

Abstract
Cyclooxygenase-1 (COX-1) and its isozyme COX-2 are key enzymes in the syntheses of prostanoids. Imaging of COX-1 and COX-2 selective radioligands with positron emission tomography (PET) may clarify how these enzymes are involved in inflammatory conditions and assist in the discovery of improved anti-inflammatory drugs. We have previously labeled the selective high-affinity COX-1 ligand, 1,5-bis(4-methoxyphenyl)-3-(2,2,2-trifluoroethoxy)-1H-1,2,4-triazole (PS13), with carbon-11 (t1/2 = 20.4 min). This radioligand ([11C]PS13) has been successful for PET imaging of COX-1 in monkey and human brain and in periphery. [11C]PS13 is being used in clinical investigations. Alternative labeling of PS13 with fluorine-18 (t1/2 = 109.8 min) is desirable to provide a longer-lived radioligand in high activity that might be readily distributed among imaging centers. However, labeling of PS13 in its 1,1,1-trifluoroethoxy group is a radiochemical challenge. Here we assess two labeling approaches based on nucleophilic addition of cyclotron-produced [18F]fluoride ion to gem-difluorovinyl precursors, either to label PS13 in one step or to produce [18F]2,2,2-trifluoroethyl p-toluenesulfonate for labeling a hydroxyl precursor. From the latter two-step approach, we obtained [18F]PS13 ready for intravenous injection in a decay-corrected radiochemical yield of 7.9% and with a molar activity of up to 7.9 GBq/μmol. PET imaging of monkey brain with [18F]PS13 shows that this radioligand can specifically image and quantify COX-1 without radiodefluorination but with some radioactivity uptake in skull, ascribed to red bone marrow. The development of a new procedure for labeling PS13 with fluorine-18 at a higher molar activity is, however, desirable to suppress occupancy of COX-1 by carrier at baseline.
Keywords: Fluorine-18, PS13, radioligand, nucleophilic addition, PET, COX-1, brain imaging
Graphical Abstract
INTRODUCTION
Cyclooxygenase-1 (COX-1) is a prostaglandin-endoperoxide synthase that is constitutively expressed throughout the body. COX-1 and its closely related isozyme, COX-2, play important roles in normal physiology and in disease processes, especially inflammation.1 These enzymes synthesize prostaglandins from arachidonic acid in response to diverse stimuli on the path to a group of important lipidic physiological mediators known as the prostanoids. COXs are targets for inflammatory drugs, notably aspirin and ibuprofen, that are widely prescribed for peripheral diseases, such as rheumatoid arthritis. Chronic inflammation in brain (neuroinflammation) is now recognized to be associated with some neuropsychiatric disorders (e.g., Alzheimer’s disease, multiple sclerosis, and clinical depression).2–4 Effective tools for using nuclear medicine to investigate these diseases and to assist in developing better drug treatments are constantly sought. Radioligands for imaging of COX-1 and COX-2 in vivo could be useful for elucidating their roles in inflammation and neuroinflammation and for developing anti-inflammatory drugs that have improved target selectivity and reduced side-effect liability.
Although COX-2 plays the larger role in peripheral inflammation, COX-1 is a key contributor to neuroinflammation.3 In healthy brain, COX-1 is almost exclusively localized to resting microglia. In response to inflammatory stimuli, these microglia become activated, and COX-1 expression increases. Consequently, proinflammatory prostanoids arising from COX-produced progenitor prostaglandins generate oxidative stress. Ultimately, oxidative stress may lead to cytotoxicity and neuronal loss.
Use of positron emission tomography (PET) with a well-designed radioligand can quantify the regional distribution of the binding of the radioligand to a protein target in vivo. Numerous proteins in brain have been imaged in this manner, including neurotransmitter receptors, transporters, plaques, and enzymes.5,6 PET radioligand design is however challenging.5–10 Several early candidate PET radioligands failed to image COX-1 in brain.11–13 In many cases, this was due to one or more factors, such as limited ability to cross the blood-brain barrier, relatively low COX-1 affinity (IC50 ≥ 20 nM), and high nonspecific binding. Some success has been achieved in rat with a prodrug approach in which a potent but poorly brain-penetrant COX-1 inhibitor [11C]ketoprofen was delivered to the brain as its methyl ester.13,14 Rapid hydrolysis in situ generated [11C]ketoprofen for binding to COX-1. Never-theless, this radiotracer was unsuccessful for imaging COX-1 in human subjects affected by Alzheimer’s disease or mild cognitive impairment.15 Moreover, a prodrug approach is not readily amenable to biomathematical analysis because of the difficulty of distinguishing hydrolysis from enzyme inhibition.
We recently produced a direct-acting, selective, and high-affinity COX-1 radioligand, namely [11C]PS13 (Figure 1A).16 PET in monkey showed that [11C]PS13 is able to enter the brain from plasma and to bind avidly to COX-1.17 A large proportion of [11C]PS13 brain uptake could be preblocked by intravenous administration of PS13 itself or S-ketoprofen methyl ester as a source of COX-1 selective ketoprofen but could not be preblocked by a selective COX-2 ligand, MC-1.17,18 The COX-1 specific signal in monkey brain is readily quantifiable without interference from radiometabolites. [11C]PS13 is similarly effective for imaging and quantifying constitutive COX-1 in human brain.19 Furthermore, [11C]PS13 shows some promise for imaging COX-1 in periphery, including tumors.20 These findings encourage further application of [11C]PS13 for the study of COX-1 expression in human inflammatory diseases and their drug treatments.
Figure 1.
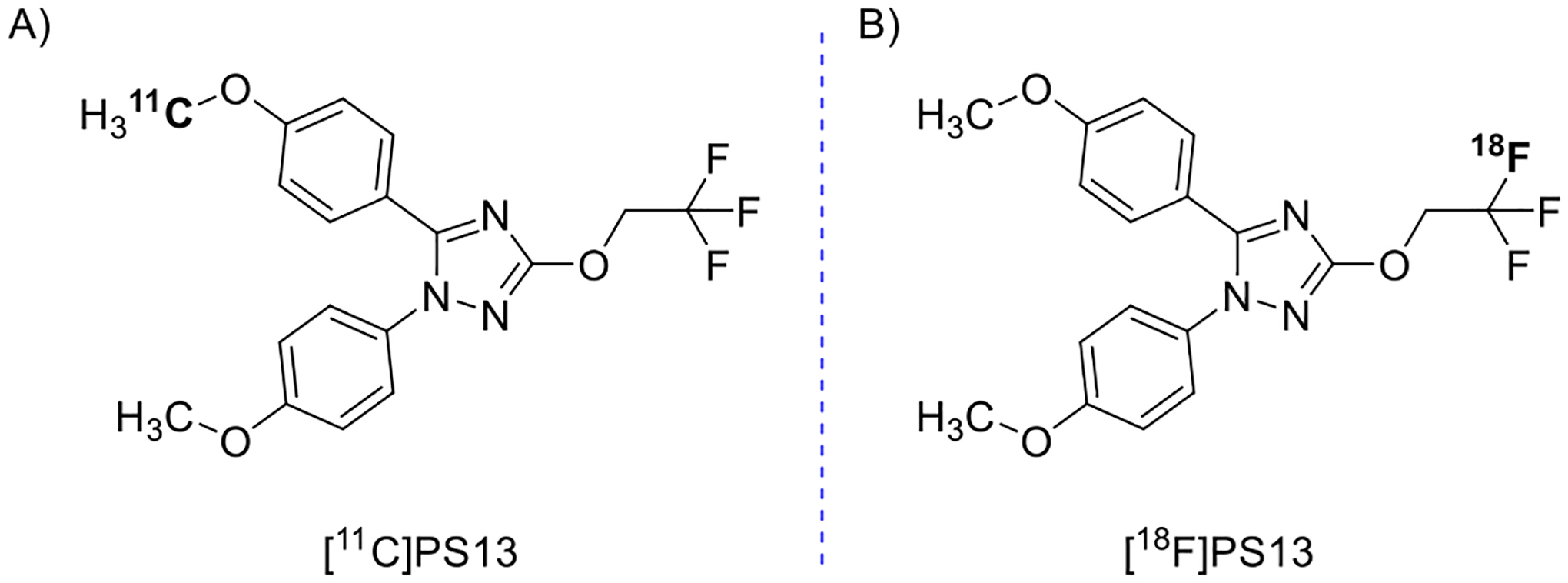
Chemical structures showing the labeling site for A [11C]PS13 and B [18F]PS13.
An 18F-labeled version of PS13 (Figure 1B) could be useful because the longer half-life of fluorine-18 (109.8 min) versus that of carbon-11 (20.4 min) would allow distribution of multiple doses to remote off-site PET imaging facilities that lack radioisotope production or radiochemistry capability. No-carrier-added fluorine-18 can be produced up to a very high activity (>1 TBq) as [18F]fluoride ion by the 18O(p,n)18F on 18O-enriched water.21 Typically, a PET radiotracer dose for PET imaging in a human participant is only about 700 MBq. Thus, efficient fluorine-18 chemistry might provide several doses of radiotracer from a single radiosynthesis. PS13 has a 1,1,1-trifluoroethoxy group (−OCH2CF3) in its structure, which is a potential site for labeling with fluorine-18.
Two main approaches have been used for introducing fluorine-18 into a 1,1,1-trifluoroethoxy group.22–28 Fawaz et al.26 reported a direct one-step method based on nucleophilic addition of the [18F]fluoride ion to a functionalized gem-difluorovinyl precursor in the presence of a proton source, e.g., isopropyl alcohol (IPA). This method was used for the synthesis of [18F]N-methyl-lansoprazole, a high-affinity PET radioligand for tau protein (Figure 2A). Kramer et al.28 recently used a very similar approach for the synthesis of [18F] N-methyl-lansoprazole for first-in-human studies. Rafique et al.27 developed a two-step procedure to produce candidate 18F-labeled radiotracers for PET imaging of neurofibrillary tangles (Figure 2B). Their procedure involved [18F]fluoride ion addition to 2,2-diifluorovinyl tosylate in the presence of a proton source (e.g., H2O) followed by reaction of the generated [18F]2,2,2-trifluoroethyl tosylate with a hydroxy precursor in the presence of cesium carbonate as base. Another example of this radiochemical approach is that of Riss et al.24 for the synthesis of an 18F-labeled astemizole derivative.
Figure 2.
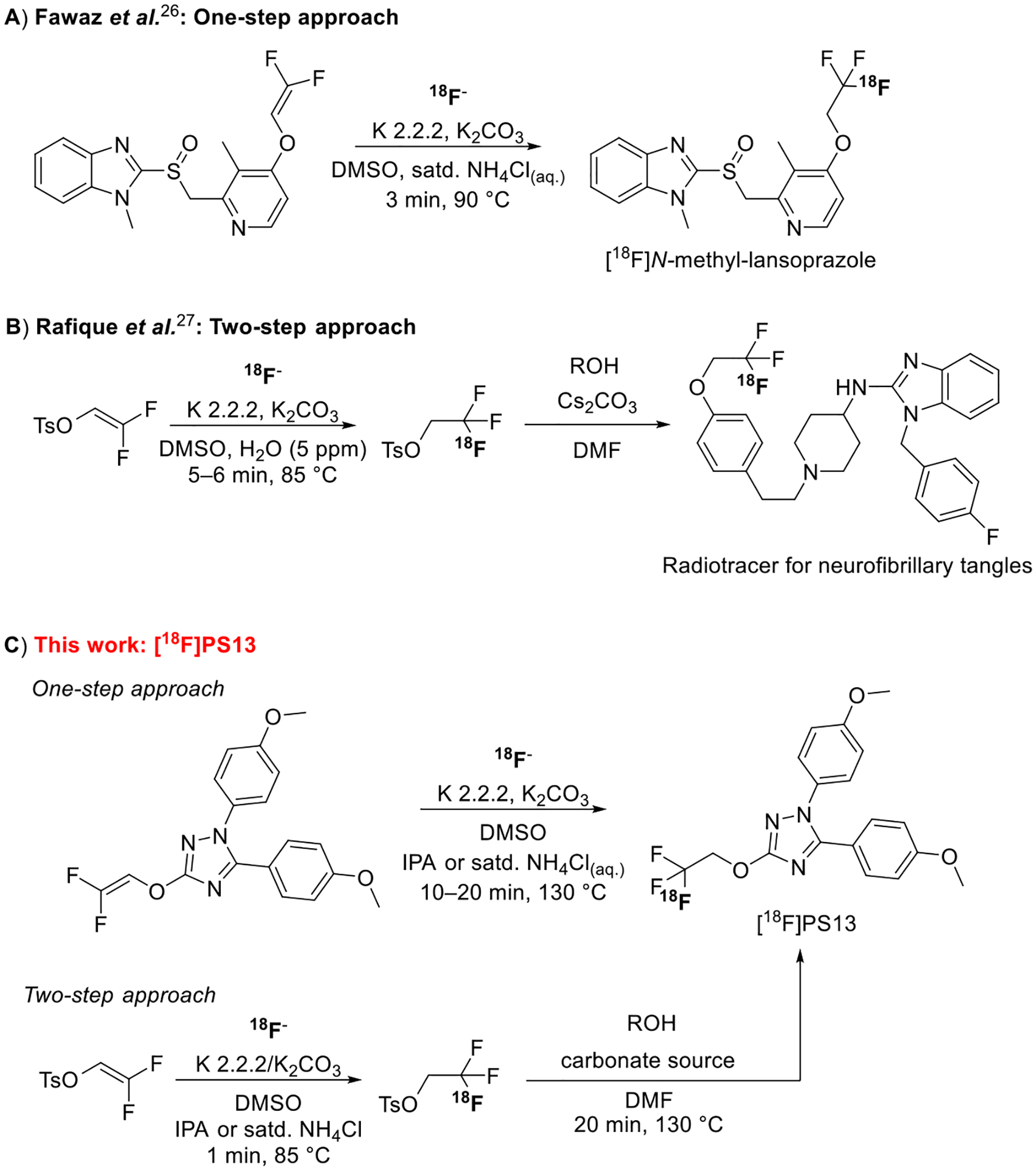
Strategies for 18F-labeling of a 1,1,1-trifluoroethoxy group: A, a one-step approach (Fawaz et al.);26 B, a two-step approach (Rafique et al.);27 and C, radiosynthesis approaches investigated in this work for the synthesis of [18F]PS13.
