

LETTER
Next-generation sequencing technology (NGS) has rapidly advanced and emerged from research applications to find growing adoption by clinical laboratories for use in patient care. With the emergence of variants, there is growing interest for SARS-CoV-2 genotyping by whole-genome sequencing (WGS) in clinical care as it enables detection of mutations that confer resistance to antiviral and monoclonal antibody therapies (1). Furthermore, we and others have demonstrated its use for infection control investigations (2–4). Despite the clinical utility of NGS assays, adoption by clinical laboratories has been limited, largely due to the molecular skills required and sufficient bioinformatics expertise needed for sequencing data analysis (5).
During our transition from a SARS-CoV-2 genotyping research endeavor to a clinical assay with reporting in the electronic medical record (EMR), we evaluated a WGS methodology using the AmpliSeq SARS-CoV-2 Insight Research Assay on Ion Torrent Genexus system (Thermo Fisher Scientific; Waltham, MA) that allowed for on-board library preparation and sequencing. This platform significantly reduces the amount of hands-on time and training needed. We compared the performance of the Ampliseq assay to generate high quality sequence data and SARS-CoV-2 variant calls against CleanPlex SARS-CoV-2 Panel (Paragon Genomics; Hayward, CA) on Illumina MiSeq. The CleanPlex and Ampliseq amplicon based WGS assays utilize 343 and 237 overlapping amplicons that span the entire SARS-CoV-2 genome, respectively (6, 7). Samples yielding sequence data that met the APHL recommended minimum criteria for submission to GISAID of >90% target base coverage and average coverage depth ≥10× were included in this study (8). Further, to ensure high confidence in lineage calls for a clinical assay we set the following thresholds for sequencing quality: average coverage depth >1,000×, base reads on-target >90%, and target base coverage at 100× >98%. A total of 75 samples met the APHL recommended criteria; of these, 91% (68/75) and 75% (56/75) of samples met our more stringent QC thresholds for the Ampliseq and CleanPlex assays, respectively (Table 1). This demonstrates that the Ampliseq assay was not inferior to the CleanPlex assay in its ability to generate high quality data. Since variants were included from different phases of the pandemic, the original CleanPlex samples had lineage calls performed at different time points using different versions of the Pangolin COVID-19 Lineage Assigner. To avoid this source of analysis variability, consensus sequences generated by both methods were analyzed using the same version of Pangolin (version 3.1.19, lineages version 2022-01-20). Lineage calls generated by the two assays were considered concordant if the same variant classification scheme defined by the SARS-CoV-2 Interagency Group (SIG) were identified (9). The lineage calls were also considered concordant when a nonvariant was identified by both methods. The two methodologies were highly concordant at 94.7% (71/75) and 98.2% (55/56) when comparing lineage calls of samples that met APHL recommended criteria and our more stringent QC criteria, respectively (Table 1). We were able to accurately detect multiple variants including alpha, epsilon, and delta. The discordant result from samples that met the more stringent QC threshold had 2-fold greater average coverage depth using the Ampliseq assay at 4,654×, compared to 1,800× for the CleanPlex.
TABLE 1.
