

Abstract
The lack of safe and effective delivery across the blood-brain barrier and the profound immune suppressive microenvironment are two main hurdles to glioblastoma (GBM) therapies. Extracellular vesicles (EVs) have been used as therapeutic delivery vehicles to GBM but with limited efficacy. We hypothesized that EV delivery to GBM can be enhanced by (i) modifying the EV surface with a brain-tumor-targeting cyclic RGDyK peptide (RGD-EV) and (ii) using bursts of radiation for enhanced accumulation. In addition, EVs were loaded with small interfering RNA (siRNA) against programmed cell death ligand-1 (PD-L1) for immune checkpoint blockade. We show that this EV-based strategy dramatically enhanced the targeting efficiency of RGD-EV to murine GBM, while the loaded siRNA reversed radiation-stimulated PD-L1 expression on tumor cells and recruited tumor-associated myeloid cells, offering a synergistic effect. The combined therapy significantly increased CD8+ cytotoxic T cells activity, halting tumor growth and prolonging animal survival. The selected cell source for EVs isolation and the presented functionalization strategy are suitable for large-scale production. These results provide an EV-based therapeutic strategy for GBM immune checkpoint therapy which can be translated to clinical applications.
Keywords: glioblastoma, extracellular vesicles, immunotherapy, radiation therapy, targeted delivery
Graphical Abstract
Glioblastoma (GBM) is the most common and deadliest tumor of the central nervous system with a median survival of less than 15 months after standard-of-care surgery, chemotherapy, and radiation therapy (RT).1,2 To date, some cancer types have responded well to immune checkpoint blockade against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or programmed cell death-ligand 1 (PD-L1).3,4 However, these therapies (nivolumab and ipilimumab) demonstrated limited to no benefit for GBM patients, presumably because of (i) the blood-brain barrier (BBB) and/or blood-brain tumor barrier (BBTB), (ii) the profound immune suppressive microenvironment in GBM, and (iii) narrow therapeutic window not allowing dose escalation with serious safety issues.5,6 Thus, there is an urgent need to develop therapeutics crossing the BBB/BBTB and targeting GBM to modulate the immune microenvironment.
Extracellular vesicles (EVs) encompassing exosomes, micro-vesicles, or apoptotic bodies, have emerged as delivery systems as well as regenerative cell-free effectors.7,8 During the past few years, many studies described the successful delivery of drugs and biologics by EVs. Indeed, EVs feature several advantages over other delivery methods such as innate stability, low immunogenicity, and intrinsic ability to cross biological barriers.9 Many efforts have been made to tailor EVs for cargo loading and targeted delivery. For instance, brain-targeting EVs were produced by engineering the donor cells to express Lamp2b fused to the neuron-specific rabies viral glycoprotein (RVG) peptide.10 We previously developed a rapid and efficient chemical method to conjugate targeting peptides onto EV surfaces.11 We used cyclo(Arg-Gly-Asp-d-Tyr-Lys) peptide [c(RGDyK)]-conjugated exosomes to deliver therapeutics to an ischemic brain by targeting integrin αvβ3 in reactive cerebral vascular endothelial cells.11 Additionally, urinary exosomes from prostate patients were used to target and deeply penetrate prostate tumors, offering a reliable mass source of tumor-targeting nanovehicles.12 As for cargo loading, popular strategies include electroporation, sonication, and incubation with hydrophobically modified compounds.13 Photosensitizers were loaded into urinary exosomes by electroporation, which successfully achieved superior photodynamic performance after delivery into cancer cells.14 The efficiency of RNA loading into EVs was enhanced using hydrophobic RNA molecules.15 Arg-Gly-Asp (RGD) peptides are well-known ligands that bind integrin αvβ3 overexpressed on different types of tumor cells and their blood vessels, including GBM. In fact, integrin αvβ3 is deferentially expressed in GBM compared with the normal brain.16 Cyclic RGD-decorated nanomaterials demonstrated successful delivery of therapeutics to the brain and tumors.17,18
RT remains the most important nonsurgical treatment against GBM. We and others have shown that short-burst radiation therapy can prime GBM for enhanced targeted delivery of nanoparticles in tumor-associated macrophage (TAM)-dependent fashion.19–21 Furthermore, RT has the potential to transform the immune landscape and sensitizes poorly immunogenic tumors to immune checkpoint blockade.22 However, tumor-associated myeloid cells (TAMCs) are largely recruited into GBM, in response to RT, with increased expression of PD-L1, leading to impairment of antitumor immunity.23 Thus, we postulated that a burst of radiation can enhance the targeting efficiency of EVs to brain tumors, while the combination of PD-L1 inhibition could provide an increased benefit for GBM treatment.
In this study, we developed c(RGDyK)-conjugated EVs (RGD-EV) and loaded them with siRNA against PD-L1. We show that short-burst radiation dramatically enhanced the targeting efficiency of RGD-EV to the brain tumor and its microenvironment, effectively attenuating radiation-induced PD-L1 expression on TAMCs as well as tumor cells. RT combined with targeted EV-based immunotherapy led to significant activation of CD8+ cytotoxic T cells, suppressing tumor growth, and extending survival of tumor-bearing mice. We provide a targeted therapeutic EVs for GBM immune checkpoint blockade primed with burst of radiation.
RESULTS
Short-Burst of Radiation Primes GBM for Targeted Delivery of RGD-EV.
We first isolated EVs from ReNcell VM (ReN) cells, a neural progenitor cell line derived from the ventral mesencephalon region of the human fetal brain, cultured in a serum-free medium for 3 days (Figure S1). These EVs were enriched for Alix and TSG101 markers (Figure 1A). Typical EV protein yield was 17–29 μg per mL of culture medium, corresponding to 3.7–6.4 × 109 particles per mL as determined by nanoparticle tracking analysis (NTA). We conjugated c(RGDyK) peptide, a brain tumor-targeting ligand,18 onto the EV surface using a fast and easy chemical modification method which we have previously described (Figure 1B).11 Briefly, the dibenzylcyclootyne (DBCO) group was introduced to the EV surface and reacted with azide-functionalized peptide to form a stable triazole linkage using copper-free click chemistry. To track EV biodistribution, Cy5.5 azide was conjugated onto their surfaces. NTA analysis and transmission electron microscopy (TEM) showed that RGD-EV were physically similar to nonfunctionalized EVs, with most particle size diameters ranging from 100 to 250 nm, with no significant differences in their morphology (Figure 1C,D). To confirm the presence of the peptide on the EV surface, we conjugated a fluorescein isothiocyanate (FITC)-labeled cyclic peptide c[RK(FITC)DyK] onto EVs following our protocol. Enzyme-linked immunosorbent assay (ELISA) against FITC revealed that c[RK(FITC)DyK] conjugated EVs produced significantly higher absorbance compared with nonconjugated EVs, while nonlabeled c(RKDyK) peptide pretreatment blocked most signals (Figure S2A,B). Further, EVs were isolated from ReN cells stably expressing tdTomato fused to palmitoylation signal (palm-tdTomato) which labels the cell membrane and corresponding EVs.24 These tdTomato-labeled EVs were also conjugated with c[RK(FITC)DyK]. Fluorescence microscopy revealed small particles corresponding to EVs where the FITC signal from the c[RK(FITC)DyK] peptide colocalized with tdTomato (Figure S2C). These data demonstrate the successful conjugation of the peptide on EVs. Further, no significant differences in fluorescence intensity (Cy5.5 or tdTomato) were observed between EVs with or without different peptide conjugation, of the same protein amount, showing that labeling efficiency was consistent among different EV pools (Figure S3A,B).
Figure 1.
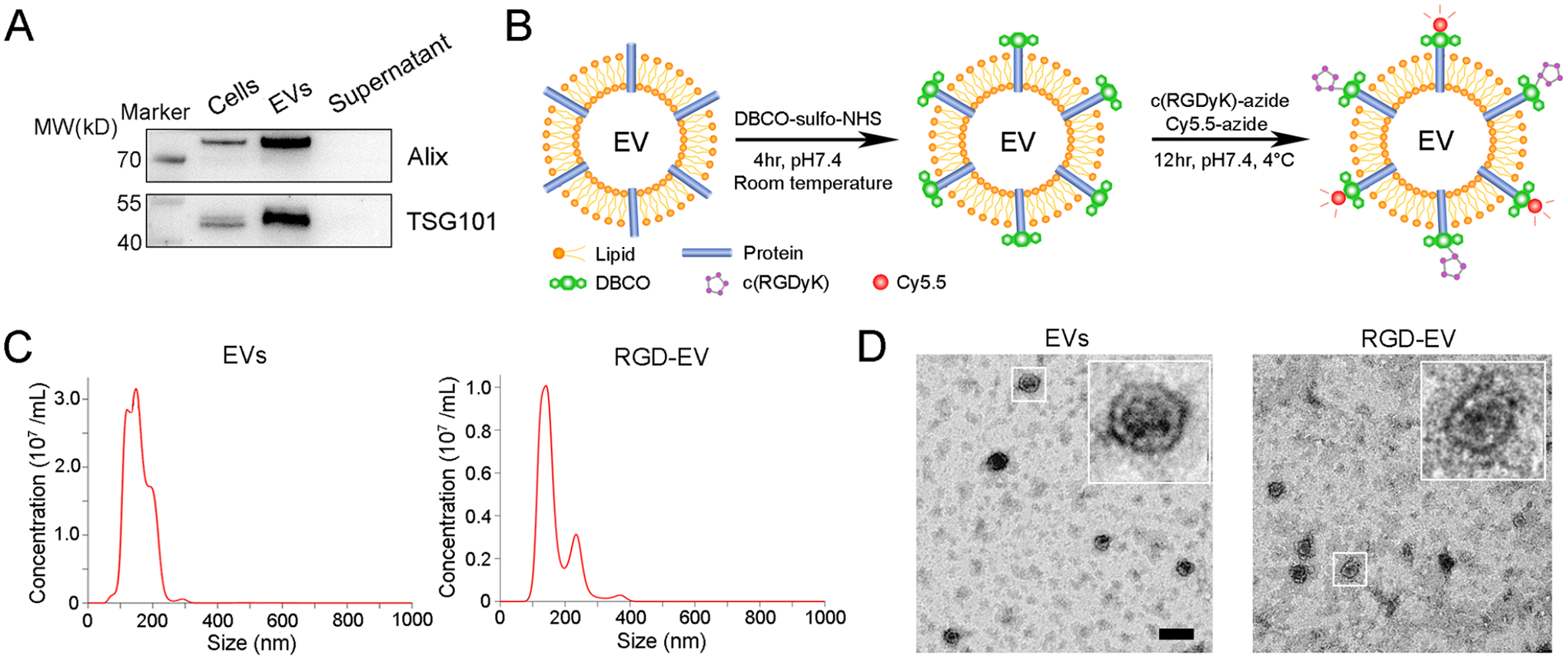
Modification and characterization of RGD-EV. (A) Western blot analysis of Alix, TSG101 in ReN cells and corresponding EVs; supernatant obtained from ultracentrifugation during EVs isolation was used as a negative control. (B) Schematic diagram summarizing conjugation of c(RGDyK) and Cy5.5 fluorophore to EV amine groups by a two-step reaction. (C,D) NTA (C) and TEM imaging (D) of unmodified EVs and RGD-EV; scale bar, 200 nm.
