

Abstract
Background:
Rheumatoid arthritis (RA) is an autoimmune disease characterised by systemic inflammation of joints. The observed complexity of RA pathogenesis and studies that have been carried out so far indicate that RA pathogenesis is regulated at multiple levels. Given the role of RNA editing in autoimmune disease, we hypothesised that RNA editing could contribute to RA pathogenesis by regulating gene expression through post-transcriptional mechanisms.
Methods:
We identified RNA editing events in synovial tissues from early and established RA compared with normal subjects from an available transcriptome data set using REDItools. To investigate the potential effect of these RNA editing events on gene expression, we carried out an analysis of differential exon usage in the vicinity of the differentially edited sites using DEXSeq. We then used STRING to identify putative interactions between differentially edited genes identified from REDItools analysis. We also investigated the possible effects of these RNA editing events on miRNA-target mRNA interactions as predicted by miRanda.
Results:
Our analysis revealed that there is extensive RNA editing in RA, with 304 and 273 differentially edited events in early RA and established RA, respectively. Of these, 25 sites were within 11 genes in early RA, and 34 sites were within 7 genes in established RA. DEXSeq analysis revealed that RNA editing correlated with differential exon usage in 4 differentially edited genes that have previously also been associated with RA in some measure: ATM, ZEB1, ANXA4, and TIMP3. DEXSeq analysis also revealed enrichment of some non-functional isoforms of these genes, perhaps at the expense of their full-length counterparts. Network analysis using STRING showed that several edited genes were part of the p53 protein-protein interaction network. We also identified several putative miRNA binding sites in the differentially edited genes that were lost upon editing.
Conclusions:
Our results suggested that the expression of genes involved in DNA repair and cell cycle, including ATM and ZEB1 which are well-known functional regulators of the DNA damage response pathway, could be regulated by RNA editing in RA synovia. This may contribute to an impaired DNA damage response in synovial tissues.
Keywords: Rheumatoid arthritis, RNA editing, differential exon usage, DNA damage response, synovium, autoimmune disease
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterised by chronic synovial inflammation that results in significant joint destruction and that in several instances leads to other systemic complications. The presence of autoantibodies such as the rheumatoid factor and antibodies against post-translational modifications in proteins like citrullination and carbamylation are hallmarks of this disease. 1 These autoantibodies form immune complexes in the joint, attracting immune cells, and causing chronic inflammation. RA is a complex, heterogeneous disease and has been categorised into multiple subtypes based on pathology, 2 pattern of cytokine expression, 3 or based on molecular signatures.4-7 Some of these subtypes show correlation with clinical manifestation and response to drugs. 2 About 60% of RA is estimated to be heritable, and several genetic loci have been linked to the onset and progression of this disease, including coding loci of many immune-related genes. 8 Multiple Disease Modifying Anti-Rheumatic Drugs (DMARDs), almost all of which target the immune response in one way or the other, are the most common treatment options to modify/delay the progression of RA. 9
RNA editing, more specifically, adenosine (A) to inosine (I) RNA editing catalysed by adenosine deaminases or ADARs on double-stranded RNA (dsRNA) substrates is a post-transcriptional modification that is known to occur at more than 100 million sites in the human genome.10,11 ADARs catalyse the deamination of adenosine to inosine through the hydrolytic deamination of the 6-position of adenosine. 12 Inosine is interpreted as guanosine by cellular machinery, and this can modify RNA function in many ways. This post-transcriptional modification contributes to the diversity of transcripts by modifying protein-coding regions, splicing sites, miRNA seed regions and their binding sites as well as by modifying the mRNA secondary structure. 11
For instance, if the editing event occurs within the coding part of a transcript, or disrupts splicing signals, or creates new splice sites, it can result in recoding to generate novel protein isoforms. An excellent example is the case of the nuclear prelamin A recognition factor (NARF) in which the exonization of a primate-specific Alu-exon exclusively depends on RNA editing. RNA editing events in NARF regulate this exonization in a tissue-dependent manner both through creation of a functional splice site and the elimination of a premature stop codon. 13 In addition, editing events in the mRNA can create or destroy microRNA (miRNA) recognition sites in the transcript that may be involved in either translational repression or mRNA degradation, thus regulating the levels of the transcript.14,15
It is now well-known that a large number of RNA editing events occur in the Alu repetitive elements which form a double-stranded RNA (dsRNA) structure. These endogenous dsRNAs can trigger an unwanted immune response as they can be potentially recognised as non-self by the melanoma differentiation–associated protein 5 (MDA5). 16 Multiple studies have suggested that ADAR-mediated RNA editing can prevent MDA5 sensing the endogenous dsRNAs transcribed from repetitive elements as non-self.17-20 Mutations in ADAR1 also cause the autoimmune disorder, Aicardi-Goutières syndrome (AGS), a rare autosomal recessive encephalopathy. 21 This syndrome is associated with upregulation of interferon-stimulated genes in the absence of infections, indicating a possible role for ADAR1 as a suppressor of type I interferon signalling. Indeed, with growing evidence on the role of RNA editing as an immune tolerance strategy for preventing an immune reaction with self-nucleic acids, 22 the idea that RNA editing may be a form of molecular immune defence mechanism has gained substantial traction.10,11
While the knockdown of ADAR1 increased markers of innate immunity reflecting the former’s role in immune tolerance, 22 in some autoimmune disorders, increased RNA editing appeared to contribute to the pathology by affecting the levels of key inflammatory molecules as well as by generating novel protein isoforms. In patients with RA, upregulation of both the expression and activity of ADARs was seen, and this was associated with higher expression of edited, Alu-enriched, pro-inflammatory genes such as cathepsin S and tumour necrosis factor (TNF) receptor-associated factors 1, 2, 3, and 5. 23 The increased levels of these genes appeared to contribute to the pathophysiology of RA. Similarly, blood samples from individuals with an autoimmune disorder, systemic lupus erythematosus (SLE), have abnormally high levels of RNA editing, some of which affect proteins and also potentially generates novel autoantigens. 24 Studies probing this have suggested that elevated RNA editing may be involved in the pathophysiology of SLE, as well as in other autoimmune diseases, by generating or increasing the autoantigen load leading to an exaggerated immune response. Dysregulated RNA editing may thus represent one of several mechanisms such as molecular mimicry for instance, which is triggered by infection or chemicals wherein similarities between foreign and self-peptides favour the activation of self-reactive T or B cells, contributing to autoimmunity. 25
While Vlachogiannis et al 23 observed increased ADAR1 expression as well as a higher expression of edited Alu-enriched, pro-inflammatory genes such as cathepsin S in RA patients, it is unclear whether A-to-I editing is deregulated at a genome-wide level in RA and what the potential consequences of this deregulation could be. To gain insights into this question, in this study, we used bioinformatic methods to identify genes differentially regulated by RNA editing in synovial tissues from RA patients. Early and established RA have distinct molecular signatures, 6 and hence could exhibit different transcriptional/post-transcriptional regulation. Therefore, we compared the predicted RNA editing events using RNA-Seq data of normal, early RA, and established RA synovial biopsies from Guo et al. 26 We then carried out further in silico studies to predict functional consequences of these differentially edited sites. Our analyses indicated that differential RNA editing events could potentially lead to impaired DNA repair response in synovial cells, thereby contributing to RA pathology.
