

Abstract
Although vaccines and monoclonal antibody countermeasures have reduced the morbidity and mortality associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, variants with constellations of mutations in the spike gene jeopardize their efficacy. Accordingly, antiviral interventions that are resistant to further virus evolution are needed. The host-derived cytokine interferon lambda (IFN-λ) has been proposed as a possible treatment based on studies in human coronavirus 2019 (COVID-19) patients. Here, we show that IFN-λ protects against SARS-CoV-2 B.1.351 (Beta) and B.1.1.529 (Omicron) variants in three strains of conventional and human ACE2 transgenic mice. Prophylaxis or therapy with nasally delivered IFN-λ2 limits infection of historical or variant SARS-CoV-2 strains in the upper and lower respiratory tracts without causing excessive inflammation. In the lung, IFN-λ is produced preferentially in epithelial cells and acts on radio-resistant cells to protect against SARS-CoV-2 infection. Thus, inhaled IFN-λ may have promise as a treatment for evolving SARS-CoV-2 variants that develop resistance to antibody-based countermeasures.
Keywords: SARS-CoV-2, immunity, interferon, omicron, pathogenesis, therapy, RNA sequencing
Graphical abstract
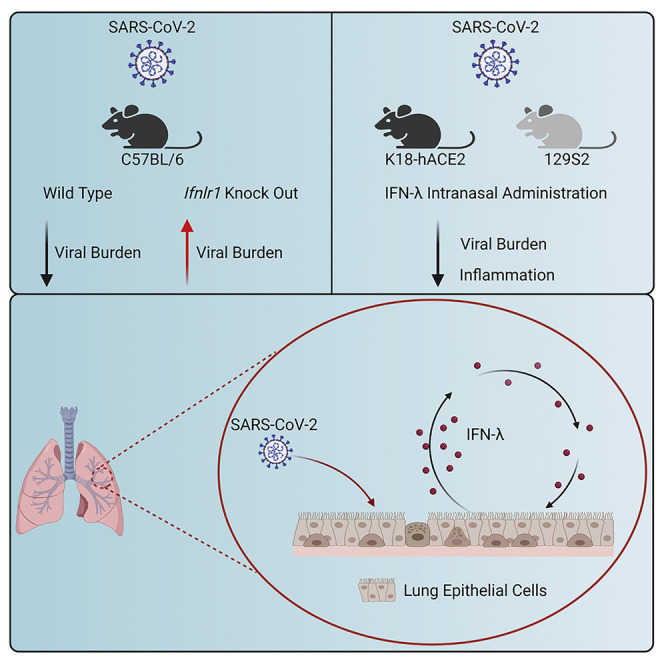
Chong et al. show that intranasally delivered murine IFN-λ2 protects mice against historical, B.1.351 (Beta), and B.1.1.529 (Omicron) SARS-CoV-2 infection in the upper and lower respiratory tracts without excessive inflammation. In the lung, IFN-λ is produced mainly by epithelial cells and acts on stromal cells to protect against of SARS-CoV-2 infection.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in 2019 and has infected more than 500 million people worldwide. The coronavirus disease 2019 (COVID-19) pandemic continues because of the evolution of highly transmissible variant strains and a failure to vaccinate large segments of the global population. SARS-CoV-2 infection causes a range of influenza-like symptoms but can progress rapidly to pneumonia, acute respiratory distress syndrome (ARDS), and death (Guan et al., 2020; Huang et al., 2020).
One hallmark of COVID-19 in some individuals is a hyper-inflammatory state with excessive production of pro-inflammatory mediators, which recruit activated immune cells that ultimately impair alveolar gas exchange and injure the lung (Mehta et al., 2020; Zhang et al., 2020). Interferons (IFNs) are pro-inflammatory cytokines that are a first line of defense against most virus infections. Type I (IFN-α subtypes and IFN-β) and type III IFNs (IFN-λ) are induced rapidly after detection by and activation of pathogen sensors (e.g., Toll-like [TLR] or RIG-I-like [RLR] receptors) and their downstream signaling pathways (Park and Iwasaki, 2020). Type I and III IFNs bind to distinct receptors on the cell surface to activate signal transducers and activators of transcription (STAT) proteins that induce expression of hundreds of antiviral IFN-stimulated genes (ISGs) (Lazear et al., 2019; Schneider et al., 2014). Cell-culture studies have shown that IFN pre-treatment can restrict SARS-CoV-2 infection in human intestinal and airway epithelia (Felgenhauer et al., 2020; Stanifer et al., 2020; Vanderheiden et al., 2020). Although type I IFNs are a potential treatment strategy for SARS-CoV-2 infection (Hoagland et al., 2021), the ubiquitous expression of the IFNAR1/IFNAR2 receptor and strong, sustained pro-inflammatory responses can have pathological consequences. In comparison, the cellular response to type III IFN-λ is thought to be less inflammatory, as it functions primarily at epithelial and barrier surfaces where its heterodimeric receptor (IFNLR1/IL10Rβ) is preferentially expressed (Andreakos and Tsiodras, 2020; Broggi et al., 2020b; Galani et al., 2017).
The role of IFN-λ in SARS-CoV-2 infection and pathogenesis remains unclear. Although patients with severe COVID-19 patients have elevated serum levels of pro-inflammatory cytokines and chemokines, generally, their type I and III IFN levels are lower (Blanco-Melo et al., 2020; Galani et al., 2021), which suggests possible virus-induced antagonism or skewing of antiviral responses. Notwithstanding this point, in one human study, higher serum IFN-λ levels were associated with less viral infection in the respiratory tract and more rapid viral clearance, and a higher IFN-λ to type I IFN ratio correlated with improved outcome (Galani et al., 2021). In the respiratory tract, IFN-λ expression varies with location, level of viral burden, and degree of disease severity and may have opposing roles at distinct anatomical sites in COVID-19 patients (Sposito et al., 2021). Thus, while IFN-λ expression appears to correlate inversely with COVID-19 severity, its mechanism(s) of protection is not well understood. Although IFN-λ has been studied in animals in the context of SARS-CoV-2 infection (Boudewijns et al., 2020; Broggi et al., 2020a; Dinnon et al., 2020; Sohn et al., 2021) and has been postulated to have a protective antiviral role, the responding cell types and targets of action have not been identified.
The emergence of SARS-CoV-2 variants (Beta, B.1.351; Gamma, B.1.1.28, Delta, B.1.617.2; and Omicron, B.1.1.529) with increasing antigenic divergence in the spike protein has highlighted a need for broad-spectrum antiviral agents that are less sensitive to viral evolution and the development of resistance. Hence, the potential benefits of host-target therapies, such as IFN-λ, have been discussed (Andreakos and Tsiodras, 2020; Prokunina-Olsson et al., 2020). Indeed, two clinical trials have explored the therapeutic effect of pegylated IFN-λ1 administered subcutaneously in COVID-19 patients, with one study showing decreased viral burden (Feld et al., 2021) but not another (Jagannathan et al., 2021). An additional trial of IFN-λ therapy for COVID-19 is ongoing (ClinicalTrials.gov: NCT04354259). Here, we investigated the potential efficacy and mechanistic actions of IFN-λ in the context of SARS-CoV-2 infection in mice. We found that Ifnlr1 −/− (also termed IL28Rα−/−) C57BL/6 mice infected with the B.1.351 or B.1.1.529 variant sustained higher viral burdens in the respiratory tract, indicating a protective role for IFN-λ against SARS-CoV-2 infection. When we administered recombinant murine IFN-λ2 by an intranasal route to K18-human (h)ACE2 transgenic mice or conventional 129S2 mice, as prophylaxis or therapy, we observed markedly reduced upper and lower respiratory tract infection and inflammation. Administration of nasally delivered IFN-λ2 several days before or after infection protected against viral infection in the lungs. IFN-λ was produced preferentially in epithelial cells and acted mainly on radio-resistant cells. Our data in mice suggest that IFN-λ has therapeutic potential as a less inflammatory, broad-spectrum antiviral agent against SARS-CoV-2 and its emerging variants.
Results
IFN-λ signaling contributes to the antiviral response against SARS-CoV-2
To assess the importance of IFN-λ signaling in protection against SARS-CoV-2 infection, we inoculated 6-week-old wild-type (WT) and congenic Ifnlr1 −/− C57BL/6 mice with 105 focus-forming units (FFU) of SARS-CoV-2 B.1.351 Beta variant, which contains K417Y, E484K, and N501Y substitutions in the spike receptor-binding domain (RBD) (Tegally et al., 2021). Prior studies have shown that the N501Y change in spike is mouse adapting and can enable binding to mouse ACE2 and infection of several laboratory strains of mice (Chen et al., 2021a; Li et al., 2021; Rathnasinghe et al., 2021; Shuai et al., 2021; Winkler et al., 2021; Zhang et al., 2021a). Ifnlr1 −/− mice showed higher viral RNA levels at 7 days post infection (dpi) in nasal washes and lung homogenates compared with WT mice (Figure 1A). Consistent with these data, we detected substantially higher levels of infectious virus by plaque assay in the lungs of Ifnlr1 −/− mice at 7 dpi (Figure 1B). Next, we investigated whether IFN-λ also had protective effects against the emerging SARS-CoV-2 B.1.1.529 Omicron variant, which has mutations that enable evasion against vaccines and therapeutic antibodies (Iketani et al., 2022; Planas et al., 2022; Zhang et al., 2021b). We inoculated 3-month-old WT and Ifnlr1 −/− mice with 105 FFU of B.1.1.529 and observed that Ifnlr1 −/− mice sustained higher levels of viral RNA in nasal turbinates, nasal washes, and lungs at 5 dpi (Figure 1C). Infectious virus titers also were greater in Ifnlr1 −/− than in WT mice in both nasal turbinates and lung homogenates (Figure 1D). Collectively, these data suggest that IFN-λ signaling has an antiviral role during SARS-CoV-2 variant infection in C57BL/6 mice.
Figure 1.

