

Abstract
Antibiotics with fundamentally new mechanisms of action such as the armeniaspirols, which target the ATP-dependent proteases ClpXP and ClpYQ, must be developed to combat antimicrobial resistance. While the mechanism of action of armeniaspirol against Gram-positive bacteria is understood, little is known about the structure–activity relationship for its antibiotic activity. Based on the preliminary data showing that modifications of armeniaspirol's N-methyl group increased antibiotic potency, we probed the structure–activity relationship of N-alkyl armeniaspirol derivatives. A series of focused derivatives were synthesized and evaluated for antibiotic activity against clinically relevant pathogens including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus. Replacement of the N-methyl with N-hexyl, various N-benzyl, and N-phenethyl substituents led to substantial increases in antibiotic activity and potency for inhibition of both ClpYQ and ClpXP. Docking studies identified binding models for ClpXP and ClpYQ that were consistent with the inhibition data. This work confirms the role of ClpXP and ClpYQ in the mechanism of action of armeniaspirol and provides important lead compounds for further antibiotic development.
Antibiotics with fundamentally new mechanisms of action such as the armeniaspirols, which target the ATP-dependent proteases ClpXP and ClpYQ, must be developed to combat antimicrobial resistance.
Introduction
With the constant threat of increasing antimicrobial resistance, antibiotics with fundamentally new mechanisms of action must be developed and brought to the clinic. The armeniaspirols discovered in 2012 are hybrid polyketide non-ribosomal peptide natural products produced by Streptomyces armeniacus1,2 that have recently been shown to possess unique pharmacology.3 The armeniaspirols are potent Gram-positive antibiotics active against methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant Streptococcus pneumoniae (PRSP), and vancomycin-resistant Enterococcus (VRE). They competitively inhibit the ATP-dependent proteases ClpXP and ClpYQ (also known as HslVU),3 whose combined activity is essential,3 leading to disruption of the divisome and cell division arrest (Fig. 1A). While inhibitors of ClpP are known antivirulence compounds, the additional targeting of ClpYQ by armeniaspirol is unprecedented and results in antibiotic activity.
Fig. 1. Analogs of armeniaspirol to probe antibiotic activity via inhibition of ClpXP and ClpYQ (A) 5-chloroarmeniaspirol inhibits the ATP-dependent proteases ClpXP and ClpYQ in Gram-positive bacteria, which dysregulates the divisome and leads to cell division arrest. (B) Summary of structure–activity relationships for armeniaspirol. Functional groups highlighted in blue are essential for antibiotic activity. R indicates sites amenable to substitution. (C) 5-chloroarmeniaspirol sites R1 and R2 were selected for detailed structure–activity relationship.

Little is known about the structure–activity relationship for antibiotic activity of armeniaspirol. Initial semi-synthesis work from the isolated natural product showed that substitution of the β-chloride with alcohols, amines, and thiols eliminated antibiotic activity as determined by minimum inhibitory concentrations (MIC).2 Similarly alkylation of the phenol and reduction of the carbonyl abolished activity (Fig. 1B).2Via total synthesis, the N-methyl moiety proved amenable to replacement with a longer N-alkyl chain without loss of activity.3 However no data is available on how structural modifications impact the activity against ClpXP and ClpYQ. Given that correctly balancing activity at each target is essential for development of effective and potent multi-target drugs, evaluating the impact of structural perturbations on each of these targets is thus essential.
Based on preliminary data showing that modification of armeniaspirol's N-methyl group increased antibiotic potency,3 we probed the structure–activity relationship of N-alkyl armeniaspirol derivatives. Fourteen armeniaspirol analogues were synthesized with varying N-alkyl groups. To compensate for the additional lipophilic character of extending the N-methyl group, the aromatic hexyl chain was simplified to a methyl substituent in a subset of the analogues. All compounds were evaluated for MIC and minimum bactericidal concentration (MBC) against clinically relevant Gram-positive pathogens including MRSA USA300, the most common community acquired MRSA. Potent analogues were evaluated for activity against ClpXP in a cell-based assay and activity against ClpYQ in a biochemical assay. Highly potent armeniaspirol analogues with improved activity against ClpXP and ClpYQ were identified. These data bode well for the further development of the armeniaspirol scaffold as a Gram-positive antibiotic.
Results
The armeniaspirols and their analogues have been isolated from the producing organism,1,4 generated through semi-synthesis,3 and accessed via total synthesis.2,3 Analogues generated from the addition of alcohols like methanol, amines such as isobutyl amine, and thiols like 2-diethylamino-ethanethiol into the Michael acceptor lack antibiotic activity against S. aureus in MIC assays.1 Both diastereomers generated from reduction of the furan ring carbonyl are inactive against S. aureus in MIC assays.1 Methylation of the aromatic phenol also produces inactive compounds.1,3 However, chlorination ortho to the phenol is tolerated with a slight reduction in antibiotic activity as measured by MIC against MRSA.1,3 Both the removal of the N-methyl4 and substitution of it with an alkyl chain3 produce active compounds, with installation of a N-hexyl chain boosting potency four fold against Bacillus subtilis.3 Lastly modification of the aromatic alkyl chain via branching produces compounds that are comparatively potent to armeniaspirol A in MIC assays with MRSA.1
Based on the known structure–activity relationships, exploring modification of the lactam through N-alkylation appeared most promising and likely to yield compounds with improved potency. Thus we set out to diversify this position by substituting the amide N with varying alkyl groups. To evaluate the overall impact of increasing lipophilicity, which typically correlates with an increase in non-selective binding,5 we planned two series of N-alkyl analogues, one with the native hexyl chain on the aromatic ring (series 1, Fig. 1CR1 = n-C6H13) and a second with a methyl substituent (series 2, Fig. 1CR1 = CH3).
