

Abstract
Predicting protein-peptide complex structures is crucial to the understanding of a vast variety of peptide-mediated cellular processes and to the peptide-based drug development. Peptide flexibility and binding mode ranking are the two major challenges for protein-peptide complex structure prediction. Peptides are highly flexible molecules, and therefore brute-force modeling of peptide conformations of interest in protein-peptide docking is beyond current computing power. Inspired by the fact that protein-peptide binding process is like protein folding, we developed a novel strategy, named as MDockPeP2, which tries to address these challenges by using physicochemical information embedded in abundant monomeric proteins with an exhaustive search strategy, in combination with an integrated global search, local flexible minimization method. Only the peptide sequence and the protein crystal structure are required. The method was systemically assessed using a newly constructed structural database of 89 non-redundant protein-peptide complexes with peptide sequence length ranging from 5 to 29, in which about half of the peptides are longer than 15 residues. MDockPeP2 yielded a total success rate of 58.4% (70.8%, 79.8%) for the bound docking (i.e., with bound receptor and fully flexible peptides) and 19.0% (44.8%, 70.7%) for the challenging unbound docking when top 10 (100, 1000) models were considered for each prediction. MDockPeP2 achieved significantly higher success rates on two other datasets, peptiDB and LEADS-PEP, which contain only short- and medium-size peptides (≤ 15 residues). For peptiDB, our method obtained a success rate of 62.0% for the bound docking and 35.9% for the unbound docking when the top 10 models were considered. For LEADS-PEP, MDockPeP2 achieved a success rate of 69.8% when the top 10 models were considered. The program is available at https://zougrouptoolkit.missouri.edu/mdockpep2/download.html.
Keywords: Protein-peptide interaction, Molecular docking, Molecular modeling, Structure prediction, Peptide-based drug
Graphical Abstract
INTRODUCTION
Protein-peptide interactions mediate a wide range of important cellular tasks, including signal transduction, immune responses, and transcriptional regulation.1–2 Peptide-based drugs are gaining increased attention due to their important advantages, such as safety, efficacy, selectivity, and specificity.3–4 Understanding interaction details between a peptide and a target protein is a key to the discovery and design of peptide-based drugs. However, it is costly and time-consuming to determine protein-peptide complex structures by experimental methods such as X-ray and NMR. In silico approaches, which complement and may even guide experimental methods, provide a cost-effective way for studying protein-peptide interactions.5–6
To predict protein-peptide complex structures from a peptide sequence and a protein structure, a big challenge is peptide flexibility. Peptides are highly flexible molecules. The existing methods for structure prediction of protein-peptide complexes focus on only short- or medium-size peptides with sequence length not longer than 15,6–20 mainly due to a peptide’s huge number of degrees of freedom. Very recently, a local docking program (for known binding sites), AutoDock CrankPep,21 increased the maximum length of the peptide to 20 amino acids for the protein-peptide complex structure prediction. Another major challenge is the scoring function for ranking. A desirable scoring function should be able to efficiently distinguish native or near-native binding modes from many decoys generated by a sampling algorithm. Unfortunately, the existing scoring functions are far from being perfect. These challenges seriously impede the application of current computational methods on peptide-based drug discovery and development.
With the fact that protein-peptide binding is like protein folding,22–23 abundant structural resources deposited in the Protein Data Bank (PDB)24 shed light on possible solutions to the challenges of protein-peptide complex structure prediction. In this study, we systemically compared structures of peptides on protein surfaces and fragments in monomeric proteins. Based on a newly constructed structural database of 89 non-redundant protein-peptide complexes with peptide sequence length ranging from 5 to 29, 25 we were always able to find at least one fragment from monomeric proteins sharing a similar structure with the peptide in a protein-peptide complex. Our results revealed that both the sequence and the environment (physicochemical interactions) are important to the conformations of peptides on a protein surface or fragments in monomeric proteins (see RESULTS for details).
Inspired by the above findings, we developed a novel strategy, MDockPeP2, for protein-peptide complex structure prediction by combining physicochemical information embedded in abundant monomeric proteins and current molecular docking methods. Our method requires only a protein structure and a peptide sequence as inputs. Figure 1 shows a flowchart of the prediction, and details of the method are available in the METHODS section. Briefly, MDockPeP2 can be divided into three stages. In Stage 1, for a query peptide, a protein structure database is searched to find fragments sharing similar sequence. These fragments are used as templates to build peptide conformers. In Stage 2, peptide conformers are generated and then independently docked to a target protein surface using a well-developed protein-protein docking program (global rigid docking). The generated models are further refined using a well-established protein-small molecule docking program (local flexible minimization). In Stage 3, peptide conformers are merged and evaluated using a hybrid scoring function, which considers both calculated binding scores and interface similarities. Our method was evaluated based on the PepPro benchmarking database, containing 89 non-redundant protein-peptide complexes and 58 unbound protein receptors. The peptide sequence length varies from 5 to 29, and 46% of them are long peptides (longer than 15 residues). MDockPeP was also tested on two other datasets, peptiDB26 and LEADS-PEP27, containing only short- and medium-size peptides (≤ 15 residues).
Figure 1.
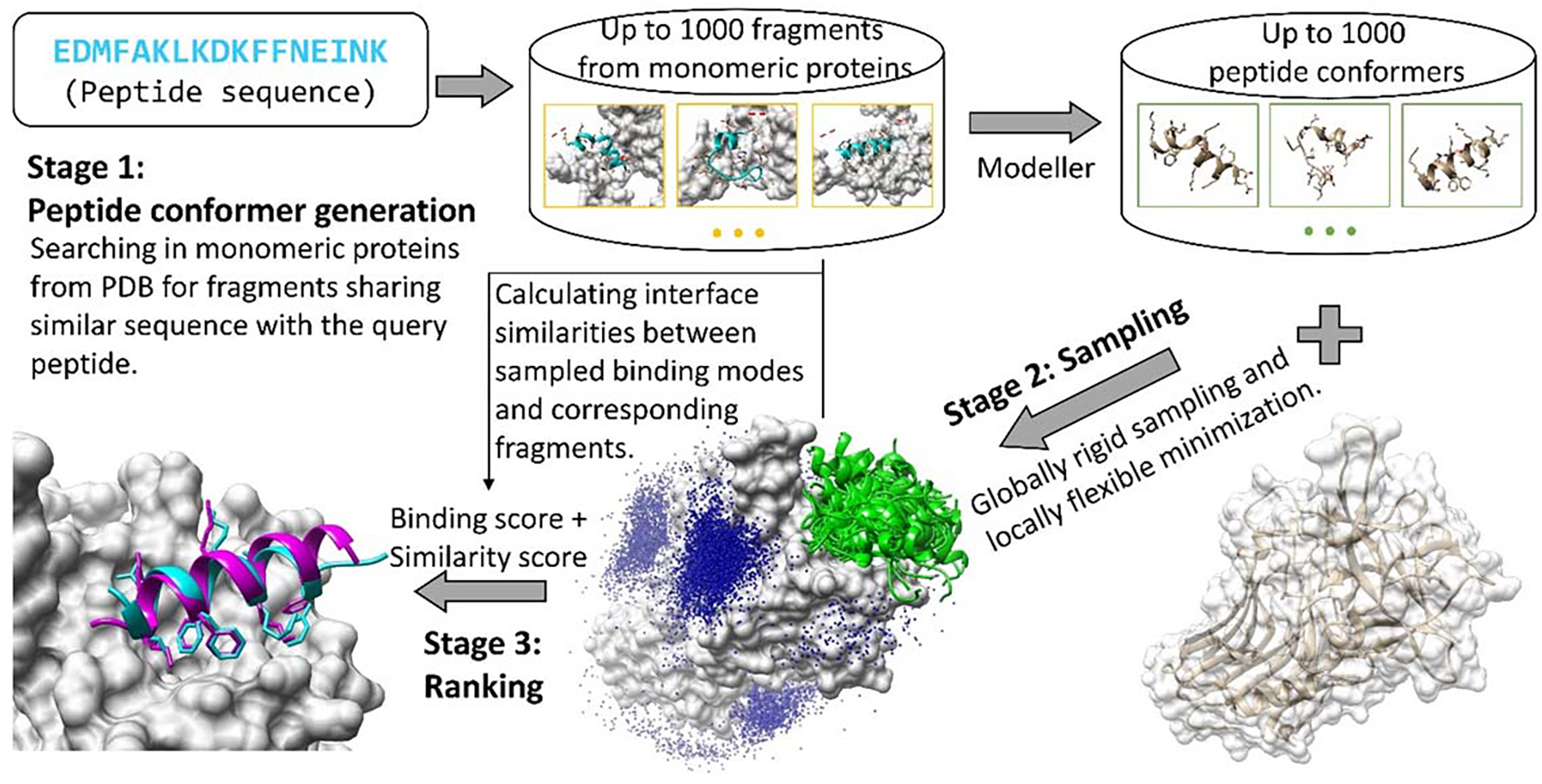
The flowchart of MDockPeP2.
METHODS
The Construction of the Peptide Conformers
For a given peptide sequence, up to 1000 peptide conformers were built based on fragment hits (sharing similar sequences with the query peptide) from a non-redundant protein database provided by MODELLER28 (pdb95.pir.gz, updated on October 20th, 2016). The sequence similarity between any two proteins in the database is lower than 95%. To ensure fragment hits were from proteins rather than peptides, proteins with sequence lengths less than or equal to 50 were removed from the database. The final protein database contains 51667 protein sequences. Here, we developed an exhaustive search strategy to find fragments with similar sequences against the query peptide. This searching strategy was implemented in the MDockPeP2 program. Specifically, the query peptide sequence worked as a sliding window and slid along each protein sequence in the protein database. The sliding process starts from the N-terminal of the protein sequence and moved only one residue for each step. The sequence similarity and the sequence identity between the query peptide and the corresponding fragment in the protein was calculated based on the BLOSUM62 matrix29. Fragments with sequence similarity less than 50%, or those containing any missing residues in PDB structures were discarded. The remaining fragments are ranked by sequence similarity. The fragments with the same sequence similarity were ordered by their sequence identities. To remove redundant fragments from the same protein, if two fragments had over half residues overlap, the fragment with higher sequence similarity (higher sequence identity if sequence similarities were the same) was kept. For the cases in the PepPro dataset, an average of 1.4×106 fragments can be found in this step for a query peptide. This average number increased to 2.3×106 when only short- and medium-size peptides (≤15 residues) were considered. The number is too large to be handled in the later modeling and docking process. Therefore, only top (up to) 3000 fragments (ranked by their sequence similarity with the query peptide) were kept and used as templates to construct peptide conformers by MODELLER. The model refinement level was set as “refine.fast” to ensure the modeled peptide structure was close to the template structure. Then, the generated peptide conformers were clustered using a heavy-atom Root-Mean-Square Deviation (hRMSD) cutoff (2.0 Å). The hRMSD between two conformers was calculated based on a one-to-one atom matching strategy (i.e., possible isomorphisms in the peptide were ignored) and the fitting was performed by a root mean-square superposition using the Kabsch algorithm30. Namely, for any two peptide conformers with their hRMSD smaller than 2.0 Å, the one with a higher sequence similarity (higher sequence identity if sequence similarities were the same) with its template fragment was kept. Finally, the top (up to) 1000 peptide conformers (ranked by sequence similarities of their templates with the query peptide sequence) were saved for docking.
