

Abstract
BRAF-inhibitor (BRAFi)-resistance compromises long term survivorship of malignant melanoma patients, and mutant NRAS is a major mediator of BRAFi-resistance. Here, employing phenotypic and transcriptomic analysis of isogenic melanoma cells that differ only by NRAS mutational status (BRAFi-sensitive A375-BRAFV600E/NRASQ61 versus BRAFi-resistant A375-BRAFV600E/NRASQ61K) we demonstrate that BRAFi (vemurafenib) treatment selectively targets BRAFV600E/NRASQ61K cells upregulating epithelial-to-mesenchymal transition (EMT) gene expression, paradoxically promoting invasiveness and metastasis in vitro and in vivo. First, NanoString nCounter™ transcriptomic analysis identified upregulation of specific gene expression networks (‘EMT’ and ‘EMT to Metastasis’) as a function of NRASQ61K-status. Strikingly, BRAFi-treatment further exacerbated upregulation of genes promoting EMT in BRAFV600E/NRASQ61K cells (with opposing downregulation of EMT-driver genes in the BRAFV600E/NRASQ61-genotype) as detected by EMT-focused RT2 Profiler™ qPCR array analysis. In BRAFV600E/NRASQ61K cells, BRAFi-treatment enhanced proliferation and invasiveness together with activation of p-AKT(Ser473), with opposing phenotypic effects observable in BRAFV600E/NRASQ61 cells displaying downregulation of p-AKT and p-ERK1/2. In a SCID mouse bioluminescent melanoma metastasis model, BRAFi treatment enhanced lung tumor burden imposed by BRAFV600E/NRASQ61K cells, while blocking BRAFV600E/NRASQ61 metastasis. These preclinical data document the BRAFi-driven enhancement of tumorigenesis and metastasis in BRAFi-resistant human BRAFV600E/NRASQ61K melanoma, a finding with potential clinical implications for patients with NRAS-driven BRAFi-resistant tumors receiving BRAFi-treatment.
INTRODUCTION
A majority of skin cancer-related deaths is caused by malignant melanoma, a tumor originating from neural crest-derived melanocytes, and the exploration of innovative molecular strategies for improved detection and treatment of melanoma is of substantial clinical significance (Curti and Faries, 2021; Jenkins and Fisher, 2021). Despite significant therapeutic progress targeting BRAF-driven melanoma, few treatment options exist for NRAS-driven melanoma known to be more aggressive and associated with poorer outcomes compared to non-NRAS-mutant melanoma (Eskandarpour et al., 2009; Nazarian et al., 2010; Jenkins and Sullivan, 2016; Munoz-Couselo et al., 2017; Yin et al., 2019). Mutational activation of NRAS (clustering at codons 12, 13, and 61) is the second most common oncogenic ‘driver’ alteration in malignant melanoma (after BRAF) (Dean, 2017).
Importantly, in treatment-naïve melanoma tumors, NRAS mutations rarely occur together with either BRAF or PTEN mutations, a phenomenon referred to as oncogene exclusion (Petti et al., 2006; Sensi et al., 2006; Heppt et al., 2017; Kumar et al., 2019). In contrast, during the course of BRAFV600E-targeted treatment employing BRAFV600E-directed kinase inhibitors (BRAFi), emergence of resistance often involves NRAS mutations [predominantly in codon 61 (Q61R, Q61K) observable in approximately 20% of patients], a unique co-occurrence of BRAF and NRAS mutations facilitated and selected for by BRAFi-based therapy (Johnson et al., 2015).
Extensive research has dissected the molecular mechanisms underlying acquired NRAS-driven BRAFi-resistance; however, little is known about the effects of BRAFi-treatment on BRAFi-resistant tumors displaying dual BRAFV600E- and NRAS-mutations, a topic of clinical relevance in the context of treatment durability and failure (Nazarian et al., 2010; Kaplan et al., 2011; Sanchez-Laorden et al., 2014; Jenkins and Sullivan, 2016). An association between long term BRAFi melanoma therapy and paradoxical oncogenesis has been established, and it has been shown that BRAFi therapy can accelerate pre-existing RAS-mutant malignancies including NRAS-mutant leukemia in melanoma patients (Callahan et al., 2012; Gibney et al., 2013). Likewise, the common clinical emergence of cutaneous squamous cell carcinoma observable in BRAFi-treated melanoma patients has been attributed to paradoxical activation of MAPK signaling confined to keratinocytes with pre-existing activational RAS-mutations (HRAS or NRAS)(Su et al., 2012).
Here, we have performed transcriptomic profiling (using NanoString nCounter™ and pathway-focused RT2-Profiler™ qPCR array approaches) and phenotypic analysis comparing BRAFi-sensitive A375-BRAFV600E/NRASQ61 and BRAFi-resistant A375-BRAFV600E/NRASQ61K melanoma cells, an accepted genetic model of BRAFi resistance. We demonstrate that BRAFi-treatment selectively targets BRAFV600E/NRASQ61K cells with induction of epithelial-to-mesenchymal transition (EMT) gene expression, accompanied by corresponding phenotypic changes and paradoxical enhancement of tumor growth and metastasis as assessed in a bioluminescent murine melanoma model.
RESULTS
NanoString nCounter™ analysis identifies pronounced gene expression changes impacting ‘EMT’ and ‘EMT to metastasis’ pathways in a genetic model of NRAS-driven BRAFi-resistance
First, NanoString nCounter™ analysis (using the ‘PanCancer Progression’ panel) compared gene expression between BRAFi-resistant (A375-BRAFV600E/NRASQ61K) and BRAFi-sensitive (A375-BRAFV600E/ NRASQ61) malignant melanoma cells (Figure 1). A heatmap depicting normalized expression data (740 genes; z-score range: −3 to 3) was generated via unsupervised hierarchical clustering (Figure 1a). Among all genes displaying statistically significant expression changes (fold change ≥ 4) as a function of NRAS-genotype, twenty-three are depicted based on the results of subsequent pathway score analysis [heatmap (Figure 1b); quantitative summary (Supplementary Table 1); nCounter™ pathway score analysis (Fig. 1c)].
Figure 1. NanoString nCounter™ profiling of isogenic malignant melanoma cells identifies pronounced gene expression changes impacting ‘EMT’ and ‘EMT to metastasis’ pathways in a genetic model of BRAFi-resistance.
