

Abstract
Homozygous mutation in arteriovenous malformation (AVM) causative genes in a fraction of endothelial cells (ECs) causes brain AVMs (bAVMs). Heterozygous mutation of an AVM causative gene, endoglin, in bone marrow (BM) caused capillary dysplasia in mouse brain. We hypothesize that homozygous mutation of activin receptor-like kinase 1 (Alk1, another AVM causative gene) in BM derived ECs (BMDECs) is sufficient to cause bAVM in brain angiogenic region, Alk1− ECs can clonally expand in bAVM, and the burden of Alk1− ECs correlates with bAVM severity. The BMs of PdgfbiCreER;Alk12f/2f;Ai14 mice with EC-specific tamoxifeninducible Cre and Alk1 floxed alleles were transplanted into wild-type mice to analyze the role of BMDECs in bAVM development. PdgfbiCreER;Alk12f/2f;confetti+/− mice with Cre-regulated confetti transgene were used to study EC clonal expansion. BAVMs were induced by intra-brain injection of an adeno-associated viral vector expressing vascular endothelial growth factor followed with intra-peritoneal injection of tamoxifen. Wild-type mice transplanted with PdgfbiCreER;Alk12f/2f;Ai14 BM developed bAVMs after bAVM induction. Recombined BMDECs were detected in bAVMs. The presence of clusters of ECs expressing same confetti color in bAVMs suggests that Alk1− ECs have been clonally expanded. Increasing tamoxifen dose increased the number of Alk1− ECs and abnormal vessels in bAVMs of PdgfbiCreER;Alk12f/2f mice. Our data indicated that homozygous mutation of Alk1 in BMDECs is sufficient to cause bAVM development in brain angiogenic region; clonal expansion of Alk1− ECs provides a likely mechanism for how a fraction of mutant ECs causes bAVM; and the burden of Alk1− ECs correlates with AVM severity.
Keywords: arteriovenous malformation, Alk1, endothelial cells, clonal expansion, bone marrow derived endothelial cells
INTRODUCTION
Arteriovenous malformations (AVMs) are tangles of abnormal vessels that shunt blood directly from arteries to veins. Vessels in AVM have abnormal wall structure and are prone to rupture causing life-threatening hemorrhage[1]. Most AVM cases are sporadic [2]. There are a few rare inherited diseases that feature AVMs, such as hereditary hemorrhagic telangiectasia (HHT), an autosomal dominant disorder caused by mutations in endoglin (ENG), activin receptor-like kinase 1 (ALK1 also known as ACVLR1), and SMAD4 [3, 4]. HHT patients have a high prevalence of telangiectasias in the skin and gastrointestinal mucosa, and AVMs in multiple organs including the brain, lung, and liver. Although HHT causal genes are mostly identified, the mechanisms of AVM development are largely unknown [2].
We have established several HHT AVM mouse models that have the AVMs in multiple organs and in the brain angiogenic region through conditional deletion of Eng or Alk1 in adult mice, combined with brain focal angiogenic stimulation [5–8]. Using these models, we found that both mutation of Alk1 or Eng in the endothelial cells (ECs) and focal angiogenic stimulation are necessary for AVM development in the brain of adult mouse [6, 7, 9]. Mutation of Alk1 or Eng in ECs is also necessary for AVM development in other organs [10]. We also found that homozygous mutation of Alk1 or Eng in a fraction of somatic ECs [5, 11] is sufficient to induce brain AVMs (bAVMs) and arteriovenous (AV) shuts in the intestines of adult mice. Bone marrow (BM) derived cells have been detected in brain angiogenic region [12]. We have shown that transplantation of BM cells with heterozygous mutation of Eng (+/−) caused capillary dysplasia in brain angiogenic regions of wild-type (WT) mice [13], suggesting a potential contribution of BM-derived cells to the pathogenesis of vascular malformation. However, it is not clear how a small fraction of mutant ECs leads to AVM, and if mutation of AVM causative genes in BM-derived ECs (BMDECs) alone can cause AVM.
EC Eng mutation in neonates increases proliferation of both Eng− and Eng+ ECs in AVM vessels [14]. We found that proliferating Alk1− ECs preferentially cluster in bAVM vessels [7]. It spurs an idea that the small numbers of ECs with biallelic loss of ALK1 can clonally overpopulate. We used the R26R-confetti reporter [15] to determine if Alk1− ECs are undergoing clonal expansion in bAVMs. Confetti mice carry four fluorescent proteins –green fluorescent protein (GFP), red fluorescent protein (RFP), yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP). Tamoxifen (TM) treatment will trigger Cre expression which will activate the expression of 1 of the 4 confetti fluorescent genes in individual cells. The progenies of these cells will express the same fluorescent protein. Thus, this reporter provides a unique way to identify a cluster of cells that are derived from a single parental cell, demonstrating clonal expansion [16].
In this paper, we found that homozygous mutation of Alk1 in BMDECs alone is sufficient for bAVM development. We also found that Alk1− ECs were clonally expanded in bAVMs, and the number of Alk1− ECs was positively correlated with AVM phenotype severity.
MATERIALS AND METHODS
Animals
The protocol and experimental procedures for using laboratory animals were approved by the Institution of Animal Care and Use Committee (IACUC) at the University of California, San Francisco. Animal husbandry was provided by the staff of the IACUC, under the guidance of supervisors who are certified Animal Technologists, and by the staff of the Animal Core Facility. Veterinary care was provided by IACUC faculties and veterinary residents located on the Zuckerberg San Francisco General Hospital campus.
PdgfbiCreER;Alk12f/2f;Ai14+/− mice that express an EC-specific TM inducible Cre recombinase, have their Alk1 gene exons 4–6 floxed [17], and carry the Ai14 reporter were used as BM donors and in other experiments. C57BL/6J and C57BL/6-Tg(CAG-EGFP)10sb/J mice (the Jackson Laboratory, Bar Harbor, ME) were used as BM recipients and donors.
PdgfbiCreER;Alk12f/2f;confetti+/− mice that carry the R26R-confetti Cre-regulated transgene were used to test the clonal expansion of Alk1− ECs. R26R-confetti transgene (Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J, the Jackson Laboratory) has a loxP-flanked STOP cassette between the CAG promoter and the Confetti sequence. Confetti transgene contains two loxP-flanked dimers; one dimer has nuclear-localized GFP and reversed cytoplasmic YFP; the other dimer has cytoplasmic RFP and a reversed membrane-tethered CFP. PdgfbiCreER;Alk12f/2f;confetti+/− mice were treated with a single intra-peritoneal (i.p.) injection of TM (2.5 mg/25g of body weight) to activate Cre expression in ECs, which deleted Alk1 gene at the same time activated the expression of 1 of the 4 confetti colors in individual ECs. The individual fluorescent color is stably propagated within the progeny of each ECs [15].
The PdgfbiCreER;Alk12f/2f;Ai14+/− mice were also treated with different doses of TM through single i.p. injections to induce different levels of Alk1 gene deletion and Ai14 expression in ECs.
Statistical Analyses
All quantifications were done by at least two researchers who were blinded to the treatment groups on images that were coded. Data were analyzed using GraphPad Prism 6 by one-way ANOVA followed with multiple comparisons and Tukey’s correction. Survival rate was analyzed using log-rank test. Data are presented as mean ± standard deviation (SD). A P value of ≤0.05 was considered significant. Sample sizes were estimated based on a α=.05 and 80% power, with effect sizes and sample variabilities estimated from our published studies [5, 7] and were indicated in the figure legends.
