

Abstract
Objective:
Thioredoxin (Trx) is a small cellular redox protein with established antioxidant and disulfide reductase properties. We hypothesized that Trx deficiency in mice would cause increased oxidative stress with consequent redox imbalance that would exacerbate obesity-induced vascular dysfunction.
Methods:
Non-transgenic (NT, C57BL/6) and dominant-negative Trx (dnTrx-Tg, low levels of redox-active protein) mice were either fed a normal diet (NC) or high fat diet plus sucrose (HFS) diet for 4 months (3-month HFD+ 1-month HFS). Weight gain, glucose tolerance test (GTT), insulin tolerance test (ITT), and other metabolic parameters were performed following NC or HFS diet. Arterial structural remodeling and functional parameters were assessed by myography.
Results:
Our study found that dnTrx mice with lower levels of active Trx exacerbated myogenic tone, inward arterial remodeling, arterial stiffening, phenylephrine-induced contraction, and endothelial dysfunction of MA. Additionally, FeTMPyP, a peroxynitrite decomposition catalyst, acutely decreased myogenic tone and contraction and normalized endothelial function in MA from dnTrx-Tg mice on HFS via increasing nitric oxide (NO)-mediated relaxation.
Conclusions:
Our results indicate that deficiency of active Trx exacerbates MA contractile and relaxing properties during diet-induced obesity demonstrating that loss of redox balance in obesity is a key mechanism of vascular endothelial dysfunction.
Keywords: arterial structure and function, diabetes, high fat diet, thioredoxin, vascular reactivity
1 |. INTRODUCTION
Obesity has been linked to the development of cardiovascular diseases1 and is a multifaceted metabolic syndrome correlated with hyperglycemia, insulin resistance, adipose tissue inflammation, endothelial dysfunction, peripheral vascular resistance, and hypertension.2–4 Obesity is also implicated in the development of arterial stiffness due to increased consumption of western diet.5 Although stiffness increases with aging, obesity accelerates the stiffening process at a much younger age, especially in females.5,6 There are several contributory factors for the development of obesity associated increase in arterial stiffness such as changes in the intimal, medial, and adventitial layers of the artery6 and due to endothelial dysfunction.7 Furthermore, increased oxidative stress in obesity could also increase arterial stiffness.8 Increased production of reactive oxygen species (ROS) due to activation of xanthine oxidase in the vasculature in obesity has been implicated in the development of arterial stiffness.6 In addition, obesity is also associated with increased oxidative stress due to superoxide anion (O2•−) produced by NADPH oxidase, activated mitochondria, and perivascular adipose tissue (PVAT).9–12
Endothelial nitric oxide synthase (eNOS) is a critical enzyme that produces nitric oxide (NO) from L-arginine.13 NO is a vasorelaxing factor that maintains flow-mediated dilation of the blood vessels and maintains endothelial function and regulates normal blood pressure.14 Progression of obesity has been linked to loss of function of eNOS resulting in decreased levels of circulating NO and increased endothelial dysfunction.6 HFD has been shown to increase arginase II activity via activation of p38 MAPK, which results in production of O2•™ instead of NO, a phenomenon known as uncoupling of eNOS.15 Additionally, increased O2•™ production by xanthine oxidase or activation of NADPH oxidase could oxidize critical eNOS co-factor tetrahydrobiopterin (BH4) resulting in uncoupling of arginine-citrulline pathway of NO production.16,17 Endothelial dysfunction due to reduce NO bioavailability resulting in impaired vasodilatation is an early biomarker for cardiovascular diseases. In obese humans, impaired microvascular dilatation has been diagnosed.18 Studies using isolated arteries from animals on a HFD have shown endothelial dysfunction in both large conduit and small resistance arteries.5,19,20 NO can react with O2•− resulting in the formation of ONOO(t) (peroxynitrite), which is a potent RNS that reacts with all biological macromolecules at a diffusion-controlled rate.13 NO and O2•− interaction could also significantly deplete NO. We have recently observed that the ONOO(t) scavenger ebselen restored endothelium-dependent relaxing responses in isolated small mesenteric arteries from high fat/high sucrose diet (HFS)-induced obese mice, indicating ONOO is involved in arterial stiffening.7
Cytosolic thioredoxin (Trx) is a multifunctional antioxidant protein that regenerates enzymes and other proteins inactivated by oxidation.21,22 The Trx system, which includes thioredoxin reductase (TrxR) in addition to Trx, utilizes NADPH as a source of reducing equivalents to function as a disulfide reductase. The active site of Trx (Trp-Cys-Gly-Pro-Cys) is highly conserved across species. Although Trx is referred to as an “antioxidant protein,” it can directly scavenge hydroxyl radicals and singlet oxygen (1O2), but not superoxide anion.23 However, it can detoxify O2•− by induction of mitochondrial superoxide dismutase (MnSOD).24 Additionally, Trx can detoxify H2O2 via peroxiredoxins, and in this case, Trx is the electron donor.22 Recent studies have shown decreased levels of Trx in subjects with type II diabetes (T2D) and impaired glucose tolerance (IGT).25 Because cells with low levels of redox-active Trx could cause redox imbalance resulting in intracellular oxidative stress, especially for beta cells due to their lower antioxidant defense,26,27 we hypothesize that decreased levels of active Trx could promote arterial stiffness and induce a T2D phenotype in conjunction with a HFS. We have generated a redox-deficient Trx mouse strain,28,29 by overexpressing the redox-inactive mutant Trx. The redox-active cys32 and cys35 of Trx perform the direct transfer of electrons to a disulfide. Mutant Trx (C32S C35S) is a competitive inhibitor of Trx for its reduction by TrxR30 Thus, overexpression of mutant protein produces a dominant-negative effect and decreases the endogenous reduced Trx levels.30,31 These transgenic mice experience enhanced oxidative stress due to decreased level of reduced Trx.28 We show here thatdnTrx-Tg mice fed HFD/HFS develop insulin resistance with glucose intolerance and show a T2D phenotype 2 weeks in advance compared with NT mice. These mice also demonstrate significant arterial stiffness and exacerbated endothelial dysfunction in small resistance arteries.
2 |. MATERIALS AND METHODS
2.1 |. Animals
We have recently created a novel dnTrx-Tg mouse strain, the details of construction of this strain and the redox state of lung and vessels of this strain was published earlier in our papers.28,29 C57BL/6J mice or dnTrx-Tg mice at 12 weeks of age (males and females) were divided into four groups and fed a normal chow (NC) diet (12% kcal from fat, Harlan Laboratories, Inc., Indianapolis, Indiana, USA) or a high fat diet (42% kcal from fat, Harlan Laboratories, Inc., Indianapolis, Indiana, USA) for a period of 3 months as reported previously.32 Sucrose (10% diluted in tap water, Sigma-Aldrich) was given in addition to the high fat diet for a period of 1 month. Thus, the groups are designated as normal chow (NC) or high fat sucrose (HFS) diet. Body weight was monitored weekly in the two young mice groups. In a subset of NT and dnTrx-Tg mice on HFS diet, the ONOO− decomposition catalyst Fe(III)-tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (FeTMPyP; 30 mg/kg) was given via tail vein injection for the duration of 1 h. All procedures were approved by the IACUC at the Texas Tech University Health Sciences Center and were consistent with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health. All animals were maintained on a standard 12-h light/12-h dark cycle, in a temperature-controlled barrier facility.
2.2 |. Fasting glucose and insulin measurement
After 4 months on the HFS diet, animals were fasted for 4–6 h, the tail was cut, and fasting blood glucose levels were measured using the OneTouch Ultra glucose monitor (LifeScan Inc., Malvern, Pennsylvania, USA). Blood was drawn from the orbital sinus and spun down at 3000 × g for 15 min, and serum was separated.
2.2.1 |. Intraperitoneal glucose and insulin tolerance tests (GTT/ITT)
Mice were anesthetized by brief inhalation of 2% isoflurane in pure oxygen, followed by an intraperitoneal injection of glucose (2.0 g/kg) or insulin (Humulin R, Eli Lilly, Indianapolis, IN, USA; 0.75 U/kg), and blood was drawn from the tail at 0 (time point taken before injection), 15, 30, 60, and 120 min after injection with the OneTouch Ultra glucose monitor (LifeScan Inc., Malvern, Pennsylvania, USA).
2.3 |. Metabolic testing
2.3.1 |. Intraperitoneal glucose tolerance test (i.p.GTT)
A GTT was performed 12 weeks after the treatment was initiated and before sucrose was given to mice. Mice were fasted for 12 h with free access to drinking water. A baseline blood sample was collected from the tails of fully conscious mice, followed by an i.p. injection of glucose (2.0 g/kg body weight), and blood was drawn from the tail at 15, 30, 60, and 120 min after injection. Glucose concentrations were determined from whole blood samples measured on a OneTouch Ultra portable glucometer (LifeScan Inc., Malvern, Pennsylvania, USA).
2.3.2 |. Intraperitoneal insulin tolerance test (i.p.ITT)
Mice were fasted for 6 h, and baseline blood samples were collected from the tails of fully conscious mice. Insulin (0.75 U/kg body weight, Humulin R, Eli Lilly, Indianapolis, IN, USA; TTUHSC pharmacy) was administered by i.p. injection, and blood samples were drawn from the tail at 15, 30, 45, and 60 min after injection. Glucose concentrations were determined from blood using a OneTouch Ultra portable glucometer (LifeScan Inc., Malvern, Pennsylvania, USA).