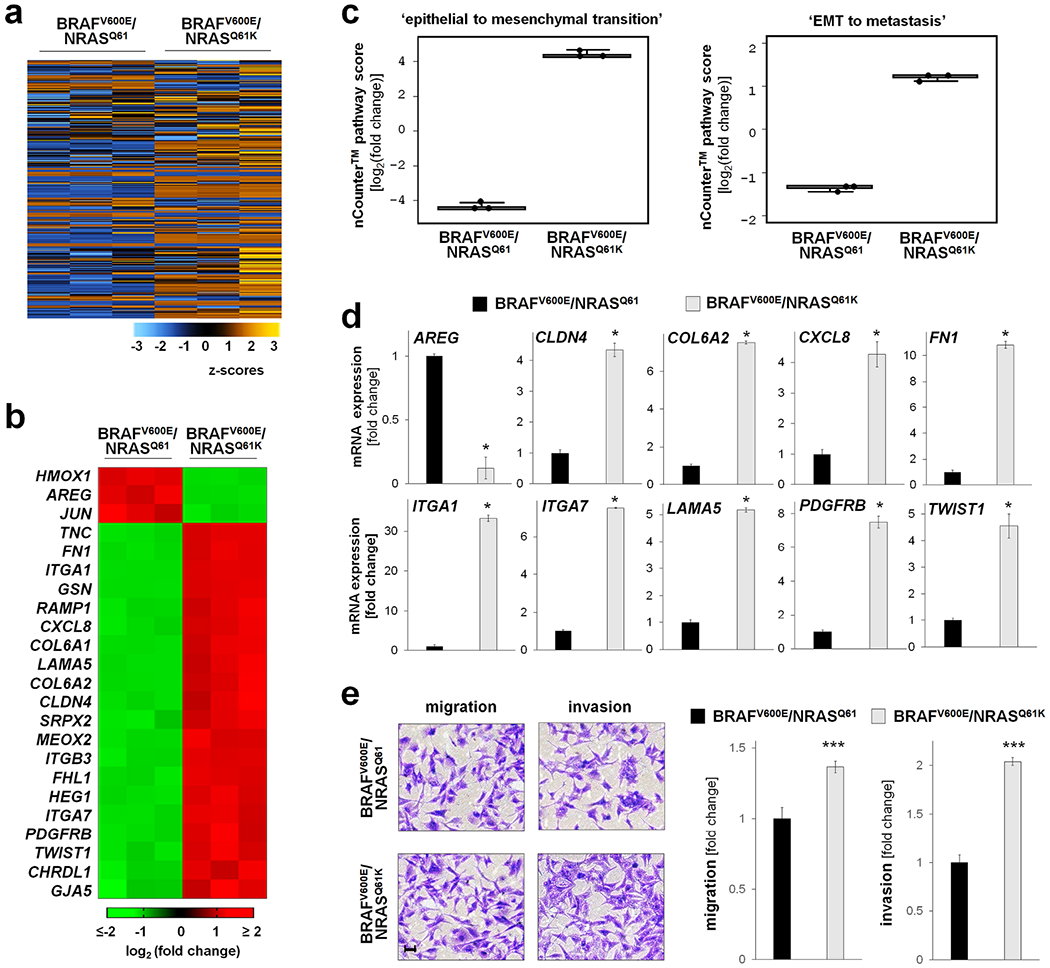
NanoString nCounter™ analysis (PanCancer Progression Panel) comparing gene expression between A375-BRAFV600E/NRASQ61K and A375-BRAFV600E/NRASQ61 cells. (a) Heatmap of normalized expression data (740 genes; z-score range: −3 to 3) generated via unsupervised hierarchical clustering; orange: high expression, blue: low expression. (b) Heatmap depicting statistically significant expression changes (fold change ≥ 4; p < 0.05; upregulated: red; downregulated: green). (c) NanoString nCounter™ gene expression ‘pathway score’ analysis as a function of NRAS-genotype identifying NRASQ61K-responsive expression networks (box plot depiction). (d) NanoString nCounter™ expression analysis of individual EMT- and metastasis-related genes (fold change ≥ 2; *p < 0.05). (e) Migration and invasion as a function of NRASQ61K-status; left panel: representative images after crystal violet staining (scale bar: 10 μm); right panels: quantitative analysis (***p < 0.001).
Next, using nCounter™ Advanced Analysis software, ‘pathway score’ profiling identified two gene expression networks characterized by differential expression patterns as a function of genotype (BRAFV600E/NRASQ61K cells versus BRAFV600E/NRASQ61): (i) ‘epithelial to mesenchymal transition’ and (ii) ‘EMT to metastasis’ (Figure 1c). NanoString nCounter™ single gene expression analysis (fold change ≥ 4; p < 0.05) identified modulation of ten genes encoding EMT- and metastasis-related transcription factors (TWIST1), extracellular matrix components (CLDN4, COL6A2, FN1, ITGA1, ITGA7, LAMA5), and components of inflammatory and growth factor signaling cascades (AREG, CXCL8, PDGFRB), all of which displayed expression changes in BRAFV600E/NRASQ61K cells consistent with EMT-promotion known to occur during melanomagenesis and progression of metastatic melanoma (Figure 1d) (Singh et al., 2010; Girotti et al., 2011; Oikawa et al., 2011; Shi et al., 2011; Li et al., 2015; Pearlman et al., 2017; Tang et al., 2020; Pommer et al., 2021).
Next, we performed a phenotypic characterization of EMT-related cell functions, assessing migration and invasion properties as a function of genotype (Figure 1e). A moderate (approximately 30%) increase of migratory activity was observed in cultured BRAFV600E/NRASQ61K as compared to isogenic BRAFV600E/NRASQ61 cells (measured in a transwell assay using uncoated Boyden chambers). Likewise, a pronounced enhancement of invasive properties (assessed in Matrigel-coated Boyden chambers) was observed as a function of genotype, with BRAFV600E/NRASQ61K displaying a two-fold increase as compared to BRAFV600E/NRASQ61 cells.
EMT-focused RT2 Profiler qPCR array analysis reveals BRAFi-induced exacerbation of gene expression promoting EMT in BRAFV600E/NRASQ61K melanoma cells with opposing downregulation in the BRAFV600E/NRASQ61-genotype
Guided by NanoString nCounter™ pathway identification (Figure 1), the potential impact of BRAFi-treatment (vemurafenib, VEM) on EMT-directed gene expression was examined as a function of NRAS-genotype. First, EMT-focused RT2 Profiler™ PCR array analysis compared BRAFi-treated BRAFV600E/NRASQ61 relative to untreated BRAFV600E/NRASQ61 cells [Figure 2a (volcano plot); Figure 2c (quantitative summary)]. Next, differential expression analysis compared BRAFi-treated BRAFV600E/NRASQ61K with untreated BRAFV600E/NRASQ61K (Figure 2b and c). Strikingly, BRAFi-induced upregulation of genes promoting EMT was detectable exclusively in BRAFV600E/NRASQ61K cells [CTNNB1, MMP2, PDGFRB, RAC1, SPARC, STAT3, TGFB2, TMEM132A, ZEB1, ZEB2 (≤ 87-fold; p < 0.05)] (Figure 2c and d). In contrast, in the isogenic variant (BRAFV600E/NRASQ61), opposing downregulation of EMT-driver genes was observed [AHNAK, CAV2, CDH2, EGFR, FN1, FGFBP1, IGFBP4, ITGB1, JAG1, KRT7, MYC, PLEK2, SERPINE1, SMAD2, TIMP1, TMEFF1, VIM, WNT5A (≤ 17-fold; p < 0.05)] with concomitant upregulation of epithelial markers (KRT19, OCLN). Remarkably, most of the genes responsive to BRAFi-treatment as a function of NRAS-genotype are known to be involved in melanoma EMT (Robert et al., 2006; Shi et al., 2011; Li et al., 2015; Pearlman et al., 2017; Tang et al., 2020).