Materials and Methods
Identification of differentially edited sites in normal and RA samples
Metadata for the RNA-Seq runs of joint synovial biopsies of normal, early RA and established RA from project PRJNA352076 was downloaded from the Sequence Read Archive (SRA).26,27 Data were available for 28 normal, 57 early RA, and 95 established RA samples. Fastq files of all the samples were downloaded from the European Nucleotide Archive (ENA). Known editing events in the samples were identified using the REDItoolknown function of the REDItools suite, and using known editing events from REDIportal. 28 At the time of download (March 2020), REDIportal data were available for the GRCh37 version of the human genome. Therefore, fastq files were aligned to GRCh37 using STAR. Aligned reads were sorted and indexed using samtools. Sorted bam files were used for identifying edited sites using the REDItoolknown.py tool and the REDIportal RNA editing database. Two samples, one normal (SRR4785826) and one early RA (SRR4785895), were omitted from further analysis due to an unusually low number of edited sites as compared with the rest of the samples reported by REDItools. Differentially edited events were identified using the get_DE_events.py script from the REDItools suite by comparing editing events in the normal sample with editing events in the early and established RA samples. Annotation of each identified differentially edited event was performed using the REDIportal database. Among several annotations available from the REDIportal database, RefSeq annotations were used for further analysis.
Differential gene expression analysis
edgeR analysis was performed to investigate whether there was a correlation between RNA editing and gene expression levels. The analysis was performed using the ReadsPerGene.out.tab output for each sample from the STAR analysis.
DEXSeq analysis
DEXSeq analysis was performed to check whether editing correlated with differential exon usage around the edited sites. As prefiltering of low-expressing isoforms is known to improve the false discovery rate (FDR) of DEXSeq runs, 29 low-expressing isoforms of differentially edited genes were filtered prior to DEXSeq analysis. To use the most current annotation of exon-intron boundaries, this analysis was performed using the GRCh38 assembly instead of GRCh37. Towards this, the coordinates of the identified differentially edited events were converted to GRCh38 coordinates using the Assembly Converter tool from Ensembl. Transcript abundance for each differentially edited gene was estimated using kallisto 30 and isoforms that showed <2% expression in both normal and RA samples were removed from the Homo_sapiens.GRCh38.101.gtf file prior to the DEXSeq run. For DEXSeq analysis, the fastq files were mapped to GRCh38 using HISAT2. The resultant bam files were sorted and indexed. DEXSeq analysis was performed using aligned reads and the modified gtf file without the low-expressing isoforms.
Identification of putative miRNA binding sites
About 300 bp long region upstream and downstream of each edited site was used for target prediction using miRanda miRNA target prediction programme. 31 Sequences were manually modified to generate the edited sequence. Putative miRNA binding sites were identified using unedited and edited sequences, and the mature miRNA sequences were downloaded from miRbase. The alignment score and energy cutoffs were set at 140 and −14, respectively. miRNAs that had putative binding sites in the unedited sequences but not in the edited sequence were identified. From these miRNA-target pairs, miRNAs that were annotated as high confidence miRNA according to miRbase were identified for further analysis.
Protein-protein interaction network in RA
Potential protein interaction networks in differentially edited genes were investigated using STRING analysis. 32 Analysis was performed using the set of all differentially edited genes in early RA and in established RA, with a confidence score of 0.4 and 5% FDR.
Results
Some identified putative RNA editing sites are differentially regulated in RA
Using data from Guo et al which described RNA-sequencing data from synovial tissues, we compared RNA editing events in normal synovial tissues (n = 28) with those from patients diagnosed with early RA (n = 57) and established RA (n = 95) using bioinformatic methods.
First, we called all known RNA editing events in each of these conditions by employing the REDIportal database of known RNA editing events and using the REDItoolknown function from REDItools. Then, using get_DE_events.py from REDItools, we identified RNA editing events that were significantly differentially edited between normal and early RA, and normal and established RA. The get_DE_events.py script applies the Mann-Whitney U test to identify significant differential RNA editing events between two samples for those editing events that have a coverage of at least 10 reads and have at least 50% of the samples per group exhibiting at least 10% frequency of the editing event. Using this analysis, we were able to identify 304 and 273 putative differentially edited sites specific to early RA and established RA, respectively. These differentially edited sites were then annotated based on the annotation information available in REDIportal (Supplemental files 1 and 2). As expected, the identified differentially edited sites were predominantly present in Alu elements (Table 1 and Figure 1A and B).
Table 1.