Please see Online Data Supplement of Detailed Materials and Methods.
RESULTS
BMDEC specific Alk1 deletion caused bAVM and Alk1− BMDECs present in bAVM lesion
We showed previously that Eng+/− BM causes cerebral capillary abnormalities of WT mice [13]. To test whether mutation of Alk1 in BMDECs alone causes bAVM, we transplanted the BM collected from PdgfbiCreER;Alk12f/2f;Ai14+/− mice to lethally irradiated 8-week-old WT mice (Fig. 1A). Brain AVM was induced by intra-brain injection of an adeno-associated viral vector expressing vascular endothelial growth factor [AAV-VEGF, 2×109 genome copies (gcs)] 4 weeks after the BM transplantation when recipients’ BMs were fully reconstituted by donors’ BMs (Supplementary Fig. 1). TM (2.5 mg/25g of body weight) was injected i.p. 2 weeks later when a fraction of BM derived cells had differentiated into ECs in brain angiogenic region [12, 13] to delete Alk1 gene in BMDECs (Fig. 1A).
Fig. 1. Mutation of Alk1 in BMDECs caused bAVMs.
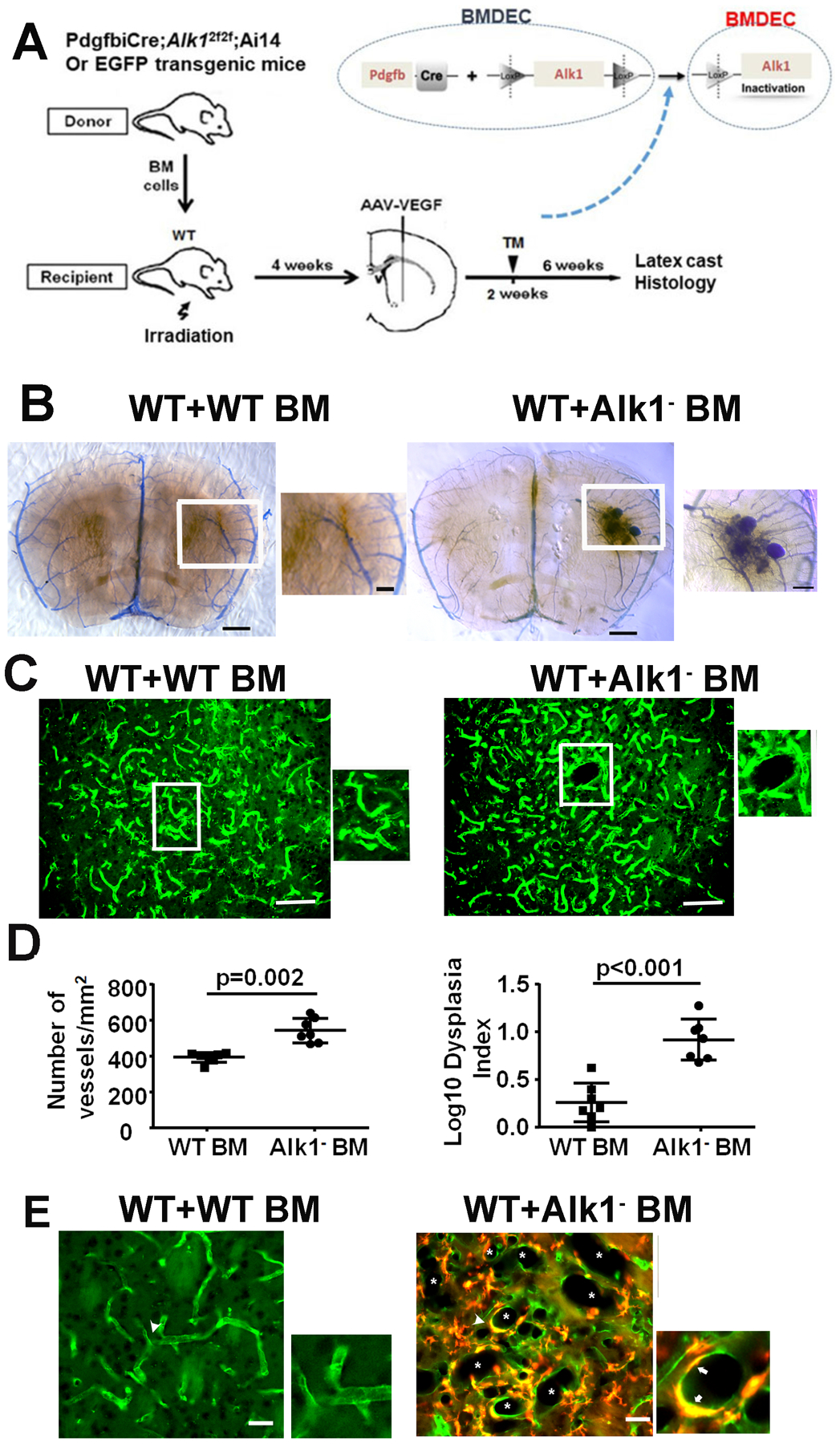
A. Study design. BM collected from PdgfbiCreER;Alk12f/2f;Ai14+/− or EGFP transgenic mice were transplanted to lethally irradiated WT mice. AAV-VEGF (2×109 gcs) was injected to the brains of recipients 4 weeks after BM transplantation followed by TM (2.5 mg/25g of body weight) treatment 2 weeks later when a fraction of BM derived cells had differentiated into ECs in the brain angiogenic region to delete Alk1 gene in BMDECs. AVM phenotype was analyzed 6 weeks after TM treatment by latex cast and histology. B. Image of latex perfused brain sections collected from a WT mouse with EGFP BM (WT+WT BM) and a WT mouse with PdgfbiCreER;Alk12f/2f;Ai14+/− BM (WT+Alk1 BM). Right: Enlarged image of the boxed area in the middle picture shows an AVM nidus. Scale bars: 1 mm for left and middle images, 200 μm for the right image. C. Representative images of brain sections. The ECs were stained by intravascular perfused lectin (green). *: dilated bAVM vessels. Scale bars: 100 μm. D. Quantification of vessel density and Dysplasia Index (number of abnormal vessels/200 vessels). Alk1 BM: WT mice transplanted with PdgfbiCreER;Alk12f/2f;Ai14+/− BM. WT BM: WT mice transplanted with EGFP BM. The numbers of dysplasia index were log transformed because they were not normally distributed. N=6. E. Microscopic image shows many dilated vessels (*) in bAVM lesion. ECs were stained by intravascular perfused lectin (green). Recombined BMDECs expressed red fluorescent Ai14 reporter, therefore, they are yellow in the image. The two images on the right show two large vessels indicate by arrowheads in the left image that have clustered BMDECs (Arrows). Scale bar: 50 μm.
