

Abstract
Sézary Syndrome (SS) is an aggressive and disseminated form of Cutaneous T-cell lymphoma (CTCL) associated with dismal prognosis in which the histone deacetylase inhibitor romidepsin, has shown remarkable activity as a single agent. However, clinical responses to romidepsin are typically transient, highlighting the need for more effective therapies. Here, we show synergistic anti-lymphoma effects of romidepsin in combination with mechlorethamine, an alkylating agent, in CTCL cell lines and primary samples with strong antitumor effects in an in vivo model of SS. Mechanistically, gene expression profiling points to abrogation of Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling as important mediator of this interaction. Consistently, the combination of mechlorethamine plus romidepsin resulted in downregulation of STAT5 phosphorylation in romidepsin-sensitive cell lines and primary SS samples, but not in romidepsin-resistant tumors. Moreover, and in further support of JAK/STAT signaling as a modulator of romidepsin activity in CTCL, treatment with romidepsin in combination with JAK inhibitors resulted in markedly increased therapeutic responses. Overall, these results support a role for romidepsin plus mechlorethamine in combination in the treatment of CTCL and uncover a previously unrecognized role for JAK-STAT signaling in the response to romidepsin and romidepsin-based combination therapies in SS.
INTRODUCTION
The diagnosis of CTCL encompasses a diverse group of lymphoid malignancies characterized by the homing of malignant T cells to the skin(Willemze et al., 2005). Mycosis Fungoides (MF), the most common form of CTCL, presents most frequently as cutaneous patches, plaques, and tumors limited to the skin. In contrast, Sézary Syndrome (SS) shows circulating tumor cells in peripheral blood frequently accompanied by diffuse skin infiltration (erythroderma) and lymph node and solid organ involvement. Genome wide mutation profiling studies in CTCL have revealed a markedly heterogeneous landscape of genetic alterations with functionally convergent recurrent mutations and copy number alterations involving genes implicated in cell cycle control, T-cell receptor signaling and epigenetic regulation of gene expression(da Silva Almeida et al., 2015, Kiel et al., 2015, McGirt et al., 2015, O’Connor et al., 2015, Prasad et al., 2016, Ungewickell et al., 2015).
Despite the lack of curative treatments topical therapies with corticosteroids, bexarotene, a RXR receptor retinoid agonist, and the alkylating agent mechlorethamine(Kim et al., 2003, Lessin et al., 2013, Talpur et al., 2014) show activity in early stages of CTCL. In addition, systemic therapies with retinoids, histone deacetylase inhibitors (HDACi) and antifolates can be transiently effective for the treatment of cases with advanced disease. In this context, romidepsin, a class I selective HDACi, shows overall response rates of about 30% and a favourable toxicity profile(Piekarz et al., 2009, Whittaker et al., 2010). Mechanistically, romidepsin treatment induces broad transcriptional changes in CTCL including the upregulation of cell cycle inhibitor and pro-apoptotic tumor suppressor genes(Aron et al., 2003, Bates et al., 2010, Piekarz and Bates, 2004). However, the specific mechanisms mediating the therapeutic activity and selectivity of HDACi therapies in CTCL remain to be fully established. Moreover, with a median response duration of about one year, romidepsin treatment as single agent offers only transient benefits while combinations with cytotoxic (doxorubicin), demethylating (azacitidine), immunomodulatory (lenalinomide), proteasome inhibitor (bortezomib) and apoptosis-inducing (venetoclax) agents still offer limited opportunity for a lasting response(Cosenza et al., 2016, Cyrenne et al., 2017, Heider et al., 2009, Moyal et al., 2016, Rozati et al., 2016), highlighting the need for mechanistically sound and more effective synergistic drug combinations.
Here, we identify and characterize a prominent synergistic antitumor activity in CTCL of romidepsin and mechlorethamine in combination. Our results point to downregulation of JAK/STAT signaling as a prominent mechanism mediating the activity of this combination and support a role for enhanced JAK/STAT signaling deregulation as driver of resistance to HDACi therapy. These results support a potential role for romidepsin-JAK1/2 combination therapies for the treatment of advanced HDACi-resistant forms of CTCL in the clinic.
RESULTS
Mechlorethamine and romidepsin induce synergistic antitumor effect in CTCL cells in vitro and in vivo
The HDACi, romidepsin, and mechlorethamine, an alkylating agent, have anticipated distinct, non-overlapping mechanisms of action and a proposed more than additive effect in CTCL(Dulmage et al., 2015). To formally evaluate their efficacy and explore the mechanisms underlying the interaction of romidepsin and mechlorethamine we first evaluated the response of Sezary Syndrome cell lines (SeAx, HH, and HuT78) to these agents alone and in combination (Figure 1a–b and Supplementary Table S1). Analysis of cell viability in these experiments revealed a strongly synergistic interaction for the romidepsin plus mechlorethamine combination in HuT78 cells (CI=0.42) and moderate synergism in SeAx (CI=0.73) and HH (CI=0.83) cells.
Figure 1. Evaluation of synergism of mechlorethamine and romidepsin in CTCL cell lines and in a mouse model of Sezary Syndrome.
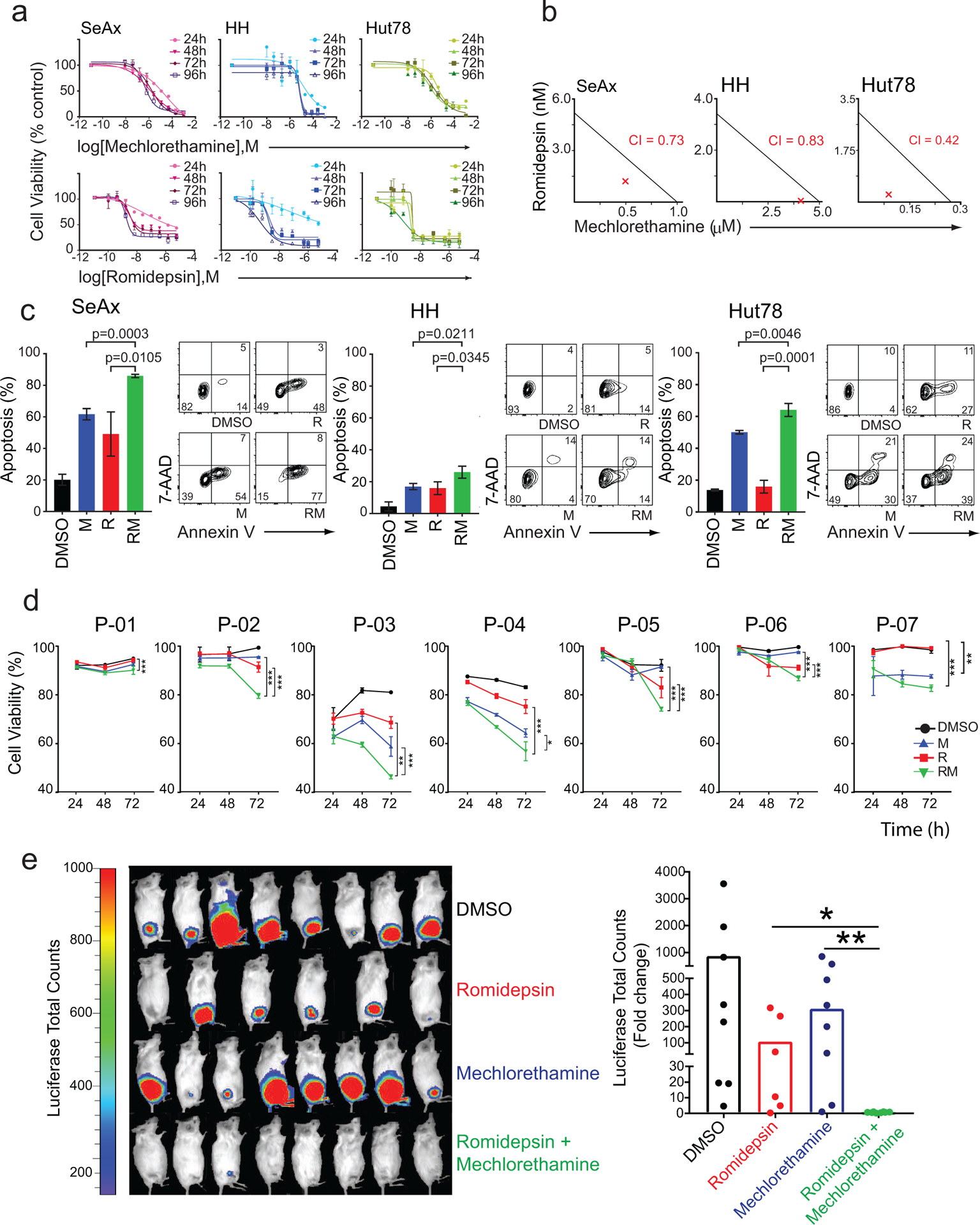
(a) Dose response cell-viability curves relative to vehicle-treated controls for each cell line at 24, 48, 72, and 96-hour treatments with mechlorethamine alone and romidepsin alone shown in log of molar concentrations (M). (b) Isobologram analysis of the effects of fixed molar ratio combinations of mechlorethamine and romidepsin during 48 hours of treatment. The red X represents the Combination index (CI). (c) Evaluation of apoptosis in CTCL cell lines SeAx, HH, and HuT78 after 24 hours of treatment with vehicle (DMSO), mechlorethamine, romidepsin, or the combination using flow cytometry analysis after annexin V/7-AAD staining. (d) Evaluation of cell viability in Sézary Syndrome primary patient samples treated in vitro with vehicle (DMSO), mechlorethamine, romidepsin, or the combination. Cell viability was assessed with thiazole orange and propidium iodide staining via flow cytometry at 24, 48, and 72 hours of treatment. (e) Quantitative analyses of changes in tumor burden measured by bioluminescence after 20 days of treatment. P values were calculated using a two-tailed Student’s t-test. Error bars, mean ± s.d. ***, p<0.001; **, p<0.01; *, p<0.05; DMSO, dimethyl sulfoxide; M, mechlorethamine; R, romidepsin. Error bars represent mean+/− SEM, n=3.