The Global Sampling/Local minimization of the Peptide Binding Modes
A hybrid docking strategy, global rigid sampling and local flexible minimization, was employed to generate putative binding modes. First, up to 1000 peptide conformers generated in stage 1 were independently docked onto the whole protein surface, using a well-developed FFT-based protein-protein docking program ZDOCK 3.031. Default settings are used for ZDOCK. For each peptide conformer, 2000 putative binding models are generated by ZDOCK and re-ranked by an in-house statistical potential-based protein-protein scoring function, ITScorePP32. For any two binding modes with ligand RMSD (L-RMSD) less than 3.0 Å, the one with a better score was kept. The top 100 models were kept for each peptide conformer, and a total up to 105 putative binding modes were generated for a protein-peptide complex.
Then, a local flexible minimization was carried out by AutoDock Vina33, using the above generated models as starting structures. The protein was treated as rigid, and the peptide was treated as fully flexible. For the local minimization, the option “local_only” was turned on, and other parameters were set as default. This local flexible minimization process was performed for all (up to 105) putative binding modes generated in global rigid sampling step.
Ranking of the sampled binding modes
To rank putative binding modes, and distinguish near-native binding modes from decoys, we introduced a hybrid scoring function for the protein-peptide complex structure prediction, named as PepProScore:
| (1) |
which combines the Vina_score (binding score) with the PC_score (physicochemical similarity score). Vina_score is calculated using the scoring function implemented in AutoDock Vina and is outputted from the local flexible sampling step for each model. PC_score is calculated by the program PCalign34, which quantifies physicochemical similarity of protein-protein interfaces. Because both protein and peptide are composed of amino acids, PCalign is also suitable for the comparison of protein-peptide interfaces. Specifically, for a putative binding mode, the protein-peptide interactions were compared to the interactions of the template fragment with the remaining part of the protein (3 adjacent residues at each terminal of the template fragment were discarded). Notably, the fragment used for calculation was the template of the peptide conformer that was used for docking to generate the corresponding putative binding mode. The PC_score values range between 0 and 1, in which 0 represents no similarity and 1 indicates identical interfaces. w, set as −9 in this study, is the weight to balance contributions from two different scores. The value of w was determined based on an average value (−8.7) of min(Vina_scores)/max(PC_scores) of all bound docking cases in PepPro dataset. For each docking case, min(Vina_scores) is the minimum value of Vina_score for all generated modes, and max(PC_scores) is the maximum value of PC_score for all generated modes.
The ranked models were then clustered with a L-RMSD cutoff (3.0 Å). Briefly, for any two binding modes with ligand L-RMSD less than 3.0 Å, the one with a better score was kept.
Benchmarking datasets
A newly constructed protein-peptide complex structure database, PepPro25, was used to evaluate MDockPeP2 for the protein-peptide complex structure prediction. The benchmark contains 89 non-redundant protein-peptide complex structures, with 58 unbound protein structures available. Each protein in the database has a unique sequence and structure. Peptides in the database cover different types of secondary structure, with sequence lengths ranging from 5 to 29. The entries of the PepPro database are available at http://zoulab.dalton.missouri.edu/PepPro_benchmark.
In addition to PepPro, MDockPeP2 was also tested on two other datasets, peptiDB26 and LEADS-PEP27. The peptide sequence length ranges from 5 to 15 in peptiDB, and ranges from 3 to 12 in LEADS-PEP. All the entries (53 bound cases) in LEADS-PEP were used in this study. For peptiDB, by carefully reviewing a total of 103 bound protein-peptide complex structures and 69 unbound protein receptor structures, 3 bound complexes and 5 unbound protein receptors were discarded from the database.15 The remaining entries, 100 bound cases and 64 unbound cases, were used to evaluate the prediction performance of the MDockPeP2.
Assessment criteria
Currently, three criteria, ligand Root-Mean-Square Deviation (L-RMSD), interface RMSD (I-RMSD), and fraction of native contact (fnat), are often used to compare a protein-peptide binding mode predicted by a docking method with the complex structure determined by experimental methods. L-RMSD is the backbone RMSD of the peptide between the predicted mode and the native structure after a structural superposition of the protein. I-RMSD is the backbone RMSD of the interface residues between the predicted mode and the native structure after a structural superposition of these interface residues. A residue is defined as the interface residue when any heavy atom of the residue in one partner of a complex is within 10 Å from the other partner. It is noted that L-RMSD and I-RMSD do not include sidechains explicitly. fnat is defined as the fraction of residue-residue contacts in the native structure that is recalled by a predicted model. A pair of residues are defined as in contact when any two heavy atoms in the two residues are within 5 Å.
Because these parameters characterize different features of a predicted protein-peptide interface, multiple combinations of L-RMSD, I-RMSD, and fnat, are often used to classify predicted binding modes. As illustrated in a recent study by Schueler-Furman and co-workers35, a predicted model can be classified into one of the following four groups: High quality: fnat [0.8 1.0] & (L-RMSD ≤ 1.0 Å || I-RMSD ≤ 0.5 Å); Medium: fnat [0.5 0.8] & (L-RMSD ≤ 2.0 Å || I-RMSD ≤ 1.0 Å) || fnat [0.8 1.0] & (L-RMSD > 1.0 Å & I-RMSD >0 .5 Å); Acceptable: fnat [0.2 0.5] & (L-RMSD ≤ 4.0 Å || I-RMSD ≤ 2.0 Å) || fnat [0.5 1.0] & (L-RMSD>2.0 Å & I-RMSD>1.0 Å); and Incorrect: the rest. We named this metric as capri_metric in this study. A drawback of the capri_metric is that it is a discrete metric and unable to further evaluate the quality of the models in each group.
Therefore, CAPRI committee recently introduced a continuous quality metric, DockQ score,36 based on the three parameters, L-RMSD, I-RMSD, and fnat. DockQ was calculated by the following equation:
with
where fnat, L-RMSD, and I-RMSD are fraction of native contacts, ligand RMSD, and interface RMSD, respectively. RMSDscaled is a scaled RMSD with a scaling factor di. As suggested in CAPRI studies, d1 = 8.5 Å for L-RMSD and d2 = 1.5 Å for I-RMSD.
It is worth mentioning that parameters like L-RMSD, I-RMSD, and fnat were originally defined for protein-protein docking evaluation, and were then used for other systems, such as protein-peptide complexes and protein-sugar complexes. However, the binding of a peptide on a protein surface is normally facilitated by a few peptide residues, known as “hot spot” residues, anchor residues, or binding motif in different studies. The sidechain conformations of these “hot spot” residues are important to the protein-peptide interactions. Unfortunately, current assessment criteria for protein-peptide interactions do not fully consider these important interactions. For example, both L-RMSD and I-RMSD consider only backbone atoms; contributions from sidechain atoms are ignored. Although fnat can partially consider sidechain conformations, it considers only the recovery of native contacts and ignores non-native contacts.
In this study, we introduced a new assessment criterion considering the sidechain conformations of peptide contact residues (including “hot spot” residues), and the peptide backbone conformation. The criterion is named as critical L-RMSD (cL-RMSD), which is the L-RMSD of the heavy atoms of peptide contact residues and the backbone atoms of peptide non-contact residues. Contact residues were determined by the relative buried surface area. The program Naccess V2.1.137 was employed and the calculation was based on the experimental protein-peptide complex structure. Here, peptide residues with relative buried surface area larger than 33.3% were identified as contact residues. For the 89 peptides in the PepPro dataset, 60.3% (815 out of the total 1352) peptide residues were defined as contact residues. Aromatic residues (Phe, Tyr, and Trp) and residues with hydrophobic side chains (e.g., Leu, Ile, Met, and Val) were identified as contact residues more frequently (> 70%) than other types of residues (see Table S1 for details). To calculate cL-RMSD, proteins in the predicted model and in the experimental complex structure were superimposed using the MatchMaker tool of UCSF Chimera38. Next, cL-RMSD was calculated based on all the heavy atoms of the contact residues and the backbone atoms of the non-contact residues in the peptide.
RESULTS
Peptides in Protein-Peptide complex vs Fragments in Monomeric Proteins
We compared conformations of peptides in protein-peptide complexes with conformations of fragments in monomeric proteins. For a given protein-peptide complex, fragments sharing sequence similarities higher than or equal to 50% with the peptide were extracted from a protein database provided by MODELLER 28(pdb95.pir.gz, updated on October 20th, 2016). Proteins with sequence lengths less than or equal to 50 were discarded, and redundant proteins were removed from the database using the program UCLUST39 with a sequence similarity cutoff of 30%. Then, bRMSDs of the peptide with the fragments were calculated. Figure 2A shows an example of a query peptide in a protein-peptide complex (PDB: 1avf) and a fragment hit in a monomeric protein (PDB: 4xgc, chain C, residues 259–280). The peptide and the fragment hit share a sequence similarity of 50%, and the PC-score of their interacting interface is 0.4. bRMSD of the peptide and the fragment is 2.3 Å.
Figure 2.
