

Abstract
Objectives
To describe a cohort of patients with arrhythmogenic left ventricular cardiomyopathy (ALVC), focusing on the spectrum of the clinical presentations.
Methods
Patients were retrospectively evaluated between January 2012 and June 2020. Diagnosis was based on (1) ≥3 contiguous segments with subepicardial/midwall late gadolinium enhancement in the left ventricle (LV) at cardiac magnetic resonance plus a likely pathogenic/pathogenic arrhythmogenic cardiomyopathy (AC) associated genetic mutation and/or familial history of AC and/or red flags for ALVC (ie, negative T waves in V4-6/aVL, low voltages in limb leads, right bundle branch block like ventricular tachycardia) or (2) pathology examination of explanted hearts or autoptic cases suffering sudden cardiac death (SCD). Significant right ventricular involvement was an exclusion criterion.
Results
Fifty-two patients (63% males, age 45 years (31–53)) composed the study cohort. Twenty-one (41%) had normal echocardiogram, 13 (25%) a hypokinetic non-dilated cardiomyopathy (HNDC) and 17 (33%) a dilated cardiomyopathy (DCM). Of 47 tested patients, 29 (62%) were carriers of a pathogenic/likely pathogenic DNA variant. Clinical contexts leading to diagnosis were SCD in 3 (6%), ventricular arrhythmias in 15 (29%), chest pain in 8 (15%), heart failure in 6 (12%) and familial screening in 20 (38%). Thirty patients (57%) had previously received a diagnosis other than ALVC with a diagnostic delay of 6 years (IQR 1–7).
Conclusions
ALVC is hidden in different clinical scenarios with a phenotypic spectrum ranging from normal LV to HNDC and DCM. Ventricular arrhythmias, chest pain, heart failure and SCD are the main clinical presentations, being familial screening essential for the affected relatives’ identification.
Keywords: Magnetic Resonance Imaging; Arrhythmogenic Right Ventricular Dysplasia; Cardiomyopathy, Dilated; Diagnostic Imaging
Key questions.
What is already known about this subject?
Arrhythmogenic left ventricular cardiomyopathy (ALVC) diagnosis is mainly based on the identification of fibro-fatty left ventricle (LV) replacement at histological analysis or demonstration of typical late gadolinium enhancement pattern with ring-like distribution at cardiac magnetic resonance (CMR) associated with positive genetic test or family history of arrhythmogenic right/left ventricular cardiomyopathy.
What does this study add?
ALVC is hidden under different clinical scenarios and degree of functional LV impairment.
Five main clinical contexts may be identified with this study: ventricular arrhythmias, chest pain, heart failure, familial screening and sudden cardiac death (SCD) as presenting event. Shared characteristics between these scenarios are represented by family history of cardiomyopathy/SCD, previous diagnosis of myocarditis, (infero)lateral negative T waves and low QRS voltages at ECG.
How might this impact on clinical practice?
We describe five diagnostic scenarios of ALVC, underlining common features that may rise clinical suspicion and guide the clinicians to request CMR and depict the typical structural involvement when clinical suspicion is high.
Introduction
The recent and rapid expansion of molecular biology and cardiac magnetic resonance (CMR) in the field of cardiomyopathies (CMP) has brought new clinical entities to the fore, whose definition goes beyond the ‘classical’ phenotype recognition. A paradigmatic situation is represented by arrhythmogenic cardiomyopathy (AC), an inherited CMP characterised by fibro-fatty infiltration, life threatening ventricular arrhythmias and sudden cardiac death (SCD). Although originally depicted as exclusive of the right ventricle—the classic arrhythmogenic right ventricle cardiomyopathy (ARVC)—in the last few years it has become clear that AC can affect either ventricles or predominantly the left ventricle (LV).1 Key features of arrhythmogenic left ventricular cardiomyopathy (ALVC) are a circumferential fibrous/fibro-fatty band between the middle and subepicardial third of the myocardium, corresponding to a ring-like late gadolinium enhancement (LGE) distribution at CMR, often associated with low QRS voltages and negative T waves in the inferior and/or lateral leads at ECG.2 Awareness for this disease is still poor, and diagnosis is often delayed or missed altogether, since this clinical entity can be concealed in different clinical settings both in terms of clinical onset and imaging phenotype. Yet, the highly malignant arrhythmogenic presentation, with ventricular arrhythmias and sudden death occurring irrespective of the degree of LV systolic dysfunction, makes prompt diagnosis essential. Therefore, the aim of this study was to describe the clinical, instrumental and genetic profile of a multicentre ALVC cohort, focusing on the spectrum of clinical presentations.
Methods
This is a retrospective observational study evaluating 62 consecutive patients with suspected ALVC between January 2012 and June 2020 at three Italian Centres (Cardiology Unit, St. Orsola Hospital, IRCCS Azienda Ospedaliero-Universitaria of Bologna (N=48); Azienda Ospedaliera Careggi, Florence (N=11) and Cardiovascular Center, University Hospital of Cona, Ferrara (=3)).