Increased susceptibility of Ifnlr1−/− mice to SARS-CoV-2 infection
(A and B) Six-week-old male and female C57BL/6 WT and Ifnlr1−/− mice were inoculated with 105 FFU of B.1.351 Beta variant.
(A) Viral RNA levels were measured from tissues at 7 dpi by qRT-PCR.
(B) Infectious virus was measured from tissues by plaque assay at 7 dpi (n = 9–11 per group, 2 experiments).
(C and D) Three-month-old female and male C57BL/6 WT and Ifnlr1−/− mice were inoculated with 105 FFU of B.1.1.529 Omicron variant.
(C) Viral RNA levels were measured at 5 dpi. Note that an earlier time point of analysis was used because B.1.1.529 is less pathogenic in mice.
(D) Infectious virus was measured at 5 dpi (n = 8–11 per group, 2 experiments).
Bars indicate median values. Data were analyzed by Mann-Whitney test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
Exogenous IFN-λ2 limits SARS-CoV-2 virus infection and inflammation in K18-hACE2 transgenic mice
We next evaluated the protective activity of exogenous IFN-λ2 against SARS-CoV-2 infection in mice. In a first set of experiments, we used K18-hACE2 transgenic mice, which express human ACE2 under regulation of the epithelial cell cytokeratin-18 promoter and are highly vulnerable to SARS-CoV-2-induced pneumonia and brain infection (Golden et al., 2020; Oladunni et al., 2020; Winkler et al., 2020). We first administered 2 μg of commercially available IFN-λ2 via intranasal or intraperitoneal route 16 h before inoculation with a historical WA1/2020 D614G SARS-CoV-2 strain. As prior experiments in K18-hACE2 mice showed peak SARS-CoV-2 infection within 2–4 days (Winkler et al., 2020), we used a day +3 time point to assess protection by IFN-λ2. Mice treated with IFN-λ2 by an intranasal route had markedly lower levels of viral RNA and infectious virus in the nasal turbinates, nasal washes, lungs, and brain at 3 dpi (Figures S1B and S1C), whereas animals treated by an intraperitoneal route did not show these reductions (Figure S1A). Based on these data, we used intranasal administration of IFN-λ2 for the remainder of our studies. We extended the window of prophylaxis in K18-hACE2 mice with a single intranasal dose of IFN-λ2 at day -2 (D-2) or -3 (D-3) before inoculation with WA1/2020 D614G. IFN-λ2 treatment at D-2 resulted in lower viral RNA levels in nasal turbinates, nasal washes, and lungs, but not in the brain, at 3 dpi (Figure 2A and S2A). Infectious virus levels in the lungs of IFN-λ2-treated animals were lower than in PBS-treated animals; however, there was no difference in the nasal turbinates of these two groups (Figure 2B). D-3 treatment with IFN-λ2 showed reduced viral RNA and infectious virus levels in the lungs at 3 dpi but not in other tissues (Figures S1D and S1E). Finally, we tested whether protection could be improved with two doses of IFN-λ2 treatment, one administered before and a second given after virus inoculation. K18-hACE2 mice were treated with 2 μg of IFN-λ2 via intranasal route at 16 h before and 8 h after intranasal inoculation with 103 FFU of WA1/2020 D614G. Notably, IFN-λ2 treatment prevented weight loss (Figure S1H) and showed reduced levels of viral RNA and infectious virus at 7 dpi in the nasal turbinates, nasal washes, lungs, and brain compared with PBS-treated mice (Figures S1I and S1J).
Figure 2.

Nasally delivered IFN-λ2 treatment protects K18-hACE2 mice against SARS-CoV-2 infection
(A–D) Eight-week-old female K18-hACE2 mice were inoculated by intranasal route with 103 FFU of WA1/2020 D614G. At D-2 (A and B) or D+1 and D+2 (C and D), mice were given a single 2-μg dose of murine IFN-λ2 or PBS by the intranasal route.
(A and C) Viral RNA levels were measured at 3 dpi.
(B and D) Infectious virus was measured at 3 dpi (A and B: n = 9 per group, 2 experiments; C and D: n = 8 per group, 2 experiments).
(E) Hematoxylin and eosin staining of lung sections from animals treated with 2-μg doses of murine IFN-λ2 or PBS by intranasal route at -16 h and +8 h relative to inoculation with WA1/2020 D614G and harvesting at 7 dpi. Low (top, scale bars, 500 μm) and high (bottom, scale bars, 100 μm) power images are shown. Representative images are from n = 5 per group except naive (n = 2).
(F) Quantitation of hematoxylin+ area as an index of cellularity in lung sections in (E).
(G) Eight-week-old female K18-hACE2 mice were treated with 2 μg of murine IFN-λ2 or PBS at -16 h and challenged with 103 FFU of WA1/2020 D614G. Heatmaps of cytokine levels in lung homogenates at 3 dpi. Fold change was calculated relative to mock-infected mice, and log2 values are plotted (2 experiments, n = 7 per group except naive [n = 4]).
(H–K) Five-month-old female K18-hACE2 mice were inoculated with 103 FFU of B.1.1529 Omicron variant. At D-1 (H and I) or D+1 and D+2 (J and K), mice were given 2 μg of murine IFN-λ2 or PBS by the intranasal route. Viral RNA (H–J) and infectious (I–K) virus levels were measured at 3 dpi (H and I: n = 7–8 per group, 2 experiments; J and K: n = 6–7 per group, 2 experiments).
Bars (A–D and G–J) indicate median values. Data were analyzed by Mann-Whitney tests (A–D and H–K) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
See also Figures S1, S2, and S3.
We next explored the therapeutic efficacy of IFN-λ2. K18-hACE2 mice were administered a single 2-μg dose of IFN-λ2 via nasal route at 8 h after infection, and animals were sacrificed at 3 dpi. IFN-λ2-treated mice showed reduced viral RNA levels in the nasal turbinates, lungs, and brain (Figure S1F) and infectious virus titers in the nasal turbinates and lungs (Figure S1G). However, therapeutic administration of IFN-λ2 did not reduce viral burden in nasal washes compared with PBS-treated animals (Figure S1F). We also administered IFN-λ2 as a two-dose therapy at 1 (D+1) and 2 (D+2) dpi, which resulted in lower viral RNA loads in nasal turbinates and lungs but not in nasal washes or the brain (Figures 2C and S2B). Infectious virus levels also were decreased in the lungs with this IFN-λ2 treatment scheme (Figure 2D).
Some COVID-19 patients develop hyper-inflammatory immune responses, which may contribute to respiratory failure (Andreakos and Tsiodras, 2020; Galani et al., 2021). Given that IFN-λ2 treatment reduced viral levels in the lung, we hypothesized that it might mitigate immune responses and lung disease. Lung tissues were collected from IFN-λ2 or PBS-treated mice at 7 dpi and sectioned for histological analysis; this time point was selected since lung pathology in K18-hACE2 mice is greater at 7 than 3 dpi. Lungs from PBS-treated, SARS-CoV-2-infected K18-hACE2 mice showed diffusely infiltrating immune cells with alveolar space consolidation consistent with pneumonia, whereas this was observed to a significantly lesser degree in IFN-λ2-treated animals (Figures 2E and 2F). Measurement of cytokine and chemokines in lung homogenates at 3 dpi showed decreased levels of granulocyte colony-stimulating factor (G-CSF), IL-1β, IL-6, CXCL10, CCL2, and tumor necrosis factor alpha (TNF-α) in IFN-λ2-treated K18-hACE2 mice (Figures 2G and S3). These results suggest that treatment with IFN-λ2 can protect mice against SARS-CoV-2 by inhibiting lung infection and inflammation.
We evaluated whether exogenous IFN-λ2 treatment could also protect K18-hACE2 mice from the B.1.1.529 Omicron variant. First, we administered mice a single 2-μg dose of IFN-λ2 at D-1. IFN-λ2-treated mice had lower levels of B.1.1.529 viral RNA in nasal turbinates, nasal washes, and lungs (Figure 2H), as well as infectious virus in lungs, than PBS-treated animals (Figure 2I). Our two-dose therapeutic regimen at D+1 and D+2 also reduced levels of B.1.1.529 viral RNA and infectious virus in the lungs but not in the nasal turbinates or washes (Figures 2J and 2K). While performing these studies, we observed an absence of viral RNA in the brain of PBS-treated B.1.1.529-infected K18-hACE2 mice (Figures S2C and S2D) and low levels of infection in nasal turbinates (Figures 2I and 2K), which is consistent with recent studies suggesting B.1.1.529 is less pathogenic in rodents (Halfmann et al., 2022). Nonetheless, our experiments demonstrate that exogenous IFN-λ2 protects against B.1.1.529 infection in K18-hACE2 mice.
Exogenous IFN-λ2 limits SARS-CoV-2 infection and inflammation in 129S2 mice
To confirm our results in another model of SARS-CoV-2 infection, we treated and challenged 129S2 mice, which are susceptible to SARS-CoV-2 strains (e.g., B.1.351) with an N501Y mouse-adapting mutation, more so than C57BL/6 mice (Chen et al., 2021a; Li et al., 2021; Rathnasinghe et al., 2021; Shuai et al., 2021; Zhang et al., 2021a). As prior studies in 129S2 mice showed high levels of SARS-CoV-2 infection at 4 dpi (Ying et al., 2022), we harvested samples at this time point. Nasal administration of IFN-λ2 at D-1 protected B.1.351-infected mice from weight loss (Figure 3A) and reduced viral burden in nasal turbinates, nasal washes, lungs, and brain (Figures 3B, 3C, and S2E). When we extended the prophylaxis window to D-3 or D-5, IFN-λ2 reduced infection-induced weight loss (Figures 3D–3G) and viral RNA and infectious virus levels in nasal turbinates and lungs but not in nasal washes (Figures 3E, 3F–3I), although the protective effect at D-5 was smaller in magnitude. Indeed, 129S2 mice treated at D-3 but not D-5 with IFN-λ2 had less viral RNA in the brain that those administered PBS (Figures S2F and S2G). We next evaluated the effect of two 2-μg doses of IFN-λ2 -16 h and +8 h infection on B.1.351 infection. Infected 129S2 mice treated with PBS showed about 15% weight loss by 4 dpi, whereas IFN-λ2-treated animals did not (Figure 3J). Levels of viral RNA and infectious virus levels were reduced in the nasal turbinates, nasal washes, lungs, and brain of IFN-λ2-treated compared with PBS-treated mice (Figures 3K, 3L, and S2H).