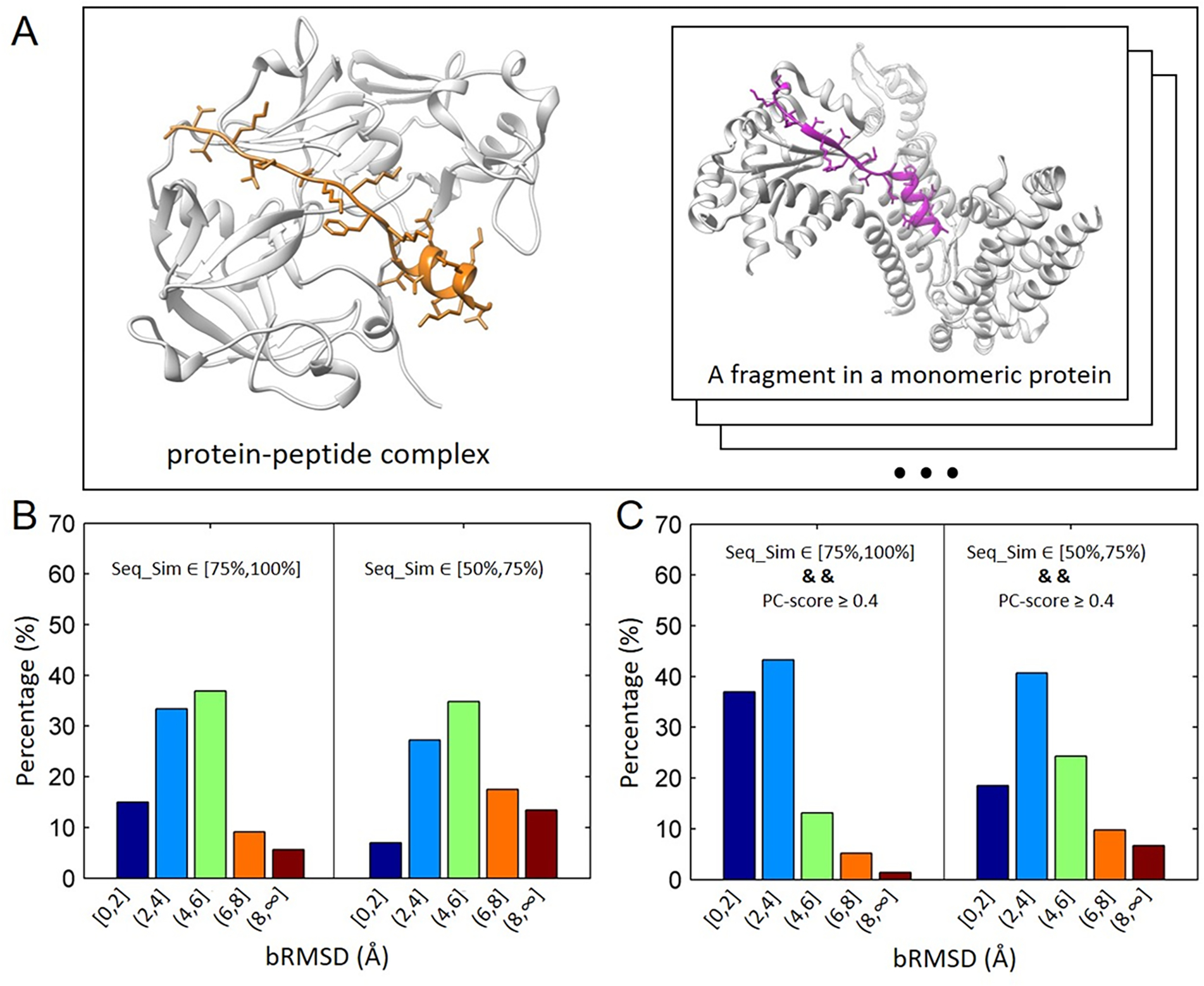
Structural comparison of peptides in protein-peptide complexes and corresponding fragments in monomeric proteins. (A) Left panel: an example of protein-peptide complex (PDB: 1avf) with the peptide sequence length 22 (color in orange); Right Panel: a fragment from a monomeric protein (PDB: 4xgc, chain C, residues 259–280). bRMSD of the peptide and the fragment hit is 2.3 Å. (B) Distributions of bRMSDs between peptide structures and corresponding fragments with two ranges of sequence similarities, [75%, 100%] and [50%, 75). The calculation was based on 89 non-redundant protein-peptide complexes in PepPro benchmark. (C) Similar to (B) except that fragments used for calculation have similar environments (or similar interacting interface, PC-score ≥ 4.0) with query peptides.
The above evaluation was performed for 89 non-redundant protein-peptide complexes from the PepPro benchmarking database25, with peptide lengths ranging from 5 to 29. Impressively, we always found a fragment with bRMSD less than 4.5 Å for all peptides, and 91% of peptides have at least one fragment with bRMSD less than 3.0 Å (Figure S1). If considering only short and medium size peptides (no more than15 residues), 47 out of 48 peptides have at least one fragment with bRMSD less than 2.0 Å. The results show that a linear sequence of amino acids can form a similar conformation either through non-covalent bonds on a protein surface (protein-peptide binding) or through folding in a monomeric protein (protein folding).
In addition to fragments sharing similar conformations with the query peptide, we also found fragments with distinct conformations in monomeric proteins. To find out factors affecting the conformation of a sequence of amino acids in both cases (protein-peptide binding and protein folding), we calculated bRMSD distributions of all fragment hits with two different ranges of the sequence similarity, [75%,100%] and [50%,75%), for the 89 non-redundant peptides. Considering that the number of fragment hits varies for each peptide, we normalized the contribution of each peptide to the final bRMSD distributions. As shown in Figure 2B, 15.0% (48.4%) of fragments with bRMSD ≤ 2.0 Å (4.0 Å) are observed for the range of [75%,100%], and the percentage decreases to 7.0% (34.2%) for fragments with lower sequence similarities [50%,75%). This indicates that the sequence is important for the peptide/fragment conformation. Although the peptide is highly flexible, fragments with higher sequence similarities tend to form more similar conformations with the peptide than those with lower sequence similarities.
Besides the sequence similarity, the environment (physicochemical interactions) of the peptide and corresponding fragments were also compared for the first time. Here, the program PCalign31 was employed to calculate similarities between the protein-peptide interacting interface and those of fragment hits with the rest of the monomeric proteins (3 adjacent residues at each terminal of fragments were discarded). The calculated similarity score, PC_score, accounted for both geometric and chemical characteristics of the two interacting interfaces. The score values range between 0 and 1, in which 0 represents no similarity and 1 indicates identical interfaces. A cutoff of 0.4 was chosen to distinguish similar and dissimilar interaction interfaces. Figure 2C shows the bRMSD distributions of fragments that share similar physicochemical interactions (PC_score ≥ 0.4) with corresponding peptides. Impressively, dramatic increases were observed in the percentage of low bRMSDs (≤ 2.0/4.0 Å) for the two different ranges of sequence similarities. Specifically, for the sequence similarity range of [75%,100%], the percentage of fragments with bRMSD ≤ 2.0 Å (4.0 Å) increased 22.0% (31.9%). While for the sequence similarity range of [50%,75%), the percentage of fragments with bRMSD ≤ 2.0 Å (4.0 Å) increased 11.5% (25.0%). Therefore, both the sequence and the environment play crucial roles in determining the structure of a peptide, with the fact that 80.3% of fragments with high sequence similarity (≥ 75%) and similar environment share a close conformation (bRMSD ≤ 4.0 Å) with corresponding peptides.
There are both exposed and buried peptides in protein-peptide complex structures. If a peptide was defined as buried when its relative buried surface area no less than 50%, 33 of 89 peptides (37.1%) in the PepPro dataset were buried in their co-bound proteins. In contrast, many more matching fragments (73.5%) were buried in monomeric proteins. To investigate the relationship between the PC_score and the relative buried surface area, we calculated the correlation coefficient (R) between PC_Scores and ΔRBSA. For each fragment of a query peptide, ΔRBSA = abs(RBSAfrag - RBSApep). Here, RBSAfrag is the ratio of the buried surface area of the fragment in the monomeric protein, and RBSApep is the ratio of the buried surface area of the query peptide in the protein-peptide complex. A correlation of R = −0.26 was observed between PC_Scores and ΔRBSA, indicating that a fragment having similar ratio of buried surface area with the query peptide tends to have a higher PC_score.
The conservation of the two processes, protein folding and protein-peptide binding, provides a cure to the protein-peptide complex structure prediction. In MDockPep2, an ensemble of peptide conformers was built based on fragments sharing similar sequence with the peptide (stage 1, see Figure 1). Then, these peptide conformers were docked to the target proteins using well-developed molecular docking methods (stage 2). Finally, generated binding models were ranked by a scoring function (stage 3), which considered both binding scores and the conservation of interacting interfaces. The results of each stage were reported as follows.
The Construction of the Peptide Conformers
For stage 1, we calculated both bRMSD and hRMSD between the constructed peptide conformers and the corresponding bound peptide structures in the PepPro database. Figure 3A shows rates for successfully building at least one near-native structure (bRMSD ≤ a preset cutoff) among the top N conformers. The results show that the larger N we set, the larger chance we can obtain near-native peptide conformers. However, the computational time for stage 2 (binding mode sampling) increases linearly with the number of peptide conformers used for docking. In this study, the maximum N was set as 1000. Figure 3B shows the bRMSD and the hRMSD of the best conformer (i.e., the conformer with the lowest bRMSD) among the top 1000 models for each peptide. Impressively, we built at least one medium quality conformer with a bRMSD less than 4.7 Å for all 89 peptides in the database. In 70.8% (94.4%) of the cases, the bRMSD of the best peptide conformer was below 2.0 Å (4.0 Å). The mean and median values of the bRMSD for all cases were 1.6 Å and 1.3 Å, respectively. Corresponding hRMSDs were normally larger than the bRMSDs. The mean and median values of hRMSD were 3.1 Å and 2.8 Å, respectively. The larger hRMSD values are most likely due to the side chains being more sensitive to the surrounding environment than the backbone atoms.
Figure 3.
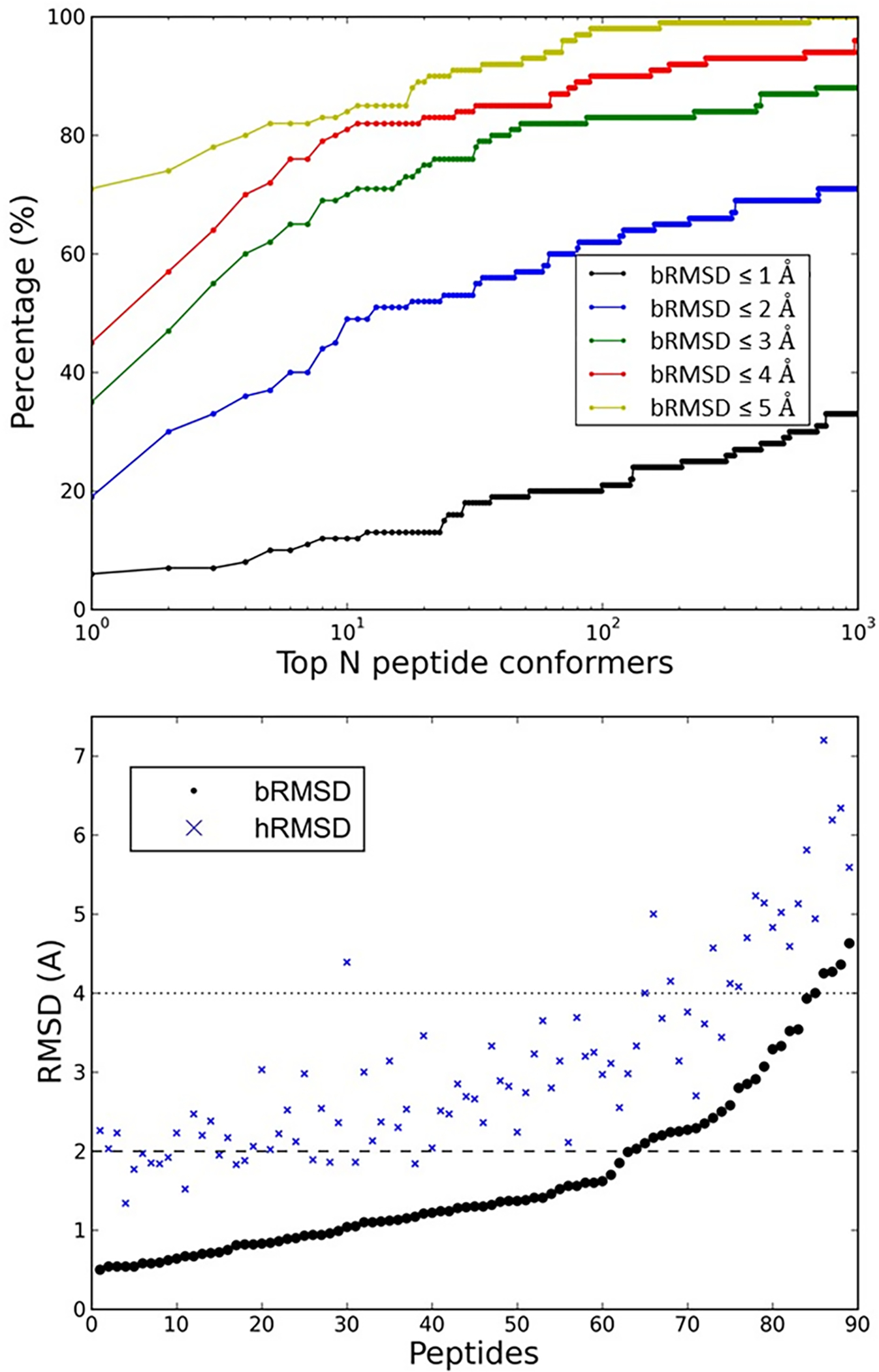
Modeling peptide conformers based on template fragments in monomeric proteins for 89 bound peptides in PepPro.