ALVC diagnosis was based on the following criteria: (1) a ring-like LGE LV pattern at CMR defined as ≥3 contiguous segments with subepicardial/midwall LGE in the same slice (isolated septal junctional LGE was not considered significant) with or without fatty infiltration plus a likely pathogenic/pathogenic AC associated genetic mutation and/or familial history of AC and/or red flags for ALVC (ie, negative T waves in V4-6/aVL, low voltages in limb leads, right bundle branch block-like ventricular tachycardia)1; (2) LV myocyte degeneration with fat and fibrosis in the same microscopic field, from subepicardial layer inward or within the mid-wall (>20% in at least two tissue blocks of 4 cm2; H&E stain) defined on microscopic analysis in explanted heart or autopsy examinations.2 3 Relatives with LV LGE were included (even if <3 consecutive segments). Significant right ventricle involvement, defined as the presence of systolic dysfunction (fractional area change (FAC) <30% at echocardiogram or EF <40% al CMR) or the presence of wall motion abnormalities (a/dyskinesia, bulging), was considered an exclusion criterion. Phenocopies (eight myocarditis and two sarcoidosis) were excluded based on a comprehensive clinical and multi-modality instrumental evaluation including myocardial biopsy and positron emission tomography (PET) when indicated (figure 1). Ischaemic heart disease was ruled out based on low pretest probability (young age, angina free, no cardiovascular risk factors) and where clinically indicated, by coronary anatomy evaluation (angiography n=29; CT scan n=2) or vasodilator stress CMR (n=2). A regional autopsy programme, integrated with molecular analysis, in patients with SCD younger than 55 years is active at St. Orsola University Hospital (Bologna) since 2018.
Figure 1.
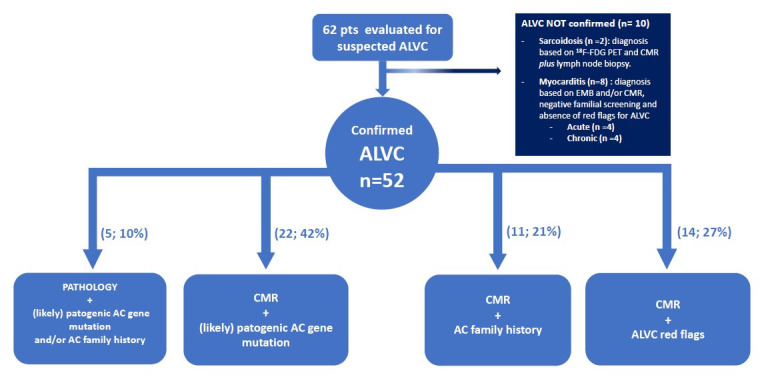
Flow chart summarising the enrolment of the study population according to the met inclusion criteria. AC, arrhythmogenic cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; CMR, cardiac magnetic resonance.
Definition of clinical scenarios
Based on the clinical setting in which diagnosis was performed, patients were divided into five groups:
SCD: a non-traumatic, unexpected fatal event occurring within 1 hour of the onset of symptoms in an apparently healthy subject (or when the victim was in good health 24 hours before the event, if death was not witnessed).4
Ventricular arrhythmias: ‘high arrhythmic burden’ defined as frequent ventricular ectopic beats (>500/24 hours)/non-sustained ventricular tachycardia (NSVT) associated with palpitations, SVT requiring emergency department admission and aborted cardiac arrest due to ventricular fibrillation.
Chest pain: patients requiring hospital admission or outpatient evaluation because of acute or chronic chest pain, respectively.
Heart failure: signs and symptoms of heart failure with evidence of reduced LV ejection fraction (LVEF).5
Familial screening: cases identified due to the family history of dilated cardiomyopathy (DCM, ALVC, ARVC or SCD.
Echocardiographic phenotypes were distinguished as normal (in terms of LV dimension and systolic function), hypokinetic non-dilated cardiomyopathy (HNDC) (LVEF <50% without LV dilatation6) and DCM (LV dilation and systolic dysfunction in the absence of abnormal loading conditions such as hypertension or valve disease, or coronary artery disease causing global systolic impairment).
CMR was carried out on 1.5 Tesla equipment at each centre using standardised protocols. The presence and location of LGE were assessed: positivity was considered when visible in two phase-encoding directions and two orthogonal planes. Intramyocardial fat replacement was reported by comparing T1-weighted and fat suppressed T1-weighted sequences.
Genetic test was performed by different sequencing technologies, using gene panels reflecting the standard practice at the time of testing in each centre. Identified variants were defined according to recommended guidance for classification.7 All patients gave written informed consent for DNA analysis.
Continuous data distribution was assessed with the Shapiro-Wilk test and expressed as median and IQR.
Results
General patient features
Fifty-two patients from 37 families (63% males, median age 45 years (IQR 31–53)) formed the final study cohort (table 1). Echocardiographic patterns were distributed as follows: normal (n=21; 41%), HNDC (n=13; 25%), DCM (n=17; 33%). Diagnosis was based on CMR plus additional criteria in 47 patients (90%). LGE involved most frequently the septum (30; 64%), the inferior (31; 66%) and the infero-lateral (30; 64%) walls, whereas a circumferential distribution was present in 14 patients (30%). Intramyocardial fat infiltration was present in 23 patients (49%) always near to the LGE areas.