Herein, we report the synthesis of [18F]PS13 through each of the two nucleophilic addition approaches (Figure 2C). Reaction variables, such as temperature, reaction time, and proton source, were investigated with attention to their effects on decay-corrected radiochemical yield (RCY) and molar activity (Am). [18F]PS13 was produced and evaluated with PET imaging in healthy Macaca mulatta monkeys. Baseline and self-blocking studies in brain and whole-body were performed showing that [18F]PS13 can be an effective radioligand without any issue of radiodefluorination or of other radiometabolites within brain. However, a new method is needed for producing [18F]PS13 at a much higher molar activity.
RESULTS AND DISCUSSION
One-Step Labeling Approach.
Initially, we investigated a one-step approach for the synthesis of [18F]PS13, resembling that taken by Fawaz et al.26 for labeling N-methyl-lansoprazole. The required gem-difluoroalkene precursor (5) was synthesized by modifying known methods16,23,26,29,30 (Figure 3). Thus, 2-(4-methoxyphenyl)hydrazine-1-carboxamide (1) was treated with 4-methoxybenzoyl chloride to yield 2-(4-methoxybenzoyl)-2-(4-methoxyphenyl)hydrazine-1-carboxamide (2). Cyclization of 2 under basic conditions gave 1,5-bis(4-methoxyphenyl)-1H-1,2,4-triazol-3-ol (3). Alkylation of 3 with 1,1,1-trifluoro-2-iodoethane yielded 1,5-bis(4-methoxyphenyl)-3-(2,2,2-trifluoroethoxy)-1H-1,2,4-triazole (4). Defluorination of 4 by treatment with n-butyllithium gave 3-(2,2-difluorovinyloxy)-1,5-bis(4-methoxyphenyl)-1H-1,2,4-triazole (5). All steps proceeded in moderate to high chemical yields.
Figure 3.
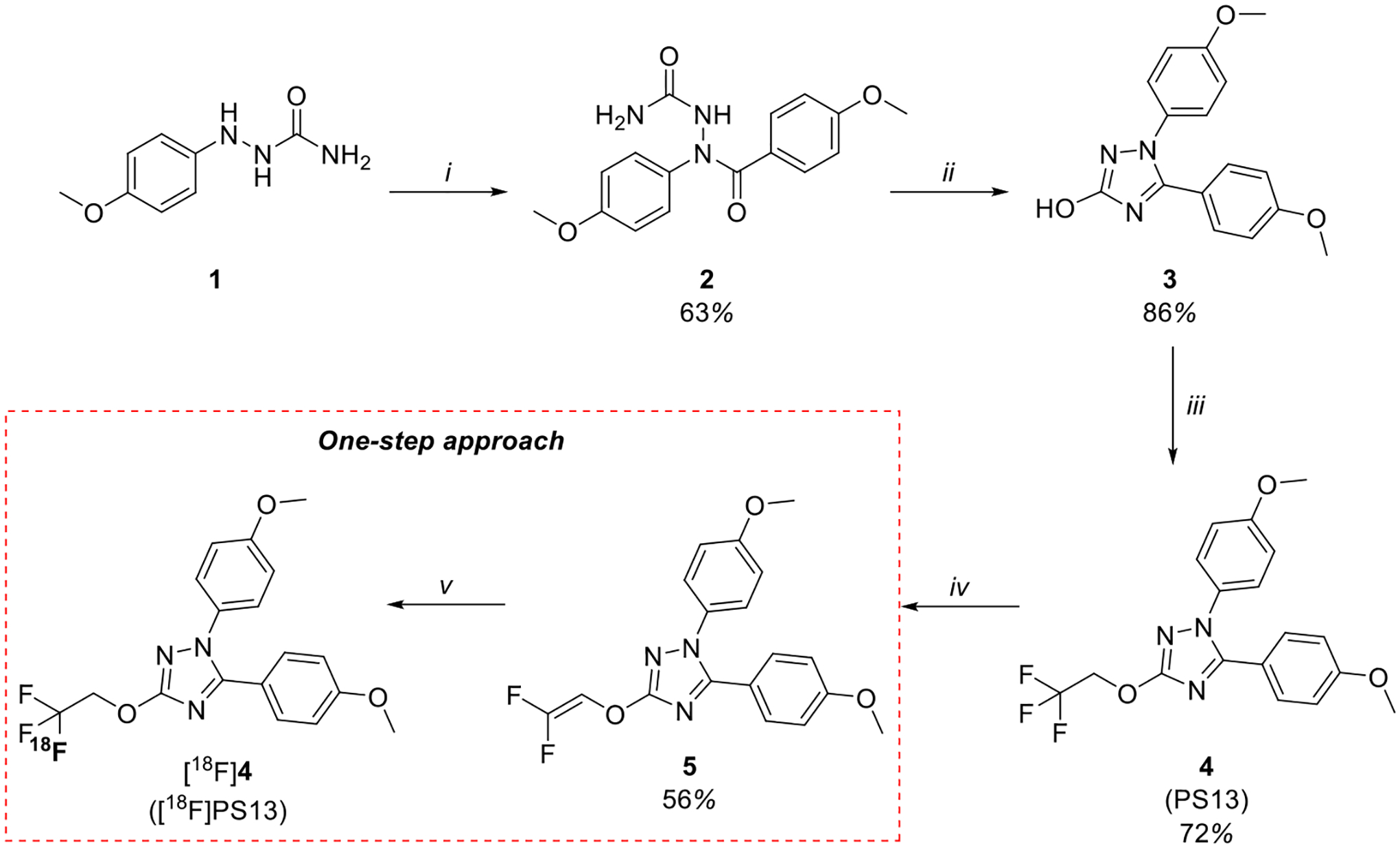
One-step approach for the synthesis of [18F]PS13 including precursor 5 synthesis. Reagents and conditions: (i) 4-methoxybenzoyl chloride, toluene, pyrimidine, 110 °C, 2.5 h; (ii) KOH (10%, aq.), EtOH, 60 °C, 1.5 h; (iii) 1. K2CO3, DMF, RT, 10 min and 2. 1,1,1-trifluoro-2-iodoethane, 100 °C, 3 h; (iv) n-BuLi (1.6 M in hexanes), THF, −78 °C, 45 min; (v) [18F]F−, K 2.2.2, K2CO3, DMSO, proton source, thermal heating, 10–20 min.
A major issue in the labeling of a 1,1,1-trifluoroethoxy group with [18F]fluoride ion is the labeling of the gem-difluoroalkene precursor through overall fluorine isotope exchange, with the extent of exchange dependent on the type of an added proton source and its concentration and other reaction conditions.22,23,27 Nucleophilic addition of no-carrier-added (NCA) [18F]fluoride ion to a gem-difluorovinyl substrate, 1,1-difluoroethene, in acetonitrile in the absence of an added proton source was reported earlier to produce [1-18F]1,1,1,2-tetrafluoroethane.31 The [18F]fluoride ion was used as its K+ complex of the cryptand 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (K+-K 2.2.2 complex). Throughout the current study, we also used the [18F]fluoride ion as its K+-K 2.2.2 complex. In our attempt to label PS13 by treating 5 with [18F]fluoride ion in the absence of an added proton source (Figure 4), unwanted [18F]5 was formed in strong preference over [18F]PS13. Fawaz et al.,26 in their study of nucleophilic addition of [18F]fluoride ion to a gem-difluorovinyl substrate, improved the ratio of the 18F-trifluoromethylated product to the 18F-labeled vinyl product and the overall yield by, for example, adding IPA as a proton source. With the aim of improving the production of [18F]PS13 over [18F]5, we explored different proton sources and reaction conditions.
Figure 4.
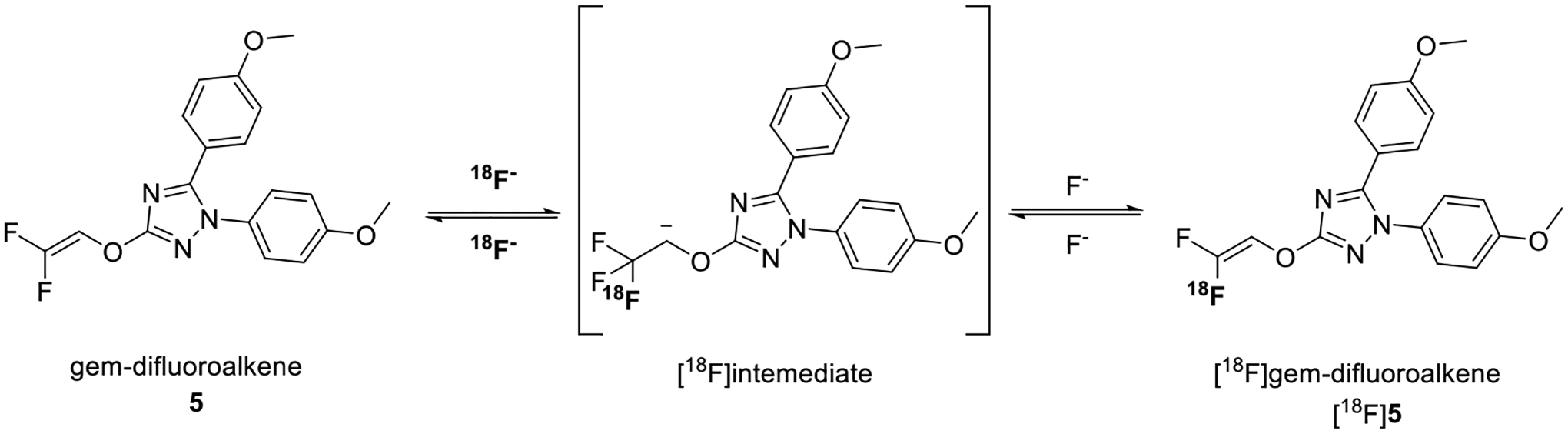
Representation of the 19F/18F addition–elimination process for 5 in the absence of an added proton source.
Initially, reaction temperature and the use of IPA or tert-butanol as a proton source were studied (Table 1, entries 1–5). [18F]PS13 and [18F]5 were obtained in a 1:5 ratio of nonisolated yields when precursor 5 (1.5 mg) was treated with [18F]fluoride ion in the presence of 10.8 μL of IPA in DMSO (300 μL) at 90 °C for 20 min (entry 1). By doubling the amount of IPA to 21.6 μL, the ratio of [18F]PS13 to [18F]5 doubled to 1:2.5 (entry 2). tert-Butanol gave no advantage over IPA as a proton source (entry 3). The ratio of [18F]PS13 to [18F]5 increased to 1:2 when the temperature was increased to 130 °C in the presence of IPA (21.6 μL) (entry 4). Use of microwave (MW) heating instead of thermal heating gave no improvement in the [18F]PS13 yield (entry 5). Therefore, the best conditions for producing [18F]PS13 using IPA as the proton source were those in entry 4.
Table 1.
Conditions Screening for the Synthesis of [18F]PS13 via the One-Step Approach
| entrya | temp (°C) | time (min) | proton source | proton source (μL)b | yield ratio of [18F]PS13 to [18F]5c | yield of [18F]PS13c (%) | Am of [18F]PS13d (GBq/μmol) |
|---|---|---|---|---|---|---|---|
| 1 | 90 | 20 | IPA | 10.8 | 1:5 | ||
| 2e | 90 | 20 | IPA | 21.6 | 1:2.5 | ||
| 3 | 90 | 20 | tert-BuOH | 21.6 | 1:5 | ||
| 4 | 130 | 20 | IPA | 21.6 | 1:2 | ||
| 5 | 130 (MW) | 20 | IPA | 21.6 | 1:2.5 | ||
| 6 | 130 | 2 | (NH4)2CO3(aq.) | 1.6 | 1:5 | ||
| 7f | 130 | 20 | NH4Cl(aq.) | 1.6 | 1:0.9 | 0.8 | 14.2 |
| 8e | 130 | 20 | NH4Cl(aq.) | 3.2 | 1:0.4 | 3.3 | 16.8 |
| 9 | 130 | 20 | NH4Cl(aq.) | 6.4 | 1:0.4 | 0.8 | 8.6 |
| 10 | 130 | 20 | NH4Cl(aq.) | 16 | 0:0 | 0 | |
| 11 | 130 (MW) | 2 | NH4Cl(aq.) | 1.6 | 1:10 | ||
| 12g | 130 | 20 | IPA | 21.6 | 5.9 | 0.6 | |
| 13g | 130 | 10 | IPA | 21.6 | 4.5 | 1.0 |
Reactions (n = 1) were performed with 5 (1.5 mg) in DMSO (300 μL), unless otherwise noted.