The synergistic effect of the romidepsin/mechlorethamine combination is also observed in skin-derived CTCL cell lines, including MyLa (CI=0.73) and HuT102 (CI= 0.73) (Supplementary Figure S1). Consistently, analysis of apoptosis showed significant increases in programmed cell death across all three cell lines tested after treatment with mechlorethamine plus romidepsin compared to vehicle only and single agent treatment controls (Figure 1c). Next, and to further investigate the relevance of the mechlorethamine and romidepsin in non-immortalized CTCL cells we tested the antitumor effects of these drugs alone and in combination against malignant CD4+ T cells isolated from seven patients with SS (Supplementary Table S2). In these experiments, treatment with mechlorethamine induced a limited therapeutic response in one patient sample only, while romidepsin induced marked and time dependent antitumor effects with decreased cell viability of up to 46% relative to control at 72 hours (Figure 1d). In addition, and most notably, treatment with mechlorethamine plus romidepsin in combination induced a significantly stronger decrease in cell viability at 72 hours in 6/7 (84%) primary samples.
Following these observations, we evaluated the therapeutic activity of mechlorethamine and romidepsin alone and in combination in vivo. Towards this goal, we xenografted luciferized HuT78 SS cells in Rag1/Il2rg double knockout (NRG) immunodeficient mice and monitored tumor engraftment and disease progression by luciferase in vivo bioimaging. Following tumor development, we segregated animals with homogeneous tumor burden into therapeutic groups for treatment with vehicle only, romidepsin, mechlorethamine, and romidepsin plus mechlorethamine in combination (n=10 animals/group) (Supplementary Figure S2a). In this setting, treatment with romidepsin effectively decreased tumor cell progression in vivo, while treatment with mechlorethamine had a minor effect in tumor burden compared with controls. In contrast, and most notably, combined treatment with mechlorethamine plus romidepsin showed strong antitumor effects and induced complete responses in 9/10 (90%) animals analyzed (Figure 1e).
Treatment with mechlorethamine plus romidepsin in combination induces downregulation of JAK-STAT signaling in CTCL
To explore the potential molecular mechanisms responsible for the synergistic anti-lymphoma effects of the mechlorethamine plus romidepsin combination we performed RNA-seq gene expression profiling of CTCL cell lines and primary CTCL samples treated for 24 hours with vehicle, romidepsin, mechlorethamine, and the romidepsin mechlorethamine combination. Dimensionality-reduction of gene expression signatures by principal component analyses segregated samples by treatment group along the first principal component (PC1), which accounted for 61% of the gene expression variance (Supplementary Figure S3a–b). In addition, supervised differential gene expression analysis showed that romidepsin treatment induced marked changes in gene expression compared with vehicle treatment controls (494 genes upregulated, 93 downregulated genes with log2 fold change >1.2 and Padj<0.01), while mechlorethamine-treated samples showed more modest transcriptional effects (63 upregulated, 16 downregulated genes, with log2 fold change >1.2 and Padj<0.01). Notably, the combination of romidepsin plus mechlorethamine induced substantially distinct gene expression programs compared with those present in single agent romidepsin-treated and mechlorethamine-treated cells, in support of an emergent and distinct mechanism of action (Supplementary Figure S3c–d). These analyses identified a total of 1,288 genes (Padj<0.01, log2 fold-change >1.2) uniquely associated with romidepsin-mechlorethamine combination of which 832 were up-regulated and 456 down-regulated (Figure 2a–b). Similarly, treatment of primary SS samples with romidepsin and mechlorethamine in combination resulted in upregulation of 525 genes and down-regulation of 56 genes (Padj<0.01, log2 fold-change >1.2) (Supplementary Figure S3 and Supplementary Table S3).
Figure 2. Analysis of expression changes induced by treatment with mechlorethamine, romidepsin and their combination in CTCL cell lines by RNAseq.
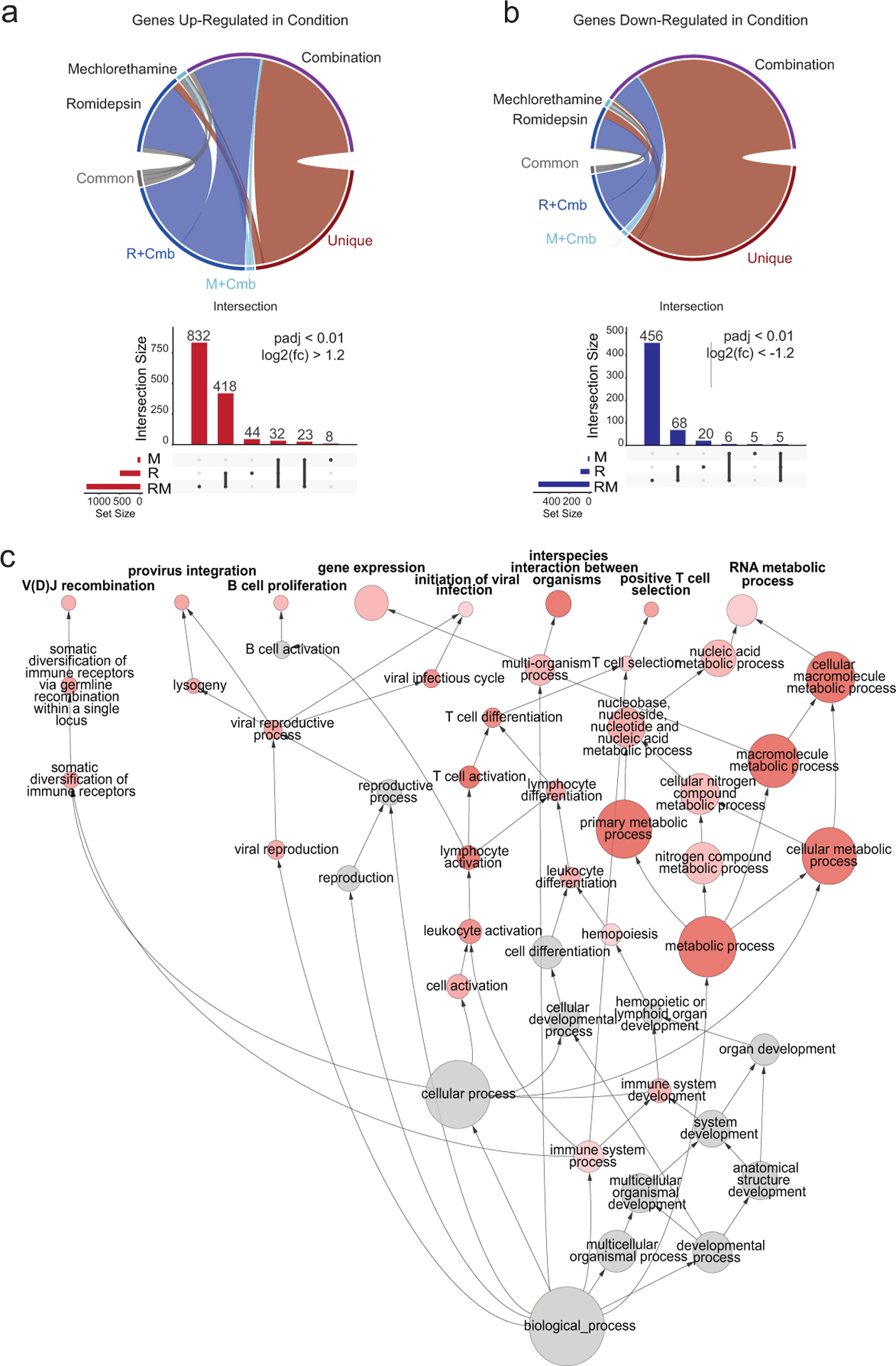
(a ,b) Circos plots indicate the relative proportions of significantly up- (a) and down-regulated (b) genes in each drug treatment condition versus vehicle control that were shared among all treatments (common), shared between the single treatments and the combination (R+Cmb or M+Cmb), or specific to the treatment (unique). Vertical bar plots display the absolute size of each intersection among the significantly up-regulated (in red) or down-regulated (in blue) genes. Horizontal bars indicate the absolute number of significantly up- or down-regulated genes among the different treatments, compared to vehicle control. (c) Directed network projection of the Gene Ontology biological process term analysis performed on genes. Node diameter indicates the relative number of genes in the GO term and the shading indicates the FDR level. R: romidepsin, M: mechlorethamine, Cmb: romidepsin plus mechlorethamine.