2.4 |. Pressure myography
Mice were killed by an overdose of CO2 inhalation, and the mesentery was removed and placed in cold KRB buffer with the following composition (in mM): 118.5 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.0 NaHCO3, and 5.5 D-glucose. From the mesentery, a segment (4 mm) of the second-order branch of the superior mesenteric artery (MA) devoid of PVAT was cannulated between two small glass micropipettes (O.D. 120–150 μm) in a pressure myograph chamber (model 110P, DMT-USA, Inc., Aarhus, Denmark). The ends of the segment were tied with two small nylon knots (17 μm thin). The chamber was filled with warm KRB and continuously gassed with carbogen (95% O2/5% CO2). Intraluminal pressure was set to 60 mmHg, and the artery was allowed to adjust in KRB containing 2.5 mM CaCl2 for 30 min. The internal lumen diameter and wall thickness were continuously monitored using the MYOVIEW II software (DMT-USA, Inc.). Viability was tested by adding the α1-adrenergic agonist PHE (0.1–1 μM). After a stable diameter was reached, cumulative concentrations of ACh (0.001–100 μM) were added to the chamber. The chamber was washed three times with KRB, and myogenic tone was assessed by monitoring internal diameter during stepwise (10 mmHg) increases in intraluminal pressures from 10 to 140 mmHg. At each pressure step, the artery was allowed to adjust its internal diameter for at least 2 min or until a stable diameter was obtained. Percentage myogenic tone was calculated as: ((Di, 0 Ca2+ − Di,2.5 mMCa2+)/Di, 0 Ca2+)) × 100%, in which Di’ 0 Ca2+ is the inner diameter at any intraluminal pressure step obtained in KRB without CaCl2 and Di, 2.5 mM Ca2+ is the inner diameter at any intraluminal pressure step obtained in KRB without CaCl2 and Di, 2.5 mM Ca2+ is the inner diameter at any intraluminal pressure step obtained in KRB with 2.5 mM CaCl2. Elastic characteristics of the vessel wall were determined under passive (calcium-free) conditions in KRB without CaCl2 and containing 2.5 mM of the Ca2+ chelator EGTA and the NO donor sodium nitroprusside (SNP; 10 μM) to allow complete relaxation of the smooth muscle. Wall-to-lumen ratio was calculated as wall thickness (NT) divided by inner diameter (Di). According to Laplace’s law, wall tension (T in mN/mm) depends upon transmural pressure (Pt in mN/mm2, where 100 mmHg equals 13.33 mN/mm2) and the inner radius of the artery (ri in mm): T = Pt · ri. Circumferential wall stress (σ in mN/mm2) is related to NT (mm) and wall tension (T): σ = T/NT. The incremental elastic modulus (Einc in mN/mm2) was calculated according to Bergel33: Einc = 1.5 · ro2 · ri · ΔP/(ro2 − ri2) · Δri, where ro = outer radius, ri inner radius, and Δp = 10 mmHg or 1.333 mN/mm2.
2.5 |. Wire myography
From the mesentery, three segments (2 mm) of MA from each mouse were carefully dissected. The femoral artery was isolated as well. Segments devoid of PVAT were mounted in a wire myograph (model 620 M; Danish Myotechnology, Aarhus, Denmark) for the recording of isometric force development. Segments were passively stretched according to a procedure first described by Halpern and Mulvany.34 In brief, segments were distended stepwise, in 50 μm increments to their optimal lumen diameter for active tension development. Arteries were stretched to a passive wall tension of 90% of the internal circumference of that achieved when they were exposed to a passive tension yielding a transmural pressure of 100 mmHg, which is referred to as its “optimal diameter.” At this passive wall tension, arteries were contracted with high K+ KRB (60 mmol/L KCl in KRB solution; replacing equimolar NaCl with KCl), thus generating a stable contraction that reached a plateau after 10–15 min. This active wall tension was set to a 100% contraction level. Contractile responses in MA were assessed by performing cumulative concentration-response curves (CRCs) to PHE (0.01–30 μM), whereas CRC to serotonin (5-HT 0.001–3 μM) were performed in femoral arteries. Relaxing responses were assessed in PHE/5-HT (single dose generated roughly 80% of K+KRB-induced contraction) contracted segments by performing CRCs to ACh (0.01–10 μM). In a subset of MA, NO-dependent relaxations were analyzed in conditions where vasorelaxing prostacyclin release and the endothelium-dependent hyperpolarization (EDH) relaxing response were pharmacologically blocked by a cocktail of the cyclooxygenase blocker indomethacin (INDO; 10 μM) and the endothelial KCa channel blockers TRAM-34 and UCL1684 (both 1 μM). EDH relaxing responses were assessed in the presence of the non-selective NO synthase inhibitor L-NAME and INDO. Endothelium-independent relaxations were assessed using the NO donor SNP (0.0001–1 μM) in arteries that were incubated with L-NAME and INDO. MA segments were incubated for 30 min with the described pharmacological blockers before assessing relaxing responses.
2.6 |. Western analysis
Protein extracts were prepared from mesenteric arteries pooled from 3 mice (each treatment) unless stated otherwise. Segments were homogenized in cold PBS buffer in a glass dounce homogenizer and treated with M-Per (Pierce, Rockford, IL, USA). The homogenate was centrifuged at 15 000 g for 15 min at 4°C. Protein concentration was determined with the BCA protein assay kit (Pierce Chemical, Rockford, IL, USA). Western analysis was performed using the Bio-Rad mini protean system. Equal amounts of protein were resolved on a 6–15% SDS-PAGE. Following electrophoresis, protein was transferred to a nitrocellulose (Hybond-ECL, GE Healthcare, Marlborough MA USA) or PVDF membrane, immunoblotted with respective primary antibodies, and visualized by the ECL system (GE Healthcare) using appropriate secondary antibodies conjugated with HRP IgG. Some of the images were acquired with G:BOX Chemi XL1.4 (Syngene, Frederick, MD, USA) using West Femto reagent from Pierce.
2.7 |. Statistical analysis
Data were analyzed using one-way analysis of variance (ANOVA) for comparison of multiple means followed by Tukey’s post hoc test; two-way ANOVA, and Student’s t test were performed for comparison of two means using Prism v 6.00 (GraphPad Prism software, La Jolla, CA, USA). Data were -presented as standard error of mean (SEM).
3 |. RESULTS
3.1 |. Trx deficiency increases weight gain in response to HFS
After a 12-week period of continuous feeding, mice fed HFS diet had statistically higher body weight compared to mice fed NC diet (Figure 1A). DnTrx-Tg mice on HFS diet gained substantial weight, which was statistically significantly higher compared to NT mice or dnTrx-Tg mice on NC (Figure 1B). The gross anatomies of NT and dn-Trx-Tg mice on NC diet were comparable (only NT shown). However, there was a noticeable change in the visceral morphology of dnTrx-Tg mice on HFS diet compared to NT mice on HFS diet (Figure 1C). DnTrx-Tg mice were significantly greater in apparent mass and had increased fat pads and a blanched liver which was increased in size with a desiccated yellowed intestine (Figure 1C). These effects were achieved in dnTrx mice after 2 weeks of 10% sucrose diluted in their water bottles in contrast to NT mice after 4 weeks of 10% sucrose in drinking water, suggesting accelerated weight gain in dnTrx-Tg mice. As shown in Figure 1D,E, the dnTrx-Tg mice showed severe deficiency of insulin in the pancreas, suggesting that these mice have basal insulin deficiency and are more likely to develop type II diabetes in response to HFS diet.
FIGURE 1.
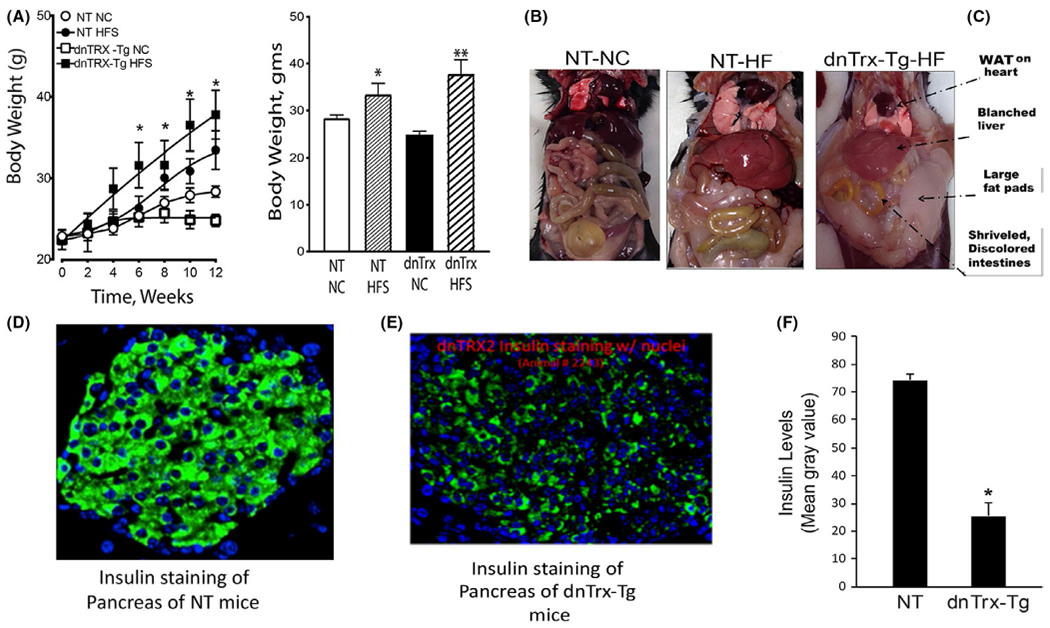
(A) dnTrx-Tg mice on HFS gained significant weight compared to NT-HFS mice: Mice were fed as mentioned in the methods. Body weight of NT-NC mice (open circle), NT-HFS (closed circle), dnTrx-Tg-NC mice (open square), and dnTrx-Tg-HFS(closed square). Values are -presented as standard error of mean (SEM). n = 6, *p < .05, NT-HFS vs. NT-NC; dnTrx-HFS vs. dnTrx-chow. (B) Final body weight of all mice groups after 12 weeks. Mice were fed similar to that described in (A). n = 6 *p < .05, NT-HFS vs. NT-NC, dnTrx-HFS vs. NT-HFS; p < .01, dnTrx-HFS vs. dnTrx-NC. (C) Visceral morphology of NT-NC, NT-HFS, and dnTrx-HFS mice; (D) Immunofluorescence staining of section of pancreas of NT and (E) dnTrx-Tg mice demonstrating the insulin levels; (F) gray scale signal intensity of NT and dnTrx-Tg mice for insulin, *p < .05 (Student’s t test), N = 3 (males and females)
3.2 |. Trx deficiency accelerates T2D phenotype in response to HFS
A glucose tolerance test (GTT) evaluation was performed in order to demonstrate the effect of weight gain on regulation of glucose metabolism. As shown in Figure 2A, 2 h after feeding, the blood glucose levels of NT or dnTrx-Tg mice fed HFS diet were significantly higher compared to NT or dnTrx-Tg mice on NC (Figure 2A). After 30 or 60 min of feeding, the dnTrx-Tg mice had significantly higher levels of blood glucose compared to NT or dnTrx mice fed NC. However, dnTrx mice demonstrated a fully developed T2D phenotype much earlier (2 weeks) compared to the NT mice that developed the T2D phenotype after 4 weeks of drinking 10% sucrose solution (Figure 2B). These data demonstrate that decreased levels of redox-active Trx cause accelerated glucose intolerance in response to HFS diet. Next, we determined insulin tolerance of NT and dnTrx-Tg mice fed HFS diet as insulin receptor has been shown to be inactivated due to continuous oxidative stress condition caused by high blood glucose levels.35 As shown in Figure 2C, all groups of mice had decreased blood glucose levels after an i.p. injection of 0.75 U/ml of insulin compared to the initial blood glucose levels before insulin was given (Figure 2C). There was no difference between the NT-HFS and the NT-chow groups; both blood glucose levels remained below 100 mg/dL for a period of 90 min. The dnTrx-Tg-chow group retained a low blood glucose level for 60 min compared to the dnTrx-Tg-HFS group that remained in that state for only 30 min. Both groups of NT mice and dnTrx-Tg mice fed a chow diet, however, remained hypoglycemic for a longer period of time than the dnTrx-Tg mice fed HFS diet, showing that the dnTrx-Tg-HFS mice may be insulin resistant.
