

Abstract
We describe a palladium catalyzed C7-acetoxylation of indolines with a range of amide directing groups. While a variety of substituents are tolerated on the indoline-core and the N1-acyl group, the acetoxylation is most sensitive to the C2- and C6-indoline substituents. The practicality of this indoline C7-acetoxylation is demonstrated using a cinnamamide substrate on mmol-scale. Several N1-acyl groups, including those present in natural alkaloids, guide C7-acetoxylation of indoline substrates over a competitive C5-oxidation. The application of this chemistry allowed for the first synthesis of N-benzoylcylindrocarine by late-stage C17-acetoxylation of N-benzoylfendleridine.
Graphical Abstract

Introduction
The indoline substructure is ubiquitous in many bioactive indole alkaloids.1 The aspidosperma family of indole alkaloids includes members of current interest for chemical synthesis given their structural complexity, boasting contiguous stereocenters and varying degrees of oxidation and substitution on a polycyclic core.1,2 Many members of this alkaloid family have a C17–O bond on the indoline substructure (Figure 1A).1b,3,4 Strategies for synthesis of C17-oxygenated aspidosperma alkaloids largely rely on the use of oxygenated starting materials that are converted into the indoline substructure.5 Notably, recent advances in transition metal catalyzed introduction of C–O bonds via arene functionalization6 have not been applied to the synthesis of C17-oxygenated aspidosperma alkaloids. Inspired by the biosynthesis of monoterpene indoline alkaloids wherein the polycyclic core undergoes enzymatic modifications including methylation, acylation, and C–H oxygenation, 7 we sought a chemoselective C17-oxygenation of aspidosperma alkaloids, serving as a platform for late-stage diversification of complex synthetic intermediates.
Figure 1.

A. Representative aspidosperma alkaloids with C17-oxygenation of their N1-acylindoline substructures. B. Evaluation of C7-acetoxylation of indoline substrates with various N1-amides and substitution.
During our total synthesis efforts directed at (+)-haplocidine (1) and (+)-haplocine (2, Figure 1A), we found an opportunity to develop a method for selective metal-catalyzed late-stage C17–O bond formation.8 Our interest in palladium catalysis for this transformation stemmed from its versatility in regioselective C7–H indoline functionalization9 and precedent for palladium-catalyzed regioselective acetoxylation of aryl and bi-aryl systems.10 Alternative contemporary strategies for this transformation required the use of toxic lead or thallium reagents.11 Subsequent to our report8 additional advances in C7-oxygenation of indolines have been reported involving the use of an N-pyrimidyl auxiliary with ruthenium, copper, and rhodium catalysts.12 The prospects of using simple N1-acyl groups already present in targets to guide C7-oxygenation of indolines, without the need for removal of an auxiliary,8 prompted further investigation of the generality of this approach to facilitate its application in complex settings. Herein we describe our findings concerning the palladium catalyzed C7-acetoxylation of indolines guided by a range of N1-acyl groups, including naturally occurring benzoyl, cinnamoyl, and acetyl groups (Figure 1A). We discuss the impact of C2- and C6-indoline substituents and the electron density of the arene, highlight this indoline oxidation on mmol-scale, and report its application in the first synthesis of N-benzoylcylindrocarine (5).
Results and Discussion
The impact of each reaction component on the overall efficiency of the directed acetoxylation was examined using acetamide 7a (Table 1). Starting with our optimized conditions (Pd(OAc)2, PhI(OAc)2, Ac2O–AcOH, 100 °C, O2, 7 h, 66% for 8a),8,13 we note that exclusion of palladium (II) acetate or stoichiometric oxidant led to no desired product and resulted in recovery of starting material. The stoichiometric oxidant alone cannot oxidize the substrate to give the desired C7-acetoxyacetamide 8a but is necessary for the oxidation of palladium during the catalytic cycle.14 We observed no advantage and more decomposition when conducting the reaction under an argon atmosphere rather than dioxygen.15 When acetic anhydride was excluded from the reaction mixture, a lower yield of the C7-acetoxyacetamide 8a (56%) was obtained, consistent with related studies that support its role in facilitating catalytic turnover.10b
Table 1.
Substrate scope for palladium catalyzed acetoxylation of indolines. Conditions: Pd(OAc)2 (15 mol %),PhI(OAc) (2.5 equiv), Ac O–AcOH (1:10, v/v), 100 °C, O, 7–8 h. Yields are average of two independent runs. a See reference 8. b Isolated yield on 1 mmol scale. c C5-acetoxylated regioisomer isolated in 28% yield, see equation 1.

|
We next evaluated various N-substituted indolines under standard conditions for directed acetoxylation. A variety of acyl directing groups are competent such as those with an aliphatic substituent providing the desired C7-acetoxylated product (8a-8f, Table 1). Conjugated amides are also effective directing groups (8g-8h, Table 1) and the C7-acetoxylated cinnamamide product 8g was obtained from the corresponding C7–H cinnamamide substrate on 1 mmol-scale (64%). We then investigated indoline substrates with varying substitution (8i-8m, Table 1). While oxidation of acetamide 7m afforded the desired C7-acetoxylated amide 8m (67%), the oxidation of the corresponding C2 des-methyl substrate was significantly less effective (ca. 40%) and returned starting material. The observed negative impact on the efficiency of C7–H oxidation in the absence of a C2-substituent is consistent with prior report concerning palladium-catalyzed C7-arylation of indolines.16
Indolines with stronger electron-withdrawing substituents at N1 (e.g., trifluoroacetyl, 2,2,2-trichloroethoxycarbonyl, methanesulfonyl, and methyl 2-oxoacetyl) led to minimal acetoxylated product (ca. 5–10%) with diminished C7 vs. C5 regioselectivity. This observation may be attributed to decreased Lewis basicity of the directing group necessary for C7–H metalation.10,13
While the C5-methyl substituted amide 7i efficiently afforded the desired C7-acetoxylated product 8i in 84% yield, the corresponding C5-chloro derivative mostly returned starting material (76%) and provided only 19% yield of the desired C7-acetoxylated product. This observation is consistent with the corresponding calculated highest occupied molecular orbital (HOMO) energy levels for these substrates where an increase in the HOMO energy level affords superior acetoxylation outcome.13 Similarly, the use of a C5-methoxylated variant of amide 7a gave the corresponding C6- and C7-acetoxylated products in 18% and 29% yield, respectively, along with recovered starting material (21%). This is consistent with the greater calculated electron density at C6 compared to C713 allowing competitive regioisomeric acetoxylation despite the directed C7-oxidation rate enhancement.10d
A similar observation for a decrease in selectivity was seen with acetamide 7j. When amide 7j was exposed to standard reaction conditions the desired acetoxylation product 8j was isolated in 31% yield along with the regioisomeric 9j in comparable yield (28%, equation 1). The non-directed C5-acetoxylation leading to product 9j is likely due to both an increased electron density, evidenced by comparable calculated atomic charge densities at C5 and C7,13 and the adverse steric impact of the C6-substituent on the directed formation of the palladacycle intermediate.
 |
(1) |
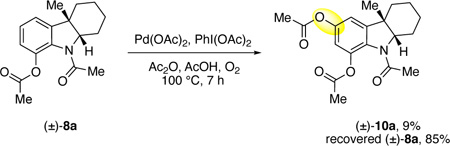
Given the presence of minor double oxidation side products in several cases (ca. 5–15%) we considered if an undesired C5-actoxylation product could lead to the these undesired over oxidation side products. The regioisomeric C5- and C7-acetoxylated acetamides 11a and 8a, respectively (equations 2 and 3), were used to evaluate their propensity for further oxidation. While exposure of amide 8a to our standard acetoxylation conditions led to slow second C5-oxidation to give product 10a in 9% yield along with significant recovery of the starting amide (85%, equation 2), amide 11a progressed faster to the bisacetoxylated product 10a (40%) under identical conditions (equation 3). These results highlight that while the directed C7-acetoxylated products may slowly oxidize further, any undirected C5-acetoxylated side-product can undergo a second oxidation rapidly leading to the minor double oxidation side-products observed in the acetoxylation of many substrates (Table 1).