Figure 2. BRAFi-treatment exacerbates upregulation of gene expression promoting EMT in BRAFV600E/NRASQ61K melanoma cells while attenuating expression in the BRAFV600E/NRASQ61-genotype as analyzed by EMT-RT2 Profiler™ qPCR array.
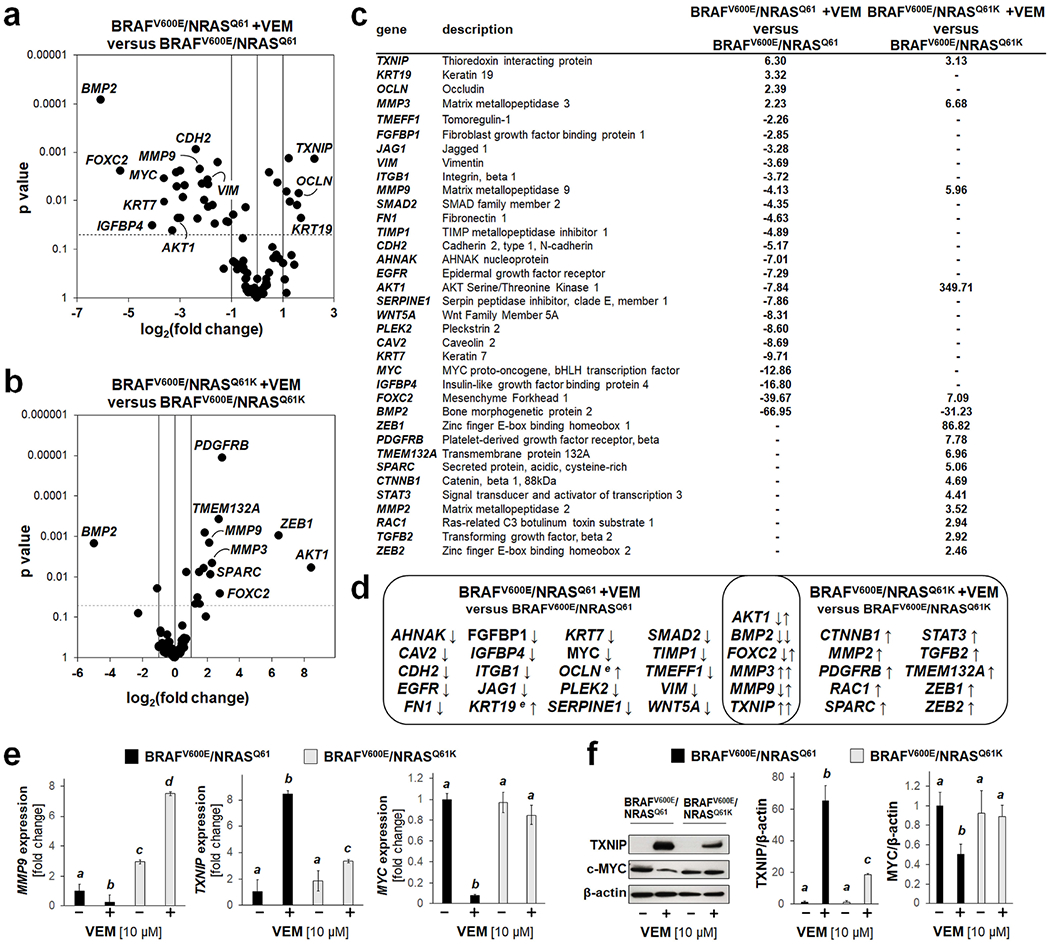
(a) RT2 Profiler™ PCR array analysis of EMT-related gene expression (VEM-treated BRAFV600/NRASQ61 relative to untreated BRAFV600E/NRASQ61). Volcano plot: differential gene expression (cut-off criteria: fold change ≥ 2; p < 0.05). (b) Expression analysis comparing VEM-treated BRAFV600E/NRASQ61K with untreated BRAFV600E/NRASQ61K cells. (c) Quantitative summary of VEM-induced expression changes comparing isogenic cell lines (cut-off criteria: fold change ≥ 2; p value < 0.05). (d) Venn diagram comparing VEM-induced expression changes. Arrows indicate up- or downregulated gene expression. For genes shared between genotypes (overlap), VEM-induced alterations are displayed as follows: BRAFV600E/NRASQ61 (left arrow); BRAFV600E/NRASQ61K (right arrow). (e) Effects of VEM on gene expression by independent RT-qPCR. (f) Immunoblot analysis (TXNIP, MYC) in response to VEM; bar graph: densitometric quantification (β-actin control).
Venn diagram depiction comparing BRAFi-induced expression changes between BRAFV600E/NRASQ61 and BRAFV600E/NRASQ61K cells indicated that six genes were responsive to BRAFi-induced modulation in both genotypes, displaying either an opposing regulation (AKT1, FOXC2, MMP9) or the same direction of expression changes (BMP2, MMP3, TXNIP) (Figure 2d) (Hargadon et al., 2019; Jandova et al., 2020).
Consistent with previous research on BRAFi-induced upregulation of the tumor suppressor TXNIP observed together with downregulation of oncogenic MYC in BRAFV600E/NRASQ61 cells (Parmenter et al., 2014), genotype-dependent expression of these EMT-related genes (including MMP9) was examined by independent RT-qPCR and immunoblot analysis (as a function of genotype) (Figure 2e and f). Indeed, dramatic BRAFi-induced TXNIP upregulation (at both the mRNA and protein levels) was observable in BRAFV600E/NRASQ61 cells, whereas only a minor increase occurred in the BRAFi-treated BRAFV600E/NRASQ61K-variant; likewise, BRAFi-treatment caused pronounced MYC downregulation in the BRAFV600E/NRASQ61 but not isogenic BRAFV600E/NRASQ61K cells
BRAFi-treatment enhances the migratory and invasive phenotype of BRAFV600E/NRASQ61K melanoma cells while causing opposing effects in the isogenic BRAFV600E/NRASQ61-genotype
After establishing the genotype-specific occurrence of BRAFi-induced EMT-related expression changes, isogenic melanoma lines were profiled for BRAFi-modulation of cellular viability, proliferation, and colony formation (Figure 3a–c). Viability remained unaffected by BRAFi irrespective of genotype (Figure 3a). Strikingly, BRAFi-treatment impacted proliferation in opposing ways as a function of genotype. Specifically, proliferation of BRAFV600E/NRASQ61 was inhibited in response to BRAFi-treatment, whereas BRAFV600E/NRASQ61K cells displayed the opposing phenotype characterized by BRAFi-induced increase in proliferation (Figure 3b), a genotype-specific effect also observed with colony formation (Figure 3c).