Genomic distribution of differential editing events in early or established RA samples compared with normal samples.
| Distribution of differential editing events | Early RA | Established RA |
|---|---|---|
| Total events | 304 | 273 |
| Alu repeats | 273 | 227 |
| Repeat regions | 24 | 32 |
| Non-repeat regions | 7 | 14 |
| Intergenic region | 166 | 148 |
| Introns | 28 | 20 |
| 5’UTR | 1 | 1 |
| 3’UTR | 12 | 21 |
| ncRNA | 56 | 42 |
Abbreviations: ncRNA, non-coding RNA; RA, rheumatoid arthritis. An overview of the genomic distribution of RA events in early and established RA samples compared with normal samples. The annotation of the differentially edited events was done using REDIportal database. From the REDIportal database, RefSeq annotation of genomic regions was used to infer their genomic distribution.
Figure 1.
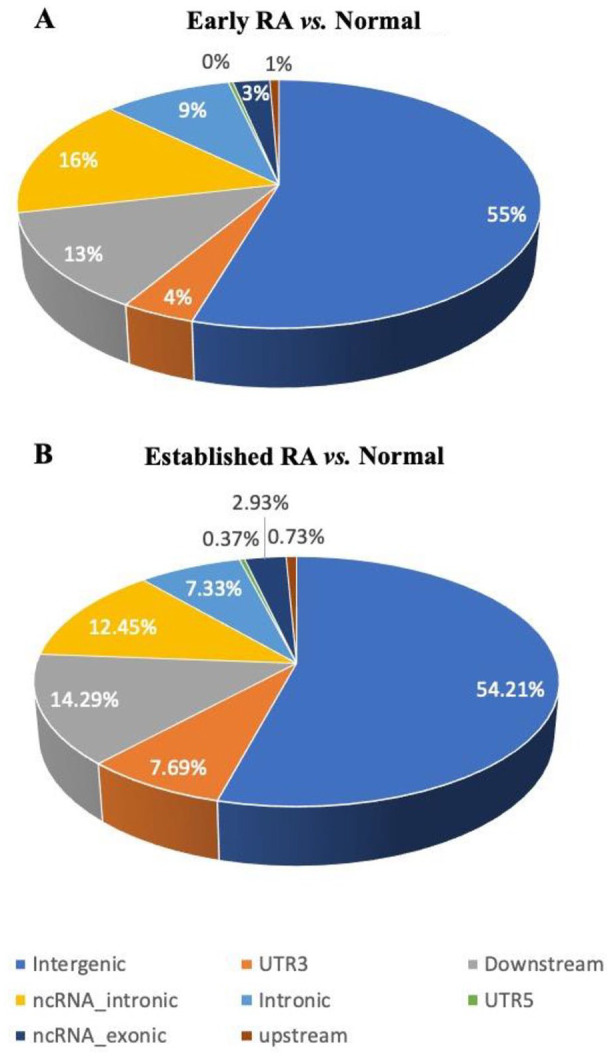
Genomic distribution of putative RNA editing sites. Pie charts represent the distribution of the identified putative RNA editing sites in different genomic regions. RNA editing events identified using REDItools were annotated using the information in REDIportal database. The percentage of editing events specific to early RA (A) and established RA (B) in the different genomic regions was plotted as pie-charts.
We focused on differential editing events that were identified within introns and UTRs, and were present on the same strand of chromosome as the gene, for further analysis. No differential editing events were observed in the protein-coding regions. We excluded RNA editing sites that were present in intergenic regions because such editing events were the least likely to exert a direct effect on gene expression. Twenty-five editing sites were identified in the introns or UTRs of 11 genes in the normal versus early RA comparison (Table 2A). Similarly, in the normal versus established RA comparison, 34 putative RNA editing sites were identified in the introns or UTRs of 7 genes (Table 2B). The identified RNA editing sites in a gene were usually clustered together. Interestingly, 4 genes, ATM, RBBP4, ZEB1, and TM6SF1, were common to both early and established RA conditions. The editing events in early and established RA in ATM, RBBP4, and TM6SF1 were clustered in the same region with some differences, but were identical in ZEB1 (Table 2A and B).
Table 2.
Differential RNA edited sites in RA, and the differential gene expression of genes harbouring these sites.
| Edited gene | Differentially edited site | Differential gene expression | |||
|---|---|---|---|---|---|
| Chromosome | Edited position | Location of the edited site | logFC | FDR | |
| A. | |||||
| APPL1 | chr3 | 57306834 | UTR3 | 0.7134 | 3.21E-06 |
| ATM | chr11 | 108236469, 108236523 | UTR3 | 1.3121 | 1.75E-14 |
| IFNGR2 | chr21 | 34734080, 34734090 | Intronic | 0.7986 | 6.56E-09 |
| OCIAD1 | chr4 | 48858537 | Intronic | 0.2954 | 0.001720587 |
| RBBP4 | chr1 |
33148623, 33149373
33149376, 33149486, 33149502 |
UTR3 | 0.5668 | 2.82E-08 |
| TM6SF1 | chr15 |
83797712, 83797764
83797765, 83799764 |
Intronic | 0.9344 | 2.67E-06 |
| ZEB1 | chr10 |
31670841, 31670900
31673050, 31673070 |
Intronic | −0.8758 | 1.14E-07 |
| ZNF146 | chr19 | 36724103, 36724207 | Intronic | 0.6606 | 1.77E-07 |
| ANXA4 | chr2 | 70020277 | Intronic | 0.1231 | 0.485437135 |
| PALMD | chr1 | 100148334 | Intronic | −0.0528 | 0.883618553 |
| TMEM51 | chr1 | 15491727, 15491751 | Intronic | 0.427 | 0.088338887 |
| B. | |||||
| ATM | chr11 |
108236469, 108236523
108237815, 108237957 |
UTR3 | 1.2092 | 0.001304199 |
| FPR3 | chr19 | 52319007 | Intronic | 2.0916 | 3.47E-09 |
| GOLGA8 N | chr15 | 32898103, 32898127, 32898133, 32898141, 32898159, 32898163 | UTR3 | −0.6114 | 0.000726014 |
| RBBP4 | chr1 |
33148196, 33148623, 33148691, 33149373, 33149376, 33149486,
33149502 |
UTR3 | 0.4565 | 0.033573174 |
| TIMP3 | chr22 |
33202030, 33202058, 33202064, 33202067, 33203193, 33203215,
33207118 |
Intronic | −0.6604 | 0.024014413 |
| TM6SF1 | chr15 | 83797712, 83797722, 83797764, 83797765, 83799764 | Intronic | 0.9192 | 0.000567265 |
| ZEB1 | chr10 |
31670841, 31670900
31673044, 31673070 |
Intronic | −0.7832 | 0.008743847 |
Abbreviation: RA, rheumatoid arthritis. Tables list genes that are differentially edited along with the location of the edited sites in early RA (A) and in established RA (B) samples, and the differential gene expression analysis of these genes. The genomic coordinates of the edited sites are as per the GRCh37 assembly. A majority of the genes in the vicinity of the identified RNA editing sites are differentially expressed in early RA and established RA and these are highlighted in bold.