 |
(2) |
 |
(3) |
Consistent with the observations described above, computational analysis of the C5- and C7-acetoxyacetamides 11a and 8a, respectively, revealed that they both have decreased atomic charge at the respective acetoxylation sites compared to amide 7a,13 leading to an overall attenuated rate of second oxidation (equations 2 and 3). Interestingly, analysis of the N1-amide bond conformers revealed that while acetamide 7a does not have a preference for the s-cis conformation (49.4% s-cis vs. 50.6% s-trans), the optimal conformation for C–H functionalization,8 the C5-acetoxyacetamide 11a preferred the s-cis conformation (74.5% s-cis vs. 25.5% s-trans), likely contributing to its susceptibility toward a second oxidation.8,13
To demonstrate the versatility of this chemistry, we utilized an N1-benzamide directed acetoxylation of a complex substrate for the first total synthesis of N-benzoylcylindrocarine (5, Scheme 1). The synthesis begins by benzoylation of synthetic fendleridine, prepared via our amide electrophilic activation chemistry,8 to give N-benzoylfendleridine (12) in 90% yield. Electrophilic opening of the C19-hemiaminal led to deactivation of the tertiary nitrogen8 and allowed benzamide directed C17-trifluoroacetoxylation which upon methanolysis provided the C17-hyxdroxy benzamide 14 in 63% yield over two steps. Synthesis of the methyl ether (80%) followed by C19-reduction gave the pentacyclic C21-alcohol 16 in 75% over two steps. A Ley Griffith oxidation17 of the C21-primary alcohol 16 afforded the corresponding aldehyde in 50% yield which upon further C21-oxidation under Pinnick conditions18 and methyl ester formation gave N-benzoylcylindrocarine (5) in 44% yield over two steps. All spectroscopic data for our synthetic (±)-N-benzoylcylindrocarine (5) was consistent with the previously reported values.4b
Scheme 1.

Synthesis of (±)-N-benzoylcylindrocarine (5).a
a Reagents and conditions: (a) TFAA, TFA, 60 °C; Pd(OAc)2 (20 mol%), PhI(OAc)2 (4 equiv), 75 °C; (b) Et3N, MeOH, 63% over 2 steps; (c) MeI, Cs2CO3, acetone, 80%; (d) TFAA, 1,2-DCE; NaBH(OMe)3, 0 → 23 °C; Et3N, MeOH, 75% over two steps; (e) TPAP, NMO, CH2Cl2, 50%; (f) NaClO2, NaH2PO4–H2O, resorcinol, H2O, DMSO, 0 °C; TMS-diazomethane, MeOH, PhH, 44% over two steps.
Conclusions
In summary, we describe a regioselective palladium catalyzed C7-acetoxylation of indolines. A variety of N1-amides, including those found in natural alkaloids, serve as an effective directing group for this C–H oxidation reaction. The overall efficiency of this transformation was demonstrated by mmol-scale synthesis of cinnamamide 8g. We also employed experimental and computational methods to correlate the observed oxidation outcome with various factors including substitution, energy and atomic charge calculations, and conformational preferences in the substrate to inform future applications. The versatility of this transformation was demonstrated through application of an N1-benzamide directed C–H oxidation en route to the first total synthesis of N-benzoylcylindrocarine (5).
EXPERIMENTAL SECTION
General Methods.
All palladium catalyzed oxidation reactions were performed in oven-dried or flame-dried modified Schlenk (Kjeldahl shape) flasks unless noted otherwise. The flasks were fitted with Teflon-wrapped glass stoppers, conducted under oxygen atmosphere, and heated in an oil bath. All other reactions were performed in oven-dried or flame-dried round-bottom flasks fitted with rubber septa and were conducted under positive argon pressure using standard Schlenk techniques. Flash column chromatography was performed as described by Still et al.19 using granular silica gel (60 Å pore size, 40−63 µm, 4−6% H2O content). Analytical thin-layer chromatography (TLC) was performed using glass plates precoated with 0.25 mm 230−400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Thin-layer chromatography plates were visualized by exposure to short wave ultraviolet light (254 nm) and irreversibly stained by treatment with an aqueous solution of ceric ammonium molybdate (CAM) followed by heating (1 min) on a hot plate (250 °C). Organic solutions were concentrated at 29−30 °C on rotary evaporators capable of achieving a minimum pressure of 2 Torr and then at 0.5 Torr (vacuum pump) unless otherwise indicated. Proton nuclear magnetic resonance (1H NMR) spectra were recorded with a 600, 500, and 400 MHz spectrometers. Chemical shifts are recorded in parts per million on the δ scale and are referenced from the residual protium in the NMR solvent (CHCl3: δ 7.26, CD2HCN: 1.94). Data are reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant(s) in Hertz, integration, assignment]. Carbon-13 nuclear magnetic resonance (13C) spectra were recorded with 150.9, 125.8, and 100.6 MHz spectrometers. Chemical shifts are recorded in parts per million on the δ scale and are referenced from the carbon resonances of the solvent (CDCl3: δ 77.16, CD3CN: 118.26). Data are reported as follows: chemical shift [assignment]. Infrared data was obtained with an FTIR and are reported as follows: [frequency of absorption (cm−1), intensity of absorption (s = strong, m = medium, w = weak, br = broad), assignment]. High resolution mass spectra (HRMS) was recorded on a Fourier transform ion cyclotron resonance mass spectrometer (FTICR-MS) using electrospray ionization (ESI) or direct analysis in real time (DART) source.
Information for Compounds.
For complete experimental procedures and full characterization data for C7-acetoxylated acetamide (±)-8a, C7-acetoxylated propionamide (±)-8b, C7-acetoxylated pivalamide (±)-8e, and C7-acetoxylated benzamide (±)-8h, please see the Supporting Information of our previous report.8 For complete experimental procedures and full characterization data for all other products reported in Table 1, please see below.
C7-Acetoxylated cyclopropylamide (±)-8c.
Diacetoxyiodobenzene (62.1 mg, 195 µmol, 2.50 equiv), palladium acetate (2.6 mg, 12 µmol, 0.15 equiv), and cyclopropylamide (±)-7c (19.9 mg, 77.9 µmol, 1 equiv) were dissolved in acetic acid (1.70 mL) followed by addition of acetic anhydride (170 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 35% ethyl acetate in hexanes) to give C7-acetoxylated cyclopropylamide (±)-8c (20.5 mg, 84%) as a colorless film. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.13 (app-t, J = 7.6 Hz, 1H, C5H), 6.99 (app-d, J = 7.4, 1.2 Hz, 1H, C4H), 6.94 (app-d, J = 8.1, 1.2 Hz, 1H, C6H), 4.24 (app-q, J = 10.6, 6.4 Hz, 1H, C2H), 2.33–2.26 (m, 1H, C3CHa), 2.25 (s, 3H, C7OC(O)CH3), 2.16–2.05 (m, 1H, C2CHa), 1.76 (app-sept, J = 7.8 Hz, 4.6 Hz, 1H, N1C(O)CH), 1.66–1.52 (m, 3H, C3CHb, C3CH2CHa, C2CH2CHa), 1.42–1.28 (m, 3H, C2CHb, C2CH2CHb), 1.16 (s, 3H, C3CH3), 1.13–1.07 (m, 1H, C3CH2CHb), 1.01–0.74 (m, 3H). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 171.5 (N1C(O)), 168.2 (C7OC(O)), 143.8 (C4a), 140.9 (C7), 132.9 (C7a), 125.6 (C5), 122.3 (C6), 119.2 (C4), 69.9 (C2), 44.8 (C3), 33.0 (C3CH2), 30.2 (C3CH3), 28.9 (C2CH2), 23.2 (C2CH2CH2), 22.1 (C3CH2CH2), 21.1 (C7OC(O)CH3), 13.2 (N1C(O)CH), 8.8 (N1C(O)CHCH2), 8.4 (N1C(O)CHCH2). FTIR (thin film) cm−1: 2926 (m), 2856 (w), 1764 (s), 1652 (s), 1608 (w), 1457 (s), 1409 (s), 1364 (m), 1206 (m), 1173 (s). HRMS (ESI) (m/z): calc’d for C19H24NO3 [M+H]+: 314.1751, found: 314.1745. TLC (25% ethyl acetate in hexanes), Rf: 0.26 (UV, CAM).
C7-Acetoxylated cyclohexylamide (±)-8d.
