prostate cancer progression (88). During tumorigenesis, stromal fibroblasts crosstalk and likely coevolve with the epithelial compartment and become CAFs. Experiments revealed that CAFs from aggressive disease are sufficient to drive the progression of prostate cancer cells with low tumorigenic potential (88). They can also contribute to castration-resistance (89). On the other hand, normal fibroblasts can slow the proliferation rate of aggressive prostate cancer models (90). The amount of tumor-associated stroma diminishes as prostate cancer becomes more aggressive (89). Although it is hard to prove causality, TME features that characterize aggressive disease include a high proportion of CAFs, low proportion of smooth muscles, high vimentin expression, asporin expression, increased manifestation of matrix metalloproteinases, increased expression of COL5A2, and decreased expression of COL4A6 (89,91-93). The increased deposition of various collagen types, such as I or III, contributes to matrix stiffness, which leads to increased tumor invasiveness and metastatic potential (88). It was also suggested that CAF-derived neuregulin 1 (NRG1) induces antiandrogen resistance, via a NRG1-HER3 axis (94). CAFs don't have the genetic alterations of the epithelial compartment. On the contrary, CAFs from aggressive prostate cancers have discrete methylation differences compared to CAFs from moderate risk disease (95). For example, epigenetic regulation of Ras activity in prostatic CAFs, was found to regulate the metabolic and neuroendocrine activity in prostate cancer that fails ADT (96). Stromal AR expression also diminishes early in prostate tumorigenesis and continues to gradually decrease as the disease evolves into a more aggressive phenotype. It has been suggested that stromal AR inhibits the growth of malignant epithelial cells (97). Neural tissue is also an active TME element in prostate cancer. Perineural invasion is a known adverse prognostic factor (98). Several neural transmission receptors are present in cancer cells (98). Moreover, it has been recently shown that the abundance of neurotrophic growth factors in the patient's urine may perform better than PSA to separate aggressive prostate tumors from indolent ones (99).
8. A senescence phenotype is associated with indolence
There are currently no reliable molecular signatures to identify prostate tumors destined to run an indolent clinical course. Emerging evidence suggests that the upregulation of pathways related to cellular aging and senescence can distinguish between indolent and aggressive disease (100) (Table I). The ‘indolence signature’ includes inactivation of Nkx3.1, and increased expression of CDKN1A (p21), FGFR1 and PMP22 genes (100) (Fig. 1). CDKN1A is a cell regulatory gene associated with senescence (101). FGFR1 is known to play a critical role in prostate development and prostate tumorigenesis (102). This suggests a potentially complex activity of FGFR1 in prostate cancer. FGF signaling plays an important role in stem cell renewal, cellular aging and senescence (103). Although PMP22 is a gene highly expressed in neurons, it has also been associated with cellular proliferation regulation in other tissues and growth arrest in fibroblasts (104,105). Increased senescence has also been associated with reduced PSA recurrence rates (106). Senescent cells not only undergo cell cycle arrest, but they also trigger an immune response that can help the clearance of neoplastic cells (107). However, they are metabolically active and their secretome can impact the surrounding non-senescent cancer cells in ways that promote cancer progression and metastasis (108,109). Senescence can also be caused by long term oncogenic signaling or DNA damage and increased oxidative stress as a result of anticancer agents or radiation (109,110). In addition, complete PTEN loss triggers a p53-dependent cellular senescence response (The overwhelming majority of patients have PTEN loss heterozygosity, which results in tumor initiation and progression) (111-113). Moreover, androgen deprivation frequently induces senescence in prostate tumor cells (114,115). In conclusion, the induction of senescent molecular signatures might contribute to the indolent clinical course in some patients with PCa, before and after treatment, especially antiandrogen therapy.
Figure 1.
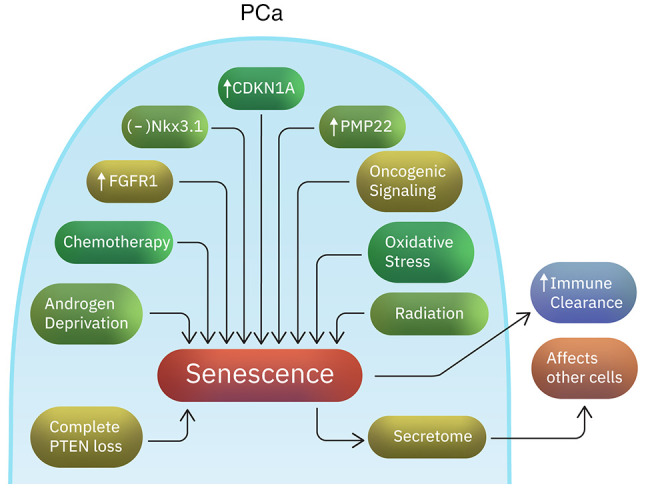
Factors associated with a ‘senescence phenotype’ in prostate cancer. Studies suggest that the upregulation of aging- and senescence-related pathways is able to distinguish between indolent and aggressive prostate tumors. The senescent signature includes Nkx3.1 inactivation and increased expression of CDKN1A, FGFR1 and PMP22 genes. Moreover, factors such as oxidative stress, oncogenic signaling, radiation, androgen deprivation or chemotherapy might induce senescence. While senescent cells can trigger immune clearance, their secretome can provide pro-tumorigenic signals to neighboring non-senescent cells.