Majority of gene loci containing the putative RNA editing sites are also differentially expressed in RA samples
It is being increasingly recognised that RNA editing changes in UTRs and intronic regions can alter gene expression and regulation.11,33 To correlate the putative RNA editing events of our interest with changes, if any, in gene expression, we used edgeR to identify transcripts that were differentially expressed between normal and early RA or established RA sample sets. At FDR < 0.05, 16 625 and 18 840 genes were identified to be differentially expressed between normal and early RA, and normal and established RA samples, respectively. Consistent with a previous report showing ADAR upregulation in RA, 23 we also observed a small but significant upregulation of ADAR in both early (log2FC = 0.465, FDR 0.0001) and established RA (log2FC = 0.396, FDR 0.0027). Interestingly, 8 genes in early RA and all 7 genes in established RA with the putative differentially edited sites that we had described in the previous section were also significantly differentially expressed between normal and early RA or established RA with an FDR < 0.05 (Table 2A and B). However, this could also simply be a reflection of the large numbers of differentially expressed genes in early and established RA samples (16 625 and 18 840 genes, respectively, out of 28 265 genes) as compared with normal samples.
Some putative RNA editing sites in the introns of genes differentially expressed in RA may influence exon usage
As many identified sites were present in introns, we also investigated whether these RNA editing events could affect exon usage and/or alternative splicing. DEXSeq was performed using 10 samples each from normal, early RA and established RA groups, in the age group 32 to 73 years. Both early and established RA samples selected for DEXSeq analysis were positive for anti-citrullinated protein antibody (ACPA) (Supplemental file 3). It has been shown that prefiltering of isoforms with low expression before DEXSeq analysis provides better control over FDR. 29 Hence, prefiltering of low-expressing isoforms of genes that showed differential editing was performed before DEXSeq analysis. For prefiltering, the expression level of all isoforms of each of the differentially edited gene was calculated using kallisto. Isoforms that accounted for less than 2% of the total expression of the gene in both RA and normal samples were removed from the Homo_sapiens.GRCh38.101.gtf file before DEXSeq analysis. The results of DEXSeq analysis were then analysed to determine changes in exon usage within the vicinity of the edited sites in RA samples. Interestingly, 2 genes, ataxia telangiectasia mutated (ATM) and zinc finger E-box binding homeobox 1 (ZEB1), showed differential exon usage around the edited sites in both early and established RA (Figure 2 and Supplemental file 4). In addition, annexin A4 (ANAX4) showed differential exon usage around the edited sites in early RA, while tissue inhibitor of matrix metalloproteinase (TIMP3) showed differential exon usage in established RA (Figure 2 and Supplemental file 4).
Figure 2.

Schematic representation of RNA editing and differential exon usage in 4 candidate genes. A schematic representation of the putative RNA editing sites identified in ATM (A), ZEB1 (B), ANXA4 (C), and TIMP3 (D) along with potential differential exon usage in regions adjacent to these RNA editing sites.
ATM
As exons might have different boundaries in different transcripts, an exon is divided into several parts or bins during DEXSeq analysis. 34 The reads mapping to each bin are counted and the regulation of each bin is analysed. As shown in Figure 2 and supplemental file 4, 2 out of the 3 DEXSeq bins encompassing the edited sites in ATM showed significant downregulation in both early and established RA samples as compared with normal samples, suggesting that these exons could be downregulated in RA. These exon bins are a part of the ATM transcripts ENST00000278616, ENST00000675843, and ENST00000452508 that translate into full-length proteins, as well as the transcript ENST00000527805 that translates into a non-functional shorter protein. This suggested that although ATM appeared to be upregulated in the RA samples according to the differential gene expression analysis, the ‘upregulation’ is likely due to an increase in the expression levels of non-functional isoforms of ATM (Figure 2A, and Supplemental file 4). In a finding unrelated to the editing of ATM transcript, we observed that exon bins encompassing the exon 108222832-108223186, which is also a part of full-length isoforms of ATM mentioned above, were also downregulated (Supplemental file 4) in both early and established RA, again suggestive of downregulation of functional ATM protein.
ZEB1
In the gene ZEB1, the edited sites lie in the intronic region (Figure 2B and Supplemental file 1). Notably, the exon following these introns, 31387124-31387266, was significantly upregulated in both early and established RA samples. As shown in supplemental file 4, the upregulated exon 31387124-31387266 was part of several isoforms of ZEB1 that gave rise to either a smaller ZEB1 protein or no protein at all. This suggested that the editing events in the intron preceding the upregulated exon may perhaps influence preferential inclusion of the exon 31387124-31387266 in ZEB1 transcripts, resulting in low levels of functional ZEB1 in RA.