Taken together, these data further support an emergent effect unique to the mechlorethamine-romidepsin combination treatment of CTCL. In order to determine which biological processes underly these emergent transcriptomic changes unique to the romidepsin-mechlorethamine combination treatment of CTCL, comprehensive Kegg pathway and Gene Ontology term analysis was performed. Intriguingly, gene ontology analysis of the genes specifically down-regulated upon romidepsin-mechlorethamine combination treatment using Biological Network Gene Ontology (BiNGO) (Maere et al., 2005) revealed a significant enrichment of GO Terms associated with lymphocyte proliferation, development, differentiation and metabolic processes (Figure 2c). In addition, KEGG pathway analysis revealed downregulation of JAK/STAT signaling-associated signatures comprised by reduced expression of prominent regulators of T-cell development, proliferation and survival including transcription factors (BCL11B), cytokines (IL10) and cytokine receptors (CSF2RB and IL2RA), signaling protein kinases (PI3K3CD and LCK), antiapoptotic factors (BCL2 and BCL2L1), and drivers of cell cycle progression (CCND2) (Figure 3a–b). In addition, SOCS3, an important negative regulator of JAK/STAT-mediated cytokine signaling was significantly upregulated in cells treated with the romidepsin-mechlorethamine combination. Consistently, gene set enrichment analysis (GSEA) demonstrated significant enrichment in signatures associated with down-regulation of JAK/STAT activity in primary CTCL samples treated with romidepsin plus mechlorethamine including gene sets derived from signatures induced by JAK inhibitor treatment and JAK2 shRNA-knockdown(Moodley et al.) (Figure 3c). Conversely we observed a negative enrichment of genes related to interferon signaling and of transcriptional programs induced by IL2(Rampal et al.) in the romidepsin plus mechlorethamine gene expression signature (Supplementary Figure S4).
Figure 3. Pathway and Gene set enrichment analysis (GSEA) supports downregulation of the JAK/STAT signaling pathway in samples treated with the romidepsin-mechlorethamine combination.
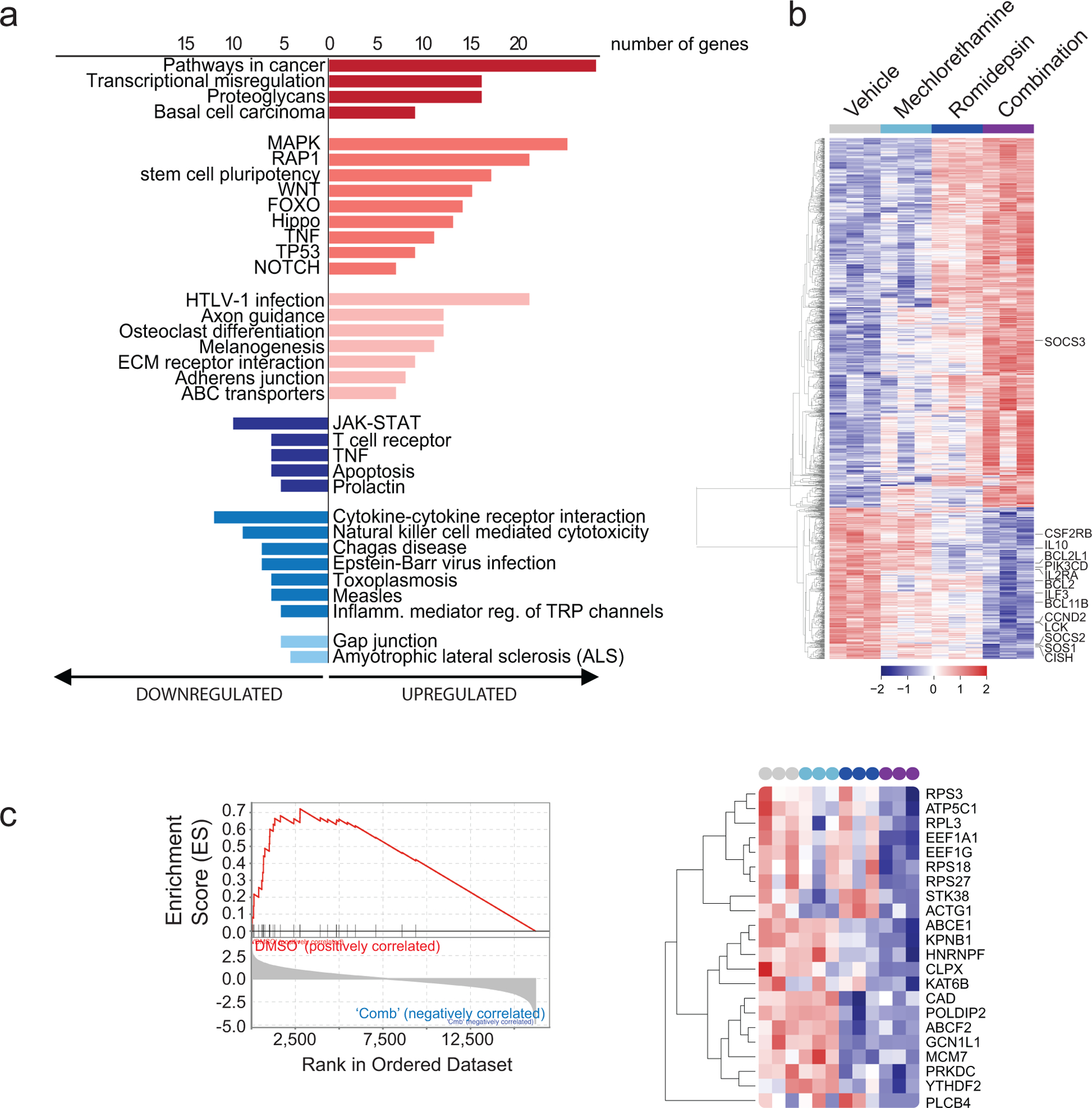
(a) KEGG pathway analysis using DAVID 6.8 on the top differentially expressed genes in cell lines treated with the combination of romidepsin and mechlorethamine (p<0.01, log2fold-change>1.5 or <−1.5). Pathways with p<0.05 are shown. (b) Heatmap of the normalized expression of all significantly de-regulated genes in cell lines treated with Mechlorethamine alone, Romidepsin alone or both in combination compared to vehicle control. Genes were clustered by the complete linkage agglomerative hierarchical clustering method on the Euclidean distances. Cutoffs used to define significance in A, B and D were; padj < 0.01, logFC > 1.2. (c) Gene Set Enrichment Analysis revealed significant enrichment of the Pathway Interaction Database IL2-STAT5 Pathway gene signature (FWER p-Value < 0.001, NES 2.53 in vehicle-combination). Enrichment plot is presented in the left panel and normalized expression of the genes comprised by the leading edge of the signature are plotted in the heatmap in the right panel.