Synthesis of chloro-armeniaspirol analogues
The synthetic route for generation of the armeniaspirol scaffold relies on a N-chlorosuccinimide (NCS)-based chlorination of the pyrrole, which forms the key spirocyclic center and the dichlorinated α,β-unsaturated lactam.2,3,6 Due to the forcing oxidative conditions, the electron rich phenyl ring is also chlorinated giving rise to 5-chloroarmeniaspirol analogues (Fig. 1C). While the 5-chloro substituent could not be selectively reduced in the presence of the dichloro-α,β-unsaturated lactam, the additional chlorination does not significantly impact the antibiotic activity as measured by MIC.2 Thus, all analogues synthesized in this study are functionalized with a chloride in the 5-position (Fig. 2).
Fig. 2. Armeniaspirol analogues. (A) Synthetic route installing the hexyl chain for the starting material used for all series 1 analogues. Reagents and conditions are as follows a) n-BuLi, Br2 b) Pd(OAc)2, SPhos, K3PO4. (B) Synthetic route generating series 1 and 2 analogues from dimethyl-2-methylresorcinol and dimethyl-2-hexylresorcinol. Reagents and conditions are as follows a) SnCl4 b) BBr3 c) NCS d) NEt3 e) NaH f) BBr3 (C) structures of series 1 and 2 analogues used in this study.

Diversification of the substituent on the phenyl ring (R1) was implemented in the start of the synthesis. The hexyl chain series of analogues (series 1) were prepared via a lithium halogen exchange followed by a Suzuki coupling with n-hexyl boronic acid (Fig. 2A), while the methyl series of analogues (series 2) were prepared from commercially available 2,6-dimethoxytoluene. The two compounds were elaborated via parallel synthetic routes (Fig. 2B). Friedel–Crafts acylation of the electron rich aromatic with the acid chloride of pyrrole carboxylic acid, followed by selective demethylation of methoxy substituent ortho to the carbonyl7 generated the key precursors for oxidative spirocyclization. NCS in acetic acid chlorinated and oxidized these intermediates generating a mixture of spirocyclic compounds with either the 2,3-dichloro-α,β-unsaturated lactam or the 2,2,3-trichloro lactam. Treatment of this mixture with triethylamine afforded the armeniaspirol scaffolds. Alkylation of the amide with a variety of electrophiles enabled diversification of each series at the R2 site. Finally, boron tribromide deprotection of the aryl ether generated the final compounds, 1–14 (Fig. 2C).
MIC evaluation of analogues
The evaluation of antibiotic activity of armeniaspirol analogues was accomplished using the microtiter broth dilution MIC and MBC assays8 against a panel of clinically relevant Gram-positive pathogens (Table 1). These include MRSA USA100, the primary lineage responsible for hospital acquired MRSA infections,9 USA200, responsible for a smaller subset of hospital acquired infections, USA300, the primary community acquired MRSA pathogen, USA400, also responsible for community acquired MRSA infections,10,11 and the high-priority pathogen VRE. MIC were also determined with the Gram-negative pathogen Pseudomonas aeruginosa PA01, to determine if any analogues had expanded activity against Gram-negative bacteria.
MIC evaluation of analogues 1–14 against a panel of Gram positive and negative bacteria.
| Minimum inhibitory concentration (μg mL−1) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
| Gram-positive | ||||||||||||||
| S. aureus IA116-USA100 | 4 | 1 | 8 | 4 | 1 | 2 | 1 | 1 | >32 | 4 | 2 | >32 | >32 | 16 |
| S. aureus MN8-USA200 | 4 | 1 | 16 | 4 | 1 | 2 | 0.5 | 0.5 | >32 | 4 | 2 | >32 | >32 | 16 |
| S. aureus LAC-Fitz-USA300 | 4 | 1 | 16 | 4 | 1 | 2 | 0.5 | 0.5 | >32 | 4 | 1 | >32 | 32 | 16 |
| S. aureus MW2-USA400 | 4 | 1 | 8 | 4 | 2 | 2 | 0.5 | 0.5 | >32 | 8 | 2 | >32 | 32 | 16 |
| E. faecalis NJ3 | 8 | 2 | 8 | 8 | 2 | 2 | 1 | 1 | >32 | 8 | 0.5 | >32 | >32 | 32 |
| Gram-negative | ||||||||||||||
| P. aeruginosa PA01 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 | >32 |
In general the series 1 hexyl chain analogues exhibited superior potency relative to the methyl derivatives from series 2. The hexyl derivative, 2, and the benzyl derivative, 5, showed two- to four-fold more potent MICs compared to 1. 6, the p-trifluorobenzyl derivative consistently showed slightly diminished potency across all strains, while the p-methylbenzyl derivative 7 showed some of the most potent activity in this study. Interesting its constitutional isomer 8 also showed comparable activity.