To evaluate the targeting ability of RGD-EV and whether short-burst radiation could enhance their uptake in GBM, we used a syngeneic mouse model by implanting GL261 cells stably expressing firefly luciferase (Fluc) and green fluorescence protein (GFP) (GL261-Fluc-GFP) in the left striatum. Tumor volume was monitored by Fluc bioluminescence imaging on day 7 postimplantation (Figure S4). Mice were then randomized in two different groups which received either 5 Gy of burst RT or control. Three days later, mice were intravenously injected with PBS or 100 μg of Cy5.5-labeled nontargeted control EVs or RGD-EVs. Brains were then removed and analyzed by near-infrared fluorescence (NIRF) imaging 24 h after administration of EVs. Interestingly, mice that received RT revealed significantly higher Cy5.5 signals in their brains, indicating that the burst of RT enhanced both EVs and RGD-EV delivery (Figure 2A–C). Particularly, RGD-EV combined with RT showed the maximum uptake by brain tumors, which produced 5.24-times higher Cy5.5 signals compared with EVs alone (Figure 2C). To confirm EVs uptake by the tumor, tdTomato-labeled RGD-EV were intravenously administered to tumor-bearing mice primed with radiation as above. Fluorescence microscopy on brain sections revealed increased tdTomato (and therefore EVs) in both GFP-positive tumor cells and CD11b-positive cells (such as TAMCs and microglia) primed with RT, compared with controls (Figure 2D; Figure S5). These results indicate that RGD-EV accumulate in GBM/microenvironment upon intravenous injection and short-burst radiation enhances their tumor targeting efficiency.
Figure 2.
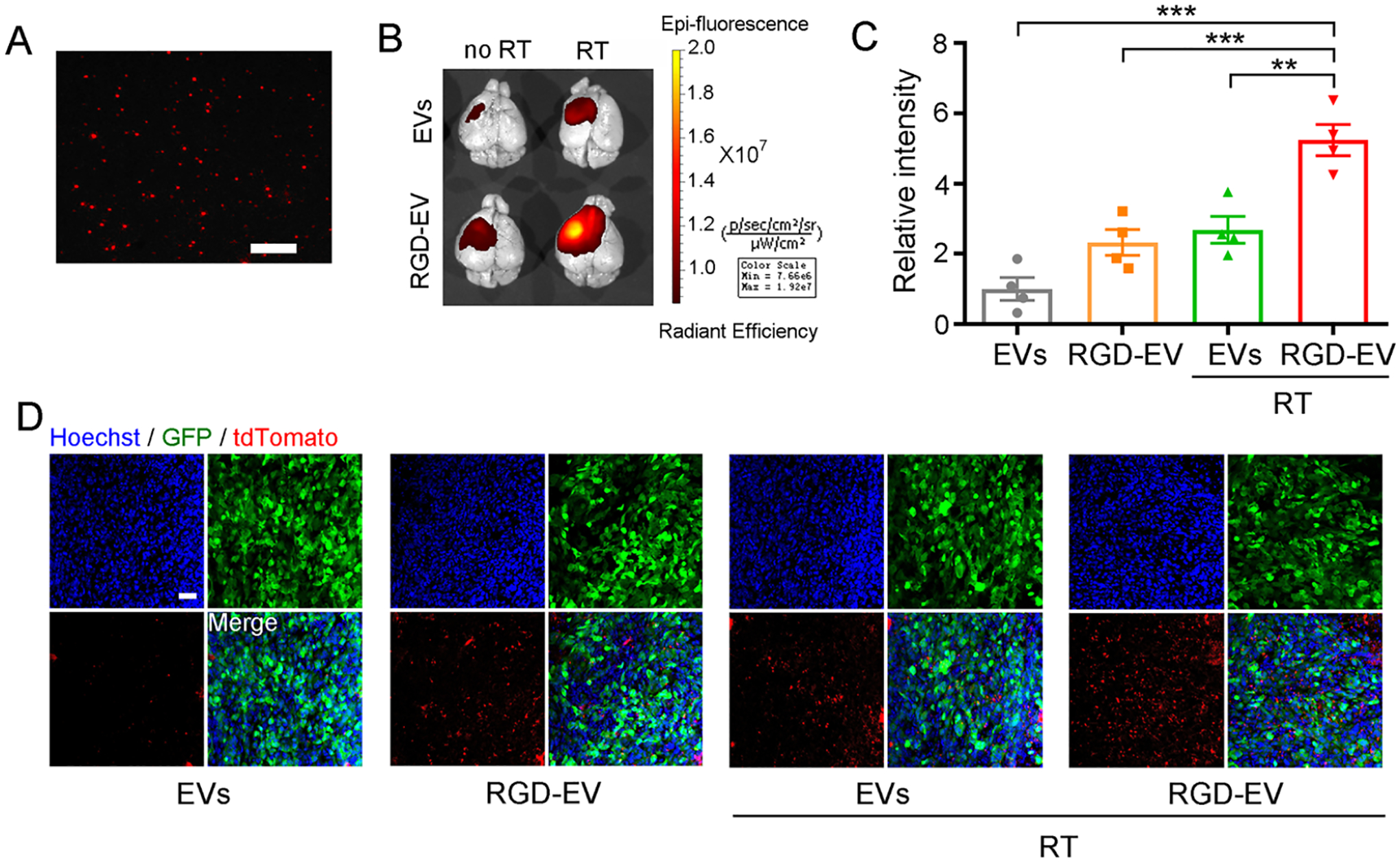
A burst of RT primes GBM for enhanced uptake of targeted RGD-EV. (A) Fluorescence image of Cy5.5-labeled RGD-EV; scale bar, 2 μm. (B,C) Mice-bearing GBM were irradiated (or not) and 3 days later injected with Cy5.5-labeled control EVs or RGD-EV. Representative NIRF images (overlaid with bright-field) of mice brain, 24 h post-EV administration (B). Quantification of fluorescence intensity in the tumor region (C); Data presented as mean ± SEM (n = 4); **P < 0.01, ***P < 0.001 by one-way ANOVA. (D) GBM-bearing mice were irradiated and injected with tdTomato-labeled EVs or RGD-EV. Representative fluorescence images of EVs (red) in GBM (GFP) 6 h post-EV administration; blue indicates nuclei; scale bar, 50 μm.
Radiation Therapy Upregulates PD-L1 in GBM and Microenvironment.
Radiation is widely used for GBM therapy to induce apoptosis through DNA damage.25 Many reports support the ability of radiation to shape TAM and host immunity, suggesting that RT can affect both tumor and immune cells.26,27 Immunofluorescence analysis on brain slices confirmed that PD-L1 was upregulated in GBM/microenvironment in response to RT (Figure 3A). We then comprehensively analyzed GBM and tumor-infiltrating immune cells in the GL261 syngeneic mouse model with and without RT. Cells were extracted from each brain and phenotypically characterized by flow cytometry (gating examples are shown in Figures S6 and S7). RT induced a significant increase in PD-L1 expression in GBM cells, TAMCs [including TAM and myeloid-derived suppressor cells (MDSCs)], and microglia but not in tumor-infiltrating lymphocytes (TILs; Figure 3B,C; Figure S8). Interestingly, as a result of RT, the abundance of tumor cells was reduced while that of TAMCs and TILs were profoundly elevated by 4 times and 3 times respectively (Figure 3D–G). These results are in line with previous reports showing that immune cells are largely recruited to GBM in response to radiation-induced DNA damage.27 In contrast, there were no significant differences in the abundance of microglia with or without RT. Taken together, given that the abundance of microglia is low (<5%), tumor cells and TAMCs were highlighted as two pivotal contributors to PD-L1-mediated immunosuppression after RT.
Figure 3.
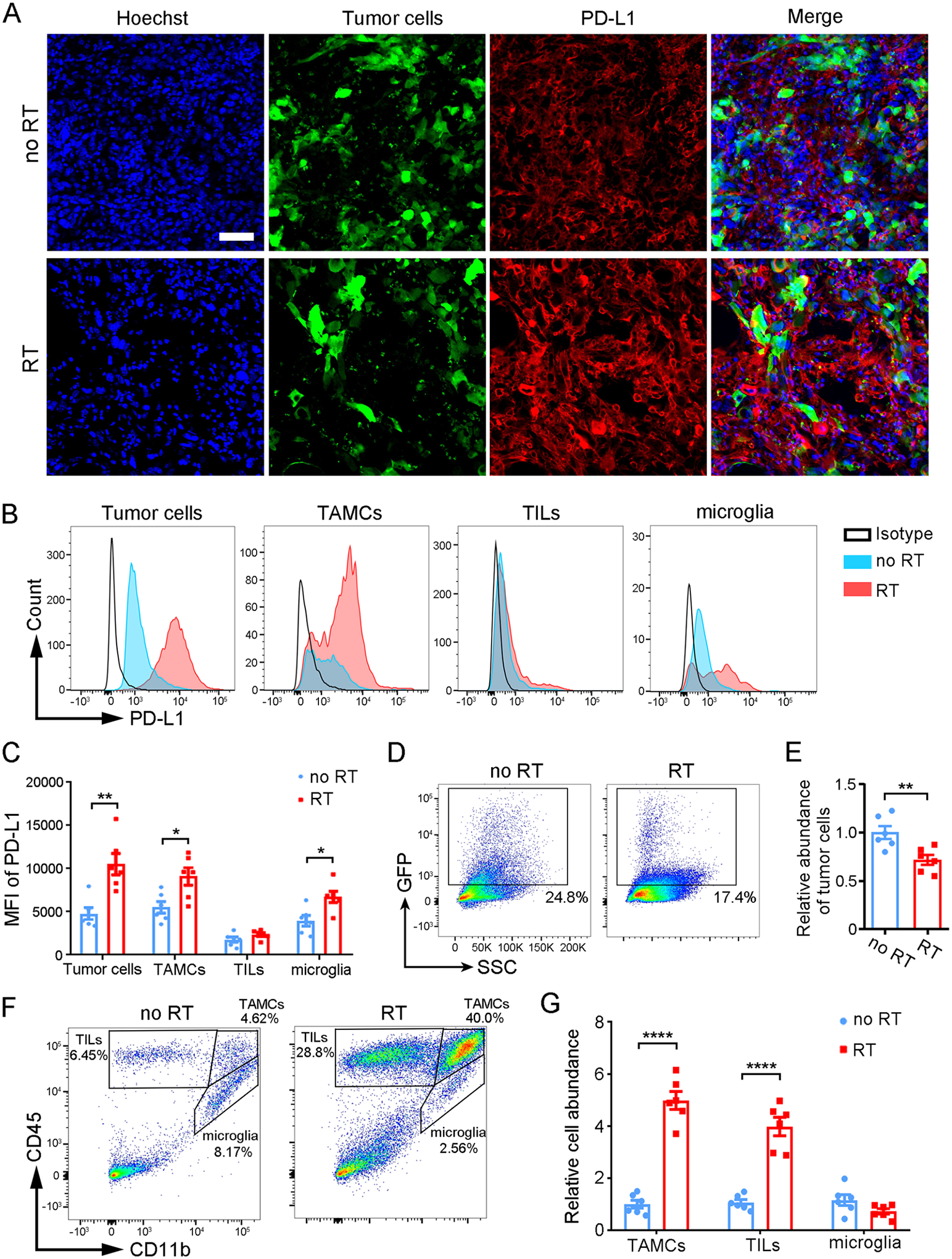
RT induces upregulation of PD-L1 in GBM and the immune microenvironment. (A) Fluorescence analysis on GL261 brain tumor (GFP) slices from irradiated and nonirradiated tumors after staining for PD-L1 (red) and Hoechst nuclei (blue); scale bar, 50 μm. (B,C) Flow cytometric analysis of PD-L1 expression among GBM cells and infiltrating immune cells (TAMC, TIL, microglia) post-RT (or control). Representative histograms of PD-L1 (B) and MFI (C). (D,E) Quantification of tumor cells abundance as determined by the percentage of GFP+ population. (F) Gating strategy for different immune cells (TILs, CD45high CD11b−; TAMCs, CD45high CD11b+; microglia, CD45int CD11b+) and percentages of immune cell subsets. (G) Relative cell abundance of TAMCs, TILs, and microglia determined by flow cytometry. Data are presented as mean ± SEM (n = 6); *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001by student’s t-test.