9. What makes high risk disease?
Data from histology and gene expression analysis can provide useful prognostic information. Luminal B tumors carry the worst outcome (69% overall survival at 10-years), followed by basal and luminal A tumors (10-year overall survival at 80% and 82% respectively) (116). It is well known that the amount of Gleason 4 disease in the primary tumor is strongly associated with clinical outcomes and disease aggressiveness (117,118). High grade localized tumors are marked by epigenetic loss of heterogeneity and common trans-regulatory signatures. They exhibit enrichment for FOXA1, CDX2 and HOXB13 transcription factor binding sites (119). A few studies compared the differential gene expression between Gleason grade 3 and Gleason grade 4 lesions (120,121). The genes exclusively expressed in Gleason 4 tumors are those that are upregulated in neuronal, embryonic and hematopoietic stem cells. Overexpression of EGFR and HER2/neu are almost exclusively confined to Gleason 4 and above cancers (120,121). These genes are associated with independent tumor cell proliferation and enhanced cell motility (117). Gleason 4 and 5 lesions have lesser frequencies of cyclin D2 methylation, which results in cyclin D2 activation and CDKN1B sequestration. This subsequently results in cell cycle entry. CDKN1B immunostaining progressively diminishes with increasing Gleason score (122-124). Gleason score is also strongly associated with expression levels of the anti-apoptotic genes DAD1 and BCL2 (125,126). Moreover, indolent cancers are more capable of subverting a brake in replication. High Gleason grade lesions also show decreased androgen signaling, suggesting dedifferentiation. The downregulation of androgen responsive genes in high grade tumors results in increased cell proliferation (127,128). In addition, poor prognosis tumors are related to increased VEGF production and microvessel density, as well as irregularity in vessel diameter (129-131). Microvessel pericyte density score is also associated with disease aggressiveness (132). Higher grade lesions overexpress elements that are permissive for tumor migration. For example, the chemokine receptor CXCR4, which is overexpressed in Gleason 4 lesions, poses a key role in the development of lymph nodes and bone metastases (133-135). Interestingly, its ligand CXCL12 is secreted in high concentrations by lymph nodes and bone marrow stroma (117).
10. The role of tumor dormancy
In PCa, metastatic disease may occur years after RP. This suggests that in some cases cancer cells undergo a long-lasting quiescent state (136). Quiescent cells are reversibly suspended in the G0 phase, but they retain the ability to re-enter the cell cycle and initiate symptomatic metastatic disease (137) (Table I; Fig. 2). Tumor dormancy might be due to intrinsic factors or due to conditions provided to the tumor cells by the surrounding microenvironment (138,139). The bone is a major site of PCa recurrence, suggesting that the bone microenvironment promotes a state of dormancy (136). In vitro studies have shown that two members of the TGFβ/BMP family, GDF10 and TGFβ2 (which are secreted from differentiated osteoblasts), induce quiescence in PCa cells (136,138). Several other osteoblast secreted factors such as Wnt5a (maintains hematopoietic stem cells in dormant state), GAS6, LIF, thrombospondin, DKK3, BMP1, vasorin, neogenin, MIA and NGAL, have also been suggested to promote dormancy in PCa (136,140-142). For example, vasorin, neogenin and DKK3 induce dormancy via activation of the p38MAPK signaling pathway (136). Besides osteoblasts, other bone marrow stromal cells secrete dormancy-inducing factors (136). The transcription factors SOX2, NANOG and the orphan receptor NR2F1 are important for the maintenance of a quiescent phenotype through epigenetic regulation (143-147). Studies suggest that dormant cells at the metastatic sites continue to acquire genomic changes (148). The receptor tyrosine kinases TYRO3 and MERTK (TAM family) were shown to promote dormancy escape (145). Norepinephrine was also hypothesized to stimulate dormancy escape in PCa through the beta-2 adrenergic signaling (149).
Figure 2.
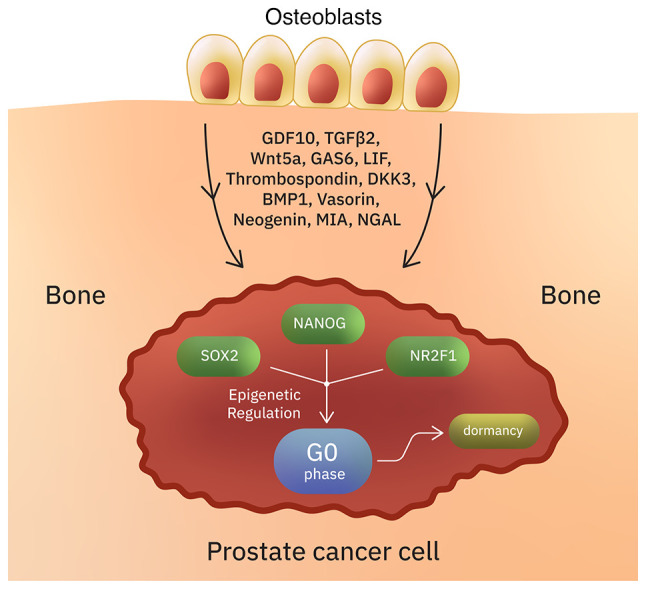
Induction of dormancy/quiescence in prostate cancer cells. Intrinsic factors and conditions provided by the TME can result in the reversible suspension of prostate cancer cells in G0 phase. In some cases, this dormant state can last for several years. Several osteoblast-secreted factors (such as NGAL, MIA, GDF10, TGFβ2, and others) have been suggested to induce quiescence in prostate cancer. The transcription factors SOX2, NANOG and NR2F1 are important for the maintenance of a quiescent phenotype through epigenetic regulation.