(A) The success rates of modeling at least one peptide conformer that closely corresponds to the bound peptide structure (bRMSD ≤ cutoff) among the top N peptide conformers.
(B) The distributions of bRMSD and hRMSD between the best modeled peptide conformer (i.e., the conformer with the lowest bRMSD) in the top 1000 models and the corresponding bound peptide structure. The dashed line and dotted line represent 2.0 Å and 4.0 Å, respectively.
Binding Mode Sampling
To evaluate interaction details of a peptides (including side chain conformations) on a protein surface, we introduced a new criterion, critical ligand RMSD (cL-RMSD, see METHODS). Figure 4A shows binding mode sampling results for both bound docking (bpro-upep) and unbound docking (upro-upep), using different values of cL-RMSD as the respective thresholds for successful predictions. The bound docking used a bound protein structure which was taken from the protein-peptide complex crystal structure. The unbound docking used an apo structure of the protein, which was more challenging than the bound docking. It is worth noting that unbound peptides (starting from peptide sequences) were used for all cases in this study. For a protein-peptide complex, the sampling was defined to be successful if the cL-RMSD of at least one model was less than or equal to the threshold value. The sampling success rates of bound docking are 28.1%, 73.0%, and 88.8% for cL-RMSD thresholds of 2.0 Å, 4.0 Å, and 6.0 Å, respectively. The sampling success rates of the more challenging unbound docking are 8.6%, 62.1%, and 82.8% for cL-RMSD thresholds of 2.0 Å, 4.0 Å, and 6.0 Å, respectively.
Figure 4.
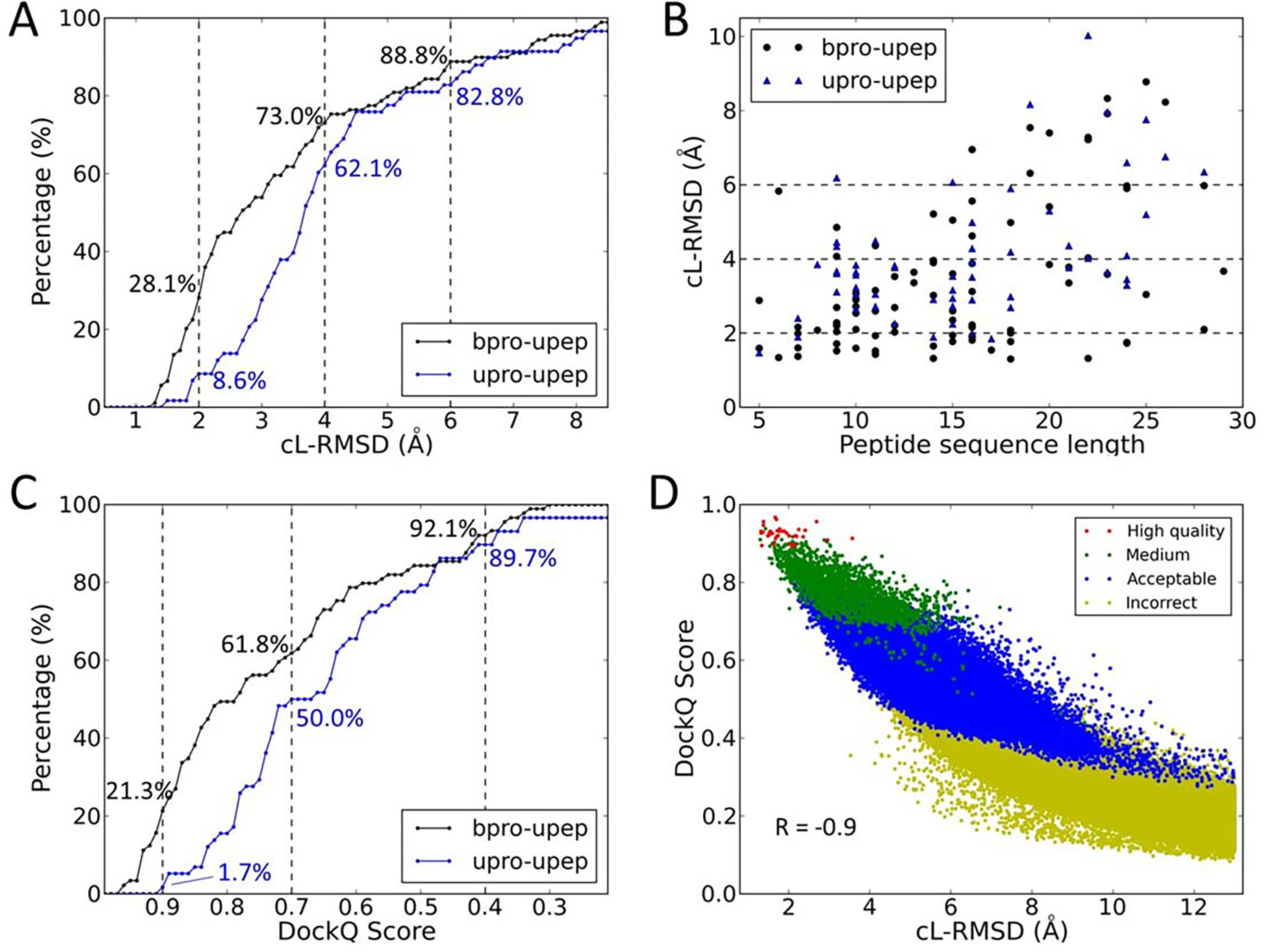
The performance of binding mode sampling for both the bound docking cases and unbound docking cases in PepPro.
(A) The success rates of peptide binding mode sampling using different values of cL-RMSD as the thresholds for the bound docking cases (bpro-upep) and unbound docking cases (upro-upep). Dashed lines represent 2.0 Å, 4.0 Å, and 6.0 Å.
(B) The lowest cL-RMSD value of sampled models for each complex for both the bound docking and the unbound docking. The peptide sequence length is shown for each complex.
(C) Sampling success rates using different values of DockQ Score as the thresholds for the bound and unbound dockings. Dashed lines represent DockQ Scores of 0.9, 0.7, and 0.4.
(D) Distributions of sampled models (bound docking) in three evaluation metrics, cL-RMSD (x-axis), DockQ Score (y-axis), and capri_metric (colors).
Figure 4B shows the cL-RMSD value of the best sampled model (the lowest cL-RMSD) for each protein-peptide complex in the PepPro dataset. For both the bound docking and the unbound docking, it is more challenging to sample a binding model with low cL-RMSD for a case containing a long peptide (> 15 residues) than a case containing a short- or medium-size peptide. If only considering cases containing short- and medium-size peptides (≤ 15 residues), sampling success rates of bound docking are 31.1%, 87.5%, and 100%, for cL-RMSD thresholds of 2.0 Å, 4.0 Å, and 6.0 Å, respectively. For the challenging unbound docking cases, sampling success rates are 9.7%, 80.6%, and 90.3%, for cL-RMSD thresholds of 2.0 Å, 4.0 Å, and 6.0 Å, respectively. For complexes containing long peptides (> 15 residues), MDockPeP2 also achieved good performances with a sampling success rate of 75.6% (cL-RMSD ≤ 6.0 Å; 56.1%% when cL-RMSD ≤ 4.0 Å; 24.4% when cL-RMSD ≤ 2.0 Å) for bound docking and 74.1% (cL-RMSD ≤ 6.0 Å; 40.7% when cL-RMSD ≤ 4.0 Å; 7.4% when cL-RMSD ≤ 2.0 Å) for unbound docking.
In addition to cL-RMSD, two other metrics, capri_metric and DockQ Score, were used to evaluate models generated by MDockPeP2 on the PepPro dataset. As described in Materials and Methods, capri_metric classifies predicted models into four classes (high quality, medium, acceptable, and incorrect) using different combinations of three parameters L-RMSD, I-RMSD, and fnat. DockQ Score is calculated based on the values of the three parameters L-RMSD, I-RMSD, and fnat. The value of DockQ Score ranges from 0 to 1, in DockQ = 0 means the two binding modes are distinct, and 1 means they are identical.
When the capri_metric was used as the criterion, MDockPeP2 successfully generated acceptable and higher quality models for 87.6% (medium and higher quality, 59.6%; high quality, 12.4%) of the bound docking cases and for 86.2% (medium and higher quality, 36.2%; high quality, 1.7%) of the unbound docking cases.
Figure 4C shows the binding mode sampling results using different values of DockQ Score as the respective thresholds for successful predictions. The sampling success rates of bound docking (bpro-upep) are 21.3%, 61.8%, and 92.1% for DockQ Score thresholds of 0.9, 0.7, and 0.4, respectively. The sampling success rates of unbound docking (upro-upep) are 1.7%, 50.0%, and 89.7% for DockQ Score thresholds of 0.9, 0.7, and 0.4, respectively.
Because both the capri_metric and the DockQ Score are based on the three parameters L-RMSD, I-RMSD, and fnat. It would be interesting to investigate the relationship between the two metrics. Here, using all the sampled models (a total of ~8.4×106 models) of the bound cases in the PepPro dataset, we analyzed the distributions of DockQ Scores for the models in different quality classes identified by the capri_metric. The results are shown in Figure S2A. The total numbers of high quality, medium, and acceptable models are 34, 2088, and 47888, respectively. Other models are in the “incorrect” class. The average values of DockQ Scores of high quality, medium, acceptable, and incorrect models are 0.93 (±0.02), 0.78 (±0.06), 0.53(±0.08), and 0.08 (±0.08), respectively. Overlaps of DockQ Scores among models with different qualities are insignificant (see Figure S2A). Therefore, we suggest that models with DockQ Scores in [0.9, 1.0], [0.7, 0.9), [0.4, 0.7), and [0, 0.4) are high-quality, medium-quality, acceptable, and incorrect models, respectively.