Volume per reaction.
Yield ratios and yields were determined from radiochromatograms acquired during HPLC analysis of the crude reaction mixture.
Decay-corrected to ERP.
n = 2.
n = 4.
Reaction used 3 mg of precursor 5.
Alternative proton sources, such as a saturated aqueous solution of ammonium carbonate [(NH4)2CO3(aq.)] or of ammonium chloride [NH4Cl(aq.)], were also investigated (entries 6–11). [18F]PS13 was formed in a low 1:5 ratio to [18F]5 when 5 (1.5 mg) was treated with the [18F]fluoride ion in the presence of 1.6 μL of (NH4)2CO3(aq.) in DMSO (300 μL) at 130 °C for 20 min (entry 6). When NH4Cl(aq.) was used, the ratio of [18F]PS13 to [18F]5 increased to 1:0.9 (entry 7). [18F]PS13 was obtained as the major 18F-labeled product by doubling the amount of NH4Cl(aq.) to 3.2 μL per reaction (entry 8). By increasing NH4Cl(aq.) to 6.4 μL, the ratio of [18F]PS13 to [18F]5 increased further to 1:0.4 (entry 9). Nonetheless, another increase of NH4Cl(aq.) to 16 μL gave no 18F-labeled products (entry 10). This was likely due to severe quenching of the nucleophilicity of the [18F]fluoride ion through a high degree of aqueous solvation.32,33 Microwave heating gave [18F]5 as the major labeled product (entry 11) and was not investigated further. Therefore, treatment of 5 (1.5 mg) in the presence of 3.2 or 6.4 μL of NH4Cl(aq.) in DMSO (300 μL) at 130 °C for 20 min (entries 8 and 9) was best for selective formation of [18F]PS13.
In summary, the use of IPA produces predominantly [18F]5 over [18F]PS13 (entries 1, 2, 4, and 5), whereas NH4Cl(aq.) favors the formation of [18F]PS13 over [18F]5 (entries 7–9). The absence of water when using IPA favors fluorine isotope exchange by addition–elimination and causes [18F]5 to prevail over [18F]PS13. Although the water in NH4Cl(aq.) is expected to suppress the nucleophilicity of the free fluoride ion (radioactive and nonradioactive) leading to a slower addition reaction, the ready availability of protons favors formation of [18F]PS13 over elimination of the fluoride ion (Figure 3).
Despite dominant formation of [18F]PS13 over [18F]5 when using NH4Cl(aq.) as a proton source, [18F]PS13 was obtained in only low nonisolated yields in the 0.8–3.3% range and with molar activities in the 8.6–16.8 GBq/μmol range (Table 1, entries 7–9). Low yields in fluorine-18 chemistry can still be useful because of the very high activities of [18F]fluoride that can be produced. However, radioligands that are intended to bind to low density binding sites with high affinity usually need to be produced with high molar activity to avoid excessive target occupancy by carrier in vivo and to avoid consequent violation of the radiotracer principle. In our laboratory, the molar activities of 18F-labeled radiotracers typically exceed 75 GBq/μmol at the end of the synthesis, as recently reported for the synthesis of [18F]LSN3316612, a radioligand for imaging O-linked-β-N-acetyl-glucosamine hydrolase in human brain.34 The highest molar activity for [18F]PS13 (16.8 GBq/μmol, corrected to the end of the radionuclide production (ERP)) was obtained when using 3.2 μL of NH4Cl(aq.) per reaction (entry 8). In this case, [18F]PS13 was obtained in only 3.3% yield. Again, this suggests that the water present in the NH4Cl(aq.) suppresses the nucleophilicity of the free [19F]/[18F]fluoride ion.
In a reaction performed with 5 (3 mg) and IPA (21.6 μL) in DMSO (300 μL) at 130 °C for 20 min, [18F]PS13 was obtained in a nonisolated yield of 5.9% with a molar activity of 0.6 GBq/μmol (decay-corrected to ERP) (entry 12). A shorter reaction time of 10 min gave [18F]PS13 in a lower nonisolated yield of 4.5% but with a higher molar activity of 1 GBq/μmol (entry 13). This indicates that fluorine isotope exchange increases over time, thereby reducing the molar activity. In summary, with this one-step approach, the molar activity of [18F]PS13 was much lower, and the yield was only slightly higher with IPA than with NH4Cl(aq.) as a proton source.
Two-Step Approach.
With the aim of increasing the yield of [18F]PS13 and its molar activity, we investigated a two-step strategy (Figure 5) resembling that reported by Rafique et al.27 (Figure 2B).
Figure 5.
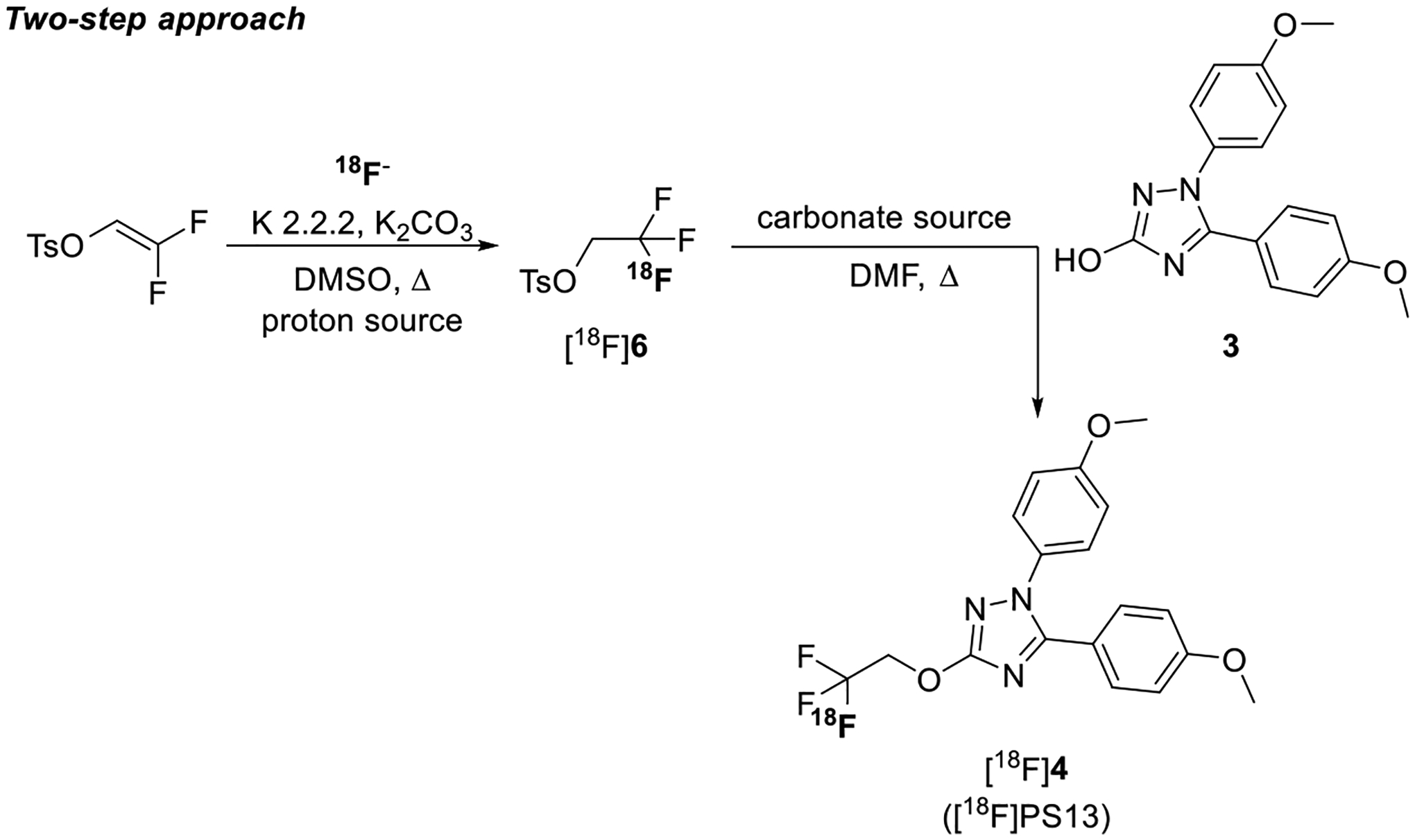
Two-step approach for the synthesis of [18F]PS13.
Step 1 Optimization.
Initially we focused on optimizing the addition of the [18F]fluoride ion to 2,2-difluorovinyl 4-methylbenzenesulfonate to give [18F]2,2,2-trifluoroethyl tosylate ([18F]6) as a labeling synthon (Figure 5).
The reaction time with IPA as a proton source was investigated (Table 2). For reactions between 1 and 10 min, yields of [18F]6 increased with time over a narrow moderate range (54–63%; entries 1–4). However, the molar activity decreased rapidly with time from 1.3 GBq/μmol after 1 min (entry 1) to 0.3 GBq/μmol after 10 min (entry 4). Therefore, a 1-min reaction time was selected for subsequent experiments aimed at optimizing temperature, type of proton source, and molar activity.
Table 2.
Time Study for Step 1 (Two-Step Approach) with IPA as a Proton Source
| entrya | time (min) | [18F]6 yield (%)b | Am of [18F]6 (GBq/μmol)c |
|---|---|---|---|
| 1 | 1 | 54 | 1.3 |
| 2 | 2.5 | 56 | 0.8 |
| 3 | 5 | 61 | 0.4 |
| 4 | 10 | 63 | 0.3 |
Reactions (n = 1) were performed with 2,2-difluorovinyl 4-methylbenzenesulfonate (2.5 mg) and IPA (38 μL) in DMSO (500 μL) at 85 °C.
Yields were determined from radiochromatograms acquired during HPLC analysis of the crude reaction mixture.
Decay-corrected to ERP.
Temperature was studied in single reactions performed in DMSO (100 μL) with 0.5 mg of 2,2-difluorovinyl 4-methylbenzenesulfonate and IPA (7.6 μL) for 1 min. At 45, 85, and 130 °C, the yields of [18F]6 were 9.4, 22, and 6.6%, respectively. Therefore, 85 °C was used for subsequent reaction optimization. Saturated NH4Cl(aq.) (1.1 μL) was compared with IPA (7.6 μL) as a proton source in triplicate reactions using 0.5 mg of the precursor at 85 °C for 1 min in DMSO (100 μL). [18F]6 was obtained in moderate yields (59 ± 4%) but with a lower molar activity (1.7 ± 0.5 GBq/μmol) when using IPA as a proton source. With NH4Cl(aq.) (1.1 μL) as a proton source, [18F]6 was obtained in much lower yield (14 ± 5%) but with higher molar activity (22 ± 1 GBq/μmol). Therefore, NH4Cl(aq.) was selected as the preferred proton source for obtaining [18F]6 in a moderately useful yield but with optimal molar activity.
Step 2 Optimization.
We proceeded to explore the second step required for the synthesis of [18F]PS13. First, we studied the reaction of [18F]6 with the alcohol precursor 3 (3 mg, 1 equiv) in DMF (300 μL) with potassium carbonate (11.6 mg, 8.4 equiv) as a base at 130 °C for 20 min (Figure 5). Conditions like those in the synthesis of reference PS13 (Figure 3, step (iii)) were applied. [18F]PS13 was obtained in a nonisolated yield of 30 ± 2.4% from [18F]fluoride ion and with a molar activity of 2.8 ± 0.5 GBq/μmol (n = 3) when IPA (7.6 μL) was used as the proton source in Step 1. A lower [18F]PS13 yield of 12 ± 1.4% and only a comparable molar activity of 3.2 ± 2 GBq/μmol were achieved by using NH4Cl(aq.) (1.1 μL) as the proton source in Step 1. It should be noted that these results were obtained using a low starting radioactivity amount (≤1.2 GBq) and that syntheses were interrupted between Step 1 and Step 2 to withdraw an aliquot for HPLC analysis of the Step 1 outcome. This may be a reason for the observed molar activity to fall from Step 1 to Step 2.
With the aim to obtain [18F]PS13 in useful activity yield with the highest achievable molar activity, use of NH4Cl(aq.) for Step 1 was chosen for producing [18F]PS13 for PET experiments in monkey.