11. Conclusions
The majority of prostate cancers follow an indolent clinical course. It is unclear whether there is one or several types of indolence in PCa. Several factors have been linked to disease aggressiveness. However, it is still largely unknown which of these factors actually have a causative role. High quality tumor analyses suggest that complex genetic aberrations in multiple critical pathways are associated with a worse phenotype. The escape from indolence into an aggressive disease is associated with a complex interplay in which genetic and epigenetic factors are likely involved. The effect of the immediate immune tumor microenvironment is also very important. Future studies will show whether halting the stepwise tumor evolution is a feasible option. Further research will also determine whether we can meaningfully prolong the life of PCa patients by inducing senescence or long-term tumor dormancy.
Acknowledgements
Not applicable.
Funding Statement
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
MS conceptualized this review, reviewed the literature and drafted and critically reviewed the final manuscript. LJF reviewed the literature, and drafted and critically reviewed the final version of the manuscript. SR reviewed the literature, and drafted and critically reviewed the manuscript. Data authentication is not applicable. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2.Wadosky KM, Koochekpour S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget. 2016;7:64447–64470. doi: 10.18632/oncotarget.10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014;33:595–606. doi: 10.1007/s10555-013-9481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koinis F, Corn P, Parikh N, Song J, Vardaki I, Mourkioti I, Lin SH, Logothetis C, Panaretakis T, Gallick G. Resistance to MET/VEGFR2 inhibition by cabozantinib is mediated by YAP/TBX5-dependent induction of FGFR1 in castration-resistant prostate cancer. Cancers (Basel) 2020;12(244) doi: 10.3390/cancers12010244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith M, De Bono J, Sternberg C, Le Moulec S, Oudard S, De Giorgi U, Krainer M, Bergman A, Hoelzer W, De Wit R, et al. Phase III study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol. 2016;34:3005–3013. doi: 10.1200/JCO.2015.65.5597. [DOI] [PubMed] [Google Scholar]
- 6.Michaelson MD, Oudard S, Ou YC, Sengeløv L, Saad F, Houede N, Ostler P, Stenzl A, Daugaard G, Jones R, et al. Randomized, placebo-controlled, phase III trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J Clin Oncol. 2014;32:76–82. doi: 10.1200/JCO.2012.48.5268. [DOI] [PubMed] [Google Scholar]
- 7.Spreafico A, Chi KN, Sridhar SS, Smith DC, Carducci MA, Kavsak P, Wong TS, Wang L, Ivy SP, Mukherjee SD, et al. A randomized phase II study of cediranib alone versus cediranib in combination with dasatinib in docetaxel resistant, castration resistant prostate cancer patients. Invest New Drugs. 2014;32:1005–1016. doi: 10.1007/s10637-014-0106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81–94. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 9.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–396. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epstein JI, Zelefsky MJ, Sjoberg DD, Nelson JB, Egevad L, Magi-Galluzzi C, Vickers AJ, Parwani AV, Reuter VE, Fine SW, et al. A contemporary prostate cancer grading system: A validated alternative to the gleason score. Eur Urol. 2016;69:428–435. doi: 10.1016/j.eururo.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson JE, Holmberg L, Johansson S, Bergström R, Adami HO. Fifteen-year survival in prostate cancer. A prospective, population-based study in Sweden. JAMA. 1997;277:467–471. [PubMed] [Google Scholar]
- 12.Simpkin AJ, Tilling K, Martin RM, Lane JA, Hamdy FC, Holmberg L, Neal DE, Metcalfe C, Donovan JL. Systematic review and meta-analysis of factors determining change to radical treatment in active surveillance for localized prostate cancer. Eur Urol. 2015;67:993–1005. doi: 10.1016/j.eururo.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Musunuru HB, Yamamoto T, Klotz L, Ghanem G, Mamedov A, Sethukavalan P, Jethava V, Jain S, Zhang L, Vesprini D, Loblaw A. Active surveillance for intermediate risk prostate cancer: Survival outcomes in the sunnybrook experience. J Urol. 2016;196:1651–1658. doi: 10.1016/j.juro.2016.06.102. [DOI] [PubMed] [Google Scholar]
- 14.Herlemann A, Huang HC, Alam R, Tosoian JJ, Kim HL, Klein EA, Simko JP, Chan JM, Lane BR, Davis JW, et al. Decipher identifies men with otherwise clinically favorable-intermediate risk disease who may not be good candidates for active surveillance. Prostate Cancer Prostatic Dis. 2020;23:136–143. doi: 10.1038/s41391-019-0167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kornberg Z, Cooperberg MR, Cowan JE, Chan JM, Shinohara K, Simko JP, Tenggara I, Carroll PR. A 17-gene genomic prostate score as a predictor of adverse pathology in men on active surveillance. J Urol. 2019;202:702–709. doi: 10.1097/JU.0000000000000290. [DOI] [PubMed] [Google Scholar]