Table 1.
Clinical and instrumental characteristics of patients with ALVC according to the clinical scenario
| Overall | SCD | Ventricular Arrhythmias | Chest Pain | Heart failure | Familiar Screening | |
| 52 | 3 (6) | 15 (29) | 8 (15) | 6 (12) | 20 (38) | |
| Age at diagnosis (years) | 45 (31–53) | 23 (19–36) | 50 (40–57) | 45 (27–57) | 47 (46–59) | 36 (30–50) |
| Males | 33 (63) | 3 (100) | 11 (73) | 4 (50%) | 5 (83) | 10 (50) |
| Probands | 36 (69) | 3 (100) | 14 (93) | 8 (100) | 5 (83) | 6 (30) |
| Family history of sudden death | 30 (58) | 0 | 10 (67) | 4 (50) | 3 (50) | 13 (65) |
| ≤40 years | 17 (33) | 0 | 3 (20) | 0 | 3 (50) | 11 (55) |
| >40 years | 13 (25) | 0 | 7 (47) | 4 (50) | 0 | 2 (10) |
| Family history of ALVC/ARVC | 19 (4) | 0 | 2 (13) | 1 (12) | 2 (33) | 14 (70) |
| Family history of DCM | 2 (4) | 0 | 1 (6) | 0 | 0 | 1 (5) |
| Any previous myocarditis diagnosis | 16 (31) | 1 (17) | 3 (21) | 6 (75) | 4 (67) | 2 (10) |
| Previous myocarditis | 9 (17 | 1 (17) | 2 (14) | 4 (50) | 1 (17) | 1 (5) |
| Instrumental diagnosis of previous myocarditis | 7 (13) | – | 1 (7) | 2 (25) | 3 (50) | 1 (5) |
| NYHA III/IV | 2 (4) | 0 | 0 | 0 | 1 (17) | 1 (5) |
| Echocardiogram | 51/52 | 2 (66) | 15 (100) | 8 (100) | 6 (100) | 20 (100) |
| Phenotype | ||||||
| Normal | 21 (41) | 2 (100) | 3 (20) | 4 (50) | 0 (0) | 12 (60) |
| HNDC | 13 (25) | 0 | 4 (26) | 3 (37) | 2 (33) | 4 (20) |
| DCM | 17 (33) | 0 | 8 (54) | 1 (12) | 4 (67) | 4 (20) |
| LVEF (%) | 50 (45–57) | 57 | 49 (43–50) | 50 (46–62) | 33 (31–47) | 53 (49–60) |
| LVEF ≤35% | 2 (4) | 0 | 0 | 0 | 2 (33) | 0 |
| LVEDV (mL/m2) | 63 (55–76) | 62 | 75 (61–80) | 59 (54–76) | 81 (70–117) | 57 (52–60) |
| Severe LV dilation | 4 (8) | 0 | 1 (6) | 0 | 2 (33) | 1 (5) |
| High arrhythmic burden | 30 (57) | 0 | 12 (80) | 2 (24) | 4 (67) | 12 (60) |
| VEB >500 | 11 (21) | 0 | 4 (25) | 1 (12) | 1 (17) | 5 (25) |
| NSVT | 19 (36) | 0 | 8 (50) | 1 (12) | 3 (50) | 7 (35) |
| Cardiac magnetic resonance | 47 | – | 15 | 8 | 4 | 20 |
| LVEF (%) | 50 (43–56) | 48 (41–52) | 49 (40–57) | 39 (36–43) | 53 (51–59) | |
| LVEDV (mL/m2) | 101 (88–110) | 103 (88–119) | 106 (98–112) | 102 (98–107) | 95 (84–109) | |
| RVEF (%) | 57 (53–61) | 56 (53–60) | 58 (56–60) | 45 (40–52) | 59 (54–61) | |
| RVEDV (mL/m2) | 83 (68–90) | 73 (66–82) | 84 (76–90) | 89 (72–105) | 85 (78–91) | |
| LV fat infiltration | 23 (49) | 12 (80) | 3 (37) | 1 (25) | 7 | |
| RV fat infiltration | 0 | 0 | 0 | 0 | 0 | |
| LGE distribution | ||||||
| Septum | 30 (64) | 9 (60) | 6 (75) | 2 (50) | 13 (65) | |
| Inferior | 31 (66) | 7 (47) | 7 (87) | 1 (25) | 16 (80) | |
| Infero-lateral | 30 (64) | 9 (60) | 7 (87) | 2 (50) | 12 (60) | |
| Lateral | 26 (55) | 6 (43) | 7 (87) | 4 (100) | 9 (45) | |
| Antero-lateral | 17 (37) | 2 (13) | 6 (75) | 2 (50) | 7 (35) | |
| Anterior | 21 (45) | 5 (33) | 5 (62) | 2 (50) | 9 (45) | |
| ECG available | 50/52 | 1 (33) | 15 (100) | 8 (100) | 6 (100) | 20 (100) |
| Normal | 4 (8) | 0 | 1 (7) | 1 (17) | 0 | 2 (10) |
| T wave inversion | ||||||
| Isolated in V1-V3 | 3 (6) | 0 | 0 | 0 | 0 | 3 (15) |
| Isolated in V4-V6 or/and D1-aVL | 12 (23) | 0 | 2 (13) | 2 (25) | 3 (50) | 5 (25) |
| Isolated in inferior leads | 2 (4) | 0 | 0 | 0 | 0 | 2 (10) |
| Infero-lateral | 6 (12) | 0 | 2 (13) | 1 (12) | 2 (33) | 1 (5) |
| Diffuse | 1 (2) | 0 | 0 | 0 | 1 (17) | 0 |
| Low QRS peripheric voltages | 23 (47) | 1 (100) | 10 (67) | 3 (37) | 2 (33) | 7 (35) |
| Q waves | 13 (52) | 0 | 6 (40) | 2 (25) | 2 (33) | 3 (15) |
| LAFB | 4 (8) | 0 | 0 | 1 (12) | 3 (50) | 0 |
| LPFB | 1 (2) | 0 | 1 (7) | 0 | 0 | 0 |
| LBBB | 0 | 0 | 0 | 0 | 0 | 0 |
| RBBB | 3 (6) | 0 | 2 (13) | 0 | 0 | 1 (5) |
| QRS duration (ms) | 100 (90–110) | 95 | 100 (90–114) | 110 (100–120) | 110 (100–120) | 97.5 (88–102) |