Diacetoxyiodobenzene (52.8 mg, 164 µmol, 2.50 equiv), palladium acetate (2.2 mg, 9.8 µmol, 0.15 equiv), and cyclohexylamide (±)-7d (19.5 mg, 65.6 µmol, 1 equiv) were dissolved in acetic acid (1.43 ml) followed by addition of acetic anhydride (143 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 35% ethyl acetate in hexanes) to give C7-acetoxylated cyclohexylamide (±)-8d (16.4 mg, 70%) as a colorless film. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.11 (app-t, J = 7.7 Hz, 1H, C5H), 6.95 (m, 2H, C4H, C6H), 3.89 (br-s, 1H, C2H), 2.45 (s, 1H, N1C(O)CH), 2.34–2.26 (m, 1H, C3CHa), 2.24 (s, 3H, C7OC(O)CH3), 2.09–2.00 (m, 1H, C2CHa), 1.88–1.75 (m, 4H, N1C(O)CHCHa, N1C(O)CH-CH2CH2CH2), 1.75–1.51 (m, 7H, C3CHb, C3CH2CHa, C2CH2CHa, N1C(O)CHCHb, N1C(O)CHCH2CHa), 1.42–1.17 (m, 4H, C2CHb, C2CH2CHb, N1C(O)CHCH2CHb), 1.14 (s, 3H, C3CH3), 1.12–1.05 (m, 1H, C3CH2CHb). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 173.3 (N1C(O)), 167.6 (C7OC(O)CH3), 143.5 (C4a), 141.1 (C7), 132.5 (C7a), 125.7 (C5), 122.2 (C6), 118.8 (C4), 69.3 (C2), 44.8 (C4), 43.4 (N1C(O)CH), 33.0 (C2CH2), 30.3 (C3CH3), 30.2 (N1C(O)CHCH2), 29.5 (N1C(O)CHCH2CH2), 29.1 (C2CH2), 25.8 (N1C(O)CHCH2CH2CH2), 23.3 (C2CH2CH2), 22.0 (C3CH2CH2), 21.0 (C7OC(O)CH3). FTIR (thin film) cm−1: 2922 (s), 2853 (s), 1761 (s), 1659 (s), 1447 (s), 1404 (m), 1363 (m), 1261 (m), 1206 (s), 1192 (s), 1174 (s). HRMS (ESI) (m/z): calc’d for C22H30NO3 [M+H]+: 356.2220, found: 356.2230. TLC (25% ethyl acetate in hexanes), Rf: 0.46 (UV, CAM).
C7-Acetoxylated adamantylamide (±)-8f.
Diacetoxyiodobenzene (33.9 mg, 105 µmol, 2.50 equiv), palladium acetate (1.4 mg, 6.3 µmol, 0.15 equiv), and adamantylamide (±)-7f (14.7 mg, 42.1 µmol, 1 equiv) were dissolved in acetic acid (0.92 mL) followed by addition of acetic anhydride (92 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 50% ethyl acetate in hexanes) to give C7-acetoxylated adamantylamide (±)-8f (11.0 mg, 63%) as a white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.12 (app-t, J = 7.7 Hz, 1H, C5H), 6.95 (m, 2H, C4H, C6H), 4.20 (app-q, J = 10.3, 5.7 Hz, 1H, C2H), 2.33–2.25 (app-d, J = 13.5 Hz, 1H, C3CHa), 2.22 (s, 3H, C7OC(O)CH3), 2.21–2.11 (m, 3H, C2CHa, N1C(O)CCH2), 2.11–2.05 (m, 3H, N1C(O)CCH2CH), 2.00–1.92 (m, 4H, N1C(O)CCH2), 1.80–1.73 (m, 6H, N1C(O)CCH2CHCH2), 1.65–1.51 (m, 3H, C2CH2CHa, C3CH2CHa, C3CHb,), 1.31–1.12 (m, 3H, C2CHb, C2CH2Hb, C3CH2CHb), 1.11 (s, 3H, C3CH3). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 176.8 (N1C(O)) 168.0 (C7OC(O)CH3), 143.2 (C4a), 142.6 (C7), 135.6 (C7a), 125.7 (C5), 121.9 (C6), 118.6 (C4), 69.3 (C2), 46.9 (C3), 43.1 (N1C(O)C), 40.0 (N1C(O)CCH2), 36.7 (N1C(O)CCHCH2CH2), 33.1 (C3CH2), 29.5 (C3CH3), 29.0 (C2CH2), 28.6 (N1C(O)CCH2CH), 23.8 (C2CH2CH2), 22.1 (C3CH2CH2), 21.2 (C7OC(O)CH3). FTIR (thin film) cm−1: 2899 (s), 2846 (m), 1766 (s), 1645 (s), 1614 (m), 1300 (m), 1446 (m), 1211 (s), 1175 (s). HRMS (ESI) (m/z): calc’d for C26H34NO3 [M+H]+: 408.2533, found: 408.2522. TLC (25% ethyl acetate in hexanes), Rf: 0.52 (UV, CAM).
C7-Acetoxylated cinnamamide (±)-8g.
Diacetoxyiodobenzene (805 mg, 2.50 mmol, 2.50 equiv), palladium acetate (34 mg, 150 µmol, 0.15 equiv), and cinnamamide (±)-7g (317 mg, 1.00 mmol, 1 equiv) were dissolved in acetic acid (21.9 mL) followed by addition of acetic anhydride (2.19 mL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (15 mL). Saturated aqueous sodium carbonate solution (30 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (30 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 35% ethyl acetate in hexanes) to give C7-acetoxylated cinnamamide (±)-8g (241 mg, 64%) as a yellow solid. Cinnamamide (±)-7g (21.0 mg, 66.1 µmol, 1 equiv) provided C7-acetoxylated cinnamamide (±)-8g (16.7 mg, 67%) as a yellow solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.78 (d, J = 15.5 Hz, 1H, N1C(O)CHCH), 7.56 (app-d, J = 7.5, 2.0 Hz, 2H, CH=CHCHar), 7.38 (m, 3H, CH=CHCHar, CH=CHCHar), 7.16 (app-t, J = 7.7 Hz, 1H, C5H), 7.03 (app-d, J = 7.4, 1.2 Hz, 1H, C4H), 7.00 (app-d, J = 8.1, 1.2 Hz, 1H, C6H), 6.76 (d, J = 15.5 Hz, 1H, N1C(O)CH), 4.29 (br-s, 1H, C2H), 2.33–2.24 (m, 1H, C3CHa), 2.24–2.16 (m, 1H, C2CHa), 2.10 (s, 3H, C7OC(O)CH3), 1.68–1.51 (m, 3H, C3CHb, C3CH2CHa, C2CH2CHa), 1.37–1.19 (m, 2H, C2CHb, C2CH2CHb), 1.16 (s, 3H, C3CH3), 1.14–1.06 (m, 1H, C3CH2CHb). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 168.4 (C7OC(O)CH3), 164.2 (N1C(O)), 144.4 (C4a), 142.7 (N1C(O)CHCH), 140.6 (C7), 135.1 (CH=CHCar), 132.5 (C7a), 130.0 (CH=CHCar), 129.0 (CH=CHCar), 129.0 (CH=CHCar), 125.9 (C5), 122.4 (C6), 119.6 (C4), 119.4 (N1C(O)CH), 70.4 (C2), 44.8 (C3), 33.0 (C3CH2), 30.0 (C3CH3), 28.7 (C2CH2), 23.1 (C2CH2CH2), 22.1 (C3CH2CH2), 21.0 (C7OC(O)CH3). FTIR (thin film) cm−1: 2923 (m), 2854 (m), 1759 (m), 1652 (s), 1616 (m), 1448 (s), 1397 (s), 1253 (w), 1206 (s), 1177 (s). HRMS (ESI) (m/z): calc’d for C24H26NO3 [M+H]+: 376.1907, found: 376.1905. TLC (25% ethyl acetate in hexanes), Rf: 0.33 (UV, CAM).
C7-Acetoxylated acetamide (±)-8i.