ANXA4
In ANXA4 gene, the edited site was part of a differentially spliced intronic region. We observed that the edited site was a part of two overlapping introns: 69781575-69803392 and 69788142-69804532 (Figure 2C and Supplemental file 4). Editing in this region correlated with upregulation of exon 69803393-69803515 which suggested preferential splicing of the intron 69781575-69803392 instead of the intron 69788142-69804532. The upregulation of exon 69803393-69803515 is likely to generate a transcript that does not translate into any protein (Figure 2 and Supplemental file 4).
TIMP3
In TIMP3 gene, the edited sites are within intron 32 802 123 to 32 849 451 which is located between exons 32801705-32802122 and exon 32849452-32849534 (Figure 2D and Supplemental file 4). Both these exons were downregulated in established RA samples, while the rest of the exons were not. TIMP3 also was downregulated according to differential gene expression analysis. Differential editing appears to correlate with TIMP3 downregulation.
DEXSeq analysis thus suggested that in RA, editing could perhaps lead to differential exon usage in some genes resulting in functional consequences which, in some instances, are very different from the consequences suggested by the differential gene expression analysis.
Protein-protein interaction network in RA
To investigate whether the set of genes that showed differential RNA editing were also part of a common interaction network, we performed protein-protein interaction analysis using the software, STRING. STRING analysis revealed that from the differentially edited gene set, several proteins from early and established RA samples belonged to a common network of the p53 pathway (Figure 3). Among the genes edited in the early RA sample set, ATM, ZEB1, ANXA4, PALMD, APPL1, and RBBP4 belonged to the p53 protein-protein interaction network (Figure 3A). In the established RA sample set, ATM, ZEB1, RBBP4, and TIMP3 were classified as part of the p53 protein-protein interaction network (Figure 3B).
Figure 3.

Network Analysis – STRING. Differentially edited genes from early RA (A) and established RA (B) were subjected to STRING network analysis to identify potential functional associations. Differentially edited genes in RA are highlighted using black circles. The type of protein-protein interaction is represented by the colour of the edge. The key for edge colour is reproduced from STRING analysis output and is shown at the bottom. Disconnected nodes in the network are not shown in the figure.
Some putative RNA editing events may affect miRNA binding
RNA editing can lead to changes in miRNA binding sites on transcripts, thereby modifying miRNA-mediated post-transcriptional regulation. Therefore, we investigated if edited and unedited sequences of the genes that showed differential editing in RA also showed differences in binding sites of miRNAs. Unedited and edited sequences were analysed using miRanda to identify putative miRNA binding sites. A comparison of these putative miRNA binding sites in unedited and edited sequences of differentially edited genes revealed that 18 miRNA-target interactions across 8 genes that were predicted in the unedited sequences were lost in their edited counterparts. As shown in supplemental file 5, 4 of these interactions were also predicted by TargetScan. Notably, in 15 of the 18 predicted miRNA binding sites, the edited nucleotide was located in the region complementary to the seed region of the corresponding miRNA.
Discussion
In this study, we identified differential editing events in early and established RA transcriptomes and investigated the potential functional consequences of these editing events. A total of 304 differentially edited sites were identified in early RA and 273 edited sites were observed in established RA. After excluding intergenic sites, differentially edited sites in introns and UTRs were observed in 11 genes in early RA, and 7 genes in established RA, and the consequences of these editing events were further investigated. In contrast, no differentially edited sites were identified in coding regions. We also observed differential exon usage in the vicinity of the edited sites in 4 genes, ATM, ZEB1, ANXA4, and TIMP3. Further analysis suggested that this differential exon usage could potentially lead to downregulation of the functional forms of ATM, ZEB1, ANXA4, and TIMP3 proteins. The identified differential RNA editing events included those that were observed in at least 50% of the samples in the group, and hence a substantial number of transcripts were likely to be edited, and to have functional consequences. Interestingly, protein-protein interaction analysis further revealed that several differentially edited candidates such as ATM, ZEB1, RBBP4, TIMP3, ANXA4, APPL1, and PALMD belonged to the protein-protein interaction network of the p53 pathway, which regulates DNA repair response and the cell cycle. The downregulation of functional forms of ATM, ZEB1, TIMP3, and ANXA4 proteins could thus lead to an impaired DNA damage response contributing to RA. In addition, we observed that RNA editing led to changes in putative miRNA binding sites in 8 differentially edited genes.
RA is a complex and heterogeneous disease with diverse modes of aetiology as well as regulation at molecular level. RA risk factors include genetic and epigenetic factors, exposure to certain chemicals, and lifestyle-related factors such as diet and smoking. 35 Our results strongly indicate that RNA editing could provide an additional level of gene regulation in RA. While RNA editing may be unlikely as the primary factor behind RA aetiology, it could contribute to disease progression and severity of the disease. These in silico studies provide a basis for further experimentation exploring and validating the role of RNA editing in RA.
ATM is a DNA damage response protein and is important for maintaining genome integrity, regulation of cell cycle check points, and DNA-damage tolerance pathways. 36 It has been reported that in RA, ATM is downregulated in T lymphocytes, 37 peripheral blood mononuclear cells, 38 and B lymphocytes. 39 Based on these findings, it has been proposed that decreased ATM activity could result in a high burden of DNA damage and accelerated cellular aging of lymphocytes, ultimately contributing to inflammation and bone erosion. In this bioinformatic analysis, although ATM mRNA was found to be upregulated in the synovial tissues of RA patients (Table 2A and B), differential exon usage analysis revealed that there could be a decrease in the levels of ATM transcripts that translate into full-length ATM protein (Figure 2A and Supplemental file 4). This would effectively reduce relative levels of functional ATM protein, potentially resulting in an effect similar to ATM downregulation such as impaired DNA repair, shorter cell cycle, and faster cell division. These effects may then contribute to dysregulated cell division and pannus formation seen in RA synovia. In another observation not related to RNA editing, we found that exon bins corresponding to the exon 108222832-108223186 in ATM were also significantly downregulated in both early and established RA. This exon is present in the ATM transcripts that translate into full-length protein, suggesting that the functional form of ATM protein may be downregulated in RA. Together, these observations reveal a new, hitherto unrecognised potential mechanism contributing to the reduced expression of functional ATM observed in RA.