RGD-EV:siPDL1 Targets and Downregulates PD-L1 Expression in GBM Primed with Radiation.
To suppress radiation-induced PD-L1 expression, RGD-EV were employed as a vehicle to deliver siRNA against PD-L1 (siPDL1) to GBM. RGD-EV were incubated with cholesterol-modified siPDL1 as we previously described.15,28 The lipophilic siPDL1 self-associates with RGD-EV, and free siRNA is removed by ultracentrifugation (Figure 4A). The produced RGD-EV:siPDL1 revealed a shift to slightly larger vesicles after siPDL1 incorporation, as analyzed by NTA and TEM (Figure 4B,C), in line with a previous report.29 To assess the targeting ability of siRNA-loaded RGD-EV, tumor-bearing mice were treated with a burst of 5-Gy RT and, 3 days later, administered with Cy5.5-labeled RGD-EV:siPDL1 intravenously, and brains were dissected after 24 h. NIRF imaging revealed a high accumulation of RGD-EV:siPDL1 in irradiated GBM, which is about 5-fold higher compared with undecorated EVs in nonirradiated mice (Figure 4D,E), similar to the results obtained with RGD-EV. At the same time, different organs (liver, lungs, kidneys, spleen, and heart) were removed, and the biodistribution of EVs was analyzed quantitatively by NIRF imaging (Figure S9A,B). EVs most predominantly accumulated in the liver, followed by the kidneys and spleen, whereas RGD-EV:siPDL1 had a significantly stronger signal in the irradiated brain. The above results indicate that siRNA loading does not affect the targeting ability of RGD-EV to GBM. To corroborate the biodistribution findings with another assay, we labeled EVs with Gaussia luciferase (Gluc) by stably expressing Gluc fused to the transmembrane domain of the platelet-derived growth factor receptor in their donor ReN cells.30 Similarly, no significant differences in Gluc activity were observed between Gluc-labeled EV:siPDL1 and RGD-EV:siPDL1 of the same protein amount showing consistency of labeling among different EV pools (Figure S3C). The two engineered EVs were systemically injected into tumor-bearing mice treated with a burst of 5-Gy RT. Twenty-four hours later, Gluc activity in different mice organs (collected following transcardial perfusion with PBS) confirmed the results obtained with Cy5.5 labeling showing that RT enhanced RGD-EV:siPDL1 uptake in brain tumors (Figure S9C).
Figure 4.
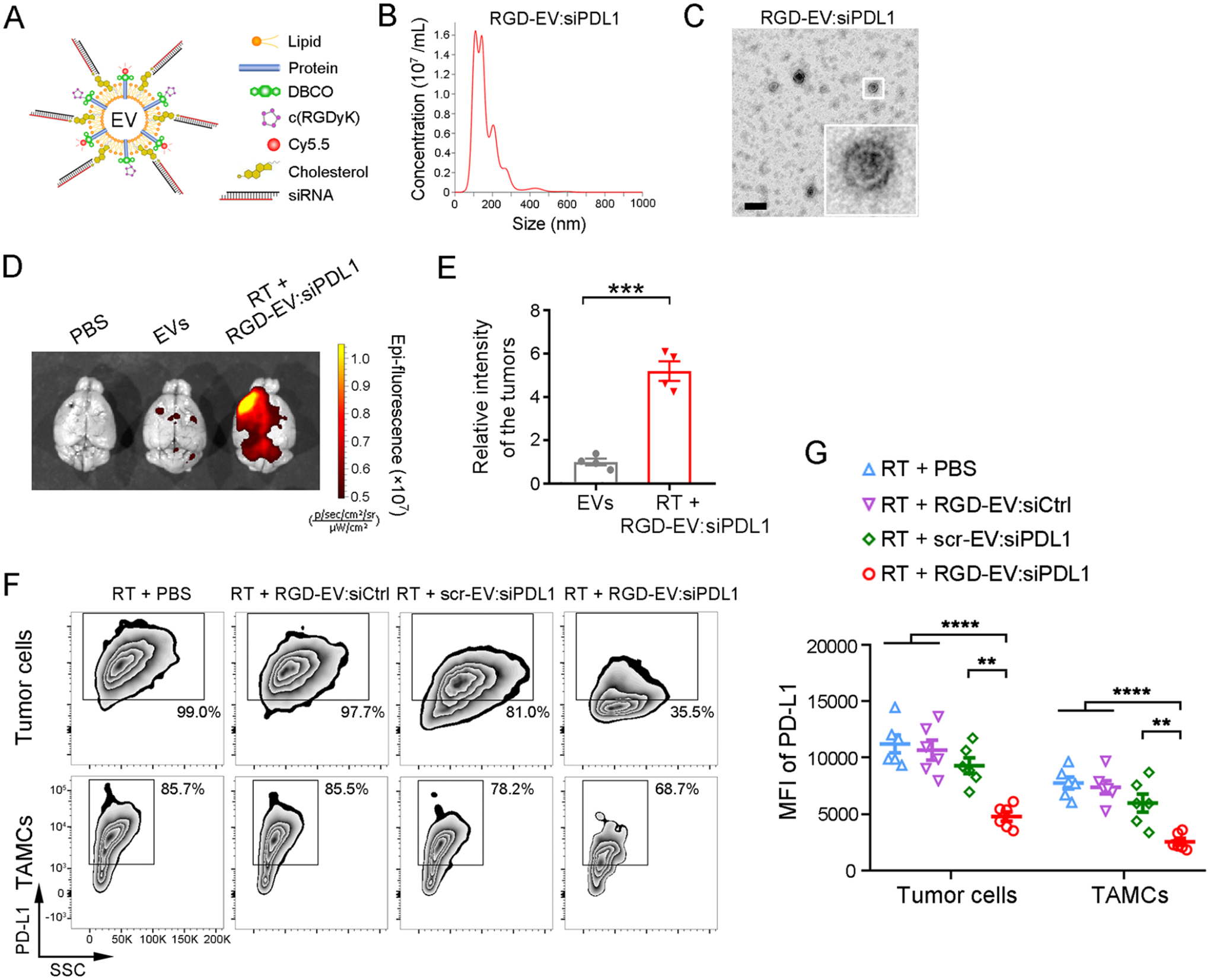
RGD-EV:siPDL1 targets and downregulates PD-L1 in GBM primed with RT. (A) Schematic of RGD-EV, Cy5.5 labeled, and loaded with cholesterol-modified siPDL1. (B,C) NTA (B) and TEM imaging (C) of RGD-EV:siPDL1; scale bar, 200 nm. (D-E) Mice-bearing GL261 tumors primed with 5-Gy RT were injected with Cy5.5-labeled EVs or RGD-EV:siPDL1 and the level of Cy5.5 was analyzed in the brain 24 h later. Representative NIRF images of brains (D) and quantification of fluorescence intensity in the tumor region (E) are shown. Data presented as mean ± SEM (n = 4); ***P < 0.001 by student’s t-test. (F,G) GBM-bearing mice were primed with RT and i.v. injected with PBS, targeted or nontargeted EVs carrying siCtrl or siPDL1. 48 h later, PD-L1 expression on GBM cells and TAMCs was analyzed by flow cytometry, as determined by the percentage of PD-L1+ population (F) and PD-L1 MFI (G). Data presented as mean ± SEM (n = 6); **P < 0.01, ****P < 0.0001 by One-way ANOVA.
To investigate the therapeutic effect of siPDL1 delivered by RGD-EV against GBM, we first verified PD-L1 knockdown in cultured GL261 cells upon treatment with different EVs (Figure S10). The dose of siRNA was estimated by loading 5-carboxyfluorescein (FAM)-labeled siRNAs (with cholesterol modification) onto scr-EV or RGD-EV and fluorescence assessment. Using fluorescent standard curves of free FAM-siCtrl or FAM-siPDL1, we calculated that 100 μg RGD-EV:siPDL1, scr-EV:siPDL1, or RGD-EV:siCtrl contained 814, 790, or 856 pmol siRNAs on average, respectively, while no significant difference of siRNA amount is observed among these groups (Figure S11). Then, different treatment regimens were evaluated in the GL261 syngeneic mouse model. Seventy-two hours after 5-Gy RT, tumor-bearing mice were intravenously injected with PBS or 100 μg (in 200 μL PBS) of either RGD-EV:siCtrl (a targeted EV carrying control siRNA), scr-EV:siPDL1 (a nontargeted EV conjugated with scrambled c(RDGyK) carrying siPDL1), or RGD-EV:siPDL1 (a targeted EV carrying siPDL1). GBM tissues were dissected and analyzed by flow cytometry 48 h after administration. RGD-EV:siPDL1 significantly reduced the level of PD-L1 on both tumor cells and TAMCs, while RGD-EV:siCtrl or scr-EV:siPDL1 groups did not show a statistically significant effect (Figure 4F,G). These results indicate that RGD-EV:siPDL1 downregulates PD-L1 on GBM cells and TAMCs, primed with RT, and such knockdown is heavily dependent on targeted delivery by RGD-EV.
RGD-EV:siPDL1 Increases TIL CD8+ Cytotoxic T Cells in GBM Primed with Radiation.
As the critical function of PD-L1 is to dampen T cell activity and induce immunosuppression, we explored the effect of RGD-EV:siPDL1 on CD8+ cytotoxic T cells in vivo. Mice-bearing GL261 GBM were locally irradiated with a burst of 5-Gy (or not irradiated as control) and 3 days later, intravenously injected with PBS (control) or 100 μg (in 200 μL PBS) of either RGD-EV:siCtrl, scr-EV:siPDL1, or RGD-EV:siPDL1. RT and EV injection were repeated according to the scheme in Figure 5A. On day 21, the proportion and proliferation of CD8+ T cells in the tumor microenvironment were assessed by flow cytometry (gating strategy shown in Figure S12A). The abundance of CD8+ T cells (the percentage of CD45+ CD3+ multiplied by the percentage of CD8+ subset) was increased with RT, reaching maximum when RGD-EV:siPDL1 was combined with RT (Figure 5D). Similarly, RT significantly increased the ratio of CD8+/CD3+ which amplified further when combined with scr-EV:siPDL1 or RGD-EV:siPDL1 (Figure 5B,E). Notably, RT + RGD-EV:siPDL1 resulted in a higher abundance of CD8+ T cells and CD8+/CD3+ ratio compared with RT + scr-EV:siPDL1, which can be attributed to targeted delivery of siPDL1. In addition, RGD-EV:siPDL1 enhanced CD8+ T cells proliferation (Ki67+), while the combination of RT and RGD-EV:siPDL1 produced the strongest effect (Figure 5C,F; Figure S12B). Moreover, immunostaining on brain slices showed superior CD8+ T cells infiltration into the tumor post-RT, with the highest amount being in the RT + RGD-EV:siPDL1 group (Figure 5G). Altogether, these results confirm that RGD-EV:siPDL1 plus RT significantly increases the number of CD8+ T cells in the GBM microenvironment and promotes their proliferation.