Figure 3.

IFN-λ2 treatment protects 129S2 mice against SARS-CoV-2 infection
(A–I) Six-week-old female 129S2 mice were inoculated by intranasal route with 105 FFU of B.1.351 Beta variant. At D-1 (A-C), D-3 (D–F), or D-5 (G–I), mice were given a single 2-μg dose of murine IFN-λ2 or PBS by intranasal route.
(A, D, and G) Weight change.
(B, E, and H) Viral RNA levels at 4 dpi.
(C, F, and I) Infectious virus levels at 4 dpi (A–C: n = 7 per group, 2 experiments; D–F: n = 6–8 per group, 2 experiments; G–I: n = 6–8 per group, 2 experiments).
(J–L) Six-week-old female 129S2 mice were inoculated by intranasal route with 105 FFU of B.1.351. At -16 h and +8 h, mice were administered 2 μg of murine IFN-λ2 or PBS by intranasal route.
(J) Weight change.
(K) Viral RNA levels at 4 dpi.
(L) Infectious virus levels at 4 dpi in (n = 8 per group, 2 experiments).
(M) Hematoxylin and eosin staining of lung sections at 4 dpi from animals treated in (J)–(L). Low (top, scale bars, 500 μm) and high (bottom, scale bars, 100 μm) power images are shown (representative of n = 5 per group).
(N) Heatmaps of cytokine levels in lung homogenates at 4 dpi from animals treated in (J)–(L). Fold change was calculated compared with mock-infected mice, and log2 values were plotted (n = 8 per group except naive [n = 4], 2 experiments).
Bars (B–C, E–F, H–I, and K–L) indicate median values. Data were analyzed by Mann-Whitney tests (B–C, E–F, H–I, and K–L) or t tests of the area under the curve (A, D, G, and J) (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
See also Figures S2 and S4.
Lung sections from B.1.351-infected, PBS-treated 129S2 mice at 4 dpi showed mild to moderate immune cell infiltration, extravasation of erythrocytes into the alveolar space, and pulmonary vascular congestion, whereas those treated with IFN-λ2 appeared more like uninfected, naive mice (Figure 3M). Consistent with these data, IFN-λ2-treated mice had reduced levels of the pro-inflammatory cytokines and chemokines that were elevated in B.1.351-infected PBS treated mice including IL-1β, IL-6, CXCL10, CCL2, CCL4, and CCL5 (Figures 3N and S4). Collectively, our data establish a protective effect of IFN-λ2 against SARS-CoV-2 infection in multiple strains of mice.
IFN-λ2 transcriptional signature in the lung
To begin to understand how IFN-λ2 protects against SARS-CoV-2 in the lung, we performed bulk RNA sequencing on tissues obtained from naive animals or animals treated IFN-λ2 via the intranasal route. Principal-component analysis showed distinct transcriptional signatures in the lungs of IFN-λ2-treated mice at 1 (D+1) or 3 (D+3) days after treatment compared with naive mice. The transcriptional signature in the lung at D+1 after IFN-λ2 was distinct from naive animals, whereas by D+3, the signature started to return to baseline (Figure 4A). We identified 1,820 and 1,317 differentially expressed genes (DEGs) in the D+1 and D+3 IFN-λ2-treated groups, respectively, and 856 DEGs were identified between the D+1 and D+3 groups (Figure 4B). We performed Metascape analysis to define biological pathways enriched in the IFN-λ2-treated groups compared with the naive group. Among the top enriched up-regulated pathways in both the D+1 and D+3 groups relative to the naive group were extracellular matrix-organization signaling (e.g., Col2a1, Col5a2, Lampb3, and Mmp15), regulation of cell-adhesion signaling (e.g., Vegfc, Jam2, and Cav1), response to wounding signaling (e.g., CD36, Timp1, and Col3a1), and negative regulation of cytokine-production signaling (e.g., Klf2, Arg2, and Foxj1) (Figures 4C, 4D, and S5). Although these pathways were enriched in both groups, expression of these genes in the D+3 group was lower (Figures 4C, 4D, and S5), suggesting that the effect of IFN-λ2 had begun to wane. In comparison, other transcriptional programs were uniquely expressed in the D+1 group, including responses to IFN-α signaling (e.g., Oas1a, Ifit2, and Bst2) and virus signaling (e.g., Cxcl10, Rsad2, Isg15, Irf7, and Ifit1) (Figures 4C and 4D), suggesting that these antiviral signals are induced quickly and decline rapidly once the stimulus is lost. Other pathways transcriptionally induced by IFN-λ2 at D+1 only included T cell-mediated cytotoxicity signaling (e.g., H2-q1, H2-q7, H2-k1, and Tap2) and morphogenesis of a branching epithelium signaling (e.g., Wnt2, Foxc2, and Myc) (Figures 4C, 4D, and S5). Biological pathways that were downregulated in D+1 and D+3 groups compared with naive samples included sodium ion transport signaling, protein citrullination signaling, and potassium ion transmembrane transport signaling. Some pathways that were downregulated only in the D+1 group included responses to xenobiotic stimulus signaling and negative regulation of lipid metabolic process signaling (e.g., Apobec1, Serpina12, and Gper1).
Figure 4.

Transcriptional signatures in the lungs of mice treated with murine IFN-λ2
(A–D) RNA sequencing of lung homogenates of naive female K18-hACE2 mice (control) or mice treated with 2 μg of murine IFN-λ2 by intranasal route for 1 (D+1) or 3 (D+3) days.
(A) Three-dimensional map from principal-component analysis. Each group is represented by an ellipse and the color-matched solid circle, which is the centroid of each group. The size of the ellipse is the centroid with one standard deviation. The dashed red lines with numbers indicate the spatial distance between centroids of the 3 groups, which is calculated by using the three-dimensional coordinates for the centroids.
(B) Venn diagrams of overlapping genes identified in differential expression analysis when comparing with control, D+1, and D+3 groups. Numbers in the parenthesis under each comparison indicate differentially expressed genes (fold change ≥ 2 at p < 0.05), followed by the proportion that are up- or down-regulated.
(C) The significantly enriched biological processes defined by a Metascape pathway analysis tool comparing control, D+1, and D+3 groups; up-regulated (brown) or down-regulated (blue) genes in the IFN-λ2-treated group (D+1 or D+3) compared with the control group or in the D+1 group compared with the D+3 group.
(D) Heatmaps of selected biological processes enriched in the D+1 group or the D+3 group versus the control group (n = 4 per group).
(E) mRNA levels of indicated target genes were measured from the lung homogenates of naive female K18-hACE2 mice or mice treated with 2 μg of murine IFN-λ2 by intranasal route for D+1 or D+3 days (n = 8–10 per group, 2 experiments).
Data in (E) were analyzed by one-way ANOVA with Dunnett’s post-test (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
See also Figure S5.
We validated our bulk RNA sequencing data by qRT-PCR by measuring expression of several ISGs including Ifit1, Isg15, and Rsad2 that can respond to IFN-λ signaling (Jilg et al., 2014; Lazear et al., 2019; Shindo et al., 2013). Notably, these ISG expression levels were upregulated at D+1 and diminished at D+3 (Figure 4E). We did not observe changes in mRNA expression of Ace2, which can be modulated by type I IFN (Ziegler et al., 2020), or Tmprss2 (Figure 4E), two key genes involved in SARS-CoV-2 attachment and entry, suggesting they do not respond to IFN-λ signals in mice. Collectively, our data demonstrate that the transcriptional program induced by IFN-λ2 is characterized by a short burst of expression of antiviral and cell-to-cell communication gene programs. However, we did not observe higher levels of nuclear factor κB (NF-κB) genes (e.g., Il6, Il1β, and Tnfα), which can be strongly induced by type I IFN (Galani et al., 2017), indicating that IFN-λ selectively induces antiviral, but not highly pro-inflammatory, genes.
IFN-λ is preferentially produced by epithelial cells during SARS-CoV-2 infection
We investigated which cell type(s) in the lung preferentially produce IFN-λ after SARS-CoV-2 infection in vivo. To determine an optimal viral dose for monitoring IFN-λ expression levels in vivo, we inoculated WT C57BL/6 mice with 105 or 106 FFU of B.1.351; we observed that Ifnl2 and Ifnl3 mRNA expression levels were upregulated at 2 dpi, with higher levels induced in mice inoculated with 106 FFU than with 105 FFU of B.1.351 (Figures 5A and S6A). Based on these results, we chose to use the 106 FFU dose for subsequent expression studies. To identify the cell types expressing IFN-λ mRNA, at 2 dpi, we sorted under BSL3 conditions lung epithelial cells and different immune cells populations (alveolar macrophages, monocytes, neutrophils, B cells, T cells, and dendritic cells [DCs]) and then performed qRT-PCR for the two IFN-λ transcripts in mice (Figures S6B–S6E). CD45-CD326+ lung epithelial cells had the highest levels of Ifnl2 and Ifnl3 mRNA expression with CD45+CD11c+ Siglec F−MHCII+ DCs showing the next highest expression; the other cell types analyzed had limited mRNA expression of Ifnl2 and Ifnl3 (Figure 5B). As expected, and based on the literature (Galani et al., 2017; Lazear et al., 2019), the Ifnlr1 receptor was expressed mainly on CD45−CD326+ epithelial cells and CD45+CD11b+Ly6G+ neutrophils (Figure 5B). To corroborate these results, we utilized Ifnl2-Egfp reporter mice (Galani et al., 2017) to evaluate IFN-λ expression. EGFP was induced at 2 dpi and localized mostly to CD326+ epithelial cells lining the bronchial walls and not the lung parenchyma (Figure 5C). The signal was specific, as we did not detect EGFP expression in non-transgenic control WT C57BL/6 mice (Figure S6F). We also investigated the tropism of SARS-CoV-2 B.1.351 after infection in the lung. Immunofluorescence microscopy staining for SARS-CoV-2 nucleocapsid protein showed that viral antigen was expressed in airway tract epithelium and co-localized with CD326+ epithelial cells (Figure 5D). This pattern suggests that epithelial cells are a dominant cell type targeted for infection by SARS-CoV-2 and a source of IFN-λ production in the lower respiratory tract. We also investigated whether IFN-λ was produced in SARS-CoV-2-infected cells. IFN-λ EGFP reporter gene expression co-localized with SARS-CoV-2 nucleocapsid protein, although we also observed an EGFP signal in non-infected cells (Figure 5E). Thus, in the lung, IFN-λ appears to be produced in both infected and non-infected cells.