Another gene that exhibited differential exon usage around the edited site was ZEB1. ZEB1 is a transcription factor that promotes epithelial-to-mesenchymal transition in cancer and is also expressed in immune cells. 40 Notably, ZEB1 was identified as a locus associated with RA in a genome-wide association study 41 and was also found to be upregulated in CD4+ T cells in RA. 42 As ZEB1 is known to repress IL2 expression, 43 ZEB1 could have an anti-inflammatory effect. Differential gene expression analysis in this study indicated that ZEB1 was downregulated in both early and established RA (Table 2A and B). Furthermore, DEXSeq analysis also showed that there could be relative enrichment of ZEB1 transcripts that translate into proteins smaller than the full-length protein or form no protein at all (Figure 2B and Supplemental File 4). Given the role of ZEB1 in suppressing IL2 production, it is possible that the downregulation of functional ZEB1 promotes inflammation in RA.
Interestingly, ZEB1 has also been shown to be a substrate of the ATM kinase. 44 In a radioresistant population of breast cancer cells, phosphorylation by ATM stabilises ZEB1, which in turn stabilises CHK1 to promote DNA damage response at the G2-M checkpoint. With previously reported observations that both ATM and ZEB1 are associated with RA pathology, and our observations in this study, it is plausible that both ATM and ZEB1 together regulate DNA damage response in synovial tissue. Downregulation of ATM and ZEB1 together could impair the DNA damage response in synovial tissue.
Two other edited genes, ANXA4 and TIMP3, displayed differential exon usage in specific stages of RA (Figure 2C and D). ANXA4 displayed differential exon usage in early RA. It is expressed in epithelial cells and regulates membrane dynamics, cell growth, and apoptosis. ANXA4 has been reported to be hyperphosphorylated in RA 45 and is also overexpressed in extracellular vesicles in another autoimmune disorder, Sjogren’s Syndrome. 46 The role of ANXA4 in these autoimmune disorders is unclear but it has been reported to regulate nuclear factor kappa B (NF-κB), a modulator of inflammation. 47 TIMP3 is another protein that is known to be involved in RA pathogenesis and showed differential exon usage in established RA. TIMP3 inhibits proteases involved in degrading the cartilaginous tissues in joints and metalloproteases that regulate the availability of inflammatory cytokines, thereby reducing inflammation in RA. 48 Overexpression of TIMP3 in RA synovial fibroblasts can reverse effects of TNFα 49 further supporting an anti-inflammatory role for TIMP3 in RA. Both decreased TIMP3 expression in RA and the observation that TIMP3 editing correlated with a downregulation of exons in the vicinity of the edited sites suggest that editing of TIMP3 could promote RA pathogenesis.
In addition, protein-protein interaction network analysis revealed that several differentially edited genes in both early and established RA, including ATM and ZEB1, were part of the p53 protein network (Figure 3). p53 is a key protein in cellular response to DNA damage and is also stabilised by ATM. 36 Downregulation of the functional form of ATM could destabilise p53. We also observed an indirect interaction between APPL1, RBBP4, and p53 in STRING analysis. APPL1 is an adaptor protein that localises to Rab5-positive endosomes. APPL1 is known to associate with the nucleosome remodelling and deacetylase (NuRD) complex, of which the histone-binding protein, RBBP4 is a component. 50 NuRD complex in turn interacts with p53. However, there is no evidence yet regarding the significance of APPL1 and RBBP4 in RA and it is not possible to predict their significance based on the current analysis. Also, APPL1 and RBBP4 did not exhibit differential exon usage in the vicinity of the edited sites.
The possible regulation of p53 interaction network by RNA editing could contribute to RA in several ways. TP53 has been reported to be mutated in fibroblast-like synoviocytes from synovial tissue in RA patients leading to the loss of p53 function. 51 This loss of p53 function could be responsible for apoptosis resistance leading to hyperplasia observed in RA. In addition, p53 loss can exert two more effects; it can enhance chronic inflammation by hyperactivation of NF-κB and can also promote angiogenesis which in turn can promote hyperplasia. 52 RNA editing–mediated effects on the p53 pathway could therefore potentially increase the severity of RA in multiple ways.
RNA editing affects transcripts in many ways. In addition to differential splicing leading to loss of full-length transcripts, it has recently been reported that absence of RNA editing can lead to unfolded protein response (UPR) and induce endoplasmic reticulum (ER) stress. 53 ER stress can lead to inflammatory response and contribute to RA. 54 It is also possible that RNA editing can also cause changes in RNA methylation, which could modulate RNA transport or translation, thereby contributing to RA.
RNA editing also led to the loss of miRNA binding sites in 18 predicted miRNA-target mRNA pairs (Supplemental file 5). Of these, 15 were sites edited in the region complementary to the miRNA seed region suggesting that these editing events are highly likely to disrupt miRNA-target mRNA interaction. Notably, all of these miRNA-target interactions are novel and have not been reported so far.
This bioinformatic study has identified novel post-transcriptional modes of gene expression regulation that could be important for RA pathogenesis. Genes that were identified as potentially regulated through RNA editing–associated post-transcriptional modification included genes previously known to be involved in RA, as well as some new genes that could be also involved in the pathogenesis of RA. This mode of regulation can now be further explored and experimentally validated to understand the diversity and complexity of RA.
Conclusions
We have identified RNA editing as a potential mode of post-transcriptional gene regulation in RA. Some editing events that were specifically observed in RA samples showed correlation with differential exon usage leading to the generation of non-functional transcripts. Interestingly, several edited genes were found to be involved in the DNA damage response and repair pathway. This study has identified new directions for RA research, which could help unravel the complex nature of RA pathogenesis.