Latex dye casting bAVMs were developed in 4 of the 10 mice with PdgfbiCreER;Alk12f/2f;Ai14+/− BM (Fig. 1B). Arteriovenous (AV) shunts were also detected in the intestines in 5 of the 9 mice with PdgfbiCreER;Alk12f/2f;Ai14+/− BM (Supplementary Fig. 2) after TM administration. Mice with PdgfbiCreER;Alk12f/2f;Ai14+/− BM had a higher vessel density (p=0.002) and more dysplasia vessels (p<0.001) than mice with WT BM (Fig. 1C &D). BMDECs were detected in bAVM lesion (Fig. 1E). About 28±10.6% ECs in bAVM lesion were BMDECs. Only 3.8±3.6% ECs in the contralateral side of brain were BMDECs (p=0.009, Supplementary Fig. 3) These data indicate that mutation of Alk1 gene in BMDECs alone is sufficient to cause bAVM development.
Alk1−/− ECs undergo clonal expansion in bAVMs
We found previously that proliferating Alk1−/− ECs are preferentially cluster in bAVM vessels [7]. This founding spurs us an idea that the small numbers of ECs with biallelic loss of ALK1 can clonally overpopulate in bAVM. Brain AVMs were induced in 8 weeks old PdgfbiCreER;Alk12f/2f;confetti+/− mice through intra-brain injection of AAV-VEGF (2×109 gcs) to induce brain focal angiogenesis followed by i.p. injection of TM (2.5 mg/25g of body weight) 2 weeks later (Fig. 2A). TM treatment will induce Alk1 gene deletion and confetti locus activation in ECs. The confetti reporter irreversibly labels individual recombined ECs with one of the four fluorescent colors. Therefore, if a single EC clonally expands, we expect to see a cluster of ECs expressing same fluorescent color. Indeed, clusters of ECs expressing the same confetti colors were detected in bAVMs (Fig. 2B), but not in the contralateral hemisphere of the same mice (Alk1−/− ECs without VEGF treatment, Fig. 2C) or in the brains of TM treated PdgfbiCreER;confetti+/− mice around AAV-VEGF injection sites (WT ECs with VEGF treatment, Fig. 3D). As it is statistically unlikely that the clusters of ECs with the same confetti colors in bAVMs were the result of multiple independent Cre recombination events, the observed pattern indicates that Alk1−/− ECs were clonally expanded in bAVMs.
Fig. 2. Alk1−/− EC clonally expanded in bAVM.
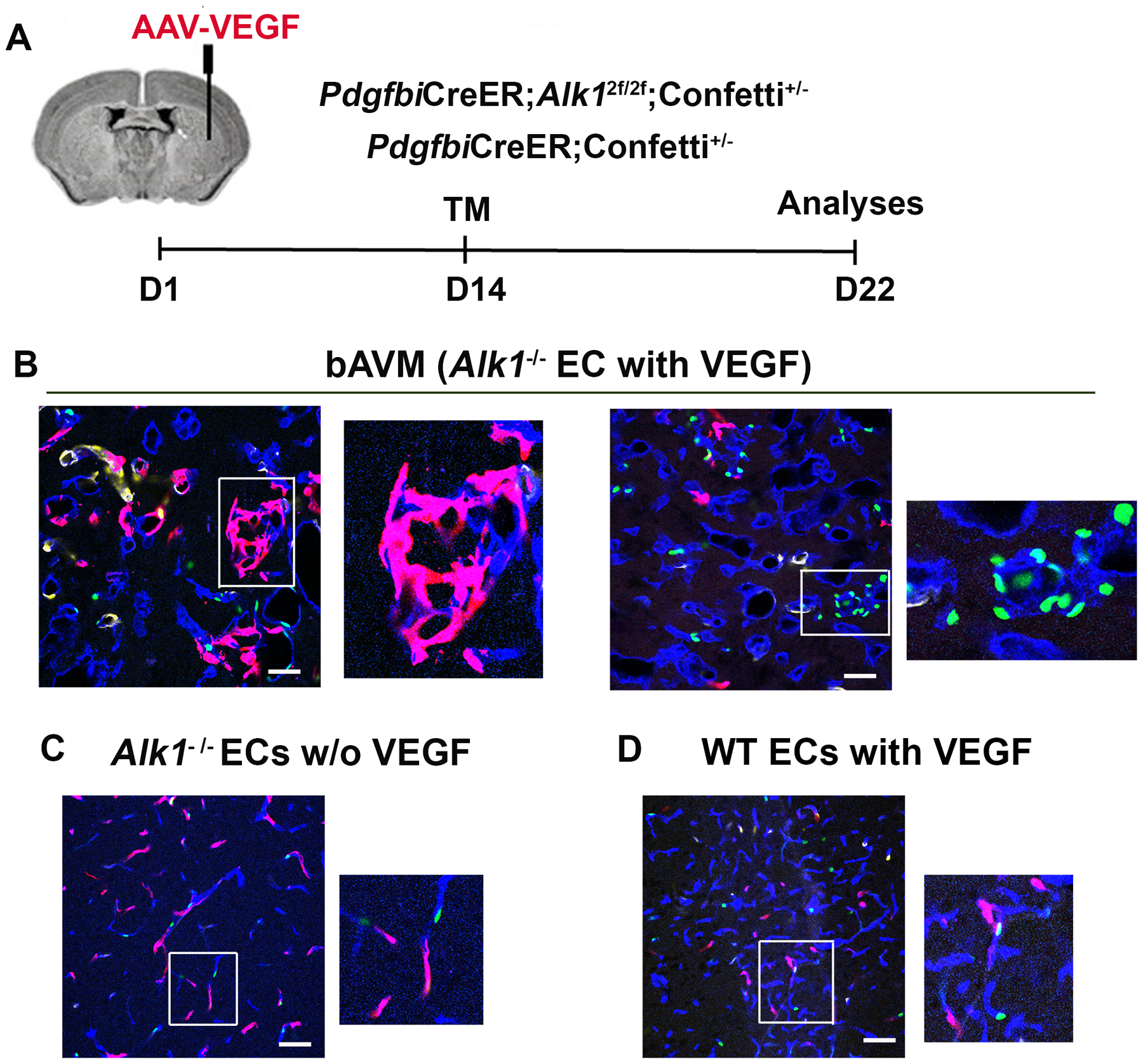
A. Design. B. Confocal images show clusters of ECs expressing same confetti colors in bAVM lesions. Vessels were stained blue using an anti-CD31 antibody. Enlarged images of the rectangle regions in the left pictures show a cluster of RFP+ ECs and a cluster of GFP+ ECs in groups of abnormal vessels. C. Mouse brain with Alk1 deleted in ECs without VEGF stimulation. D. WT mouse brain with VEGF stimulation. Enlarged images are the squared regions in D and E showing individual Confetti+ ECs scattered in vessels. Scale bars: 50 μm.
Fig. 3. Equal numbers of Alk1+ and Alk1− ECs were proliferating in the established bAVMs.