FIGURE 2.
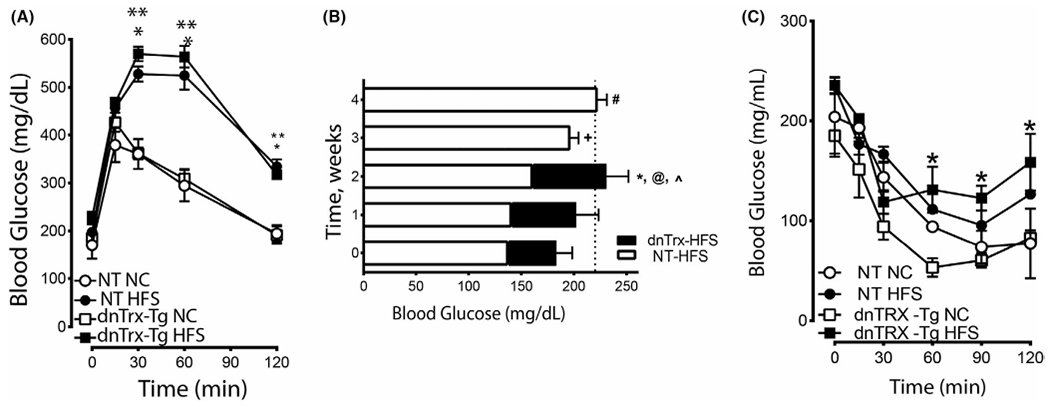
Loss of redox-active Trx accelerates T2D phenotype in response to HFS diet: (A) Glucose tolerance test: Mice were fed NC diet or HFS diet as described in the methods. Blood glucose levels of mice were measured after they were injected with glucose as described in the methods. NT-NC (open circle), NT-HFS (closed circle), dnTrx-NC (open square), and dnTrx-HFS(closed square) were injected with glucose. Values are -presented as standard error of mean (SEM); (B) Blood glucose levels in NT or dnTrx-Tg mice in HFS diet. NT or dnTrx-Tg mice (n = 6) were on HFS for 4 weeks. Blood glucose levels were measured each week for 4 weeks. *, @, ^ p < .05 dnTrx at 2 weeks vs. NT-HFS. (C) Insulin tolerance test: Blood glucose levels of mice after being injected with insulin as described in the methods in NT-NC, NT-HFS, dnTrx-Tg-NC and dnTrx-Tg-HFS mice. Values are -presented as standard error of mean (SEM). (n = 6, males and females) p < .05, (*) dnTrx-HFS vs. dntrx-chow at time points 60, 90 and 120 min
3.3 |. HFS diet severely impairs pressure-induced autoregulation and myogenic tone of small mesenteric artery in dnTrx-Tg mice
Isolated second-order mesenteric arteries (MA) from NT or dnTrx-Tg on NC developed active tone in response to step increases in intraluminal pressure (10–140 mmHg) in the presence of 2.5 mM CaCl2 and without the use of any vasoactive agent (Figure 3A,B). At low intravascular pressures (10 to 40 mmHg), diameters increased in MA from NT and dnTrx-Tg on NC. Beyond this point, further increases in pressure until 100 mmHg resulted in maintenance of inner diameter, the so-called “autoregulation” range. At higher pressures, the diameter could not be maintained at this range, but the active diameters were still statistically significantly lower compared to their passive diameters (0 mM CaCl2). In MA from HFS NT and dnTrx-Tg mice, active diameters were lower compared to their NC counterparts (Figure 3A,B). This was especially apparent at lower pressures (Figure 3A,B). Strikingly, the characteristic autoregulation plateau is severely impaired in MA from HFS NT and dnTrx-Tg mice, as active lumen diameters progressively and near linearly increased with higher intravascular pressures. Consequently, the percentage of myogenic tone was increased in MA from HFS diet at lower intravascular pressures (Figure 3C,D). We further probed into the mechanism of the loss of autoregulatory capacity of MA by evaluating the role of ROS in this phenomenon. We had previously shown that ebselen, a peroxynitrite scavenger,7 was effective in restoring HFS diet-mediated endothelial dysfunction.7 Therefore, we sought to determine whether ONOO− decomposition catalyst FeTMPyP would correct the autoregulatory response of MA. As shown in Figure 3C,D, FeTMPyP injection improved autoregulation and significantly lowered the myogenic tone (Figure 3C,D) of MA in HFS diet, suggesting a dysfunctional eNOS is likely involved in the impairment of autoregulatory response of MA in HFS diet.
FIGURE 3.
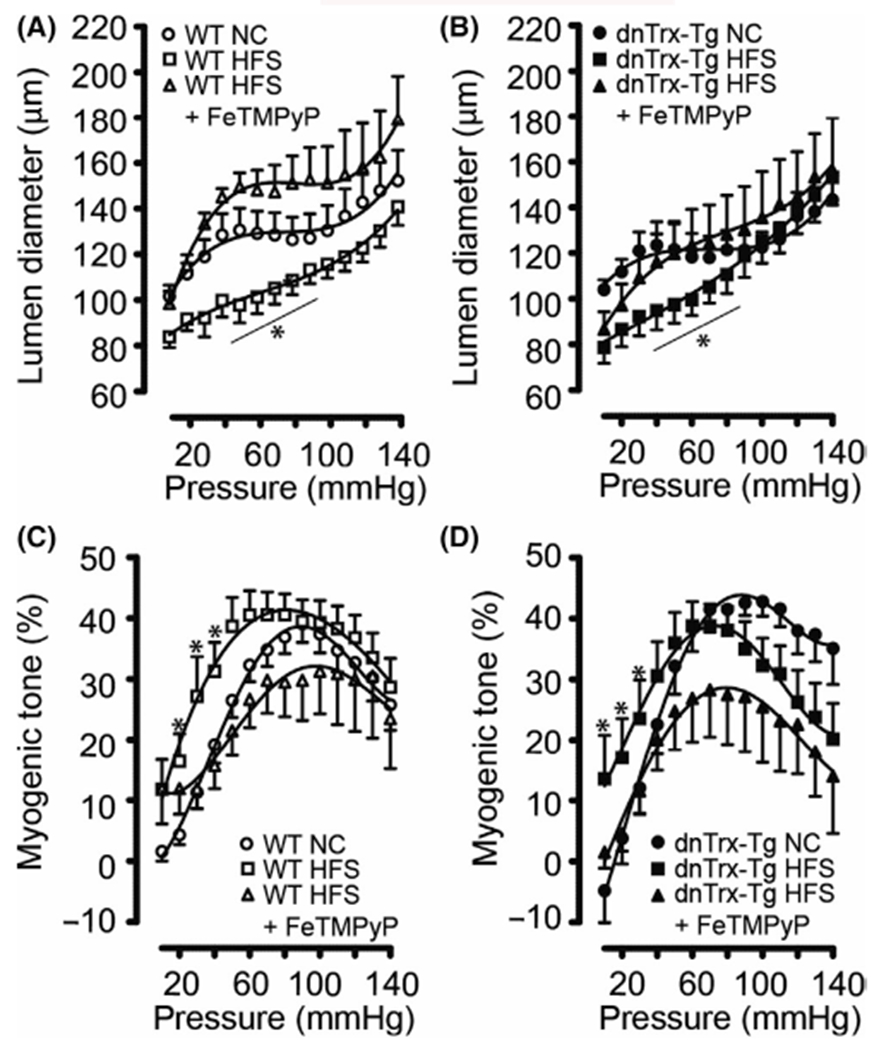
Pressure-induced autoregulation and myogenic tone of small mesenteric arteries (A–D): Active (2.5 mM CaCl2) pressure-diameter curves for pressurized 2nd-order mesenteric arteries isolated from NT (A) and dnTrx-Tg (B) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Percentage myogenic tone (see methods) was plotted for NT (C) and dnTrx-Tg (D) mice fed a NC diet (circles), HFS diet (squares), and HFS +FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 6, males and females) used are shown in brackets
3.4 |. Trx deficiency promotes structural remodeling of small mesenteric artery in dnTrx-Tg mice in HFS diet
Passive lumen diameters were comparable for MA from both NT and dnTrx-Tg mice on a NC diet (Figure 4A,B). However, diet-induced obesity resulted in inward remodeling, which was markedly higher in MA from dnTrx-Tg mice (Figure 4B) compared with NT mice (Figure 4A). Wall thickness did not change in MA from NT mice on HFS diet (Figure 4C), but was significantly increased in dnTrx-Tg mice on a HFS diet (Figure 4D). From these passive inner diameters and wall thicknesses, structural arterial remodeling and elasticity parameters were calculated. Wall-to-lumen ratios represent a degree of remodeling with higher ratios associated with hypertrophic inward/eutrophic remodeling. No changes in wall-to-lumen ratios were observed between MA from NC and HFS NT mice (Figure 5A). In contrast, wall-to-lumen ratios were statistically significantly larger in MA from dnTrx-Tg on HFS diet compared to their NC counterparts, indicating an inward hypertrophic remodeling during obesity in MA from dnTrx-Tg mice (Figure 5B). As expected, short-term injection of FeTMPyP did not result in structural arterial changes in MA from NT and dnTrx-Tg mice on HFS diet (Figure 5A,B). Arterial elasticity was calculated by plotting circumferential wall stress (CWS) values versus incremental elastic modulus (Einc) values (see Methods). No changes in the CWS versus Einc curves were observed between MA on a NC and HFS diet (Figure 5C). However, a marked leftward shift in these curves (representing decreased elasticity) was observed in MA of dnTrx-Tg mice on HFS diet, irrespective of FeTMPyP injection, compared to MA of dnTrx-Tg mice on a NC diet (Figure 5D). These data indicate that structural arterial remodeling under HFS diet was significantly altered in mice lacking redox-active Trx.
FIGURE 4.