Figure 5.

IFN-λ expression in vivo
(A) Six-week-old male and female C57BL/6 mice were inoculated with 106 FFU of B.1.351 Beta variant. Ifnl2 and Ifnl3 mRNA levels from lungs were measured at indicated days post-infection by qRT-PCR (n = 4–9 per group, 2 experiments) (ND, not detectable; qRT-PCR Ct value >40).
(B) Six-week-old male and female C57BL/6 mice were inoculated with 106 FFU of B.1.351. Ifnl2, Ifnl3, and Ifnlr1 mRNA expression levels were measured at 2 dpi (n = 4 per group, each dot represents 4 mice pooled together, 2 experiments).
(C–E) Six-week-old C57BL/6 or Ifnl2-Egfp reporter mice were inoculated with 106 FFU of SARS-CoV-2 B.1.351.
(C) Localization of EGFP and epithelial cells (ECs; CD326) in the lungs of Ifnl2-Egfp reporter mice at 2 dpi. Frozen sections stained for GFP (green), CD326 (magenta), and Hoechst (blue) are shown.
(D) Localization of SARS-CoV-2 nucleocapsid protein (NP) antigen and ECs in the lung of mice at 2 dpi. Frozen sections stained for SARS-CoV-2 NP (green), CD326 (magenta), and Hoechst (blue) are shown.
(E) Localization of EGFP and SARS-CoV-2 NP antigen in cells of lungs of Ifnl2-Egfp reporter mice at 2 dpi. Frozen sections stained for GFP (green), NP antigen (magenta), and Hoechst (blue) are shown.
Scale bars in (C)–(E): 50 μm.
(F and G) Six-week-old male and female WT, Mavs−/−, cGas−/−, or Myd88−/− C57BL/6 mice were inoculated with 106 FFU of B.1.351. Viral RNA levels (F) or Ifnl2 and Ifnl3 mRNA expression levels (G) from lungs were measured at 2 dpi (n = 6–10, 2 experiments). Bars in (F) indicate median values. Data in (F) and (G) were analyzed by one-way ANOVA with Dunnett’s post-test (ns, not significant; ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001).
See also Figure S6.
We next evaluated which pathogen recognition receptor signaling pathways induced IFN-λ expression. Since IFNs can be activated though TLRs, RLRs, or cGAS-STING pathways after viral infections (Park and Iwasaki, 2020), we repeated B.1.351 infection experiments in Mavs −/−, cGas −/−, and Myd88 −/− C57BL/6 mice. In naive WT, Mavs −/−, cGas −/−, and Myd88 −/− mice, IFN-λ expression was very low, as transcripts were below our threshold of detection (qRT-PCR cycle threshold [Ct] value >40) (Figure S6G). At 2 dpi, levels of Ifnl2 and Ifnl3 mRNA in the lung were remarkably decreased in both Mavs −/− and Myd88 −/− mice, but not in cGas −/− mice, compared with WT mice (Figure 5G). Viral RNA levels were relatively equivalent among different mouse genotypes at this early time point (Figure 5F), suggesting that the differences in IFN-λ expression levels were not skewed by viral burden and that the antiviral effect conferred by IFN-λ in the lung requires several days to manifest. Overall, our data suggest that in the lungs of mice after SARS-CoV-2 infection, IFN-λ is produced though both MAVS- and MyD88-dependent signaling pathways.
IFN-λ signaling in radio-resistant cells controls SARS-CoV-2 infection in the lung
As our qRT-PCR data demonstrated, in the lung, IFN-λ receptors (IFNLR1/IL10Rβ) are expressed in epithelial cells and some immune cells, including neutrophils (Broggi et al., 2017; Lazear et al., 2019). To determine which cell type contributed to the protective effect mediated by IFN-λ against SARS-CoV-2 in vivo, we first depleted circulating neutrophils with anti-Ly6G (1A8 monoclonal antibody [mAb]) in the context of IFN-λ2 treatment (Figures S7A–S7C). Depletion of neutrophils in the blood and lung had no impact on the reduction in weight loss or viral burden conferred by IFN-λ2 (Figures 6A–6C). We next generated reciprocal sets of chimeric animals in which the radio-resistant compartment or radio-sensitive hematopoietic cells lacked that capacity for IFN-λ signaling using donor WT or Inflr1 −/− bone narrow and sublethally irradiated WT or Inflr1 −/− recipient mice (Figures 6D and S7D). Animals lacking IFN-λ signaling in the radio-resistant compartment sustained similar levels of infection in the nasal washes as fully Ifnlr1 −/− mice, whereas animals lacking IFN-λ signaling in hematopoietic cells had similar levels of viral RNA as mice with intact IFN-λ signaling in all cells (Figure 6E). In the lungs, similar trends were observed with higher levels of viral RNA in animals lacking Ifnlr1 in the radio-resistant cell compartment (Figure 6E). In the nasal turbinates, the data were more nuanced, where both radio-resistant and -sensitive Ifnlr1 signaling cell populations appear to contribute to IFN-λ-dependent control of SARS-CoV-2 infection (Figure 6E). To determine which cells contributed to the protective response after exogenous IFN-λ treatment, we repeated experiments with bone marrow chimeric mice in the setting of treatment at D-1 with IFN-λ2. Mice lacking IFN-λ signaling only in the radio-sensitive cell compartment were protected against SARS-CoV-2 infection, whereas animals lacking IFN-λ signaling in the radio-resistant cell compartment lost the protective effect of IFN-λ2 and sustained higher viral burden similar to that seen in Ifnlr1 −/− mice (Figures 6D, 6F, and 6G). Overall, our data suggest that IFN-λ signaling protects mice against SARS-CoV-2 infection and depends dominantly on signaling in radio-resistant cells in the lung.
Figure 6.

IFN-λ signaling in radio-resistant cells protects against SARS-CoV-2 infection
(A–C) Six-week-old female 129S2 mice received anti-Ly6G or isotype control antibodies by intraperitoneal injection at D-1, D+1, and D+3 relative to B.1.351 Beta variant infection (105 FFU). Mice also were treated with 2 μg of murine IFN-λ2 or PBS at -16 h and +8 h by the intranasal route.
(A) Weight change.
(B) Viral RNA levels at 4 dpi.
(C) Infectious virus levels at 4 dpi (n = 8 per group, 2 experiments).
(D) Experimental scheme for generating of WT and Ifnlr1−/− bone marrow chimeric mice. Ten weeks after irradiation, mice were inoculated by the intranasal route with 105 FFU of B.1.351.
(E) Viral RNA levels at 7 dpi (n = 13–15 per group, 3 experiments). Bars indicate median values.
(F and G) Chimeric mice from (D) were given 2 μg of murine IFN-λ2 at D-1 by intranasal route and then inoculated with 105 FFU of B.1.351. Viral RNA levels at 3 dpi (F). Infectious virus levels at 3 dpi (G) (n = 8–9 per group).
Data were analyzed by one-way ANOVA with Dunnett’s post-test of the area under the curve (A) and Kruskal-Wallis test with Dunnett’s post-test (B–C and E–G) (ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
See also Figure S7.
Discussion
In humans and other animals, SARS-CoV-2 targets the respiratory tract, which can result in the development of pneumonia, ARDS, and death (Guan et al., 2020; Huang et al., 2020). While existing neutralizing antibodies and vaccines against SARS-CoV-2 have conferred protection for many individuals, their efficacy is jeopardized by emerging variants that have increasing numbers of amino-acid substitutions in the spike protein (Baum et al., 2020; Chen et al., 2021b; Hoffmann et al., 2021; Liu et al., 2021). Thus, therapeutic approaches are needed that can overcome viral resistance. IFN-λ induces hundreds of ISGs and has protective functions against many different virus infections, at least in cell-culture and animal models (Lazear et al., 2019; Park and Iwasaki, 2020). Also, IFN-λ preferentially functions at mucosal sites, including the respiratory tract, because of the selected cellular expression of IFNLR1, a subunit of its receptor (Broggi et al., 2020b; Lazear et al., 2019). While type I IFN is also antiviral and has greater potency, treatment is often associated with collateral systemic effects and inflammation. For these reasons, we investigated the potential of IFN-λ in preventing and treating SARS-CoV-2 infection. Our data in mice show that IFN-λ can protect against infection by two variants (B.1.351 and B.1.1.529) and diminish inflammatory responses in the lung. In the context of SARS-CoV-2 infection, IFN-λ in the lung was produced primarily by epithelial cells and acted on radio-resistant cells to confer protection.
Host-derived innate immune responses have the potential to limit the impact of viral evolution since multiple genes and pathways contribute to inhibitory responses. Nonetheless, virus-mediated attenuation of innate immune antiviral response occurs and is linked to SARS-CoV-2 disease severity (Blanco-Melo et al., 2020; Galani et al., 2021; Sposito et al., 2021). Indeed, serum IFN-λ levels are low in patients with severe COVID-19, yet those with higher levels have better outcomes (Blanco-Melo et al., 2020; Galani et al., 2021). Related to this observation, high levels of IFN-λ in the upper respiratory tract were associated with higher viral burden but less disease severity, whereas patients with severe COVID-19 had elevated IFN-λ levels in the lower respiratory tract (Sposito et al., 2021). In mice, we detected IFN-λ gene expression in the lung within days of SARS-CoV-2 infection, and Ifnlr1 −/− mice lacking IFN-λ signaling sustained higher viral burden in the upper and lower respiratory tracts, suggesting that IFN-λ can protect against SARS-CoV-2 infection in vivo.