Figure 5.
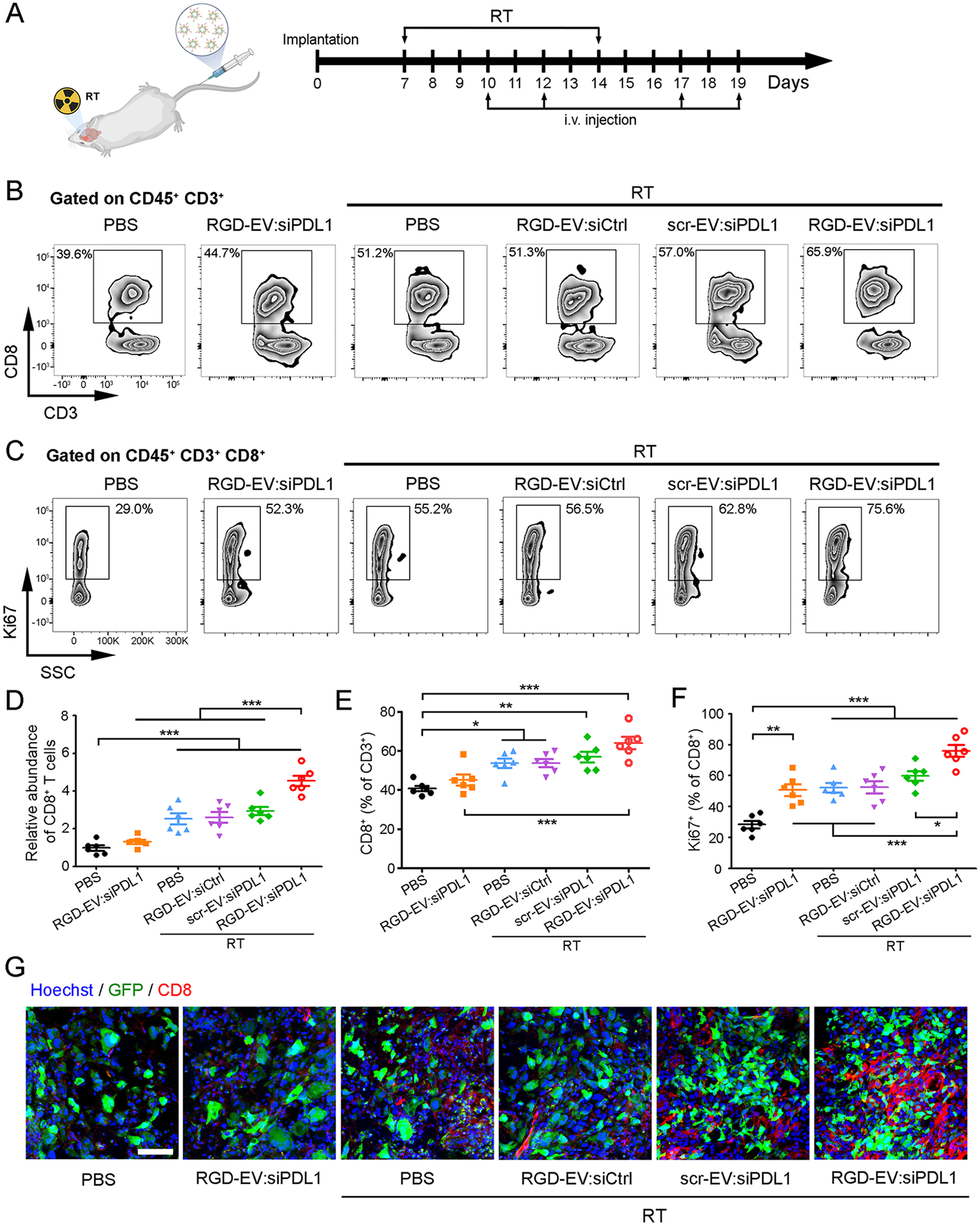
RGD-EV:siPDL1 increases TIL CD8+T cells in GBM microenvironment after a burst of RT. (A) Schematic of the experimental workflow for RT and EV treatment in GBM-bearing mice. Brain tumor samples were collected on day 21. (B,C) Representative zebra plots for CD8+/CD3+ ratios (B) and Ki67 staining on CD8+ T cells (C). (D) CD8+ T cells abundance determined as the percentage of CD8+ subset in CD3-CD8 plots multiplied by the percentage of CD45+ CD3+, normalized to PBS control group. (E) Quantification of CD8+/CD3+ ratios. (F) Quantification of the percentages of proliferating (Ki67+) CD8+ T cells. (G) Immunostaining on brain tumor (GFP) slices for CD8 (red) and nuclei (blue); scale bar, 50 μm. Data presented as mean ± SEM (n = 6); *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA.
RGD-EV:siPDL1 Synergizes with RT to Promote Antitumor Response.
To confirm whether the combination of RGD-EV:siPDL1 with RT can enhance the effector function of CD8+ T cells, RT and different EV treatment regimens were carried out in GBM-bearing mice according to the scheme in Figure 5A. On day 21, infiltrating lymphocytes were extracted from the GBM microenvironment and cultured under stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin. Flow cytometry analysis showed that RGD-EV:siPDL1 alone or RT (with PBS or RGD-EV:siCtrl) led to an increase in the percentage of CD8+ T cells producing IFN-γ, TNF-α, and Granzyme B, which was further increased in groups treated with either targeted or nontargeted EVs carrying siPDL1 (Figure 6A,B; Figure S13). Among all regimens, RGD-EV:siPDL1 combined with RT induced the maximum amount of effector CD8+ T cells. Additionally, RT + RGD-EV:siPDL1 resulted in statistically higher MFI of all three functional markers (IFN-γ, TNF-α, and Granzyme B) of CD8+ T cells (Figure 6A,B).
Figure 6.
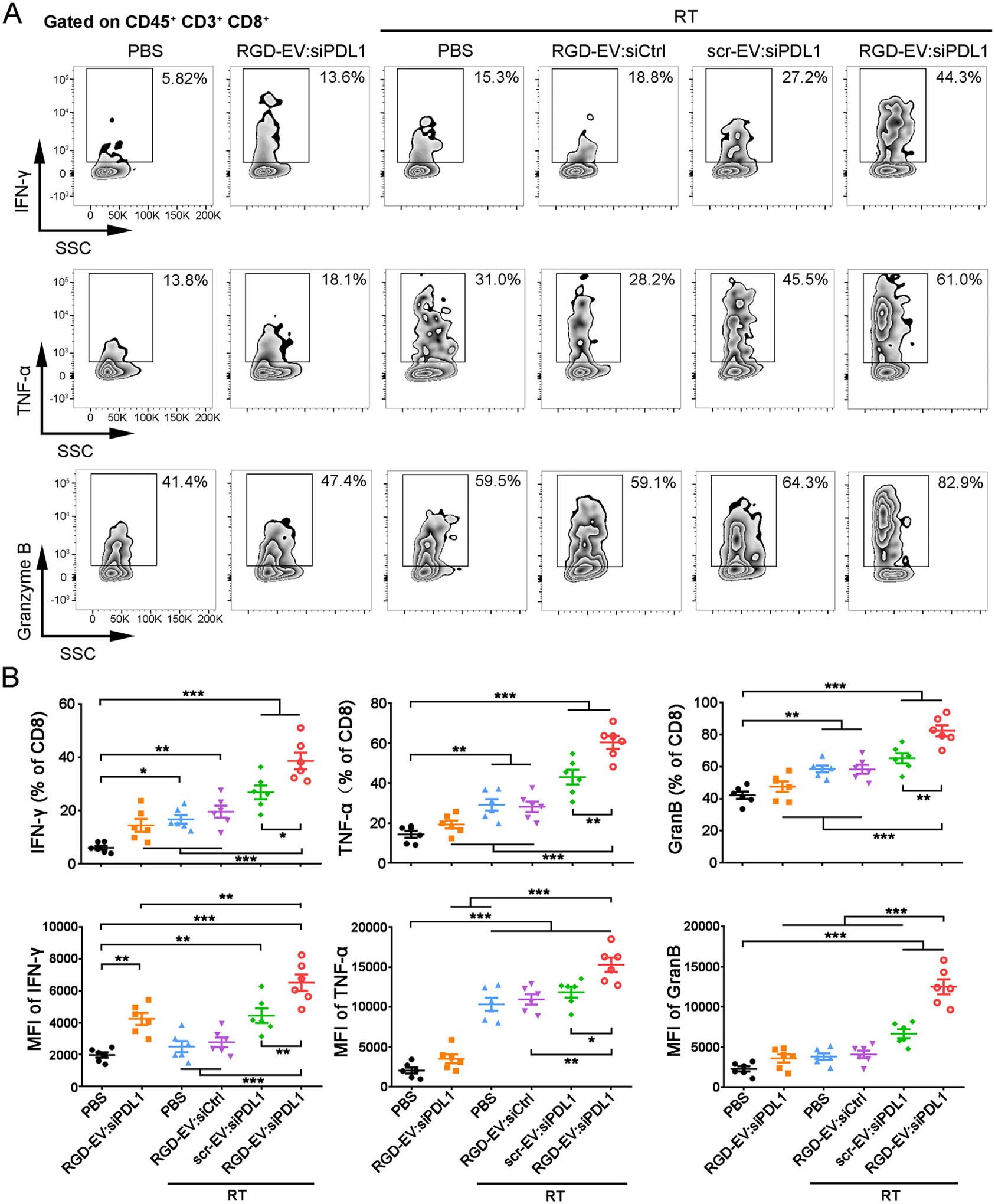
RGD-EV:siPDL1synergizes with RT to promote CD8+T cell effector function in GBM. GBM-bearing mice were treated with RT and/or different EV formulations, and infiltrating lymphocytes were extracted from the GBM microenvironment and cultured under stimulation with PMA and ionomycin. (A) Representative zebra plots depicting IFN-γ, TNF-α, and Granzyme B expression on TIL CD8+ T cells in GBM. (B) Quantification of TIL CD8+ T cell effector function as determined by the percentage of IFN-γ+, TNF-α+, and Granzyme B+ population and their MFI. Data presented as mean ± SEM (n = 6); *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA.