| QRS≥120 ms | 6 (12) | 0 | 3 (20) | 2 (25) | 2 (33) | 0 |
| LVH | 2 (4) | 0 | 1 (7) | 1 (12) | 0 | 0 |
| ICD implantation | 29 (56) | – | 9 (60) | 3 (37) | 4 (66) | 13 (65) |
| Primary prevention | 20 (38) | – | 2 (13) | 3 (37) | 4 (66) | 11 (55) |
| Secondary prevention | 9 (27) | – | 7 (47) | 0 | 0 | 2 (10) |
Values are expressed as n, n (%) or median (IQR).
Severe LV dilatation: >100 mL/m2 for male and >80 mL/m2 for female (J Am Soc Echocardiogr 2015;28:1–39).
ALVC, arrhythmogenic left ventricle cardiomyopathy; ARVC, arrhythmogenic right ventricle cardiomyopathy; DCM, dilated cardiomyopathy; HNDC, hypokinetic non-dilated cardiomyopathy; ICD, implantable cardioverter defibrillator; LAFB, left anterior fascicular block; LBBB, left bundle branch block; LVEDV, left ventricle end-diastolic volume; LVEF, left ventricle ejection fraction; LVH, left ventricular hypertrophy (one of Sokolow-Lyon, Cornell, Romhilt-Estes, R wave in aVL); NSVT, non-sustained ventricular tachyarrhythmia; NYHA, New York Heart Association; RBBB, right bundle branch block; SCD, sudden cardiac death; TWI, T wave inversion; VEB, ventricular ectopic beats.
Diagnosis was based on whole heart examination in five patients: three patients suffering SCD and two heart transplant patients affected by end stage heart failure. Pathology revealed LV fibro-fatty replacement predominantly in the postero-lateral wall in the three cases of SCD, while RV was only mildly involved. Regarding the two explanted hearts, a diffuse LV fibro-fatty replacement involving the septum, anterior, postero-lateral wall and the right ventricle was present in first one whereas a fibro-fatty replacement of the LV anterior wall with septal and RV outflow tract extensive fibrosis was detected in the second one. In both of them, RV was not significantly involved in the first clinical evaluation.
Applying the 2010 Task Force Criteria in the group of 47 patients clinically diagnosed (ie, excluding the 5 patients with pathological diagnosis), 11 (23%) had a definite diagnosis, while 3 (6%) had a borderline diagnosis and 20 (42%) a possible diagnosis. One minor criterion was present in 13/47 (28%) patients.
Notably, 29 of the 52 patients (56%) had previously received a diagnosis other than ALVC: 12 idiopathic DCM, 5 post-myocarditis DCM, 7 acute myocarditis, 3 idiopathic sustained ventricular tachyarrhythmias, 1 frequent ventricular ectopic beats and 1 non-ST-elevated myocardial infarction. In particular, in 15 patients ALVC was diagnosed following the introduction of MRI in the diagnostic work-up of patients with DCM and in two cases by pathology examination after heart transplantation. The diagnostic delay was 6 years (IQR 1–7) with a maximum of 21 years.
Genetic testing
Forty-seven (90%) patients underwent genetic test (table 2). All were screened for the exons and flanking intronic sequences of the following desmosomal genes: desmoplakin (DSP), plakophilin-2 (PKP2), desmoglein-2 (DSG2), desmocollin-2 (DSC2) and plakoglobin (JUP). Of them, 43 (91%) were screened for phospholamban (PLN), TMEM43, desmin (DES), lamin A/C (LMNA) and SCN5A as well, while 11 patients (23%) for filamin C (FLNC). Two patients with negative next-generation sequencing (NGS) panel were tested for multiplex ligation probe amplification and no copy number variation was detected. Pathogenic/likely pathogenic mutations were identified in 29 (62%) patients and DSP accounted for the most frequently involved gene (n=23; 49%). It should be noted that a relevant subgroup of patients had a double sequence variant, with VUS/likely benign variation in addition to a pathogenic/likely pathogenic mutation, raising the hypothesis of a digenic contribution or a modifier role (online supplemental table 1).