The series 2 analogues, which all possess a methyl substituent at R1, showed little to no inhibition of bacterial growth except for 10 and 11. 10 is the constitutional isomer of 1 and shows near identical potency across all six strains. The dodecyl derivative 11 was the most potent of all series 2 compounds and exhibited comparable MIC values to several of the best series 1 derivatives. While derivatives from series 1 were generally more potent than the derivatives from series 2, consistent with the increase in lipophilicity, the N-dodecyl derivative in series 2, 11, was substantially more potent than 3. In addition to having unique potency in series 2, 11 proved to be bactericidal differentiating it from all the other analogues. The parent compound and all other analogues were bacteriostatic as defined by having MBC greater than four times the MIC (ESI† Table S1).12–14
None of the compounds showed activity against the Gram-negative pathogen P. aeruginosa. Previous work has shown that the armeniaspirols are not active against many Gram-negative pathogens including Acinetobacter baumannii, Klebsiella pneumonia, Salmonella typhimurium, Shigella dysenteriae.4 Interestingly while 1 does not inhibit the growth of wild-type Escherichia coli, it does inhibit growth of an E. coli ΔtolC mutant (ESI† Table S4), suggesting efflux may be limiting activity in Gram-negative bacteria.15,16
Inhibition of recombinant purified ClpYQ
Mechanistically, armeniaspirol functions via inhibition of both the ATP-dependent proteases ClpXP and ClpYQ.3 We thus evaluated the ability of analogues with enhanced MIC potency relative to 1 for their ability to inhibit peptide hydrolysis by recombinant purified ClpYQ (Table 2, ESI† Fig. S1, ESI† Table S2). Given that ClpYQ from S. aureus was inactive in our in vitro biochemical assays, inhibition kinetics were performed on the characterized B. subtilis ClpYQ.3
Kinetic parameters of potent analogues.
| Analogue | R 1 group | R 2 group | MECClpXP (μg mL−1) | K I ClpYQ (μM) | MIC (USA300 μg mL−1) |
|---|---|---|---|---|---|
| (± std dev) | |||||
| 1 | –n-C6H13 | –CH3 | 1 | 3.2 ± 0.2 | 4 |
| 2 | –n-C6H13 | –n-C6H13 | 0.067 | 1.3 ± 0.2 | 1 |
| 5 | –n-C6H13 |
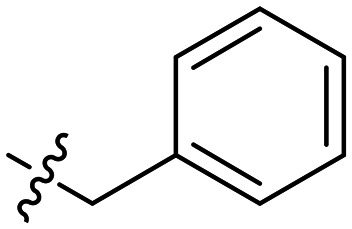
|
0.25 | 0.58 ± 0.05 | 1 |
| 6 | –n-C6H13 |
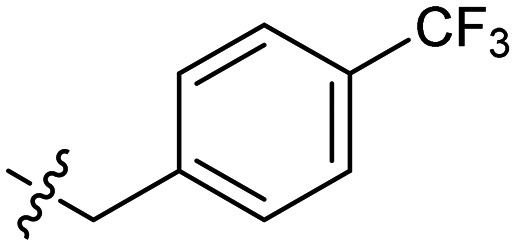
|
1 | 0.57 ± 0.10 | 2 |
| 7 | –n-C6H13 |
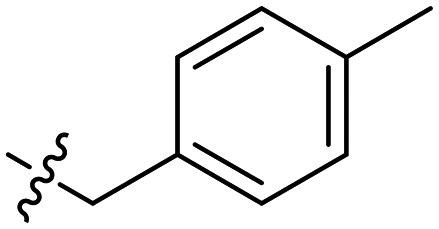
|
0.25 | 0.15 ± 0.02 | 0.5 |
| 8 | –n-C6H13 |
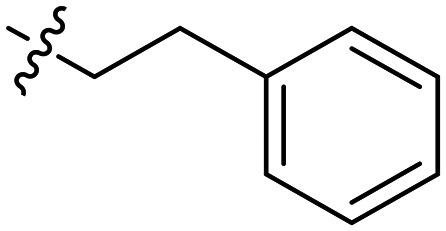
|
0.25 | 0.41 ± 0.07 | 0.5 |
| 11 | –CH3 | –n-C12H25 | 0.5 | 1.0 ± 0.2 | 1 |
A clear dose response curve was observed with all compounds for ClpYQ hydrolysis of 100 μM fluorogenic substrate Cbz-GGL-AMC. While all compounds exhibited more potent inhibition of ClpYQ, 7 showed the most potent inhibition of ClpYQ, consistent with its highly potent MICs. Given that 1 was shown to be competitive inhibitor of ClpYQ proteolysis activity,3 we applied the Cheng–Prusoff relationship to determine KI. 7 inhibits ClpYQ with a KI = 150 ± 20 nM.