Finally, we evaluated the potential of using RGD-EV:siPDL1 as a combination therapeutic strategy with a burst of RT. GBM-bearing mice were treated with PBS, RGD-EV-siPDL1, RT, or a combination of RT and different EV formulations, according to the schematic presented in Figure 5A. Tumor growth was monitored over time by Fluc bioluminescence imaging (Figure 7A,B). RGD-EV:siPDL1 alone had a slight but not significant effect on tumor growth compared to the PBS group. RT alone or in combination with RGD-EV:siCtrl or scr-EV:siPDL1 produced a moderate antitumor effect. Importantly, mice that received RT plus RGD-EV:siPDL1 had the most significant effect on tumor growth. Furthermore, GL261 is known as an aggressive murine glioma model leading to a short median survival of 22.5 days (Figure 7C). RGD-EV:siPDL1 monotherapy showed only marginal benefit (median survival 24 days). RT alone or in combination with RGD-EV:siCtrl had a strong effect on mouse survival (median survival around 30 days). RT + scr-EV:siPDL1 showed an even stronger effect with a median survival of 34 days. Notably, the combination of RT and RGD-EV:siPDL1 had the greatest effect extending the median survival to 47 days, with 20% of mice surviving over 60 days. Collectively, these data confirm that RGD-EV:siPDL1 can synergize with RT to promote antitumor response, inhibit tumor growth, and increase animal survival.
Figure 7.
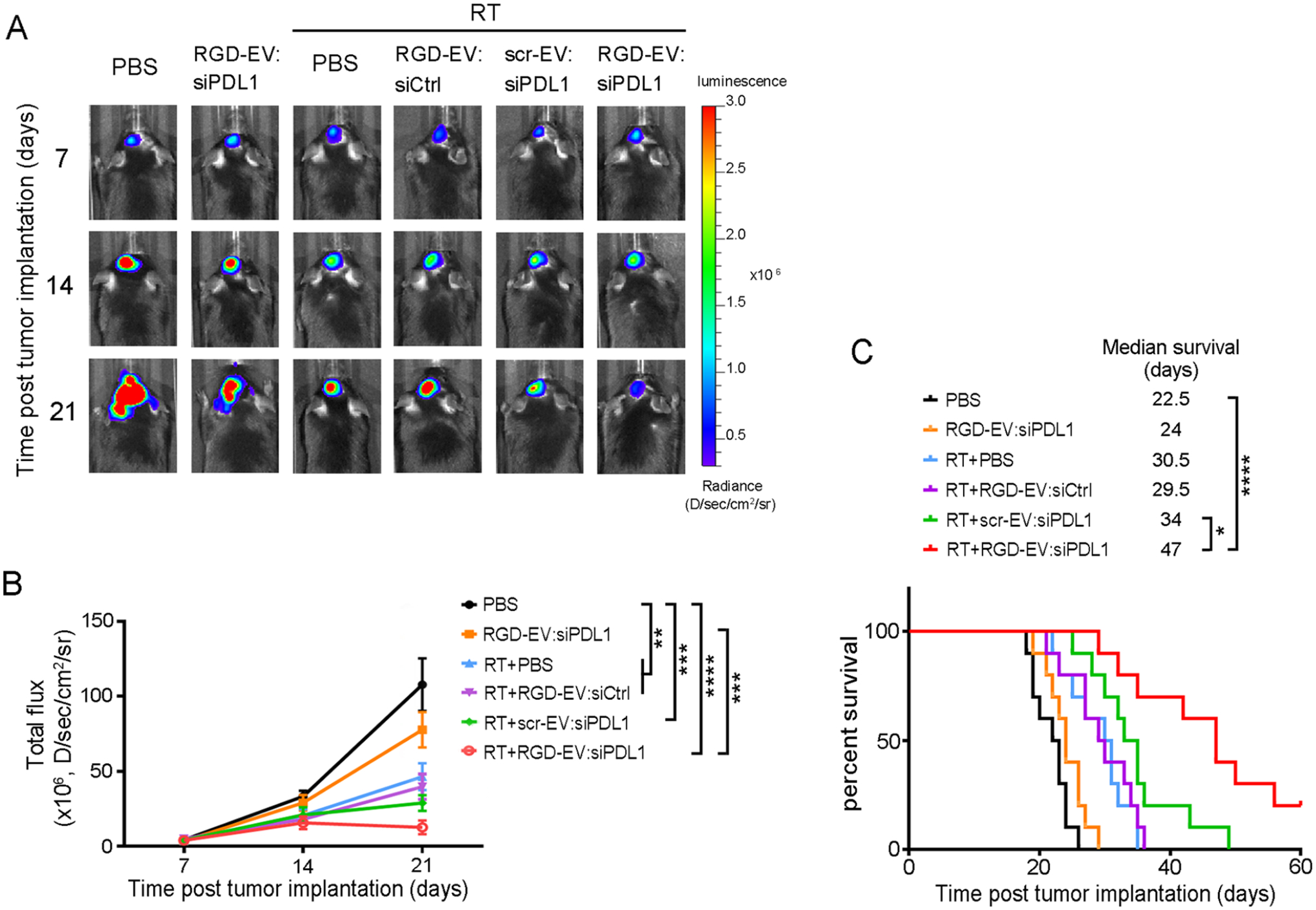
RGD-EV:siPDL1 synergizes with RT to enhance immune checkpoint blockade of GBM. GBM-bearing mice were primed with RT (or no radiation as a control) and 72 h later, i.v. injected with PBS or different EVs (RGD-EV:siPDL1, RGD-EV-siCtrl, scr-EV-siPDL1). (A) Fluc bioluminescence imaging in a representative mouse from each group is shown over time. (B) Quantification of tumor-associated Fluc radiance intensity with data presented as mean ± SEM; **P < 0.01, ***P < 0.001, ****P < 0.0001, by One-way ANOVA. (C) Kaplan–Meier survival curves are shown (n = 10). *P < 0.05, ****P < 0.0001 by Mantel–Cox (log-rank) test.
DISCUSSION
Although different therapeutic approaches have been developed against GBM, the possibility of using these in the clinic is limited due to the lack of safe and effective drug delivery systems (DDS) able to cross the BBB/BBTB and to deliver the therapeutic to the tumor and/or microenvironment.31 EVs have been recently demonstrated to be a safe and efficient DDS.7 These natural DDS are an appealing alternative to the more established synthetic DDS, avoiding toxicity and rapid clearance, as well as intrinsic ability to cross the BBB.9,32 Primary cells such as dendritic cells or mesenchymal stem cells are typically used to produce EVs;10,33 however, the high cost impedes their clinical translation, as large-scale EV isolation requires cell replenishment (limited expansion ability of primary cells) and validation.34 In this study, we selected ReN cells, a human neural progenitor cell line retaining a normal diploid karyotype even after prolonged passage (>45 passages),35–37 as a robust source for EVs. Our previous work has shown successful delivery of ReNcell-derived EVs to the ischemic brain, and no obvious liver toxicity or tissue damage was observed in mice treated with these EVs.38 Second, EVs were decorated with c(RGDyK) to enhance targeting to GBM across the BBTB. It is known that high levels of integrin αvβ3, which binds to RGD peptides, are overexpressed on active endothelial cells in the tumor but not on quiescent endothelial cells.17,39 Furthermore, cyclic RGD peptides such as c(RGDyK) showed higher affinity and selectivity for integrin αvβ3 than linear RGD peptides.39 Thus, we used a chemical strategy for EV conjugation rather than the popular cell engineering method which cannot display cyclic peptides on the EV membrane. In addition, the chemical strategy based on bio-orthogonal copper-free click chemistry is easy, rapid, and scalable. Third, siPDL1 was loaded into RGD-EV. Electroporation is one of the most popular strategies for loading RNAs into EVs, however, it can lead to aggregation and precipitation of the siRNA.40 Another RNA-loading method based on sonication and incubation with permeabilization agents can deform EVs and disrupt EV integrity.41 Here, we loaded RGD-EV with cholesterol-conjugated siPDL1 using a quick hydrophobic association. As analyzed by NTA and TEM, the shape and size distribution remained constant after c(RGDyK)-conjugation, and siPDL1 loading suggesting that the integrity of EVs is not affected. Furthermore, the targeting ability of RGD-EV:siPDL1 was confirmed indicating that incorporation of cholesterol-modified siPDL1 did not affect RGD-EV tropism to GBM.
Therapeutic suppression of immune checkpoint molecules elicits a tumor immune response and improves long-term survival in several types of cancers.42,43 Unfortunately, little effect has been observed with checkpoint inhibitors against GBM thus far, which is attributed not only to the lack of DDS but also to the profound immune suppressive microenvironment and narrow therapeutic window.44 Our data revealed that treatment with RGD-EV:siPDL1 alone did not significantly increase CD8+ T cell abundance and CD8+/CD3+ ratio, and promoted effector production of CD8+ T cells only to a small extent. These phenomena may be caused by a poorly immunogenic or “cold” GBM microenvironment.45 RT has been shown to counteract the immunosuppressive GBM microenvironment by enhancing the presentation of normally suppressed tumor-associated antigens, increasing the expression of MHC-1 and proinflammatory cytokines, promoting dendritic cell maturation and CD8+ T cell recruitment.46 However, RT also enhances the expression of PD-L1 on tumor cells and microenvironment mainly through the release of IFN-γ from TILs. Interferon regulatory factor 1 (IRF1) is the central player for PD-L1 induction in response to IFN-γ.47 Importantly, IRF1 binding to PD-L1 promoter in the JAK-STAT-IRF1 signaling pathway mediates the enhanced PD-L1 expression by IFN-γ.48,49 Therefore, the combination of radiation and checkpoint inhibition therapy holds a promise to reverse this effect.50 Furthermore, delivery of siRNAs to block genes responsible for tumor proliferation or immunosuppression is a promising strategy for cancer therapy. CD8+ T cell proliferation was enhanced by PD-1/PD-L1 blockade through CD28 costimulation signaling.51 We hypothesized that siPDL1 can restore preexisting CD8+ T cell activation suppressed by PD-1 signaling. Here, we employed RGD-EV to deliver siPDL1 to GBM and showed that RT-induced PD-L1 was reversed, while combination therapy with a burst RT plus RGD-EV-based checkpoint inhibition significantly increased CD8+ T cell number and cytotoxic activity.
Previous studies by our group and others have shown that TAMs serve as a nanoparticle drug depot, and local short-burst radiation increases TAMs relative to tumor cells and, thus, prime tumors for enhanced distribution of nanotherapeutics.20,21 Here, we expand on these generic observations and show that a similar phenomenon also occurs for EVs delivery to GBM. Both NIRF and bioluminescence EV-tracking analysis revealed that RGD-EVs had significantly stronger signals in the irradiated brain than that of nontargeted EVs in the nonirradiated brain; however, some biodistribution variations between the two tracking systems were observed which can be explained by the absorption and scattering of photons in tissues associated with fluorescence imaging. Such variation is in line with previous reports suggesting that EV distribution is heterogeneous with varying methodologies.52 Importantly, combining EV-based targeted delivery system with radiation bursts led to robust activation of CD8+ T cells and survival benefit in GBM-bearing mice. These data strongly support that our RGD-EV-based strategy could be used as a combination therapy to synergize with burst of RT (akin sterotactic radiosurgery). Given their nanoscale size, EVs can traverse the blood-brain barrier (BBB).53,54 Surface proteins inherited from donor cells help EVs to penetrate the BBB and enter into the brain parenchyma.41,55 EVs’ subpopulations with high density and smaller size can breach the BBB through transcytosis.56 Additionally, small EVs are internalized preferentially by cells compared with larger EVs.57 Thus, further dissection of EV heterogeneity will improve the EV-based delivery system. An advantage of our therapeutic strategy for GBM therapy is the high efficiency to deliver siPDL1 across the BBB/BBTB, especially in tumors primed with radiation. According to previous reports, the brain delivery of antibody-based drugs was associated with delivery rates of ~0.1%.58,59 Our strategy combining burst of RT with engineered EVs achieved significant targeting to GBM. Another advantage is the synergistic antitumor effect of RGD-EV-based immunotherapy with RT. To date, more than half of all cancer patients undergo RT at some point. EVs have emerged as delivery systems beyond academic study to enter the pipeline for pharmaceutical companies. Thus, rapid translation into clinical practice could be expected.