The use of potassium carbonate as a base in Step 2 posed problems in scale-up of the process to a higher starting activity. After Step 1, the reagents for Step 2 were added together to the reaction vial through a needle controlled with an automated robotic arm (see Methods section). The reagents had to be in an almost homogeneous solution to allow smooth addition without compromising the apparatus function through the robot needle blockade. Ammonium carbonate [(NH4)2CO3] has higher solubility in organic solvents (e.g., DMF) than potassium carbonate. Therefore, ammonium carbonate was tested as a base for Step 2. [18F]PS13 was obtained in a higher nonisolated yield (17%) when using (NH4)2CO3 rather than K2CO3 (12 ± 1.4%, n = 3). We therefore selected (NH4)2CO3 as a base for Step 2 in scale-up of the synthesis of [18F]PS13 for PET imaging experiments in monkey. Separation of [18F]PS13 was achieved by reversed phase HPLC (Supporting Information, Figure S1). Radiochemically pure [18F]PS13 was reproducibly obtained (Supporting Information, Figure S2) and formulated ready for intravenous injection in an isolated decay-corrected yield of 7.9 ± 2.7% from the starting [18F]fluoride ion with a molar activity of 7.9 ± 2.2 GBq/μmol (n = 6).
Stability of Formulated [18F]PS13.
The radiochemical purity of formulated [18F]PS13 kept at RT for 1 and 4 h was analyzed with radio-HPLC. [18F]PS13 maintained a radiochemical purity of greater than 98% with no evidence of radiodefluorination.
PET Experiments in Monkey.
In this study, [18F]PS13 was only obtainable for intravenous injection with a moderate molar activity. This implies that a dose administered to monkey in a PET experiment would have a moderately high amount of carrier, with a risk of appreciable occupancy of the target COX-1, and therefore of a diminished COX-1 specific PET signal. Nonetheless, we performed experiments in monkey to characterize [18F]PS13 as a PET radioligand. One reason was to assess whether further radiochemical research would be warranted to find a method of labeling that could deliver adequate yield with a reliably higher molar activity. The spectrum of radiometabolites that is generated from a PET radioligand depends on the molecular position of the radiolabel.35 One position of the radiolabel may give troublesome radiometabolites, whereas another position may not. We especially wished to know whether [18F]PS13 is extensively defluorinated in monkey in vivo, because avid bone uptake of the [18F]fluoride ion in skull can be problematic for quantification of radioligand binding in nearby brain regions.
We performed baseline and self-blocking PET experiments on brain in two monkeys. In many respects, results in the two monkeys were quite comparable and are exemplified here for monkey A (see the Supporting Information for imaging results from monkey B). In the baseline experiment, [18F]PS13 binding was highest in the prefrontal and parietal cortices (Figure 6A). The whole brain time-activity curve at baseline peaked early at 3.79 SUV and then declined smoothly to the end of the scan (Figure 6B). In the preblock experiment, peak radioactivity uptake was similar, but subsequent radioactivity decline was faster, indicating the presence of some COX-1 specific binding in the baseline experiment. These time-activity curves are compared with those of [11C]PS13 of higher molar activity (~105 GBq/μmol) for another monkey, as published previously.17 The baseline whole brain radioactivity curve for [18F]PS13 is very similar to that for [11C]PS13. An intravenous PS13 dose of 0.3 mg/kg was used in the preblock experiment with [18F]PS13. In the experiment with [11C]PS13, the preblocking PS13 dose was 1.0 mg/kg, i.v., and gave a somewhat faster decline in radioactivity after peak but down to a similar level to that for [18F]PS13 at 90 min.
Figure 6.
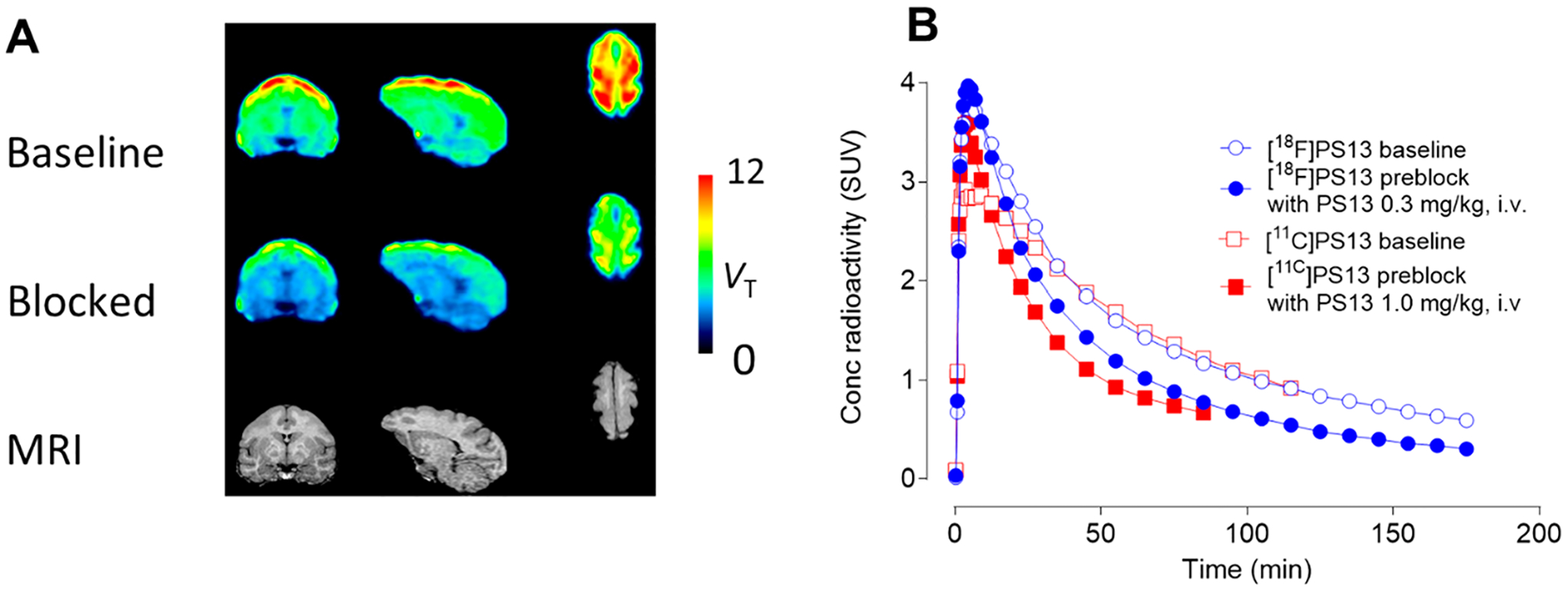
PET studies of brain in monkey A with [18F]PS13 (carrier PS13:6.6 nmol/kg at baseline). Panel A: regional VT images from baseline scan (top row) and blocked scans (with PS13, 0.3 mg/kg, i.v.) (middle row), and corresponding MRI images (bottom row). Left column: coronal images; middle column: sagittal images; right column: horizontal images. Highest radioactivity uptake occurred in the prefrontal and parietal cortices. Panel B: whole brain time-activity curves at baseline and after preblock obtained with [18F]PS13. Corresponding curves obtained in a different monkey with [11C]PS13 from the study in ref 17 with a much lower PS13 carrier dose (0.19 nmol/kg at baseline) are shown for comparison.
In our study of monkey A, the dose of [18F]PS13 was 23.6 MBq/kg, and the carrier dose was 6.6 nmol/kg. 18F-Labeled ligands are typically administered at lower doses of radioactivity per kg in humans than in nonhuman primates for imaging low density targets in brain. For example, in the study of [18F]LSN3316612, the radioactive dose was, on average, about 190 MBq in subjects that on average weighed 75 kg, equivalent to an average dose of 2.53 MBq/kg.34 Thus, by using a similar radioactive dose of [18F]PS13 in humans, the dose of carrier (in nmol/kg) might be reduced almost 10-fold, to about 0.66 nmol/kg, and the possible issue of enzyme occupancy at baseline might be reduced. By comparison, the typical dose of carrier in our human experiments with [11C]PS13 has been at 0.11 ± 0.06 nmol/kg.19
In these experiments with [18F]PS13, radioactivity cleared rapidly from plasma. Several radiometabolites emerged in plasma that were less hydrophobic than [18F]PS13, as judged by their faster elution in reversed phase HPLC analysis (Figure 7A). Negligible radioactivity eluted at the solvent front, indicating the absence of [18F]fluoride ion as a radiometabolite. The time for radiometabolites to reach 50% of the radioactivity in plasma (~20 min) was similar to that for [11C]PS13 (28 min).17 The time-courses for unchanged radioactivity and [18F]PS13 in plasma in the baseline and preblock experiments were similar (Figure 7B and Figure 7C). Plasma free fractions (f P) for [18F]PS13 in these PET experiments were low (1.14–2.79%) but accurately measurable (Supporting Information, Table S1).
Figure 7.
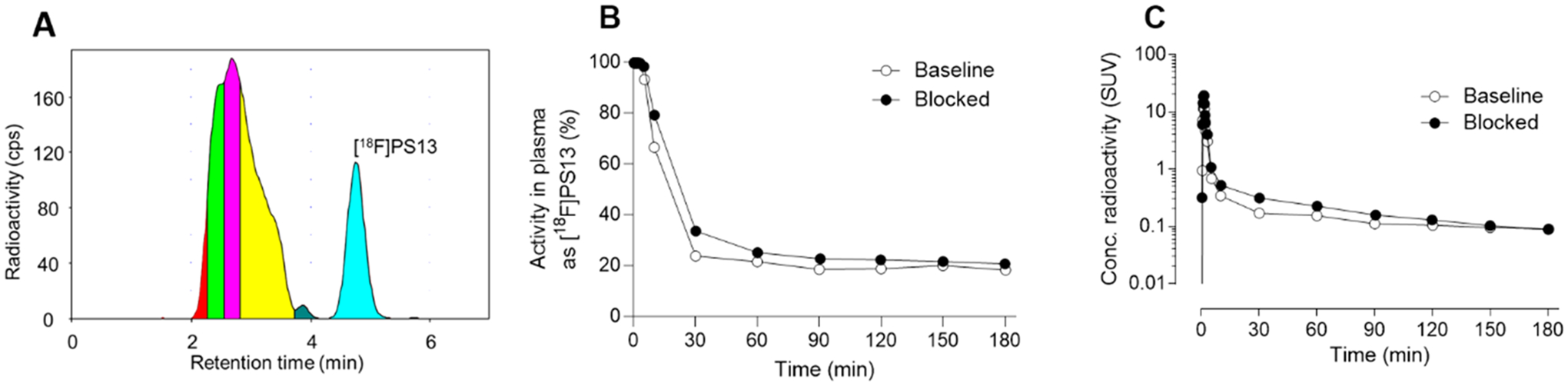
Plasma clearance and metabolism of [18F]PS13 in monkey A. Panel A: reversed phase HPLC analysis of radiometabolites in plasma at 180 min after intravenous radioligand injection at baseline. Panel B: time-course of the percentage of radioactivity in plasma represented by unchanged [18F]PS13 under baseline and preblocked conditions. Panel C: time-course for unchanged [18F]PS13 in plasma under baseline and preblocked conditions.
Brain time-activity curves from [18F]PS13 were well fitted with the two-tissue compartment model and gave regional total distribution volumes (VT’s) (Figure 8A) and their adjustments for plasma free fractions (VT/f P’s) (Figure 8B). These were consistent with those obtained in our previous study with [11C]PS13.17 VT ranged from 4.47 in the limbic region to 8.03 in the frontal cortex at baseline. In the preblock experiment, they decreased between 25 and 55% across brain regions, as did VT/f P values. Moreover, at baseline whole brain and regional VT values rapidly reached stable values with respect to the duration of PET data used in their determination (Figure 8C), indicating that radioactivity within brain was not contaminated by radiometabolites. The occupancy of COX-1 achieved with PS13 as a preblocking agent may be estimated graphically with a Lassen plot where the X-axis is VT at baseline and the Y-axis is the decrease in VT from baseline for several brain regions under the preblock conditions. Major assumptions of the Lassen plot are that the radioligand and blocking agent bind selectively to the target, in this case COX-1, and that target occupancy and nonspecific binding are uniform across brain. The Lassen plot for monkey A showed the occupancy of COX-1 (slope of curve) by the 0.3 mg/kg intravenous dose of PS13 to be 83% and the nondisplaceable volume of distribution, VND (X-axis intercept), to be 2.70 mL/cm3. By comparison, in the study17 of [11C]PS13 using a higher intravenous blocking dose of 1.0 mg/kg, occupancy was 87 ± 4%, based on the slopes of Lassen plots, and VND was 2.5 mL/cm3. The strong correlation coefficient (Figure 8D) indicates that the major assumptions of the Lassen plots were well satisfied for [18F]PS13, as they were for [11C]PS13.17
Figure 8.