Figure 3. BRAFi-treatment enhances proliferation and invasiveness of BRAFV600E/NRASQ61K melanoma cells with opposing effects in the BRAFV600E/NRASQ61 isogenic variant.

(a) Cellular viability in response to VEM (≤ 40 μM, 72 h continuous exposure) as monitored by flow cytometry (annexin V-PI staining). (b) Cellular proliferation in response to VEM (≤ 40 μM, 72 h continuous exposure). (c) Colony formation as a function of treatment and genotype; representative images with quantitative analysis (scale bar: 200 μm). (d) Migration; representative images with quantitative analysis (scale bar: 10 μm). (e) Invasion through Matrigel-coated Boyden chambers; representative images with quantitative analysis (scale bar: 10 μm). (f-g) Immunoblot analysis of (f) ERK-phosphorylation (over total ERK1/2) or (g) AKT-phosphorylation (Ser473; over total ART) in response to VEM (exposure time: 0-90 min) as a function of genotype with densitometric quantification.
Next, guided by transcriptomic profiling of BRAFi-induced expression changes impacting EMT (Figure 2), we performed phenotypic EMT-related characterization assessing migration and invasion properties as a function of NRAS- genotype in cells exposed to BRAFi (Figure 3d and e). Strikingly, BRAFi-attenuation of migratory properties observable in BRAFV600E/NRASQ61 cells was contrasted by increased BRAFV600E/NRASQ61K migration (Figure 3d). Likewise, BRAFi-modulation of invasive properties (in Matrigel-coated Boyden chambers) followed a similar trend, with BRAFV600E/NRASQ61K displaying an almost three-fold BRAFi-induced increase (Figure 3e).
Next, differential effects of BRAFi-treatment on AKT- and ERK1/2- signaling as a function of NRAS-genotype were explored, inspired by earlier studies examining signaling in BRAF- or NRAS-driven melanoma (Figure 3f and g) (Davies et al., 2009; Kaplan et al., 2011; Lidsky et al., 2014). Immunoblot analysis revealed pronounced inhibition of ERK1/2-phosphorylation in response to BRAFi-treatment in BRAFV600E/NRASQ61 cells, whereas BRAFV600E/NRASQ61K cells displayed a diminished sensitivity to BRAFi-induced blockade of p-ERK1/2. Strikingly, BRAFi-treatment upregulated AKT-phosphorylation (p-AKT; Ser473) in BRAFV600E/NRASQ61K cells, whearas p-AKT was almost undetectable in BRAFi-treated BRAFV600E/NRASQ61 cells.
In a bioluminescent SCID mouse model of human malignant melanoma, BRAFi-treatment enhances BRAFV600E/NRASQ61K lung metastasis and tumor growth while displaying inhibitory effects in the BRAFV600E/NRASQ61 isogenic variant
Next, the impact of BRAFi-treatment on tumor metastasis and growth was examined in a bioluminescent SCID mouse model comparing BRAFV600E/NRASQ61K and BRAFV600E/NRASQ61 melanoma cells (Figure 4a). In BRAFV600E/NRASQ61K-recipient mice, BRAFi-treatment caused a pronounced attenuation of lung tumor burden (by more than 80% as quantified by tumor bioluminescence versus carrier control); in contrast, tumor burden increased approximately fivefold in BRAFi-treated BRAFV600E/NRASQ61K-recipient mice over control mice receiving carrier only (Figure 4b). Visual examination followed by immunohistochemical analysis of lung tissue indicated that no metastases were present in BRAFi-treated BRAFV600E/NRASQ61-recipient mice, whereas BRAFi-treated BRAFV600E/NRASQ61K-recipient mice displayed pronounced metastatic tumor burden that exceeded that of BRAFV600E/NRASQ61K-recipient mice receiving carrier only (Figure 4c–e). In mock-treated recipient mice, immunohistochemical (IHC) analysis indicated an equal presence of PCNA-positive cells in lung metastasis specimens irrespective of genotype. However, consistent with BRAFi-enhancement of proliferation in cultured BRAFV600E/NRASQ61K cells (Figure 3b and c), in response to BRAFi-treatment, an enhanced proportion of PCNA-positive cells was observed only in BRAFV600E/NRASQ61K-recipent mice, whereas the complete absence of metastases occurred in BRAFV600E/NRASQ61-recipents (Figure 4d). MMP9-IHC staining (H-score) was of equal intensity among tumor specimens irrespective of NRAS-genotype; however, in contrast to BRAFi-induced MMP9 upregulation observed in the corresponding in vitro model (Figure 2e), BRAFi-treatment did not increase MMP9 H-score in tumors of BRAFV600E/NRASQ61K-recipient mice.
Figure 4. BRAFi-treatment enhances lung metastasis and tumor growth in a bioluminescent SCID mouse model of NRAS-driven BRAFi-resistance.