All transcriptomic analyses support prominent downregulation of JAK/STAT signaling in CTCL cells treated with the combination of mechlorethamine and romidepsin. In agreement with the observed transcriptional changes, phospho-flow analysis of JAK/STAT signaling in HH and HuT78 CTCL cells showed strong phosphorylation of STAT5 protein in response to IL2. Interestingly, HUT78 cells showed elevated STAT5 phosphorylation in basal conditions (Figure 4a). Conversely, treatment with romidepsin and mechlorethamine individually resulted in reduced levels of pSTAT5 and blunted IL2-induced JAK/STAT5 activation (Figure 4a). Moreover, and most significantly, combination treatment with mechlorethamine plus romidepsin induced a markedly stronger abrogation of STAT5 phosphorylation indicative of a non-epistatic convergent interaction (Figure 4a). Analysis of JAK1 in HUT78 cells revealed a marked decrease in protein levels in response to mechloretamine, romidepsin and even more after treatment with the drug combination (Supplementary Figure S5). We also evaluated the effect on STAT5 phosphorylation of mechlorethamine and romidepsin alone and in combination in vivo using our luciferized HuT78 CTCL mouse model. Following tumor development, we segregated animals with homogeneous tumor burden into therapeutic groups for treatment with vehicle only, romidepsin, mechlorethamine, and romidepsin plus mechlorethamine in combination (n=3 animals/group) and treated them for 7 days (Supplementary Figure S2b). At this time point, treatment with romidepsin or mechlorethamine had a minor effect in tumor burden compared with controls, but allowed the isolation of lymphoma cells for analysis of phosphor-STAT5 by flow cytometry. Our data indicates that the combination of mechlorethamine plus romidepsin decreases phosphorylation of STAT5 also in vivo (Figure 4b). Similarly, STAT5 phosphorylation in romidepsin-sensitive primary patient samples (P-02 and P-03) revealed modest reduction in JAK/STAT5 signaling following single-agent treatments with mechoretamine and romidepsin (Figure 4c), which was markedly deeper after treatment with mechlorethamine and romidepsin in combination. Of note, the effects of mechlorethamine, romidepsin and the combination of romidepsin plus mechlorethamine in JAK/STAT5 signaling were markedly less pronounced in cells from Sezary Syndrome primary patient samples resistant to romidepsin (P-01 and P-07) (Figure 4c).
Figure 4. Analysis of STAT5 phosphorylation in CTCL cell lines in vitro, in a mouse model of Sezary Syndrome in vivo, and in primary samples after treatment with romidepsin, mechlorethamine or their combination.
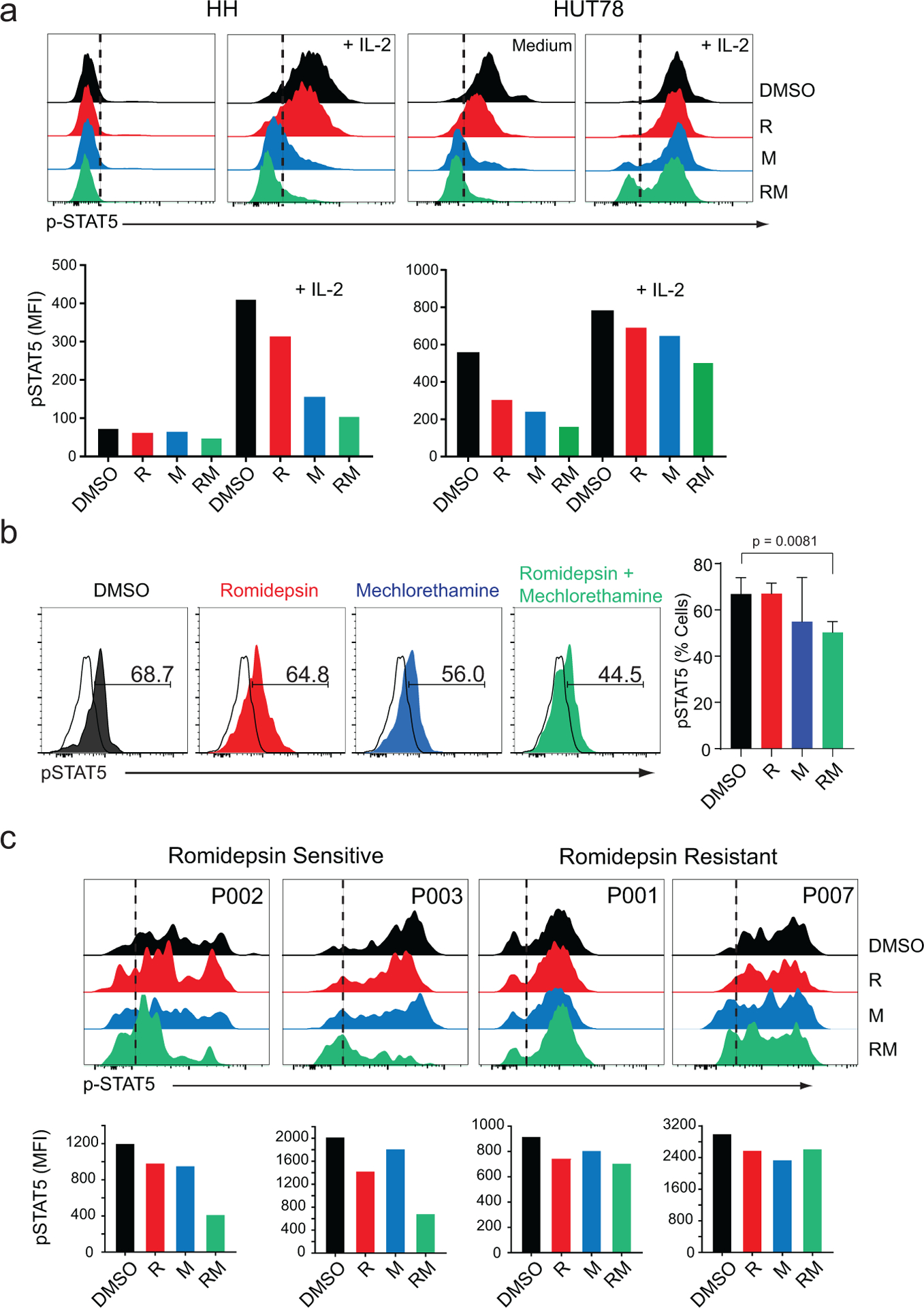
(a) Flow cytometry analysis of phosphor-STAT5 in HH and HUT78 Sezary syndrome cell lines after treatment with vehicle (DMSO), mechlorethamine, romidepsin, or the combination for 48 hours, in the absence or presence of IL-2. (b) Flow cytometry analysis of phosphor-STAT5 in lymphoma cells isolated from a mouse model of Sezary Syndrome after in vivo treatment with vehicle (DMSO), mechlorethamine, romidepsin, or the combination for 7 days. (c) Analysis of STAT5 phosphorylation in primary patient samples cultured in vitro in the presence of IL-2 and treated as described above. Patient samples are representative cases of romidepsin-sensitive and romidepsin-resistant individuals. Data are representative of at least two independent experiments. DMSO: dimethyl sulfoxide, M: mechlorethamine, R: romidepsin. P value was calculated using a two-tailed Student’s t-test. Error bars, mean ± s.d.
Reversal of romidepsin resistance by JAK inhibitors
In all, our gene expression and signaling analyses support a role for suppression of JAK/STAT5 signaling in the response of romidepsin sensitive cells to co-tretament with romidepsin plus mechlorethamine and point to a therapeutically relevant association between romidepsin resistance and constitutively active/sustained JAK/STAT activation. A corollary of these observations is that direct pharmacologic inhibition of JAK/STAT signaling could enhance the therapeutic activity of HDACi therapy in CTCL and potentially abrogate resistance to romidepsin therapy.
To test this hypothesis, we evaluated the response of CTCL lines to JAK inhibitor treatment alone and in combination with romidepsin. These experiments revealed significant antitumor effects of momelotinib and ruxolitinib in CTCL (Supplementary Figure S6). Moreover, co-treatment with romidepsin plus these JAK inhibitors revealed increased antitumor responses in cells HuT78 cells treated with romidepsin plus momelotinib and romidepsin plus ruxolitinib in combination (Supplementary Figure S7). In addition, and most notably, treatment of romidepsin-resistant P-01 and P-07 Sezary primary samples with romidepsin-momelotinib and romidepsin-ruxolitinib in combination showed effective reversal of romidepsin resistance resulting in marked antitumor responses (Figure 5a). Across these experiments, we observed effective abolishment of STAT5 phosphorylation by momelotinib and ruxolitinib alone and in combination with romidepsin (Figure 5b).
Figure 5. Analysis of apoptosis and STAT5 phosphorylation in romidepsin-resistant CTCL primary patient samples after treatment with romidepsin, JAK inhibitors momelotinib and ruxolitinib or their combination.