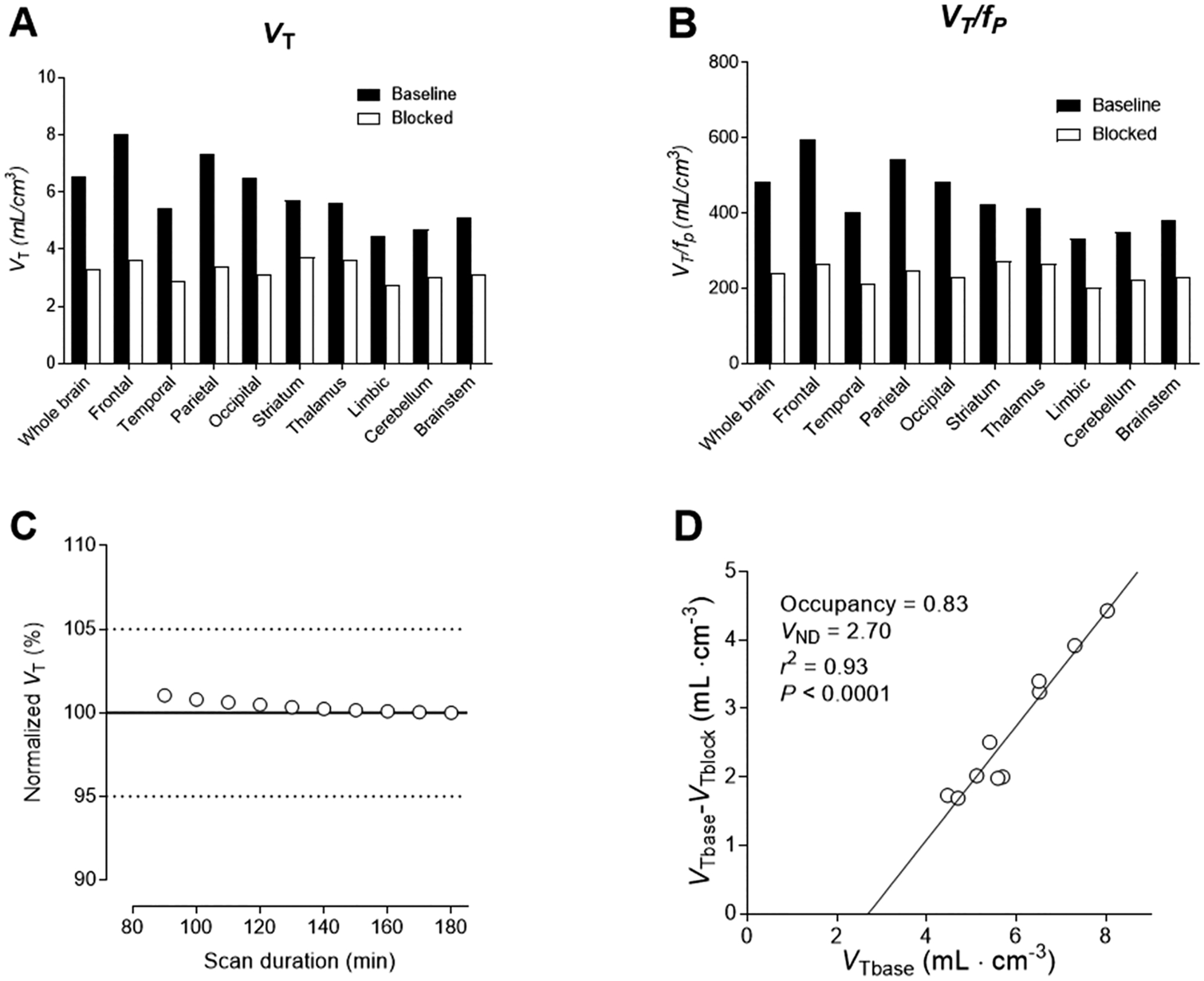
Analysis of PET data from monkey A under baseline and preblock conditions. Panel A: regional VT values. Panel B: regional VT values adjusted for normalized plasma free fraction (Supporting Information, Table S1). Panel C: VT values determined from PET data for different durations of scan data from time of radioligand injection, normalized to the value at the end of scanning (180 min). Panel D: Lassen plot showing VND as the X-axis intercept and occupancy (slope of plot) of the available COX-1 (that not already occupied by carrier at baseline) by the blocking agent PS13 (0.3 mg/kg, i.v.).
The study in monkey B gave results comparable to those in monkey A with respect to radioligand brain uptake and distribution (Supporting Information, Figure S3) and the emergence of radiometabolites in plasma at baseline (Supporting Information, Figure S4A) and under preblock conditions (Supporting Information, Figures S4B and 4C). However, in this monkey, much less reduction of total binding (VT) was achieved in the preblocking experiment with the same preblocking regimen used for monkey A. Reductions in VT were absent in cerebellum, limbic region, thalamus, and striatum. Only modest reductions in VT (≤35%) were seen for whole brain and frontal, parietal, temporal, and occipital cortices (Supporting Information, Figure S5A). These regional reductions in VT were much lower than in a control experiment in the same monkey with [11C]PS13 of much higher molar activity (221 GBq/μmol) and much lower carrier dose (0.1 nmol/kg) when administered with the same blocking dose (0.3 mg/kg) at 15 to 5 min before the scan (Supporting Information, Table S2). Reduction in VT for whole brain was 2-fold higher at 44% for [11C]PS13 and 22% for [18F]PS13 (Supporting Information, Table S2). These data suggest that occupancy of COX-1 at baseline was already substantial in the experiment with [18F]PS13 (although the carrier dose was lower than in the baseline experiment for monkey A). Nonetheless, as for monkey A, VT from [18F]PS13 in monkey B showed stability with respect to the duration of PET data needed for its calculation (Supporting Information, Figure S5B).
In both baseline experiments and in one self-blocking study (monkey B), some radioactivity uptake in skull was observed (Supporting Information, Figure S6). At first, this was suspected to be due to radiodefluorination of [18F]PS13. However, a summed whole body maximum-intensity projection (MIP) PET image of monkey at baseline showed radioactivity uptake in the areas of red marrow, such as vertebrae, pelvic bones, skull, and the proximal parts of the humeri, but not in rapidly metabolizing peripheral bone (Figure 9). This indicated that no radiodefluorination of [18F]PS13 had occurred and that the distribution of uptake followed the areas of red marrow. The molecular identity of this radioactivity uptake however remains unknown. The distribution of [18F]PS13 uptake in major organs was quite consistent with that previously reported for [11C]PS1318, as well as with constitutive COX-1 expression measured in postmortem human studies.36,37
Figure 9.
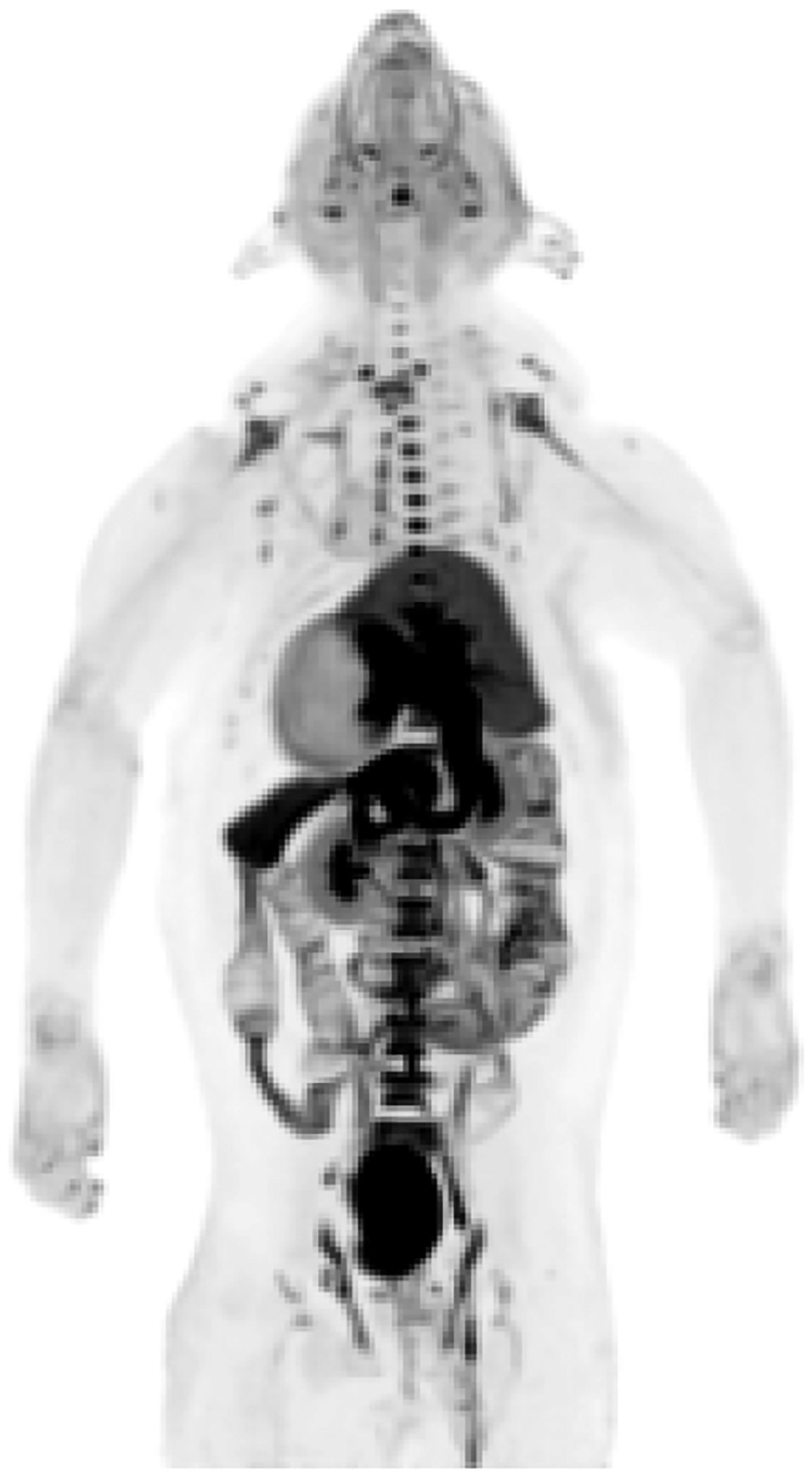
Summed (0–180 min) whole-body MIP PET image obtained with [18F]PS13 in monkey.
In view of the unusually low COX-1 specific signal found with PET in the brain of monkey B, an improved procedure or method for synthesizing [18F]PS13 at an appreciably higher molar activity is needed to avoid concerns over COX-1 occupancy at baseline. Certain strategies might be applied with the current radiochemistry to improve molar activity. One strategy would be to perform the radiochemistry with a very high amount of the cyclotron-produced [18F]fluoride ion. In our setting, we usually start with a relatively low radioactivity (~10 GBq) having a moderate molar activity (75–200 GBq/μmol), but other facilities are capable of producing and using a much higher activity of the [18F]fluoride ion in a safe manner. Generally, the amount of the carrier fluoride ion accompanying a cyclotron production of [18F]fluoride ion does not increase greatly with the activity produced. Hence, exceptionally high molar activity can be achieved under high-level radioactivity production conditions. For example, there are reports of 18F-labeled products having molar activities at the end of synthesis of >740 GBq/μmol from 185 GBq of [18F]fluoride ion38,39 and an unusually high molar activity of 4.7–5.9 TBq/μmol from 220 GBq of [18F]fluoride ion.40 The dominant source of carrier in our method for producing [18F]PS13 is from the release of the fluoride ion from the gem-difluorovinyl precursor during the labeling reaction, and this quantity of carrier will be a quite constant quantity. Therefore, the amount of carrier in a labeling reaction will be relatively fixed, and the molar activity should increase with starting radioactivity. This approach could be successful for producing [18F]PS13 as an efficacious PET radioligand in high activity, as has been achieved for other radiotracers. Technical improvement, such as performing the radiolabeling in a low microvolume with a smaller amount of the precursor, might also foster higher molar activity.41
CONCLUSIONS
Of the two approaches explored for labeling PS13 with fluorine-18, the two-step approach gave the best compromise between overall radiochemical yield and molar activity. Despite the low molar activity obtained in producing [18F]PS13 for evaluation with PET in monkey, this radioligand gave a sizable COX-1 specific signal in the brain of one monkey with time-stable VT values, indicating absence of radiometabolites. Some low radioactivity uptake was seen in skull in some instances but not due to radiodefluorination. Brain regional VT values and VND were comparable with those previously measured in monkey with [11C]PS13. Nonetheless, an improved procedure or method for labeling PS13 with the NCA [18F]fluoride ion remains desirable to ensure that the molar activity can be reliably higher to avoid any risk of unacceptable occupancy of COX-1 at baseline. Radioactivity uptake in red marrow of skull may remain an issue for quantitative PET imaging in human subjects, especially for quantification of radioligand uptake in regions near skull.
METHODS
General Materials and Methods.
Water from a purification apparatus (Milli-Q; Waters Corp; Columbia, MD) was used in syntheses and radiosyntheses, unless otherwise stated. Other solvents and chemicals were purchased from Aldrich Chemical Co. (Milwaukee, WI), Acros Organics BVBA (Geel, Belgium), and Enamine Ltd. (Kiev, Ukraine) and used as received. [11C]PS13 for a control experiment in monkey B (see the Supporting Information) was produced as described previously.17
1H- (400 MHz), 13C- (100 MHz), and 19F-NMR (376.49 MHz) spectra were recorded at RT on an Avance-400 spectrometer (Bruker; Billerica, MA). 1H and 13C chemical shifts are reported in δ units (ppm) downfield relative to the chemical shift for tetramethylsilane and 19F chemical shifts relative to that for CFCl3. Abbreviations br, s, d, t, and m denote broad, singlet, doublet, triplet, and multiplet, respectively. LC-MS for compound characterization was performed on an LCQ Deca instrument (Thermo Fisher Scientific Corp.; Waltham, MA) equipped with a Synergi Fusion-RP column (4 μm, 150 × 2 mm; Phenomenex; Torrance, CA). Flash chromatography was performed on a semiautomated apparatus (CombiFlash Rf + UV; Teledyne ISCO Inc.; Lincoln, NE).