Inhibition of ClpXP activity in S. aureus
To evaluate the inhibition of ClpXP, a cell-based assay was developed. Although we were able to express and purify S. aureus ClpXP and the closely related B. subtilis ClpXP, we could only achieve single turnover for proteolysis under all conditions investigated. Recombinant purified E. coli ClpXP was active and inhibition kinetics could be readily obtained,3 however the orthologs from Gram-negative E. coli are distantly related to the S. aureus proteins. Thus E. coli ClpXP inhibition data was expected to be of limited relevance to inhibition of S. aureus ClpXP. Because direct ClpP inhibition increases urease activity,3,17 we were able to determine the minimum effective concentration (MEC) of a ClpXP inhibitor required to increase urease activity above background in MRSA USA300. Thus in Christensen's media supplemented with phenol red pH indicator, inhibition of ClpXP activity in MRSA USA300 will increase urease activity, hydrolyzing the urea in the media, releasing ammonia, and leading to a colour change from yellow to red as the pH of the media is increased.3,17
Compounds 1–14 were analyzed for their MEC for urease activation from concentrations of 0.067 μg mL−1 to 1 μg mL−1 (ESI† Table S3). As concentrations approached the MIC (within two fold), bacterial growth slowed. In all cases however, activation of urease by ClpXP inhibition could be detected prior to growth inhibition. The potent benzyl derivatives 5–8 showed similar but improved ClpXP activity compared to 1. Compound 2 containing a hexyl group at both R1 and R2 showed the most potent ClpXP inhibition with a MEC of 0.067 μg mL−1.
Discussion
A series of focused derivatives of the natural product antibiotic armeniaspirol were synthesized and evaluated for antibiotic activity against clinically relevant pathogens including MRSA and VRE. Analogues that were more potent than the parent compound 1 were evaluated for their ability to inhibit ClpYQ and ClpXP, the biochemical targets of armeniaspirol.3 The derivatives focused on diversifying the N-methyl moiety (R2), which proved to be highly amenable to modification. Replacement of the N-methyl with N-hexyl, various N-benzyl, and N-phenethyl substituents lead to substantial increases in antibiotic activity and potency for inhibition of both ClpYQ and ClpXP. Replacement of the hexyl chain on the aryl core of armeniaspirol (R1) with a methyl substituent lead to a series of analogues substituted on the amide nitrogen (series 2) that were less active than the corresponding hexyl analogues (series 1), with the exception of 11.
Armeniaspirol targets both ClpXP and ClpYQ.3 While single target inhibitors of SaClpXP are known, without the additional ClpYQ inhibition activity, these compounds are not antibiotic.17–24 Thus this dual target action is required to intervene sufficiently in the redundant proteolytic pathways that regulate the divisome and cell division. Over half of approved antibiotics since 2015 have multitarget mechanisms of action,25 likely due to their natural product origins.26 Optimizing activity of compounds that inhibit multiple targets is challenging. The relevant substrate, cofactor, and allosteric regulator concentrations in the cell can impact the level of inhibition required at each target. Thus it is typically not possible to predict the optimal level of inhibition at each target for maximal effect. Structure–activity relationships at each target are needed as is a structure–activity relationship from an assay that integrates the function of both targets together. In the case of armeniaspirol analogues, the MIC assays provide the data integrating activity at both ClpYQ and ClpXP.
In general, increasing potency at either target appears to correlate with improved overall antibiotic activity. For example series 1 analogues 1, 2, and 7 show a steady ≈20-fold decrease in KI for ClpYQ inhibition, which correlates with the 8-fold increase in potency as measured by MIC. Similarly, analogues 1, 11 and 8 show a steady decrease in MEC, representative of inhibition of ClpXP, which correlates to a decrease in MIC against MRSA USA300. This data is consistent with armeniaspirol antibiotic activity being derived from both ClpXP and ClpYQ inhibition. In addition, the data suggests that inhibitory activity against both targets is balanced, since analogues that possess increased potency against ClpXP or ClpYQ, increase antibiotic activity.
Some analogues however show highly potent inhibition of one of the targets over the other. For example, 7 is a sub-micromolar inhibitor of ClpYQ (KI = 150 ± 20 nM), however it is no more active in the MIC assays than the less potent 8 (KI = 410 ± 70 nM) in MIC assays (MIC = 0.5 μg mL−1). This is even more apparent with 2, which shows ClpXP inhibition at 0.067 μg mL−1 but is no more active in MIC assays (MIC = 1.0 μg mL−1) than 5, which inhibits ClpXP at 0.25 μg mL−1. These data suggest that as analogues become highly potent towards one of the targets, without significantly increasing the potency against the other, little increase in antibiotic activity is obtained.