Supplemental Material
Supplemental material, sj-pdf-1-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-pdf-2-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Acknowledgments
The authors thank all the researchers and patients associated with the project PRJNA352076 for making the sequencing data available for free. We thank Ernesto Picardi both for his help with interpreting REDItools results and for his inputs on the REDItools analysis. We also thank Mihir Mahajan for help.
Footnotes
Author Contributions: The project was conceptualised by YG and SS. REDI tools and DEXSeq analysis was performed by YG and SS. Differential gene expression, miRNA analysis, and STRING analysis was performed by YG. YG and SS prepared the manuscript.
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs: Yashoda Ghanekar  https://orcid.org/0000-0001-5413-2875
https://orcid.org/0000-0001-5413-2875
Subhashini Sadasivam
https://orcid.org/0000-0002-7875-7180
Supplemental material: Supplemental material for this article is available online.
References
- 1. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. J Autoimmun. 2020;110110:102392. doi: 10.1016/j.jaut.2019.102392. [DOI] [PubMed] [Google Scholar]
- 2. Dennis G, Jr, Holweg CT, Kummerfeld SK, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. 2014;16:R90. doi: 10.1186/ar4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klimiuk PA, Goronzy JJ, Björnsson J, Beckenbaugh RD, Weyand CM. Tissue cytokine patterns distinguish variants of rheumatoid synovitis. Am J Pathol. 1997;151:1311-1319. https://pubmed.ncbi.nlm.nih.gov/9358757 [PMC free article] [PubMed] [Google Scholar]
- 4. van der Pouw Kraan TC, van Gaalen FA, Huizinga TW, Pieterman E, Breedveld FC, Verweij CL. Discovery of distinctive gene expression profiles in rheumatoid synovium using cDNA microarray technology: evidence for the existence of multiple pathways of tissue destruction and repair. Genes Immun. 2003;4:187-196. doi: 10.1038/sj.gene.6363975. [DOI] [PubMed] [Google Scholar]
- 5. Tsubaki T, Arita N, Kawakami T, et al. Characterization of histopathology and gene-expression profiles of synovitis in early rheumatoid arthritis using targeted biopsy specimens. Arthritis Res Ther. 2005;7:R825-R836. doi: 10.1186/ar1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lequerré T, Bansard C, Vittecoq O, et al. Early and long-standing rheumatoid arthritis: distinct molecular signatures identified by gene-expression profiling in synovia. Arthritis Res Ther. 2009;11:R99. doi: 10.1186/ar2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Baarsen LG, Wijbrandts CA, Timmer TC, van der Pouw Kraan TC, Tak PP, Verweij CL. Synovial tissue heterogeneity in rheumatoid arthritis in relation to disease activity and biomarkers in peripheral blood. Arthritis Rheum. 2010;62:1602-1607. doi: 10.1002/art.27415. [DOI] [PubMed] [Google Scholar]
- 8. Okada Y, Eyre S, Suzuki A, Kochi Y, Yamamoto K. Genetics of rheumatoid arthritis: 2018 status. Ann Rheum Dis. 2019;78:446-453. doi: 10.1136/annrheumdis-2018-213678. [DOI] [PubMed] [Google Scholar]
- 9. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376-381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakahama T, Kawahara Y. Adenosine-to-inosine RNA editing in the immune system: friend or foe? Cell Mol Life Sci. 2020;77:2931-2948. doi: 10.1007/s00018-020-03466-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eisenberg E, Levanon EY. A-to-I RNA editing – immune protector and transcriptome diversifier. Nat Rev Genet. 2018;19:473-490. doi: 10.1038/s41576-018-0006-1. [DOI] [PubMed] [Google Scholar]
- 12. Walkley CR, Li JB. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017;18:1-13. doi: 10.1186/s13059-017-1347-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lev-Maor G, Sorek R, Levanon EY, Paz N, Eisenberg E, Ast G. RNA-editing-mediated exon evolution. Genome Biol. 2007;8:R29. doi: 10.1186/gb-2007-8-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakano M, Fukami T, Gotoh S, Nakajima M. A-to-I RNA editing up-regulates human dihydrofolate reductase in breast cancer. J Biol Chem. 2017;292:4873-4884. doi: 10.1074/jbc.M117.775684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakano M, Fukami T, Gotoh S, Takamiya M, Aoki Y, Nakajima M. RNA editing modulates human hepatic aryl hydrocarbon receptor expression by creating MicroRNA recognition sequence. J Biol Chem. 2016;291:894-903. doi: 10.1074/jbc.M115.699363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Volkman HE, Stetson DB. The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol. 2014;15:415-422. doi: 10.1038/ni.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. George CX, Ramaswami G, Li JB, Samuel CE. Editing of cellular self-RNAs by adenosine deaminase ADAR1 suppresses innate immune stress responses. J Biol Chem. 2016;291:6158-6168. doi: 10.1074/jbc.M115.709014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015;43:933-944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mannion NM, Greenwood SM, Young R, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482-1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liddicoat BJ, Piskol R, Chalk AM, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous DsRNA as nonself. Science. 2015;349:1115-1120. http://science.sciencemag.org/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crow YJ, Rehwinkel J. Aicardi-Goutiè res syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. doi: 10.1093/hmg/ddp293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chung H, Calis JJA, Wu X, et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell. 2018;172:811-824.e14. doi: 10.1016/j.cell.2017.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vlachogiannis NI, Gatsiou A, Silvestris DA, et al. Increased adenosine-to-inosine RNA editing in rheumatoid arthritis. J Autoimmun. 2020;106. doi: 10.1016/j.jaut.2019.102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roth SH, Danan-Gotthold M, Ben-Izhak M, et al. Increased RNA editing may provide a source for autoantigens in systemic lupus erythematosus. Cell Rep. 2018;23:50-57. doi: 10.1016/j.celrep.2018.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rojas M, Restrepo-Jiménez P, Monsalve DM, et al. Molecular mimicry and autoimmunity. J Autoimmun. 2018;95:100-123. doi: 10.1016/j.jaut.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 26. Guo Y, Walsh AM, Fearon U, et al. CD40L-dependent pathway is active at various stages of rheumatoid arthritis disease progression. J Immunol. 2017;198:4490LP-4501. doi: 10.4049/jimmunol.1601988. [DOI] [PubMed] [Google Scholar]