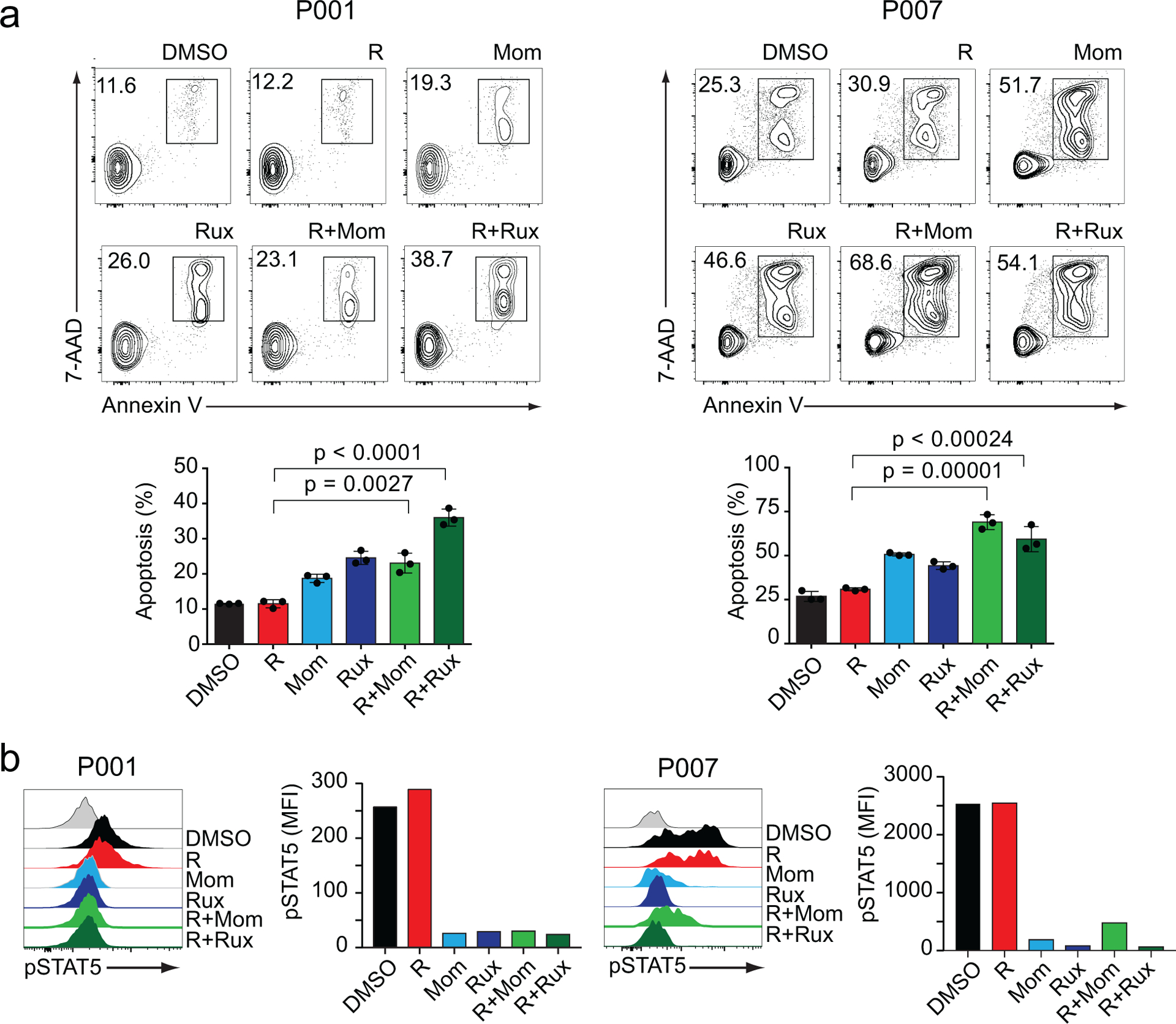
(a) Apoptosis analysis determined by flow cytometry after Annexin V/7AAD staining in romidepsin-resistant P001 and P007 primary patient samples after treatment with vehicle (DMSO), romidepsin, momelotinib, ruxolitinib or the combination for 48 hours in presence of IL-2. (b) Analysis of STAT5 phosphorylation in primary patient samples cultured in vitro in the presence of IL-2 and treated as described above. Data are representative of at least two independent experiments. R: romidepsin, Rux: ruxolitinib, Mom: momelotinib. P values were calculated using a two-tailed Student’s t-test. Error bars, mean ± s.d.
To formally test whether combination therapies including romidepsin plus JAK/STAT inhibition can deliver an effective in vivo antitumor response we evaluated the efficacy of romidepsin and ruxolitinib in our SS HuT78 xenograft model (Supplementary Figure S2b). In these experiments, HuT78 xenograft tumors showed markedly increased responses to treatment with romidepsin-ruxolitinib in combination compared with those observed in mice from single agent treatment groups (Figure 6a–c), which was accompanied by a strong reduction in STAT5 phosphorylation (Supplementary Figure S8). Taken together, these results support a prominent role for JAK/STAT signaling as mediator of the therapeutic efficacy of romidepsin plus mechlorethamine combination and as modulator of the therapeutic response to romidepsin calling for the clinical testing of HDACi plus JAK/STAT inhibition treatments in romidepsin refractory cases.
Figure 6. Romidepsin combined with ruxolitinib treatment decreases tumor load in a mouse model of Sezary syndrome.
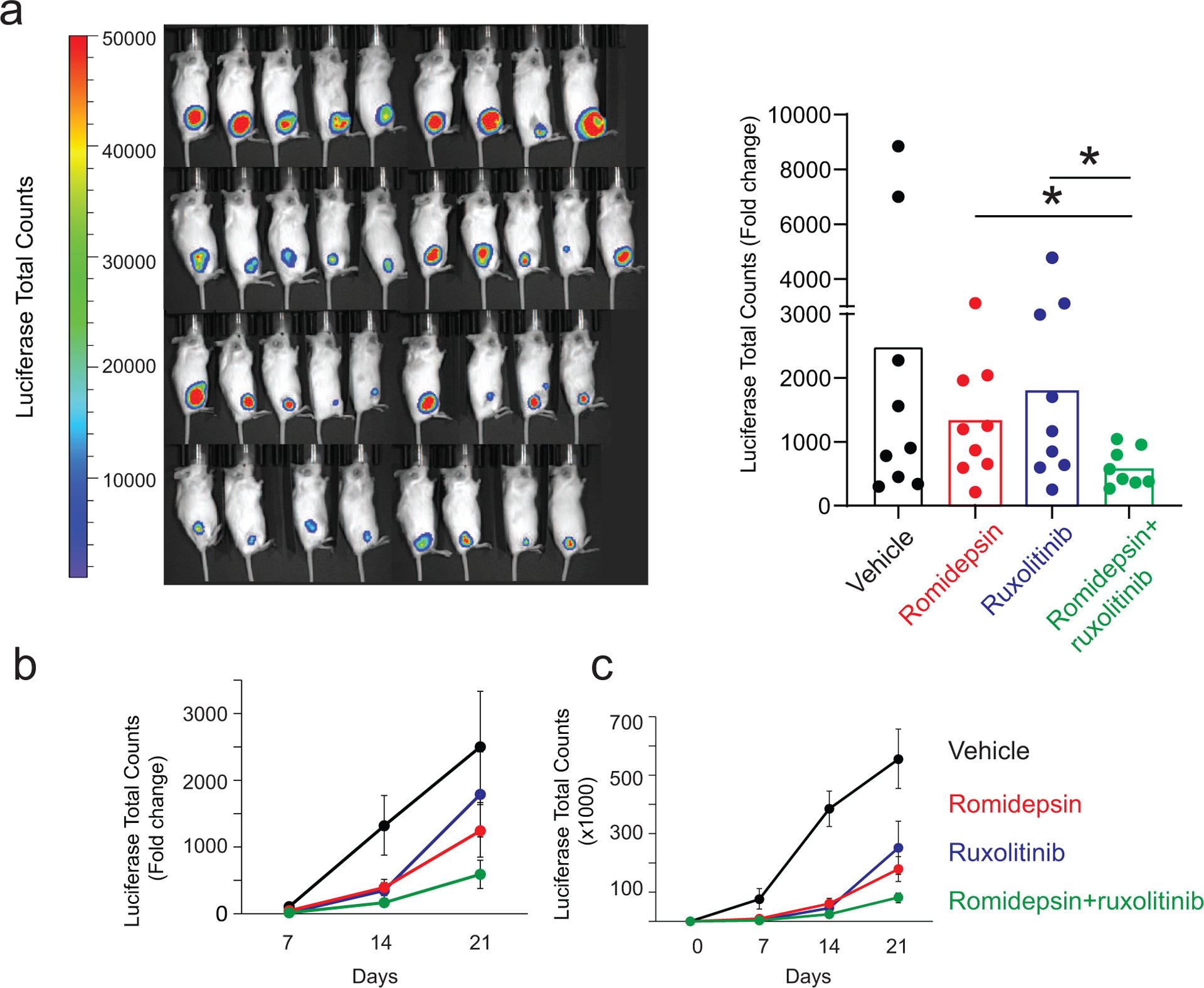
(a) Quantitative analyses of changes in tumor burden measured by bioluminescence at the endpoint analysis. (b, c) Quantitative analyses of changes in tumor burden measured by bioluminescence at different time points represented as fold change (b) or total counts (c). P values were calculated using a two-tailed Student’s t-test. Error bars, mean ± s.d. *p ≤ 0.05.