The enhanced activity of the more lipophilic series 1 analogues over series 2 compounds raised concerns that activity was either a function of aggregation or non-specific lipophilicity-mediated affinity. To evaluate if aggregation of the analogues played a significant role in activity, the MICs for the parent compound 1 and the highly lipophilic 2 were determined in the presence of detergent. Addition of 0.01% v/v Triton X-100 had minimal impact on the MICs of either compounds against MRSA USA300 (ESI† Table S4). In further support of aggregation playing a limited role in activity, fitting of the ClpYQ does response data to an IC50 model where the slope could vary provided Hill coefficients of −1 to −1.5. In cases where aggregation plays a significant role in inhibition, steep slopes are frequently obtained, corresponding to Hill coefficients well below −1.5.27 As such aggregation was not considered a major source of the increased potency observed for the more lipophilic analogues.
Careful examination of the data shows that activity is not simply a function of increased lipophilicity as the most lipophilic compounds 3 and 6 (highest cLog Ps) were not the most potent in any of the assays. Though there does appear to be a correlation between low activity and low lipophilicity since 9 and 12, two of the library members with the lowest clog P are some of the least active compounds. As the proteolytic chambers of both the ClpYQ and ClpXP must accommodate their unfolded protein clients whose hydrophobic cores are exposed, it stands to reason that moderately lipophilic inhibitors could be well suited to inhibition.
Models rationalizing the observed KI and MEC data were generated by docking armeniaspirol into ClpP and ClpQ high resolution structures using AutoDock Vina.28 Armeniaspirol showed two preferred binding site on the active conformation of the S. aureus ClpP heptamer (3V5E),29 both of which were in the proteolytic cavity (Fig. 3 and ESI† Fig. S2A). The most frequent pose from the lowest energy docked structures showed armeniaspirol bound in the same site as seen for the binding of the ClpXP inhibitor bortezomib to Thermus thermophilus ClpP (6HWM, Fig. 3C).30 Armeniaspirol, like bortezomib, H-bonds to backbone NHs at the C-terminal end of αC (ref. 30) via its ketone. In addition, our model shows the ketone H-bonding to the side chain of the active site Ser70. The hexyl chain engages in van der Waals interactions with the hydrophobic face of αE. Lastly the phenol of armeniaspirol H-bonds to His142 and Thr146 of αE. The second pose showed armeniaspirol bound closer to the N-terminus of ClpP, though still in the proteolytic chamber (ESI† Fig. S2A).
Fig. 3. Structure and docking results of S. aureus ClpP (3V5E). (A) Side and top views of S. aureus ClpP (SaClpP) tetradecamer. A single monomer per heptameric ring (transparent cartoons) are highlighted in cyan and dark cyan respectively. (B) Cartoon representation of the SaClpP monomer bound to armeniaspirol. Secondary structures are coloured in rainbow, helices are named by letters while strands are indicated by numbers. The dashed-line box indicates the armeniaspirol binding site predicted by molecular docking. (C) Zoom-in of armeniaspirol docking site in SaClpP. Possible residues involved in armeniaspirol binding are shown as sticks with proposed hydrogen bonds shown as grey dashed lines (inset 1). Overlay of Thermus thermophilus ClpP (TtClpP) bound to bortezomib (PDB: 6HWM) and SaClpP bound to armeniaspirol is shown (inset 2).

This ClpP binding model is in excellent accordance with the bortezomib TtClpP structure (Fig. 3C, inset 2) and is consistent with all current structure–activity relationship data. For example, both the phenol and ketone are required for antibiotic activity and in this model they both have discrete H-binding interactions with the target. Similarly, our current work shows that the hexyl chain of series 1 analogues is preferred over the methyl chain of series 2 analogues, consistent with the model showing the hexyl to be buried between αE and the loop between β6 and β7. Lastly the model shows significant space available for the N-methyl group to be replaced with varying N-alkyl groups, consistent with these N-alkyl analogues displaying potent inhibition of ClpP. While experimental evidence validating this model is clearly needed, it does provide for multiple testable atomic level hypothesis for inhibitor binding.
A model of armeniaspirol binding to the dodecamer of S. aureus ClpQ (6KUI) was also generated (Fig. 4),31 with two main poses being observed. In the first pose (Fig. 4B) armeniaspirol is positioned at the active site, with the phenol hydrogen bonding to the active site Thr1 backbone NH and Thr29 side-chain. This pose is similar to that seen in the high resolution structure of the NLVS inhibitor bound to Haemophilus influenzae ClpQ (1KYI, Fig. 4C).32 For example, Ser21 of HiClpQ, analogous to Thr29 of SaClpQ, H-bonds with the inhibitor. The armeniaspirol hexyl chain fits into the same S1 pocket as the aliphatic iPr group from NLVS and packs against SaClpQ Val28, in an analogous fashion to HiClpQ Val20. The ketone of armeniaspirol engages with Gln32, and the amide carbonyl with Glu101, anchoring armeniaspirol in front of the active site. The second pose for armeniaspirol shows the compound bound at the interface between two protomers of one of the hexameric rings (ESI† Fig. S2B). While the ClpQ binding model agrees with known structure–activity data and the data generated from this study, it remains to be experimentally validated.