Table 2.
Genetic findings in tested patients (n=47)
| Pathogenic | Likely pathogenic | VUS | Likely benign/benign | |
| 24 (52%) | 5 (10%) | 8 (17%) | 3 (6%) | |
| Single desmosome mutation | ||||
| DSP | 18 | 1 | 1 | – |
| DSG2 | – | – | 2 | – |
| Single non-desmosome mutation | ||||
| FLNC | 3 | – | – | – |
| RYR2 | – | – | 1 | – |
| DES | – | 1 | – | – |
| Double mutation | ||||
| DSP+FLNC | 2 (DSP) | – | 2 (FLNC) | – |
| DSP+TMEM43 | – | 1 (DSP) | 1 (TMEM43) | – |
| PKP2+TMEM43 | 1 (PKP2) | – | – | 1 (TMEM43) |
| SCN5A+ANK2 | – | 2 (SCN5A) | – | 2 (ANK2) |
ANK2, ankirin2; DES, desmin; DSG2, desmoglein 2; DSP, desmoplakin; FLNC, filamin C; PKP2, plakophillin; RYR2, ryanodine receptor 2; SCN5A, sodium voltage-gated channel alpha subunit 5; TMEM43, transmembrane protein 43; VUS, variant of unknown significance.
openhrt-2021-001914supp001.pdf (77.2KB, pdf)
Clinical presentations
Clinical presentations leading to ALVC diagnosis were: SCD in 3 (6%) patients, ventricular arrhythmias in 15 (29%), chest pain in 8 (15%), heart failure in 6 (12%). Moreover, ALVC was identified through familial screening in 20 (38%). Clinical and instrumental characteristics of the patients according to the clinical context are reported in table 1 and figures 2 and 3.
Figure 2.

Echocardiographic phenotype according to the clinical scenario. Distribution of normal (grey), hypokinetic non-dilated cardiomyopathy (HNDC, white) and dilated cardiomyopathy phenotypes (DCM, black) among the different diagnostic pathways are shown by stacked bar chart.
Figure 3.
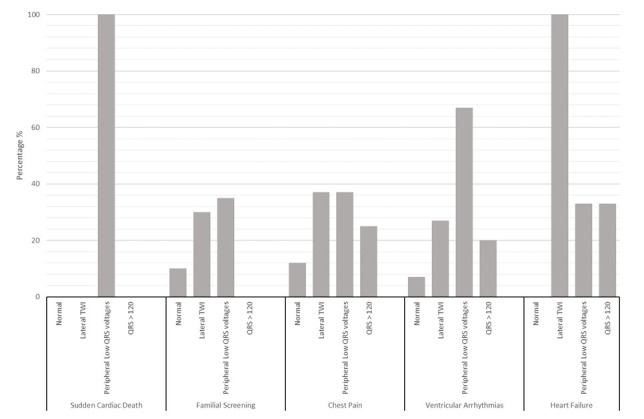
ECG findings according to the clinical scenario. The prevalence of the main ECG characteristics for each clinical scenario shown by histograms. Lateral T wave inversion (TWI) includes: isolated lateral, infero-lateral and diffuse TWI.
Sudden cardiac death
Of 50 cases of SCD referred to St. Orsola University Hospital of Bologna from 2018 to 2020, 3 (6%) received a diagnosis of ALVC. They were all males and had no family history of SCD or CMP.
The first was a 49-year-old man who died during physical exertion. Normal ECG and echocardiogram were recorded the year before. A likely pathogenic mutation in DSP (c.3533 T>G) and a VUS in TMEM43 (c.1150 C>G) were identified by molecular analysis. Familial screening led to ALVC diagnosis in his 19-year-old child with mid-wall LGE in the infero-lateral wall at CMR despite a normal ECG and echocardiogram; the same DSP gene mutation was identified. The second patient died at the age of 30 years during recreational sport activity. He previously received a diagnosis of myocarditis (chest pain and elevated plasmatic troponins) at the age of 26 years. Low QRS voltages were present on ECG whereas LV volumes and EF were normal at echocardiogram, with no segmental wall motion abnormalities. Familial screening performed after death revealed a DCM in his sister and an HNDC in his mother with a likely pathogenic mutation in SCN5A (c.2244G>A) and a VUS in ANK (c.10054G>C) in both family members. The last patient was a 15-year-old boy who had SCD at rest. A pathogenic FLNC variant (c.8034delC) determining a truncated protein was found by molecular autopsy. Family screening revealed the same FLNC variant in his father who presented an HNDC with LVEF 45% and a subepicardial circumferential LGE at CMR.