Because of the potential of IFN-λ as a broadly acting therapy, we evaluated its antiviral activity in vivo. Notably, equivalent doses of IFN-λ2 delivered by a nasal, but not systemic, route could limit SARS-CoV-2 infection. The basis for this disparity remains uncertain, although higher doses given by a peripheral route might have protective effects, as was seen by others after subcutaneous administration of pegylated forms of IFN-λ (Dinnon et al., 2020). Post-exposure therapy with IFN-λ2 also conferred protection in the lung in mice, but the antiviral effects in other tissues were diminished, suggesting that once infection is established in the upper airway and viral evasion mechanisms are induced, IFN-λ2 therapy may have less benefit. Randomized clinical trials in COVID-19 patients administered pegylated IFN-λ by a subcutaneous route within 3 to 7 days of symptom onset or diagnosis have been mixed: one showed benefit but the other did not (Feld et al., 2021; Jagannathan et al., 2021). Thus, the treatment route, dosing, and formulation of IFN-λ may require further optimization. By testing two key variants (B.1.351 and B.1.1.529), we established that intranasally delivered IFN-λ2 could protect broadly against antigenically distinct SARS-CoV-2 isolates in mice and thus may be less susceptible to immune escape than monoclonal- or serum-derived antibodies (Baum et al., 2020; Chen et al., 2021a; Hoffmann et al., 2021; Liu et al., 2021).
Even a single dose of IFN-λ at D-5 conferred partial protection in mice, demonstrating a persistent antiviral effect. The basis for this durable inhibitory effect remains uncertain especially in light of our transcriptional profiling data in the lung, which showed a rapid induction and then dampening of gene induction. We speculate that the half-life of certain inhibitory ISG products may be longer or that transcriptional activation downstream of IFN-λ signaling may have distinct kinetics in the upper airway. Alternatively, immune cells might become “trained” by IFN-λ to protect against subsequent viral infection, as described for other innate immune stimuli and bacterial or viral infections (Netea et al., 2020; Zhang et al., 2014). We used soluble IFN-λ in our administration scheme. It remains possible that the window of prevention and clinical utility could be extended by administration of longer-acting (e.g., pegylated) forms of IFN-λ.
Type I IFNs have been used to treat several viral diseases including chronic hepatitis C virus (HCV) and human papillomavirus (HPV) (Lazear et al., 2019). Although type I IFNs have garnered interest as a treatment strategy in COVID-19 (Palermo et al., 2021; Park and Iwasaki, 2020; Schreiber, 2020), their ability to exacerbate inflammation has tempered enthusiasm. One group tried to overcome this limitation by administering type I IFN by an intranasal route; in hamsters, they showed that nasally delivered type I IFN could reduce viral burden, prevent virus transmission, and lower inflammation in vivo (Hoagland et al., 2021). In our mouse models, administration of IFN-λ protected mice from infection, weight loss, lung inflammation, and lung disease, suggesting that the less pro-inflammatory nature of IFN-λ (Lazear et al., 2019) may have advantages as a therapeutic strategy. Our RNA sequencing data also showed that IFN-λ treatment induced genes that are associated with tissue repair, which contrasts with some studies showing that persistent type I or type III IFN signaling can disrupt lung epithelial barriers and prevent tissue repair (Broggi et al., 2020a; Major et al., 2020). Nonetheless, administration of IFN-λ later in the course of SARS-CoV-2 infection, when most of the disease is caused by the host response and not by viral replication, could be detrimental and warrants further study.
By leveraging flow cytometry, qRT-PCR, and Ifnl2-gfp reporter mice, we found that IFN-λ was produced predominantly in lung epithelial cells after SARS-CoV-2 infection. This observation agrees with experiments after influenza A virus infection (Galani et al., 2017). We also showed IFN-λ acted primarily on radio-resistant cells in the lung to confer protection against SARS-CoV-2 infection, which is consistent with recent findings (Broggi et al., 2020a). Nonetheless, others have shown that IFN-λ can be produced by DCs in the lung via a TLR3 signaling pathway (Broggi et al., 2020a); that study used poly(I:C), rather than SARS-CoV-2 infection, as the inflammatory stimulus in vivo. The differences in cell-type-specific production of IFN-λ between studies could be due in part to the preferential expression of TLR3 in immune cells (Wu and Chen, 2014). Others have suggested that IFN-λ signaling in neutrophils is required for optimal antifungal or antiviral defenses or limiting tissue damage (Broggi et al., 2017; Espinosa et al., 2017; Galani et al., 2017); our neutrophil-depletion studies showed no effect on IFN-λ-mediated protection against SARS-CoV-2 infection or weight loss in mice. The basis for the difference is uncertain but could be due to the disparate models of pathogen infection or inflammation.
Limitations of the study
Although our experiments establish a role for IFN-λ in protecting against infection by SARS-CoV-2 strains including B.1.1.529, we acknowledge several limitations to our study. (1) We used female mice in our IFN-λ treatment models, so studies in male animals are needed to exclude sex-based differences in therapeutic effects. Notwithstanding this point, another group recently showed protective effects of IFN-λ against SARS-CoV-2-induced death in male K18-hACE2 mice (Sohn et al., 2021). (2) The relationship between induction of IFN-λ responses in mice and COVID-19 patients is unclear, especially given that many patients with severe disease have blunted IFN responses. While some of the diminished type I IFN response may be due to autoantibodies (Bastard et al., 2020; van der Wijst et al., 2021), the presence of such inhibitors against IFN-λ has not been described. (3) Although our neutrophil-depletion and bone marrow chimera studies suggest that radio-resistant cells respond to IFN-λ to confer a protective antiviral effect, the precise cell type was not defined. Future studies with Ifnlr1 fl/fl conditional knockout mice are required to fully address this question. (4) Our studies are restricted to mice. IFN-λ treatment experiments in other animals (e.g., hamsters, ferrets, or nonhuman primates) and ultimately humans are needed for corroboration and determination of clinical utility.
In summary, we present evidence that nasal administration of IFN-λ confers pre- and post-exposure protection against several SARS-CoV-2 strains including key variants of concern without causing extensive inflammation. In the lung, IFN-λ is induced in a MAVS- and MyD88-dependent manner primarily in epithelial cells, and acts upon radio-resistant cells to control infection. Additional studies are warranted to evaluate further the potential of IFN-λ as a broadly acting antiviral agent against SARS-CoV-2.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC/Cyanine7 anti-mouse CD45 Antibody | BioLegend | 103115RRID: AB_312980 |
| APC anti-mouse/human CD11b Antibody | BioLegend | 101212RRID: AB_312795 |
| Brilliant Violet 421™ anti-mouse Ly-6G Antibody | BioLegend | 127627RRID: AB_10897944 |
| Brilliant Violet 421 anti-mouse CD11c Antibody | BioLegend | 117329RRID: AB_10897814 |
| PE anti-mouse CD170 Antibody (Siglec F) | BioLegend | 155506RRID: AB_2750235 |
| PE anti-mouse CD19 Antibody | BioLegend | 115508RRID: AB_313643 |
| PE anti-mouse CD326 Antibody (Ep-CAM) | BioLegend | 118205RRID: AB_1134176 |
| PE anti-mouse Ly6C Antibody | BioLegend | 128007RRID: AB_1186133 |
| Brilliant Violet 421™ anti-mouse CD3 Antibody | BioLegend | 100228RRID: AB_2562553 |
| eBioscience™ Fixable Viability Dye eFluor™ 506 | Invitrogen | 65-0866-14 |
| TruStain FcX™ anti-mouse CD16/32 Antibody | BioLegend | 101320RRID: AB_1574975 |
| Alexa Fluor® 700 anti-mouse I-A/I-E Antibody (MHC II) | BioLegend | 107622RRID: AB_493727 |
| FITC anti-mouse CD45.1 Antibody | BioLegend | 110705RRID: AB_313494 |
| APC anti-mouse CD45.2 Antibody | BioLegend | 109813RRID: AB_389210 |
| Brilliant Violet 421™ anti-mouse CD45 Antibody | BioLegend | 103134RRID: AB_2562559 |
| Alexa Fluor® 700 anti-mouse Ly-6C Antibody | BioLegend | 128024RRID: AB_256255 |
| PE/Cyanine7 anti-mouse Ly-6G Antibody | BioLegend | 127618RRID: AB_1877261 |
| FITC anti-mouse Ly6B antibody | Abcam | ab53453RRID: AB_881408 |
| CD326 (EpCAM) Monoclonal Antibody (G8.8) | eBioscience | 14-5791-81RRID: AB_953624 |
| SARS-CoV-2 Nucleocapsid Antibody | Sino Biological | 40143-R001RRID: AB_2827974 |
| Anti-Green Fluorescent Protein Antibody | Aves Labs | GFP-1010RRID: AB_2307313 |
| Goat anti-Chicken IgY (H + L) Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-11039RRID: AB_2534096 |
| Chicken anti-Rat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | A-21472RRID: AB_2535875 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-21206RRID: AB_2535792 |
| Critical Commercial Assays | ||
| MagMAX-96 Viral RNA Isolation Kit | Thermo Fisher | AM1836 |
| TaqMan RNA-to-Ct 1-Step Kit | Thermo Fisher | 4392939 |
| Mouse Cytokine Array/Chemokine Array 31-Plex (MD31) | Eve Technologie | MD31 |
| Collagenase, Type 1 | Worthington | LS004194 |
| DNase I | Roche | 10104159001 |
| Dead Cell Removal Kit | STEMCELL | 17899 |
| RNeasy Plus Mini Kit | QIAGEN | 74134 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Bacterial and Virus strains | ||
| SARS-CoV-2 WA/20 D614G | Chen et al., 2021b | N/A |
| SARS-CoV-2 B.1.351 | Chen et al., 2021b | N/A |
| SARS-CoV-2 B.1.1.529 | VanBlargan et al., 2022 | N/A |
| Experimental Models: Cell Lines | ||
| Vero-TMPRSS2 | Chen et al., 2021b | N/A |
| Vero-TMPRSS2-ACE2 | Chen et al., 2021b | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | 000664 |
| RRID:IMSR_JAX:000664 | ||
| 129S2/SvPasCrl | Charles River | 476 |
| RRID:IMSR_CRL:476 | ||
| B6. Cg-Tg(K18-ACE2)2Prlmn/J | Jackson Laboratory | 034860 |
| RRID:IMSR_JAX:034860 | ||
| Oligonucleotides | ||
| SARS-CoV-2 N F: 50-ATGCTGCAATCGTGCTACAA-30 | 32838945 | N/A |
| SARS-CoV-2 N R: 50-GACTGCCGCCTCTGCTC-30 | 32838945 | N/A |
| SARS-CoV-2 N Probe: 50-/56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/-30 | 32838945 | N/A |
| Gapdh qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.39a.1 |
| Isg15 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.41476392.g |
| Rsad2 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.11280480 |
| Ifit1 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.32674307 |
| Ace2 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.8312550 |
| Tmprss2 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.28840201 |
| Ifnl2 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.31485549 |
| Ifnl3 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.8956530 |
| Ifnl1 qPCR primer + probe | IDT PrimeTime Assay | Mm.PT.58.10781457 |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | v10 |
| GraphPad Prism | GraphPad | v8 |
| ImageJ | National Institute of Health | V1.53 |
| Nanozoomer Digital Pathology | Hamamatsu | v2 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Michael S. Diamond (mdiamond@wustl.edu).