- 27. Walsh AM, Wechalekar MD, Guo Y, et al. Triple DMARD treatment in early rheumatoid arthritis modulates synovial T cell activation and plasmablast/plasma cell differentiation pathways. Plos One. 2017;12:e0183928. doi: 10.1371/journal.pone.0183928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lo Giudice C, Tangaro MA, Pesole G, Picardi E. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat Protoc. 2020;15:1098-1131. doi: 10.1038/s41596-019-0279-7. [DOI] [PubMed] [Google Scholar]
- 29. Soneson C, Matthes KL, Nowicka M, Law CW, Robinson MD. Isoform prefiltering improves performance of count-based methods for analysis of differential transcript usage. Genome Biol. 2016;17:12. doi: 10.1186/s13059-015-0862-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525-527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 31. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in drosophila. Genome Biol. 2003;5:R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607-D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Licht K, Jantsch MF. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol. 2016;213:15-22. doi: 10.1083/jcb.201511041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008-2017. doi: 10.1016/j.jaut.2019.102329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romão VC, Fonseca JE. Etiology and risk factors for rheumatoid arthritis: a state-of-the-art review. Front Med (Lausanne). 2021;8:689698. doi: 10.3389/fmed.2021.689698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801-817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 37. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206:1435-1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang H-H, Liu G-Y, Dwivedi N, et al. A molecular signature of preclinical rheumatoid arthritis triggered by dysregulated PTPN22. JCI Insight. 2016;1. doi: 10.1172/jci.insight.90045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mensah KA, Chen JW, Schickel J-N, et al. Impaired ATM Activation in B Cells Is Associated with Bone Resorption in Rheumatoid Arthritis. Vol. 11. http://stm.sciencemag.org/. Updated 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott CL, Omilusik KD. ZEBs: novel players in immune cell development and function. Trends Immunol. 2019;40:431-446. doi: 10.1016/j.it.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 41. Prasad P, Kumar A, Gupta R, Juyal RC, Thelma BK. Caucasian and Asian specific rheumatoid arthritis risk loci reveal limited replication and apparent allelic heterogeneity in North Indians. PLoS ONE. 2012;7:e31584. doi: 10.1371/journal.pone.0031584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ye H, Zhang J, Wang J, et al. CD4 T-cell transcriptome analysis reveals aberrant regulation of STAT3 and Wnt signaling pathways in rheumatoid arthritis: evidence from a case–control study. Arthritis Res Ther. 2015;17:76. doi: 10.1186/s13075-015-0590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang J, Lee S, Teh CE-Y, Bunting K, Ma L, Shannon MF. The transcription repressor, ZEB1, cooperates with CtBP2 and HDAC1 to suppress IL-2 gene activation in T cells. Int Immunol. 2009;21:227-235. doi: 10.1093/intimm/dxn143. [DOI] [PubMed] [Google Scholar]
- 44. Zhang P, Wei Y, Wang L, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864-875. doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Katano M, Kurokawa MS, Matsuo K, et al. Phosphoproteome analysis of synoviocytes from patients with rheumatoid arthritis. Int J Rheum Dis. 2017;20:708-721. doi: 10.1111/1756-185X.12997. [DOI] [PubMed] [Google Scholar]
- 46. Aqrawi LA, Galtung HK, Guerreiro EM, et al. Proteomic and histopathological characterisation of sicca subjects and primary Sjögren’s syndrome patients reveals promising tear, saliva and extracellular vesicle disease biomarkers. Arthritis Res Ther. 2019;21:181. doi: 10.1186/s13075-019-1961-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yao H-S, Sun C, Li X-X, et al. Annexin A4-nuclear factor-κB feedback circuit regulates cell malignant behavior and tumor growth in gallbladder cancer. Sci Rep. 2016;6:31056. doi: 10.1038/srep31056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mohammed FF, Smookler DS, Khokha R. Metalloproteinases, inflammation, and rheumatoid arthritis. Ann Rheum Dis. 2003;62:ii43. doi: 10.1136/ard.62.suppl_2.ii43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Drynda A, Quax PHA, Neumann M, et al. Gene transfer of tissue inhibitor of metalloproteinases-3 reverses the inhibitory effects of TNF-α on fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. J Immunol. 2005;174:6524. doi: 10.4049/jimmunol.174.10.6524. [DOI] [PubMed] [Google Scholar]
- 50. Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445-456. doi: 10.1016/S0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 51. Yamanishi Y, Boyle DL, Green DR, et al. P53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R12-R18. doi: 10.1186/ar1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taghadosi M, Adib M, Jamshidi A, Mahmoudi M, Farhadi E. The p53 status in rheumatoid arthritis with focus on fibroblast-like synoviocytes. Immunol Res. 2021;69:225-238. doi: 10.1007/s12026-021-09202-7. [DOI] [PubMed] [Google Scholar]
- 53. Guallar D, Fuentes-Iglesias A, Souto Y, et al. ADAR1-dependent RNA editing promotes MET and iPSC reprogramming by alleviating ER stress. Cell Stem Cell. 2020;27:300-314e11. doi: 10.1016/j.stem.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rahmati M, Moosavi MA, McDermott MF. ER stress: a therapeutic target in rheumatoid arthritis. Trends Pharmacol Sci. 2018;39:610-623. doi: 10.1016/j.tips.2018.03.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-pdf-2-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322221088725 for RNA Editing–Associated Post-Transcriptional Gene Regulation in Rheumatoid Arthritis by Yashoda Ghanekar and Subhashini Sadasivam in Bioinformatics and Biology Insights