Fig. 4. Structure and docking results of S. aureus ClpQ (6KUI). (A) Side and top views of S. aureus ClpQ (SaClpQ) dodecamer. A single monomer per hexameric ring (transparent cartoons) are highlighted in sandstone and magenta respectively. (B) Cartoon representation of the SaClpQ monomer bound to armeniaspirol. Secondary structures are coloured in rainbow, helices are named by letters while strands are indicated by numbers. The dashed-line box indicates the armeniaspirol binding site predicted by molecular docking. (C) Overlay of Haemophilus influenzae ClpQ (HiClpQ) covalently bound to a vinyl sulfone inhibitor (PDB: 1KYI) and SaClpQ bound to armeniaspirol (inset 1). (D) Zoom-in of armeniaspirol docking site in SaClpQ. Possible residues involved in armeniaspirol binding are shown as sticks with proposed hydrogen bonds shown as grey dashed lines (inset 2).

While both bortezomib and NLVS are covalent inhibitors of their respective targets ClpP and ClpQ,30,321 has been shown to be a competitive inhibitor of the peptide substrate in purified enzyme assays.3 In the models of armeniaspirol bound to ClpP and ClpQ the electrophilic β-carbon of the α,β-unsaturated lactam is well removed from the active site nucleophiles, consistent with a competitive model for inhibition of proteolysis.
In addition to target binding, lipophilicity can impact off-target interactions.5,33,34 In particular membrane targeting may be enhanced with increased lipophilicity. As such the hemolytic activity of 1 and two of the most potent and lipophilic analogues 2 and 8 was determined (Fig. S3†).35 At 200 μM 1 showed less than 15% lysis of sheep erythrocytes whereas the more lipophilic 2 and 8 showed less than 5% hemolysis. Thus while lypophilic, the armeniaspirols do not destabilize mammalian red blood cell membranes at pharmacologically relevant concentrations. The toxicity of 1, 2, and 8 was also evaluated against human lung epithelial carcinoma (A549) cells. The cell culture cells were incubated with up to 100 μM compound, stained with calcein-AM and ethidium homodimer-1, and evaluated by both by confocal microscopy and flow cytometry to determine the fraction of viable and dead cells (Fig. S4†). All three compounds at 100 μM where indistinguishable from the vehicle control and showed a greater than 95% viable cell population.
Lipophilicity is also an important driver of protein binding, which can impact antibiotic pharmacokinetics and pharmacodynamics.36 To evaluate the effect of protein binding on antimicrobial activity, we determined the MIC of 1 and the highly lipophilic 2 against MRSA USA300 in the presence of bovine serum albumin (Table S4†). Addition of serum suppressed antibiotic activity, consistent with a high degree of protein binding expected for these lipophilic compounds. For example, the MIC of the parent compound exceeded 32 μg mL−1 in the presence of BSA. However armeniaspirol is known to be active in an in vivo mouse model of MRSA septicaemia with 10 mg kg−1 i.p. delivery. Thus while this assay confirms that protein binding is a significant factor in the antibiotic activity of the armeniaspirols, the addition of serum albumin alone does not adequately model the pharmacodynamics behaviour of the drug in vivo.37
Conclusion
This study has provided new highly potent Gram-positive antibiotics active against clinically relevant strains of MRSA, including USA300 the most common community acquired MRSA infection. Our work has shown that the increased potency of analogues against ClpXP and ClpYQ correlates with increased antibiotic activity, strongly supporting the mechanism of action. We show that activity against both targets is balanced, such that increasing activity at either target increases antibiotic activity. Furthermore, while potency tracks loosely with lipophilicity, this is likely due to binding to the hydrophobic proteolysis chamber rather than non-selective binding interactions. Docking models support this hypothesis and show armeniaspirol may bind at the active site, but in a configuration that does not enable covalent modification of the active site nucleophiles, consistent with competitive inhibition of ClpXP and ClpYQ.
Author contributions
C. N. B and M. G. D. conceived of the study. M. G. D, M. H. D. and A. R. P. synthesized and characterized compounds. M. G. D., P. L. and C. N. collected MIC and MBC data. T. D. L. and P. L. collected ClpYQ inhibition data. M. G. D. collected ClpXP inhibition data. J. T. B.-H. performed docking studies. A. R. P. and M. D. collected hemolysis data. N. D. C. collected live human cell data. C. N. B. supervised and managed the study. All authors contributed to analysing data and writing and editing the manuscript.
Conflicts of interest
There are no conflicts of interest.