- 16.Spratt DE, Zhang J, Santiago-Jiménez M, Dess RT, Davis JW, Den RB, Dicker AP, Kane CJ, Pollack A, Stoyanova R, et al. Development and validation of a novel integrated clinical-genomic risk group classification for localized prostate cancer. J Clin Oncol. 2018;36:581–590. doi: 10.1200/JCO.2017.74.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spratt DE, Yousefi K, Deheshi S, Ross AE, Den RB, Schaeffer EM, Trock BJ, Zhang J, Glass AG, Dicker AP, et al. Individual patient-level meta-analysis of the performance of the decipher genomic classifier in high-risk men after prostatectomy to predict development of metastatic disease. J Clin Oncol. 2017;35:1991–1998. doi: 10.1200/JCO.2016.70.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA. 1999;281:1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 19.Boorjian SA, Thompson RH, Tollefson MK, Rangel LJ, Bergstralh EJ, Blute ML, Karnes RJ. Long-term risk of clinical progression after biochemical recurrence following radical prostatectomy: The impact of time from surgery to recurrence. Eur Urol. 2011;59:893–899. doi: 10.1016/j.eururo.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 20.Martin NE, Chen MH, Beard CJ, Nguyen PL, Loffredo MJ, Renshaw AA, Kantoff PW, D'Amico AV. Natural history of untreated prostate specific antigen radiorecurrent prostate cancer in men with favorable prognostic indicators. Prostate Cancer. 2014;2014(912943) doi: 10.1155/2014/912943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trock BJ, Han M, Freedland SJ, Humphreys EB, DeWeese TL, Partin AW, Walsh PC. Prostate cancer-specific survival following salvage radiotherapy vs observation in men with biochemical recurrence after radical prostatectomy. JAMA. 2008;299:2760–2769. doi: 10.1001/jama.299.23.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stone L. Prostate cancer: ADT after radical prostatectomy-when and how? Nat Rev Urol. 2016;13(367) doi: 10.1038/nrurol.2016.97. [DOI] [PubMed] [Google Scholar]
- 23.Muralidhar V, Mahal BA, Nguyen PL. Conditional cancer-specific mortality in T4, N1, or M1 prostate cancer: Implications for long-term prognosis. Radiat Oncol. 2015;10(155) doi: 10.1186/s13014-015-0470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 25.James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, Ritchie AWS, Amos CL, Gilson C, Jones RJ, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–351. doi: 10.1056/NEJMoa1702900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandrasekar T, Yang JC, Gao AC, Evans CP. Targeting molecular resistance in castration-resistant prostate cancer. BMC Med. 2015;13(206) doi: 10.1186/s12916-015-0457-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fine SW. Neuroendocrine tumors of the prostate. Mod Pathol. 2018;31 (S1):S122–S132. doi: 10.1038/modpathol.2017.164. [DOI] [PubMed] [Google Scholar]
- 28.The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. Cancer Genome Atlas Research Network. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA. 2014;111:11139–11144. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paris PL, Andaya A, Fridlyand J, Jain AN, Weinberg V, Kowbel D, Brebner JH, Simko J, Watson JE, Volik S, et al. Whole genome scanning identifies genotypes associated with recurrence and metastasis in prostate tumors. Hum Mol Genet. 2004;13:1303–1313. doi: 10.1093/hmg/ddh155. [DOI] [PubMed] [Google Scholar]
- 32.Chu LW, Troncoso P, Johnston DA, Liang JC. Genetic markers useful for distinguishing between organ-confined and locally advanced prostate cancer. Genes Chromosomes Cancer. 2003;36:303–312. doi: 10.1002/gcc.10171. [DOI] [PubMed] [Google Scholar]
- 33.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 34.Sboner A, Habegger L, Pflueger D, Terry S, Chen DZ, Rozowsky JS, Tewari AK, Kitabayashi N, Moss BJ, Chee MS, et al. FusionSeq: A modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11(R104) doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38(e178) doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomlins SA, Bjartell A, Chinnaiyan AM, Jenster G, Nam RK, Rubin MA, Schalken JA. ETS gene fusions in prostate cancer: From discovery to daily clinical practice. Eur Urol. 2009;56:275–286. doi: 10.1016/j.eururo.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 37.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tzelepi V, Logotheti S, Efstathiou E, Troncoso P, Aparicio A, Sakellakis M, Hoang A, Perimenis P, Melachrinou M, Logothetis C, Zolota V. Epigenetics and prostate cancer: Defining the timing of DNA methyltransferase deregulation during prostate cancer progression. Pathology. 2020;52:218–227. doi: 10.1016/j.pathol.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Macedo-Silva C, Benedetti R, Ciardiello F, Cappabianca S, Jerónimo C, Altucci L. Epigenetic mechanisms underlying prostate cancer radioresistance. Clin Epigenetics. 2021;13(125) doi: 10.1186/s13148-021-01111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ngollo M, Dagdemir A, Karsli-Ceppioglu S, Judes G, Pajon A, Penault-Llorca F, Boiteux JP, Bignon YJ, Guy L, Bernard-Gallon DJ. Epigenetic modifications in prostate cancer. Epigenomics. 2014;6:415–426. doi: 10.2217/epi.14.34. [DOI] [PubMed] [Google Scholar]
- 41.Kumaraswamy A, Welker Leng KR, Westbrook TC, Yates JA, Zhao SG, Evans CP, Feng FY, Morgan TM, Alumkal JJ. Recent advances in epigenetic biomarkers and epigenetic targeting in prostate cancer. Eur Urol. 2021;80:71–81. doi: 10.1016/j.eururo.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weinhold B. Epigenetics: The science of change. Environ Health Perspect. 2006;114:A160–A167. doi: 10.1289/ehp.114-a160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugiura M, Sato H, Kanesaka M, Imamura Y, Sakamoto S, Ichikawa T, Kaneda A. Epigenetic modifications in prostate cancer. Int J Urol. 2021;28:140–149. doi: 10.1111/iju.14406. [DOI] [PubMed] [Google Scholar]
- 44.Dawson MA. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science. 2017;355:1147–1152. doi: 10.1126/science.aam7304. [DOI] [PubMed] [Google Scholar]