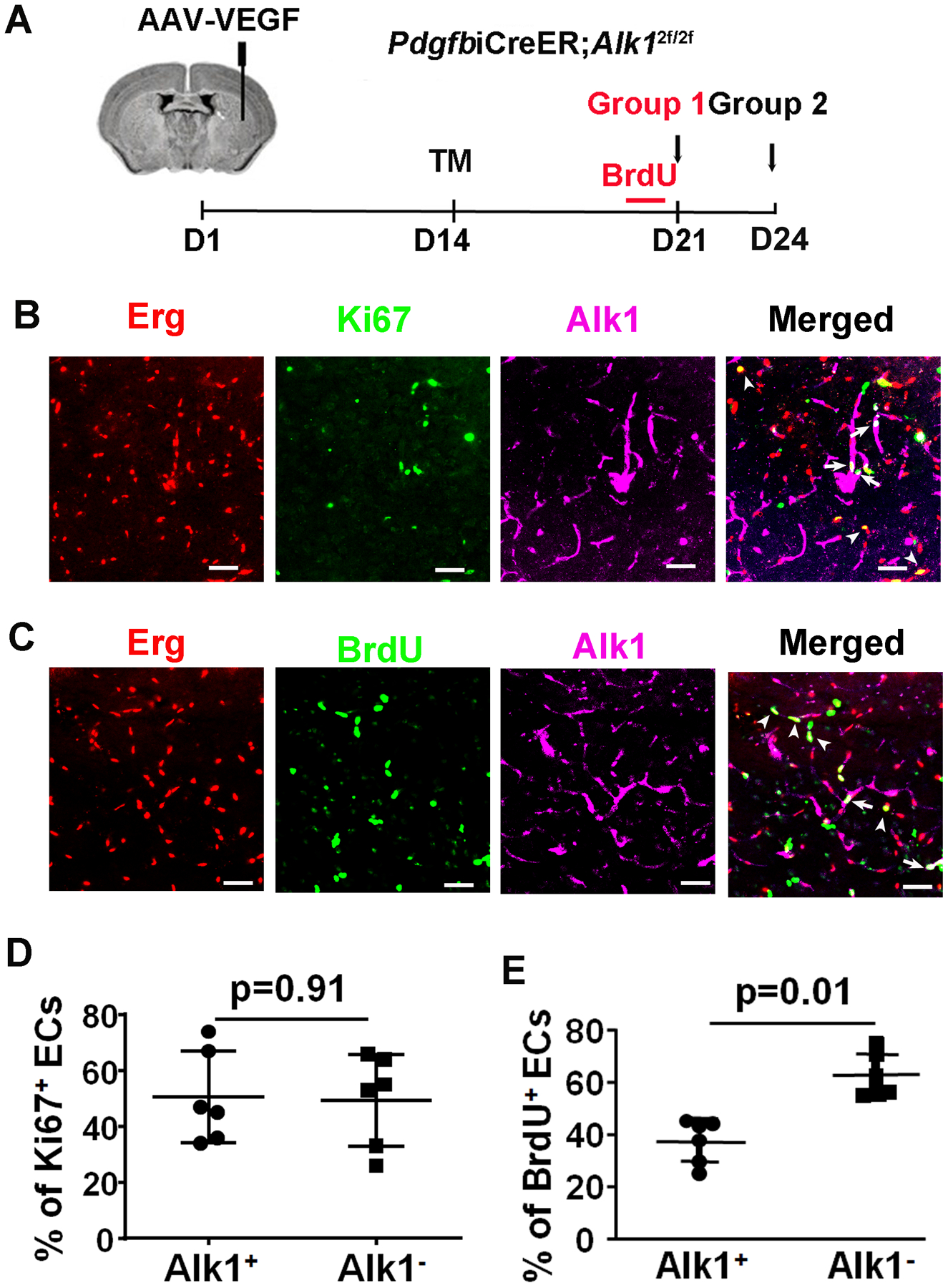
A. Design. B. Images of bAVM sections. The nuclei of ECs were stained by an Erg antibody (red). Proliferating cells were stained by a Ki67 antibody (green). Alk1 expression was stained by an Alk1 antibody (purple). Arrow: a Ki67+ Alk1+ EC. Arrowhead: a Ki67+ Alk1− EC. Scale bars=50 μm. B. Quantification of Ki67+ Alk1− and Alk1+ ECs. N=6.
Although more Confetti+ ECs were found in the bAVM lesions than in the contralateral Alk1−/− brains or WT brain angiogenic regions, there were about 60% of ECs lacking any confetti color (Fig. 2B). It is most likely due to ineffective recombination of the confetti allele induced by 2.5 mg/25g TM, because a half of this dose (1.25 mg/25g) had induced near 100% recombination of Alk1 floxed alleles as evidenced by the lack of Alk1 protein expression in brain ECs and the development of bAVMs (Supplementary Fig. 4A & B).
Equal numbers of Alk1+ and Alk1− ECs were proliferating in bAVMs
We showed previously that more ECs were proliferating in bAVMs that have Alk1 deletion in near 100% ECs than in brain angiogenic regions of WT mice [7]. However, it is not clear if Alk1+ ECs are also proliferating in bAVMs. To answer this question, we created a mosaic of Alk1+ and Alk1− ECs in bAVM by treating PdgfbiCreER;Alk12f/2f mice with a low dose of TM (0.01 mg/25g of body weight) 14 days after intra-brain injection of AAV-VEGF. Brain samples were collected at 14 days after TM treatment (Fig. 3A), sectioned and stained for Alk1 (to assess the degree of gene knockout) and CD31 (to mark all ECs) to validate the mosaic status. Alk1 expression in ECs was identified by co-localization of Alk1 and CD31 expression. Mice treated with this dose of TM had about 48.0±16.8% ECs were Alk1+ ECs (Supplementary Fig. 5).
To analyze EC-proliferation in bAVM lesions with mosaic Alk1+ and Alk1− ECs in mice treated with 0.01 mg/25g of body weight TM, we co-labelled brain sections with the proliferation marker Ki67, Erg (a pan-EC transcription factor), and Alk1 (to identify Alk1− and Alk1+ ECs). We observed that 45±13.3% of Ki67+ ECs were Alk1− ECs and 55±13.3% Ki67+ ECs were Alk1+ ECs (p=0.42, Fig. 3), indicating that Alk1+ and Alk1− ECs have similar proliferation capacity in bAVMs.
The burden of ALK1− ECs correlates with bAVM severity and mouse mortality
To determine whether there is an association between the number of Alk1− ECs and bAVM severity, PdgfbiCreER;Alk12f/2f;Ai14+/− mice were treated with a high dose (1.25 mg/25g of body weight) or a low dose of TM (0.01 mg/25g of body weight) 14 days after intra-brain injection of AAV-VEGF (Fig. 4A). As expected, high dose TM treatment resulted in a greater degree of Cre recombination and Alk1 deletion in ECs (based on the numbers of Ai14+ and Alk1− ECs). Mice treated with the low dose TM had fewer Ai14+ ECs than mice treated with the high dose TM in the bAVM lesions (61±20% vs. 87%±5%, P=0.01, Fig. 4B and C). The TM-vehicle (corn oil) treated mice also showed a low (but not zero) degree of recombination (29±10%), possibly due to Cre leakiness (Fig. 4B and C). The mice in the low TM group have more Alk1+ ECs identified by immunostaining in bAVM lesions (48.0±16.8%) than mice in the high TM group (2.3±4.34%, P<0.001). Almost all ECs in the vehicle treated mice were Alk1+ (99 ±8.25%, Fig. 4D & E). Mice in the high TM group also expressed a lower level of Alk1 protein in bAVM lesions (Supplementary Fig. 6).
Fig. 4: Increase of TM dose increased Ai14+ and decreased Alk1+ ECs.