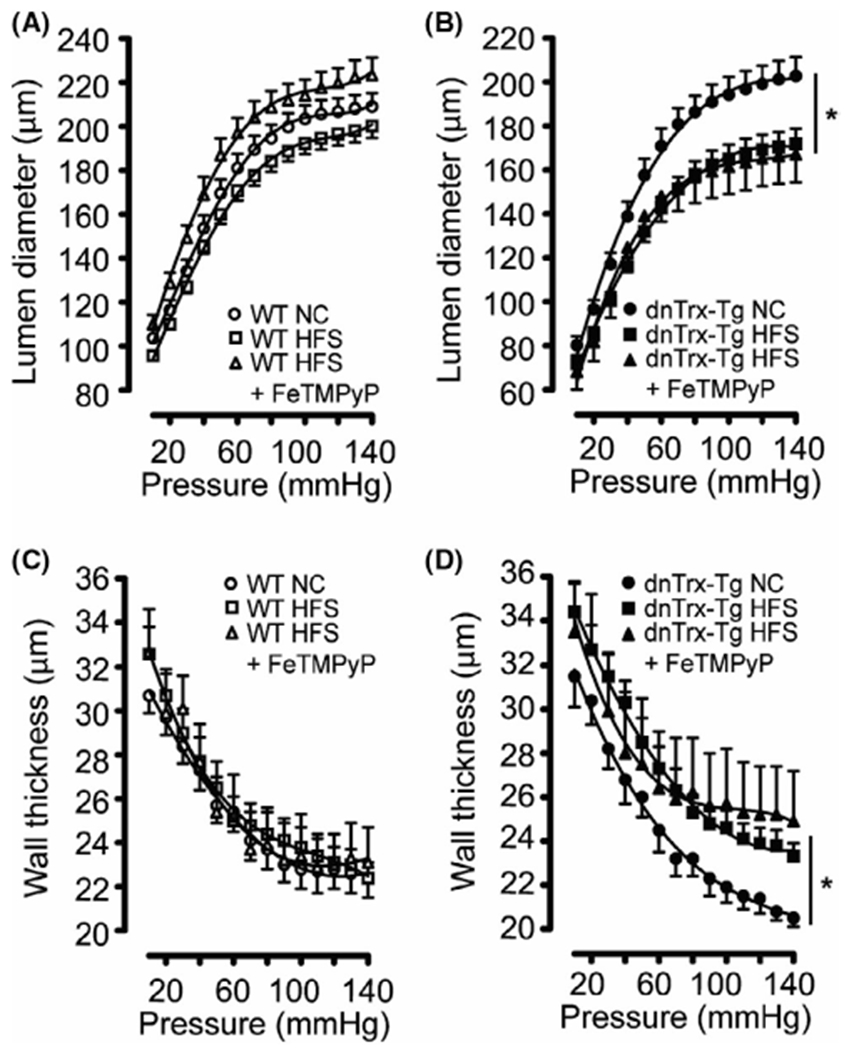
Passive pressure-diameter and pressure-wall thickness relationships of small mesenteric arteries (A–D): Passive (0 mM CaCl2 + 10 μM SNP + 2.5 mM EGTA) pressure-diameter curves (A and B) and pressure-wall thickness curves (C and D) for pressurized 2nd-order mesenteric arteries isolated from NT (A and C) and dnTrx-Tg (B and D) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 5, males and females)
FIGURE 5.
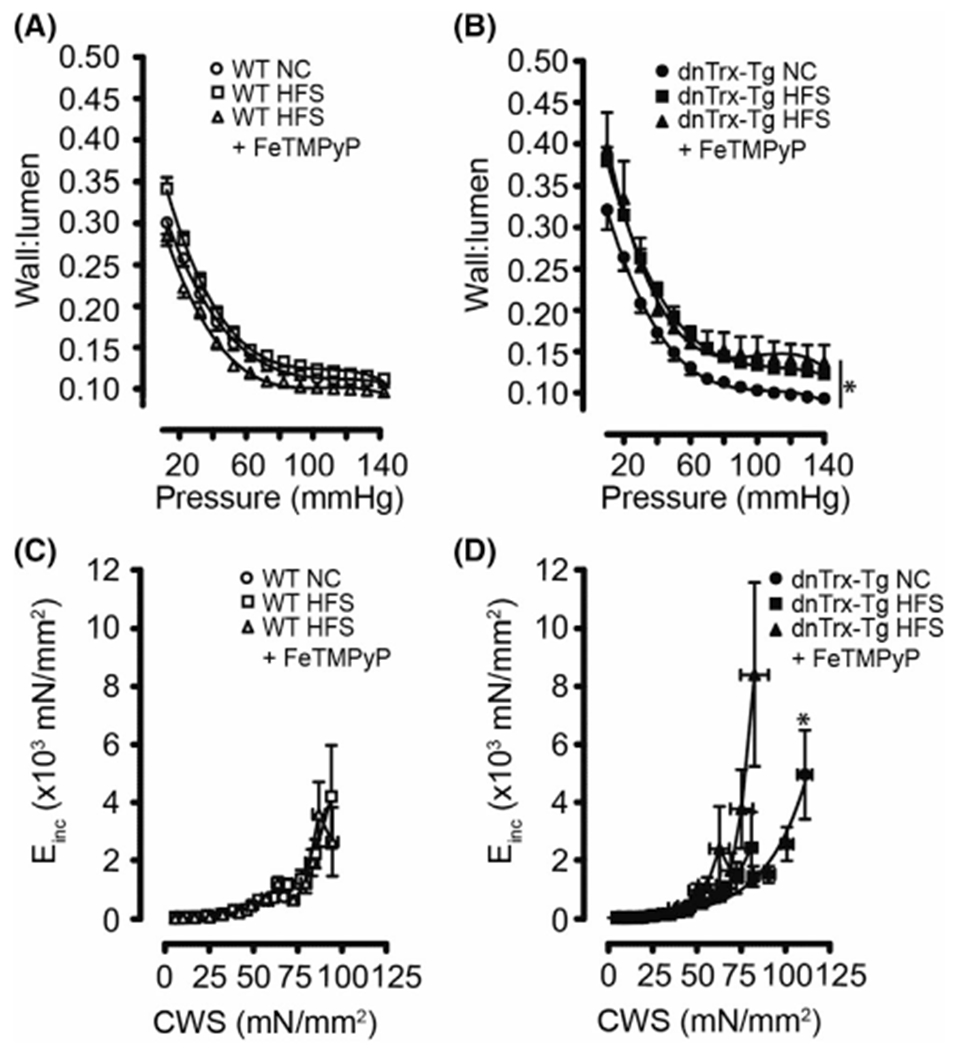
Passive wall-to-lumen ratios and elasticity of small mesenteric arteries (A–D): Passive (0 mM CaCl2 + 10 μM SNP + 2.5 mM EGTA) wall-to-lumen curves (A and B) and circumferential wall stress (CWS) versus incremental elastic modulus (Einc) curves (C and D) for pressurized 2nd-order mesenteric arteries isolated from NT (A and C) and dnTrx-Tg (B and D) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 7, males, and females)
3.5 |. Trx deficiency induces endothelial dysfunction in MA of dnTrx-Tg mice in HFS diet
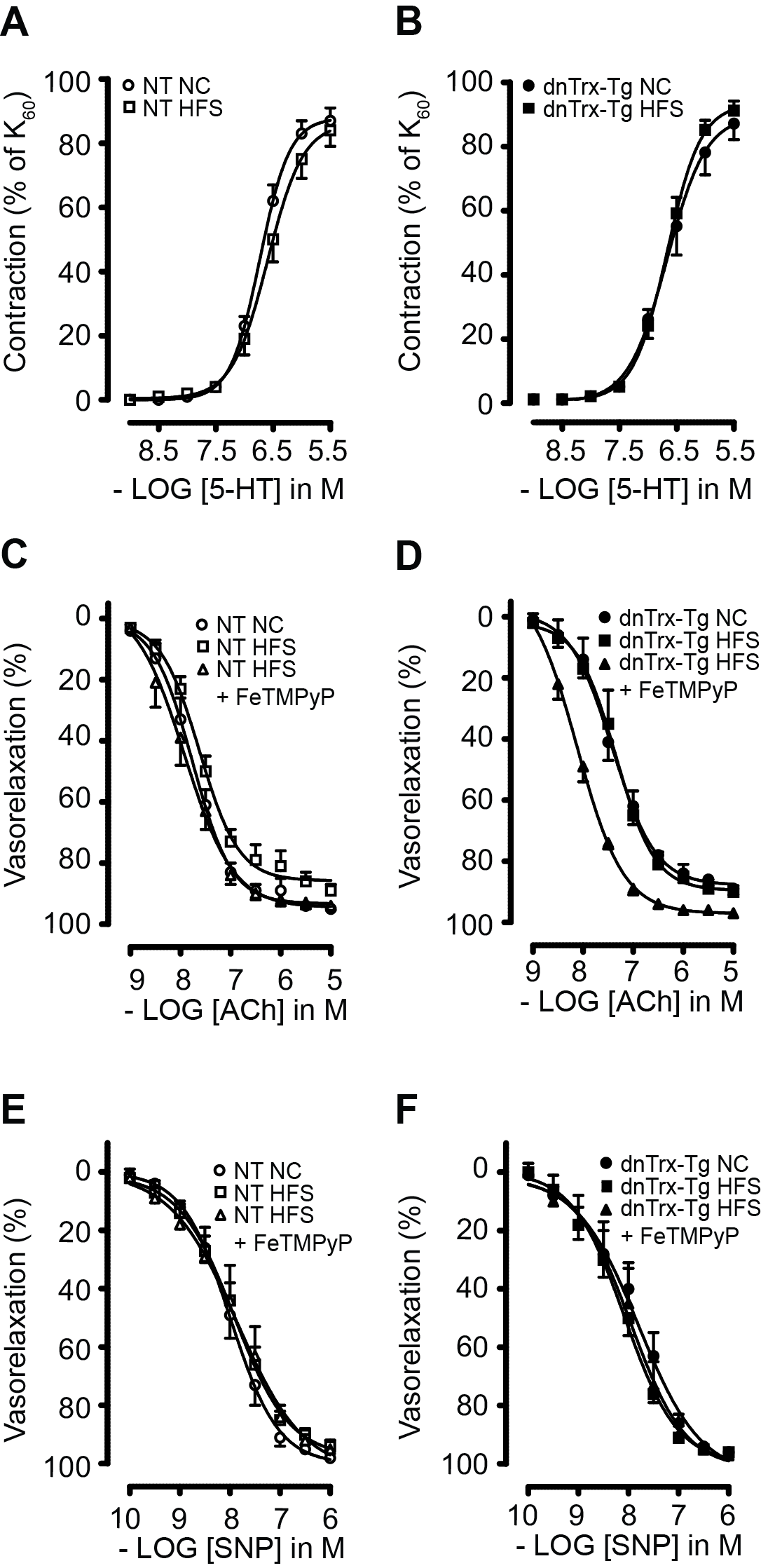
Since HFS diet caused significant decrease in MA elasticity in dnTrx-Tg mice, we reasoned that these arteries may experience significant endothelial dysfunction. We assessed ACh-induced endothelium-dependent vasodilatations in pressurized MA. In a dose-dependent manner, ACh (0.001–100 μM) elicited significantly greater dilatations in MA from mice on a NC diet than those from HFS diet (Figure 6A). Additionally, MA from dnTrx-Tg mice on HFS showed more pronounced endothelial dysfunction (Emax 21 ± 9%; Figure 6B) compared to NT mice (Emax 38 ± 11; Figure 6A). These data demonstrate that loss of redox active of MA accentuates endothelial dysfunction in response to HFS diet.