We further analyzed the relationship between cL-RMSD and DockQ Score, and the relationship between cL-RMSD and capri_metric based on all the sampling models of bound docking in the PepPro dataset. The results are shown in Figure 4D. For each generated model, its cL-RMSD value is displayed on the x-axis and its DockQ Score is displayed on the y-axis. The classification based on cpari_metric is shown by different colors. The correlation coefficient (R) between cL-RMSDs and DockQ Scores is −0.9, which shows that cL-RMSD is a reasonable metric for the evaluation of protein-peptide complex structures. The distributions of cL-RMSD values of models in different classes defined by the capri_metric are shown in Figure S2B. The average values of cL_RMSD of high quality, medium, acceptable, and incorrect models are 1.9 (±0.5), 3.5 (±1.0), 6.0 (±1.3), and 26.5 (±13.0), respectively. Interestingly, we observed significant overlaps of cL-RMSD values among the models with different qualities defined by the capri_metric (see Figure S2). For example, 15.6% of the medium-quality models (capri_metric) have cL-RMSD values larger than 4.5 Å, while 13.6% of the acceptable models (capri_metric) have cL-RMSDs lower than 4.5 Å. This is mainly because the capri_metric does not directly consider peptide sidechain conformations, which are important to peptide binding.
Ranking
The sampled models were evaluated and ranked using a hybrid scoring function, PepProScore, which was composed of the calculated protein-peptide binding score and the physicochemical similarity between the predicted protein-peptide interface and interacting interface of the template fragment in the monomeric protein (see METHODS).
Figure 5A shows the rates of successfully ranking at least one model with cL-RMSD ≤ a threshold (2.0 Å, 4.0 Å, or 6.0 Å) among the top N models for both bound docking (bpro-upep) and unbound docking (bpro-upep). Not surprisingly, bound docking achieved significantly higher success rates than the challenging unbound docking. For bound docking, MDockPeP2 yielded a total success rate (cL-RMSD ≤ 6.0 Å) of 58.4% (cL-RMSD ≤ 4.0 Å: 36.0%; cL-RMSD ≤ 2.0 Å: 5.6%) when only top 10 models were considered for each prediction. A significantly higher success rate of 70.8% (79.8%) was achieved if the top 100 (1000) models were considered for each prediction. Unbound docking yielded a relatively low success rate of 19.0% (cL-RMSD ≤ 4.0 Å: 8.0%; cL-RMSD ≤ 2.0 Å: 1.7%) when only top 10 models were considered for each case. When top 100 and 1000 models were considered for each case, the total success rates increased to 44.8% and 70.7%, respectively. The above performances were based on the ranking results of the hybrid scoring function (PepProScore). If only the Vina_Score was used for ranking, a total success rates of the bound docking and the unbound docking were 34.8% (60.7%, and 76.4%) and 8.6% (29.3%, and 60.3%), respectively, when top 10 (100, and 1000) models were considered for each case.
Figure 5.
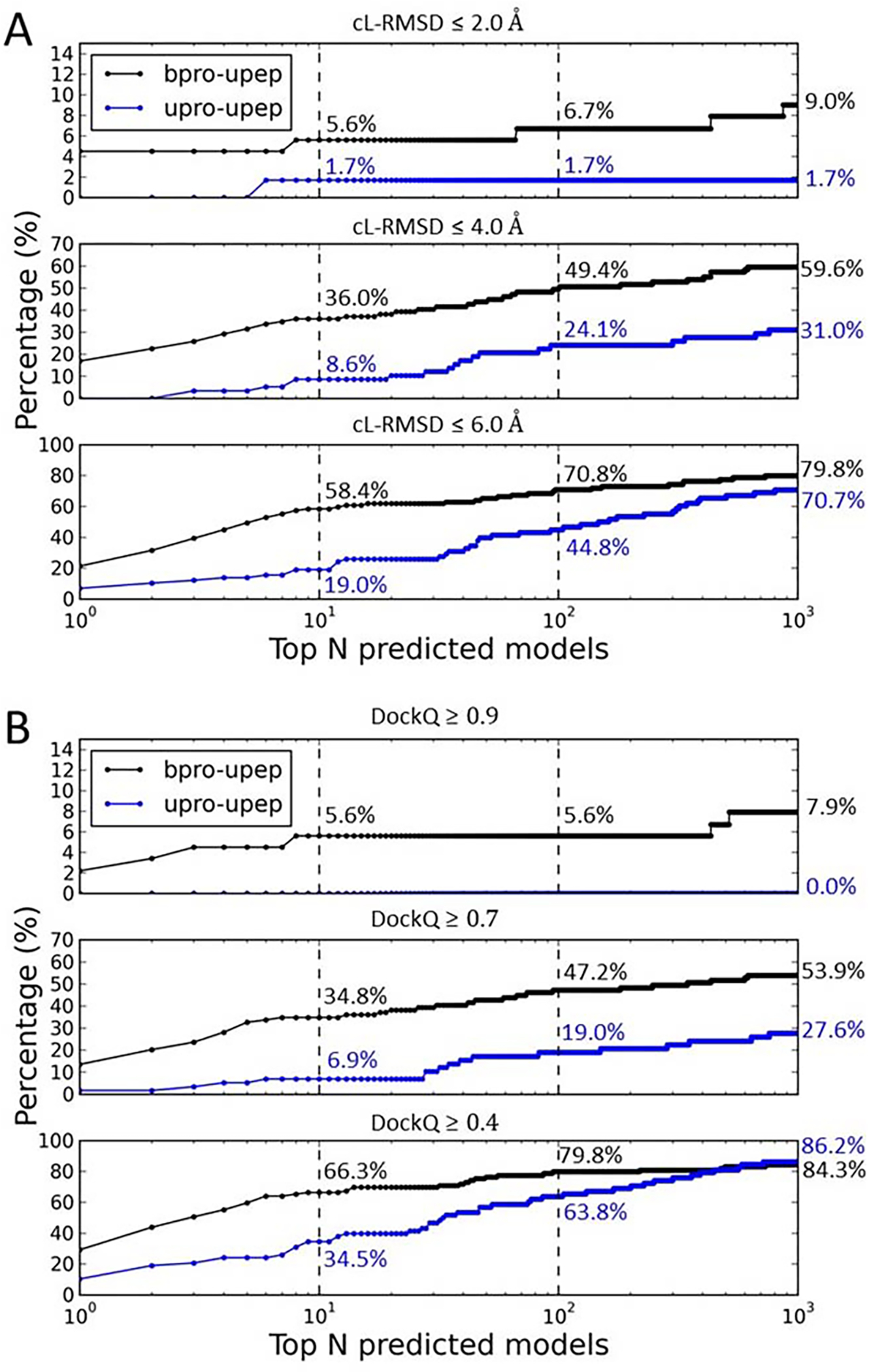
The ranking of the sampled peptide binding modes for both the bound docking cases (bpro-upep) and the unbound docking cases (upro-upep) in PepPro based on the evaluation metrics cL-RMSD (A) and DockQ Score (B). Subpanels show ranking performances using different thresholds of cL-RMSD (2.0 Å, 4.0 Å, and 6.0 Å) and DockQ Scores (0.9, 0.7, and 0.4). Numbers (%) reported in the figures are ranking success rates when top 10, 100, or 1000 models were considered for each prediction.
Figure 5B shows ranking success rates when the DockQ Score was used as the evaluation metric. For the bound docking, MDockPeP2 achieved a total success rate (DockQ ≥ 0.4) of 66.3% (DockQ ≥ 0.7: 34.8%; DockQ ≥ 0.9: 5.6%) when top 10 models were considered for each prediction. The total success rate increased to 79.8% (84.3%) when top 100 (1000) models were considered for each case. For the challenging unbound docking, our docking method yielded a total success rate (DockQ ≥ 0.4) of 34.5% (DockQ ≥ 0.7: 6.9%; DockQ ≥ 0.9: 0.0%) when top 10 models were considered for each prediction. The success rate increased to 63.8% (86.2%) when top 100 (1000) models were considered for each case.
If the capri_metric was used as the evaluation metric, the bound docking yielded a total ranking success rate (acceptable and higher quality) of 60.7% (high- and medium-quality: 33.7%; high-quality: 2.2%) when top 10 models were considered for each prediction. The total success rate increased to 75.3% (79.8%) when top 100 (1000) models were considered for each case. The unbound docking achieved a total success rate (acceptable and higher quality) of 29.3% (high- and medium-quality: 6.9%; high-quality: 0.0%) when top 10 models were considered for each prediction. The success rate increased to 48.3% (67.2%) when top 100 (1000) models were considered for each prediction.
As mentioned above, about half of cases in the PepPro database containing peptides with sequence length longer than 15. If considering only cases containing short- and medium-size peptides (less than or equal to 15 residues), significantly higher success rates were achieved. Using cL-RMSD as the evaluation metric, the bound docking achieved total success rates of 64.6%, 81.2%, and 91.7% when top 10, 100, and 1000 models were considered for each case, respectively. For unbound docking, MDockPeP2 yielded total success rates of 29.0%, 51.6%, and 83.9% when top 10, 100, and 1000 models were considered for each case, respectively.
Figure 6 presents six examples for bound docking cases (Figure 6A–C) and unbound docking cases (Figure 6D–F). The peptides in experimental structures are colored cyan, and those in predicted models are colored magenta. Bound protein structures are colored light gray, and unbound protein structures are colored blue.
Figure 6.