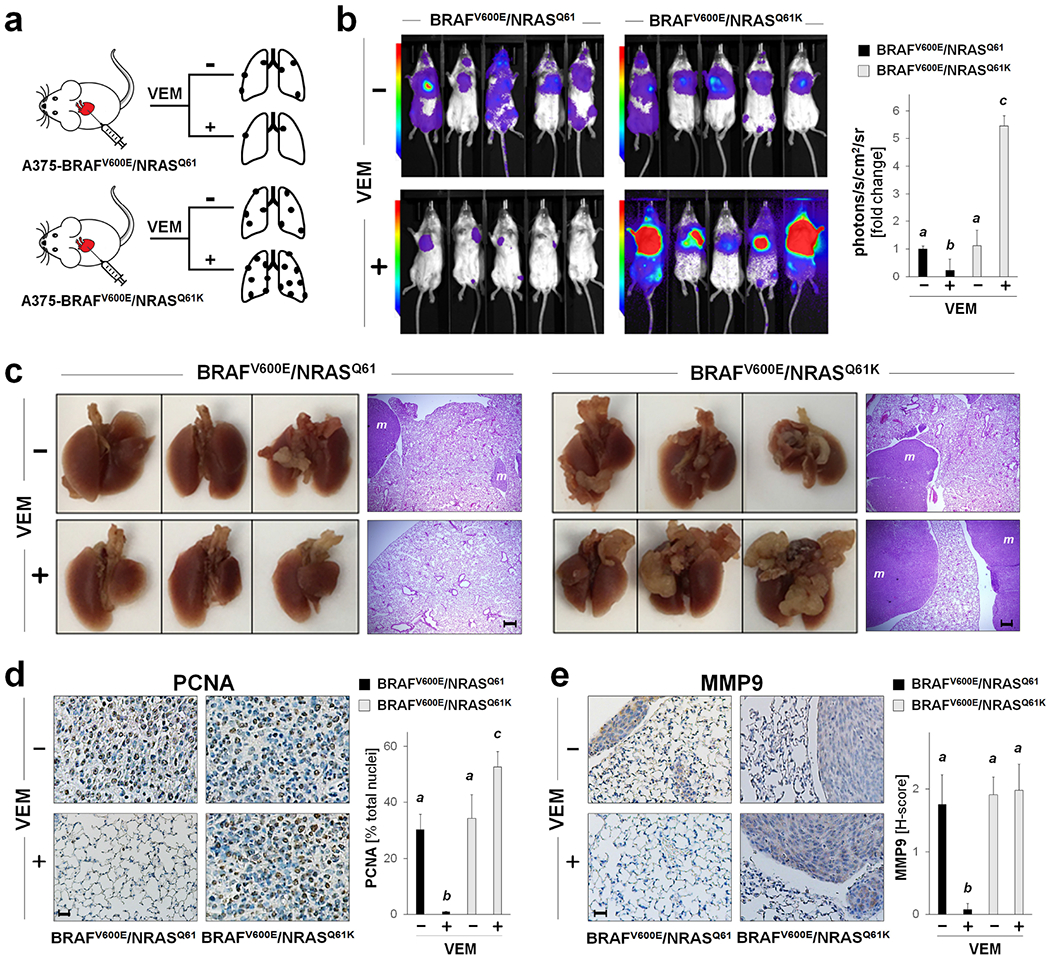
(a) Experimental scheme: Luciferase A375 isogenic melanoma cells (BRAFV600E/NRASQ61K versus BRAFV600E/NRASQ61) were injected intracardially (n = 10 per group). After pair-matching, lung metastasis and tumor growth were monitored by bioluminescent image analysis over a 14 d period. Starting at time of pair matching (d 0) until end of experiment (d 14), mice received VEM-treatment (q.d.; in corn oil, via gavage) or carrier only. (b) Bioluminescent tumor imaging (d 14; representative images displaying 5 mice per group with quantitative image analysis. (c) Lung pathology (photography); exemplary lung specimens (3 per group; d 14) with metastases (scale bar: 3 mm) with H&E-stained tumor cross-sections (‘m’ indicates metastasis; scale bar: 200 μm). (d-e) Tumor cross-sections stained for PCNA [scale bar: 25 μm (d)] and MMP-9 [scale bar: 25 μm (e)] with summary quantification.
DISCUSSION
The role of mutated NRAS as a major driver of non-BRAF mutant melanoma is well established (Vanni et al., 2020). Moreover, an additional critical role of mutated NRAS in conferring BRAFi-resistance in BRAFV600E-driven melanoma has been established (Johnson et al., 2015). Importantly, BRAFi-treatment has been shown before to enhance proliferation and survival of human BRAF-wildtype/NRAS-mutant melanoma cells attributed to transactivation of BRAF signaling upstream of MAPK (Kaplan et al., 2011; Jenkins and Sullivan, 2016; Jenkins and Fisher, 2021). Moreover, it is well documented that BRAFi-treatment induces metastasis in KRASG12D mouse melanoma and enhances invasiveness of RAS mutated (NRAS or KRAS) mouse and human melanoma cells with BRAF-wildtype (Sanchez-Laorden et al., 2014). Strikingly, it has been shown before that BRAFi-therapy can accelerate pre-existing RAS-mutant malignancies (including NRAS-mutant leukemia and HRAS-mutant squamous cell carcinoma) in melanoma patients (Callahan et al., 2012; Fedorenko et al., 2013; Gibney et al., 2013), and BRAFi-treatment is known to confer proliferation and survival advantage in NRAS-mutant/BRAF-wildtype melanoma (Kaplan et al., 2011). However, little is known about the effects of sustained BRAFi-exposure on BRAFi-resistant NRAS-mutant/ BRAFV600E melanoma cells.
Here, we have examined the effects of BRAFi-treatment on dual BRAF/NRAS-mutated BRAFi-resistant melanoma, employing a genetic model of NRASQ61K-driven BRAFi-resistance in isogenic BRAFV600E-driven melanoma cell lines. Consistent with prior research that documented the increased metastatic potential of NRAS-driven melanoma (Eskandarpour et al., 2009; Fedorenko et al., 2013), our comprehensive NanoString™-based expression profiling substantiated the differential upregulation of EMT- and metastasis-related gene expression networks as a crucial hallmark associated with the NRASQ61K-genotype (Figure 1). To further explore the differential expression of EMT-related genes as a function of genotype and BRAFi-exposure, we employed focused PCR array profiling demonstrating that BRAFi paradoxically enhanced EMT-related gene expression in BRAFV600E/NRASQ61K melanoma cells while displaying opposing effects in the BRAFV600E/NRASQ61-genotype (Figure 2).
The role of EMT in melanoma progression is well established, and mechanisms regulating EMT in melanomagenesis are now considered valid therapeutic targets (Li et al., 2015; Pearlman et al., 2017; Tang et al., 2020). Moreover, BRAFi-resistance has been associated with the emergence of EMT-directed rewiring of transcription factors, and BRAFi-sensitivity is restored in BRAFi-resistant melanoma cells by targeting EMT-related extracellular matrix remodeling (Cohen-Solal et al., 2018; Marusak et al., 2020; Tabolacci et al., 2021). BRAFi-induced enhancement of invasive behavior of BRAFV600E melanoma cells displaying acquired resistance (elicited by long term culture) has been described before, but occurrence of NRAS mutation was either excluded (based on whole exosome sequencing) or remained unexplored (Caporali et al., 2016; Molnar et al., 2019).
NRAS activational mutations are established key mediators of acquired BRAFi-resistance in BRAF-driven melanoma compromising therapeutic efficacy (Nazarian et al., 2010; Sanchez-Laorden et al., 2014; Luebker and Koepsell, 2019), and EMT-upregulation has been observed in the general context of BRAFi-resistance without detailed exploration of NRAS-involvement (Caporali et al., 2016; Cohen-Solal et al., 2018; Molnar et al., 2019; Marusak et al., 2020; Tabolacci et al., 2021). Our study employs a stringent genetic model of NRAS-driven BRAFi-resistant (BRAFV600E/NRASQ61K) melanoma demonstrating BRAFi-enhancement of EMT gene expression, invasiveness, and lung metastasis imposed by BRAFV600E/NRASQ61K cells (while inactivating the BRAFV600E/ NRASQ61-genotype), a preclinical finding relevant to patients developing NRAS-driven resistance to BRAFi-treatment. However, the specific molecular mechanism underlying BRAFi-enhancement of BRAFV600E/NRASQ61K melanoma remains to be elucidated since it might differ from the mechanism already established in BRAF-wildtype/NRAS-mutant tumors (Kaplan et al., 2011; Jenkins and Sullivan, 2016; Jenkins and Fisher, 2021).