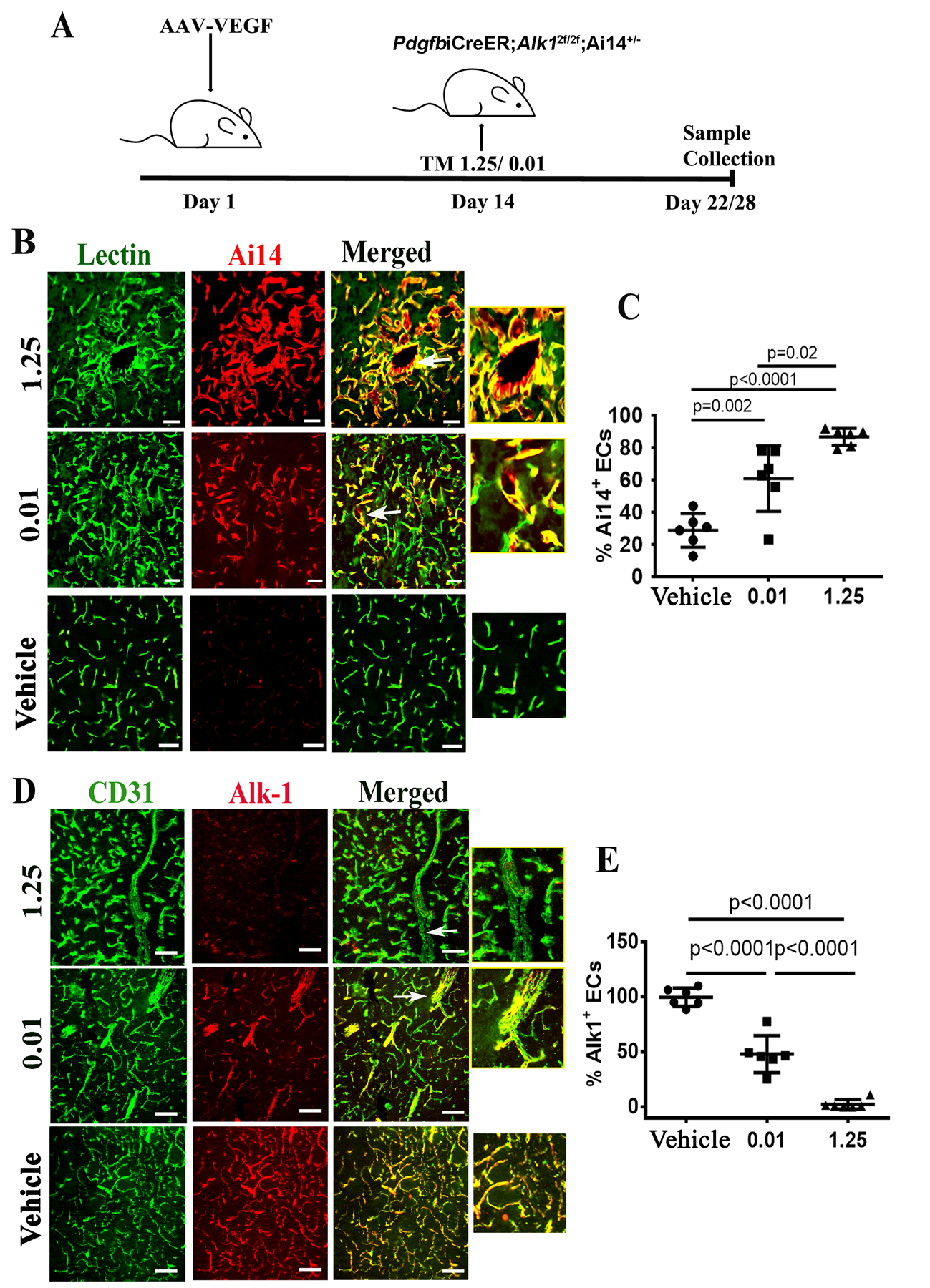
A. Design. TM 0.01/1.25: mice treated with 0.01 or 1.25 mg/25g of body weight TM. Day 22: samples were collected 22 days after intra-brain injection of AAV-VEGF from mice treated with high dose TM (1.25 mg/25g); Day 28: samples were collected 28 days after intra-brain injection of AAV-VEGF from mice treated with the low dose TM (0.01 mg/25g). B. Representative images of brain sections collected from lectin perfused mice. ECs were stained by intravascular perfused lectin (green). Cre activation in ECs was indicated by the expression of Ai14 reporter (red). Arrows indicate dysplastic vessels. Scale bars: 50 μm. The right-side pictures show enlarged images of abnormal vessels indicated by arrows in the pictures on their left side. C. Quantification of Ai14+ ECs in the bAVM lesions. D. Representative images of brain sections co-stained with anti-CD31 (green, ECs) and anti-Alk1 (red) antibodies. Arrows indicate abnormal vessels. Scale bar: 80 μm. The right-side pictures show enlarged images of abnormal vessels indicated by arrows in the pictures on their left side. E. Quantification of Alk1+ ECs. WT: corn oil treated mice; 0.01 or 1.25: mice treated with 0.01 mg/25g or 1.25 mg/25g TM. N=6.
We quantified dysplastic vessels (expressed as dysplasia index: number of vessels with lumens >15 mm per 200 vessels) in bAVM lesions and counted the number of intestinal arteriovenous (AV) shunts using latex dye casting. We found that mice in the low TM dose group (4.5±1.1) have fewer dysplastic vessels than mice in the high TM dose group (7.9±1.2, P=0.001, Fig. 5A and B). No dysplastic vessel was found in the vehicle treated mice. Vessel densities were similar among groups (Fig. 5C). Large bAVM vessels (detected by latex dye casting) were present in 100% of mice treated with high TM dose, but only in 50% of low dose TM treated mice. No large bAVM vessels was detected in vehicle treated mice. The phenotype of low dose TM treated mice were analyzed 28 days after intra-brain injection of AAV-VEGF, which was 6 days later than mice in the high dose TM group. Increase of TM dose also increased the number of AV shunts and AV shunt penetrance in the intestines (Fig. 6A & B, and Table 1) and mouse mortality (Fig. 6C). Together, these data indicate that the burden of Alk1− ECs correlates with AVM severity.
Fig. 5. Reduction of the TM dose reduced the number of dysplastic vessels in bAVM lesions.