γ-Radioactivity from 18F was measured with a calibrated dose calibrator (Atomlab 300; Biodex Medical Systems, USA) or for low levels (<40 kBq) with a well-type γ-counter (model 1080 Wizard; PerkinElmer; Boston, MA) having an electronic window set between 360 and 1,800 keV. 18F Radioactivity measurements were corrected for background and physical decay. All radiochemistry with fluorine-18 was performed in lead-shielded hot-cells for personnel radiation protection.
A semiautomated apparatus (Synthia)42 was used for all fluorine-18 radiochemistry. Dedicated recipes were created in Autorad software and followed step by step for the syntheses of [18F]PS13. The HPLC apparatus for [18F]PS13 separation comprised a pump (P4.1S; Knauer; Berlin, Germany), a UV absorbance detector (UVD2.1S; Knauer), and a radioactivity detector (flow-count; Eckert & Ziegler; Berlin, Germany). Clarity Chromatography Station software (Data-Apex; Prague, Czech Republic) was used to record the chromatograms. Radio-HPLC equipment for analyses comprised a pump (DGU-20A3R; Shimadzu; Columbia, MD), a UV absorbance detector (CBM-20A; Shimadzu), and a radioactivity detector (flow-count; Eckert & Ziegler).
All animals used in this study were handled in accordance with the Guide for the Care and Use of Laboratory Animals43 and the National Institute of Mental Health Animal Care and Use Committee.
Results of statistical analyses are presented as mean ± SD.
Syntheses.
1,5-Bis(4-methoxyphenyl)-1H-1,2,4-triazol-3-ol (3).
The alcohol precursor 3 was prepared in two steps according to a similar literature procedure.16 2-(4-Methoxyphenyl)hydrazine-1-carboxamide (1) (2 g, 11.04 mmol, 1 equiv) was placed in an oven-dried argon-flushed round-bottomed flask (100 mL) with anhydrous toluene (22 mL). Pyrimidine (1.12 mL, 13.81 mmol, 1.25 equiv) and a solution of 4-methoxybenzoyl chloride (2.36 g, 13.81 mmol, 1.25 equiv) in anhydrous toluene (11 mL) were added slowly to the flask under an argon atmosphere (balloon) and then refluxed (~110 °C) for 1.5 h with magnetic stirring. The solution was cooled, poured into a conical flask containing a mixture of EtOAc–THF (9:1 v/v; 450:50 mL) and water (100 mL), and left under vigorous magnetic stirring for 2 h. The resultant precipitate was filtered off to give 2-(4-methoxybenzoyl)-2-(4-methoxyphenyl)hydrazine-1-carboxamide (2) (2.19 g, 6.96 mmol) in 63% yield. 1H NMR and LC-MS (ESI) analyses of 2 agree with literature values;16,30 1H NMR (d6-DMSO): δ 8.87 (br, 1H), 7.49 (br, 2H), 7.28 (d, 2H), 6.89 (m, 4H), 3.77 (s, 3H), 3.73 (s, 3H); LC-MS(ESI): m/z = 316.2 [M]+. Compound 2 was used without further purification for the synthesis of 3, as follows.
Precursor 2 (2 g, 6.34 mmol, 1 equiv) was added to a solution of KOH(aq.) (10% w/v; 17 mL) and ethanol (8.5 mL) in a round-bottomed flask (100 mL). This mixture was heated to 60 °C and left for 1.5 h under an argon atmosphere (balloon) with magnetic stirring. The solvent was then removed by rotary evaporation. Cold water (5 mL) was added, and the pH of the mixture was adjusted to 2 with 1 M HCl (~25 mL) under magnetic stirring. The whitish precipitate was filtered off, washed with cold water (3 × 30 mL), and desiccated to give 3 (1.62 g, 5.44 mmol) in 86% yield. Compound 3 was stored under desiccation until future use. 1H NMR and LC-MS(ESI) analyses of 3 agree with literature values:16,30 1H NMR (d6-DMSO): δ 11.25 (br, 1H), 7.33 (d, 2H), 7.29 (d, 2H), 7.02 (d, 2H), 6.94 (d, 2H), 3.77 (s, 3H), 3.73 (s, 3H); LC-MS(ESI): m/z = 298.2 [M]+.
(2,2-Difluorovinyloxy)-1,5-bis(4-methoxyphenyl)-1H-1,2,4-triazole (5).
This compound was prepared according to similar literature procedures.16,22,26 The alcohol precursor 3 (400 mg, 1.34 mmol, 1 equiv) was placed in an oven-dried argon-flushed round-bottomed flask (25 mL) with anhydrous DMF (4 mL) and K2CO3 (929 mg, 6.72 mmol, 5 equiv). This mixture was stirred for 10 min at RT. Then 1,1,1-trifluoro-2-iodoethane (663 μL, 6.72 mmol, 5 equiv) was added slowly under an argon atmosphere (balloon). The mixture was heated to 100 °C, left for 3 h under magnetic stirring, and then cooled. EtOAc (140 mL) and water (30 mL) were then poured into the reaction flask. The organic phase was separated off and washed with water (1 × 60 mL) and brine (1 × 60 mL). The organic layers were collected and dried (MgSO4). The solvent was removed by rotary evaporation. Silica gel flash chromatography (hexanes/EtOAc) of the crude product gave 1,5-bis(4-methoxyphenyl)-3-(2,2,2-trifluoroethoxy)-1H-1,2,4-triazole (4) (366 mg, 0.96 mmol) in 72% yield. 1H NMR, 13C NMR, 19F NMR, and LC-MS(ESI) analyses of 4 agree with literature values:16 1H NMR (CDCl3): δ 7.36−7.34 (d, 2H), 7.19−7.18 (d, 2H), 6.87−6.85 (d, 2H), 6.77−6.74 (d, 2H), 4.70−4.64 (q, 2H), 3.78−3.73 (d, 6H); 13C NMR (CDCl3): δ 166.22, 161.03, 159.87, 153.46, 131.09, 130.30, 127.07, 124.36, 121.61, 119.66, 114.64, 113.98, 66.27−65.16, 55.59−55.34; 19F NMR (CDCl3), δ 74.2; LC-MS(ESI): m/z = 380.1 [M]+.
Compound 4 (200 mg, 0.53 mmol, 1 equiv) was placed in an oven-dried argon-flushed round-bottomed flask (25 mL) with anhydrous THF (2 mL). The flask was placed in a dry-ice/acetone cooling bath (~−78 °C). Then n-BuLi (450 μL, 1.1 mmol, 2.1 equiv) was added dropwise under an argon atmosphere (balloon) with magnetic stirring. The reaction was left at −78 °C under magnetic stirring for 45 min. Then the reaction was quenched with water–THF (1:1 v/v; 5 mL) and left to warm to RT. The organic phase was extracted from the aqueous phase with EtOAc (2 × 20 mL). The combined organic layers were washed with brine (1 × 30 mL), dried (MgSO4), and concentrated under vacuum. The crude product was purified with silica gel flash column chromatography (hexanes/EtOAc) to give 5 (106 mg, 0.30 mmol) in 56% yield. 1H NMR (CDCl3): δ 7.46−7.44 (d, 2H), 7.31−7.28 (d, 2H), 6.97−6.94 (d, 2H), 6.86−6.84 (d, 2H), 6.80−6.75 (q, 1H), 3.87−3.83 (d, 3H); 13C NMR (CDCl3): δ 130.30, 127.47, 114.56, 113.90, 105.47−105.02, 55.37, 55.13; 19F NMR (CDCl3): δ 96.04, 116.94; LC-MS(ESI): m/z = 360.1 [M]+.
Production of [18F]Fluoride Ion.
The NCA [18F]fluoride ion was produced with the 18O(p,n)18F reaction by irradiation of [18O]water (95 atom %; 1.8 mL) with protons (14.1 MeV, 20–25 μA) generated with a PETtrace cyclotron (GE; Milwaukee, WI). Labeling experiments for radiochemistry optimization started with about 0.2–1.2 GBq of [18F]fluoride ion, whereas radiotracer productions for monkey studies started with about 10–15 GBq.
One-Step Synthesis of [18F]PS13.
Reagent Preparation and Apparatus Setup.
A stock solution of K 2.2.2 (37.64 mg, 0.1 mmol, 2 equiv) and K2CO3 (6.91 mg, 0.05 mmol, 1 equiv) in water (50 μL) plus anhydrous acetonitrile (450 μL) was loaded into a glass screw-neck 1 mL V-vial (12 × 32 mm, part # 186002802; Waters Corp.) and sealed with a bonded PTFE-silicone septum (12 × 32 mm, part # 186000274; Waters Corp.). An aliquot of this stock solution (100 μL) was placed in an oven-dried 3 mL V-vial (part # 95030; Alltech Associates Inc., Deerfield, IL) and sealed with a PTFE-silicone septum (Alltech Associates Inc.). This vial was then placed in a lead-shielded pot and prepared for receiving the cyclotron-produced [18F]fluoride ion in 18O-enriched water. The remaining K 2.2.2/K2CO3 stock solution was stored at −20 °C for not more than one month.
The gem-difluoroalkene 5 (1.5 mg, 4.2 μmol, 1 equiv), proton source (IPA or NH4Cl(aq.)), and anhydrous DMSO (300 μL) were placed in a glass screw-cap V-vial (1 mL, 12 × 32 mm, part # 186002802 Waters Corp.). The vial was sealed with a bonded PTFE-silicone septum (part # 186000274; Waters Corp.) and placed in the Synthia apparatus.
An empty oven-dried 5 mL V-vial (part # 95050; Alltech Associates Inc.) was sealed with a PTFE-silicone septum and screw cap (Alltech) and placed in the oven of the Synthia apparatus that would be used for drying the [18F]fluoride ion and subsequent reaction.
Synthesis of [18F]PS13.
The V-vial containing the cyclotron-produced [18F]fluoride ion was measured for radioactivity upon receipt. This vial was then placed in position in the Synthia apparatus. The recipe for the one-step synthesis of [18F]PS13 was started with a prompt command in the Autorad software. After programmed washing of the apparatus with acetonitrile, the desired volume (50–500 μL) was withdrawn from the [18F]fluoride ion vial by the robotic arm and placed in the reaction vial present in the apparatus oven. This solution was taken to dryness by three additions of acetonitrile and evaporation at 110 °C under nitrogen flow and reduced pressure. Then the radioactivity in the reaction vial was measured. The solution containing the reagents for the synthesis of [18F]PS13 was then added to the vial containing the dried [18F]fluoride ion and heated at 130 °C for 20 min. Then the reaction was quenched with acetonitrile–water (50:50 v/v; 0.5 mL). The activity in the reaction vial was measured. An aliquot of the crude reaction mixture was analyzed with radio-HPLC to evaluate the radiochemical yield, radiochemical purity, and molar activity of [18F]PS13.
Analysis of [18F]PS13.
[18F]PS13 was analyzed on a Luna PFP(2) column (5 μm 100 Å, 250 × 4.6 mm; Phenomenex, Torrance, CA) eluted with acetonitrile–water (45:55 v/v) at 1.8 mL/min with eluate monitored for radioactivity and absorbance at 254 nm. After each use, the HPLC column was washed for at least 30 min with acetonitrile–water (65:35 v/v).
Two-Step Synthesis of [18F]PS13.
Reagent Preparation and Apparatus Setup.
An oven-dried 3 mL V-vial was loaded with stock K 2.2.2/K2CO3 solution (100 μL), as described above for the one-step method, and the Synthia apparatus was prepared in readiness for receipt of the cyclotron-produced [18F]fluoride ion in 18O-enriched water.
A stock solution of the precursor for Step 1 was prepared from 2-difluorovinyl 4-methylbenzenesulfonate (4 μL, 22.2 μmol, 1 equiv), saturated NH4Cl(aq.) (10.6 μL), and anhydrous DMSO (1 mL). This was then placed in an oven-dried glass screw-cap 1 mL V-vial (12 × 32 mm; part # 186002802; Waters Corp.) and sealed with a bonded PTFE-silicone septum (part # 186000274; Waters Corp.). An aliquot of this solution (100 μL) was placed in an oven-dried glass screw-cap 1 mL V-vial (12 × 32 mm; part # 186002802; Waters Corp.) and sealed with a bonded PTFE-silicone septum (part # 186000274; Waters Corp.). This vial was then loaded into the Synthia apparatus. The remaining stock solution for Step 1 was stored at −20 °C up to one month.
For Step 2, the alcohol precursor 3 (2.5–3 mg, 8.41–10.09 μmol, 1 equiv), anhydrous DMF (300 μL), and (NH4)2CO3 (12.5–13 mg, 130–135 mmol, 15 equiv) were placed in an oven-dried tapered crimp-capped V-vial (0.9 mL; Thermo Scientific, Waltham, MA). This vial was sealed with a PTFE-silicone septum and placed in the Synthia apparatus.