Future research must examine if BRAFi-enhancement of BRAFV600E/NRASQ61K melanoma is representative of other NRAS mutations conferring BRAFi-resistance in malignant melanoma (Grill and Larue, 2016; Dean, 2017; Tanda et al., 2020). Also, due to the pharmacological diversity of clinically relevant BRAFi-therapeutics it remains to be explored if BRAFV600E/NRASQ61K-directed effects (observed here with VEM) represent a BRAFi group effect that applies to all members of this drug class (Menzies et al., 2013; Luebker and Koepsell, 2019). Importantly, in spite of the clinical availability and standard of care status of BRAFi-based combination therapeutics [including FDA-approved MEK inhibitors (MEKi: tramatenib, binimetinib, cobimetinib) and triple combinations with immune checkpoint inhibitors (such as atezolizumab plus vemurafenib/cobimetinib)] (Larkin et al., 2014; Dummer et al., 2018; Sullivan et al., 2019), the occurrence of drug resistance and paradoxical oncogenesis limits treatment success of current combination therapeutics (Arozarena and Wellbrock, 2019; Hartman et al., 2020; Subbiah et al., 2020). Thus, therapy-induced emergence of NRAS-based resistance as well as a potential for BRAFi-enhancement of BRAFV600E/NRASQ61K melanoma remain to be characterized for individual BRAFi/MEKi combination therapeutics, and it is expected that a molecular profiling strategy as pursued in this vemurafenib-directed preclinical study might inform future clinical decisions (Subbiah et al., 2020). Lastly, it will also be interesting to explore the therapeutic implications of our findings in the context of the potential occurrence of double-mutant BRAF/NRAS treatment-naive melanoma reported sporadically in the literature (should this co-occurrence be confirmed at the single cell level) (Chiappetta et al., 2015; Heppt et al., 2017; Kumar et al., 2019).
Taken together, these preclinical data document the BRAFi-driven enhancement of tumorigenesis and metastasis in BRAFi-resistant human BRAFV600E/NRASQ61K melanoma, a finding with potential clinical implications for melanoma patients with NRAS-driven BRAFi-resistant tumors receiving BRAFi-treatment.
MATERIALS AND METHODS
Chemicals
Vemurafenib (PLX4032) was obtained from MedChemExpress (HY-12057; MCE; Monmouth Junction, NJ). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO).
Cell culture
Parental A375-BRAFV600E/ NRASQ61 human malignant melanoma cells were CRISPR/Cas9 gene-edited generating an isogenic variant containing both BRAFV600E and mutated NRASQ61K, followed by transduction using a lentiviral vector encoding firefly luciferase (LUC2) under control of the EF-1alpha promoter. BRAFV600E/NRASQ61 (ATCC® CRL-1619-LUC2™, ATCC, Manassas, VA) and BRAFV600E/NRASQ61K (ATCC® CRL-1619IG-2-LUC2™) cells were maintained (DMEM; 10% FBS) as published (Jandova et al., 2021a).
NanoString nCounter™ gene expression analysis
Total RNA from both isogenic variants of A375 cells was isolated (RNeasy Mini kit, Qiagen, Valencia, CA) followed by NanoString nCounter™ analysis (PanCancer Progression Panel, NanoString Technologies, Seattle, WA), monitoring expression of 770 genes (Jandova and Wondrak, 2021). A set of 30 housekeeping genes was used for normalization; pathway score analysis employed nCounter™ Advanced Analysis software (version 2.0.115). For heatmap depiction, a ‘z-score’ for a specific gene indicates the number of standard deviations away from the mean of expression in the reference sample. Numerical ‘pathway score’ represents average fold expression change (log2 scale) for all genes associated with the specific pathway (Jandova and Wondrak, 2021).
Boyden chamber transwell migration and invasion
Uncoated (assessing migration) or Matrigel™-coated (assessing invasion) transwell-inserts (BD Biosciences, San Jose, CA) were used evaluating migration and invasion of isogenic variants in the presence/ absence of vemurafenib (10 μM, 16 h) (Jandova et al., 2020; Jandova and Wondrak, 2021).
Human EMT RT2 Profiler™ PCR Expression Array analysis
The RT2 PCR expression array technology was used as published (Davis et al., 2015; Park et al., 2015; Jandova et al., 2020). Total RNA was isolated and reverse transcribed (RT2 First Strand kit, Qiagen, Valencia, CA), followed by human EMT RT-qPCR array analysis (PAHS090ZA, Qiagen) covering 84 EMT-related and 5 housekeeping genes (ACTB, B2M, GAPDH, HPRT1, RPLP0).
Individual gene RT-qPCR
Human primer/probes [TXNIP (Hs_01006900_m1), MYC (Hs_00153408_m1), MMP9 (Hs_00234579_m1), and housekeeping genes RSP18 (Hs_01375212_g1)] were used to quantify the amplification of target genes by RT-qPCR using the ABI7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) (Jandova et al., 2020; Jandova and Wondrak, 2021).
Immunoblot analysis
Analysis was performed using primary antibodies as published: TXNIP (NBP2-75692, Novus Biologicals, Centennial, CO); c-MYC (5605), p-ERK1/2 (4370), ERK1/2 (4695), p-AKT (4060), ART (4691, Cell Signaling, Danvers, MA); β-actin (A5441, Sigma Aldrich). Image Studio™ Lite quantification software (LI-COR Biosciences, Lincoln, NE) was used for densitometric analysis (Perer et al., 2020; Jandova and Wondrak, 2021).
Cell proliferation
Cells were seeded (1,000 / 35mm dish) in normal growth medium with or without vemurafenib (≤ 40 μM; 72 h), and proliferation was quantified using a Cell Viability Analyzer (Beckman Coulter, Fullerton, CA) (Park et al., 2015).
Colony formation assay
Cells (500 / well) were seeded in 6-well plates in growth medium with or without vemurafenib (10 μM, 14 d). Colonies were fixed and stained (0.1% crystal violet, 20% methanol). Individual colonies (≥ 70 μm) were counted by light microscopy (Jandova and Wondrak, 2021).
Cell viability
Cell viability was analyzed by flow cytometry (annexin V-FITC/PI double staining) using a commercial apoptosis detection kit (APO-AF, Sigma, St. Louis, MO) (Davis et al., 2015).
Bioluminescent melanoma metastasis model in SCID mice
Bioluminescent monitoring of melanoma lung metastasis was performed as published (Jandova et al., 2021a). Female SCID mice [UA Cancer Center in-house colony; age: 9 weeks; average weight: 20 g] underwent intracardial injection of 5×105 A375 melanoma cells (100 μL HBSS; BRAFV600E/NRASQ61-Luc2 and BRAFV600E/NRASQ61K-Luc2). Vemurafenib (1 mg/ 200 μL corn oil/mouse) administration (via gavage) started on the day of cell injection [5 d per week (Mon-Fri regimen), 14 d total duration] with administration of com oil serving as carrier control (5 mice per treatment group; a total of 10 per genotype). Lung metastasis and tumor growth were monitored by quantitative bioluminescent imaging, and lung tissue was harvested at the end of the experiment. This study was performed in accordance with the recommendations of the National Institutes of Health (University of Arizona Institutional Animal Care and Use Committee; protocol number: IACUC 17–298).