Diacetoxyiodobenzene (67.5 mg, 0.210 mmol, 2.50 equiv), palladium acetate (2.8 mg, 13 µmol, 0.15 equiv), and acetamide (±)-7i (20.4 mg, 83.0 µmol, 1 equiv) were dissolved in acetic acid (1.83 mL) followed by addition of acetic anhydride (183 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 8 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 40% ethyl acetate in hexanes) to give C7-acetoxylated acetamide (±)-8i (20.6 mg, 81%) as an off-white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 45 °C): δ 6.78 (app-s, 1H, C4H), 6.75 (app-s, 1H, C6H), 3.96 (br-s, 1H, C2H), 2.33 (s, 3H, C5CH3), 2.26 (s, 3H, C7OC(O)CH3), 2.25–2.21 (m, 1H, C3CHa), 2.20 (s, 3H, N1C(O)CH3), 2.08–1.99 (m, 1H, C2CHa), 1.65–1.49 (m, 3H, C3CHb, C3CH2CHa, C2CH2CHa), 1.36–1.15 (m, 2H, C2CHb, C2CH2CHb), 1.14 (s, 3H, C3CH3), 1.13–1.04 (m, 1H, C3CH2CHb). 13C NMR (125.8 MHz, CDCl3, 45 °C): δ 168.0 (2C, N1C(O), C7OC(O)CH3), 143.6 (C4a), 140.6 (C7), 136.1 (C5), 130.3 (C7a), 122.7 (C6), 120.0 (C4), 70.6 (C2), 44.7 (C3), 33.0 (C3CH2), 30.2 (C3CH3), 28.5 (C2CH2), 23.2 (C2CH2CH2), 22.7 (N1C(O)CH3), 22.1 (C3CH2CH2), 21.2 (C5CH3 or C7OC(O)CH3), 21.1 (C5CH3 or C7OC(O)CH3). FTIR (thin film) cm−1: 2927 (s), 2858 (m), 1770 (s), 1662 (s), 1476 (m), 1393 (m), 1364 (m), 1202 (s), 1185 (w), 1137 (m). HRMS (ESI) (m/z): calc’d for C18H24NO3 [M+H]+: 302.1751, found: 302.1744. TLC (25% ethyl acetate in hexanes), Rf: 0.11 (UV, CAM).
C7-Acetoxylated acetamide (±)-8j and C5-acetoxylated acetamide (±)-9j.
Diacetoxyiodobenzene (48.2 mg, 0.150 mmol, 2.50 equiv), palladium acetate (2.0 mg, 9.0 µmol, 0.15 equiv), and acetamide (±)-7j (15.4 mg, 59.8 µmol, 1 equiv) were dissolved in acetic acid (1.31 mL) followed by addition of acetic anhydride (131 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 1 → 15% acetone in dichloromethane) to give C7-acetoxylated acetamide (±)-8j (6.0 mg, 31%) as a colorless film and C5-acetoxylated acetamide (±)-9j (5.3 mg, 28%) as a colorless film. Structural assignments were made using additional information from gCOSY, gHSQC, gHMBC and NOESY experiments. 1H NMR (400 MHz, CDCl3, 50 °C): δ 6.73 (s, 1H, C5H), 3.93 (m, 1H, C2H), 2.55 (app-d, J = 14.6, 4.3, 2.6 Hz, 1H, C3CHa), 2.29 (s, 3H, C4CH3), 2.26 (s, 3H, C7OC(O)CH3), 2.19 (s, 3H, N1C(O)CH3), 2.16 (s, 3H, C6CH3), 2.02–1.92 (m, 1H, C2CHa), 1.67–1.53 (m, 2H, C2CH2CHa, C3CH2CHa), 1.52–1.42 (m, 1H, C3CHb), 1.42–1.20 (m, 2H, C2CHb, C2CH2CHb), 1.18 (s, 3H, C3CH3), 1.17–1.08 (m, 1H, C3CH2CHb). 13C NMR (100.6 MHz, CDCl3, 50 °C): δ 168.2 (N1C(O)), 167.7 (C7OC(O)CH3), 138.5 (C7), 137.5 (C4a), 134.1 (C4 or C6), 131.0 (C4 or C6), 130.4 (2C, C5, C7a), 71.0 (C2), 47.0 (C3), 33.5 (C3CH2), 28.2 (C2CH3), 27.3 (C3CH3), 23.4 (C2CH2CH2), 22.8 (N1C(O)CH3), 22.0 (C3CH2CH2), 20.8 (C7OC(O)CH3), 18.7 (C4CH3), 16.5 (C6CH3). FTIR (thin film) cm−1: 2957 (m), 2924 (s), 2856 (m), 1760 (s), 1669 (s), 1415 (s), 1369 (s), 1304 (m), 1266 (w), 1184 (s). HRMS (ESI) (m/z): calc’d for C19H26NO3 [M+H]+: 316.1907, found: 316.1911. TLC (10% acetone in dichloromethane), Rf: 0.44 (UV, CAM).
C5-Acetoxylated acetamide (±)-9j.
Structural assignments were made using additional information from gCOSY, gHSQC, gHMBC and NOESY experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.94 (br-s, 1H, C7H), 3.68 (br-s, 1H, C2H), 2.53–2.42 (m, 1H, C2CHa), 2.33 (s, 3H, C5OC(O)CH3), 2.25 (br-s, 3H, N1C(O)CH3), 2.12 (s, 3H, C6CH3), 2.11 (s, 3H, C4CH3), 1.95 (s, 1H, C2CHa), 1.65–1.48 (m, 3H, C3CHb, C2CH2CHa, C3CH2CHa), 1.38–1.21 (m, 2 H, C2CH2CHb, C3CH2CHb), 1.19 (s, 3H, C3CH3). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 169.2 (C5OC(O)CH3), 168.1 (N1C(O)), 144.6 (C5), 139.0 (C7a), 134.5 (C4a), 129.0 (C6), 126.2 (C4), 118.3 (C7), 69.1 (C2), 46.2 (C3), 33.1 (C3CH2), 28.5 (C2CH2), 28.5 (C3CH3), 23.5 (N1C(O)CH3), 22.5 (C2CH2CH2 or C3CH2CH2), 21.4 (C2CH2CH2 or C3CH2CH2), 20.6 (C5OC(O)CH3), 17.1 (C6CH3), 11.9 (C4CH3). FTIR (thin film) cm−1: 2927 (m), 2857 (m), 1759 (s), 1657 (s), 1470 (m), 1448 (m), 1415 (m), 1371 (m), 1309 (m), 1215 (m). HRMS (ESI) (m/z): calc’d for C19H26NO3 [M+H]+: 316.1907, found: 316.1893. TLC (10% acetone in dichloromethane), Rf: 0.50 (UV, CAM).
C7-Acetoxylated acetamide (±)-8k.
Diacetoxyiodobenzene (44.9 mg, 0.140 mmol, 2.50 equiv), palladium acetate (1.9 mg, 8.4 µmol, 0.15 equiv), and acetamide (±)-7k (15.6 mg, 55.8 µmol, 1 equiv) were dissolved in acetic acid (1.22 mL) followed by addition of acetic anhydride (122 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 1 → 12% acetone in dichloromethane) to give C7-acetoxylated acetamide (±)-8k (9.6 mg, 51%) as tan solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 8.05 (app-d, J = 8.4, 1.3 Hz, 1H, C4CH), 7.81 (app-d, J = 7.8, 1.7 Hz, 1H, C5CH), 7.50 (s, 1H, C6H), 7.41 (m, 2H, C4CHCH, C5CHCH), 4.04 (br-s, 1H, C2H), 2.84 (d, J = 14.5 Hz, 1H, C3CHa), 2.32 (s, 3H, C7OC(O)CH3), 2.26 (s, 3H, N1C(O)CH3), 2.07–1.99 (m, 1H, C2CHa), 1.80 (app-t, J = 14.6, 11.9, 3.0 Hz, 1H, C3CHb), 1.67–1.44 (m, 3H, C2CHb, C2CH2CHa, C3CH2CHa), 1.40 (s, 3H, C3CH3), 1.39–1.32 (m, 1H, C2CH2CHb), 1.23–1.09 (m, 1H, C3CH2CHb). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 168.7 (2C, N1C(O), C7OC(O)CH3), 140.2 (C7), 136.5 (C4a), 133.2 (C7a), 132.9 (C4), 129.1 (C5CH), 128.3 (C5), 126.2 (C5CHCH or C4CHCH), 125.1 (C5CHCH or C4CHCH), 122.4 (C4CH), 121.0 (C6), 71.1 (C2), 48.1 (C3), 34.2 (C3CH2), 27.7 (C2CH2), 27.6 (C3CH3), 22.9 (N1C(O)CH3), 22.7 (C2CH2CH2), 21.4 (C7OC(O)CH3), 21.3 (C3CH2CH2). FTIR (thin film) cm−1: 2933 (m), 2863 (w), 1765 (s), 1664 (s), 1446 (m), 1395 (s), 1368 (m), 1338 (w), 1305 (m), 1194 (s). HRMS (ESI) (m/z): calc’d for C21H24NO3 [M+H]+: 338.1751, found: 338.1748. TLC (10% acetone in dichloromethane), Rf: 0.56 (UV, CAM).
C7-Acetoxylated acetamide (±)-8l.