Ventricular arrhythmias
This was the most common clinical presentation, being present in a third of the study population (n=15, 29%). Aborted cardiac arrest was documented in 2 (4%) patients, SVT in 7 (13%) and high arrhythmic burden in 6 (11%). Family history of SCD was present in 10 patients (67%). Eight patients (54%) had DCM, four (26%) HNDC and three (20%) normal LV volumes and EF. Most (80%) where in New York Heart Association (NYHA) functional class I. A normal ECG was rare (7%) whereas most tracings were characterised by low QRS voltages in limb leads (67%). Three patients (21%) had a previous clinical or instrumental diagnosis of myocarditis. Of 13 patients tested, 3 had a truncating DSP mutation (in 1 case associated with a VUS on FLNC), 1 had a truncating FLNC mutation, 1 DSG2, 1 RYR2.
Chest pain
Eight (15%) patients were included in this group characterised by two main clinical scenarios: acute and chronic chest pain. The first setting involved six patients (median age 36 years (27–44); 50% males) accessing the emergency department for acute chest pain with elevated troponins consistent with acute myocardial injury but no coronary arteries obstruction on angiography. The initial diagnosis was acute myocarditis in five and myocardial infarction with non-obstructed coronary arteries (MINOCA) in one. Three of them had been previously hospitalised for similar episodes interpreted as myocarditis (N=2) or MINOCA (N=1). The final diagnosis of ALVC was supported by typical CMR findings and six were successfully genotyped (4 DSP). Main clinical and instrumental characteristics in this subgroup were a family history of sudden death (50%), prior acute myocarditis (50%), (infero)lateral T wave inversion (TWI) (33%) and low QRS voltages in limb leads (37%).
In the second setting two outpatients with chronic chest pain atypical for angina, mild LV dysfunction and abnormal ECG at rest were included (a 60-year-old man and a 57-year-old woman). ALVC diagnosis originated from a stress perfusion CMR, performed to exclude an ischaemic aetiology, which unexpectedly showed non-ischaemic ring-like subepicardial fibrosis plus fatty infiltration in the septum and lateral wall.
Heart failure
Six patients (12%) were referred due to exertional dyspnoea (NYHA class II or III) and impaired LVEF. None experienced acute heart failure. The echocardiographic phenotype was characterised by DCM in four patients (67%) and HNDC in two (33%), with a median LVEF of 33% (IQR 31–47) and a median indexed end-diastolic volume of 81 mL/m2 (IQR 70–117 mL/m2). The ECG showed prolonged QRS (ie, ≥120 ms) in 2 patients (33%), TWI in the (infero)lateral leads in 6 (100%) and low peripheral QRS voltages in 2 (33%). No patient had either left bundle branch block or LV hypertrophy. Three patients had truncating DSP mutations and three had a negative genetic test.
Familial screening
Twenty patients (38%) were identified by screening performed due to family history of ALVC (n=10), ARVC (n=2), DCM (n=2) or SCD (n=6). A family history of SCD was frequent (65%) and patients were mainly asymptomatic (90%). Two patients (10%) had normal ECG whereas six (30%) had (infero)lateral TWI and seven (35%) low QRS voltages in limb leads. The echocardiogram was normal in 12 (60%) whereas DCM and HNDC phenotypes were evenly distributed. Frequent ventricular ectopic beats and/or NSVT were recorded in 12 patients (60%) by ECG Holter monitoring. DSP variants were reported in 11 patients (55%), alone or with mutations in other genes. It is worth noting that the relatives identified by familial screening had clinical features that might have led to ALVC diagnosis through a different way, as shown in figures 4 and 5.
Figure 4.

Central illustration: family tree. Black filled symbols stand for affected carriers. The 54-year-old woman proband (patient I, arrow) showed T wave inversion in inferolateral leads at ECG (IA) and a dilated cardiomyopathy at echocardiogram (IB–IC). Her 61-year-old sister (patient II) had a high premature ventricular ectopic beats burden (IB), a hypokinetic non-dilated cardiomyopathy (HNDC) phenotype and an initial subepicardial fatty replacement (IIB) with a semi-circumferential mid-wall distribution of late gadolinium enhancement (LGE) (IIC) at cardiac magnetic resonance (CMR). Patient III had a history of hospital admission at the age of 21 years for chest pain and suspected myocarditis with an HNDC phenotype. During the hospitalisation, non-sustained ventricular tachycardia occurred (IIIA) and CMR showed an extensive intramyocardial oedema at T2-weighted sequences (IIIB) with a mutual mid-wall distribution of LGE (IIIC).
Figure 5.

Central illustration: family tree. Affected male (squares) and female subjects (circles) are shown as solid black. The 49-year-old woman proband (patient I, arrow), affected by dilated cardiomyopathy with diffuse T wave inversion at ECG (IA), underwent heart transplantation due to severe heart failure. The histopathological examination of the explanted heart showed extensive mid-wall fibrosis with fibrofatty replacement on the right ventricular side of the septum (IB), hypertrophy and sarcoplasmic vacuolation of cardiomyocytes (IC). Her 47-year-old sister (patient II) showed normal echocardiographic phenotype, frequent premature ventricular beats at ECG, infero-lateral T wave inversion and peripheral low voltages (IIA); at cardiac magnetic resonance (CMR) T1-weighted sequences detected a linear fatty infiltration of mid-wall interventricular septum (IIB) and a diffuse mid-wall/subendocardial and circumferential late gadolinium enhancement (LGE) on anterior, septal and inferior walls of left ventricle (IIC). Patient III was asymptomatic, ECG and echocardiogram were normal (IIIA), but CMR revealed a small area of subepicardial fat on infero-basal septum (IIIB) and a focal mid-wall/subendocardial distribution of LGE on inferior wall (IIIC).