Immunohistochemistry
Lung tissue was analyzed for detection of PCNA (2586, Cell Signaling) and MMP-9 (13667, Cell Signaling) as published (Perer et al., 2020; Jandova and Wondrak, 2021). Nuclear staining was performed using Hematoxylin QS counterstain (H3404; Vector Laboratories, Burlingame, CA). Images were captured using an Olympus BX50 and Spot (Model 2.3.0) camera. Quantitative tissue scoring (H-score) was performed as published (Bair et al., 2010).
Statistical analysis
Statistical analysis was performed as published (Jandova et al., 2021b). All data from in vitro experiments represent at least three independent biological replicates (n ≥ 3). Unless specified otherwise, ANOVA with Tukey’s posthoc test was used for data set analysis (Prism 8.4.3 software, Prism Software Corp., Irvine, CA); in bar graphs comparing more than two groups, means without a common letter differ (p < 0.05). NanoString nCounter™ gene expression analysis was performed using the nSolver analysis software (version 4.0 containing nCounter™ Advanced Analysis software version 2.0.115) for p-value adjustment (Benjamini-Yekutieli false discovery rate) (Jandova and Wondrak, 2021). RT2 Profiler™ PCR gene expression array data were analyzed using two-tailed Student’s t-test. Nonparametric Mann–Whitney test was used to analyze data from murine experiments. Differences with p < 0.05 were considered statistically significant.
Data availability statement
All gene array expression data generated in this study are being disclosed and presented in the manuscript. NanoString nCounter™-derived large data sets were deposited in the NCBI gene expression omnibus (GEO) repository database (public on September 23, 2021): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184633 According to GEO accession display, data were generated using the GPL30655 nCounter PanCancer Progression panel: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL30655
Supplementary Material
ACKNOWLEDGMENTS
Preliminary data from this research were accepted for presentation at the following meetings: Annual Meeting of the Society for Investigative Dermatology (SID), May 3-8, 2021; Third RAS Initiative Symposium, June 8-10, 2021, Frederick National Laboratory for Cancer Research (NCI). This study was supported in part by grants from the National Institutes of Health (1R01CA229418; ES007091; ES006694; UA Cancer Center Support Grant CA023074).
Abbreviations:
- BRAFi
BRAF inhibitor
- EMT
epithelial-to-mesenchymal transition
- VEM
vemurafenib
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no relevant conflicts of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper.
REFERENCES
- Arozarena I, Wellbrock C, Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer 2019;19:377–91. [DOI] [PubMed] [Google Scholar]
- Bair WB 3rd, Cabello CM, Uchida K, Bause AS, Wondrak GT, GLO1 overexpression in human malignant melanoma. Melanoma Res 2010;20:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med 2012;367:2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporali S, Alvino E, Lacal PM, Levati L, Giurato G, Memoli D, et al. Targeting the PI3K/AKT/mTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int J Oncol 2016;49:1164–74. [DOI] [PubMed] [Google Scholar]
- Chiappetta C, Proietti I, Soccodato V, Puggioni C, Zaralli R, Pacini L, et al. BRAF and NRAS mutations are heterogeneous and not mutually exclusive in nodular melanoma. Appl Immunohistochem Mol Morphol 2015;23:172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Solal KA, Kaufman HL, Lasfar A, Transcription factors as critical players in melanoma invasiveness, drug resistance, and opportunities for therapeutic drug development. Pigment Cell Melanoma Res 2018;31:241–52. [DOI] [PubMed] [Google Scholar]
- Curti BD, Faries MB, Recent Advances in the Treatment of Melanoma. N Engl J Med 2021;384:2229–40. [DOI] [PubMed] [Google Scholar]
- Davies MA, Stemke-Hale K, Lin E, Tellez C, Deng W, Gopal YN, et al. Integrated Molecular and Clinical Analysis of AKT Activation in Metastatic Melanoma. Clin Cancer Res 2009;15:7538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AL, Qiao S, Lesson JL, Rojo de la Vega M, Park SL, Seanez CM, et al. The quinone methide aurin is a heat shock response inducer that causes proteotoxic stress and Noxa-dependent apoptosis in malignant melanoma cells. J Biol Chem 2015;290:1623–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean L, Vemurafenib Therapy and BRAF and NRAS Genotype. In: Pratt VM, Scott SA, Pirmohamed M, et al. , editors. Medical Genetics Summaries. Bethesda (MD): NCBI (US); 2017; available from: https://www.ncbi.nlm.nih.gov/books/NBK447416. [PubMed] [Google Scholar]
- Dummer R, Ascierto PA, Gogas HJ, Arance A, Mandala M, Liszkay G, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2018;19:603–15. [DOI] [PubMed] [Google Scholar]
- Eskandarpour M, Huang F, Reeves KA, Clark E, Hansson J, Oncogenic NRAS has multiple effects on the malignant phenotype of human melanoma cells cultured in vitro. Int J Cancer 2009;124:16–26. [DOI] [PubMed] [Google Scholar]
- Fedorenko IV, Gibney GT, Smalley KS, NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene 2013;32:3009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS, Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol 2013;10:390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girotti MR, Fernandez M, Lopez JA, Camafeita E, Fernandez EA, Albar JP, et al. SPARC promotes cathepsin B-mediated melanoma invasiveness through a collagen I/alpha2beta1 integrin axis. J Invest Dermatol 2011;131:2438–47. [DOI] [PubMed] [Google Scholar]
- Grill C, Larue L, NRAS, NRAS, Which Mutation Is Fairest of Them All? J Invest Dermatol 2016;136:1936–38. [DOI] [PubMed] [Google Scholar]
- Hargadon KM, Gyorffy B, Strong EW, Tarnai BD, Thompson JC, Bushhouse DZ, et al. The FOXC2 Transcription Factor Promotes Melanoma Outgrowth and Regulates Expression of Genes Associated With Drug Resistance and Interferon Responsiveness. Cancer Genomics Proteomics 2019;16:491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ML, Sztiller-Sikorska M, Gajos-Michniewicz A, Czyz M, Dissecting Mechanisms of Melanoma Resistance to BRAF and MEK Inhibitors Revealed Genetic and Non-Genetic Patient- and Drug-Specific Alterations and Remarkable Phenotypic Plasticity. Cells 2020;9:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppt MV, Siepmann T, Engel J, Schubert-Fritschle G, Eckel R, Mirlach L, et al. Prognostic significance of BRAF and NRAS mutations in melanoma: a German study from routine care. BMC Cancer 2017;17:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandova J, Hua AB, Fimbres J, Wondrak GT, Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers (Basel) 2021a;13:605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandova J, Perer J, Hua A, Snell JA, Wondrak GT, Genetic Target Modulation Employing CRISPR/Cas9 Identifies Glyoxalase 1 as a Novel Molecular Determinant of Invasion and Metastasis in A375 Human Malignant Melanoma Cells In Vitro and In Vivo. Cancers (Basel) 2020;12:1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandova J, Snell J, Hua A, Dickinson