Diacetoxyiodobenzene (59.5 mg, 185 µmol, 2.50 equiv), palladium acetate (2.5 mg, 11 µmol, 0.15 equiv), and acetamide (±)-7l (17.1 mg, 73.9 µmol, 1 equiv) were dissolved in acetic acid (1.61 mL) followed by addition of acetic anhydride (161 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 35% ethyl acetate in hexanes) to give C7-acetoxylated acetamide (±)-8l (15.5 mg, 72%) as a yellow oil. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 50 °C): δ 7.11 (app-t, J = 8.2, 7.3 Hz, 1H, C5H), 6.96 (app-d, J = 9.8, 7.8, 1.2 Hz, 2H, C4H, C6H), 4.12 (br-s, 1H, C2H), 2.29 (s, 3H, C7OC(O)CH3), 2.24 (s, 3H, N1C(O)CH3), 1.97 (app-t, J = 6.8, 3.8 Hz, 1H, C2CH), 1.40 (s, 3H, C3CH3), 1.19 (s, 3H, C3CH3), 1.06 (d, J = 6.8 Hz, 3H, C2CHCH3), 0.57 (d, J = 6.8 Hz, 3H, C2CHCH3). 13C NMR (100.6 MHz, CDCl3, 50 °C): δ 171.0 (N1C(O)), 168.2 (C7OC(O)CH3), 146.8 (C4a), 140.4 (C7), 134.3 (C7a), 126.2 (C5), 122.6 (C6), 118.6 (C4), 79.2 (C2), 44.6 (C3), 31.1 (C3CH3), 29.9 (C2CH), 22.7 (N1C(O)CH3), 21.9 (C2CHCH3), 21.2 (C7OC(O)CH3), 20.3 (C3CH3), 18.0 (C2CHCH3). FTIR (thin film) cm−1: 2962 (m), 1769 (s), 1662 (s), 1460 (m), 1369 (s), 1297 (m), 1247 (m), 1207 (m), 1179 (s), 1012 (w). HRMS (ESI) (m/z): calc’d for C17H24NO3 [M+H]+: 290.1751, found: 290.1749.
C7-Acetoxylated acetamide (±)-8m.
Diacetoxyiodobenzene (64.9 mg, 202 µmol, 2.50 equiv), palladium acetate (2.7 mg, 12 µmol, 0.15 equiv), and acetamide (±)-7m (19.7 mg, 80.6 µmol, 1 equiv) were dissolved in acetic acid (1.76 mL) followed by addition of acetic anhydride (176 µL) at 23 °C. The reaction was sparged with dioxygen for 10 minutes, sealed with a Teflon wrapped glass stopper, and heated to 100 °C. After 7 h, the reaction mixture was cooled to 23 °C and diluted with ethyl acetate (5 mL). Saturated aqueous sodium carbonate solution (10 mL) was carefully added. The resulting mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 10 → 35% ethyl acetate in hexanes) to give C7-acetoxylated acetamide (±)-8m (15.5 mg, 64%) as a white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (400 MHz, CDCl3, 25 °C): δ 7.11 (app-t, J = 7.8 Hz, 1H, C5H), 7.01 (app-d, J = 7.4 Hz, 1H, C4H), 6.94 (app-d, J = 8.1 Hz, 1H, C6H), 4.39 (br-s, 1H, C2H), 2.27 (s, 3H, C7OC(O)CH3), 2.23 (s, 3H, N1C(O)CH3), 1.87–1.56 (m, 4H), 1.56–1.41 (m, 2H), 1.41–1.22 (m, 4H), 1.18 (d, J = 6.5 Hz, 3H, C2CH3). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 168.1 (2C, N1C(O), C7OC(O)CH3), 144.8 (C4a), 140.3 (C7), 131.8 (C7a), 125.9 (C5), 122.5 (C6), 120.0 (C4), 64.9 (C2), 48.0 (C3), 36.9 (C3CH2), 29.2 (C3CH2), 25.7 (C3CH2CH2CH2), 23.5 (C3CH2CH2), 23.4 (C3CH2CH2), 22.8 (N1C(O)CH3), 21.1 (C7OC(O)CH3), 14.9 (C2CH3). FTIR (thin film) cm−1: 2921 (m), 2842 (w), 1754 (s), 1663 (s), 1456 (m), 1400 (m), 1310 (m), 1265 (w), 1212 (s), 1192 (s). HRMS (ESI) (m/z): calc’d for C18H24NO3 [M+H]+: 302.1751, found: 302.1738. TLC (25% ethyl acetate in hexanes), Rf: 0.43 (UV, CAM).
C5,C7-Diacetoxylated acetamide (±)-10a:
Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (600 MHz, CDCl3, 25 °C): δ 6.77 (d, J = 2.2 Hz, 1H, C4H), 6.75 (d, J = 2.2 Hz, 1H, C6H), 3.95 (br-s, 1H, C2H), 2.27 (s, 3H, C5OC(O)CH3 or C7OC(O)CH3), 2.26 (s, 3H, C5OC(O)CH3 or C7OC(O)CH3), 2.21 (s, 3H, N1C(O)CH3), 2.19–2.17 (m, 1H, C3CHa), 2.09–2.02 (m, 1H, C2CHa), 1.66–1.60 (app-d, 1H, C2CH2CHa), 1.59–1.51 (m, 2H, C3CHb,C3CH2CHa), 1.36–1.17(m, 2H, C2CHb, C2CH2CHb), 1.16 (s, 3H, C3CH3), 1.15–1.07 (m, 1H, C3CH2CHb). 13C NMR (150.9 MHz, CDCl3, 25 °C): δ 169.2 (C5OC(O)CH3 or C7OC(O)CH3), 167.6 (2C, N1C(O), C5OC(O)CH3 or C7OC(O)CH3), 148.3 (C5 or C7), 144.4 (C4a), 140.8 (C5 or C7), 130.2 (C7a), 115.6 (C6), 112.9 (C4), 70.6 (C2), 44.9 (C3), 32.9 (C3CH2), 30.0 (C3CH3), 28.5 (C2CH2), 23.1 (C2CH2CH2), 22.8 (N1C(O)CH3), 22.0 (C3CH2CH2), 21.3 (C5OC(O)CH3 or C7OC(O)CH3), 21.0 (C5OC(O)CH3 or C7OC(O)CH3). FTIR (thin film) cm−1: 2926 (m), 2857 (w), 1769 (m), 1666 (m), 1614 (w), 1468 (m), 1397 (m), 1367 (m), 1198 (s). HRMS (ESI) (m/z): calc’d for C19H24NO5 [M+H]+: 346.1649, found: 346.1652. TLC (50% ethyl acetate in hexane), Rf: 0.30 (UV, CAM).
C17-Hydroxy benzamide (±)-14.
(±)-N-Benzoylfendleridine8 (12, 4.7 mg, 12 µmol, 1 equiv) was dissolved in trifluoroacetic anhydride (33 µL) and trifluoroacetic acid (330 µL) in a pressure tube. The reaction was sealed and heated to 60 °C. After 4 h, the reaction mixture was cooled to 23 °C. Palladium acetate (0.5 mg, 2 µmol, 0.2 equiv) and diacetoxyiodobenzene (15 mg, 47 µmol, 4.0 equiv) were added. The reaction vessel was sealed and heated to 75 °C. After 2 h, the reaction was concentrated under reduced pressure and subsequently dissolved in methanol (0.65 mL) and triethylamine (130 µL). After 12 h, the reaction was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 3% triethylamine and 0 → 30% ethyl acetate in hexanes) to give C17-hydroxy benzamide (±)-14 (3.1 mg, 63%) as a white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (500 MHz, CDCl3, 25 °C): δ 10.18 (s, 1H, OH), 7.54–7.48 (m, 5H, N1C(O)C6H5), 7.20 (app-d, J = 7.6, 1.3 Hz, 1H, C14H), 7.15 (t, J = 7.8 Hz, 1H, C15H), 6.91 (app-d, J = 8.1, 1.2 Hz, 1H, C16H), 4.16 (t, J = 8.5 Hz, 1H, C21Ha), 4.06 (app-q, J = 10.9, 8.2, 6.2 Hz, 1H, C21Hb), 3.73 (app-d, J = 11.2, 5.2 Hz, 1H, C2H), 2.87 (t, J = 7.5 Hz, 2H, C10H2), 2.71 (app-t, J = 11.6, 2.8 Hz, 1H, C8Ha), 2.52 (app-d, J = 11.3, 2.7, 2.1 Hz, 1H, C8Hb), 2.10–2.01 (m, 1H, C11Ha), 1.94–1.82 (m, 3H, C3Ha, C11Hb, C20Ha), 1.82–1.74 (m, J = 13.5, 4.1 Hz, 1H, C3Hb), 1.70–1.56 (m, 3H, C4Ha, C6Ha, C7Ha), 1.48–1.42 (m, 1H, C7Hb), 1.35–1.24 (m, 2H, C4Hb, C6Hb), 1.21 (app-q, J = 12.0, 6.1 Hz, 1H, C20Ha). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 170.2 (N1C(O)), 147.3(C17), 141.4 (C13), 136.3 (N1C(O)C6H5), 130.4 (N1C(O)C6H5), 129.1 (N1C(O)C6H5), 128.9 (C15), 127.9 (C18), 126.3 (N1C(O)C6H5), 118.2 (C16), 116.1 (C14), 101.9 (C19), 70.8 (C2), 65.3 (C21), 58.7 (C12), 48.8 (C10), 43.8 (C8), 39.8 (C5), 35.6 (C11), 34.6 (C20), 32.9 (C6), 26.4 (C4), 25.7 (C3), 20.9 (C7). FTIR (thin film) cm−1: 2937 (br-s), 2861 (m), 2832 (m), 2760 (m), 1620 (s), 1598 (s), 1566 (s), 1495 (m), 1465 (s), 1445 (s), 1428 (s). HRMS (ESI) (m/z): calc’d for C26H29N2O3 [M+H]+: 417.2173, found: 417.2176. TLC (3% triethylamine and 30% ethyl acetate in hexanes), Rf: 0.38 (UV, CAM).