An empty oven-dried 5 mL V-vial (part # 95050; Alltech) sealed with a screw-cap (Alltech Associates Inc.) and PTFE-silicone septum was placed in the oven of the Synthia apparatus that would be used for the azeotropic drying of the [18F]fluoride ion and subsequent reaction.
Synthesis [18F]PS13.
The V-vial containing the cyclotron-produced [18F]fluoride ion was measured for radioactivity. The vial was then placed in position in the Synthia apparatus. The recipe for the two-step synthesis of [18F]PS13 was started by selecting the prompt command in the Autorad software. After a programmed acetonitrile wash of the Synthia apparatus, the desired activity volume (50–500 μL) was withdrawn from the [18F]fluoride ion vial by the robotic arm, placed in the reaction vial present in the apparatus oven, and then subjected to three azeotropic dryings with acetonitrile at 110 °C under with nitrogen flow at reduced pressure. Then the activity in the reaction vial was measured, and the solution containing the reagents for the synthesis of ([18F]6) was added to the reaction vial and heated at 85 °C for 1 min. Reagents for Step 2 were added immediately to the reaction vial, which was then heated to 130 °C for 20 min. Then the radioactivity in the reaction vial was measured.
HPLC Separation of [18F]PS13.
The HPLC apparatus for purification of [18F]PS13 was started with a prompt command of the Autorad software. The crude reaction mixture was quenched with water (~1 mL), mixed in the robotic arm syringe, and injected onto a Luna PFP(2) column (5 μm, 100 Å, 250 × 10 mm; Phenomenex), eluted with acetonitrile–water (65:35 v/v) at 2 mL/min with eluate monitored for radioactivity and absorbance at 254 nm (Supporting Information, Figure S1). The retention time of [18F]PS13 was about 23 min. After each use, the HPLC column was washed for at least 30 min with acetonitrile–water (65:35 v/v).
Formulation of [18F]PS13 for Intravenous Injection.
The pure [18F]PS13 was collected in a sterile round-bottomed flask within a TRACERlab FX2 N apparatus (GE Healthcare; Chicago, IL) and diluted with sterile water (~40 mL; USP grade; Hospira; Lake Forest, IL) before being passed through into a C18 Sep-Pak cartridge that had been previously eluted with ethanol (USP grade; 10 mL; Warner Graham; Cockeysville, MD) followed by water (HPLC grade; 10 mL; EMD Millipore Corp.; Burlington, MA). The Sep-Pak cartridge was then washed with sterile water (USP grade; 10 mL), before eluting off the adsorbed [18F]PS13 with ethanol (USP grade; 1 mL) followed with saline for injection (0.9% w/v, USP grade, 10 mL; Fresenius Kabi; Lake Zurich, IL) into a round-bottomed flask. This solution was then passed through a Millex LG sterile filter (0.2 μm; EMD Millipore) into a sealed sterile vial (Hospira). A small aliquot of this formulated product (~500 μL) was withdrawn into a sterile syringe (1 mL; Air-Tite Products Co. Inc.; Virginia Beach, VA), placed in a separate vial, and measured for radioactivity. This aliquot was used for HPLC analysis.
Analysis of [18F]PS13.
[18F]PS13 was analyzed on a Luna PFP(2) column (5 μm, 100 Å, 250 × 4.6 mm; Phenomenex), eluted with acetonitrile–water (1:1 v/v) at 2 mL/min with eluate monitored for absorbance at 254 nm (Supporting Information, Figure S2). After each use, the HPLC column was washed for at least 30 min with acetonitrile–water (65:35 v/v).
Measurement of Molar Activity (Am) of [18F]PS13.
Aliquots (20 μL) of reference PS13 solution at five different known concentrations (9.8 × 10−3−9.8 × 10−2 mM) were each analyzed twice with radio-HPLC to obtain a calibration curve of average peak area (mAU × s) versus concentration.
At the end of synthesis, an aliquot (20 μL) of the formulated [18F]PS13 was analyzed with radio-HPLC. The peak area of PS13 eluting with [18F]PS13 was used to determine the concentration of carrier in the formulated [18F]PS13 dose from the calibration curve. The value (μmol/mL) obtained was multiplied by the total volume of the formulated [18F]PS13 dose to determine the total amount of carrier (μmol). The activity of the [18F]PS13 dose at the end of synthesis divided by the total amount of carrier gave the molar activity of [18F]PS13. This value was then decay-corrected to ERP.
Stability of [18F]PS13 in Buffer and in Monkey Whole Blood and Plasma in Vitro.
The stability of [18F]PS13 to incubation in sodium phosphate buffer (0.15 M, pH 7.4) for 1 and 4 h at RT was assessed by reversed phase HPLC on an Xterra column (7.8 × 300 mm, 10 μm; Waters Corp.), eluted at 4.0 mL/min with methanol–water–triethylamine (85:20:0.1, by vol.).
The stability of [18F]PS13 in monkey whole blood and plasma was also measured with HPLC, essentially as previously described for [11C]PS13.17
Plasma Free Fraction Determinations.
Plasma free fractions (f p) of [18F]PS13 in monkeys studied with PET were determined by an ultrafiltration method,44 as previously described17 for [11C]PS13.
PET Experiments in Monkey.
For each PET scanning session, the monkey was immobilized with ketamine and maintained under anesthesia with 1.6% isoflurane in oxygen. An intravenous perfusion line, filled with saline (0.9% w/v), was used for bolus injection of [18F]PS13. PET serial dynamic images were obtained on a Focus 220 PET camera (Siemens Medical Solutions; Knoxville, TN) or a Biograph mCT camera (Siemens Healthneers; Erlangen, Germany).
Brain Scans.
Two male rhesus (Macaca mulatta) monkeys (monkey A, 12.2 kg; monkey B, 13.8 kg) were scanned at baseline for up to 180 min to measure uptake of radioactivity into brain and to determine activity distribution after bolus intravenous injection of [18F]PS13 (monkey A, 287 MBq, PS13 carrier 6.2 nmol/kg; monkey B, 128 MBq, PS13 carrier 3.0 nmol/kg) using a Focus 220 PET camera (Siemens Medical Solutions; Knoxville, TN). Preblock experiments were also performed in the same monkeys in which PS13 (0.3 mg/kg, i.v.) was administered intravenously between 15 and 5 min before [18F]PS13 (monkey A, 266 MBq; PS13 carrier 6.6 nmol/kg; monkey B, 281 MBq; PS13 carrier, 6.6 nmol/kg). All monkeys had T1-weighted magnetic resonance imaging (TR/TE/x = 24 ms/3 ms/300), acquired on a 1.5-T Horizon instrument (GE Medical Systems; Waukesha, WI).
Whole-Body Scan.
A third rhesus monkey (Macaca mulatta) (monkey C; 10.3 kg, female) was injected with [18F]PS13 (166 MBq; PS13 carrier 5.0 nmol/kg, i.v.) for whole-body PET imaging on a Biograph mCT camera (Siemens Healthneers; Erlangen, Germany) for 180 min.
Image Analysis.
PET brain images were reconstructed using Fourier rebinning plus two-dimensional filtered back-projection. PET images were initially coregistered to each monkey’s T1-weighted magnetic resonance image (MRI) and then to a standardized monkey MRI template using the Fuse It module of PMOD 3.9 (PMOD Technologies; Zurich, Switzerland). A set of 33 predefined brain regions of interest from the template was then applied to the coregistered PET image to obtain regional decay-corrected time–activity curves for frontal cortex, temporal cortex, parietal cortex, occipital cortex, striatum, thalamus, limbic region, cerebellum, and brain stem. Data reported for whole brain data are from gray matter. All PET images were corrected for attenuation and scatter. Radioactivity concentrations were expressed as standardized uptake value (SUV), which normalizes for subject weight and injected radioactivity, according to the equation
Whole-body PET images were reconstructed with ordered subset expectation maximization (OSEM) with time-of-flight and resolution recovery.
Analysis of [18F]PS13 Radiometabolites in Monkey Plasma.
To determine a radiometabolite-corrected arterial input function for brain PET scans, blood samples (0.5–1 mL each) were drawn from the monkey femoral artery into heparin-treated Vacutainer tubes at 15 s intervals until 120 s, followed by 0.5–4 mL samples at 3, 5, 10, 30, 60, 90, 120, 150, and 180 min. The concentration of parent radioligand was measured with reversed phase HPLC on an Xterra column (7.8 × 300 mm, 10 μm; Waters Corp.), eluted at 4.0 mL/min (for monkey A) or 3.5 mL/min (for monkey B) with methanol–water–triethylamine (85:20:0.1, by vol.), after separating plasma from whole blood, as previously described.45
PET Brain Data Analysis.
Total distribution volume (VT), a sum of a specific binding component known as the specific volume of distribution (VS) and a nondisplaceable volume of distribution (VND), may serve as an index of binding site density that equals the ratio at equilibrium of the concentration of the radioligand in tissue to that in plasma.46 VT values, uncorrected for radioactivity in blood (5% of brain volume), were estimated for different brain regions with Logan graphical analysis47,48 by using the PET brain time–activity curves and the measured radiometabolite-corrected arterial input functions. Kinetic analyses were performed at the voxel level, so that the resulting VT values for [18F]PS13 could be shown as parametric PET images. The temporal stabilities of VT in whole brain in the baseline experiments were assessed by estimating VT from progressively time-truncated data sets. Standard Error (i.e.,% SE or identifiability) was expressed as a percentage. A smaller percentage indicates better identifiability.48 Additionally, regional brain VT data were used to generate a Lassen plot49,50 to estimate COX-1 occupancy in the preblock conditions and to estimate VND. The PET image analysis, including kinetic modeling, was performed with PMOD 3.9 (PMOD Technologies).
The time-stability of VT values was evaluated by fitting regional time-activity curves for PET data with truncated acquisition times using the two-tissue compartment model, ranging from 180 to 60 min. The ratio of the regional VT values from the truncated scan to that from the 180 min measurement was computed for each region of interest.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH Clinical PET Center (Director: Dr. Peter Herscovitch) for fluorine-18 production. The authors thank Dr. Prachi Singh and Dr. Stal Shrestha for their previous related work on [11C]PS13 and Dr. Shuiyu Lu for his technical assistance in the radiosynthesis of [18F]PS13.
Funding
This study was supported by the Intramural Research Program of NIH (National Institute of Mental Health; project number ZIA-MH002793 to V.W.P. and project number ZIA-MH002795 to R.B.I.).
ABBREVIATIONS
- A m
molar activity
- COX-1
cyclooxygenase-1
- COX-2
cyclooxygenase-2
- ERP
end of radioisotope production
- f p
plasma free fraction
- IPA
isopropyl alcohol
- K 2.2.2
4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane
- MW
microwave
- MIP
maximum intensity projection
- MRI
magnetic resonance image
- PET
positron emission tomography
- PTFE
polytetra-fluoroethylene
- RCY
radiochemical yield
- RT
room temperature
- V S
specific volume of distribution
- SUV
standardized uptake value
- V ND
nondisplaceable volume of distribution
- V T
total distribution volume
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.0c00737.
NMR spectra, radiochromatograms for radioligand separations and analyses, monkey PET, and metabolism data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.0c00737
The authors declare no competing financial interest.