Concordance between Ion AmpliSeq SARS-CoV-2 and reference method
| Specimen number | Ct value | Paragon Genomics CleanPlexa | Ion AmpliSeq SARS-CoV-2 | Avg base coverage depth | Base reads on target | Target base coverage at 100× | Concordance |
|---|---|---|---|---|---|---|---|
| 1 | 29.38 | B.1.2 | B.1.2 | 3,165 | 99.58% | 99.83% | Yes |
| 2 | 27.38 | B.1.427 | B.1.427 | 3,418 | 99.76% | 99.87% | Yes |
| 3 | 26.57 | B.1.240 | B.1.240 | 4,114 | 99.81% | 99.91% | Yes |
| 4 | 25.6 | B.1.1 | B.1.1 | 4,060 | 99.94% | 99.84% | Yes |
| 5 | 27.87 | B.1.429 | B.1.429 | 3,619 | 99.75% | 99.68% | Yes |
| 6 | 24.27 | B.1.1 | B.1.1 | 4,809 | 99.45% | 99.65% | Yes |
| 7 | 23.88 | B.1.1.7 | B.1.1.7 | 4,189 | 99.89% | 99.71% | Yes |
| 8 | 22.83 | P.1.17 | P.1.17 | 4,374 | 99.91% | 99.65% | Yes |
| 9 | 27.33 | AY.20 | AY.20 | 3,834 | 99.55% | 99.32% | Yes |
| 10 | 29.88 | AY.103 | AY.103 | 1,875 | 99.81% | 99.57% | Yes |
| 11 | 28.49 | AY.100 | AY.100 | 2,431 | 99.43% | 98.70% | Yes |
| 12 | 29.11 | AY.26 | AY.26 | 2,248 | 98.54% | 99.41% | Yes |
| 13 | 31.82 | B.1.2 | B.1.2 | 2,631 | 98.52% | 99.82% | Yes |
| 14 | 31.27 | B.1.2 | B.1.2 | 994 | 96.61% | 98.36% | Yes |
| 15 | 30.34 | B.1.429 | B.1.429 | 9,205 | 98.23% | 99.78% | Yes |
| 16 | 30.17c | B.1.429 | B.1.429 | 623 | 94.90% | 95.47% | Yes |
| 17 | 31.45 | B.1.429 | B.1.361 | 4,654 | 99.69% | 99.87% | No |
| 18 | 30.8 | None | B.1.429 | 2,090 | 97.93% | 99.73% | No |
| 19 | 30.34 | B.1.427 | B.1.429 | 454 | 78.76% | 88.26% | Yesb |
| 20 | 18.87 | B.1.427 | B.1.427 | 7,644 | 99.93% | 99.87% | Yes |
| 21 | 25.55 | B.1.427 | B.1.427 | 2,168 | 99.08% | 99.70% | Yes |
| 22 | 31.36c | B | B.1.429 | 3,340 | 91.18% | 99.57% | No |
| 23 | 28.5c | B.1.427 | B.1.429 | 1,690 | 93.30% | 99.05% | Yesb |
| 24 | 15 | B.1.526 | B.1.526 | 1,949 | 99.95% | 99.68% | Yes |
| 25 | 20.64 | P.1 | P.1 | 3,692 | 99.96% | 99.67% | Yes |
| 26 | 27.04 | AY.44 | AY.44 | 2,725 | 99.93% | 99.76% | Yes |
| 27 | 10.82 | BB.2 | BB.2 | 8,006 | 99.95% | 99.57% | Yes |
| 28 | 18.57 | P.1 | P.1 | 10,070 | 99.94% | 99.87% | Yes |
| 29 | 28.11 | B.1.241 | B.1.241 | 1,949 | 99.87% | 99.68% | Yes |
| 30 | 26.81c | B.1.1.239 | B.1.1.239 | 498 | 94.46% | 96.82% | Yes |
| 31 | 15.1 | B.1.1.291 | B.1.1.291 | 9,327 | 99.93% | 99.82% | Yes |
| 32 | 16.62 | B.1.526 | B.1.526 | 8,781 | 99.95% | 99.83% | Yes |
| 33 | 21.33 | B.1.1.7 | B.1.1.7 | 2,633 | 99.04% | 99.23% | Yes |
| 34 | 29.75c | B | B.1.429 | 327 | 79.92% | 90.43% | No |
| 35 | 14.45 | B.1.1.7 | B.1.1.7 | 2,998 | 99.80% | 99.68% | Yes |
| 36 | 19.37 | P.1 | P.1 | 5,640 | 99.77% | 99.62% | Yes |