FIGURE 6.
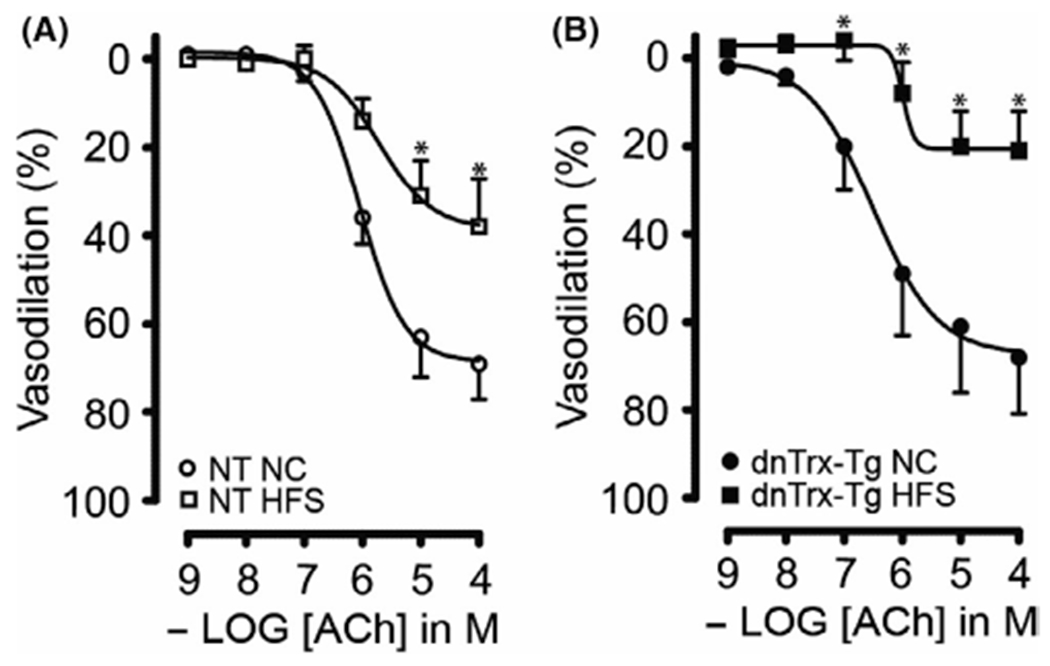
Endothelium-dependent ACh-induced vasodilatation of small mesenteric arteries: Acetylcholine (ACh; 0.001–100 μmol/L)-induced relaxations in pressurized and contracted (pressure- and phenylephrine-induced) 2nd-order mesenteric arteries isolated from NT (A) and dnTrx-Tg (B) mice fed a normal chow (NC) diet (circles) or high fat/high sucrose (HFS) diet (squares). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 5, males and females)
3.6 |. Trx deficiency increases contractile responses to PHE in MA due to increased OONO− production
We further determined the effect of loss of active Trx on contractile response of MA in dnTrx-Tg mice. We assessed contractile responses to the α1-adrenergic agonist PHE (0.01–30 μM) in the wire myograph. MAs were incubated with the cyclooxygenase blocker indomethacin (10 μM) to rule out vasoactive contribution of prostaglandins. Contractions were expressed as percentage to the tension observed in response to 60 mM KCl. Contractile responses were comparable for MA derived from NT and dnTrx-Tg mice on a NC diet (Figure 7A,B). Sensitivity (pEC50) to PHE averaged at 5.45 ± 0.03 in NT and 5.55 ± 0.06 in dnTrx-Tg mice on a NC diet. Maximal contraction to 30 μM PHE (Emax) was similar for NT and dnTrx-Tg mice on a NC diet (109 ± 9% vs. 102 ± 7%, respectively; Figure 7A,B). HFS diet statistically significantly increased sensitivity to PHE to a greater extent in dnTrx-Tg mice than NT mice (5.75 ± 0.04 vs. 5.54 ± 0.01, respectively; p < .05; Figure 7A,B). However, Emax values were comparable for NT and dnTrx-Tg on HFS diet (135 ± 6% vs. 134 ± 8%, respectively; Figure 7A,B). We further determined whether enhanced contractile response in dnTrx-Tg mice is due to increased OONO− production in response to HFS. Injection with FeTMPyP statistically significantly reduced pEC50 values in MA from both NT and dnTrx-Tg mice (5.31 ± 0.01 and 5.51 ± 0.01, respectively; (Figure 7A,B). Similarly, FeTMPyP statistically significantly lowered Emax in obese NT (100 ± 9%); (Figure 7A) and obese dnTrx-Tg mice (116 ± 20%; Figure 7B).
FIGURE 7.
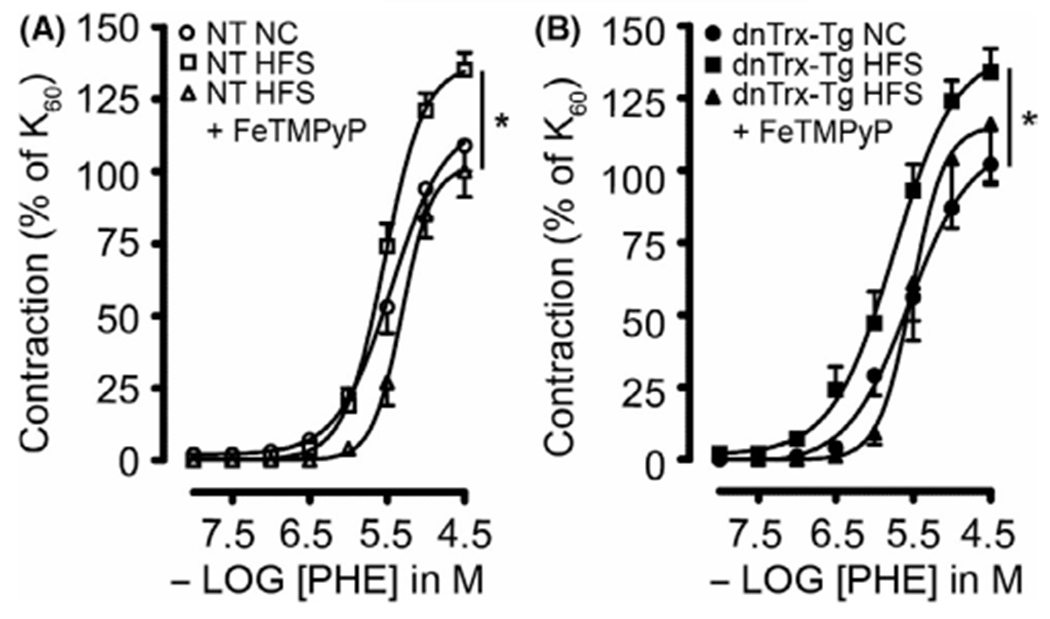
α1-adrenergic contractions in small mesenteric arteries. Phenylephrine (PHE; 0.01–30 μmol/L)-induced contractions of stretched 2nd-order mesenteric arteries isolated from NT (A) and dnTrx-Tg (B) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 5, males and females)
3.7 |. Trx deficiency exacerbated HFS-induced endothelial dysfunction via production OONO−
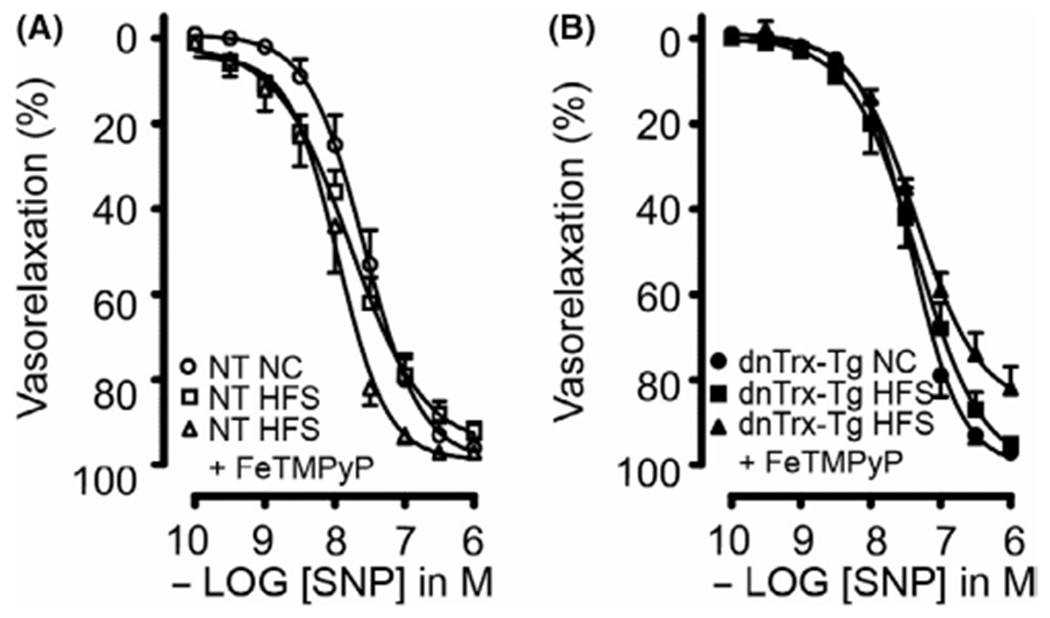
In order to rule out vascular effects of vasoactive prostaglandins, all MA segments were incubated with indomethacin (10 μM). Pressurized arteries treated with ACh resulted in dose-dependent relaxations (Figure 8). Sensitivity and maximal responses to 10 μM ACh were greater in MA mounted in the wire myograph compared to pressurized arteries, presumably because of direct contact of ACh in wire-mounted arteries instead of the diffusion through the arterial wall to the endothelium in pressurized arteries. MA from NT mice on HFS diet did not result in a statistically significant impairment in ACh-induced relaxations (Figure 8A) in sharp contrast to obese dnTrx-Tg mice (Figure 8B). NO-dependent relaxations were assessed under conditions in which endothelium-dependent hyperpolarization (EDH) was pharmacologically blocked. This was achieved by the incubation of inhibitors of the endothelial calcium-activated K+ channels responsible for initiating the EDH response in the mesenteric arterial bed, TRAM-34 (KCa3.1 blocker; 1 μM), and UCL1684 (KCa3.2 blocker; 1 μM). NO-mediated relaxations were not significantly altered in MA from NT mice on HFS diet (Figure 8C), whereas a markedly reduced relaxation was observed in MA from dnTrx-Tg on HFS diet (Figure 8D). EDH relaxing responses were measured in conditions in which NO production was pharmacologically blocked with L-NAME. In agreement with our previously published observations, EDH-mediated relaxations were statistically significantly reduced in MA from dnTrx-Tg mice on a NC diet compared to NT on a NC diet (Figure 8E,F). However, HFS diet did not compromise EDH responses in MA from obese NT and dnTrx-Tg mice (Figure 8E,F). This suggests that the endothelial dysfunction was mainly caused by reduced NO signaling. In our previous study, we have observed that incubation of the ONOO− inhibitor ebselen improved endothelial dysfunction in isolated MA from NT mice on a year-long high fat diet.7 In order to assess the role of ONOO− in inhibiting endothelial function during HFS in dnTrx-Tg mice, we injected mice on HFS diet with FeTMPyP via tail vein injection. In MA from both NT and dnTrx-Tg mice on HFS diet FeTMPyP statistically significantly improved overall endothelium-dependent relaxations (Figure 8A,B) and NO-mediated relaxing responses (Figure 8C,D), but not EDH (Figure 8E,F). We assessed endothelium-independent relaxing responses to the NO donor sodium nitroprusside (SNP; 0.1 nM–1 μM). SNP resulted in comparable relaxations in MA from NT and dnTrx-Tg mice irrespective of their diets, suggesting that the diet-induced changes were endothelial-specific (Figure 9). These observations indicate that FeTMPyP improves NO bioavailability and suggest that ONOO− does not inhibit endothelial KCa channels that initiate the EDH response.