- 45.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 47.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 48.Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, Huang K, Massari F, Lu X, Cimadamore A, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann Oncol. 2020;31:470–479. doi: 10.1016/j.annonc.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, Irizarry RA, Morgan J, Hicks J, DeWeese TL, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res. 2008;68:8954–8967. doi: 10.1158/0008-5472.CAN-07-6088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, Weinstein AS, Friedl V, Zhang C, Witte ON, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J Clin Oncol. 2018;36:2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies A, Zoubeidi A, Selth LA. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat Cancer. 2020;27:R35–R50. doi: 10.1530/ERC-19-0420. [DOI] [PubMed] [Google Scholar]
- 52.Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, Smith BA, Cheng C, Tsai BL, Cheng D, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science. 2018;362:91–95. doi: 10.1126/science.aat5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCabe MT, Davis JN, Day ML. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 2005;65:3624–3632. doi: 10.1158/0008-5472.CAN-04-2158. [DOI] [PubMed] [Google Scholar]
- 54.Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, Lu YY, Tang YA, Yang YC, Yang PC, Wang YC. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010;70:5807–5817. doi: 10.1158/0008-5472.CAN-09-4161. [DOI] [PubMed] [Google Scholar]
- 55.Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 56.Shan J, Al-Muftah MA, Al-Kowari MK, Abuaqel SWJ, Al-Rumaihi K, Al-Bozom I, Li P, Chouchane L. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discov. 2019;5(139) doi: 10.1038/s41420-019-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30:563–577. doi: 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah N, Wang P, Wongvipat J, Karthaus WR, Abida W, Armenia J, Rockowitz S, Drier Y, Bernstein BE, Long HW, et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife. 2017;6(e27861) doi: 10.7554/eLife.27861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, et al. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol. 2006;26:8122–8135. doi: 10.1128/MCB.00289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang X, Coleman IM, Brown LG, True LD, Kollath L, Lucas JM, Lam HM, Dumpit R, Corey E, Chéry L, et al. SRRM4 expression and the loss of REST activity may promote the emergence of the neuroendocrine phenotype in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:4698–4708. doi: 10.1158/1078-0432.CCR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Y, Donmez N, Sahinalp C, Xie N, Wang Y, Xue H, Mo F, Beltran H, Gleave M, Wang Y, et al. SRRM4 drives neuroendocrine transdifferentiation of prostate adenocarcinoma under androgen receptor pathway inhibition. Eur Urol. 2017;71:68–78. doi: 10.1016/j.eururo.2016.04.028. [DOI] [PubMed] [Google Scholar]
- 62.Nam RK, Benatar T, Amemiya Y, Wallis CJD, Romero JM, Tsagaris M, Sherman C, Sugar L, Seth A. MicroRNA-652 induces NED in LNCaP and EMT in PC3 prostate cancer cells. Oncotarget. 2018;9:19159–19176. doi: 10.18632/oncotarget.24937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, Licht JD, Zhao JC, Yu J. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018;25:2808–2820.e4. doi: 10.1016/j.celrep.2018.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bilusic M, Madan RA, Gulley JL. Immunotherapy of prostate cancer: Facts and hopes. Clin Cancer Res. 2017;23:6764–6770. doi: 10.1158/1078-0432.CCR-17-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vitkin N, Nersesian S, Siemens DR, Koti M. The tumor immune contexture of prostate cancer. Front Immunol. 2019;10(603) doi: 10.3389/fimmu.2019.00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–734. doi: 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- 68.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, Stagg J. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res. 2016;22:158–166. doi: 10.1158/1078-0432.CCR-15-1181. [DOI] [PubMed] [Google Scholar]
- 71.Ness N, Andersen S, Valkov A, Nordby Y, Donnem T, Al-Saad S, Busund LT, Bremnes RM, Richardsen E. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate. 2014;74:1452–1461. doi: 10.1002/pros.22862. [DOI] [PubMed] [Google Scholar]
- 72.Petitprez F, Fossati N, Vano Y, Freschi M, Becht E, Lucianò R, Calderaro J, Guédet T, Lacroix L, Rancoita PMV, et al. PD-L1 expression and CD8+ T-cell infiltrate are associated with clinical progression in patients with node-positive prostate cancer. Eur Urol Focus. 2019;5:192–196. doi: 10.1016/j.euf.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 73.Zhao SG, Lehrer J, Chang SL, Das R, Erho N, Liu Y, Sjöström M, Den RB, Freedland SJ, Klein EA, et al. The immune landscape of prostate cancer and nomination of PD-L2 as a potential therapeutic target. J Natl Cancer Inst. 2019;111:301–310. doi: 10.1093/jnci/djy141. [DOI] [PubMed] [Google Scholar]