Materials availability
All requests for resources and reagents should be directed to the lead contact author. This includes mice, antibodies, viruses, and proteins. All reagents will be made available on request after completion of a Materials Transfer Agreement (MTA).
Experimental model and subject details
Cells and viruses
Vero-TMPRSS2 and Vero-TMPRSS2-ACE2 cells (Chen et al., 2021b) were cultured at 37°C in Dulbecco’s Modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES pH 7.3, and 100 U/mL of penicillin–streptomycin. The SARS-CoV-2 WA1/2020 D614G virus was produced from an infectious clone and has been described previously (Chen et al., 2021b). The B.1.351 and B.1.1529 viruses were isolated from infected individuals (Chen et al., 2021a; VanBlargan et al., 2022). Infectious stocks were propagated in Vero-TMPRSS2 cells as described (Case et al., 2020). All work with infectious SARS-CoV-2 was performed in approved BSL3 and A-BSL3 facilities at Washington University School of Medicine using appropriate positive pressure air respirators and protective equipment.
Mice
Animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine. Virus inoculations were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering. WT C57BL/6J (#000664) mice were obtained from The Jackson Laboratory or bred in a pathogen-free animal facility at Washington University. Ifnlr1 −/− (Ank et al., 2008) and Ifnl2-gfp reporter mice (Galani et al., 2017) (generated by Evangelos Andreakos and kindly provided by Megan Baldridge, Washington University) were bred and housed in a pathogen-free animal facility at Washington University. Heterozygous K18-hACE C57BL/6J mice (strain: 2B6.Cg-Tg(K18-ACE2)2Prlmn/J) were obtained from The Jackson Laboratory. 129S2 mice were obtained from Charles River. Animals were housed in groups and fed standard chow diets. The ages and sex of mice used in this study were as follows: (a) six-week-old or three-month-old male and female WT and Ifnlr1 −/− C57BL/6 mice; (b) eight-week-old or five-month-old female K18-hACE2 mice; (c) six-week-old female 129S2 mice; and (d) six-week-old male and female Mavs −/−, cGas −/−, Myd88 −/− or WT C57BL/6 mice.
Method details
Mouse infection, immune cell depletion, and bone marrow chimeric mice studies
For mouse infections, virus inoculations were performed under anesthesia that was induced and maintained with ketamine hydrochloride and xylazine, and all efforts were made to minimize animal suffering. Depending on the experimental design, mice were administered 103, 105, or 106 FFU of SARS-CoV-2 WA1/2020 D614G, B.1.351 (Beta) or B.1.1.529 (Omicron) strains by intranasal route in 50 μL of PBS. For neutrophil depletions, anti-Ly6G (BioXCell; clone 1A8) or an isotype control (BioXCell; clone 2A3) was administered to mice by intraperitoneal injection at D-1 (500 μg), D+1 (200 μg) and D+3 (200 μg) relative to B.1.351 inoculation. For bone marrow chimeric mice, six-week-old male and female WT (CD45.1) and Ifnlr1 −/− (CD45.2) recipient mice were irradiated with 9 Gy (X-ray) total body irradiation. One day later, mice were injected with 5×106 sex-matched bone marrow cells from donor WT (CD45.2) or Ifnlr1 −/− (CD45.2) mice. Ten weeks later, peripheral blood cell from recipient chimeric mice were analyzed by flow cytometry as described below.
Plaque assay
Vero-TMPRSS2-ACE2 cells were seeded at a density of 1.25 x 105 cells per well in flat-bottom 24-well tissue culture plates. The following day, media was removed and replaced with 200 μL of 10-fold serial dilutions of sample, diluted in DMEM with 2% FBS. One hour later, 1 mL of methylcellulose overlay was added. Plates were incubated for 72 h, then fixed with 4% paraformaldehyde (final concentration) in PBS for 1 h. Plates were stained with 0.05% (w/v) crystal violet in 20% methanol and washed twice with distilled, deionized water. Plaques were counted, and titers were calculated according to a previously described method (Case et al., 2020).
Measurement of viral RNA
Mice were euthanized and tissues were collected. Nasal washes were collected in 0.5 mL of PBS. Tissues were weighed and homogenized with zirconia beads in a MagNA Lyser instrument (Roche Life Science) in 1 mL of DMEM media supplemented with 2% FBS. Tissue homogenates were clarified by centrifugation at 10,000 rpm for 5 min and stored at −80°C. Viral RNA from homogenized tissues or nasal washes was isolated using the MagMAX Viral RNA Isolation Kit (ThermoFisher) and measured by TaqMan one-step quantitative reverse-transcription PCR (RT-qPCR) on an ABI 7500 Fast Instrument. Copies of SARS-CoV-2 N gene RNA in samples were determined using a previously published assay (Case et al., 2020). Briefly, a TaqMan assay was designed to target a highly conserved region of the N gene (Forward primer: ATGCTGCAATCGTGCTACAA; Reverse primer: GACTGCCGCCTCTGCTC; Probe:/56-FAM/TCAAGGAAC/ZEN/AACATTGCCAA/3IABkFQ/). This region was included in an RNA standard to allow for copy number determination down to 10 copies per reaction. The reaction mixture contained final concentrations of primers and probe of 500 and 100 nM, respectively.
Cytokine and chemokine protein measurements
Lung homogenates were incubated with Triton X-100 (1% final concentration) for 1 h at room temperature to inactivate SARS-CoV-2. Homogenates were analyzed for cytokines and chemokines by Eve Technologies Corporation (Calgary, AB, Canada) using their Mouse Cytokine Array/Chemokine Array 31-Plex (MD31) platform.
Lung histology
Animals were euthanized before harvest and fixation of tissues. Lungs were inflated with 1.2 mL of 10% neutral buffered formalin using a 3-mL syringe and catheter inserted into the trachea. Tissues were embedded in paraffin, and sections were stained with hematoxylin and eosin. Images were captured using the Nanozoomer (Hamamatsu) at the Alafi Neuroimaging Core at Washington University. Hematoxylin+ area as an index of cellularity was quantified in whole lung sections using a NanoZoomer slide scanner (Hamamatsu) and ImageJ software as described previously (Wu et al., 2021).
Flow cytometry analysis of peripheral blood and lung
For analysis of immune cell depletion, peripheral blood cells were collected, and erythrocytes were lysed with ACK lysis buffer (Gibco) and resuspended in RPMI supplemented with 10% FBS. Single cell suspensions were preincubated with Fc Block antibody (BD PharMingen) in PBS with 2% FBS for 10 min at room temperature before staining. Cells were incubated with antibodies against the following markers: BV421 anti-CD45, AF700 anti-Ly6C, FITC anti-Ly6B, PE-CY7 anti-Ly6G and APC anti-CD11b. All antibodies were used at a dilution of 1:200. Cells were stained for 20 min at 4°C, washed with PBS, fixed with 4% PFA for 15 min, washed with PBS and resuspended with FACS (PBS, 2% FBS, and 2 mM EDTA) buffer. For the lungs, single cells suspensions were collected as described below and stained in the same way as peripheral blood.
Lung digestion and cell sorting by flow cytometry
Lungs were collected and digested at 37°C with 5 mg/mL of collagenase I (Worthington) and 1 mg/mL of DNase I (Roche) for 45 min in HBSS buffer. Digested lung tissues were minced, passed through a 40 μm strainer, and centrifuged at 500 g for 10 min. Red blood cells were lysed with ACK lysis buffer. Dead cells were removed by Dead Cell Removal Kit (STEMCELL) according manufacturer’s protocol. Single cell suspensions were incubated with APC-CY7 anti-CD45, APC anti-CD11b, BV421 anti-Ly6G, BV-421 anti-CD11c, PE anti-Siglec F (CD170), AF-700 anti-MHC II (I-A/I-E), BV421 anti-CD3, PE anti-CD19, APC anti-CD11b, PE anti-CD326 and PE anti-Ly6C antibodies as described above following Fc antibody (BD PharMingen) blockade. AM (CD45+ SiglecFhi CD11chi), DCs (CD45+ SiglecF− CD11c+ MHCII+), B cells (CD45+ CD19+), T cells (CD45+ CD3+), Nφ (CD45+CD11b+Ly6G+), ECs (CD45- CD326+) and Mo (CD45+ CD11b+ Ly6Chi) were sorted by flow cytometry (Sony SH800Sorter) under BSL3 conditions. RNA was extracted with RNeasy Micro Kit (QIAGEN) according to manufacturer’s protocol and then Ifnl2, Ifnl3, and Ifnlr1 mRNA levels were measured by qRT-PCR as described above.