FIGURE 8.
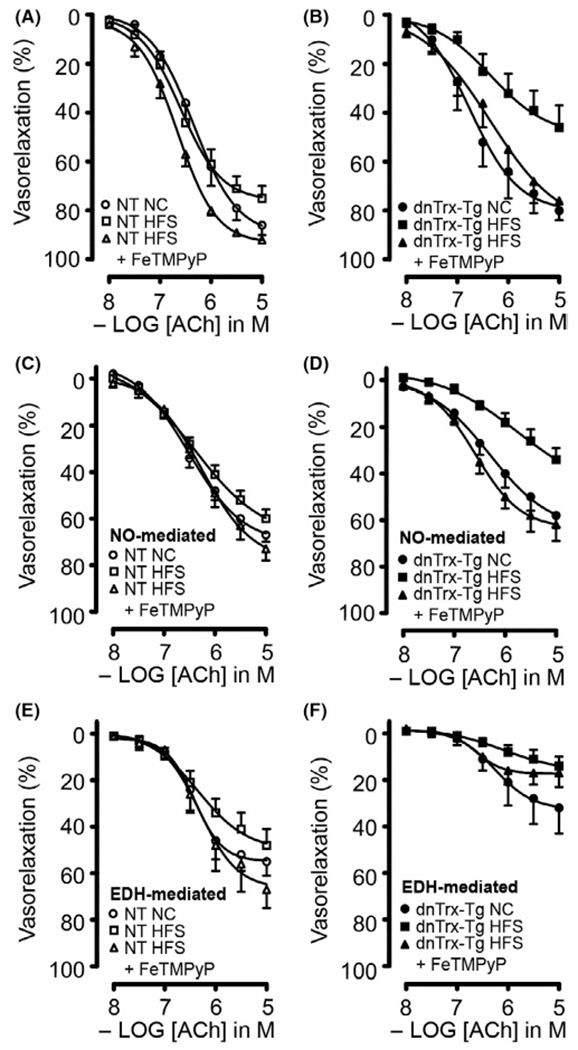
Endothelium-dependent ACh-induced vasorelaxations of small mesenteric arteries. Endothelium-dependent ACh-mediated relaxations in the presence of the cyclooxygenase blocker indomethacin (10 μM) (A and B), NO-dependent ACh-mediated relaxations in the presence of indomethacin, TRAM-34 (1 μM) and UCL 1684 (1 μM) (C and D), and endothelium-dependent hyperpolarizing (EDH) relaxing responses in the presence of indomethacin and the non-selective NOS blocker L-NAME (E and F) for small mesenteric arteries isolated from NT (A, C and E) and dnTrx-Tg (B, D and F) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 7, males and females)
FIGURE 9.
Endothelium-independent relaxing responses in small mesenteric arteries. Endothelium-independent sodium nitroprusside (SNP; 0.1 nM-1 μM)-mediated relaxations in the presence of L-NAME and indomethacin for small mesenteric arteries isolated from NT (A) and dnTrx-Tg (B) mice fed a normal chow (NC) diet (circles), high fat/high sucrose (HFS) diet (squares), and HFS followed by tail vein injection of the ONOO− decomposition compound FeTMPyP (triangles). Values are shown as means ± SEM. *p < .05 versus NC. Number of mice (n = 5, males and females)
3.8 |. Trx deficiency increased arterial stiffness in dnTrx mice with NC or HFD
dnTrx-Tg mice showed significant endothelial dysfunction compared to NT mice with HFS diet. In addition, dnTrx-Tg in HFD also demonstrated significant structural arterial remodeling suggesting structural components of arteries might have been modulated. We evaluated the expression of collagen-1 in the small mesenteric artery as increased collagen deposition has been correlated with arterial stiffness and decreased endothelial dysfunction. As shown in Figure 10A,B, the expression of collagen-1 was increased about 3-fold in dnTrx-Tg mice in NC diet compared to NT mice in NC diet, indicating that deficiency of Trx alone could cause structural remodeling of small mesenteric artery. Additionally, dnTrx-Tg mice with HFD had 2-fold higher expression of collagen-1 compared to dnTrx-Tg mice in NC diet and NT mice with HFD.
FIGURE 10.
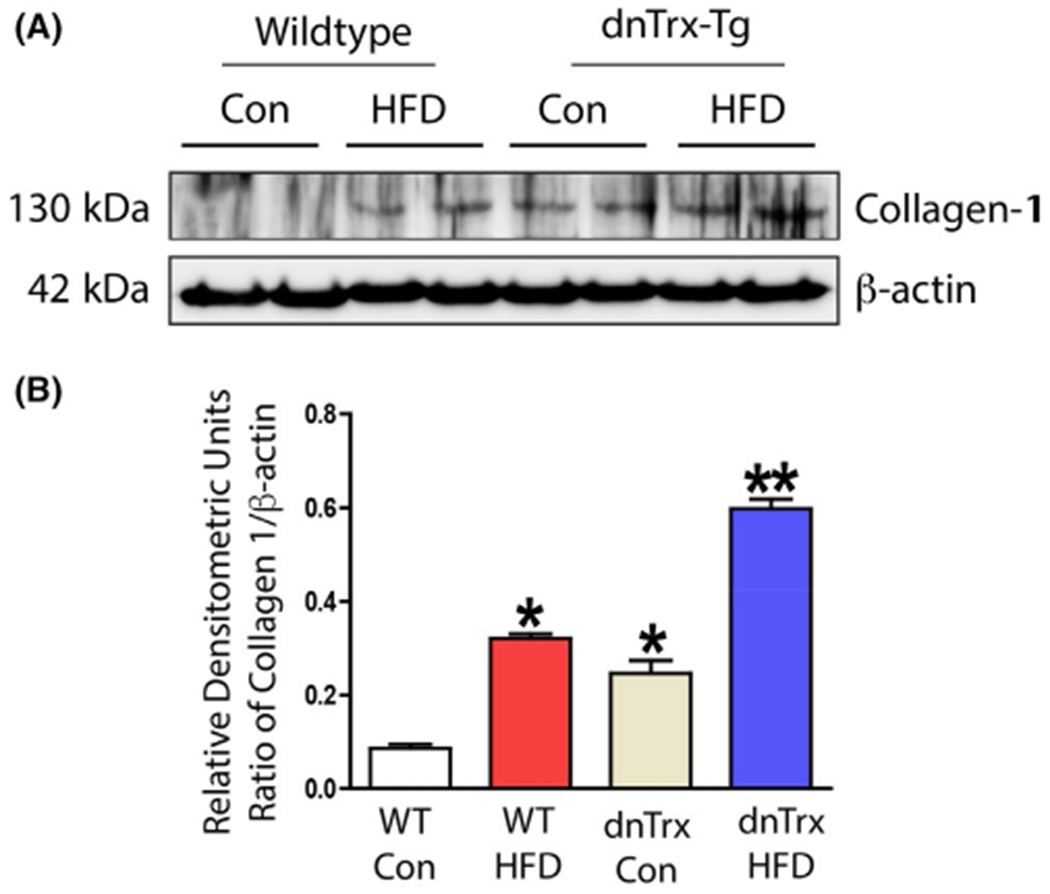
Increased expression of collagen in dnTrx-Tg mice fed NC or HFD: Small mesenteric arteries were isolated from NT or dnTrx-Tg mice fed NC or HFD. Pooled arterial lysates were prepared as described in the methods and western analysis was performed as described in the methods. (A) Analysis of collagen and β-actin, (B) Densitometry of A. Number of mice (pooled samples of n = 3, males and females)
3.9 |. FeTMPyP ameliorates impairment of femoral artery endothelial function in HFS-fed mice.
To assess whether the observed effects of FeTMPyP tail vein injection were specific to the mesenteric vascular bed, we also isolated femoral arteries and studied vascular reactivity via wire myography. HFS diet did not result in increased sensitivity to 5-HT in femoral arteries from NT and dnTrx-Tg mice (Figure S1A,B), but resulted in impaired ACh-induced relaxation in NT mice (Figure S1C). Sensitivity to ACh was statistically significantly lower in femoral arteries from dn-Trx-Tg mice on a NC and did not differ between those derived from dnTrx-Tg mice on HFS diet (Figure S1D), indicating that endothelial dysfunction was already apparent in dnTrx-Tg mice on a regular diet. One-hour injection of FeTMPyP statistically significantly improved ACh-induced relaxing responses in femoral arteries from both NT and dnTrx-Tg mice on HFS diet (Figure S1E,F).