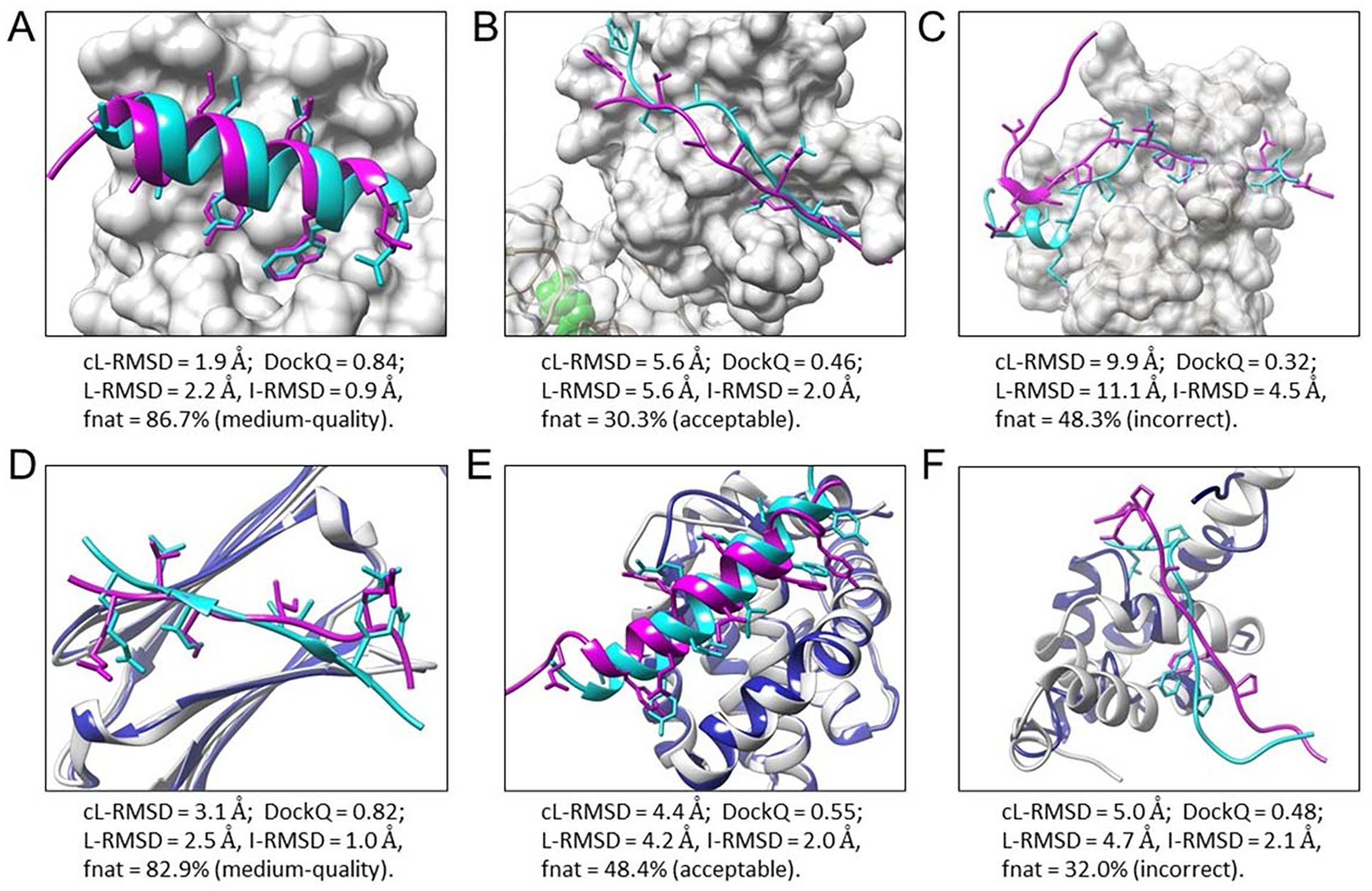
Six examples of peptide binding modes predicted by MDockPeP2. Experimental bound peptide structures are colored cyan, and predicted peptide binding modes are colored magenta. Side chains of contact peptide residues are represented by the stick model. Proteins in the bound docking cases (A-C) are displayed by the surface and colored light gray. For the unbound docking cases (D-F), unbound protein structures (colored blue) are superimposed on bound protein structures (colored light gray). (A) The top predicted model of PDB 1t0j. (B) A predicted model (no. 289) of PDB 2whx. (C) A low-quality model of PDB 4ext. (D) A predicted model (no. 8) of PDB 4m5s, using the unbound protein structure PDB 3utk. (E) A predicted model (no. 260) of PDB 3kj0, using the unbound protein structure PDB 2mhs. (F) A predicted model (no. 228) of PDB 3kut, using the unbound protein structure PDB 1g9l.
Figure 6A shows the top predicted binding mode for the protein-peptide complex PDB 1t0j. This prediction is a high-quality model using cL-RMSD (1.9 Å) as the metric. It is a medium-quality model using the capri_metric (L-RMSD = 2.2 Å, I-RMSD = 0.9 Å, fnat = 86.7%) or DockQ Score (0.84) Figure 6B presents a predicted model of PDB 2whx (being ranked no. 289). This prediction is an acceptable model in the three different evaluation metrics: cL-RMSD = 5.6 Å; capri_metric, L-RMSD = 5.6 Å, I-RMSD = 2.0 Å, fnat = 30.3%; DockQ = 0.46. The low ranking in this case is due to the receptor being a multi-domain protein, and many predicted modes fall into the large domain (non-peptide binding domain) which contains a ligand binding pocket (colored green). Figure 6C shows a low-quality model of PDB 4ext: cL-RMSD = 9.9 Å; incorrect model using capri_metric (L-RMSD = 11.1 Å, I-RMSD = 4.5 Å, fnat = 48.3%); DockQ = 0.32. MDockPeP2 correctly sampled the peptide binding mode for the coil region but failed for the helix region.
Figure 6D shows a correctly predicted model (no.8) of PDB 4m5s when an unbound protein structure (PDB 3utk) was used. The prediction is a medium-quality model using the three different evaluation metrics: cL-RMSD = 3.1 Å; capri_metric, L-RMSD = 2.5 Å, I-RMSD = 1.0 Å, fnat = 82.9%; DockQ = 0.82. Figure 6E presents one of the best sampled models of PDB 3kj0, using the unbound protein structure PDB 2mhs. This is an acceptable model: cL-RMSD = 4.4 Å; capri_metric, L-RMSD = 4.2 Å, I-RMSD = 2.0 Å, fnat = 48.4%; DockQ = 0.55. Interestingly, the bound docking case resulted in a high-quality model (cL-RMSD = 1.8 Å) that was sampled and ranked no. 1. This indicates that the quality of the models is important to the scoring study. Figure 6F shows one of the best sampled models (no. 228) of PDB 3kut, using the unbound protein structure PDB 1g9l. Large conformational changes were observed between bound and unbound protein structures in this case. This prediction is an incorrect model using the capri_metic (L-RMSD = 4.7 Å, I-RMSD = 2.1 Å, fnat = 32.0%), while its cL-RMSD and DockQ Score are 5.0 Å and 0.48, respectively.
Accuracy vs Efficiency
The above results are based on using up to 1000 peptide conformers for each protein-peptide complex. As already mentioned, the computing cost of sampling increases linearly with the number of peptide conformers used for docking. By testing on 20 Intel Xeon cores (Intel(R) Xeon(R) Gold 6148 CPU @ 2.40GHz), an average run time of MDockPeP2 on one case in the PepPro dataset is about 8 hours when 1000 peptide conformers were used for docking. The average run time decreases to less an hour (~50 minutes) when 100 peptide conformers were used for docking. On the other hand, the rate of successfully generating a high-quality peptide conformer (low bRMSD compared with the bound peptide structure) is also in proportion to the number of peptide conformers, as shown in Figure 3C. Therefore, it is meaningful to find a balance between accuracy and efficiency, namely the balance between the performance of the binding mode prediction and the number of peptide conformers used for docking.
The results on the binding mode sampling and ranking show that similar performances were obtained by using different metrics, cL-RMSD, capri_metric, and DockQ. For simplicity purposes, cL-RMSD will be used as the primary metric in the following studies.
Figure 7A shows the success rates for the peptide binding mode sampling using different numbers of peptide conformers for both the bound docking and the unbound docking. The total success rate increased with the increasing number of peptide conformers for both bound and unbound dockings when different thresholds of cL-RMSD (2.0 Å, 4.0 Å, and 6.0 Å) were used. Using the threshold of cL-RMSD 4.0 Å for the bound docking as an example, the success rates were 36.0%, 53.9%, 60.7%, and 73.0% when top 10, 100, 200, and 1000 peptide conformers were used for docking, respectively. If a threshold of cL-RMSD 6.0 Å was used, the success rates of unbound docking were 58.6%, 75.9%, 77.6%, and 82.8% for top 10, 100, 200, and 1000 peptide conformers, respectively. Here, a logarithmic scale is used on the horizontal axis in the figure. Although the success rate continuously increases in the whole range (from 1 to 1000) of the number of peptide conformers, the success rates after 100 increases much less than those before 100.
Figure 7.
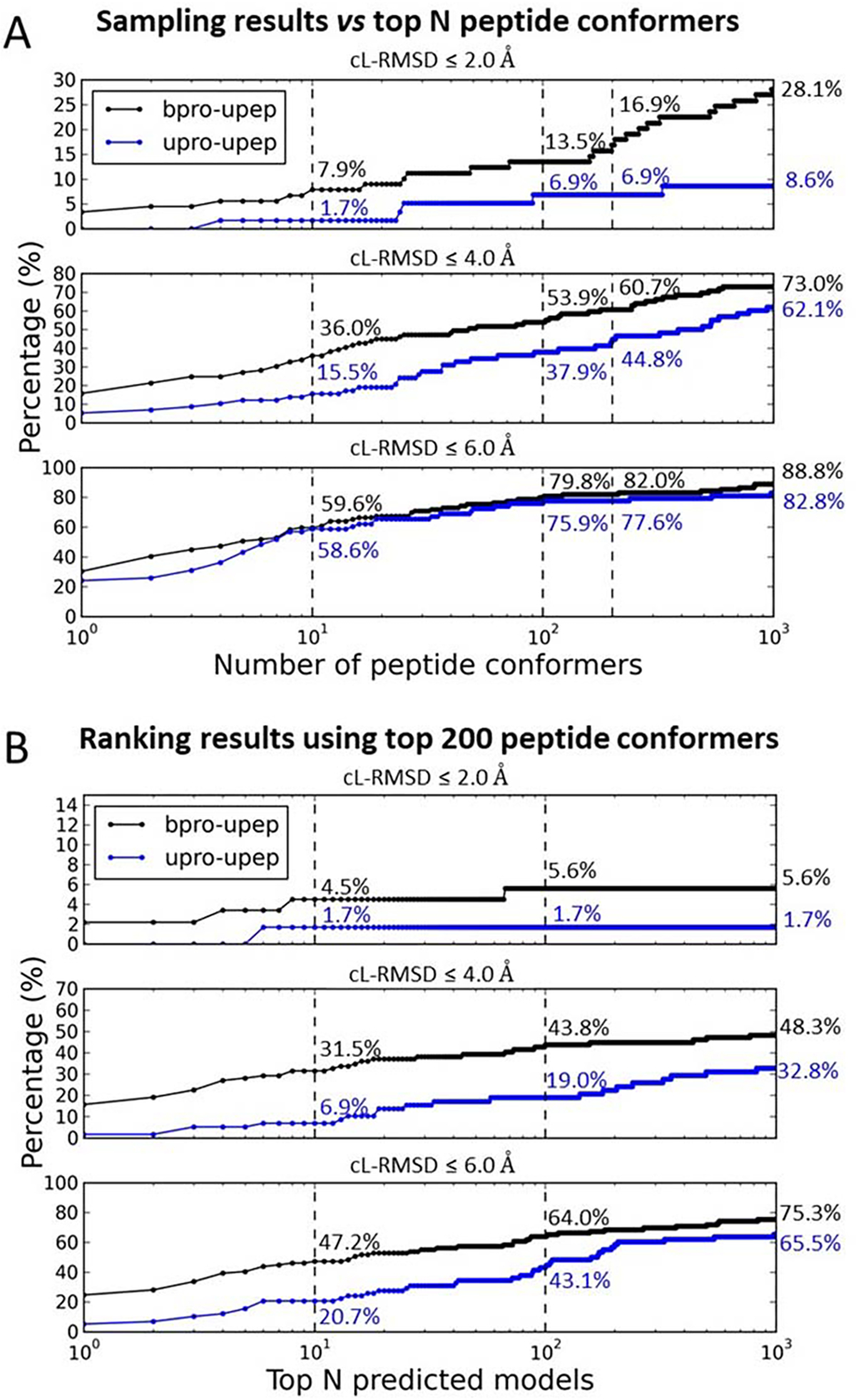
The performance using different number of peptide conformers for docking. (A) The performances of binding mode sampling for both the bound docking (bpro-upep) and the unbound docking (upro-upep). Subpanels show sampling performances using different thresholds of cL-RMSD (2.0 Å, 4.0 Å, and 6.0 Å). Numbers (%) reported in the figures are sampling success rates when top 10, 100, 200, or 1000 peptide conformers were used for docking in each prediction. (C) The performance of ranking the peptide binding modes generated using top 200 peptide conformers in the sampling stage. Numbers (%) reported in the figures are ranking success rates when top 10, 100, 1000 models were considered for each case.