- 74.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nardone V, Botta C, Caraglia M, Martino EC, Ambrosio MR, Carfagno T, Tini P, Semeraro L, Misso G, Grimaldi A, et al. Tumor infiltrating T lymphocytes expressing FoxP3, CCR7 or PD-1 predict the outcome of prostate cancer patients subjected to salvage radiotherapy after biochemical relapse. Cancer Biol Ther. 2016;17:1213–1220. doi: 10.1080/15384047.2016.1235666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lundholm M, Hägglöf C, Wikberg ML, Stattin P, Egevad L, Bergh A, Wikström P, Palmqvist R, Edin S. Secreted factors from colorectal and prostate cancer cells skew the immune response in opposite directions. Sci Rep. 2015;5(15651) doi: 10.1038/srep15651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Heninger E, Krueger TE, Thiede SM, Sperger JM, Byers BL, Kircher MR, Kosoff D, Yang B, Jarrard DF, McNeel DG, Lang JM. Inducible expression of cancer-testis antigens in human prostate cancer. Oncotarget. 2016;7:84359–84374. doi: 10.18632/oncotarget.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanda MG, Restifo NP, Walsh JC, Kawakami Y, Nelson WG, Pardoll DM, Simons JW. Molecular characterization of defective antigen processing in human prostate cancer. J Natl Cancer Inst. 1995;87:280–285. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martini M, Testi MG, Pasetto M, Picchio MC, Innamorati G, Mazzocco M, Ugel S, Cingarlini S, Bronte V, Zanovello P, et al. IFN-gamma-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine. 2010;28:3548–3557. doi: 10.1016/j.vaccine.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 81.Chen L, Guo D. The functions of tumor suppressor PTEN in innate and adaptive immunity. Cell Mol Immunol. 2017;14:581–589. doi: 10.1038/cmi.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pencik J, Schlederer M, Gruber W, Unger C, Walker SM, Chalaris A, Marié IJ, Hassler MR, Javaheri T, Aksoy O, et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat Commun. 2015;6(7736) doi: 10.1038/ncomms8736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, Sharma P. Differences in tumor microenvironment dictate T helper lineage polarization and response to immune checkpoint therapy. Cell. 2019;179:1177–1190.e13. doi: 10.1016/j.cell.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 86.Stultz J, Fong L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021;24:697–717. doi: 10.1038/s41391-021-00340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bonollo F, Thalmann GN, Kruithof-de Julio M, Karkampouna S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers (Basel) 2020;12(1887) doi: 10.3390/cancers12071887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blom S, Erickson A, Östman A, Rannikko A, Mirtti T, Kallioniemi O, Pellinen T. Fibroblast as a critical stromal cell type determining prognosis in prostate cancer. Prostate. 2019;79:1505–1513. doi: 10.1002/pros.23867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Suhovskih AV, Kashuba VI, Klein G, Grigorieva EV. Prostate cancer cells specifically reorganize epithelial cell-fibroblast communication through proteoglycan and junction pathways. Cell Adh Migr. 2017;11:39–53. doi: 10.1080/19336918.2016.1182292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma JB, Bai JY, Zhang HB, Gu L, He D, Guo P. Downregulation of collagen COL4A6 is associated with prostate cancer progression and metastasis. Genet Test Mol Biomarkers. 2020;24:399–408. doi: 10.1089/gtmb.2020.0009. [DOI] [PubMed] [Google Scholar]
- 92.Ren X, Chen X, Fang K, Zhang X, Wei X, Zhang T, Li G, Lu Z, Song N, Wang S, Qin C. COL5A2 promotes proliferation and invasion in prostate cancer and is one of seven gleason-related genes that predict recurrence-free survival. Front Oncol. 2021;11(583083) doi: 10.3389/fonc.2021.583083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bahmad HF, Jalloul M, Azar J, Moubarak MM, Samad TA, Mukherji D, Al-Sayegh M, Abou-Kheir W. Tumor microenvironment in prostate cancer: Toward identification of novel molecular biomarkers for diagnosis, prognosis, and therapy development. Front Genet. 2021;12(652747) doi: 10.3389/fgene.2021.652747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Z, Karthaus WR, Lee YS, Gao VR, Wu C, Russo JW, Liu M, Mota JM, Abida W, Linton E, et al. Tumor microenvironment-derived NRG1 promotes antiandrogen resistance in prostate cancer. Cancer Cell. 2020;38:279–296.e9. doi: 10.1016/j.ccell.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lawrence MG, Pidsley R, Niranjan B, Papargiris M, Pereira BA, Richards M, Teng L, Norden S, Ryan A, Frydenberg M, et al. Alterations in the methylome of the stromal tumour microenvironment signal the presence and severity of prostate cancer. Clin Epigenetics. 2020;12(48) doi: 10.1186/s13148-020-00836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mishra R, Haldar S, Placencio V, Madhav A, Rohena-Rivera K, Agarwal P, Duong F, Angara B, Tripathi M, Liu Z, et al. Stromal epigenetic alterations drive metabolic and neuroendocrine prostate cancer reprogramming. J Clin Invest. 2018;128:4472–4484. doi: 10.1172/JCI99397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singh M, Jha R, Melamed J, Shapiro E, Hayward SW, Lee P. Stromal androgen receptor in prostate development and cancer. Am J Pathol. 2014;184:2598–2607. doi: 10.1016/j.ajpath.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sejda A, Sigorski D, Gulczyński J, Wesołowski W, Kitlińska J, Iżycka-Świeszewska E. Complexity of neural component of tumor microenvironment in prostate cancer. Pathobiology. 2020;87:87–99. doi: 10.1159/000505437. [DOI] [PubMed] [Google Scholar]