4 |. DISCUSSION
In this study, we have shown that loss of redox-active Trx accelerates the progression of T2D due to HFS diet, demonstrating a critical role of redox balance in the onset of metabolic diseases. In addition, there was significant structural remodeling and functional changes in the small resistance arteries in these mice pointing to a far-reaching consequence of high fat ingestion on the development of an adverse milieu that would accelerate the occurrence of cardiovascular disorders. We have further shown that the loss of redox-active Trx accelerates vascular endothelial dysfunction brought about by HFS diet was not limited to mesenteric artery, but also occurred in the femoral arteries demonstrating that all vascular beds are affected. The small mesenteric artery dysfunction was manifested by (1) greater inward hypertrophic remodeling, (2) arterial stiffening, (3) increased myogenic tone, (4) loss of pressure-induced autoregulation, (5) increased α1-adrenergic contractions, and (6) reduced NO bioavailability due to increased ONOO− production. These observations suggest that obesity-induced endothelial dysfunction is exaggerated by redox imbalance due to lack of functional Trx.
Myogenic arteries, including small mesenteric arteries, constrict in response to physiological pressures and dilate at physiologically lower and higher pressures due to autoregulation of blood flow for a constant perfusion of the tissue.36 This autoregulation is shown in Figure 3A reflecting maintenance of near constant lumen diameter over a broad pressure range (40–100 mmHg). Obesity increases myogenic tone in small mesenteric arteries from db/db mice,37 skeletal muscle arterioles from obese Zucker rats,38 and cerebral and skeletal muscle arterioles from streptozotocin-induced diabetic type I rats.39,40 However, our data in dnTrx-Tg mice show that progression of obesity with concomitant loss of redox balance (increased oxidant load) would exacerbate arterial remodeling and accelerate the onset of metabolic disorders. We have also shown that pressure-dependent autoregulation in small mesenteric arteries is lost in mice fed HFS diet. Although it has been suggested that pressure flow autoregulation in the SMA is not myogenic,41 we found increased sensitivity to pressure characterized by a leftward shift in pressure-myogenic tone curves (Figure 3C,D). Consequently, myogenic tone was significantly enhanced at lower pressures. ONOO− has been demonstrated to increase myogenic tone by inhibiting large-conductance Ca2+-activated K+channels (BKCa) in rat cerebral arteries.42 However, mesenteric arteries do not express BKCa channels, but ONOO− has been shown to increase myogenic tone via alternate mechanisms.43 Interestingly, the inhibitory effect of ONOO− could be restored by reduced glutathione suggesting thiol oxidation of gate-modulating cysteine residues on the BKCa channel.42 ONOO− results in coronary artery dysfunction during diabetes.44 FeTMPyP injection has been shown to exert beneficial effects in diabetes.44–46 Our observation that acute administration of FeTMPyP normalized myogenic tone and restored autoregulation confirms this hypothesis. Increased NO has been shown to attenuate SMA autoregulation, as treatment of SMA with L-NAME increased autoregulatory response.47 However, NO effect on autoregulation is organ specific47; it is pro-autoregulatory in coronary vasculature, but in kidney NO has been shown to regulate autoregulation.48
Chronic increases in myogenic tone would result in high blood pressure, and the artery wall would adapt to this increase in circumferential wall stress by structural inward hypertrophic remodeling.49–52 Our data show that NT mice on four-month HFS diet failed to develop significant structural arterial remodeling of MA compared with mice on a regular diet. These findings are in disagreement with other studies using small mesenteric arteries derived from young C57Bl/6 mice on a short-term high fat diet, who either measured a reduced medial cross-sectional area or a greater wall thickness.53,54 In our study, we used the pressure myograph system that allowed simultaneous recording of lumen diameter and arterial wall thickness. Hence, morphological analysis of formalin-fixed and paraffin-embedded mesenteric artery cross sections was not necessary. Formalin fixation results in dehydration and will result in underestimation of wall thickness. Discrepancies in structural arterial measurements may result from differences in techniques and experimental approach. In contrast, dnTrx-Tg mice on a high fat diet resulted in marked inward hypertrophic remodeling of small mesenteric arteries as evidenced by a greater wall-to-lumen ratio (Figure 5B). This inward hypertrophic structural remodeling was chronic and could not be restored by FeTMPyP injection. Inward hypertrophic remodeling of resistance-sized arteries is characteristic of that observed during hypertension.55–57 Whether obese dnTrx-Tg mice are hypertensive needs to be further elucidated. It is anticipated that elevated ROS derived from NADPH oxidases are causative of this inward hypertrophy as reviewed elsewhere.58 In addition, eNOS, which normally is “coupled” and produces NO, under some conditions, such as in the presence of excess oxidative stress or decreased tetrahydrobiopterin, is “uncoupled” and generates ·O2−. This leads to the production of ONOO−, as a result of the action of ·O2− on NO.59 In New Zealand, obese mice second-order mesenteric arteries showed increased wall-to-lumen ratio, uncoupled eNOS, and enhanced production of ONOO−.60 We also observed decreased arterial elasticity or stiffening of mesenteric arteries from obese dnTrx-Tg. This is in agreement with the observed hypertrophic inward remodeling and the stiffening of resistance arteries during obesity.53,61 Besides increased myogenic tone, obesity is characterized by augmented α1-adrenergic contraction.62 Here we report enhanced phenylephrine-induced contraction in small mesenteric arteries from obese NT and dnTrx-Tg mice. Acute infusion of FeTMPyP was able to significantly reduced sensitivity and maximal contraction to phenylephrine suggesting that elevated ONOO− signaling accounts for the enhanced contraction.
Uncoupling of eNOS and elevated ROS production has been associated with reduced NO bioavailability.63,64 An impaired NO bioavailability should manifest itself by a blunted endothelium-dependent ACh-induced vasorelaxation. Although we observed blunted ACh-induced dilatations using pressure myography, there was no significant impairment of ACh-induced vasorelaxations in MA from NT mice on HFS diet compared to NC diet. This is in disagreement with the study by Tian and colleagues, who observed endothelial dysfunction in small mesenteric arteries from mice placed on a high fat diet for 3 months,65 but in agreement with Ellis and colleagues.66 Endothelial dysfunction was observed in obese dn-Trx-Tg mice, which was due most likely to a blunted NO-mediated signaling, as FeTMPyP administration restored endothelial function in MA from both obese dnTrx-Tg. DnTrx-Tg mice express a mutant redox-inactive Trx in a dominant-negative manner that lowers the level of active Trx due to its competition with TrxR.28,29 Previously, we reported that dnTrx-Tg mice predominantly express the oxidized form of Trx (Trx-S2).29 Since Trx-(SH)2 is an electron donor for peroxiredoxins (Prx) that are known to function as a peroxynitrite reductase,67 we believe that loss of redox-active Trx could compromise the reduction of peroxynitrite resulting in significant impairment of NO production by eNOS. Our data also indicate that structural remodeling of small mesenteric arteries due to HFD could also account for the endothelial dysfunction and myogenic tone, which is accentuated in dnTrx-Tg mice. Loss of redox-active Trx induced high levels of collagen deposition in the arteries suggesting arterial stiffness could be an important mechanism for vascular dysfunction in HFD in dnTrx-Tg mice. Further, NT mice with HFD also showed high expression of collagen-1 expression indicating HFD-dependent redox state alteration could have induced collagen deposition causing increased arterial stiffness and endothelial dysfunction. These reasonings could provide a rationale for the enhanced myogenic tone, contraction, and severely blunted NO bioavailability in obese dnTrx-Tg mice.
5 |. PERSPECTIVE
Our study demonstrates that loss of redox homeostasis in obesity cause microvascular dysfunction with arterial remodeling. Increased oxidant burden in obesity and diabetes impacts microvascular redox imbalance with consequent microcirculatory abnormalities, which may result in functional impairment of many organs including heart, lung, kidney, or brain. Our study provides an important underlying mechanistic understanding of contractile dysfunction impacted by redox deficiency in the context of type II diabetes.
Supplementary Material
{kind=link}
ACKNOWLEDGMENT
The authors gratefully acknowledge the technical help by Dr. Venkatesh Kundumani-Sridharan and Dr. Kumar Subramani.
FUNDING INFORMATION
Research reported in this manuscript was supported by the National Heart Lung and Blood Institute of the National Institutes of Health under Award number R01 HL 107885, R01 HL132953, and 3R01 HL107885-05S1 to KCD.