| 37 | 20.98 | P.1.17 | P.1.17 | 6,679 | 99.79% | 99.83% | Yes |
| 38 | 24.9 | AY.103 | AY.103 | 2,730 | 98.69% | 99.28% | Yes |
| 39 | 19.81 | AY.119 | AY.119 | 3,383 | 99.56% | 98.81% | Yes |
| 40 | 16.67 | AY.116.1 | AY.116.1 | 6,547 | 99.76% | 99.82% | Yes |
| 41 | 17.77 | B.1.637 | B.1.637 | 4,703 | 99.70% | 99.77% | Yes |
| 42 | 18.34 | B.1.526 | B.1.526 | 3,220 | 99.78% | 99.59% | Yes |
| 43 | 20.18 | B.1.1.7 | B.1.1.7 | 3,749 | 99.80% | 99.97% | Yes |
| 44 | 19.59 | B.1.1.7 | B.1.1.7 | 4,018 | 99.79% | 99.83% | Yes |
| 45 | 27.37 | B.1.421 | B.1.421 | 2,039 | 98.76% | 99.66% | Yes |
| 46 | 24.48 | B.1.421 | B.1.421 | 3,585 | 99.73% | 99.40% | Yes |
| 47 | 19.75 | B.1.421 | B.1.421 | 5,402 | 99.79% | 99.80% | Yes |
| 48 | 14.42 | B.1.421 | B.1.421 | 10,677 | 99.75% | 99.91% | Yes |
| 49 | 28.81 | B.1.404 | B.1.404 | 1,315 | 96.18% | 99.62% | Yes |
| 50 | 27.86 | B.1.404 | B.1.404 | 4,447 | 99.80% | 99.54% | Yes |
| 51 | 28.03 | B.1 | B.1.438.4 | 1,136 | 95.45% | 98.43% | Yesb |
| 52 | 27.03 | B.1.399 | B.1.399 | 4,699 | 99.76% | 98.96% | Yes |
| 53 | 17.42 | B.1.399 | B.1.399 | 20,056 | 99.96% | 99.09% | Yes |
| 54 | 14.77 | B.1.399 | B.1.399 | 5,681 | 99.96% | 99.74% | Yes |
| 55 | 25.64 | B | B.1.324 | 1,016 | 98.23% | 99.37% | Yesb |
| 56 | 25.53 | B | B.1.609 | 2,834 | 97.45% | 99.37% | Yesb |
| 57 | 18.71 | B.1.324 | B.1.324 | 8,486 | 99.96% | 99.84% | Yes |
| 58 | 14.06 | B.1.298 | B.1.298 | 4,901 | 99.97% | 100% | Yes |
| 59 | 19.54 | B.1 | B.1.265 | 6,085 | 99.94% | 99.44% | Yesb |
| 60 | 25.76 | B.1.232 | B.1.232 | 3,925 | 99.77% | 99.13% | Yes |
| 61 | 24.19 | B.1.232 | B.1.232 | 3,213 | 97.54% | 99.39% | Yes |
| 62 | 25.9 | B.1.126 | B.1.126 | 3,699 | 99.78% | 98.30% | Yes |
| 63 | 16.35 | B.1.429 | B.1.429 | 9,198 | 99.97% | 99.82% | Yes |
| 64 | 23.84 | B.1.1.7 | B.1.1.7 | 6,214 | 99.95% | 99.98% | Yes |
| 65 | 27.82 | B.1.1.228 | B.1.1.228 | 2,319 | 99.87% | 99.02% | Yes |
| 66 | 29.39 | AY.54 | AY.54 | 2,607 | 99.77% | 99.44% | Yes |
| 67 | 21.89 | BB.2 | BB.2 | 10,089 | 99.89% | 99.78% | Yes |
| 68 | 21.34 | B.1.526 | B.1.526 | 10,482 | 99.97% | 99.73% | Yes |
| 69 | 26.24 | B.1.126 | B.1.126 | 6,545 | 99.71% | 98.51% | Yes |
| 70 | 13.94 | AY.103 | AY.103 | 5,381 | 99.96% | 99.78% | Yes |
| 71 | 14.06 | P.1 | P.1 | 8,572 | 99.95% | 99.93% | Yes |
| 72 | 17.4 | B.1.1.7 | B.1.1.7 | 6,248 | 99.95% | 99.99% | Yes |
| 73 | 14.02 | B.1.1.7 | B.1.1.7 | 17,717 | 99.94% | 100% | Yes |
| 74 | 16.35 | B.1.429 | B.1.429 | 4,951 | 99.95% | 99.79% | Yes |
| 75 | 16.35 | B.1.429 | B.1.429 | 19,243 | 99.94% | 99.96% | Yes |
Underlined CleanPlex lineage calls did not pass more stringent QC thresholds.