C17-Methyl ether (±)-15.
Cesium carbonate (58 mg, 0.17 mmol, 10 equiv) and iodomethane (10 µL, 0.2 mmol, 10 equiv) were added to a solution of C17-hydroxy benzamide (±)-14 (6.9 mg, 17 µmol, 1 equiv) in acetone (530 µL) at 23 °C. After 3 h, a solution of saturated aqueous ammonium chloride (5 mL) and dichloromethane (5 mL) were added and the layers were separated. The aqueous layer was extracted with dichloromethane (2 × 5 mL) and the combined organic layers were dried with anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 3% triethylamine and 5 → 25% ethyl acetate in hexanes) to give C17-methyl ether (±)-15 (5.7 mg, 80%) as a white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (500 MHz, CDCl3, 25 °C): δ 7.52–7.42 (m, 2H, N1C(O)C6H5), 7.39 (t, J = 7.4 Hz, 1H, N1C(O)C6H5), 7.35–7.26 (m, 3H, C14H, N1C(O)C6H5), 7.05 (t, J = 7.9 Hz, 1H, C15H), 6.62 (d, J = 8.1 Hz, 1H, C16H), 4.22 (br-s, 1H, C2H), 4.17–4.03 (m, 2H, C21H2), 3.25 (br-s, 3H, OCH3), 3.04–2.88 (m, 1H, C10H2), 2.77 (t, J = 11.1 Hz, 1H, C8Ha), 2.61 (d, J = 11.2 Hz, 1H, C8Hb), 2.23 (br-s, 1H, C11Ha), 2.16–2.07 (m, 1H, C3Ha), 1.99–1.83 (m, 3H, C4Ha, C11Hb, C20Ha), 1.79–1.66 (m, 3H, C3Hb, C6Ha, C7Ha), 1.49 (m, 1H, C7Hb), 1.44–1.29 (m, 2H,C4Hb, C6Hb), 1.21 (app-d, J = 12.3, 6.1 Hz, 1H, C20Hb). 13C NMR (125.8 MHz, CDCl3, 25 °C): δ 169.7 (N1C(O)), 149.1 (C17), 141.8 (C13), 137.7 (N1C(O)C6H5), 130.5 (N1C(O)C6H5), 130.3 (C18), 128.1 (N1C(O)C6H5), 127.2 (N1C(O)C6H5), 126.4 (C15), 117.3 (C14), 111.0 (C16), 102.3 (C19), 70.9 (C2), 65.2 (C21), 59.8 (C12), 54.8 (OCH3), 49.0 (C10), 43.9 (C8), 39.8 (C5), 35.1 (C11), 34.7 (C20), 33.1 (C6), 26.5 (C4), 24.5 (C3), 21.1 (C7). FTIR (thin film) cm−1: 2936 (s), 2834 (m), 1634 (s), 1579 (m), 1487 (s), 1454 (s), 1384 (s), 1338 (m), 1295 (m). HRMS (DART) (m/z): calc’d for C27H31N2O3 [M+H]+: 431.2329, found: 431.2340. TLC (3% triethylamine and 40% ethyl acetate in hexanes), Rf: 0.47 (UV, CAM).
C21-Alcohol (±)-16.
C17-methyl ether (±)-15 (36.9 mg, 0.100 mmol, 1 equiv) was azeotropically dried by concentration from anhydrous benzene (3 × 200 µL) and the residue was dissolved in 1,2-dichloroethane (0.75 mL). Trifluoroacetic anhydride (0.7 M in 1,2-dichloroethane, 50 µL, 40 µmol, 5 equiv) was added dropwise at 23 °C. After 1 h, the reaction mixture was cooled to 0 °C and sodium trimethoxyborohydride (9.8 mg, 77 µmol, 10 equiv) was added as a solid. After 5 min, the reaction was warmed to 23 °C. After 30 min, a solution of saturated aqueous sodium bicarbonate (5 mL) and dichloromethane (5 mL) were added and the layers were separated. The aqueous layer was extracted with dichloromethane (3 × 5 mL) and the combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The residue was used directly in the next step. Triethylamine (150 µL) was added to a solution of the residue in methanol (0.75 mL) at 23 °C. After 1 h, the reaction was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 3% triethylamine and 10 → 50% ethyl acetate in hexanes) to give C21-alcohol (±)-16 (2.5 mg, 75%) as a white solid. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (600 MHz, CD3CN, 60 °C): δ 7.46 (d, J = 7.4 Hz, 3H, N1C(O)CCH, N1C(O)CCHCHCH), 7.39 (app-t, J = 8.3, 7.0 Hz, 2H, N1C(O)CCHCH), 7.09 (t, J = 7.9 Hz, 1H, C15H), 6.94 (app-d, J = 7.5, 1.0 Hz, 1H, C14H), 6.75 (d, J = 8.2 Hz, 1H, C16H), 4.13 (app-q, J = 11.4, 5.8 Hz, 1H, C2H), 3.51–3.44 (m, 1H, C21Ha), 3.44–3.37 (m, 1H, C21Ha), 3.35 (s, 3H, C17OCH3), 3.02 (app-t, J = 9.0, 3.5 Hz, 1H, C10Ha), 2.96 (app-d, J = 10.9, 4.0, 1.8 Hz, 1H, C8Ha), 2.35 (s, 1H, C19H), 2.26 (app-q, J = 10.6, 8.9, 7.3 Hz, 1H, C10Hb), 2.15–2.06 (m, 2H, C3Ha, C11Ha), 2.03–1.95 (m, 1H, C4Ha, C8Hb,), 1.75–1.54 (m, 4H, C3Hb, C6Ha, C7Ha, C11Hb), 1.53–1.42 (m, 2H, C7Hb, C20Hb), 1.24 (app-t, J = 13.6, 4.7 Hz, 1H, C6Hb), 1.14–1.05 (m, 2H, C4Hb, C20Hb). 13C NMR (150.9 MHz, CD3CN, 60 °C): δ 171.1 (N1C(O)), 151.1 (C17), 143.7 (C13), 139.1 (N1C(O)C), 131.6 (1C or 2C, N1C(O)CCH or N1C(O)CCHCHCH), 131.4 (C18), 129.3 ((N1C(O)CCHCH), 128.1 (1C or 2C, N1C(O)CCH or N1C(O)CCHCHCH), 127.4 (C15), 116.8 (C14), 112.7 (C16), 72.1 (C2), 71.7 (C19), 58.3 (C21), 55.9 (C17OCH3), 55.6 (C12), 54.4 (C8), 53.1 (C10), 41.6 (C20), 38.7 (C11), 36.4 (C5), 36.2 (C6), 26.6 (C3), 25.3 (C4), 22.6 (C7). FTIR (thin film) cm−1: 3402 (br-s), 2932 (s), 2868 (m), 2783 (m), 2725 (w), 1620 (s), 1578 (m), 1489 (s), 1458 (s), 1385 (s), 1323 (m), 1274 HRMS (DART) (m/z): calc’d for C27H33N2O3 [M+H]+: 433.2486, found:433.2492. TLC (3% triethylamine and 60% ethyl acetate in hexanes), Rf: 0.34 (UV, CAM).
(±)-N-Benzoylcylindrocarine (5).