Footnotes
CONFLICT OF INTEREST
None.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY STATEMENT
Data used in this manuscript are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. [DOI] [PubMed] [Google Scholar]
- 2.Prieto D, Contreras C, Sanchez A. Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol. 2014;12:412–426. [DOI] [PubMed] [Google Scholar]
- 3.Rao A, Pandya V, Whaley-Connell A. Obesity and insulin resistance in resistant hypertension: implications for the kidney. Adv Chronic Kidney Dis. 2015;22:211–217. [DOI] [PubMed] [Google Scholar]
- 4.Xia N, Li H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br J Pharmacol. 2017;174(20):3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender SB, Castorena-Gonzalez JA, Garro M, et al. Regional variation in arterial stiffening and dysfunction in Western diet-induced obesity. Am J Physiol Heart Circ Physiol. 2015;309:H574–H582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aroor AR, Jia G, Sowers JR. Cellular mechanisms underlying obesity-induced arterial stiffness. Am J Physiol Regul Integr Comp Physiol. 2018;314:R387–R398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn SM, Hilgers RH, Das KC. Decreased EDHF-mediated relaxation is a major mechanism in endothelial dysfunction in resistance arteries in aged mice on prolonged high-fat sucrose diet. Physiol Rep. 2017;5:e13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lastra G, Manrique C, Jia G, et al. Xanthine oxidase inhibition protects against Western diet-induced aortic stiffness and impaired vasorelaxation in female mice. Am J Physiol Regul Integr Comp Physiol. 2017;313:R67–R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gil-Ortega M, Condezo-Hoyos L, Garcia-Prieto CF, et al. Imbalance between pro and anti-oxidant mechanisms in perivascular adipose tissue aggravates long-term high-fat diet-derived endothelial dysfunction. PLoS One. 2014;9:e95312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noronha BT, Li JM, Wheatcroft SB, Shah AM, Kearney MT. Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes. 2005;54:1082–1089. [DOI] [PubMed] [Google Scholar]
- 12.Winters B, Mo Z, Brooks-Asplund E, et al. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice. J Appl Physiol (1985). 2000;89:2382–2390. [DOI] [PubMed] [Google Scholar]
- 13.McCall TB, Boughton-Smith NK, Palmer RM, Whittle BJ, Moncada S. Synthesis of nitric oxide from L-arginine by neutrophils. Release and interaction with superoxide anion. Biochem J. 1989;261:293–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilgers RH, Kundumani-Sridharan V, Subramani J, et al. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci Transl Med. 2017;9:eaaf6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y, Rajapakse AG, Montani JP, Yang Z, Ming XF. p38 mitogen-activated protein kinase is involved in arginase-II-mediated eNOS-uncoupling in obesity. Cardiovasc Diabetol. 2014;13:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han CY. Roles of reactive oxygen species on insulin resistance in adipose tissue. Diabetes Metab J. 2016;40:272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ J. 2010;74:1479–1487. [DOI] [PubMed] [Google Scholar]
- 18.de Jongh RT, Serne EH, IJzerman RG, de Vries G, Stehouwer CD. Impaired microvascular function in obesity: implications for obesity-associated microangiopathy, hypertension, and insulin resistance. Circulation. 2004;109:2529–2535. [DOI] [PubMed] [Google Scholar]
- 19.Erdei N, Toth A, Pasztor ET, et al. High-fat diet-induced reduction in nitric oxide-dependent arteriolar dilation in rats: role of xanthine oxidase-derived superoxide anion. Am J Physiol Heart Circ Physiol. 2006;291:H2107–H2115. [DOI] [PubMed] [Google Scholar]
- 20.Woodman CR, Thompson MA, Turk JR, Laughlin MH. Endurance exercise training improves endothelium-dependent relaxation in brachial arteries from hypercholesterolemic male pigs. J Appl Physiol (1985). 2005;99:1412–1421. [DOI] [PubMed] [Google Scholar]
- 21.Holmgren A, Bjornstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995;252:199–208. [DOI] [PubMed] [Google Scholar]
- 22.Holmgren A, Johansson C, Berndt C, Lonn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005;33:1375–1377. [DOI] [PubMed] [Google Scholar]
- 23.Das KC, Das CK. Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: redox independent functions. Biochem Biophys Res Commun. 2000;277:443–447. [DOI] [PubMed] [Google Scholar]
- 24.Das KC, Lewis-Molock Y, White CW. Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am J Respir Cell Mol Biol. 1997;17:713–726. [DOI] [PubMed] [Google Scholar]
- 25.Venojarvi M, Korkmaz A, Aunola S, et al. Decreased thioredoxin-1 and increased HSP90 expression in skeletal muscle in subjects with type 2 diabetes or impaired glucose tolerance. Biomed Res Int. 2014;2014:386351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stern MP, Haffner SM. Body fat distribution and hyperinsulinemia as risk factors for diabetes and cardiovascular disease. Arteriosclerosis. 1986;6:123–130. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto M, Yamato E, Toyoda S, et al. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal. 2008;10:43–49. [DOI] [PubMed] [Google Scholar]
- 28.Das KC. Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;308:L429–L442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilgers RH, Das KC. Role of in vivo vascular redox in resistance arteries. Hypertension. 2015;65:130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oblong JE, Berggren M, Gasdaska PY, Powis G. Site-directed mutagenesis of active site cysteines in human thioredoxin produces competitive inhibitors of human thioredoxin reductase and elimination of mitogenic properties of thioredoxin. J Biol Chem. 1994;269:11714–11720. [PubMed] [Google Scholar]
- 31.Yamamoto M, Yang G, Hong C, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest. 2003;112:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunn SM, Hilgers R, Das KC. Decreased EDHF-mediated relaxation is a major mechanism in endothelial dysfunction in resistance arteries in aged mice on prolonged high-fat sucrose diet. Physiol Rep. 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergel DH. The static elastic properties of the arterial wall. J Physiol. 1961;156:445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halpern W, Mulvany MJ. Tension responses to small length changes of vascular smooth muscle cells [proceedings]. J Physiol. 1977;265:21P–23P. [PubMed] [Google Scholar]
- 35.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson PC. Autoregulation of blood flow. Circ Res. 1986;59:483–495. [DOI] [PubMed] [Google Scholar]
- 37.Lagaud GJ, Masih-Khan E, Kai S, van Breemen C, Dube GP. Influence of type II diabetes on arterial tone and endothelial function in murine mesenteric resistance arteries. J Vase Res. 2001;38:578–589. [DOI] [PubMed] [Google Scholar]
- 38.Frisbee JC, Maier KG, Stepp DW. Oxidant stress-induced increase in myogenic activation of skeletal muscle resistance arteries in obese Zucker rats. Am J Physiol Heart Circ Physiol. 2002;283:H2160–H2168. [DOI] [PubMed] [Google Scholar]
- 39.Ungvari Z, Pacher P, Kecskemeti V, Papp G, Szollar L, Koller A. Increased myogenic tone in skeletal muscle arterioles of diabetic rats. Possible role of increased activity of smooth muscle Ca2+ channels and protein kinase C. Cardiovasc Res. 1999;43:1018–1028. [DOI] [PubMed] [Google Scholar]
- 40.Zimmermann PA, Knot HJ, Stevenson AS, Nelson MT. Increased myogenic tone and diminished responsiveness to ATP-sensitive K+ channel openers in cerebral arteries from diabetic rats. Circ Res. 1997;81:996–1004. [DOI] [PubMed] [Google Scholar]
- 41.Lautt WW. Autoregulation of superior mesenteric artery is blocked by adenosine antagonism. Can J Physiol Pharmacol. 1986;64:1291–1295. [DOI] [PubMed] [Google Scholar]
- 42.Brzezinska AK, Gebremedhin D, Chilian WM, Kalyanaraman B, Elliott SJ. Peroxynitrite reversibly inhibits Ca(2+)-activated K(+) channels in rat cerebral artery smooth muscle cells. Am J Physiol Heart Circ Physiol. 2000;278:H1883–H1890. [DOI] [PubMed] [Google Scholar]
- 43.Maneen MJ, Cipolla MJ. Peroxynitrite diminishes myogenic tone in cerebral arteries: role of nitrotyrosine and F-actin. Am J Physiol Heart Circ Physiol. 2007;292:H1042–H1050. [DOI] [PubMed] [Google Scholar]
- 44.Drel VR, Pacher P, Vareniuk I, et al. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int J Mol Med. 2007;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- 45.Cassuto J, Dou H, Czikora I, et al. Peroxynitrite disrupts endothelial caveolae leading to eNOS uncoupling and diminished flow-mediated dilation in coronary arterioles of diabetic patients. Diabetes. 2014;63:1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drel VR, Pacher P, Vareniuk I, et al. A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur J Pharmacol. 2007;569:48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Macedo MP, Lautt WW. Autoregulatory capacity in the superior mesenteric artery is attenuated by nitric oxide. Am J Physiol. 1996;271:G400–G404. [DOI] [PubMed] [Google Scholar]
- 48.Kiyomoto H, Matsuo H, Tamaki T, et al. Effect of L-NG-nitroarginine, inhibitor of nitric oxide synthesis, on autoregulation of renal blood flow in dogs. Jpn J Pharmacol. 1992;58:147–155. [DOI] [PubMed] [Google Scholar]
- 49.Jimenez-Altayo F, Martin A, Rojas S, et al. Transient middle cerebral artery occlusion causes different structural, mechanical, and myogenic alterations in normotensive and hypertensive rats. Am J Physiol Heart Circ Physiol. 2007;293:H628–H635. [DOI] [PubMed] [Google Scholar]
- 50.Prewitt RL, Rice DC, Dobrian AD. Adaptation of resistance arteries to increases in pressure. Microcirculation. 2002;9:295–304. [DOI] [PubMed] [Google Scholar]
- 51.Sonoyama K, Greenstein A, Price A, Khavandi K, Heagerty T. Vascular remodeling: implications for small artery function and target organ damage. Ther Adv Cardiovasc Dis. 2007;1:129–137. [DOI] [PubMed] [Google Scholar]
- 52.Trask AJ, Katz PS, Kelly AP, et al. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J Appl Physiol (1985). 2012;113:1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foote CA, Castorena-Gonzalez JA, Ramirez-Perez FI, et al. Arterial stiffening in western diet-fed mice is associated with increased vascular elastin, transforming GroNTh factor-beta, and plasma neuraminidase. Front Physiol. 2016;7:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu H, Garver H, Fernandes R, et al. BKchannel beta1-subunit deficiency exacerbates vascular fibrosis and remodelling but does not promote hypertension in high-fat fed obesity in mice. J Hypertens. 2015;33:1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amiri F, Virdis A, Neves MF, et al. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation. 2004;110:2233–2240. [DOI] [PubMed] [Google Scholar]
- 56.Ko EA, Amiri F, Pandey NR, et al. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292:H1789–H1795. [DOI] [PubMed] [Google Scholar]
- 57.Pistea A, Bakker EN, Spaan JA, Hardeman MR, van Rooijen N, VanBavel E. Small artery remodeling and erythrocyte deformability in L-NAME-induced hypertension: role of transglutaminases. J Vase Res. 2008;45:10–18. [DOI] [PubMed] [Google Scholar]
- 58.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. [DOI] [PubMed] [Google Scholar]
- 59.Schiffrin EL. Oxidative stress, nitric oxide synthase, and superoxide dismutase: a matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension. 2008;51:31–32. [DOI] [PubMed] [Google Scholar]
- 60.Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL. Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension. 2009;54:1384–1392. [DOI] [PubMed] [Google Scholar]
- 61.Seifalian AM, Filippatos TD, Joshi J, Mikhailidis DP. Obesity and arterial compliance alterations. Curr Vase Pharmacol. 2010;8:155–168. [DOI] [PubMed] [Google Scholar]
- 62.Stepp DW, Frisbee JC. Augmented adrenergic vasoconstriction in hypertensive diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol. 2002;282:H816–H820. [DOI] [PubMed] [Google Scholar]
- 63.Lee DY, Wauquier F, Eid AA, et al. Nox4 NADPH oxidase mediates peroxynitrite-dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem. 2013;288:28668–28686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11:89–97. [DOI] [PubMed] [Google Scholar]
- 65.Tian XY, Wong NT, Xu A, et al. Uncoupling protein-2 protects endothelial function in diet-induced obese mice. Circ Res. 2012;110:1211–1216. [DOI] [PubMed] [Google Scholar]
- 66.Ellis A, Cheng ZJ, Li Y, et al. Effects of a Western diet versus high glucose on endothelium-dependent relaxation in murine micro- and macro-vasculature. Eur J Pharmacol. 2008;601:111–117. [DOI] [PubMed] [Google Scholar]
- 67.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data used in this manuscript are available from the corresponding author upon reasonable request.