Discussion
ALVC diagnosis is challenging since this clinical entity is hidden in different clinical contexts and is characterised by a wide phenotypic spectrum ranging from normal LV, to HNDC and classic DCM. In fact, differently from the traditional CMPs phenotypes, ALVC diagnosis mainly relies on typical tissue characterisation and distribution of fibro-fatty replacement, regardless of the degree of LV functional impairment or dilatation. In the present study, we describe five clinical scenarios leading to ALVC diagnosis with the aim to underline common features and discrepancies that may prompt clinical suspicion.
Sudden cardiac death
SCD is not uncommon in AC, especially among male young individuals and athletes, and can be the first disease presentation. We reported three young males experiencing SCD, two of them during moderate effort. In a large cohort of 5205 SCD cases, Miles et al3 collected 202 cases (4%) of AC including 35 (17%) ALVC, whose death occurred during effort or at rest/sleep in equal proportion and generally without preceding symptoms or relevant family history. At initial inspection 20% of the AC cases were macroscopically normal regardless of LV involvement suggesting a concealed phase of the disease. This is in line with the hypothesis that in ALVC, differently from ARVC and biventricular AC, the risk of major cardiac events could be relevant even in absence of evident functional impairment.8 Additionally, this finding emphasises the importance of dedicated regional or national cardiac pathology programmes for young SCD victims. When appropriate pathological expertise is lacking, autopsies are often not performed or reach a non-specific diagnosis of ‘myocarditis’ which does not trigger family screening and hence fails to identify other affected members.
Ventricular arrhythmias
In our population, the most frequent clinical presentation was characterised by symptoms associated with ventricular arrhythmias. Half of the patients had a DCM whereas normal echocardiogram and HNDC were equally distributed in the other half. On the ECG, low QRS voltages in limb leads were highly prevalent (67%). DSP was the most frequent gene involved. In line with our findings, in a cohort of DSP-related CMP, ventricular arrhythmias occurred with an LVEF between 35% and 55%, occasionally >55%, confirming that the established LVEF threshold <35% for ICD implantation is a poor event predictor in this setting.9 The not negligible risk, even with preserved EF, may be the result of early gap junction remodelling, recurrent inflammatory episodes or extensive LV fibro-fatty replacement that can precede or be discrepant with respect to the degree of LV dysfunction.
Chest pain
Chest pain, mainly with acute onset and troponin release without coronary obstruction on angiography, was the second most frequent clinical presentation. Accordingly, Sen-Chowdhry et al1 reported that almost one-third of 42 patients with ALVC had recurrent chest pain and a previous diagnosis of presumed viral myocarditis characterised by acute chest pain and angiographically normal coronary arteries. Similarly, in 107 patients with pathogenic DSP mutations Smith et al9 reported chest pain with troponin elevation and normal coronary angiography in 15%, associated with a non-ischaemic LGE mainly in the LV subepicardium. Therefore, when presenting with acute chest pain, ALVC may be misdiagnosed with a first step label of myocarditis or MINOCA. Clinicians should keep in mind AC as possible genetic cause when facing these diagnostic ‘boxes’. In this perspective, both CMR and family history play an important role. Piriou et al10 suggested that the presence of a family history of CMP or SCD, in case of acute myocarditis, should always prompt genetic testing to search for desmosomal mutations.
Heart failure
Patients in this subset were typically middle-aged males, coming to medical attention due to HF symptoms and a moderate–severe reduction of LVEF but mildly dilated LV. Awareness of desmosome gene-related aetiologies among the broad DCM spectrum has gained momentum in the last decade, beyond mere nosography, because of the implications for SCD prevention.11–13 Augusto et al2 compared patients with DSP/FLNC mutations to those without desmosomal gene variants in 89 DCM patients and identified the subepicardial ring-like LGE pattern at CMR together with LV regional wall motion abnormalities as the most defining elements in DSP/FLNC. Notably, LVEF and global longitudinal strain were on average higher in the DSP-FLNC group. This peculiar LGE pattern, confined to the outer layers of the LV wall, has been associated with milder degrees of LV systolic dysfunction in ALVC, differently from classic, non-desmosomal DCM where LV dysfunction is mainly the expression of a primary myocyte contractile dysfunction.14 Two patients in our study, with a previous diagnosis of post-myocarditis DCM, received ALVC diagnosis by pathological evaluation after heart transplantation. Similarly, in a recent autoptic series by Miles et al3 more than half of the decedents with pathological features of AC at autopsy were diagnosed with DCM in life. High arrhythmic burden, low voltages in limb leads, TWI in the infero-lateral leads and the absence of left bundle branch block at ECG might be useful red flags for ALVC diagnosis.14
Familiar screening
AC is most commonly inherited by an autosomal dominant pattern and a thorough familial screening is mandatory. Our study underlines the relevance of this strategy to identify affected relatives (being the largest group in the cohort) and contributes to the awareness that AC can express with different clinical presentations and different phenotypes even within the same family. Moreover, the high proportion of patients with normal LV function among relatives remarks the need of tissue characterisation through CMR, that should be considered part of the first level imaging approach to not miss ALVC diagnosis.