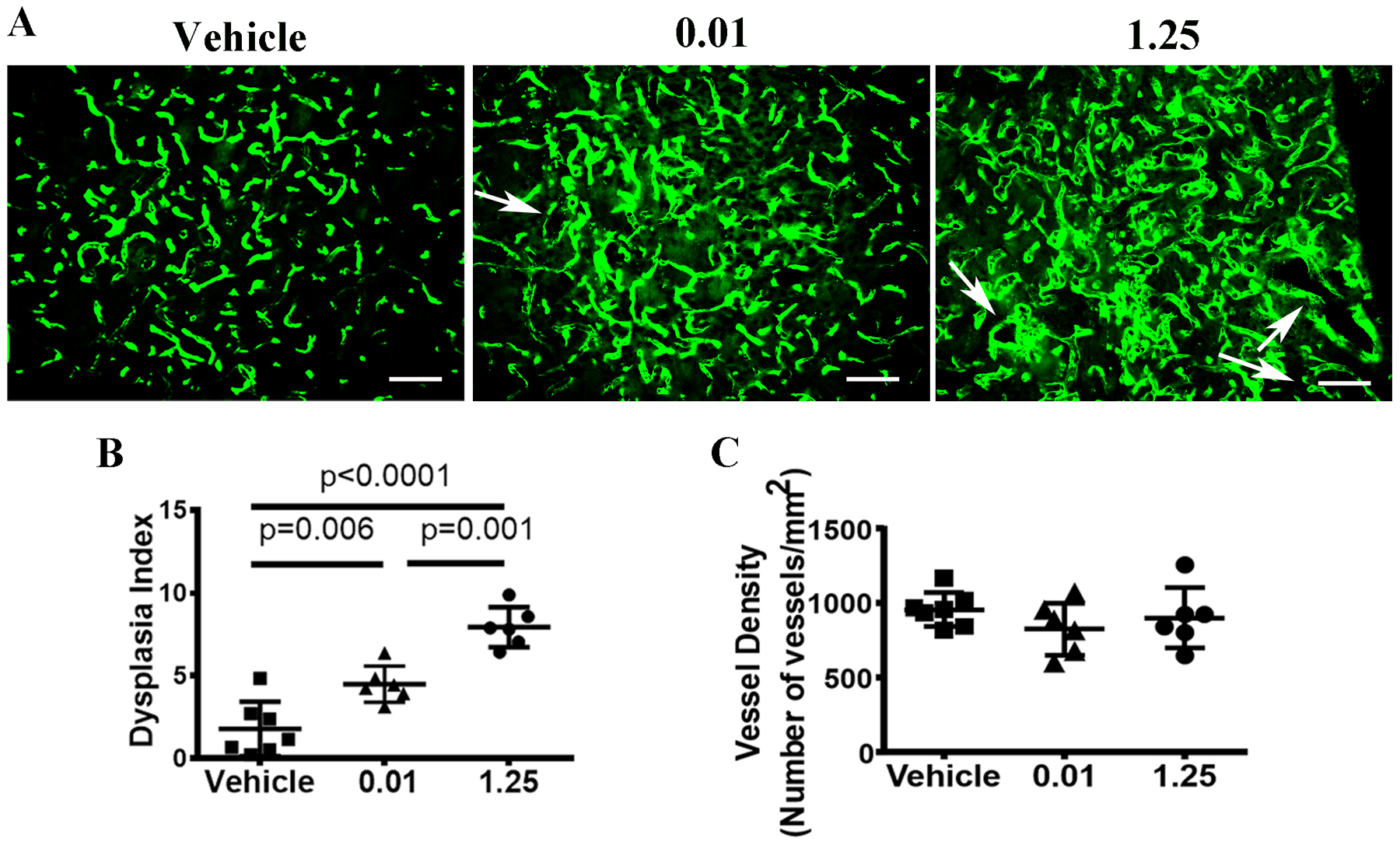
A. Representative images of brain sections. ECs were stained by intravascular perfused lectin (green). Arrows indicate dysplastic vessels. Scale bar: 80 μm. B. Quantification of the Dysplasia Index. C. Quantification of vessel density. Vehicle: corn oil treated mice; 0.01 or 1.25: mice treated with 0.01 mg/25g or 1.25mg/25g TM. N=7 for the Vehicle group; N=6 for the 0.01 and 1.25 TM groups.
Fig. 6. Reduction of TM dose reduced the number of AV shunts in the intestines and mouse mortality.
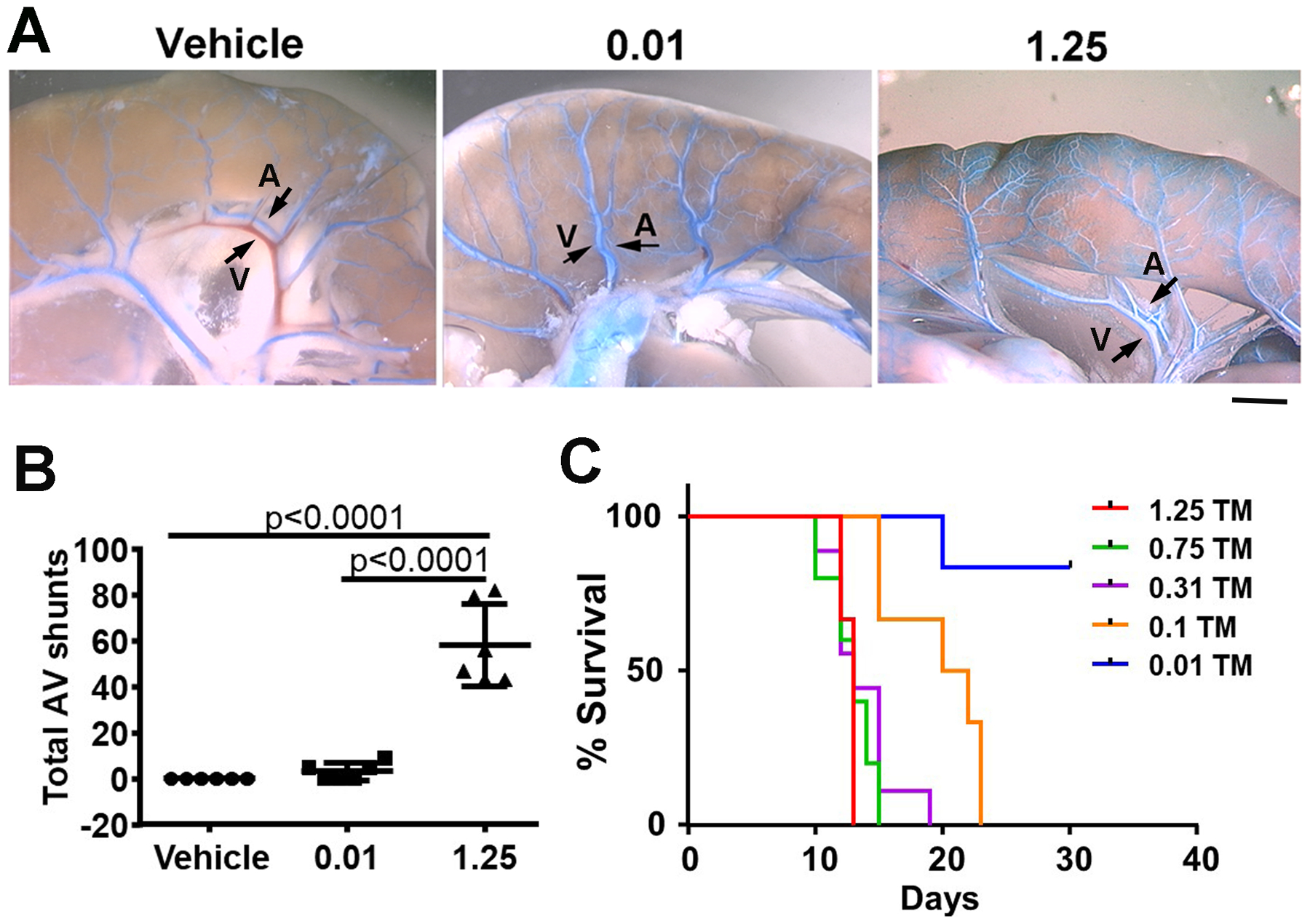
A. Representative images show AV shunts in the intestines. The vessels were casted with latex dye (blue). Latex dye entered some veins of mice treated with 0.01 and 1.25 mg/25g TM after being injected into intra-left cardiac ventricles. Arrows indicate an artery (A) and a vein (V). Scale bar: 1 mm. B. Quantification of AV shunts. 0.01 and 1.25: mice treated with 0.01 or 1.25 mg/25g TM. Vehicle: mice treated with corn oil. C. The survival curve. Mice were treated with one i.p. injection of 1.25, 0.75, 0.3, or 0.1 mg/25g of body weight TM. N=6.