Figure 7C shows ranking results using the top 200 peptide conformers in each protein-peptide docking process. When only the top 10 predicted models were considered for each prediction, a total success rate (cL-RMSD ≤ 6.0 Å) of 47.2% (cL-RMSD ≤ 4.0 Å: 31.5%; cL-RMSD ≤ 2.0 Å: 4.5%) was achieved for the bound docking. This is about 10% lower than the success rate when 1000 peptide conformers were used for docking (Figure 4C). Interestingly, for unbound docking, a slightly higher success rate 20.7% was achieved, compared to that of using 1000 peptide conformers for docking (19.0%). When the top 100 (1000) predicted models were considered for each case, the success rates were 64.0% (75.3%) and 43.1 (65.5%) for bound and unbound docking, respectively. The performances are comparable to those using 1000 peptide conformers for docking. Ranking results using 100 and 500 peptide conformers for docking are also presented in Figure S3. Generally, the more peptide conformers that are used for docking, the higher the success rate can be achieved in the ranking stage. However, the computing cost also increases as the number of the peptide conformers increases.
peptiDB and LEADS-PEP datasets
MDockPeP2 was further tested on two other datasets of protein-peptide complex structures, peptiDB and LEADS-PEP. For each case in a dataset, up to 1000 peptide conformers were used for docking. Performances of our method based on peptiDB and LEADS-PEP are presented in Figure 8A and 8B, respectively.
Figure 8.
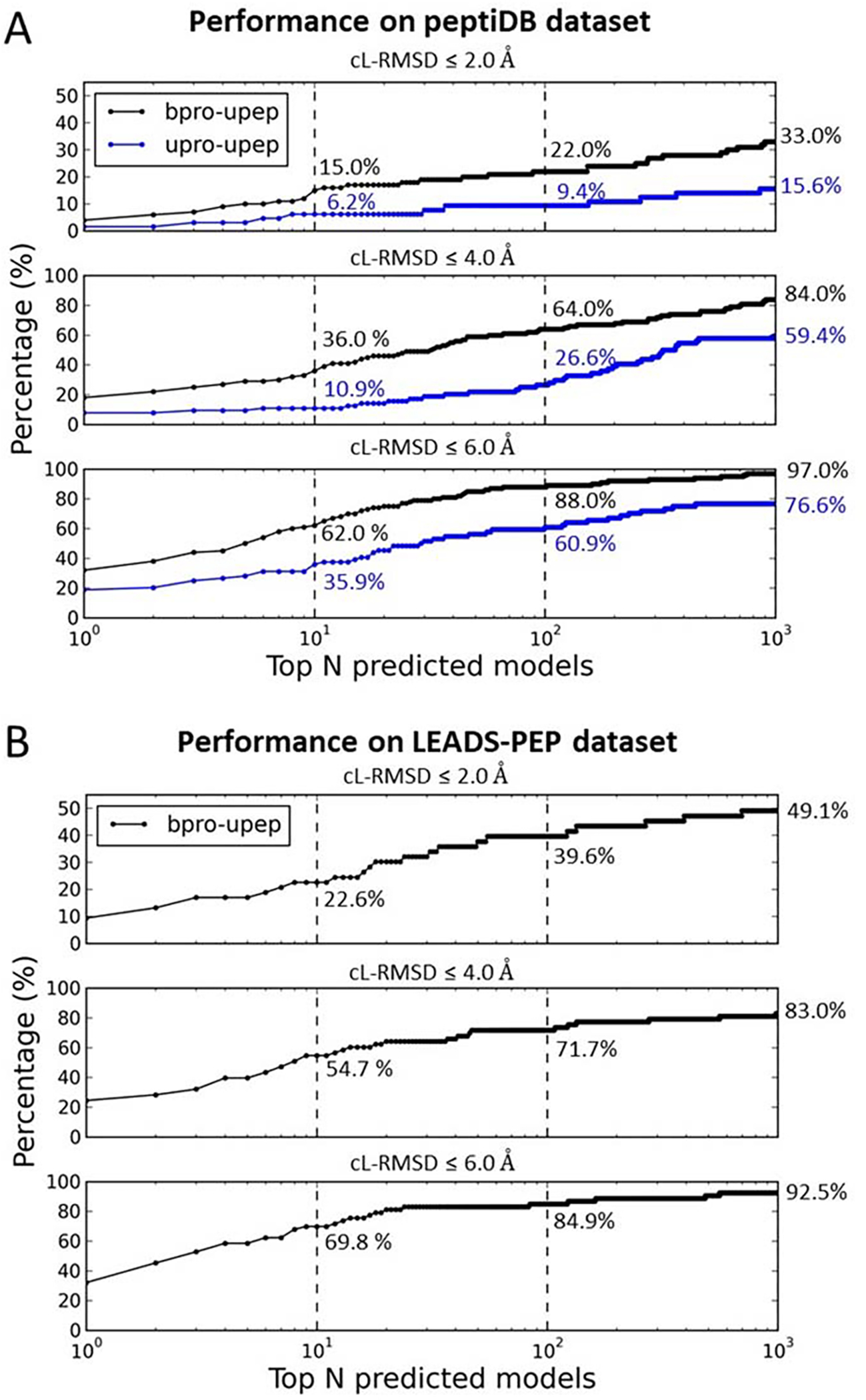
Performance of MDockPeP2 on the peptiDB dataset (A) and the LEADS-PEP dataset (B). peptiDB contains both bound docking cases and unbound docking cases. LEADS-PEP contains only bound docking cases. Subpanels show ranking results using different thresholds of cL-RMSD (2.0 Å, 4.0 Å, and 6.0 Å). Numbers (%) reported in the figures are ranking success rates when top 10, 100, 1000 models were considered for each case.
Figure 8A shows the rates of successfully ranking at least one model with cL-RMSD ≤ a threshold (2.0 Å, 4.0 Å, or 6.0 Å) among the top N models for both bound docking (bpro-upep) and unbound docking (bpro-upep). Like the PepPro dataset, bound docking achieved significant higher success rates than the challenging unbound docking. For bound docking, MDockPeP2 yielded a total success rate (cL-RMSD ≤ 6.0 Å) of 62.0% (cL-RMSD ≤ 4.0 Å: 36.0%; cL-RMSD ≤ 2.0 Å: 15.0%) when only top 10 models were considered for each prediction. The total success rates (cL-RMSD ≤ 6.0 Å) increased to 88.0% (97.0%) when top 100 (1000) models were considered for each docking. Unbound docking yielded a relatively low success rate of 35.9% (cL-RMSD ≤ 4.0 Å: 10.9%; cL-RMSD ≤ 2.0 Å: 6.2%) when only top 10 models were considered for each case. When top 100 and 1000 models were considered for each case, the total success rates (cL-RMSD ≤ 6.0 Å) increased to 60.9% and 76.6%, respectively.
peptiDB was widely used as a benchmark dataset by other global docking methods. By testing on this dataset, one of the most powerful protein-peptide docking methods, CABS-dock yielded a total success rate of 53.0% for bound docking and 37.0% for unbound docking when top 10 models were considered for each prediction11. Slightly better performances were achieved by our previous developed docking method, with a total success rate of 54.0% for bound docking and 37.5% for unbound docking.15 In these studies, ligand RMSD (L-RMSD) was used as the primary criterion, and a cutoff of 5.5 Å was used to distinguish near-native binding modes from decoys. Using the same criterion, MDockPeP2 achieved significantly better performances than our previous method MDockPeP, with a total success rate of 62.0% for bound docking and 40.0% for unbound docking. In addition, HPEPDOCK18 reported a total success rate (I-RMSD ≤ 2.0 Å) of 33.3% for the unbound cases in peptiDB. Our method achieved a similar performance (32.8%) when the same criterion (i.e., I-RMSD ≤ 2.0 Å) was applied.
LEADS-PEP dataset contains only bound cases. Prediction results are shown in Figure 8B. A total success rate (cL-RMSD ≤ 6.0 Å) of 69.8% (cL-RMSD ≤ 4.0 Å: 54.7%; cL-RMSD ≤ 2.0 Å: 22.6%) was achieved when top 10 models were considered for each prediction. The success rate increased to 84.9% (92.5%) when top 100 (1000) models were considered for each case.
Very recently, Zhang and Sanner developed a protein-peptide local docking method (known binding site), AutoDock CrankPep (ADCP)21, which achieved impressive performance on the LEADS-PEP dataset. Using L-RMSD as the evaluation metric, ADCP yielded a success rate of 85.7% (L-RMSD ≤ 2.5 Å) when top 10 models were considered for each prediction. Our global-docking MDockPeP2 method achieved a success rate of 41.5% (L-RMSD ≤ 2.5 Å) when top 10 models were considered for each case.
DISCUSSION
In this study, a new criterion, cL-RMSD, was introduced to compare a predicted protein-peptide binding mode with the experimentally determined complex structure. Prediction results based on the new metric were compared to those based on two other metrics, capri_metric and DockQ Score (see sections of “Assessment criteria” and “Binding mode sampling”). As shown in Figure 4D, the results based on cL-RMSD and those based on DockQ have a good correlation. However, large overlaps of cL-RMSD values among the models with different qualities defined by capri_metric were observed (as shown in Figure S3B). We observed many cases which were defined as acceptable or even medium-quality models by capri_metric but had cL-RMSD larger than 6 Å (see Figure 4D). Figure S4A shows an example of this case. The predicted model was defined as medium-quality due the large contribution from fnat (fraction of native contacts). However, the predicted peptide conformation was incorrect. The peptide adopted a compact β-hairpin conformation in the crystal structure but was predicted as an extended structure in the docking model. The incorrectly predicted extended region of the peptide resulted in many non-native contacts, which were not considered in neither capri_metric nor DockQ Score. The DockQ Score of this model was 0.44, indicating it as an acceptable model. However, cL-RMSD of this model was as large as 9.3 Å, which means this model was incorrect. Figure S4B shows an opposite case, in which the predicted model was defined as incorrect by capri_metric but was an acceptable model according to cL-RMSD (4.4 Å) or DockQ (0.51). In this predicted model, the peptide conformation was well-modeled. Contacting residues in the modeled structure were close to those in the crystal structure. It is more like a near-native model rather than an incorrect model.
As described in the section of “Assessment criteria”, both capri_metric and DockQ Score are based on three parameters, L-RMSD, I-RMSD, and fnat, in which only fnat partially considers peptide sidechain conformations. It is worth mentioning that fnat considers only the recovery of native contacts in a modeled structure. Both capri_metric and DockQ Score ignore non-native contacts in the predicted models. In fact, the sidechain conformations of some peptide residues (e.g. “hot spot” residues, anchor residues, or binding motif) are crucial to the protein-peptide binding. Therefore, we do need a parameter like cL-RMSD which can directly analyze peptide sidechain conformations. Another advantage of cL-RMSD is that it is a single variable. Capri_metric involves complex combinations of three different parameters, while DockQ Score depends on the scaling factor used for each parameter.