Confocal microscopy
Lung tissues were collected as described above and fixed for 7 days. Tissues then were washed three time with PBS and placed into 30% sucrose in PBS overnight until sinking to the bottom of the tube. Tissues were placed into O.C.T. medium in cryomolds on dry ice, wrapped in aluminum foil, and stored in −80C. Sections were cut and embedded on superfrost glass slides. Slides were rinsed three times with PBS, blocked with 5% FBS, 1% BSA and 0.3% Triton X-100 in PBS, and incubated with rat anti-CD326 (1: 500), rabbit anti-nucleocapsid protein (1: 500), and chicken anti-GFP (1: 1000) primary antibodies at 4°C overnight. The next day, slides were stained with goat anti-chicken (1: 500), donkey anti-rabbit (1: 500) and donkey anti-rat (1: 500) secondary antibodies for 1 h at room temperature and with Hoechst dye (1:10,000) for 5 min. Slides were washed with PBS once, mounted with AquaPoly, and stored in the dark at 4°C until imaged.
RNA sequencing
RNA from lung tissues was extracted by RNeasy Pls Mini Kit (QIAGEN) according to manufacturer’s protocol. cDNA libraries were constructed starting with 10 ng of total RNA. cDNA was generated using the Seqplex kit (Sigma-Aldrich, St. Louis, MO) with amplification of 20 cycles. Library construction was performed using 100 ng of cDNA undergoing end repair, A-tailing, ligation of universal TruSeq adapters, and 8 cycles of amplification to incorporate unique dual index sequences. Libraries were sequenced on the NovaSeq 6000 (Illumina, San Diego, CA) targeting 40 million read pairs and extending 150 cycles with paired end reads. RNA-seq reads were aligned to the mouse Ensembl GRCh38.76 primary assembly with STAR program (version 2.5.1a) (Dobin et al., 2013). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount (version 1.4.6-p5) (Liao et al., 2014). The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC (version 2.6.2) (Liao et al., 2014). All gene counts were preprocessed with the R package EdgeR (Robinson et al., 2010) to adjust samples for differences in library size using the trimmed mean of M values (TMM) normalization procedure. Viral and ribosomal genes and genes not expressed in at least five samples (the smallest group size) at a level greater than or equal to 1 count per million reads were excluded, resulting 19,280 unique genes in further analysis. The R package limma (Ritchie et al., 2015) with voomWithQualityWeights function (Liu et al., 2015) was utilized to calculate the weighted likelihoods for all samples, based on the observed mean-variance relationship of every gene and sample. Differentially expressed genes were defined as those with at least 2-fold difference between two individual groups at p < 0.05.
Quantification and statistical analysis
Statistical significance was assigned when p values were <0.05 using Prism version 8 (GraphPad). Tests, number of animals (n), median values, and statistical comparison groups are indicated in the Figure legends. Analysis of weight change was determined by t test or one-way ANOVA with Dunnett’s post-test of the area under the curve depending on the number of comparison groups. Viral burden was analyzed by Mann-Whitney test when comparing two groups, or one-way ANOVA or Kruskal-Wallis test with Dunnett’s post-test when comparing three or more groups. Cytokine data were analyzed by one-way ANOVA with Tukey’s multiple comparison test. qRT-PCR data were analyzed by one-way ANOVA with Dunnett’s post-test.
Acknowledgments
This study was supported by grants and contracts from the NIH: R01 AI157155, U01 AI151810, and 75N93019C00051 (all to M.S.D.), 75N93021C00014 (to Y.K.), F30 AI152327 (to E.S.W.), T32GM139774-01 (to C.E.K.), and R35 HL145242 (to M.J.H.).
Author contributions
Z.C. and C.E.K. performed the infection experiments in mice. Z.C. and E.S.W. titrated virus in tissues. Z.C., S.P.K., and M.J.H. analyzed lung histopathology. J.Y. analyzed the RNA sequencing data. P.J.H. and Y.K. isolated and propagated the B.1.1.529 isolate. M.S.D. obtained funding and supervised research. Z.C. and M.S.D. wrote the initial draft, with all other authors providing editorial comments.
Declaration of interests
M.S.D. is a consultant for Inbios, Vir Biotechnology, and Carnival Corporation and is on the Scientific Advisory Boards of Moderna and Immunome. The M.S.D. laboratory has received unrelated funding support in sponsored research agreements from Moderna, Vir Biotechnology, Immunome, and Emergent BioSolutions. Y.K. has received unrelated funding support from Daiichi Sankyo Pharmaceutical, Toyama Chemical, Tauns Laboratories, Inc., Shionogi & Co. Ltd., Otsuka Pharmaceutical, KM Biologics, Kyoritsu Seiyaku, Shinya Corporatoin, and Fuji Rebio. M.J.H. is founder and president of NuPeak Therapeutics, Inc., a member of a Data Safety Monitoring Board for AstraZeneca, and a scientific advisor for Lonza-Bend Pharma.
Published: April 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110799.
Supplemental information
Data and code availability
-
•
All data supporting the findings of this study are available within the paper and or upon request from the corresponding author. RNA sequencing datasets are available for analysis (GEO accession number GSE193990).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andreakos E., Tsiodras S. COVID-19: lambda interferon against viral load and hyperinflammation. EMBO Mol. Med. 2020;12:e12465. doi: 10.15252/emmm.202012465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N., Iversen M.B., Bartholdy C., Staeheli P., Hartmann R., Jensen U.B., Dagnaes-Hansen F., Thomsen A.R., Chen Z., Haugen H., et al. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008;180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Beziat V., et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Moller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudewijns R., Thibaut H.J., Kaptein S.J.F., Li R., Vergote V., Seldeslachts L., Van Weyenbergh J., De Keyzer C., Bervoets L., Sharma S., et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 2020;11:5838. doi: 10.1038/s41467-020-19684-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A., Ghosh S., Sposito B., Spreafico R., Balzarini F., Lo Cascio A., Clementi N., De Santis M., Mancini N., Granucci F., Zanoni I. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369:706–712. doi: 10.1126/science.abc3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A., Granucci F., Zanoni I. Type III interferons: balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020;217:e20190295. doi: 10.1084/jem.20190295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A., Tan Y., Granucci F., Zanoni I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 2017;18:1084–1093. doi: 10.1038/ni.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J.B., Bailey A.L., Kim A.S., Chen R.E., Diamond M.S. Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology. 2020;548:39–48. doi: 10.1016/j.virol.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.E., Winkler E.S., Case J.B., Aziati I.D., Bricker T.L., Joshi A., Darling T.L., Ying B., Errico J.M., Shrihari S., et al. In vivo monoclonal antibody efficacy against SARS-CoV-2 variant strains. Nature. 2021;596:103–108. doi: 10.1038/s41586-021-03720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.E., Zhang X., Case J.B., Winkler E.S., Liu Y., VanBlargan L.A., Liu J., Errico J.M., Xie X., Suryadevara N., et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021;27:717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinnon K.H., 3rd, Leist S.R., Schafer A., Edwards C.E., Martinez D.R., Montgomery S.A., West A., Yount B.L., Jr., Hou Y.J., Adams L.E., et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature. 2020;586:560–566. doi: 10.1038/s41586-020-2708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa V., Dutta O., McElrath C., Du P., Chang Y.J., Cicciarelli B., Pitler A., Whitehead I., Obar J.J., Durbin J.E., et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci. Immunol. 2017;2:eaan5357. doi: 10.1126/sciimmunol.aan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld J.J., Kandel C., Biondi M.J., Kozak R.A., Zahoor M.A., Lemieux C., Borgia S.M., Boggild A.K., Powis J., McCready J., et al. Peginterferon lambda for the treatment of outpatients with COVID-19: a phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021;9:498–510. doi: 10.1016/s2213-2600(20)30566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgenhauer U., Schoen A., Gad H.H., Hartmann R., Schaubmar A.R., Failing K., Drosten C., Weber F. Inhibition of SARS-CoV-2 by type I and type III interferons. J. Biol. Chem. 2020;295:13958–13964. doi: 10.1074/jbc.ac120.013788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galani I.E., Rovina N., Lampropoulou V., Triantafyllia V., Manioudaki M., Pavlos E., Koukaki E., Fragkou P.C., Panou V., Rapti V., et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021;22:32–40. doi: 10.1038/s41590-020-00840-x. [DOI] [PubMed] [Google Scholar]