Table 1.
Penetrance of AV shunt in the intestines
| 1.25 | 0.01 | Vehicle |
| 100% (6/6) | 50% (3/6) | 0% (0/6) |
Note: 1.25 and 0.01: mice treated with 1.25 or 0.01 mg/25g TM. Vehicle: mice treated with corn oil.
DISCUSSION
In this paper, we show that mutation of Alk1 gene in BMDECs caused bAVM and Alk1− ECs were clonally expanded in mouse bAVM. Increasing TM dose increased the number of Alk1− ECs in PdgfbicreER;Alk12f/2f;Ai14+/− mice and the bAVM severity.
Several studies, including our own, support a role of BM-derived cells in bAVM development [13]. After VEGF stimulation in the brain, WT mice with Eng+/− BM developed a similar degree of cerebral capillary dysplasia as mice with Eng+/− somatic and BM cells, suggesting that loss of even one allele of Eng in BM-derived cells results in cerebrovascular dysplasia [13]. Conversely, Eng+/− mice transplanted with WT BM had fewer dysplastic capillaries than Eng+/− mice with Eng+/− BM, suggesting that BM-derived cells contribute to vascular malformations and that the tendency to form vascular malformation might be mitigated by transplantation of normal BM cells. However, it is not clear which type of BM cells play the central role in AVM development. More than 70% of BM derived cells differentiated into macrophages in brain angiogenic region while fewer than 7% of them differentiated into ECs [12, 13]. Previous studies showed that mutation of Eng or Alk1 in ECs causes AVM development, and mutation of these genes in macrophage will not initiate AVM development [6, 7, 10]. Therefore, in this study, we tested if BMDECs are responsible for transmission of abnormal cerebrovascular phenotype. We transplanted the BM cells isolated from PdgfbicreER;Alk12f/2f;Ai14+/− mice to lethally irradiated WT mice. Brain angiogenesis were induced after the BM of WT mice were reconstituted by PdgfbicreER;Alk12f/2f;Ai14+/− BM. Alk1 deletion was induced two weeks after the induction of brain angiogenesis when BM cells had homed to brain angiogenic region and a fraction of homed BM cells had differentiated into ECs (Fig. 1A). AVMs in the brains and AV shunts in the intestines developed in WT mice with PdgfbicreER;Alk12f/2f;Ai14+/− BM. Our data demonstrate that BMDEC is an important cellular contributor among all BM cells in AVM development.
Analysis of human brain and lung AVMs in HHT patients indicated that haploinsufficiency of the HHT causative gene is not sufficient to cause AVM development [18]. Inactivation of the remaining WT allele appears to be required for lesion formation, irrespective of the mechanism by which it is inactivated, e.g., loss-of-function somatic mutation of the WT allele or loss of protein during inflammation [19, 20]. In mice, the loss of a single allele of Eng or Alk1 recapitulates certain aspects of the human disease, primarily in older animals [21, 22]. Brain AVM does not commonly develop in mice with haploinsufficiency of these genes. Loss of both alleles of any HHT-causative gene is embryonically lethal in mice [23, 24] and conditional (tissue/time-specific) homozygous deletion of Eng [19] or Alk1 [25, 26] results in striking vascular malformations resembling the AVMs found in HHT.
Although it is known that deletion of AVM causative genes in ECs is essential for AVM development [6, 7, 10] and gene deletion in a small fraction of ECs can cause bAVM [5, 11], currently, it is not clear how a small fraction of ECs with Alk1 or Eng deletion leads to bAVM. It is also not clear if all ECs with Alk1 or Eng deletion in a bAVM are descended from one or a few ECs with Alk1 or Eng deletion. Using mice with confetti reporter transgene, we observed that ECs expressing the same confetti color clustered in bAVM lesions. Each bAVM lesion had several EC-clusters that express different confetti reporters, suggesting that a bAVM can be developed by several ECs with Alk1 deletion. This phenotype was not detected in brain angiogenic regions of WT mice, nor in the brains of mice with Alk1 gene deletion alone. Our data suggest that Alk1 mutant ECs in bAVMs undergo clonal expansion, which could partially explain why a fraction of Alk1 mutant ECs can lead to bAVM development. Due to the high recombination rate of Alk1 floxed allele (near 100%) and low recombination rate of confetti transgene in response to 2.5 mg/25g of body weight TM treatment, we could not analyze if WT ECs are also clonally expanding in our AVMs model. We will consider using a different reporter line that is more effective in recombination to address this question in the future [27].
We showed previously that deletion of Alk1 gene in ECs in the adult brain increased EC-proliferation in response to VEGF stimulation [7]. Alk1 gene was deleted in almost all ECs in that study. AVM vessels had more proliferating ECs than the surrounding normal capillaries. Jin et al. showed that deletion of Eng in ECs in neonates resulted in more proliferating Eng− ECs than WT ECs in bAVM lesions [14]. In our study, using an adult mouse model with a mosaic pattern of Alk1− and Alk1+ ECs, we found that equal numbers of Alk1+ and Alk1− ECs were proliferating in bAVM lesions. Our data suggest that the Alk1− ECs may promote the growth of surrounding Alk1+ ECs. Similarly, the clonal analysis of the mosaic retinal vasculature of Eng knockout mice revealed an equal proliferation rate of Eng+ and Eng− ECs in AVMs vessels in the retina, although more Eng− ECs were proliferating in the vessels outside of AVMs, suggesting that non-cell autonomous mechanisms contributes to the expansion of AVMs [14]. It has been shown previously that Alk1−/− embryos expressed higher levels of angiogenic factors than Alk1+/− and Alk1+/+ embryos [28]. Therefore, Alk1− ECs in bAVM could stimulate surrounding WT EC-proliferation through increasing the levels of angiogenic factors in the environment. The increased flow in AVM could also contribute to the increase of EC-proliferation and vascular remodeling in the lesions [29, 30]. However, the role of WT ECs in bAVM pathogenesis remains to be elucidated.
We also evaluated the relationship between the number of Alk1− ECs and AVM severity. By titrating the TM dose, we were able to reduce the number of Alk1− ECs in PdgfbicreER;Alk12f/2f;Ai14+/− mice, and found that mice treated with a high dose of TM had more severe AVM phenotype than mice treated with a low dose of TM. Since most of the mice will die within 10 days after the high dose TM treatment, we collected tissues 8 days after TM treatment from this group to ensure that most of the treated mice were still alive at the time of sample collection. However, the phenotype of the low dose TM treated mice develops slower and is less severe than that of the high TM treated mice. We postponed the sample collection time from the low dose TM treated mice to ensure that significant AVM phenotype can be detected. Although the samples were collected at different time points, our conclusion was not affected, because even the low dose TM treated mice had 6 more days than the high dose TM treated mice to develop AVM phenotype, these mice still showed less severe phenotype than that in the high dose group. It is consistent with our conclusion that reduction of TM dose reduced the AVM severity. These data suggest that novel therapeutic strategies, such as transfusion of normal circulating ECs or overexpression of Alk1 gene [31], that can reduce the burden of mutant ECs could potentially alleviate AVM phenotype severity in HHT patents.