DISCUSSION
Multiple genomic studies have improved our understanding of the genetic basis of CTCL over the past few years. The mutational landscape inferred from these studies depicts a highly heterogeneous disease with no clear dominant drivers, which affects the development of effective targeted therapies for CTCL patients.(da Silva Almeida et al., 2015, Kiel et al., 2015, McGirt et al., 2015, O’Connor et al., 2015, Prasad et al., 2016, Ungewickell et al., 2015) Histone deacetylases (HDACs) are enzymes capable of removing acetylation marks in histone and nonhistone proteins. HDACs modulate multiple pathways and processes, many of which are dysregulated in cancer, including cell cycle, apoptosis, and protein folding. HDACis are small molecules that prevent the removal of acetyl groups by HDACs, thus effectively counteracting their effects on protein acetylation (Su et al., 2021). HDACis have demonstrated significant clinical activity in hematologic malignancies.(Chun, 2015, O’Connor et al., 2015, Robey et al., 2011, San-Miguel et al., 2014) Multiple HDACs, including HDAC 1, 2, and 6, were shown to be overexpressed in T-cell lymphoma compared with normal lymphoid tissue, making this disease a potential target for the use of HDACi (Marquard et al., 2009). Indeed, several HDACi have been approved for treatment of T-cell lymphomas based on the remarkable therapeutic effects achieved in the context of clinical trials(Bates et al., 2015) . Romidepsin was approved for the treatment of CTCL in 2009 for patients who fail at least one prior systemic therapy. Approval based on two phase II open-label, multicenter clinical trials, showing that romidepsin treatment elicited a 34% overall response rates and 6% complete response, as well as improvement in the duration of response and pruritus over conventional therapies(Piekarz et al., 2009) . Though romidepsin is now a mainstay treatment for CTCL, response rates and duration of response are variable(Martinez-Escala et al., 2016, Piekarz et al., 2009). Indeed, despite the initial promising results, the duration of response to romidepsin in lymphoma patients is still limited, and eventually most of the patients relapse. The characterization of biomarkers associated with high probability of response would increase the therapeutic potential of HDACi, however, biomarkers of response have yet to be identified (Bates et al., 2015). In addition, mechanisms of resistance to romidepsin have not been well characterized (Bates et al., 2015).
To improve the therapeutic effect of romidepsin, combination therapies are currently being tested in the context of clinical trials. As more drugs with clinical activity become available, the need to identify rational drug combinations with therapeutic activity in lymphoma becomes essential.
While empirical approaches may offer clinical benefits, identifying the mechanisms that underlie response and biomarkers for personalized therapy might offer the best opportunity. Here we report two active romidepsin combinations with increased therapeutic activity in preclinical models of CTCL.
Our data demonstrate that the combination of mechlorethamine and romidepsin is highly synergistic in CTCL cell lines and primary patient samples in vitro. The synergism of romidepsin and mechloretamine has been validated in both leukemic and skin-derived CTCL cell lines, supporting the relevance of this romidepsin combination with a topical skin directed agent as mechlorethamine. Moreover, the combination of romidepsin and mechlorethamine demonstrates a strong anti-tumor activity in vivo with complete responses in a mouse model of SS. This combination utilizes a skin-directed therapy together with a systemic therapy and is therapeutically advantageous because both agents are already FDA-approved as monotherapies(Bates and Geskin, 2016). In depth analysis of the molecular mechanisms underlying the synergistic effect of the mechlorethamine-romidepsin combination using RNAseq and gene expression profiling on cell lines and primary samples revealed a unique set of pathways specifically regulated by the combination. Our gene expression profiling analysis shows that romidepsin is the major determinant of gene expression changes in combination treatment, compared to mechlorethamine. Most notably, we specifically identified inhibition of the JAK/STAT signaling pathway by the mechlorethamine-romidepsin combination.
The JAK/STAT pathway transduces the signals initiated by the engagement of cytokine receptors, and it is essential for the immune response. Deregulation of the JAK/STAT pathway is implicated in the pathogenesis of hematologic malignancies, and is generally associated with constitutive activation of members of the STAT family of transcription factors, normally mediated by JAK-mediated tyrosine phosphorylation, leading to increased proliferation and survival. In CTCL, constitutive activation of the JAK/STAT pathway is a highly recurrent event which can be achieved by multiple mechanisms, including up-regulation of cytokine signaling and recurring gain-of-function alterations predicted to hyperactivate JAK1, JAK3, STAT3, and STAT5B.(Damsky and Choi, 2016) Our data indicate that the JAK/STAT pathway plays an important role in the response to romidepsin and mechlorethamine in CTCL. We have observed a marked downregulation of JAK1 protein levels in the presence of mechlorethamine and romidepsin. Mechlorethamine is a highly reactive alkylating agent that acts by crosslinking guanine residues in DNA but also can induce direct alkylation of protein cysteine residues (Loeber et al., 2009, Noort et al., 2002), a modification that could facilitate protein degradation, therefore, we cannot exclude alkylation-induced degradation as a potential mechanism for JAK/STAT pathway regulation. Additionally, we had also detected significant upregulation of SOCS3, a negative regulator of JAK/STAT pathway in the combination treatment in our RNAseq data, which normally targets phosphor-JAK1 for degradation by the proteasome (Boyle et al., 2009).Analysis of STAT proteins phosphorylation after treatment with mechlorethamine and romidepsin in cell lines showed a decrease of STAT5 phosphorylation consistent with an additive effect of the drug combination on the regulation of the activity of STAT5. Upregulation of STAT5 in CTCL has been associated with lymphomagenesis because of induction of expression of oncogeneic miR- 155 and miR- 21, inhibition of apoptosis and secretion of Th2 cytokines (Kopp et al., 2013, Lindahl et al., 2016, Litvinov et al., 2013).
More interestingly, our data showed that the romidepsin-mechlorethamine combination failed to inhibit the phosphorylation of STAT5 in romidepsin-resistant samples. These results suggest that the JAK/STAT pathway could modulate the response to romidepsin, and potentially mediate resistance to romidepsin treatment. Indeed, activation of JAK/STAT signaling has been previously suggested to play a role on resistance to HDAC inhibitors in the B-cell lymphoma cell lines(Smith et al., 2010) and in resistance to glucocorticoids in T-cell acute lymphoblastic leukemia (De Smedt et al., 2019). Importantly, our data demonstrate in vivo and in vitro, that the combination of JAK inhibitors with romidepsin induces increased therapeutic response compared to single agents even in romidepsin resistant samples. As JAK inhibitors are making their way into the therapeutic arsenal for hematologic malignancies, our data suggest that CTCL patients might benefit from treatment with these inhibitors in combination with romidepsin.
Analysis of STAT5 phosphorylation after romidepsin treatment should be evaluated as a potential biomarker predictive of therapeutic activity; high and persistant p-STAT5 could predict lack of response and potential benefit of a romidepsin plus JAK inhibitor combination. Pharmacological targeting of these pathways could potentially represent a unique approach to treat resistance to romidepsin in CTCL.
MATERIALS AND METHODS
Cell Lines
We obtained the HH (CRL-2105), MJ (CRL-8294) , HuT102 (TIB-162.1) and HuT78 (TIB-161) CTCL cell lines from the ATCC(Starkebaum et al., 1991) and purchased the mycosis fungoides My-La cell line (95051032) from Millipore-Sigma (St. Louis, MO, USA). The SeAx Sezary syndrome cell line(Gazdar et al., 1980, Kaltoft et al., 1987) was generously provided by Dr. Maarten Vermeer at Leiden University Medical Center, the Netherlands.