Lineage calling was interpreted as concordant when Pango nomenclatures (e.g., B.1.617.2, AY.X) classified as the same variant based on SIG classification (i.e., Delta) or both Pango nomenclatures are considered a nonvariant that will only be reported out as nonvariant.
Samples did not meet QC cutoffs on first attempt. These samples were repeated and pooled with other low titer samples. Low titer was considered Ct >25.5 or approximately 1,000 copies/mL. QC metrics were improved and lineage calls were unchanged. Results are from low titer pooled repeat testing run.
Following implementation, all clinical specimens positive for SARS-CoV-2 by RT-PCR with sufficient viral titer (cycle threshold (Ct) <30) are automatically reflexed to WGS. Samples were batched into full runs of 14 patient samples plus 2 controls, twice weekly, to allow for turnaround time (TAT) range of 24–96 h. While the Ampliseq assay has about half the throughput of the CleanPlex assay on a weekly basis, it requires less than a tenth of the hands-on time (Table 2). Weekly testing throughput quickly expanded as we transitioned to Monday–Friday testing once a case of S-gene dropout detected by the TaqPath COVID-19 RT-PCR assay suspicious for the omicron variant (lineage B.1.1.529) was identified in December 2021. The automated features of the Ion Torrent Genexus system allowed us to quickly reflex to SARS-CoV-2 WGS. Within 30 h of initial SARS-CoV-2 detection by RT-PCR, the omicron variant was reported in the EMR. The analysis was significantly simplified through software plugins, which automatically list mutations and produce SARS-CoV-2 variant calls (Table 2). The time to genotyping result was approximately 44 h from the time specimen was received by the laboratory. Such rapid TATs can facilitate clinically actionable results, enabling critical outbreak investigations and therapeutic management within days rather than weeks. Since implementation, the genomic epidemiology has rapidly transitioned to omicron predominance (Fig. 1).
TABLE 2.
Logistical differences between assays
| Category | Ampliseq assay | CleanPlex assay |
|---|---|---|
| Platform | Ion Torrent Genexus | Illumina MiSeq |
| Library prep | Automated | Manual |
| Wet lab hands-on time | 10–20 mins | 245 mins |
| Analysis hands-on time | Fully automated | 95 mins |
| Cost per testa | $$$ | $$ |
| TATb | 1 run/24 hrs | 1 run/96 hrs |
| Throughput | 14 samples/24 hrs | 144 samples/wk |
Includes costs for reagents and consumables only.
Estimated without overnight or weekend staffing.
FIG 1.
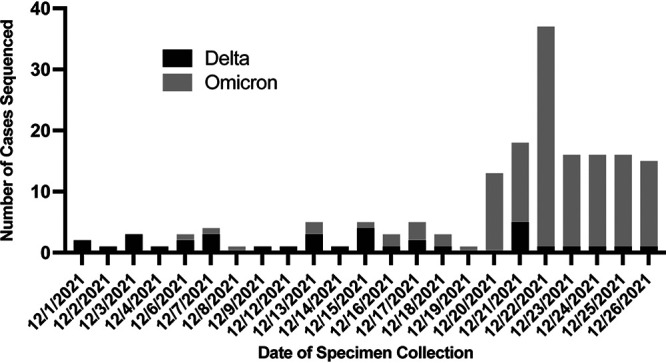
Number of cases of delta and omicron variants that were sequenced at our institution during the month of December 2021. Specimens included were positive by RT-PCR and had sufficient viral titer for sequencing (Ct <30) (n = 171).