- Galani I.E., Triantafyllia V., Eleminiadou E.E., Koltsida O., Stavropoulos A., Manioudaki M., Thanos D., Doyle S.E., Kotenko S.V., Thanopoulou K., Andreakos E. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46:875–890.e6. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- Golden J.W., Cline C.R., Zeng X., Garrison A.R., Carey B.D., Mucker E.M., White L.E., Shamblin J.D., Brocato R.L., Liu J., et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight. 2020;5:e142032. doi: 10.1172/jci.insight.142032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/nejmoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfmann P.J., Iida S., Iwatsuki-Horimoto K., Maemura T., Kiso M., Scheaffer S.M., Darling T.L., Joshi A., Loeber S., Singh G., et al. Consortium Mount Sinai Pathogen Surveillance PSP study group SARS-CoV-2 Omicron virus causes attenuated disease in mice and hamsters. Nature. 2022;603:687–692. doi: 10.1038/s41586-022-04441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoagland D.A., Moller R., Uhl S.A., Oishi K., Frere J., Golynker I., Horiuchi S., Panis M., Blanco-Melo D., Sachs D., et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity. 2021;54:557–570.e5. doi: 10.1016/j.immuni.2021.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Arora P., Groß R., Seidel A., Hornich B.F., Hahn A.S., Kruger N., Graichen L., Hofmann-Winkler H., Kempf A., et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell. 2021;184:2384–2393.e12. doi: 10.1016/j.cell.2021.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/s0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iketani S., Liu L., Guo Y., Liu L., Chik K.K.H., Yuen T.T.T., Yin M.T., Sobieszczyk M.E., Yuen K.Y., Wang H.H., et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature. 2022 doi: 10.1038/s41586-022-04594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan P., Andrews J.R., Bonilla H., Hedlin H., Jacobson K.B., Balasubramanian V., Purington N., Kamble S., de Vries C.R., Quintero O., et al. Peginterferon Lambda-1a for treatment of outpatients with uncomplicated COVID-19: a randomized placebo-controlled trial. Nat. Commun. 2021;12:1967. doi: 10.1038/s41467-021-22177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilg N., Lin W., Hong J., Schaefer E.A., Wolski D., Meixong J., Goto K., Brisac C., Chusri P., Fusco D.N., et al. Kinetic differences in the induction of interferon stimulated genes by interferon-alpha and interleukin 28B are altered by infection with hepatitis C virus. Hepatology. 2014;59:1250–1261. doi: 10.1002/hep.26653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear H.M., Schoggins J.W., Diamond M.S. Shared and distinct functions of type I and type III interferons. Immunity. 2019;50:907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Nie J., Wu J., Zhang L., Ding R., Wang H., Zhang Y., Li T., Liu S., Zhang M., et al. SARS-CoV-2 501Y.V2 variants lack higher infectivity but do have immune escape. Cell. 2021;184:2362–2371.e9. doi: 10.1016/j.cell.2021.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu R., Holik A.Z., Su S., Jansz N., Chen K., Leong H.S., Blewitt M.E., Asselin-Labat M.L., Smyth G.K., Ritchie M.E. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 2015;43:e97. doi: 10.1093/nar/gkv412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., VanBlargan L.A., Bloyet L.M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021;29:477–488.e4. doi: 10.1016/j.chom.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major J., Crotta S., Llorian M., McCabe T.M., Gad H.H., Priestnall S.L., Hartmann R., Wack A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369:712–717. doi: 10.1126/science.abc2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P., McAuley D.F., Brown M., Sanchez E., Tattersall R.S., Manson J.J., HLH Across Speciality Collaboration, UK COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/s0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Dominguez-Andres J., Barreiro L.B., Chavakis T., Divangahi M., Fuchs E., Joosten L.A.B., van der Meer J.W.M., Mhlanga M.M., Mulder W.J.M., et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oladunni F.S., Park J.G., Pino P.A., Gonzalez O., Akhter A., Allue-Guardia A., Olmo-Fontanez A., Gautam S., Garcia-Vilanova A., Ye C., et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun. 2020;11:6122. doi: 10.1038/s41467-020-19891-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo E., Di Carlo D., Sgarbanti M., Hiscott J. Type I interferons in COVID-19 pathogenesis. Biology. 2021;10:829. doi: 10.3390/biology10090829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A., Iwasaki A. Type I and type III interferons - induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe. 2020;27:870–878. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas D., Saunders N., Maes P., Guivel-Benhassine F., Planchais C., Buchrieser J., Bolland W.H., Porrot F., Staropoli I., Lemoine F., et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature. 2022;602:671–675. doi: 10.1038/s41586-021-04389-z. [DOI] [PubMed] [Google Scholar]
- Prokunina-Olsson L., Alphonse N., Dickenson R.E., Durbin J.E., Glenn J.S., Hartmann R., Kotenko S.V., Lazear H.M., O'Brien T.R., Odendall C., et al. COVID-19 and emerging viral infections: the case for interferon lambda. J. Exp. Med. 2020;217:e20200653. doi: 10.1084/jem.20200653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnasinghe R., Jangra S., Cupic A., Martinez-Romero C., Mulder L.C.F., Kehrer T., Yildiz S., Choi A., Mena I., De Vrieze J., et al. The N501Y mutation in SARS-CoV-2 spike leads to morbidity in obese and aged mice and is neutralized by convalescent and post-vaccination human sera. medRxiv. 2021 doi: 10.1101/2021.01.19.21249592. Preprint at. [DOI] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider W.M., Chevillotte M.D., Rice C.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G. The role of type I interferons in the pathogenesis and treatment of COVID-19. Front. Immunol. 2020;11:595739. doi: 10.3389/fimmu.2020.595739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo H., Maekawa S., Komase K., Miura M., Kadokura M., Sueki R., Komatsu N., Shindo K., Amemiya F., Nakayama Y., et al. IL-28B (IFN-λ3) and IFN-α synergistically inhibit HCV replication. J. Viral Hepat. 2013;20:281–289. doi: 10.1111/j.1365-2893.2012.01649.x. [DOI] [PubMed] [Google Scholar]
- Shuai H., Chan J.F.W., Yuen T.T.T., Yoon C., Hu J.C., Wen L., Hu B., Yang D., Wang Y., Hou Y., et al. Emerging SARS-CoV-2 variants expand species tropism to murines. EBioMedicine. 2021;73:103643. doi: 10.1016/j.ebiom.2021.103643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn S.Y., Hearing J., Mugavero J., Kirillov V., Gorbunova E., Helminiak L., Mishra S., Mackow E., Hearing P., Reich N.C., Kim H.K. Interferon-Lambda intranasal protection and differential sex pathology in a murine model of SARS-CoV-2 infection. mBio. 2021;12:e0275621. doi: 10.1128/mbio.02756-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sposito B., Broggi A., Pandolfi L., Crotta S., Clementi N., Ferrarese R., Sisti S., Criscuolo E., Spreafico R., Long J.M., et al. The interferon landscape along the respiratory tract impacts the severity of COVID-19. Cell. 2021;184:4953–4968.e16. doi: 10.1016/j.cell.2021.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanifer M.L., Kee C., Cortese M., Zumaran C.M., Triana S., Mukenhirn M., Kraeusslich H.G., Alexandrov T., Bartenschlager R., Boulant S. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep. 2020;32:107863. doi: 10.1016/j.celrep.2020.107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592:438–443. doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- van der Wijst M.G.P., Vazquez S.E., Hartoularos G.C., Bastard P., Grant T., Bueno R., Lee D.S., Greenland J.R., Sun Y., Perez R., et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 2021;13:eabh2624. doi: 10.1126/scitranslmed.abh2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBlargan L.A., Errico J.M., Halfmann P.J., Zost S.J., Crowe J.E., Jr., Purcell L.A., Kawaoka Y., Corti D., Fremont D.H., Diamond M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022;28:490–495. doi: 10.1038/s41591-021-01678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderheiden A., Ralfs P., Chirkova T., Upadhyay A.A., Zimmerman M.G., Bedoya S., Aoued H., Tharp G.M., Pellegrini K.L., Manfredi C., et al. Type I and type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J. Virol. 2020;94:e00985-20. doi: 10.1128/jvi.00985-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler E.S., Bailey A.L., Kafai N.M., Nair S., McCune B.T., Yu J., Fox J.M., Chen R.E., Earnest J.T., Keeler S.P., et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020;21:1327–1335. doi: 10.1038/s41590-020-0778-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler E.S., Chen R.E., Alam F., Yildiz S., Case J.B., Uccellini M.B., Holtzman M.J., Garcia-Sastre A., Schotsaert M., Diamond M.S. SARS-CoV-2 causes lung infection without severe disease in human ACE2 knock-in mice. J. Virol. 2021;96:e0151121. doi: 10.1128/JVI.01511-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Chen Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- Wu K., Kamimoto K., Zhang Y., Yang K., Keeler S.P., Gerovac B.J., Agapov E.V., Austin S.P., Yantis J., Gissy K.A., et al. Basal epithelial stem cells cross an alarmin checkpoint for postviral lung disease. J. Clin. Invest. 2021;131:e149336. doi: 10.1172/jci149336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying B., Whitener B., VanBlargan L.A., Hassan A.O., Shrihari S., Liang C.Y., Karl C.E., Mackin S., Chen R.E., Kafai N.M., et al. Protective activity of mRNA vaccines against ancestral and variant SARS-CoV-2 strains. Sci. Transl. Med. 2022;14:eabm3302. doi: 10.1126/scitranslmed.abm3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Chassaing B., Shi Z., Uchiyama R., Zhang Z., Denning T.L., Crawford S.E., Pruijssers A.J., Iskarpatyoti J.A., Estes M.K., et al. Prevention and cure of rotavirus infection via TLR5/NLRC4–mediated production of IL-22 and IL-18. Science. 2014;346:861–865. doi: 10.1126/science.1256999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Cui Z., Li Q., Wang B., Yu Y., Wu J., Nie J., Ding R., Wang H., Zhang Y., et al. Ten emerging SARS-CoV-2 spike variants exhibit variable infectivity, animal tropism, and antibody neutralization. Commun. Biol. 2021;4:1196. doi: 10.1038/s42003-021-02728-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Zhao Y., Zhang F., Wang Q., Li T., Liu Z., Wang J., Qin Y., Zhang X., Yan X., et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the Perspectives of clinical immunologists from China. Clin. Immunol. 2020;214:108393. doi: 10.1016/j.clim.2020.108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wu S., Wu B., Yang Q., Chen A., Li Y., Zhang Y., Pan T., Zhang H., He X. SARS-CoV-2 Omicron strain exhibits potent capabilities for immune evasion and viral entrance. Signal Transduct. Target. Ther. 2021;6:430. doi: 10.1038/s41392-021-00852-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler C.G.K., Allon S.J., Nyquist S.K., Mbano I.M., Miao V.N., Tzouanas C.N., Cao Y., Yousif A.S., Bals J., Hauser B.M., et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181:1016–1035. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data supporting the findings of this study are available within the paper and or upon request from the corresponding author. RNA sequencing datasets are available for analysis (GEO accession number GSE193990).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.