Primary CTCL samples
We obtained de-identified peripheral blood mononuclear cells (PBMCs) isolated from patients who met WHO-EORTC criteria for Sezary Syndrome from the leukemia and lymphoma tumour repository bank at Columbia University Irving Medical Center (CUIMC). Patient samples were collected with written informed consent and analysis was conducted under the supervision of the CUIMC Institutional Review Board. We isolated CD4+ T cells via negative selection using the Human CD4+ T cell isolation kit (Miltenyi, Auburn, CA, USA).
Cell culture
We cultured all cell lines in RPMI 1640 medium with 2.05 mM L-Glutamine (Hyclone) supplemented with penicillin (100 μg Ml−1), streptomycin (50 μg mL−1) and 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2 and checked periodically for mycoplasma contamination using MycoSensor qPCR Assay Kits (Agilent Technologies, Santa Clara, CA, USA). We cultured primary lymphoma cells RPMI 1640 medium with 2.05 mM L-Glutamine supplemented with penicillin (100 μg mL−1), streptomycin (50 μg mL−1), 10% human serum (Sigma-Aldrich, St. Louis, MO, USA), 10 μg mL−1 IL-2 and 10 μg mL−1 IL-7 (PeproTech, Rocky Hill, NJ, USA). Prior to drug treatments, the cells were stimulated for 48 hours with 10 μg mL−1 plated anti-CD3 (clone HIT3a) and 2 μg mL−1 soluble anti-CD28 (clone CD28.2) both from Thermo Fisher (Waltham, MA, USA)
Drugs
We purchased mechlorethamine HCl (122564) and romidepsin (SML1175) from Sigma-Aldrich (St. Louis, MO, USA) and the JAK kinase inhibitors baricitinib (S2851), momelotinib (S2219), ruxolitinib (S1378), tofacitinib (S5001), FM-381 (S8541) and PF-06651600 (S8538) from SelleckChem (Houston, TX, USA). We dissolved all drugs as stock solutions in dimethyl sulfoxide (DMSO) and stored them at −20°C.
Flow cytometry analyses
We immunophenotyped primary CTCL tumour cells by staining single cell suspensions with fluorochrome-conjugated antibodies against CD4 (VIT4)(Miltenyi) and antibodies against CD3 (HIT3a), CD7 (eBio124-1D1), CD8 (SK1) and CD26 (2A6) (all from Thermo Fisher) using standard procedures. We performed flow cytometry analyses using a FACS Canto II Flow Cytometer (BD, San Jose, CA, USA) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA). For flow cytometry detection of phosphorylated intracellular proteins we fixed single cell suspensions with 4% formaldehyde and permeabilized them with 90% ice cold methanol and then incubated them with a primary antibody against pSTAT5 (Cat #9314) followed by anti-rabbit Alexa-647 secondary antibody (Cat #4414) staining, all from Cell Signaling Technology (Danvers, MA, USA).
Cell proliferation and drug response analyses
We plated cells at a density of 3–4 105 cells mL−1 in 96-well plates and treated in triplicate with the different drug combinations for 24, 48, 72, and 96 hours a TecanD300e digital dispenser (Tecan, Männedorf, Switzerland). We measured cell growth and viability by a colorimetric 3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay using the Cell Proliferation Kit I (Roche, Penzberg, Germany). We used GraphPad Prism 7.0 to generate non-linear fit dose-response curves and calculate the mean inhibitory concentration (IC50) for each drug at each time point in each cell line after 48 hours of treatment. We evaluated synergism using the median effect equation of Chou and Talalay to generate a combination index and isobologram at 50% effect level.(Chou and Talalay, 1984) We calculated the combination index with the CompuSyn software (ComboSyn, INC., Paramus, NJ, USA) and considered a combination index less than 1 as indicative of synergism, equal to 1 indicative additive effects, and greater than 1 indicative of antagonism.
Apoptosis and cell viability analyses
For analysis of apoptosis, we treated CTCL cell lines with vehicle, single drugs and drug combinations for 24 hours and analyse them by flow cytometry after staining with APC-conjugated Annexin V and 7-AAD (eBioscience Annexin V Apoptosis Detection Kit APC, Thermo Fisher). We quantified cell viability by flow cytometry after thiazole orange and propidium iodide staining using the BD Cell Viability Kit (BD Biosciences, San Jose, CA), according to the manufacturer’s protocol.
RNA isolation
We extracted and purified total RNA from cells by guanidinium thiocyanatephenol-chloroform extraction (TriZol, Sigma-Aldrich) and the RNeasy mini kit (QIAGEN, Germantown, MD, USA). We analysed and quantified RNA using an Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA).
RNAseq and gene expression profiling
We generated RNAseq libraries at the Sulzberger Genome Center at Columbia University Medical Center using the TruSeq Stranded RNA Library Prep kit with RiboZero gold (Illumina, San Diego, CA, USA). We multiplexed sequencing libraries and sequenced them on an Illumina HiSeq2500 using 100 bp paired-end read sequencing and a total depth of 30 million reads per sample. We performed base calling using RTA (Illumina) and converted to fastq format using bcl2fastq (version 2.17). We aligned RNA-sequencing reads to GRCh38 using STAR v2.5 with default parameters(Dobin et al., 2013). We normalized read counts were normalized using the DESeq2 Bioconductor package for R (Love et al., 2014) and performed differential expression analysis using a negative binomial model including treatment condition as the covariate. We performed variance stabilization using the regularized log transformation on the read count matrices and the first two principal components for each sample were plotted in R. We performed Gene Set Enrichment Analysis(Subramanian et al., 2005) using Gene Pattern (Broad Institute)(Reich et al., 2006) to test for the enrichment of specific JAK/STAT pathway signatures(Moodley et al., 2016, Rampal et al., 2014). Gene Ontology term enrichment analysis was performed in Cytoscape using the BINGO application (Maere et al., 2005). Heatmaps of normalized gene expression levels were plotted in R.
RNAseq data is available in Gene Expression Omnibus (accession# GSE110248).
Protein isolation and western blot
Protein lysates were prepared in RIPA buffer (20mM Tris-HCl pH 7.4, 150mM NaCl, 1% Triton-X-100, 0.1% SDS, 0.5% sodium deoxycholate). Western blot analysis was performed on HUT78 cell lysates separated by SDS-PAGE and transferred to nitrocellulose membranes using standard procedures. For immunoblot detection, we used the following antibodies: JAK1 (dilution 1:1000) (Cell Signaling Technology, Cat. No. 3332), JAK2 (dilution 1:1/500) (Santa Cruz Biotechnology , Cat. No. sc390539) or GAPDH (D16H11) (dilution 1:1000)(Cell Signaling Technology, Cat. No. 5174). The blots were developed with Thermo Scientific™ SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Cat. No. 34080) or SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Cat. No. 34094) according to the manufacturer’s instructions.
Generation of luciferase expressing Sezary Syndrome cell lines
We transfected the FUW-mCherry-Puro-Luc lentiviral vector carrying the mCherry and luciferase genes with the plasmid vectors encoding the Gag-Pol (pCMV ΔR8.91) and V-SVG (pMD.G VSVG) viral proteins into HEK293T cells using the JetPEI transfection reagent (Polyplus Transfection, Illkirch-Graffenstaden, France). We used viral-particle containing supernatants for infection of HuT78 cells by spinoculation using standard procedures. We assessed the efficiency of infection by detection of mCherry expressing cells by flow cytometry.
Mouse husbandry and procedures
We maintained all animals in specific pathogen-free facilities at the Irving Cancer Research Center at CUIMC. All animal procedures are approved by the Institutional Animal Care and Use Committee (IACUC) at CUIMC.
For tumor transplantation assays, we injected 2×106 luciferized HuT78 T-cells subcutaneously into Rag1/Il2rg double knockout recipient mice (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ, Stock No: 007799, Jackson Laboratory). We monitored tumor development by luciferase bioimaging with the In Vivo Imaging System (IVIS, Xenogen, Alameda, CA).
Statistical analyses
We evaluated statistical significance in apoptosis and cell viability assays using two-tailed Student’s t-test.