The system currently has some limitations. The throughput is only 16 samples per run for this assay, including any controls. There is potential for reagent waste if processing less than 16 samples per run, as one of four lanes of the Genexus chip is consumed for each automated run. When the omicron case was discovered, the throughput was sufficient as SARS-CoV-2 positivity at our institution was 5.47%, but as case positivity increases, the resulting backlog could increase TATs or require prioritization of samples. On the other hand, the low throughput may be more beneficial in some laboratories. There is also the flexibility to increase throughput by switching to manual library prep. While this does increase the wet lab hands-on time, it still takes advantage of the automated analysis. Sequencing data quality is reduced when loading mixed viral titer samples onto the instrument in the same run but can be improved by batching samples of similar titers together.
Overall, this methodology provides a significantly simplified workflow that makes it possible to generate real-time in-house WGS results and allows for a routine clinical laboratory to pursue genomic surveillance. These technological advances could have lasting benefit by offering clinical microbiology laboratories realistic tools to explore other infectious diseases NGS assays, from genotyping other viruses for the purposes of detecting resistance markers to inform antiviral therapy to identification of bacterial and fungal pathogens from clinical specimens.
Contributor Information
Jennifer Dien Bard, Email: jdienbard@chla.usc.edu.
Yi-Wei Tang, Cepheid.
REFERENCES
- 1.Greninger AL, Bard JD, Colgrove RC, Graf EH, Hanson KE, Hayden MK, Humphries RM, Lowe CF, Miller MB, Pillai DR, Rhoads DD, Yao JD, Lee FM. 2022. Clinical and infection prevention applications of severe acute respiratory syndrome coronavirus 2 genotyping: an Infectious Diseases Society of America/American Society for Microbiology consensus review document. J Clin Microbiol 60:e01659-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryutov A, Gai X, Ostrow D, Maglinte DT, Flores J, Salas EJ, Glucoft M, Smit M, Bard JD. 2021. Utility of viral whole-genome sequencing for institutional infection surveillance during the coronavirus disease 2019 (COVID-19) pandemic. Infect Control Hosp Epidemiol 1–2. doi: 10.1017/ice.2021.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meredith LW, Hamilton WL, Warne B, Houldcroft CJ, Hosmillo M, Jahun AS, Curran MD, Parmar S, Caller LG, Caddy SL, Khokhar FA, Yakovleva A, Hall G, Feltwell T, Forrest S, Sridhar S, Weekes MP, Baker S, Brown N, Moore E, Popay A, Roddick I, Reacher M, Gouliouris T, Peacock SJ, Dougan G, Török ME, Goodfellow I. 2020. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis 20:1263–1271. doi: 10.1016/S1473-3099(20)30562-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellingford JM, George R, McDermott JH, Ahmad S, Edgerley JJ, Gokhale D, Newman WG, Ball S, Machin N, Black GC. 2021. Genomic and healthcare dynamics of nosocomial SARS-CoV-2 transmission. Elife 10:e65453. doi: 10.7554/eLife.65453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graf EH. 2021. Finding the middle ground with the clinical laboratory’s role in SARS-CoV-2 genomic surveillance. J Clin Microbiol 59:e01816-21. doi: 10.1128/JCM.01816-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charre C, Ginevra C, Sabatier M, Regue H, Destras G, Brun S, Burfin G, Scholtes C, Morfin F, Valette M, Lina B, Bal A, Josset L. 2020. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol 6:veaa075. doi: 10.1093/ve/veaa075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plitnick J, Griesemer S, Lasek-Nesselquist E, Singh N, Lamson DM, St. George K. 2021. Whole-genome sequencing of SARS-CoV-2: assessment of the Ion Torrent AmpliSeq panel and comparison with the Illumina MiSeq ARTIC Protocol. J Clin Microbiol 59:e00649-21. doi: 10.1128/JCM.00649-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Association of Public Health Laboratories (APHL). 2021. Recommendations for SARS-CoV-2 sequence data quality & reporting. https://www.aphl.org/programs/preparedness/Crisis-Management/Documents/APHL-SARS-CoV-2-Sequencing.pdf.
- 9.Centers for Disease Control and Prevention (CDC). 2021. SARS-CoV-2 variant classifications and definitions. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html.