CONCLUSIONS
In summary, we have developed a targeted EV delivery system for GBM across the BBB/BBTB by modifying their surface with c(RGDyK) peptide and show that short-burst RT can prime GBM and microenvironment for enhanced delivery of targeted EVs. Delivery of siRNA by RGD-EV reversed radiation-induced PD-L1 expression and activated antitumor immunity. These results provide a therapeutic EV-based approach for GBM immune checkpoint blockade primed with a burst of radiation (akin sterotactic radiosurgery). We believe that ReN cells as an EV source, the EV conjugation approach, and siRNA loading strategy are suitable for large-scale production of functionalized EVs, which would allow clinical translation of these technologies to different fields.
METHODS/EXPERIMENTAL
Cell Culture.
Human neural progenitor cell line ReN Cells (Millipore, U.S.A.) were cultured in DMEM/F12 medium (Life Technologies, U.S.A.) containing 2% B27 (Life Technologies), bFGF (10 ng/mL; Abm, Canada) and EGF (20 ng/mL; Abm) under 5% CO2 at 37 °C. GL261 murine glioma cell line was maintained in DMEM/F12 containing 10% fetal bovine serum (FBS) (PAN, Germany) in a CO2 incubator. To label EVs with tdTomato or Gluc, ReN cells were transduced with a lentivirus vector to stably express palm-tdTomato or Gluc-TM which labels cell membrane and thus EV membrane as we described.24,30 For bioluminescence and fluorescence imaging studies, GL261 cells were stably transduced with a lentivirus vector carrying expression cassettes for Fluc and GFP.
EV Isolation and siRNA Incorporation.
Every 1.5–2.0 × 106 ReN cells were cultured in a 100 mm dish for 72 h before the supernatant was collected. EV isolation was performed based on our previous study.24 In brief, cell-free conditioned media were centrifuged at 300g for 10 min, 1200g for 20 min, and 10 000g for 30 min at 4 °C to remove cells and debris. Subsequently, the supernatant was ultracentrifuged at 140 000g for 90 min at 4 °C in a Type SW32Ti rotor using an L-80XP ultracentrifuge (Beckman Coulter, U.S.A.). The pellet was resuspended and ultracentrifuged at 140 000g for 90 min. EV pellets were resuspended with double-0.22 μm-filtered PBS and analyzed using a BCA Protein Assay kit (Pierce, U.S.A.). EV markers were analyzed by Western blotting using anti-Alix and anti-TSG101 antibodies (Abcam, U.K.).
The siPDL1 and siCtrl were synthesized, modified with 2′Ome, and conjugated with cholesterol on the 3′ terminus by GenePharma (China). The sequences were as follows: for siPDL1:5′-AGACGUAAGCAGUGUUGAAdTdT-3′; for siCtrl: 5′-UUCUCCGAACGUGUCACGUdTdT-3′. siRNAs were loaded into EVs following a previous protocol.28 Cholesterol-conjugated siPDL1 or siCtrl (1 nmol) were incubated with EVs, RGD-EV, or scr-EV (100 μg) in PBS. siRNAs were inserted into the EV membrane through a hydrophobic interaction by incubation at 37 °C for 1 h. For each independent experiment, the same type of EVs was pooled together. After they were washed with PBS and centrifuged at 140 000g for 90 min, the modified EVs were resuspended and stored at −80 °C prior to use. To estimate the extent of siRNAs incorporated onto EVs, FAM-labeled and cholesterol-conjugated siPDL1 or siCtrl were synthesized (GenePharma) and loaded onto EVs. Then 1 μg EVs were solubilized by 0.5% Triton X-100 (in 100 μL PBS) to ensure that the fluorescence was not being quenched, and subjected to fluorescence analysis. Standard curves of free FAM-labeled siPDL1 or siCtrl (20–140 nM) were used to calculate the concentration of siRNAs in each EV sample.
TEM and NTA.
EV samples were fixed with glutaraldehyde, dropped onto a carbon-coated copper grid, stained with 1% phosphotungstic acid, and analyzed using a Tecnai G2 transmission electron microscope (FEI, U.S.A.). NTA was performed using a NanoSight NS300 system (Malvern Instruments, U.K.) to track the Brownian motion of EVs in PBS, and size distribution was calculated using the Stokes–Einstein equation.
Conjugation of Ligands to EVs.
Following our previous study,11 dibenzocyclooctyne-sulfo-N-hydroxysuccinimidyl ester (3 nmol; DBCO-sulfo-NHS; Sigma, U.S.A.) was incubated with EVs (500 μg) in PBS for 4 h at room temperature and then filtered with 100-kDa ultrafiltration tubes (Millipore) to remove unconjugated DBCO-sulfo-NHS. The DBCO-conjugated EVs (DBCO-EV) were ready for conjugation to azide-containing molecules via copper-free click chemistry. c(RGDyK) and scrambled c(RDGyK) peptide with an azide group were synthesized by SciLight Biotechnology Co. (China). Next, each peptide (0.3 nmol) was mixed with DBCO-EV (500 μg) in PBS, and Cy5.5 azide (0.3 nmol; Lumiprobe, U.S.A.) was subsequently added for imaging studies. The reaction was allowed to incubate on a rotating mixer at 4 °C for 12 h. To remove unincorporated ligands, EVs were washed by centrifugation at 140 000 g for 90 min in PBS. The modified EVs were resuspended in PBS. For each independent experiment, the same type of EVs was pooled together. Then 10 μg EVs were solubilized with 0.5% Triton X-100 (in 100 μL PBS) to ensure that the fluorescence was not being quenched and subjected to fluorescence analysis.
To confirm peptide conjugation on EVs, c(RKDyK) with or without FITC on the first lysine and an azide group on the last lysine was synthesized (NJPeptide Co., China). The c[RK(FITC)DyK] was conjugated onto EVs using the same method for RGD-EV and analyzed by ELISA (for FITC). ELISA plates (Biolegend, U.S.A.) were coated with CD63 antibody (0.5 μg per well; Cosmo Bio, Japan) overnight at 4 °C. Free binding sites were blocked with 4% BSA (200 μL) for 1 h at room temperature. EV samples (10 μg in 100 μL PBS) were added to each well and incubated for 2 h. Then FITC antibody (1:200; Life Technologies) was added and incubated for 1 h, plates were washed, and horseradish peroxidase-conjugated secondary antibody was added for 1 h. Plates were developed with tetramethylbenzidine (Life Technologies) and stopped with 0.5N H2SO4. Absorbance at 450 nm was analyzed by a BioTek plate reader.
Syngeneic Glioma Model.
All animal experiments were carried out in compliance with institutional guidelines and were approved by the Animal Care and Use Committee of the Massachusetts General Hospital and Nanjing Medical University. To establish orthotopic glioblastoma xenograft model, C57BL/6 mice were anesthetized with 3% isoflurane and stereotactically injected with 1 × 105 GL261-Fluc-GFP cells in PBS (2 μL) using a 30-gauge syringe (Model 701RN, Hamilton, U.S.A.) and the following coordinates: 2.0 mm lateral, 0.5 mm anterior to bregma, 2.5 mm depth from the skull surface.
Bioluminescence and NIRF Imaging.
A Xenogen IVIS Spectrum imaging system (PerkinElmer, U.S.A.) was used for bioluminescence and NIRF imaging. Mice were injected intraperitoneally with D-luciferin (100 μg per gram body weight) and then transferred to the imaging chamber. Imaging was acquired 10 min after luciferin injection. The signal intensity was quantified using the Living Image software (PerkinElmer). For the biodistribution study, Cy5.5-labeled EVs or RGD-EV (100 μg) in PBS (200 μL) were administered via tail vein 72 h after RT (5-Gy), and PBS was injected as a control. Twenty-four h later, mice were anesthetized and sacrificed. The brain, heart, lungs, liver, spleen, and kidneys were dissected and fixed in 4% paraformaldehyde. Cy5.5 fluorescence signals in the dissected organs were captured using the IVIS system and living Image software to distinguish the fluorescence signal associated with Cy5.5 from the autofluorescence signal.
Immunofluorescence Staining and Confocal Imaging.
To study cellular localization of EVs in the brain, mice-bearing tumors primed with radiation were i.v. injected with tdTomato-labeled EVs, scr-EV, or RGD-EV (100 μg). Six hours later, mice were perfused with PBS (25 mL) followed by 4% paraformaldehyde (25 mL). Brains were removed, cryosectioned in 40 μm slices, treated with 0.3% Triton X-100, blocked with 3% BSA, and then stained overnight at 4 °C with anti-PD-L1, anti-CD8, or anti-CD11b (Abcam). Samples were then washed with PBST (PBS containing 0.1% Triton X-100) and incubated with Alexa 594 or Alexa 647-conjugated secondary antibody (Life Technologies) at room temperature. After washing with PBST, samples were stained with Hoechst 33342 and ProLong Antifade Reagents (Life Technologies), and tissue slides were imaged using an FV-1200 confocal microscope (Olympus, Japan). Images were processed and analyzed by ImageJ software (NIH). All settings of imaging and processing were kept constant. The percentage of EV-positive (tdTomato-labeled) tumor cells (GFP) and CD11b+ cells were calculated from six random imaging fields for each independent experiment, and counts were averaged.
In Vivo Analysis of Therapeutic EVs Combined with Radiation Therapy.
One hundred thousand GL261-Fluc-GFP cells were stereotactically injected in the striatum of C57BL/6 mice. On day 7 and day 14 postimplantation, mice were exposed to a single dose of 5-Gy of radiation using a Biology X-ray Irradiator (RadSource, U.S.A.) using a lead shield around the body except for the head. Mice were i.v. injected with PBS or different EV formulations (100 μg of all formulation in 200 μL PBS) on days 10, 12, 17, and 19 post-tumor implantation. Nonirradiated and PBS-treated mice served as controls. Bioluminescence imaging was carried out once a week to monitor tumor growth, and survival was recorded.
Flow Cytometry Analysis.