Contributor Information
Carlotta Taddei, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Cheryl L. Morse, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
Min-Jeong Kim, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Jeih-San Liow, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Jose Montero Santamaria, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Andrea Zhang, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Lester S. Manly, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
Paolo Zanotti-Fregonara, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Robert L. Gladding, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
Sami S. Zoghbi, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
Robert B. Innis, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
Victor W. Pike, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
REFERENCES
- (1).Smyth EM, Grosser T, Wang M, Yu Y, and FitzGerald GA (2009) Prostanoids in health and disease. J. Lipid Res 50 (April Suppl), S423–S428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Aïd S, and Bosetti F (2011) Targeting cyclooxygenases-1 and −2 in neuroinflammation: therapeutic implications. Biochimie 93 (1), 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Choi S-H, Aid S, and Bosetti F (2009) The distinct roles of cyclooxygenase-1 and −2 in neuroinflammation: implications for translational research. Trends Pharmacol. Sci 30 (4), 174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Frank-Cannon TC, Alto LT, McAlpine FE, and Tansey MG (2009) Does neuroinflammation fan the flame in neuro-degenerative diseases? Mol. Neurodegener 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pike VW (2016) Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem 23 (18), 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).McCluskey SP, Plisson C, Rabiner EA, and Howes O (2020) Advances in CNS PET: the state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 47, 451–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pike VW (1997) The status of PET radiochemistry for drug development and evaluation. Drug Inf. J 31, 997–1013. [Google Scholar]
- (8).Kuchar M, and Mamat C (2015) Methods to increase the metabolic stability of 18F-radiotracers. Molecules 20 (9), 16186–16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).van der Born D, Pees A, Poot AJ, Orru RVA, Windhorst AD, and Vugts DJ (2017) Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev 46 (15), 4709–4773. [DOI] [PubMed] [Google Scholar]
- (10).Deng X, Rong J, Wang L, Vasdev N, Zhang L, Josephson L, and Liang SH (2019) Chemistry for positron emission tomography: recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew. Chem., Int. Ed 58 (9), 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McCarthy TJ, Sheriff AU, Graneto MJ, Talley JJ, and Welch MJ (2002) Radiosynthesis, in vitro validation, and in vivo evaluation of 18F-Labeled COX-1 and COX-2 inhibitors. J. Nucl. Med 43 (1), 117–124. [PubMed] [Google Scholar]
- (12).Fujisaki Y, Kawamura K, Wang W-F, Ishiwata K, Yamamoto F, Kuwano T, Ono M, and Maeda M (2005) Radiosynthesis and in vivo evaluation of 11C-labeled 1,5-diary-lpyrazole derivatives for mapping cyclooxygenases. Ann. Nucl. Med 19 (7), 617–625. [DOI] [PubMed] [Google Scholar]
- (13).Takashima-Hirano M, Shukuri M, Takashima T, Goto M, Wada Y, Watanabe Y, Onoe H, Doi H, and Suzuki M (2010) General method for the 11C-labeling of 2-arylpropionic acids and their esters: construction of a PET tracer library for a study of biological events involved in COXs expression. Chem. - Eur. J 16 (14), 4250–4258. [DOI] [PubMed] [Google Scholar]
- (14).Shukuri M, Mawatari A, Ohno M, Suzuki M, Doi H, Watanabe Y, and Onoe H (2016) Detection of cyclooxygenase-1 in activated microglia during amyloid plaque progression: PET studies in Alzheimer’s disease model mice. J. Nucl. Med 57 (2), 291–296. [DOI] [PubMed] [Google Scholar]
- (15).Ohnishi A, Senda M, Yamane T, Mikami T, Nishida H, Nishio T, Akamatsu G, Ikari Y, Kimoto S, Aita K, Sasaki M, Shinkawa H, Yamamoto Y, Shukuri M, Mawatari A, Doi H, Watanabe Y, and Onoe H (2016) Exploratory human PET study of the effectiveness of 11C-ketoprofen methyl ester, a potential biomarker of neuroinflammatory processes in Alzheimer’s disease. Nucl. Med. Biol 43 (7), 438–444. [DOI] [PubMed] [Google Scholar]
- (16).Singh P, Shrestha S, Cortes-Salva MY, Jenko KJ, Zoghbi SS, Morse CL, Innis RB, and Pike VW (2018) 3-Substituted 1,5-diaryl-1H-1,2,4-triazoles as prospective PET radioligands for imaging brain COX-1 in monkey. Part 1: Synthesis and pharmacology. ACS Chem. Neurosci 9 (11), 2610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Shrestha S, Singh P, Cortes-Salva MY, Jenko KJ, Ikawa M, Kim M-J, Kobayashi M, Morse CL, Gladding RL, Liow J-S, Zoghbi SS, Fujita M, Innis RB, and Pike VW (2018) 3-Substituted 1,5-diaryl-1H-1,2,4-triazoles as prospective PET radioligands for imaging brain COX-1 in monkey. Part 2: selection and evaluation of [11C]PS13 for quantitative imaging. ACS Chem. Neurosci 9 (11), 2620–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kim M-J, Shrestha SS, Cortes M, Singh P, Morse C, Liow J-S, Gladding RL, Brouwer C, Henry K, Gallagher E, Tye GL, Zoghbi SS, Fujita M, Pike VW, and Innis RB (2018) Evaluation of two potent and selective PET radioligands to image COX-1 and COX-2 in rhesus monkeys. J. Nucl. Med 59 (12), 1907–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kim M-J, Lee J-H, Anaya FJ, Hong J, Miller W, Telu S, Singh P, Cortes MY, Henry K, Tye GL, Frankland MP, Montero Santamaria JA, Liow J-S, Zoghbi SS, Fujita M, Pike VW, and Innis RB (2020) First-in-human evaluation of [11C]PS13, a novel PET radioligand, to quantify cyclooxygenase-1 in the brain. Eur. J. Nucl. Med. Mol. Imaging 47, 3143–3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Boyle AJ, Tong J, Zoghbi SS, Pike VW, Innis RB, and Vasdev N (2020) Repurposing [11C]PS13 for PET imaging of cyclooxygenase-1 (COX-1) in ovarian cancer xenograft mouse models. J. Nucl. Med, DOI: 10.2967/jnumed.120.249367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eberl S, Eriksson T, Svedberg O, Norling J, Henderson D, Lam P, and Fulham M (2012) High beam current operation of a PETtrace™ cyclotron for 18F production. Appl. Radiat. Isot 70 (6), 922–930. [DOI] [PubMed] [Google Scholar]
- (22).Riss PJ, and Aigbirhio FI (2011) A simple, rapid procedure for nucleophilic radiosynthesis of aliphatic [18F]trifluoromethyl groups. Chem. Commun 47 (43), 11873–11875. [DOI] [PubMed] [Google Scholar]
- (23).Riss PJ, Ferrari V, Brichard L, Burke P, Smith R, and Aigbirhio FI (2012) Direct, nucleophilic radiosynthesis of [18F]trifluoroalkyl tosylates: improved labelling procedures. Org. Biomol. Chem 10 (34), 6980–6986. [DOI] [PubMed] [Google Scholar]
- (24).Riss PJ, Brichard L, Ferrari V, Williamson DJ, Fryer TD, Hong YT, Barona J-C, and Aigbirhio FI (2013) Radiosynthesis and characterization of astemizole derivatives as lead compounds toward PET imaging of τ-pathology. MedChemComm 4 (5), 852–855. [Google Scholar]
- (25).Lien VT, and Riss PJ (2014) Radiosynthesis of [18F]trifluoroalkyl groups; scope and limitations. BioMed. Res. Int 2014, 380124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fawaz MV, Brooks AF, Rodnick ME, Carpenter GM, Shao X, Desmond TJ, Sherman P, Quesada CA, Hockley BG, Kilbourn MR, Albin RL, Frey KA, and Scott PJH (2014) High affinity radiopharmaceuticals based upon lansoprazole for PET imaging of aggregated tau in Alzheimer’s disease and progressive supranuclear palsy: synthesis, preclinical evaluation, and lead selection. ACS Chem. Neurosci 5 (8), 718–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rafique W, Kramer V, Pardo T, Smits R, Spilhaug MM, Hoepping A, Savio E, Engler H, Kuljs R, Amaral H, and Riss PJ (2018) Image-guided development of heterocyclic sulfoxides as ligands for tau neurofibrillary tangles: from first-in-man to second-generation ligands. ACS Omega 3 (7), 7567–7579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kramer V, Brooks AF, Haeger A, Kuljis RO, Rafique W, Koeppe RA, Raffel DM, Frey KA, Amaral H, Scott PJH, and Riss PJ (2020) Evaluation of [18F]-N-Methyl lansoprazole as a tau PET imaging agent in first-in-human studies. ACS Chem. Neurosci 11 (3), 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Aoki S, Nagagawa T, Nakamura K, Omori H, Kubota A, and Hashimoto N Triazole derivatives. PCT/JP02/11314. 2002, U.S. Patent Appl. 2003 2003019155 (A1).
- (30).Takahashi F, Nakagawa T, Matsushima Y, and Nakamura K Imidazole and triazole derivatives useful as selective COX-1 inhibitors. PCT/JP2003/015921, Patent 2004 WO 2004060367.
- (31).Aigbirhio FL, Pike VW, Waters SL, Makepeace J, and Tanner RJN (1993) Efficient and selective labelling of the CFC alternative, 1,1,1,2-tetrafluoroethane, with 18F in the 1-position. J. Chem. Soc., Chem. Commun, 1064–1065. [Google Scholar]
- (32).Cai L, Pike VW, and Lu S (2008) Chemistry with [18F]fluoride ion. Eur. J. Org. Chem 2008, 2853–2873. [Google Scholar]
- (33).Landini D, Maia A, and Rampoldi A (1989) Dramatic effect of the specific solvation on the reactivity of quaternary ammonium fluorides and poly(hydrogen fluorides), (HF)n•F−, in media of low polarity. J. Org. Chem 54 (2), 328–332. [Google Scholar]
- (34).Lu S, Haskali MB, Ruley KM, Dreyfus NJ-F, DuBois SL, Paul S, Liow J-S, Morse CL, Kowalski A, Gladding RL, Gilmore J, Mogg AJ, Morin SM, Lindsay-Scott PJ, Ruble JC, Kant NA, Shcherbinin S, Barth VN, Johnson MP, Cuadrado M, Jambrina E, Mannes AJ, Nuthall HN, Zoghbi SS, Jesudason CD, Innis RB, and Pike VW (2020) PET ligands [18F]LSN3316612 and [11C]LSN3316612 quantify O-linked—N-acetyl-glucosamine hydrolase in the brain. Sci. Transl. Med 12 (543), No. eaau2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Pike VW (2009) PET radiotracers: crossing the blood-brain barrier and avoiding metabolism. Trends Pharmacol. Sci 30 (8), 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Yasojima K, Schwab C, McGeer EG, and McGeer PL (1999) Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res 830 (2), 226–236. [DOI] [PubMed] [Google Scholar]
- (37).Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, and Stajer D (2009) Cyclooxygenase in normal human tissues – is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J. Cell. Mol. Med 13 (9B), 3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Libert LC, Franci X, Plenevaux AR, Ooi T, Maruoka K, Luxen AJ, and Lemaire CF (2013) Production at the Curie level of no-carrier-added 6-[18F]fluoro-L-DOPA. J. Nucl. Med 54 (7), 1154–1161. [DOI] [PubMed] [Google Scholar]
- (39).Lemaire C, Libert L, Franci X, Genon J-L, Kuci S, Giacomelli F, and Luxen A (2015) Automated production at the Curie level of no-carrier-added 6-[18F]fluoro-L-DOPA and 2-[18F]-fluoro-L-tyrosine on a FASTlab synthesizer. J. Labelled Compd. Radiopharm 58 (7), 281–290. [DOI] [PubMed] [Google Scholar]
- (40).Ravert HT, Holt DP, Chen Y, Mease RC, Fan H, Pomper MG, and Dannals RF (2016) An improved synthesis of the radiolabeled prostate-specific membrane antigen inhibitor, [18F]DCFPyL. J. Labelled Compd. Radiopharm 59 (11), 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sergeev M, Lazari M, Morgia F, Collins J, Javed MR, Sergeeva O, Jones J, Phelps ME, Lee JT, Keng PY, and van Dam RM (2018) Performing radiosynthesis in microvolumes to maximize molar activity of tracers for positron emission tomography. Commun. Chem 1, 10 Article 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bjurling P, Reineck R, Westerburg G, Gee AD, Sutcliffe J, and Långström B Synthia, a compact radiochemistry system for automated production of radiopharmaceuticals. In Link JM, and Ruth TJ, Eds. Proc. - Sixth Workshop on Targetry and Target Chemistry; TRIUMF, Vancouver, 1995. 282–284. [Google Scholar]
- (43).Clark JD, Baldwin RL, Bayne KA, Brown MJ, Gebhart GF, Gonder JC, Gwathmey JK, Keeling ME, Kohn DF, Robb JW, Smith OA, Steggerda J-AD, and Van de Ber JL Guide for the Care and Use of Laboratory Animals; National Academy Press, Washington DC, 1996. [Google Scholar]
- (44).Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, and Innis RB (1994) Evaluation of ultrafiltration for the free-fraction detection of single photon emission computed tomography (SPECT) radiotracers: β-CIT, IBF, and iomazenil. J. Pharm. Sci 83 (7), 1014–1019. [DOI] [PubMed] [Google Scholar]
- (45).Zoghbi SS, Shetty UH, Ichise M, Fujita M, Imaizumi M, Liow J-S, Shah J, Musachio JL, Pike VW, and Innis RB (2006) PET Imaging of the dopamine transporter with 18F-FECNT: a polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med 47 (3), 520–527. [PubMed] [Google Scholar]
- (46).Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, and Carson RE (2007) Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab 27, 1533–1539. [DOI] [PubMed] [Google Scholar]
- (47).Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, and Christman DR (1990) Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl]-(−)-cocaine PET studies in human subjects. J. Cereb. Blood Flow Metab 10, 740–747. [DOI] [PubMed] [Google Scholar]
- (48).Carson RE Parameter estimation in positron emission tomography. In Positron Emission Tomography and Autoradiography: Principles and Applications for the Brain and Heart (Phelps ME, Mazziotta JC, and Schelbert HR, Eds.), Raven Press, New York, 1986. pp 347–390, Ch. 8. [Google Scholar]
- (49).Cunningham VJ, Rabiner EA, Slifstein M, Laruelle M, and Gunn RN (2010) Measuring drug occupancy in the absence of a reference region: the Lassen plot re-visited. J. Cereb. Blood Flow Metab 30, 46–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lassen NA, Bartenstein PA, Lammertsma AA, Prevett MC, Turton DR, Luthra SK, Osman S, Bloomfield PM, Jones T, Patsalos PN, O’Connell MT, Duncan JS, and Andersen JV (1995) Benzodiazepine receptor quantification in vivo in humans using [11C]flumazenil and PET: application of the steady-state principle. J. Cereb. Blood Flow Metab 15, 152–165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.