In the sampling step of MDockPeP2, 2000 binding modes generated by ZDOCK were ranked by ITScorePP, and then top 100 models were kept for each peptide conformer in each docking case. If all the models sampled by ZDOCK were considered, higher sampling success rates could be achieved. Using bound docking of PepPro dataset as an example, the total sampling success rate (cL-RMSD ≤ 6.0 Å) was 95.5% (cL-RMSD ≤ 4.0 Å: 79.8%; cL-RMSD ≤ 2.0 Å: 28.1%) when all the ZDOCK poses (without local minimization; 2000 poses for each peptide conformer) were considered. The sampling success rate (cL-RMSD ≤ 6.0 Å) decreased to 89.9% (cL-RMSD ≤ 4.0 Å: 74.2%; cL-RMSD ≤ 2.0 Å: 24.7%) when top 100 models (ranked by ITScorePP) were used for each peptide conformer. This indicates that the performance of our method can be further improved by keeping more models in the rigid docking step at the expense of computing time.
In this study, peptides were treated as fully flexible, but protein structures were treated as rigid. This should be responsible for low success rates of the unbound docking comparing to the bound docking. In the real protein-peptide binding process, both peptide and protein are flexible. Unfortunately, it is difficult to address the protein flexibility issue in current large-scale studies. However, our method can provide reasonable starting structures for other accurate but time-consuming methods such as molecular dynamics (MD) simulations, which can improve the results by treating proteins as flexible.
Another challenge is the scoring function. As shown in Figure 4A, MDockPeP2 successfully generated at least one near-native binding mode (cL-RMSD ≤ 6.0 Å) for 88.8% bound cases in the PepPro dataset. However, our current scoring function ranked at least one near-native binding mode into top 10 models only for 58.4% bound cases (Figure 5A). The ranking success rates decreased to 21.3% and 39.3% when the top model or the top 3 models were considered. This indicates that there is a large room to improve the scoring function in our future studies.
CONCLUSION
We developed a novel method MDockPeP2 for the protein-peptide complex structure prediction, using only the protein structure and the peptide sequence as inputs. By testing on PepPro benchmark with peptide sequence length ranging from 5 to 29, MDockPeP2 yielded excellent performances for binding mode sampling, with total success rates of 88.8% and 82.8% for bound cases and unbound cases, respectively. when only short- and medium-size peptides (less than or equal to 15 residues) were considered, the success rates increased to 100% and 90.3% for bound cases and unbound cases, respectively. Good performances were also achieved for binding mode ranking. The overall success rate after ranking is 58.4% for bound docking and 19.0% for unbound docking for the top 10 models. When top 100 (1000) models were considered for each case, total success rates increased to 70.8% (79.8) and 44.8% (70.7%) for bound docking and unbound docking, respectively. Significantly higher success rates were achieved for the datasets peptiDB and LEADS-PEP, which contain only short- and medium-size peptides (≤ 15 residues). For peptiDB, the ranking success rates (top 10 models) were 62.0% and 35.9% for bound and unbound docking, respectively. For LEADS-PEP, MDockPeP2 achieved a success rate of 69.8% when the top 10 models were considered. Therefore, our method is suitable for both protein-peptide complex structure prediction and initial-stage sampling of the protein-peptide binding modes for other docking or simulation methods.
Data and Software Availability
The MDockPeP2 program and docking modes for the three datasets, PepPro, peptiDB, and LEADS-PEP are available at https://zougrouptoolkit.missouri.edu/mdockpep2/download.html.
Supplementary Material
Acknowledgements
We are grateful to Benjamin Ryan Merideth for editing the article. This work was supported by NIH R01GM109980 and R35GM136409 (PI: XZ), NIH R01HL126774 (PI: Jianmin Cui) and NIH R01HL142301 (PI: Jonathan R. Silva) to XZ. The computations were performed on the high-performance computing infrastructure supported by NSF CNS-1429294 (PI: Chi-Ren Shyu) and the HPC resources supported by the University of Missouri Bioinformatics Consortium (UMBC).
Footnotes
ASSOCIATED CONTENT
Supporting Information Available: Percentages of the contact residues in the peptides from the PepPro dataset (Table S1), distribution of the bRMSD values (Figure S1), distributions of DockQ Scores (A) and cL-RMSD values (Figure S2), ranking of the peptide binding modes (Figure S3), and two examples of peptide binding models (Figure S4).
References
- 1.Petsalaki E; Russell RB, Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol 2008, 19 (4), 344–50. [DOI] [PubMed] [Google Scholar]
- 2.Dyson HJ; Wright PE, Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Bio 2005, 6 (3), 197–208. [DOI] [PubMed] [Google Scholar]
- 3.Fosgerau K; Hoffmann T, Peptide therapeutics: current status and future directions. Drug Discov Today 2015, 20 (1), 122–8. [DOI] [PubMed] [Google Scholar]
- 4.Bruzzoni-Giovanelli H; Alezra V; Wolff N; Dong CZ; Tuffery P; Rebollo A, Interfering peptides targeting protein-protein interactions: the next generation of drugs? Drug Discov Today 2018, 23 (2), 272–285. [DOI] [PubMed] [Google Scholar]
- 5.London N; Raveh B; Schueler-Furman O, Peptide docking and structure-based characterization of peptide binding: from knowledge to know-how. Curr Opin Struct Biol 2013, 23 (6), 894–902. [DOI] [PubMed] [Google Scholar]
- 6.Ciemny M; Kurcinski M; Kamel K; Kolinski A; Alam N; Schueler-Furman O; Kmiecik S, Protein-peptide docking: opportunities and challenges. Drug Discovery Today 2018, 23 (8), 1530–1537. [DOI] [PubMed] [Google Scholar]
- 7.Antes I, DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins 2010, 78 (5), 1084–104. [DOI] [PubMed] [Google Scholar]
- 8.London N; Raveh B; Cohen E; Fathi G; Schueler-Furman O, Rosetta FlexPepDock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res 2011, 39 (Web Server issue), W249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verschueren E; Vanhee P; Rousseau F; Schymkowitz J; Serrano L, Protein-Peptide Complex Prediction through Fragment Interaction Patterns. Structure 2013, 21 (5), 789–797. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Shimon A; Niv MY, AnchorDock: Blind and Flexible Anchor-Driven Peptide Docking. Structure 2015, 23 (5), 929–940. [DOI] [PubMed] [Google Scholar]
- 11.Kurcinski M; Jamroz M; Blaszczyk M; Kolinski A; Kmiecik S, CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Research 2015, 43 (W1), W419–W424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H; Heo L; Lee MS; Seok C, GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res 2015, 43 (W1), W431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindler CEM; de Vries SJ; Zacharias M, Fully Blind Peptide-Protein Docking with pepATTRACT. Structure 2015, 23 (8), 1507–1515. [DOI] [PubMed] [Google Scholar]
- 14.Obarska-Kosinska A; Iacoangeli A; Lepore R; Tramontano A, PepComposer: computational design of peptides binding to a given protein surface. Nucleic Acids Research 2016, 44 (W1), W522–W528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan CF; Xu XJ; Zou XQ, Fully Blind Docking at the Atomic Level for Protein-Peptide Complex Structure Prediction. Structure 2016, 24 (10), 1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alam N; Goldstein O; Xia B; Porter KA; Kozakov D; Schueler-Furman O, High-resolution global peptide-protein docking using fragments-based PIPER-FlexPepDock. Plos Comput Biol 2017, 13 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Porter KA; Xia B; Beglov D; Bohnuud T; Alam N; Schueler-Furman O; Kozakov D, ClusPro PeptiDock: efficient global docking of peptide recognition motifs using FFT. Bioinformatics 2017, 33 (20), 3299–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou P; Jin BW; Li H; Huang SY, HPEPDOCK: a web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Research 2018, 46 (W1), W443–W450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou P; Li BT; Yan YM; Jin BW; Wang LB; Huang SY, Hierarchical Flexible Peptide Docking by Conformer Generation and Ensemble Docking of Peptides. J Chem Inf Model 2018, 58 (6), 1292–1302. [DOI] [PubMed] [Google Scholar]
- 20.Johansson-Åkhe I, Mirabello C and Wallner B, InterPep2: global peptide–protein docking using interaction surface templates. Bioinformatics 2020, 36(8), 2458–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y and Sanner MF, AutoDock CrankPep: combining folding and docking to predict protein–peptide complexes. Bioinformatics, 2019, 35(24), 5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanhee P; Stricher F; Baeten L; Verschueren E; Lenaerts T; Serrano L; Rousseau F; Schymkowitz J, Protein-Peptide Interactions Adopt the Same Structural Motifs as Monomeric Protein Folds. Structure 2009, 17 (8), 1128–1136. [DOI] [PubMed] [Google Scholar]
- 23.Wright PE; Dyson HJ, Linking folding and binding. Curr Opin Struct Biol 2009, 19 (1), 31–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu X and Zou X, PepPro: A Nonredundant Structure Data Set for Benchmarking Peptide–Protein Computational Docking. J Comput Chem 2020, 41(4), 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.London N; Movshovitz-Attias D; Schueler-Furman O, The structural basis of peptide-protein binding strategies. Structure 2010, 18 (2), 188–99. [DOI] [PubMed] [Google Scholar]
- 27.Hauser AS; Windshügel B, LEADS-PEP: a benchmark data set for assessment of peptide docking performance. J Chem Inf Model, 2016, 56(1), 188–200. [DOI] [PubMed] [Google Scholar]
- 28.Webb B; Sali A, Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics 2014, 47, 5.6.1–5.6.32. [DOI] [PubMed] [Google Scholar]
- 29.Henikoff S; Henikoff JG, Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 1992, 89 (22), 10915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kabsch W, A discussion of the solution for the best rotation to relate two sets of vectors. Acta Cryst 1978, 34, 827–828. [Google Scholar]
- 31.Pierce BG; Hourai Y; Weng ZP, Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. Plos One 2011, 6 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang SY; Zou X, An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008, 72 (2), 557–79. [DOI] [PubMed] [Google Scholar]
- 33.Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31 (2), 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng S; Zhang Y; Brooks CL 3rd, PCalign: a method to quantify physicochemical similarity of protein-protein interfaces. BMC Bioinformatics 2015, 16, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcu O; Dodson EJ; Alam N; Sperber M; Kozakov D; Lensink MF; Schueler-Furman O, FlexPepDock lessons from CAPRI peptide-protein rounds and suggested new criteria for assessment of model quality and utility. Proteins 2017, 85 (3), 445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lensink MF; Nadzirin N; Velankar S; Wodak SJ, Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 2020, 88(8), 916–938. [DOI] [PubMed] [Google Scholar]
- 37.Lee B; Richards FM, The interpretation of protein structures: estimation of static accessibility. J Mol Biol 1971, 55 (3), 379–400. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25 (13), 1605–12. [DOI] [PubMed] [Google Scholar]
- 39.Edgar RC, Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26 (19), 2460–2461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MDockPeP2 program and docking modes for the three datasets, PepPro, peptiDB, and LEADS-PEP are available at https://zougrouptoolkit.missouri.edu/mdockpep2/download.html.