- 99.March B, Lockhart KR, Faulkner S, Smolny M, Rush R, Hondermarck H. ELISA-based quantification of neurotrophic growth factors in urine from prostate cancer patients. FASEB Bioadv. 2021;3:888–896. doi: 10.1096/fba.2021-00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Irshad S, Bansal M, Castillo-Martin M, Zheng T, Aytes A, Wenske S, Le Magnen C, Guarnieri P, Sumazin P, Benson MC, et al. A molecular signature predictive of indolent prostate cancer. Sci Transl Med. 2013;5(202ra122) doi: 10.1126/scitranslmed.3006408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. López-Domínguez JA, Rodríguez-López S, Ahumada-Castro U, Desprez PY, Konovalenko M, Laberge RM, Cárdenas C, Villalba JM, Campisi J. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging (Albany NY) 2021;13:13380–13392. doi: 10.18632/aging.203110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang F, Zhang Y, Ressler SJ, Ittmann MM, Ayala GE, Dang TD, Wang F, Rowley DR. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013;73:3716–3724. doi: 10.1158/0008-5472.CAN-12-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Coutu DL, Galipeau J. Roles of FGF signaling in stem cell self-renewal, senescence and aging. Aging (Albany NY) 2011;3:920–33. doi: 10.18632/aging.100369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–280. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 105.Suter U, Snipes GJ. Peripheral myelin protein 22: Facts and hypotheses. J Neurosci Res. 1995;40:145–151. doi: 10.1002/jnr.490400202. [DOI] [PubMed] [Google Scholar]
- 106.Wagner J, Damaschke N, Yang B, Truong M, Guenther C, McCormick J, Huang W, Jarrard D. Overexpression of the novel senescence marker β-galactosidase (GLB1) in prostate cancer predicts reduced PSA recurrence. PLoS One. 2015;10(e0124366) doi: 10.1371/journal.pone.0124366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Prata LGPL, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin Immunol. 2018;40(101275) doi: 10.1016/j.smim.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang B, Kohli J, Demaria M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer. 2020;6:838–857. doi: 10.1016/j.trecan.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 109.Hwang HJ, Jung SH, Lee HC, Han NK, Bae IH, Lee M, Han YH, Kang YS, Lee SJ, Park HJ, et al. Identification of novel therapeutic targets in the secretome of ionizing radiation-nduced senescent tumor cells. Oncol Rep. 2016;35:841–850. doi: 10.3892/or.2015.4473. [DOI] [PubMed] [Google Scholar]
- 110.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536–1546. doi: 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Parisotto M, Grelet E, El Bizri R, Metzger D. Senescence controls prostatic neoplasia driven by Pten loss. Mol Cell Oncol. 2018;6(1511205) doi: 10.1080/23723556.2018.1511205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jung SH, Hwang HJ, Kang D, Park HA, Lee HC, Jeong D, Lee K, Park HJ, Ko YG, Lee JS. mTOR kinase leads to PTEN-loss-induced cellular senescence by phosphorylating p53. Oncogene. 2019;38:1639–1650. doi: 10.1038/s41388-018-0521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Blute ML Jr, Damaschke N, Wagner J, Yang B, Gleave M, Fazli L, Shi F, Abel EJ, Downs TM, Huang W, Jarrard DF. Persistence of senescent prostate cancer cells following prolonged neoadjuvant androgen deprivation therapy. PLoS One. 2017;12(e0172048) doi: 10.1371/journal.pone.0172048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pernicová Z, Slabáková E, Kharaishvili G, Bouchal J, Král M, Kunická Z, Machala M, Kozubík A, Souček K. Androgen depletion induces senescence in prostate cancer cells through down-regulation of Skp2. Neoplasia. 2011;13:526–536. doi: 10.1593/neo.11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao SG, Chang SL, Erho N, Yu M, Lehrer J, Alshalalfa M, Speers C, Cooperberg MR, Kim W, Ryan CJ, et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017;3:1663–1672. doi: 10.1001/jamaoncol.2017.0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ahmed HU, Arya M, Freeman A, Emberton M. Do low-grade and low-volume prostate cancers bear the hallmarks of malignancy? Lancet Oncol. 2012;13:e509–e517. doi: 10.1016/S1470-2045(12)70388-1. [DOI] [PubMed] [Google Scholar]
- 118.Sharma M, Miyamoto H. Percent Gleason pattern 4 in stratifying the prognosis of patients with intermediate-risk prostate cancer. Transl Androl Urol. 2018;7 (Suppl 4):S484–S489. doi: 10.21037/tau.2018.03.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eksi SE, Chitsazan A, Sayar Z, Thomas GV, Fields AJ, Kopp RP, Spellman PT, Adey AC. Epigenetic loss of heterogeneity from low to high grade localized prostate tumours. Nat Commun. 2021;12(7292) doi: 10.1038/s41467-021-27615-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ross AE, Marchionni L, Vuica-Ross M, Cheadle C, Fan J, Berman DM, Schaeffer EM. Gene expression pathways of high grade localized prostate cancer. Prostate. 2011;71:1568–1577. doi: 10.1002/pros.21373. [DOI] [PubMed] [Google Scholar]
- 121.Skacel M, Ormsby AH, Pettay JD, Tsiftsakis EK, Liou LS, Klein EA, Levin HS, Zippe CD, Tubbs RR. Aneusomy of chromosomes 7, 8, and 17 and amplification of HER-2/neu and epidermal growth factor receptor in Gleason score 7 prostate carcinoma: A differential fluorescent in situ hybridization study of Gleason pattern 3 and 4 using tissue microarray. Hum Pathol. 2001;32:1392–1397. doi: 10.1053/hupa.2001.29676. [DOI] [PubMed] [Google Scholar]
- 122.Susaki E, Nakayama KI. Multiple mechanisms for p27(Kip1) translocation and degradation. Cell Cycle. 2007;6:3015–3020. doi: 10.4161/cc.6.24.5087. [DOI] [PubMed] [Google Scholar]
- 123.Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, Minna JD, Gazdar AF. Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin Cancer Res. 2003;9:4730–4734. [PubMed] [Google Scholar]
- 124.Guo Y, Sklar GN, Borkowski A, Kyprianou N. Loss of the cyclin-dependent kinase inhibitor p27(Kip1) protein in human prostate cancer correlates with tumor grade. Clin Cancer Res. 1997;3:2269–2274. [PubMed] [Google Scholar]