Tumors were harvested from the brains, chopped into small pieces, and digested in Hank’s balanced salt solution (HBSS; 2 mL) supplemented with collagenase IV (1.5 mg/mL; Life Technologies) and DNase I (200 U/mL; Yeasen Biotechnology, China) at 37 °C for 30 min with pipetting the samples every 10 min. Single-cell suspension was obtained by filtration using a 70 μm cell strainer (BD Biosciences, U.S.A.). Cells were then resuspended in 30% Percoll (Sigma-Aldrich) for myelin removal. Red blood cells were lysed with RBC lysis buffer (1.5 mL; Biolegend) for 2 min at room temperature. For cytokine analysis, Percoll-purified infiltrating immune cells were cultured in RPMI 1640 medium supplemented with Brefeldin A (5 μg/mL; Biolegend) and monensin (2 μM; Biolegend). PMA (0.25 μM; Sigma) and ionomycin (1 μg/mL; Sigma) were added to the culture medium to stimulate the lymphocytes for 5 h. Cells were washed with Cell Staining Buffer (Biolegend) and counted. 106 cells in Cell Staining Buffer (100 μL) were blocked with TruStainFcX PLUS (0.25 μg; Biolegend) for 10 min on ice. Cells were then stained with antibodies against different surface antigens for 60 min on ice. Dead cells were excluded using LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Life Technologies). For intracellular staining, cells were fixed and permeabilized by Foxp3/Transcription Factor Staining Buffer Set (Life Technologies) and then stained for intracellular proteins in Permeabilization Buffer (Life Technologies) for 60 min on ice. After washing with Permeabilization Buffer, cells were analyzed using FACSVerse Flow Cytometer (BD Biosciences). Three thousand cells from each group were collected for analysis of cytokines in CD8+ T cells, while 10 000 cells were used from each group for all other analyses. The following antibodies were purchased from Biolegend: anti-CD45 PE/Cy7, anti-CD11b APC, anti-CD3 APC, anti-CD8 PerCP/Cy5.5, anti-PD-L1 PE, anti-IFN-γ PE, anti-TNF-α PE, anti-Granzyme B PE, anti-Ly6G APC/Cy7, and anti-Ly6C BV510. anti-Ki67 PE was obtained from Life Technologies.
Gluc Activity Assays.
The protocol of sample preparation and Gluc activity measurement has been previously described.30 Briefly, organs were harvested from mice after transcardial perfusion with PBS. Then 100 mg of each organ was homogenized, and 20 μL of organ lysates was diluted by PBS and plated in triplicates into a white 96-well luminometer plate. Gluc activity was measured by a GloMax luminometer (Promega) with an automated injection of 50 μL of 50 ng/mL coelenterazine (Nanolight, U.S.A.) followed by photoncounts for 10 s. Total RLU (relative luminescence units) per organ was counted as follows and adjusted to baseline signal: (RLU/20 μL) × (500 μL lysis buffer/100 mg of organ) × (organ weight in mg).
Statistical Analysis.
Data were presented as mean ± SEM from at least four independent experiments (n values are included in each figure legend). Three technical replicates were collected from each independent experiment to account for variability in the sample. Statistical analysis was accomplished using GraphPad Prism software (GraphPad Sofware, U.S.A.). Comparison between two groups was performed by Student’s t-test. Significance among multiple groups was determined by one-way Analysis of Variance (ANOVA) followed by a Tukey test. A P value <0.05 was considered statistically significant. Survival was analyzed using Kaplan–Meier curves and the log-rank (Mantel–Cox) test.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from NIH/NCI P01CA069246 (to B.A.T., E.A.C., and R.W.), NIH/NINDS R21NS111922 (to B.A.T.), and the National Natural Science Foundation of China 81972364 (to T.T.). We Thank Dr. Semer Maksoud (MGH) for critical reading of the manuscript.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c05505.
Bright-field images of ReN cells cultured for 3 days (Figure S1). Detection of FITC on EVs after conjugation of FITC-labeled c(RKDyK) peptide (Figure S2). Evaluation of reporters on the EVs (Figure S3). Fluc bioluminescence imaging was employed to confirm tumor formation on day 7 postimplantation (Figure S4). Fluorescence images of EVs in CD11b+ cells post-EV administration and corresponding statistics (Figure S5). Sorting strategy for tumor cells in the GL261 syngeneic mouse GBM model by flow cytometric analysis (Figure S6). Phenotyping of immune cells in the GL261 syngeneic mouse GBM model by flow cytometric analysis (Figure S7). Flow cytometric analysis of TAMC subsets in GBM post-RT or control (Figure S8). Biodistribution of engineered EVs analyzed by NIRF imaging and bioluminescence (Figure S9). The expression levels of PD-L1 in cultured GL261 cells were determined by Western blot analysis after treatment of EV:siPDL1 or control (Figure S10). Estimation of siRNA amount incorporated onto the EVs (Figure S11). Sorting strategy for CD8+ T cells in the GL261 syngeneic mouse GBM model and analysis of Ki67 expression by flow cytometric analysis (Figure S12). Flow cytometric analysis of IFN-γ, TNF-α, and Granzyme B expression in CD8+ T cells from mouse GBM model (Figure S13) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.1c05505
The authors declare no competing financial interest.
Contributor Information
Tian Tian, Experimental Therapeutics and Molecular Imaging Unit, Department of Neurology, Neuro-Oncology Division, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02129, United States; Department of Neurobiology, Key Laboratory of Human Functional Genomics of Jiangsu, Nanjing Medical University, Nanjing, Jiangsu 211166, China.
Ruyu Liang, Department of Neurobiology, Key Laboratory of Human Functional Genomics of Jiangsu, Nanjing Medical University, Nanjing, Jiangsu 211166, China.
Gulsah Erel-Akbaba, Experimental Therapeutics and Molecular Imaging Unit, Department of Neurology, Neuro-Oncology Division, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02129, United States; Department of Pharmaceutical Biotechnology, Faculty of Pharmacy, Izmir Katip Celebi University, Izmir 35620, Turkey.
Lorenzo Saad, Experimental Therapeutics and Molecular Imaging Unit, Department of Neurology, Neuro-Oncology Division, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02129, United States.
Pierre J. Obeid, Department of Chemistry, University of Balamand, Deir El-Balamand, Tripoli, Lebanon
Jun Gao, Department of Neurobiology, Key Laboratory of Human Functional Genomics of Jiangsu, Nanjing Medical University, Nanjing, Jiangsu 211166, China.
E. Antonio Chiocca, Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States.
Ralph Weissleder, Center for Systems Biology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02129, United States.
Bakhos A. Tannous, Experimental Therapeutics and Molecular Imaging Unit, Department of Neurology, Neuro-Oncology Division, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02129, United States.
REFERENCES
- (1).Stupp R; Mason WP; van den Bent MJ; Weller M; Fisher B; Taphoorn MJ; Belanger K; Brandes AA; Marosi C; Bogdahn U; Curschmann J; Janzer RC; Ludwin SK; Gorlia T; Allgeier A; Lacombe D; Cairncross JG; Eisenhauer E; Mirimanoff RO; et al. European Organisation for, R. Radiotherapy plus concomitant and adjuvant Temozolomide for glioblastoma. N. Engl. J. Med 2005, 352, 987–996. [DOI] [PubMed] [Google Scholar]
- (2).Stupp R; Hegi ME; Mason WP; van den Bent MJ; Taphoorn MJ; Janzer RC; Ludwin SK; Allgeier A; Fisher B; Belanger K; Hau P; Brandes AA; Gijtenbeek J; Marosi C; Vecht CJ; Mokhtari K; Wesseling P; Villa S; Eisenhauer E; Gorlia T; et al. Effects of radiotherapy with concomitant and adjuvant Temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009, 10, 459–466. [DOI] [PubMed] [Google Scholar]
- (3).Ribas A; Wolchok JD Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sharpe AH; Pauken KE The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol 2018, 18, 153–167. [DOI] [PubMed] [Google Scholar]
- (5).Lim M; Xia Y; Bettegowda C; Weller M Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol 2018, 15, 422–442. [DOI] [PubMed] [Google Scholar]
- (6).Galstyan A; Markman JL; Shatalova ES; Chiechi A; Korman AJ; Patil R; Klymyshyn D; Tourtellotte WG; Israel LL; Braubach O; Ljubimov VA; Mashouf LA; Ramesh A; Grodzinski ZB; Penichet ML; Black KL; Holler E; Sun T; Ding H; Ljubimov AV; Ljubimova JY; et al. Blood-brain barrier permeable nano immunoconjugates induce local immune responses for glioma therapy. Nat. Commun 2019, 10, 3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Escude Martinez de Castilla P; Tong L; Huang C; Marios Sofias A; Pastorin G; Chen X; Storm G; Schiffelers RM; Wang JW Extracellular vesicles as a drug delivery system:A systematic review of preclinical studies. Adv. Drug Delivery Rev 2021, 175, 113801. [DOI] [PubMed] [Google Scholar]
- (8).Liu B; Qiao G; Cao W; Li CH; Pan SH; Wang L; Liu Y; Ma L; Cui D Proteomics Analyses Reveal Functional Differences between Exosomes of Mesenchymal Stem Cells Derived from The Umbilical Cord and Those Derived from The Adipose Tissue. Cell J 2021, 23, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Elsharkasy OM; Nordin JZ; Hagey DW; de Jong OG; Schiffelers RM; Andaloussi SE; Vader P Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Delivery Rev 2020, 159, 332–343. [DOI] [PubMed] [Google Scholar]
- (10).Alvarez-Erviti L; Seow Y; Yin H; Betts C; Lakhal S; Wood MJ Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol 2011, 29, 341–345. [DOI] [PubMed] [Google Scholar]
- (11).Tian T; Zhang HX; He CP; Fan S; Zhu YL; Qi C; Huang NP; Xiao ZD; Lu ZH; Tannous BA; Gao J Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [DOI] [PubMed] [Google Scholar]
- (12).Pan S; Zhang Y; Huang M; Deng Z; Zhang A; Pei L; Wang L; Zhao W; Ma L; Zhang Q; Cui D Urinary exosomes-based Engineered Nanovectors for Homologously Targeted Chemo-Chemodynamic Prostate Cancer Therapy via abrogating EGFR/AKT/NF-kB/IkB signaling. Biomaterials 2021, 275, 120946. [DOI] [PubMed] [Google Scholar]
- (13).Armstrong JP; Holme MN; Stevens MM Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano 2017, 11, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pan S; Pei L; Zhang A; Zhang Y; Zhang C; Huang M; Huang Z; Liu B; Wang L; Ma L; Zhang Q; Cui D Passion fruit-like exosome-PMA/Au-BSA@Ce6 nanovehicles for real-time fluorescence imaging and enhanced targeted photodynamic therapy with deep penetration and superior retention behavior in tumor. Biomaterials 2020, 230, 119606. [DOI] [PubMed] [Google Scholar]