Data Availability Statement
RNAseq datasets related to this article can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110248, hosted at is available in Gene Expression Omnibus data base (accession# GSE110248).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Maarten Vermeer and Dr. Cornelis Tensen from Leiden University Medical Center for providing the SeAx and HuT78 cell lines. We thank Dr. Alberto Ambesi-Impiombato for assistance with RNAseq analysis. This work was supported by the National Cancer Institute (R01 CA197945 and NCI 5P50CA192937-03 DRP to T.P.), the Leukemia & Lymphoma Society (TRP-6507-17 to T.P.); a Velocity Cancer Research Award (T.P.) and an Actelion Pharmaceuticals US, Inc. research grant (to L.J.G.). This research was also funded in part through the NIH/NCI Cancer Center Support Grant P30CA013696. C.C.P. and Y.G. were supported by the Columbia College of Physicians and Surgeons Dean’s Research Fellowship and J.R.C. was supported by a postdoctoral fellowship from the Lady Tata Memorial Trust.
CONFLICT OF INTEREST
Dr. Geskin serves on the Advisory Board for Therakos, Inc., Merck & Co., Helsinn, and Celgene Corporation and is an investigator for Actelion Pharmaceuticals US, Inc., Merck & Co., Inc., Kyowa Kirin Pharmaceutical Development, Inc. and Celgene Corporation. Dr. Palomero is the recipient of a research grant from Kura Technologies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aron JL, Parthun MR, Marcucci G, Kitada S, Mone AP, Davis ME, et al. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood 2003;102(2):652–8. [DOI] [PubMed] [Google Scholar]
- Bates SE, Eisch R, Ling A, Rosing D, Turner M, Pittaluga S, et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. Br J Haematol 2015;170(1):96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates SE, Geskin LJ. Romidepsin Therapy Over 5 Years in a Clinical Setting-Real-world Experience. JAMA Oncol 2016;2(6):794–5. [DOI] [PubMed] [Google Scholar]
- Bates SE, Zhan Z, Steadman K, Obrzut T, Luchenko V, Frye R, et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol 2010;148(2):256–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle K, Zhang JG, Nicholson SE, Trounson E, Babon JJ, McManus EJ, et al. Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal 2009;21(3):394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- Chun P Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res 2015;38(6):933–49. [DOI] [PubMed] [Google Scholar]
- Cosenza M, Civallero M, Fiorcari S, Pozzi S, Marcheselli L, Bari A, et al. The histone deacetylase inhibitor romidepsin synergizes with lenalidomide and enhances tumor cell death in T-cell lymphoma cell lines. Cancer Biol Ther 2016:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrenne BM, Lewis J, Weed J, Carlson K, Mirza FN, Foss F, et al. Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients. Blood 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet 2015;47(12):1465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky WE, Choi J. Genetics of Cutaneous T Cell Lymphoma: From Bench to Bedside. Curr Treat Options Oncol 2016;17(7):33. [DOI] [PubMed] [Google Scholar]
- De Smedt R, Morscio J, Goossens S, Van Vlierberghe P. Targeting steroid resistance in T-cell acute lymphoblastic leukemia. Blood Rev 2019;38:100591. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulmage BO, Story SK, Falo LD Jr., Geskin LJ. Novel therapeutic combination demonstrates more than additive effects in cutaneous T-cell lymphoma. Leuk Lymphoma 2015;56(7):2225–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Carney DN, Bunn PA, Russell EK, Jaffe ES, Schechter GP, et al. Mitogen requirements for the in vitro propagation of cutaneous T-cell lymphomas. Blood 1980;55(3):409–17. [PubMed] [Google Scholar]
- Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur J Haematol 2009;82(6):440–9. [DOI] [PubMed] [Google Scholar]
- Kaltoft K, Bisballe S, Rasmussen HF, Thestrup-Pedersen K, Thomsen K, Sterry W. A continuous T-cell line from a patient with Sezary syndrome. Arch Dermatol Res 1987;279(5):293–8. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun 2015;6:8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Martinez G, Varghese A, Hoppe RT. Topical nitrogen mustard in the management of mycosis fungoides: update of the Stanford experience. Arch Dermatol 2003;139(2):165–73. [DOI] [PubMed] [Google Scholar]
- Kopp KL, Ralfkiaer U, Nielsen BS, Gniadecki R, Woetmann A, Odum N, et al. Expression of miR-155 and miR-126 in situ in cutaneous T-cell lymphoma. APMIS 2013;121(11):1020–4. [DOI] [PubMed] [Google Scholar]
- Lessin SR, Duvic M, Guitart J, Pandya AG, Strober BE, Olsen EA, et al. Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol 2013;149(1):25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl LM, Fredholm S, Joseph C, Nielsen BS, Jonson L, Willerslev-Olsen A, et al. STAT5 induces miR-21 expression in cutaneous T cell lymphoma. Oncotarget 2016;7(29):45730–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvinov IV, Pehr K, Sasseville D. Connecting the dots in cutaneous T cell lymphoma (CTCL): STAT5 regulates malignant T cell proliferation via miR-155. Cell Cycle 2013;12(14):2172–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeber RL, Michaelson-Richie ED, Codreanu SG, Liebler DC, Campbell CR, Tretyakova NY. Proteomic analysis of DNA-protein cross-linking by antitumor nitrogen mustards. Chem Res Toxicol 2009;22(6):1151–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005;21(16):3448–9. [DOI] [PubMed] [Google Scholar]
- Marquard L, Poulsen CB, Gjerdrum LM, de Nully Brown P, Christensen IJ, Jensen PB, et al. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009;54(6):688–98. [DOI] [PubMed] [Google Scholar]
- Martinez-Escala M, Kuzel TM, Kaplan JB, et al. Durable responses with maintenance dose-sparing regimens of romidepsin in cutaneous t-cell lymphoma. JAMA Oncology 2016;2(6):790–3. [DOI] [PubMed] [Google Scholar]
- McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015;126(4):508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci U S A 2016;113(35):9852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyal L, Feldbaum N, Goldfeiz N, Rephaeli A, Nudelman A, Weitman M, et al. The Therapeutic Potential of AN-7, a Novel Histone Deacetylase Inhibitor, for Treatment of Mycosis Fungoides/Sezary Syndrome Alone or with Doxorubicin. PLoS One 2016;11(1):e0146115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noort D, Hulst AG, Jansen R. Covalent binding of nitrogen mustards to the cysteine-34 residue in human serum albumin. Arch Toxicol 2002;76(2):83–8. [DOI] [PubMed] [Google Scholar]
- O’Connor OA, Horwitz S, Masszi T, Van Hoof A, Brown P, Doorduijn J, et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 2015;33(23):2492–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piekarz R, Bates S. A review of depsipeptide and other histone deacetylase inhibitors in clinical trials. Curr Pharm Des 2004;10(19):2289–98. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol 2009;27(32):5410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad A, Rabionet R, Espinet B, Zapata L, Puiggros A, Melero C, et al. Identification of Gene Mutations and Fusion Genes in Patients with Sezary Syndrome. J Invest Dermatol 2016;136(7):1490–9. [DOI] [PubMed] [Google Scholar]
- Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014;123(22):e123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet 2006;38(5):500–1. [DOI] [PubMed] [Google Scholar]
- Robey RW, Chakraborty AR, Basseville A, Luchenko V, Bahr J, Zhan Z, et al. Histone deacetylase inhibitors: emerging mechanisms of resistance. Mol Pharm 2011;8(6):2021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozati S, Cheng PF, Widmer DS, Fujii K, Levesque MP, Dummer R. Romidepsin and Azacitidine Synergize in their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clin Cancer Res 2016;22(8):2020–31. [DOI] [PubMed] [Google Scholar]
- San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014;15(11):1195–206. [DOI] [PubMed] [Google Scholar]
- Smith J, Walsh KJ, Jacobs CL, Liu Q, Fan S, Patel A, et al. Upregulated JAK/STAT Signaling Represents a Major Mode of Resistance to HDAC Inhibition In Lymphoma and Provides a Rationale for Novel Combination Therapy. Blood 2010;116(21):434-. [Google Scholar]
- Starkebaum G, Loughran TP Jr., Waters CA, Ruscetti FW. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int J Cancer 1991;49(2):246–53. [DOI] [PubMed] [Google Scholar]
- Su M, Gong X, Liu F. An update on the emerging approaches for histone deacetylase (HDAC) inhibitor drug discovery and future perspectives. Expert Opin Drug Discov 2021:1–17. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talpur R, Venkatarajan S, Duvic M. Mechlorethamine gel for the topical treatment of stage IA and IB mycosis fungoides-type cutaneous T-cell lymphoma. Expert Rev Clin Pharmacol 2014;7(5):591–7. [DOI] [PubMed] [Google Scholar]
- Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat Genet 2015;47(9):1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J Clin Oncol 2010;28(29):4485–91. [DOI] [PubMed] [Google Scholar]
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105(10):3768–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq datasets related to this article can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110248, hosted at is available in Gene Expression Omnibus data base (accession# GSE110248).