Supplementary Material
Acknowledgments
This work was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC) (A. J. S. RGPIN 2021-03387; C. N. B RGPIN 2019-06859). The authors thank the John L. Holmes Mass Spectrometry Facility for assistance in acquiring mass spectrometry data and the University of Ottawa Faculty of Science NMR facility for assistance in collecting NMR spectra.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00355k
Notes and references
- Dufour C. Wink J. Kurz M. Kogler H. Olivan H. Sablé S. Heyse W. Gerlitz M. Toti L. Nußer A. et al., Isolation and structural elucidation of armeniaspirols A-C: Potent antibiotics against gram-positive pathogens. Chem. – Eur. J. 2012;18(50):16123–16128. doi: 10.1002/chem.201201635. [DOI] [PubMed] [Google Scholar]
- Couturier C. Bauer A. Rey A. Schroif-Dufour C. Broenstrup M. Armeniaspiroles, a new class of antibacterials: Antibacterial activities and total synthesis of 5-chloro-Armeniaspirole A. Bioorg. Med. Chem. Lett. 2012;22(19):6292–6296. doi: 10.1016/j.bmcl.2012.06.107. [DOI] [PubMed] [Google Scholar]
- Labana P. Dornan M. H. Lafrenière M. Czarny T. L. Brown E. D. Pezacki J. P. Boddy C. N. Armeniaspirols inhibit the AAA+ proteases ClpXP and ClpYQ leading to cell division arrest in Gram-positive bacteria. Cell Chem. Biol. 2021;28(12):1703–1715. doi: 10.1016/j.chembiol.2021.07.001. [DOI] [PubMed] [Google Scholar]
- Qiao Y. Yan J. Jia J. Xue J. Qu X. Hu Y. Deng Z. Bi H. Zhu D. Characterization of the biosynthetic gene cluster for the antibiotic armeniaspirols in streptomyces armeniacus. J. Nat. Prod. 2019;82(2):318–323. doi: 10.1021/acs.jnatprod.8b00753. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery. 2007;6(11):881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Durham D. G. Rees A. H. Chlorination of Pyrroles. Part I. Can. J. Chem. 1971;49(1):136–138. doi: 10.1139/v71-021. [DOI] [Google Scholar]
- Schäfer W. Franck B. Selektive Ätherspaltung von 4-Hydroxy-methoxy-chinolincarbonsäureestern. Chem. Ber. 1966;99(1):160–164. doi: 10.1002/cber.19660990127. [DOI] [Google Scholar]
- Wiegand I. Hilpert K. Hancock R. E. W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008;3(2):163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- Chen Y. Crosby H. A. Oosthuysen W. F. Diekema D. J. Kelley S. T. Horswill A. R. Draft genome sequence of usa100 methicillin-resistant Staphylococcus aureus strain 209. Genome Announc. 2018;6(1):e01399-17. doi: 10.1128/genomeA.01399-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber J. T. Community-associated methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2005;15:S269–S272. doi: 10.1086/430788. [DOI] [PubMed] [Google Scholar]
- King J. M. Kulhankova K. Stach C. S. Vu B. G. Salgado-Pabón W. Phenotypes and Virulence among Staphylococcus aureus USA100, USA200, USA300, USA400, and USA600 Clonal Lineages. mSphere. 2016;1(3):e00071-16. doi: 10.1128/mSphere.00071-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison M. E. Pharmacodynamics of antimicrobial drugs. Infect. Dis. Clin. North Am. 2004:451–465. doi: 10.1016/j.idc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- French G. L. Bactericidal agents in the treatment of MRSA infections-the potential role of daptomycin. J. Antimicrob. Chemother. 2006;58:1107–1117. doi: 10.1093/jac/dkl393. [DOI] [PubMed] [Google Scholar]
- Pankey G. A. Sabath L. D. Clinical relevance of bacteriostatic versus bactericidal mechanisms of action in the treatment of gram-positive bacterial infections. Clin. Infect. Dis. 2004;38(6):864–870. doi: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- Paulsen I. T. Park J. H. Choi P. S. Saier M. H. A family of gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from gram-negative bacteria. FEMS Microbiol. Lett. 1997;156(1):1–8. doi: 10.1016/S0378-1097(97)00379-0. [DOI] [PubMed] [Google Scholar]
- Koronakis V. Eswaran J. Hughes C. Structure and function of TolC: the bacterial exit duct for proteins and drugs. Annu. Rev. Biochem. 2004;73:467–489. doi: 10.1146/annurev.biochem.73.011303.074104. [DOI] [PubMed] [Google Scholar]
- Gao P. Ho P. L. Yan B. Sze K. H. Davies J. Kao R. Y. T. Suppression of Staphylococcus aureus virulence by a small-molecule compound. Proc. Natl. Acad. Sci. U. S. A. 2018;115(31):8003–8008. doi: 10.1073/pnas.1720520115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher T. Sieber S. A. β-lactones as specific inhibitors of ClpP attenuate the production of extracellular virulence factors of Staphylococcus aureus. J. Am. Chem. Soc. 2008;130(44):14400–14401. doi: 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]