C21-alcohol (±)-16 (6.8 mg, 16 µmol, 1 equiv) was azeotropically dried by concentration from anhydrous benzene (3 × 200 µL) and the residue was dissolved in dichloromethane (1.70 mL). N-Methylmorpholine N-oxide (NMO, 5.4 mg, 47 µmol, 3.0 equiv) was added followed by tetrapropylammonium perruthenate (TPAP, 0.1 M in dichloromethane, 40 µL, 2 µmol, 0.1 equiv) at 23 °C. After 20 min, the reaction mixture was diluted with dichloromethane (10 mL) and filtered through a pad of celite. The filter cake was rinsed with dichloromethane (2 × 10 mL) and the combined organic liquid was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 3% triethylamine and 10 → 40% ethyl acetate in hexanes) to give C21-aldehyde S1 (3.4 mg, 50%) as a thin film.
C21-aldehyde S1 (3.4 mg, 7.9 µmol, 1 equiv) was dissolved in dimethylsulfoxide (DMSO) (1.30 mL) and water (0.10 mL) then cooled to 0 °C. Resorcinol (4.3 mg, 39 µmol, 5.0 equiv) was added followed by a solution of sodium phosphate monobasic (NaH2PO4–H2O, 5.4 mg, 39 µmol, 5.0 equiv) in water (50 µL). Sodium chlorite (NaClO2, 3.6 mg, 39 µmol, 5.0 equiv) in water (50 µL) was added dropwise at 0 °C. After 10 min, a solution of saturated aqueous ammonium chloride (5 mL) and diethyl ether (5 mL) were added and warmed to 23 °C. The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined organic extracts were further washed with saturated aqueous sodium chloride solution (10 mL), dried with anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The yellow residue was used directly in the next step.
The yellow residue was dissolved in methanol (260 µL) and benzene (1.0 mL). Trimethylsilyldiazomethane20 (TMS-diazomethane, 2 M in hexanes, 40 µL, 80 µmol, 10 equiv) was added at 23 °C. After 30 min, the reaction mixture was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography on silica gel (eluent: 3% triethylamine and 0 → 30% ethyl acetate in hexanes) to give (±)-N-benzoylcylindrocarine (5) (1.6 mg, 44%) as a thin film. Structural assignments were made using additional information from gCOSY, gHSQC, and gHMBC experiments. 1H NMR (600 MHz, CD3CN, 60 °C): δ 7.50–7.44 (m, 3H, N1C(O)CCHCHCH, N1C(O)CCH), 7.39 (app-t, J = 8.3, 6.9 Hz, 2H, N1C(O)CCHCH), 7.09 (t, J = 7.9 Hz, 1H, C15H), 6.94 (d, J = 8.5 Hz, 1H, C14H), 6.75 (d, J = 8.2 Hz, 1H, C16H), 4.15 (app-q, J = 11.4, 6.0 Hz, 1H, C2H), 3.53 (s, 3H, C21OCH3), 3.34 (s, 3H, C17OCH3), 3.05 (app-t, J = 9.1, 3.6 Hz, 1H, C10Ha), 2.97 (app-d, J = 12.8, 4.0, 1.8 Hz, 1H, C8Ha), 2.52 (s, 1H, C19H), 2.30 (app-q, J = 10.8, 8.9, 7.0 Hz, 1H, C10Hb), 2.20 (d, J = 14.3 Hz, 1H, C20Ha), 2.18–2.08 (m, 3H, C3Ha, C4Ha, C11Ha, ), 2.02–1.98 (m, 1H, C8Hb), 1.96–1.92 (m, 1H, C20Hb),21 1.77–1.55 (m, 4H, C3Hb, C6Ha, C7Ha, C11Hb), 1.49 (app-d, J = 13.2, 4.9, 2.4 Hz, 1H, C7Hb), 1.42–1.28 (m, 1H, C6Hb), 1.28–1.22 (m, 1H, C4Hb). 13C NMR (150.9 MHz, CD3CN, 60 °C): δ 172.8 (C21), 171.0 (N1C(O)), 151.2 (C17), 142.9 (C13), 139.0 (N1C(O)C), 131.6 (1C or 2C, N1C(O)CCH or N1C(O)CCHCHCH), 131.5 (C18), 129.4 (2C, N1C(O)CCHCH), 128.1 (1C or 2C, N1C(O)CCH or N1C(O)CCHCHCH), 127.5 (C15), 116.6 (C14), 113.0 (C16), 71.6 (C2), 70.4 (C19), 56.0 (C17OCH3), 55.5 (C12), 54.2 (C8), 53.0 (C10), 51.7 (C21OCH3), 43.2 (C20), 38.3 (C11), 37.1 (C5), 35.9 (C6), 26.5 (C3), 25.4 (C4), 22.6 (C7). FTIR (thin film) cm−1: 2943 (s), 2871 (w), 1733 (s), 1636 (s), 1579 (w), 1489 (s), 1459 (s), 1386 (s), 1334 (m), 1277 (m), 1204 (m), 1157 (m), 1042 (w), 922 (w), 889 (w), 793 (m), 782 (m), 720 (m), 703 (m).22 HRMS (DART) (m/z): calc’d for C28H33N2O4 [M+H]+: 461.2435, found: 461.2431. TLC (3% triethylamine and 40% ethyl acetate in hexanes), Rf: 0.37 (UV, CAM).
Energy and Atomic Charge Calculations.
The geometries (gas phase) in the ground state of indoline amides (6–10 depending on substrate) were optimized with Merck Molecular Force Field (MMFF)23 followed by density functional theory at B3LYP level with 6–311+g(d,p) as basis set (Gaussian09, by Gaussian, Inc.).24 The electrostatic potential (ESP) derived charge were calculated using the lowest energy s-cis conformation. The total percent s-cis and s-trans were calculated using the Boltzmann distribution to determine the average contribution of each conformation (6–8 depending on substrate) at 23 °C and the percent s-cis was added up independently to s-trans.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support by NIH-NIGMS (GM-074825 and GM-141963). We thank the NSF CCI Center for Selective C–H Functionalization (CHE-1700982). K.M.F. acknowledges an MIT Dean of Science graduate fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Copies of 1H and 13C NMR spectra of all compounds described in the experimental section. Coordinates and computational data of compounds discussed are described in the supporting information.