In summary, we showed that Alk1 deletion in BMDECs causes bAVM development and that Alk1− ECs were clonally expanded in the bAVMs. The proportion of Alk1− ECs is positively correlated with AVM phenotype severity. Together, our data suggest that reduction of Alk1− ECs in bAVM lesions can be a target for developing new therapies to alleviate the severity of bAVMs in HHT patients.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Ludmila Pawlikowska for assistance with manuscript preparation.
Funding
This study was supported by grants to H.S. from the National Institutes of Health (R01 HL122774, NS027713 and NS112819) and from the Michael Ryan Zodda Foundation. N.S. is supported by AHA fellowship (20POST35120371).
Footnotes
Conflicts of interests
The authors do not have competing interests.
Ethics approval
The protocol and experimental procedures for using laboratory animals were approved by the Institution of Animal Care and Use Committee (IACUC) at the University of California, San Francisco.
Availability of data and material
The authors declare that all data supporting the findings of this study are available in the paper and its Supplementary Information.
REFERENCES
- 1.Gauden AJ, McRobb LS, Lee VS, Subramanian S, Moutrie V, Zhao Z, et al. Occlusion of Animal Model Arteriovenous Malformations Using Vascular Targeting. Transl Stroke Res. 2020;11(4):689–99. [DOI] [PubMed] [Google Scholar]
- 2.Pan P, Weinsheimer S, Cooke D, Winkler E, Abla A, Kim H, et al. Review of treatment and therapeutic targets in brain arteriovenous malformation. J Cereb Blood Flow Metab. 2021:271678X211026771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bharatha A, Faughnan ME, Kim H, Pourmohamad T, Krings T, Bayrak-Toydemir P, et al. Brain arteriovenous malformation multiplicity predicts the diagnosis of hereditary hemorrhagic telangiectasia: quantitative assessment. Stroke. 2012;43(1):72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim H, Su H, Weinsheimer S, Pawlikowska L, Young WL. Brain arteriovenous malformation pathogenesis: a response-to-injury paradigm. Acta Neurochir Suppl. 2011;111:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker EJ, Su H, Shen F, Choi EJ, Oh SP, Chen G, et al. Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann Neurol. 2011;69(6):954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi EJ, Chen W, Jun K, Arthur HM, Young WL, Su H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One. 2014;9(2):e88511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen W, Sun Z, Han Z, Jun K, Camus M, Wankhede M, et al. De novo cerebrovascular malformation in the adult mouse after endothelial Alk1 deletion and angiogenic stimulation. Stroke. 2014;45(3):900–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu W, Saw D, Weiss M, Sun Z, Wei M, Shaligram S, et al. Induction of Brain Arteriovenous Malformation Through CRISPR/Cas9-Mediated Somatic Alk1 Gene Mutations in Adult Mice. Transl Stroke Res. 2019;10(5):557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen W, Choi EJ, McDougall CM, Su H. Brain arteriovenous malformation modeling, pathogenesis, and novel therapeutic targets. Transl Stroke Res. 2014;5(3):316–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrido-Martin EM, Nguyen HL, Cunningham TA, Choe SW, Jiang Z, Arthur HM, et al. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler Thromb Vasc Biol. 2014;34(10):2232–6. [DOI] [PubMed] [Google Scholar]
- 11.Choi EJ, Walker EJ, Shen F, Oh SP, Arthur HM, Young WL, et al. Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc Dis. 2012;33(6):540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hao Q, Liu J, Pappu R, Su H, Rola R, Gabriel RA, et al. Contribution of bone marrow-derived cells associated with brain angiogenesis is primarily through leukocytes and macrophages. Arterioscler Thromb Vasc Biol. 2008;28(12):2151–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi EJ, Walker EJ, Degos V, Jun K, Kuo R, Pile-Spellman J, et al. Endoglin deficiency in bone marrow is sufficient to cause cerebrovascular dysplasia in the adult mouse after vascular endothelial growth factor stimulation. Stroke. 2013;44(3):795–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C, et al. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat Cell Biol. 2017;19(6):639–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450(7166):56–62. [DOI] [PubMed] [Google Scholar]
- 16.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337(6095):730–5. [DOI] [PubMed] [Google Scholar]
- 17.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, et al. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2 (HHT2). Blood. 2008;111(2):633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bourdeau A, Cymerman U, Paquet ME, Meschino W, McKinnon WC, Guttmacher AE, et al. Endoglin expression is reduced in normal vessels but still detectable in arteriovenous malformations of patients with hereditary hemorrhagic telangiectasia type 1. Am J Pathol. 2000;156(3):911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, et al. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106(8):1425–33. [DOI] [PubMed] [Google Scholar]
- 20.Snellings DA, Gallione CJ, Clark DS, Vozoris NT, Faughnan ME, Marchuk DA. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet. 2019;105(5):894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP, et al. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. 2003;12(5):473–82. [DOI] [PubMed] [Google Scholar]
- 22.Bourdeau A, Faughnan ME, Letarte M. Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med. 2000;10(7):279–85. [DOI] [PubMed] [Google Scholar]
- 23.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26(3):328–31. [DOI] [PubMed] [Google Scholar]
- 24.Sorensen LK, Brooke BS, Li DY, Urness LD. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFbeta coreceptor. Dev Biol. 2003;261(1):235–50. [DOI] [PubMed] [Google Scholar]
- 25.Milton I, Ouyang D, Allen CJ, Yanasak NE, Gossage JR, Alleyne CH Jr., et al. Age-dependent lethality in novel transgenic mouse models of central nervous system arteriovenous malformations. Stroke. 2012;43(5):1432–5. [DOI] [PubMed] [Google Scholar]
- 26.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119(11):3487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontes-Quero S, Heredia L, Casquero-Garcia V, Fernandez-Chacon M, Luo W, Hermoso A, et al. Dual ifgMosaic: A Versatile Method for Multispectral and Combinatorial Mosaic Gene-Function Analysis. Cell. 2017;170(4):800–14 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, et al. Activin receptor-like kinase 1 modulates transforming growth factor- beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97(6):2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corti P, Young S, Chen CY, Patrick MJ, Rochon ER, Pekkan K, et al. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development. 2011;138(8):1573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Ma L, Yang S, Burkhardt JK, Lu J, Ye X, et al. Quantitative Angiographic Hemodynamic Evaluation After Revascularization Surgery for Moyamoya Disease. Transl Stroke Res. 2020;11(5):871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YH, Phuong NV, Choe SW, Jeon CJ, Arthur HM, Vary CP, et al. Overexpression of Activin Receptor-Like Kinase 1 in Endothelial Cells Suppresses Development of Arteriovenous Malformations in Mouse Models Of Hereditary Hemorrhagic Telangiectasia. Circ Res. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available in the paper and its Supplementary Information.