Limitations
A third of the study cohort had a negative genetic test, but the pick-up rate was similar to previous ALVC/ARVC studies.1 15 Additionally, molecular analyses mostly relied on next-generation DNA sequencing that does not detect copy number variants of large DNA segments. Finally, although in the recently proposed ‘Padua criteria’ a pathogenic AC-causing mutation is deemed necessary for ALVC diagnosis, phenocopies were excluded based on a comprehensive clinical and multi-modality instrumental evaluation.16
Conclusions
ALVC diagnosis is challenging since this clinical entity is hidden in different clinical scenarios with a phenotypic spectrum ranging from normal LV, to HNDC and classic DCM. Five main clinical scenarios leading to ALVC diagnosis may be identified including ventricular arrhythmias, chest pain, heart failure, familial screening and, in 6% of patients, SCD as the presenting event. Family history of CMP/SCD, previous diagnosis of myocarditis, ECG infero-lateral negative T waves and low QRS voltages are common features that may increase clinical suspicion. Normal ECG and echocardiograms cannot rule out ALVC diagnosis and CMR is crucial to exclude typical structural involvement when suspicion is high or in familial screening.
Footnotes
Twitter: @MatteoBertini5
MG and RD contributed equally.
Contributors: MG and RD contributed equally to this work. Study design: EB and CR. Collection of data: MG, RD, VP, MM, VF, MB, GT. Data analysis: MG, RD, EB. Statistical analysis: RD, MG. Drafting of the manuscript: MG, RD and EB. Critical revision for important intellectual content: IO, CR. Revision, editing and approval of the final manuscript: all authors. Author responsible for the overall content as the guarantor: MG, RD and EB.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s).
Ethics approval
This study involves human participants and was approved by CMPBO-19CmpiBo. Participants gave informed consent to participate in the study before taking part.
References
- 1.Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 2008;52:2175–87. 10.1016/j.jacc.2008.09.019 [DOI] [PubMed] [Google Scholar]
- 2.Augusto JB, Eiros R, Nakou E, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging 2020;21:326–36. 10.1093/ehjci/jez188 [DOI] [PubMed] [Google Scholar]
- 3.Miles C, Finocchiaro G, Papadakis M, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation 2019;139:1786–97. 10.1161/CIRCULATIONAHA.118.037230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of cardiology (ESC). endorsed by: association for European paediatric and congenital cardiology (AEPC). Eur Heart J 2015;36:2793–867. 10.1093/eurheartj/ehv316 [DOI] [PubMed] [Google Scholar]
- 5.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–200. 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 6.Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC Working group on myocardial and pericardial diseases. Eur Heart J 2016;37:1850–8. 10.1093/eurheartj/ehv727 [DOI] [PubMed] [Google Scholar]
- 7.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aquaro GD, De Luca A, Cappelletto C, et al. Prognostic value of magnetic resonance phenotype in patients with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2020;75:2753–65. 10.1016/j.jacc.2020.04.023 [DOI] [PubMed] [Google Scholar]
- 9.Smith ED, Lakdawala NK, Papoutsidakis N, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020;141:1872–84. 10.1161/CIRCULATIONAHA.119.044934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piriou N, Marteau L, Kyndt F, et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail 2020;7:1520–33. 10.1002/ehf2.12686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott P, O'Mahony C, Syrris P, et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2010;3:314–22. 10.1161/CIRCGENETICS.110.937805 [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Pavia P, Syrris P, Salas C, et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 2011;97:1744–52. 10.1136/hrt.2011.227967 [DOI] [PubMed] [Google Scholar]
- 13.Disertori M, Quintarelli S, Mazzola S, et al. The need to modify patient selection to improve the benefits of implantable cardioverter-defibrillator for primary prevention of sudden death in non-ischaemic dilated cardiomyopathy. Europace 2013;15:1693–701. 10.1093/europace/eut228 [DOI] [PubMed] [Google Scholar]
- 14.Cipriani A, Bauce B, De Lazzari M, et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J Am Heart Assoc 2020;9:e014628. 10.1161/JAHA.119.014628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Romano S, Judd RM, Kim RJ, et al. Feature-Tracking Global Longitudinal Strain Predicts Death in a Multicenter Population of Patients With Ischemic and Nonischemic Dilated Cardiomyopathy Incremental to Ejection Fraction and Late Gadolinium Enhancement. JACC Cardiovasc Imaging 2018;11:1419–29. 10.1016/j.jcmg.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corrado D, Perazzolo Marra M, Zorzi A, et al. Diagnosis of arrhythmogenic cardiomyopathy: the Padua criteria. Int J Cardiol 2020;319:106–14. 10.1016/j.ijcard.2020.06.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
openhrt-2021-001914supp001.pdf (77.2KB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.