- (15).Haraszti RA; Miller R; Didiot MC; Biscans A; Alterman JF; Hassler MR; Roux L; Echeverria D; Sapp E; DiFiglia M; Aronin N; Khvorova A Optimized Cholesterol-siRNA Chemistry Improves Productive Loading onto Extracellular Vesicles. Mol. Ther 2018, 26, 1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ellert-Miklaszewska A; Poleszak K; Pasierbinska M; Kaminska B Integrin Signaling in Glioma Pathogenesis: From Biology to Therapy. Int. J. Mol. Sci 2020, 21, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Niu W; Xiao Q; Wang X; Zhu J; Li J; Liang X; Peng Y; Wu C; Lu R; Pan Y; Luo J; Zhong X; He H; Rong Z; Fan JB; Wang Y A Biomimetic Drug Delivery System by Integrating Grapefruit Extracellular Vesicles and Doxorubicin-Loaded Heparin-Based Nanoparticles for Glioma Therapy. Nano Lett 2021, 21, 1484–1492. [DOI] [PubMed] [Google Scholar]
- (18).Chen C; Duan Z; Yuan Y; Li R; Pang L; Liang J; Xu X; Wang J Peptide-22 and Cyclic RGD Functionalized Liposomes for Glioma Targeting Drug Delivery Overcoming BBB and BBTB. ACS Appl. Mater. Interfaces 2017, 9, 5864–5873. [DOI] [PubMed] [Google Scholar]
- (19).Stapleton S; Jaffray D; Milosevic M Radiation effects on the tumor microenvironment: Implications for nanomedicine delivery. Adv. Drug Delivery Rev 2017, 109, 119–130. [DOI] [PubMed] [Google Scholar]
- (20).Miller MA; Chandra R; Cuccarese MF; Pfirschke C; Engblom C; Stapleton S; Adhikary U; Kohler RH; Mohan JF; Pittet MJ; Weissleder R Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci. Transl. Med 2017, 9, eaal0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Erel-Akbaba G; Carvalho LA; Tian T; Zinter M; Akbaba H; Obeid PJ; Chiocca EA; Weissleder R; Kantarci AG; Tannous BA Radiation-Induced Targeted Nanoparticle-Based Gene Delivery for Brain Tumor Therapy. ACS Nano 2019, 13, 4028–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang X; Schoenhals JE; Li A; Valdecanas DR; Ye H; Zang F; Tang C; Tang M; Liu CG; Liu X; Krishnan S; Allison JP; Sharma P; Hwu P; Komaki R; Overwijk WW; Gomez DR; Chang JY; Hahn SM; Cortez MA; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res 2017, 77, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang P; Miska J; Lee-Chang C; Rashidi A; Panek WK; An S; Zannikou M; Lopez-Rosas A; Han Y; Xiao T; Pituch KC; Kanojia D; Balyasnikova IV; Lesniak MS Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 23714–23723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lai CP; Kim EY; Badr CE; Weissleder R; Mempel TR; Tannous BA; Breakefield XO Visualization and tracking of tumour extracellular vesicle delivery and RNA translation using multiplexed reporters. Nat. Commun 2015, 6, 7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mann J; Ramakrishna R; Magge R; Wernicke AG Advances in Radiotherapy for Glioblastoma. Front. Neurol 2018, 8, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kalbasi A; June CH; Haas N; Vapiwala N Radiation and immunotherapy: a synergistic combination. J. Clin. Invest 2013, 123, 2756–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Deng L; Liang H; Xu M; Yang X; Burnette B; Arina A; Li XD; Mauceri H; Beckett M; Darga T; Huang X; Gajewski TF; Chen ZJ; Fu YX; Weichselbaum RR STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang H; Wu J; Wu J; Fan Q; Zhou J; Wu J; Liu S; Zang J; Ye J; Xiao M; Tian T; Gao J Exosome-mediated targeted delivery of miR-210 for angiogenic therapy after cerebral ischemia in mice. J. Nanobiotechnology 2019, 17, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).O’Loughlin AJ; Mager I; de Jong OG; Varela MA; Schiffelers RM; El Andaloussi S; Wood MJA; Vader P Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther 2017, 25, 1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lai CP; Mardini O; Ericsson M; Prabhakar S; Maguire C; Chen JW; Tannous BA; Breakefield XO Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Quader S; Liu X; Chen Y; Mi P; Chida T; Ishii T; Miura Y; Nishiyama N; Cabral H; Kataoka K cRGD peptide-installed epirubicin-loaded polymeric micelles for effective targeted therapy against brain tumors. J. Controlled Release 2017, 258, 56–66. [DOI] [PubMed] [Google Scholar]
- (32).O’Brien K; Breyne K; Ughetto S; Laurent LC; Breakefield XO RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol 2020, 21, 585–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Karbasiafshar C; Sellke FW; Abid MR Mesenchymal stem cell-derived extracellular vesicles in the failing heart: past, present, and future. Am. J. Physiol. Heart Circ. Physiol 2021, 320, H1999–H2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lai RC; Yeo RW; Padmanabhan J; Choo A; de Kleijn DP; Lim SK Isolation and Characterization of Exosome from Human Embryonic Stem Cell-Derived C-Myc-Immortalized Mesenchymal Stem Cells. Methods Mol. Biol 2016, 1416, 477–494. [DOI] [PubMed] [Google Scholar]
- (35).Pires F; Ferreira Q; Rodrigues CA; Morgado J; Ferreira FC Neural stem cell differentiation by electrical stimulation using a cross-linked PEDOT substrate: Expanding the use of biocompatible conjugated conductive polymers for neural tissue engineering. Biochim. Biophys. Acta 2015, 1850, 1158–1168. [DOI] [PubMed] [Google Scholar]
- (36).Donato R; Miljan EA; Hines SJ; Aouabdi S; Pollock K; Patel S; Edwards FA; Sinden JD Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 2007, 8, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Oh JH; Jung CR; Lee MO; Kim J; Son MY Comparative analysis of human embryonic stem cellderived neural stem cells as an in vitro human model. Int. J. Mol. Med 2017, 41, 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tian T; Cao L; He C; Ye Q; Liang R; You W; Zhang H; Wu J; Ye J; Tannous BA; Gao J Targeted delivery of neural progenitor cell-derived extracellular vesicles for anti-inflammation after cerebral ischemia. Theranostics 2021, 11, 6507–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Haubner R; Wester HJ; Burkhart F; Senekowitsch-Schmidtke R; Weber W; Goodman SL; Kessler H; Schwaiger M Glycosylated RGD-containing peptides: tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J. Nucl. Med 2001, 42, 326–336. [PubMed] [Google Scholar]
- (40).Kooijmans SAA; Stremersch S; Braeckmans K; de Smedt SC; Hendrix A; Wood MJA; Schiffelers RM; Raemdonck K; Vader P Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Controlled Release 2013, 172, 229–238. [DOI] [PubMed] [Google Scholar]
- (41).Haney MJ; Klyachko NL; Zhao Y; Gupta R; Plotnikova EG; He Z; Patel T; Piroyan A; Sokolsky M; Kabanov AV; Batrakova EV Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Controlled Release 2015, 207, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Del Paggio JC Immunotherapy: Cancer immunotherapy and the value of cure. Nat. Rev. Clin. Oncol 2018, 15, 268–270. [DOI] [PubMed] [Google Scholar]
- (43).Hwang WL; Pike LRG; Royce TJ; Mahal BA; Loeffler JS Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol 2018, 15, 477–494. [DOI] [PubMed] [Google Scholar]
- (44).Kurz SC; Cabrera LP; Hastie D; Huang R; Unadkat P; Rinne M; Nayak L; Lee EQ; Reardon DA; Wen PY PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology 2018, 91, e1355–e1359. [DOI] [PubMed] [Google Scholar]
- (45).Sharma P; Allison JP The future of immune checkpoint therapy. Science 2015, 348, 56–61. [DOI] [PubMed] [Google Scholar]
- (46).Teng F; Kong L; Meng X; Yang J; Yu J Radiotherapy combined with immune checkpoint blockade immunotherapy: Achievements and challenges. Cancer Lett 2015, 365, 23–29. [DOI] [PubMed] [Google Scholar]
- (47).Garcia-Diaz A; Shin DS; Moreno BH; Saco J; Escuin-Ordinas H; Rodriguez GA; Zaretsky JM; Sun L; Hugo W; Wang X; Parisi G; Saus CP; Torrejon DY; Graeber TG; Comin-Anduix B; Hu-Lieskovan S; Damoiseaux R; Lo RS; Ribas A Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 2017, 19, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Shin DS; Zaretsky JM; Escuin-Ordinas H; Garcia-Diaz A; Hu-Lieskovan S; Kalbasi A; Grasso CS; Hugo W; Sandoval S; Torrejon DY; Palaskas N; Rodriguez GA; Parisi G; Azhdam A; Chmielowski B; Cherry G; Seja E; Berent-Maoz B; Shintaku IP; Le DT; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discovery 2017, 7, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lee SJ; Jang BC; Lee SW; Yang YI; Suh SI; Park YM; Oh S; Shin JG; Yao S; Chen L; Choi IH Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett 2006, 580, 755–762. [DOI] [PubMed] [Google Scholar]
- (50).Dovedi SJ; Cheadle EJ; Popple AL; Poon E; Morrow M; Stewart R; Yusko EC; Sanders CM; Vignali M; Emerson RO; Robins HS; Wilkinson RW; Honeychurch J; Illidge TM Fractionated Radiation Therapy Stimulates Antitumor Immunity Mediated by Both Resident and Infiltrating Polyclonal T-cell Populations when Combined with PD-1 Blockade. Clin. Cancer Res 2017, 23, 5514–5526. [DOI] [PubMed] [Google Scholar]
- (51).Kamphorst AO; Wieland A; Nasti T; Yang S; Zhang R; Barber DL; Konieczny BT; Daugherty CZ; Koenig L; Yu K; Sica GL; Sharpe AH; Freeman GJ; Blazar BR; Turka LA; Owonikoko TK; Pillai RN; Ramalingam SS; Araki K; Ahmed R Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kang M; Jordan V; Blenkiron C; Chamley LW Biodistribution of extracellular vesicles following administration into animals: A systematic review. J. Extracell. Vesicles 2021, 10, e12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Shi J; Zhang Y; Yao B; Sun P; Hao Y; Piao H; Zhao X Role of Exosomes in the Progression, Diagnosis, and Treatment of Gliomas. Med. Sci. Monit 2020, 26, e924023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Heidarzadeh M; Gursoy-Ozdemir Y; Kaya M; Eslami Abriz A; Zarebkohan A; Rahbarghazi R; Sokullu E Exosomal delivery of therapeutic modulators through the blood-brain barrier; promise and pitfalls. Cell Biosci 2021, 11, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Grapp M; Wrede A; Schweizer M; Huwel S; Galla HJ; Snaidero N; Simons M; Buckers J; Low PS; Urlaub H; Gartner J; Steinfeld R Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun 2013, 4, 2123. [DOI] [PubMed] [Google Scholar]
- (56).Morad G; Carman CV; Hagedorn EJ; Perlin JR; Zon LI; Mustafaoglu N; Park TE; Ingber DE; Daisy CC; Moses MA Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Choi D; Montermini L; Jeong H; Sharma S; Meehan B; Rak J Mapping Subpopulations of Cancer Cell-Derived Extracellular Vesicles and Particles by Nano-Flow Cytometry. ACS Nano 2019, 13, 10499–10511. [DOI] [PubMed] [Google Scholar]
- (58).Zuchero YJ; Chen X; Bien-Ly N; Bumbaca D; Tong RK; Gao X; Zhang S; Hoyte K; Luk W; Huntley MA; Phu L; Tan C; Kallop D; Weimer RM; Lu Y; Kirkpatrick DS; Ernst JA; Chih B; Dennis MS; Watts RJ Discovery of Novel Blood-Brain Barrier Targets to Enhance Brain Uptake of Therapeutic Antibodies. Neuron 2016, 89, 70–82. [DOI] [PubMed] [Google Scholar]
- (59).Johnsen KB; Burkhart A; Melander F; Kempen PJ; Vejlebo JB; Siupka P; Nielsen MS; Andresen TL; Moos T Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci. Rep 2017, 7, 10396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.