REFERENCES
- (1).(a) Siegler DS Indole Alkaloids. In Plant Secondary Metabolism; Springer Science+Business: New York, 1998; pp 628–654. [Google Scholar]; (b) Saxton JE Alkaloids of the Aspidospermine Group. In the Alkaloids, Chemistry and Biology; Cordell GA, Ed.; Academic Press: San Diego, 1998; Vol. 51, pp 1–197. [Google Scholar]; (c) O’Connor SE Alkaloids. In Comprehensive Natural Products II; Mander L, Liu H-W, Eds.; Elsevier: Amsterdam, 2010; Vol. 1, pp 977–1007. [Google Scholar]
- (2).(a) Lopchuk JM Recent Advances in the Synthesis of Aspidosperma-Type Alkaloids. In Progress in Heterocyclic Chemistry; Gribbe GW, Joule JA, Eds.; Elsevier: Amsterdam, 2011; Vol. 23, pp 1–25. [Google Scholar]; (b) Saya JM; Ruijter E; Orru RV Total Synthesis of Aspidosperma and Strychnos Alkaloids Through Indole Dearomatization. Chem. Eur. J 2019, 25, 8916–8935. [DOI] [PubMed] [Google Scholar]
- (3).(a) Cava MP; Talapatra SK; Nomura K; Weisbach JA; Douglas B; Shoop EC Haplocine and Haplocidine: New Aspidospermine-Type Akaloids from Haplophyton cimicidum. Chem. and Ind 1963, 1242–1243. [Google Scholar]; (b) Cava MP; Nomura K; Talapatra SK The Stereochemistry of Limaspermine, Haplocine and Haplocidine. Tetrahedron 1964, 20, 581–583. [Google Scholar]; (c) Walser A; Djerassi C Alkaloid-Studien LII. Die Alkaloide aus Vallesia dichotoma RUIZet PAV. Helv. Chim. Acta 1965, 48, 391–404. [Google Scholar]; (d) Dastoor NJ; Gorman AA; Schmid H Über die Alkaloide von Aspidosperma discolor A. DC. Helv. Chim. Acta 1967, 50, 213–231. [DOI] [PubMed] [Google Scholar]
- (4).(a) Dejrassi C; Archer AAPG; George T; Gilbert B; Shoolery JN; Johnson LF Alkaloid Studies XXV. The Structures of the Aspidosperma Alkaloids Cylindrocarpine and Cylindrocarpidine. Experientia 1960, 16, 532–534. [DOI] [PubMed] [Google Scholar]; (b) Milborrow V; Djerassia C Alkaloid Studies. Part LXI. The Structure of Twelve New Alkaloids from Aspidosperma cylindrocarpon. J. Chem. Soc. C 1969, 417–424. [Google Scholar]
- (5).(a) Lajiness JP; Jiang W; Boger DL Divergent Total Synthesis of (–)-Aspidospermine and (+)-Spegazzine. Org. Lett 2012, 14, 2078–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Satoh H; Ueda H; Tokuyama H Divergent Total Synthesis of (–)-Asidophytine and its Congeners via Fischer Indole Synthesis. Tetrahedron 2013, 69, 89–95. [Google Scholar]; (c) Shen X-L; Zhao R-R; Mo M-J; Peng F-Z; Zhang H-B; Shao Z-H Catalytic Enantioselective and Divergent Total Synthesis of (+)-10-Oxocylindrocarpinol, (+)-Cylindrocarpidine, (–)- N-Acetylclindrocarpinol, and (+)-Apsidospermine. J. Org. Chem 2014, 79, 2473–2480. [DOI] [PubMed] [Google Scholar]; (d) Wang N; Du S; Li D; Jiang X Divergent Asymmetric Total Synthesis of (+)-Vincadifformine, (–)-Quebrachamine, (+)-Aspidospermidine, (–)-Aspidospermine, (–)-Pyrifolidine, and Related Natural Products. Org. Lett 2017, 19, 3167–3170. [DOI] [PubMed] [Google Scholar]
- (6).Evano G; Wang J; Nitelet A Metal-Mediated C–O Bond Forming Reactions in Natural Product Synthesis. Org. Chem. Front 2017, 4, 2480–2499. [Google Scholar]
- (7).(a) O’Connor SE; Maresh JJ Chemistry and Biology of Monoterpene Indole Alkaloid Biosynthesis. Nat. Prod. Rep 2006, 23, 532–547. [DOI] [PubMed] [Google Scholar]; (b) El-Sayed M; Verpoorte R Catharanthus Terpenoid Indole Alkaloids: Biosynthesis and Regulation. Phytochem. Rev 2007, 6, 277–305. [Google Scholar]
- (8).White KL; Movassaghi M Concise Total Syntheses of (+)-Haplocidine and (+)-Haplocine via Late-Stage Oxidation of (+)-Fendleridine Derivatives. J. Am. Chem. Soc 2016, 138, 11383–11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jiao L-Y; Oestreich M C–H Bond Functionalization at the Benzene Core of Indoles and Indolines (Excluding C-2 and C-3). In Strategies for Palladium-Catalyzed Non-Directed and Directed C-H Bond Functionalization; Kapdi AR, Maiti D, Eds.; Elsevier: Amsterdam, 2017; pp 205–232. [Google Scholar]
- (10).(a) For selected examples of directed acetoxylation, seeDick AR; Hull KL; Sanford MS A Highly Selective Catalytic Method for the Oxidative Functionalization of C–H Bonds. J Am. Chem. Soc 2004, 126, 2300–2301. [DOI] [PubMed] [Google Scholar]; (b) Giri R; Liang J; Lei J-G; Li J-J; Wang D-H; Chen X; Naggar IC; Guo C; Foxman BM; Yu J-Q Pd-Catalyzed Stereoselective Oxidation of Methyl Groups by Inexpensive Oxidants under Mild Conditions: A Dual Role for Carboxylic Anhydrides in Catalytic C–H Bond Oxidation. Angew. Chem. Int. Ed 2005, 44, 7420–7424. [DOI] [PubMed] [Google Scholar]; (c) Wang G-W; Yuan T-T; Wu X-L Direct Ortho-Acetoxylation of Anilides via Palladium-Catalyzed sp2 C–H Oxidative Activation. J. Org. Chem 2008, 73, 4717–4720. [DOI] [PubMed] [Google Scholar]; (d) Li G; Wan L; Zhang G; Leow D; Spangler J; Yu J-Q Pd(II)-Catalyzed C–H Functionalizations Directed by Distal Weekly Coordinating Functional Groups. J. Am. Chem. Soc 2015, 137, 4391–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Taniguchi M; Yamamoto I; Nakagawa M; Hino T Cyclic Tautomers of Tryptophans and Tryptamines. VIII. Cyclic Tautomers of Cyclo-L-Prolyl-L-tryptophyl and Related Compounds. Chem. Pharm. Bull 1985, 33, 4783–4791. [Google Scholar]; (b) Somei M; Kawasaki T; Ohta T A Simple Synthesis of 7-Substituted 1-Acetyl-2–3-Dihydroindoles. Heterocycles 1988, 27, 2363–2365. [Google Scholar]
- (12).(a) De PB; Banerjee S; Pradhan S; Punniyamurthy T Ru(II)-Catalyzed C7-Acyloxylation of Indolines with Carboxylic Acids. Org. Biomol. Chem 2018, 16, 5889–5898. [DOI] [PubMed] [Google Scholar]; (b) Zhai W; Li B; Wang B RhIII-Catalyzed Directed Selective C7-Hydroxylation and Acetoxylation of Indolines. ChemistrySelect 2018, 3, 8035–8039. [Google Scholar]; (c) Gao S; Xu X; Tang H; Wu J-Q; Luo J Cu-Mediated C7 Acetoxylation of Indolines. ChemistrySelect 2019, 4, 5835–5838. [Google Scholar]
- (13).See Supplementary Information for details.
- (14).(a) Yoneyama T; Crabtree RH Pd(II) Catalyzed Acetoxylation of Arenes with Iodosyl Acetate. J. of Mol. Cat. A 1996, 108, 35–40. [Google Scholar]; (b) Cook AK; Sanford MS Mechanism of the Palladium-Catalyzed Arene C–H Acetoxylation: A Comparison of Catalysts and Ligand Effects. J. Am. Chem. Soc 2015, 137, 3109–3118. [DOI] [PubMed] [Google Scholar]
- (15).(a) Wang D-H; Mei T-S; Yu J-Q Versatile Pd(II)-Catalyzed C–H Activation/Aryl-Aryl Coupling of Benzoic and Phenyl Acetic Acids. J. Am. Chem. Soc 2008, 130, 17676–17677. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle KM; Wang D-H; Yu J-Q Ligand-Accelerated C–H Activation Reactions: Evidence for a Switch of Mechanism. J. Am. Chem. Soc 2010, 132, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jiao L-Y; Oestreich M Oxidative Palladium(II)-Catalyzed Dehydrogenative C–H/C–H Cross-Coupling of 2,3-Substituted Indolines with Arenes at the C7 Position. Chem. Eur. J 2013, 19, 10845–10848. [DOI] [PubMed] [Google Scholar]
- (17).Griffith WP; Ley SV; Whitcombe GP; White AD Preparation and Use of Tetra-n-butylammonium Per-ruthenate (TBAP reagent) and Tetra-n-propylammonium Per-ruthenate (TPAP reagent) as New Catalytic Oxidants for Alcohols. J. Chem. Soc., Chem. Commun 1987, 1625–1627. [Google Scholar]
- (18).(a) Lindgren BO; Nilsson T Preparation of Carboxylic Acids from Aldehydes (Including Hydroxylated Benzaldehydes) by Oxidation with Chlorite. Acta Chem. Scand 1973, 27, 888–890. [Google Scholar]; (b) Bal BS; Childers WE Jr.; Pinnick HW; Oxidation of α,β-Unsaturated Aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar]
- (19).Still WC; Kahn M; Mitra A Rapid Chromatographic Technique for Preparative Separations with Moderate Resolution. J. Org. Chem 1978, 43, 2923–2925. [Google Scholar]
- (20).All operations involving trimethylsilyldiazomethane were carried out in a well-ventilated fume hood. This includes but is not limited to: measuring the reagent, execution of the transformation, work-up of the reaction mixture, and concentration of the crude reaction mixture.
- (21).This resonance overlaps with CD2HCN; the exact position of resonance was determined by gHSQC correlation (1H,13C) in ppm: (2.20, 43.2) and (1.96–1.92, 43.2).
- (22).Literature value: 1740 (s), 1490, 1380, 793 (w), 782 (s), 742, 720 (w), and 705 (s), see reference 4c.
- (23).Halgren TA Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- (24).Frisch MJT, G. W.; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam MJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ; Gaussian, Inc.: Wallingford CT, 2009; Vol. Revision D.01.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.