- 125.True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R, Beer TM, Gelmann E, Datta M, Mostaghel E, et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci USA. 2006;103:10991–10996. doi: 10.1073/pnas.0603678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fleischmann A, Huland H, Mirlacher M, Wilczak W, Simon R, Erbersdobler A, Sauter G, Schlomm T. Prognostic relevance of Bcl-2 overexpression in surgically treated prostate cancer is not caused by increased copy number or translocation of the gene. Prostate. 2012;72:991–997. doi: 10.1002/pros.21504. [DOI] [PubMed] [Google Scholar]
- 127.Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, Kalyana-Sundaram S, Wei JT, Rubin MA, Pienta KJ, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 128.Hendriksen PJ, Dits NF, Kokame K, Veldhoven A, van Weerden WM, Bangma CH, Trapman J, Jenster G. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66:5012–5020. doi: 10.1158/0008-5472.CAN-05-3082. [DOI] [PubMed] [Google Scholar]
- 129.West AF, O'Donnell M, Charlton RG, Neal DE, Leung HY. Correlation of vascular endothelial growth factor expression with fibroblast growth factor-8 expression and clinico-pathologic parameters in human prostate cancer. Br J Cancer. 2001;85:576–583. doi: 10.1054/bjoc.2001.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Erbersdobler A, Isbarn H, Dix K, Steiner I, Schlomm T, Mirlacher M, Sauter G, Haese A. Prognostic value of microvessel density in prostate cancer: A tissue microarray study. World J Urol. 2010;28:687–692. doi: 10.1007/s00345-009-0471-4. [DOI] [PubMed] [Google Scholar]
- 131.Mucci LA, Powolny A, Giovannucci E, Liao Z, Kenfield SA, Shen R, Stampfer MJ, Clinton SK. Prospective study of prostate tumor angiogenesis and cancer-specific mortality in the health professionals follow-up study. J Clin Oncol. 2009;27:5627–5633. doi: 10.1200/JCO.2008.20.8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Killingsworth MC, Wu X. Vascular pericyte density and angiogenesis associated with adenocarcinoma of the prostate. Pathobiology. 2011;78:24–34. doi: 10.1159/000322739. [DOI] [PubMed] [Google Scholar]
- 133.Lin D, Bayani J, Wang Y, Sadar MD, Yoshimoto M, Gout PW, Squire JA, Wang Y. Development of metastatic and non-metastatic tumor lines from a patient's prostate cancer specimen-identification of a small subpopulation with metastatic potential in the primary tumor. Prostate. 2010;70:1636–1644. doi: 10.1002/pros.21199. [DOI] [PubMed] [Google Scholar]
- 134.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 136.Yu-Lee LY, Lee YC, Pan J, Lin SC, Pan T, Yu G, Hawke DH, Pan BF, Lin SH. Bone secreted factors induce cellular quiescence in prostate cancer cells. Sci Rep. 2019;9(18635) doi: 10.1038/s41598-019-54566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. 2020;20:398–411. doi: 10.1038/s41568-020-0263-0. [DOI] [PubMed] [Google Scholar]
- 138.Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, Yu KJ, Liu B, Creighton CJ, Rodriguez-Canales J, et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFβRIII-p38MAPK-pS249/T252RB pathway. Cancer Res. 2018;78:2911–2924. doi: 10.1158/0008-5472.CAN-17-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Esposito M, Guise T, Kang Y. The biology of bone metastasis. Cold Spring Harb Perspect Med. 2018;8(a031252) doi: 10.1101/cshperspect.a031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, Huang S, Chen X, Lai Y, Du H, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med. 2019;216:428–449. doi: 10.1084/jem.20180661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, Wang J, Zalucha S, Loberg RD, Pienta KJ, Taichman RS. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12:116–127. doi: 10.1593/neo.91384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Singh DK, Patel VG, Oh WK, Aguirre-Ghiso JA. Prostate cancer dormancy and reactivation in bone marrow. J Clin Med. 2021;10(2648) doi: 10.3390/jcm10122648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sosa MS. Dormancy programs as emerging antimetastasis therapeutic alternatives. Mol Cell Oncol. 2015;3(e1029062) doi: 10.1080/23723556.2015.1029062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cackowski FC, Heath EI. Prostate cancer dormancy and recurrence. Cancer Lett. 2022;524:103–108. doi: 10.1016/j.canlet.2021.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cackowski FC, Eber MR, Rhee J, Decker AM, Yumoto K, Berry JE, Lee E, Shiozawa Y, Jung Y, Aguirre-Ghiso JA, Taichman RS. Mer tyrosine kinase regulates disseminated prostate cancer cellular dormancy. J Cell Biochem. 2017;118:891–902. doi: 10.1002/jcb.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang J, Si J, Gan L, Di C, Xie Y, Sun C, Li H, Guo M, Zhang H. Research progress on therapeutic targeting of quiescent cancer cells. Artif Cells Nanomed Biotechnol. 2019;47:2810–2820. doi: 10.1080/21691401.2019.1638793. [DOI] [PubMed] [Google Scholar]
- 147.Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun. 2015;6(6170) doi: 10.1038/ncomms7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Recasens A, Munoz L. Targeting cancer cell dormancy. Trends Pharmacol Sci. 2019;40:128–141. doi: 10.1016/j.tips.2018.12.004. [DOI] [PubMed] [Google Scholar]
- 149.Decker AM, Jung Y, Cackowski FC, Yumoto K, Wang J, Taichman RS. Sympathetic signaling reactivates quiescent disseminated prostate cancer cells in the bone marrow. Mol Cancer Res. 2017;15:1644–1655. doi: 10.1158/1541-7786.MCR-17-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.