- Zeiler E. Korotkov V. S. Lorenz-Baath K. Böttcher T. Sieber S. A. Development and characterization of improved β-lactone-based anti-virulence drugs targeting ClpP. Bioorg. Med. Chem. 2012;20(2):583–591. doi: 10.1016/j.bmc.2011.07.047. [DOI] [PubMed] [Google Scholar]
- Krysiak J. Stahl M. Vomacka J. Fetzer C. Lakemeyer M. Fux A. Sieber S. A. Quantitative Map of β-Lactone-Induced Virulence Regulation. J. Proteome Res. 2017;16(3):1180–1192. doi: 10.1021/acs.jproteome.6b00705. [DOI] [PubMed] [Google Scholar]
- Hackl M. W. Lakemeyer M. Dahmen M. Glaser M. Pahl A. Lorenz-Baath K. Menzel T. Sievers S. Böttcher T. Antes I. et al., Phenyl Esters Are Potent Inhibitors of Caseinolytic Protease P and Reveal a Stereogenic Switch for Deoligomerization. J. Am. Chem. Soc. 2015;137(26):8475–8483. doi: 10.1021/jacs.5b03084. [DOI] [PubMed] [Google Scholar]
- Pahl A. Lakemeyer M. Vielberg M. T. Hackl M. W. Vomacka J. Korotkov V. S. Stein M. L. Fetzer C. Lorenz-Baath K. Richter K. et al., Reversible Inhibitors Arrest ClpP in a Defined Conformational State that Can Be Revoked by ClpX Association. Angew. Chem., Int. Ed. 2015;54(52):15892–15896. doi: 10.1002/anie.201507266. [DOI] [PubMed] [Google Scholar]
- Gersch M. Kolb R. Alte F. Groll M. Sieber S. A. Disruption of oligomerization and dehydroalanine formation as mechanisms for ClpP protease inhibition. J. Am. Chem. Soc. 2014;136(4):1360–1366. doi: 10.1021/ja4082793. [DOI] [PubMed] [Google Scholar]
- Moreno-Cinos C. Goossens K. Salado I. G. Van Der Veken P. De Winter H. Augustyns K. ClpP protease, a promising antimicrobial target. Int. J. Mol. Sci. 2019;20(9):2232. doi: 10.3390/ijms20092232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel C. Lonneman M. Wu C. Polypharmacological drug actions of recently FDA approved antibiotics. Eur. J. Med. Chem. 2021:112931. doi: 10.1016/j.ejmech.2020.112931. [DOI] [PubMed] [Google Scholar]
- Ho T. T. Tran Q. T. Chai C. L. The polypharmacology of natural products. Future Med. Chem. 2018;10(11):1361–1368. doi: 10.4155/fmc-2017-0294. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006;49(25):7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- Trott O. Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gersch M. List A. Groll M. Sieber S. A. Insights into structural network responsible for oligomerization and activity of bacterial virulence regulator caseinolytic protease P (ClpP) protein. J. Biol. Chem. 2012;287(12):9484–9494. doi: 10.1074/jbc.M111.336222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix J. Weinhäupl K. Chipot C. Dehez F. Hessel A. Gauto D. F. Morlot C. Abian O. Gutsche I. Velazquez-Campoy A. et al., Mechanism of the allosteric activation of the ClpP protease machinery by substrates and active-site inhibitors. Sci. Adv. 2019;5(9):eaaw3818. doi: 10.1126/sciadv.aaw3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong S. Ahn J. Kwon A. R. Ha N. C. Cleavage-Dependent Activation of ATP-Dependent Protease HslUV from Staphylococcus aureus. Mol. Cells. 2020;43(8):694–704. doi: 10.14348/molcells.2020.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M. C. Kessler B. M. Overkleeft H. S. McKay D. B. Crystal Structure of HslUV Complexed with a Vinyl Sulfone Inhibitor: Corroboration of a Proposed Mechanism of Allosteric Activation of HslV by HslU. J. Mol. Biol. 2002;318(3):779–785. doi: 10.1016/S0022-2836(02)00145-6. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Mason J. S. Overington J. P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006;16(1):127–136. doi: 10.1016/j.sbi.2006.01.013. doi: 10.1016/j.sbi.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Peters J.-U. Schnider P. Mattei P. Kansy M. Pharmacological Promiscuity: Dependence on Compound Properties and Target Specificity in a Set of Recent Roche Compounds. ChemMedChem. 2009;4(4):680–686. doi: 10.1002/cmdc.200800411. [DOI] [PubMed] [Google Scholar]
- Evans B. C. Nelson C. E. Yu S. S. Beavers K. R. Kim A. J. Li H. Nelson H. M. Giorgio T. D. Duvall C. L. Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J. Visualized Exp. 2013;9(73):e50166. doi: 10.3791/50166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger M. A. Derendorf H. Mouton J. W. Cars O. Craig W. A. Andes D. Theuretzbacher U. Protein binding: do we ever learn? Antimicrob. Agents Chemother. 2011;55(7):3067–3074. doi: 10.1128/AAC.01433-10. doi: 10.1128/AAC.01433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer J. Wagner C. C. Zeitlinger M. Protein Binding of Antimicrobials: Methods for Quantification and for Investigation of its Impact on Bacterial Killing. AAPS J. 2009;11(1):1–12. doi: 10.1208/s12248-008-9072-1. doi: 10.1208/s12248-008-9072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.