S, Fimbres J, Wondrak GT, Topical hypochlorous acid (HOCl) blocks inflammatory gene expression and tumorigenic progression in UV-exposed SKH-1 high risk mouse skin. Redox Biol 2021b;45:102042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandova J, Wondrak GT, Genomic GLO1 deletion modulates TXNIP expression, glucose metabolism, and redox homeostasis while accelerating human A375 malignant melanoma tumor growth. Redox Biol 2021;39:101838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Fisher DE, Treatment of Advanced Melanoma in 2020 and Beyond. J Invest Dermatol 2021;141:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Sullivan RJ, NRAS mutant melanoma: an overview for the clinician for melanoma management. Melanoma Manag 2016;3:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 2015;51:2792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan FM, Shao Y, Mayberry MM, Aplin AE, Hyperactivation of MEK-ERK1/2 signaling and resistance to apoptosis induced by the oncogenic B-RAF inhibitor, PLX4720, in mutant N-RAS melanoma cells. Oncogene 2011;30:366–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Njauw CN, Reddy BY, Ji Z, Rajadurai A, Klebanov N, et al. Growth suppression by dual BRAF(V600E) and NRAS(Q61) oncogene expression is mediated by SPRY4 in melanoma. Oncogene 2019;38:3504–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76. [DOI] [PubMed] [Google Scholar]
- Li FZ, Dhillon AS, Anderson RL, McArthur G, Ferrao PT, Phenotype switching in melanoma: implications for progression and therapy. Front Oncol 2015;5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidsky M, Antoun G, Speicher P, Adams B, Turley R, Augustine C, et al. Mitogen-activated protein kinase (MAPK) hyperactivation and enhanced NRAS expression drive acquired vemurafenib resistance in V600E BRAF melanoma cells. J Biol Chem 2014;289:27714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebker SA, Koepsell SA, Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front Oncol 2019;9:268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusak C, Thakur V, Li Y, Freitas JT, Zmina PM, Thakur VS, et al. Targeting Extracellular Matrix Remodeling Restores BRAF Inhibitor Sensitivity in BRAFi-resistant Melanoma. Clin Cancer Res 2020;26:6039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Kefford RF, Long GV, Paradoxical oncogenesis: are all BRAF inhibitors equal? Pigment Cell Melanoma Res 2013;26:611–5. [DOI] [PubMed] [Google Scholar]
- Molnar E, Garay T, Donia M, Baranyi M, Rittler D, Berger W, et al. Long-Term Vemurafenib Exposure Induced Alterations of Cell Phenotypes in Melanoma: Increased Cell Migration and Its Association with EGFR Expression. Int J Mol Sci 2019;20:4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Couselo E, Adelantado EZ, Ortiz C, Garcia JS, Perez-Garcia J, NRAS-mutant melanoma: current challenges and future prospect. Onco Targets Ther 2017;10:3941–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010;468:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa Y, Hansson J, Sasaki T, Rousselle P, Domogatskaya A, Rodin S, et al. Melanoma cells produce multiple laminin isoforms and strongly migrate on alpha5 laminin(s) via several integrin receptors. Exp Cell Res 2011;317:1119–33. [DOI] [PubMed] [Google Scholar]
- Park SL, Justiniano R, Williams JD, Cabello CM, Qiao S, Wondrak GT, The Tryptophan-Derived Endogenous Aryl Hydrocarbon Receptor Ligand 6-Formylindolo[3,2-b]Carbazole Is a Nanomolar UVA Photosensitizer in Epidermal Keratinocytes. J Invest Dermatol 2015;135:1649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov 2014;4:423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman RL, Montes de Oca MK, Pal HC, Afaq F, Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett 2017;391:125–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perer J, Jandova J, Fimbres J, Jennings EQ, Galligan JJ, Hua A, et al. The sunless tanning agent dihydroxyacetone induces stress response gene expression and signaling in cultured human keratinocytes and reconstructed epidermis. Redox Biology 2020;36:101594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti C, Molla A, Vegetti C, Ferrone S, Anichini A, Sensi M Coexpression of NRASQ61R and BRAFV600E in human melanoma cells activates senescence and increases susceptibility to cell-mediated cytotoxicity. Cancer Res 2006;66:6503–11. [DOI] [PubMed] [Google Scholar]
- Pommer M, Kuphal S, Bosserhoff AK, Amphiregulin Regulates Melanocytic Senescence. Cells 2021;10:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert G, Gaggioli C, Bailet O, Chavey C, Abbe P, Aberdam E, et al. SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res 2006;66:7516–23. [DOI] [PubMed] [Google Scholar]
- Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal 2014;7:ra30. [DOI] [PubMed] [Google Scholar]
- Sensi M, Nicolini G, Petti C, Bersani I, Lozupone F, Molla A, et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006;25:3357–64. [DOI] [PubMed] [Google Scholar]
- Shi H, Kong X, Ribas A, Lo RS Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res 2011;71:5067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Singh AP, Sharma B, Owen LB, Singh RK, CXCL8 and its cognate receptors in melanoma progression and metastasis. Future Oncol 2010;6:111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012;366:207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah V, Baik C, Kirkwood JM, Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020;6:797–810. [DOI] [PubMed] [Google Scholar]
- Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med 2019;25:929–35. [DOI] [PubMed] [Google Scholar]
- Tabolacci C, Cordella M, Mariotti S, Rossi S, Senatore C, Lintas C, et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines 2021;9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanda ET, Vanni I, Boutros A, Andreotti V, Bruno W, Ghiorzo P, et al. Current State of Target Treatment in BRAF Mutated Melanoma. Front Mol Biosci 2020;7:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Durand S, Dalle S, Caramel J, EMT-Inducing Transcription Factors, Drivers of Melanoma Phenotype Switching, and Resistance to Treatment. Cancers (Basel) 2020;12:2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanni I, Tanda ET, Dalmasso B, Pastorino L, Andreotti V, Bruno W, et al. Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front Mol Biosci 2020;7:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Zhu B, Zhang T, Liu T, Chen S, Liu Y, et al. Pharmacological Targeting of STK19 Inhibits Oncogenic NRAS-Driven Melanomagenesis. Cell 2019;176:1113–27 e16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All gene array expression data generated in this study are being disclosed and presented in the manuscript. NanoString nCounter™-derived large data sets were deposited in the NCBI gene expression omnibus (GEO) repository database (public on September 23, 2021): https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184633 According to GEO accession display, data were generated using the GPL30655 nCounter PanCancer Progression panel: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL30655