

CONSPECTUS:
Titanium is an attractive metal for catalytic reaction development: it is earth abundant, inexpensive, and generally nontoxic. However—like most early transition metals—catalytic redox reactions with Ti are difficult, owing to the stability of the high-valent TiIV state. Understanding the fundamental mechanisms behind Ti redox processes is key for making progress toward potential catalytic applications. This Account will detail recent progress in Ti-catalyzed (and mediated) oxidative amination reactions that proceed through formally TiII/TiIV catalytic cycles.
This class of reactions is built off of our initial discovery into Ti-catalyzed [2+2+1] pyrrole synthesis from alkynes and azobenzene, where detailed mechanistic studies have revealed important factors that allow for catalytic turnover despite the inherent difficulty of Ti redox. Two important conclusions from mechanistic studies are that (1) low valent Ti intermediates in catalysis can be stabilized through coordination of π-acceptor substrates or products, where they can act as “redox-noninnocent” ligands through metal-to-ligand π-backdonation; and (2) reductive elimination processes with Ti proceed through π-type electrocyclic (or pericyclic) reaction mechanisms rather than direct σ-bond coupling.
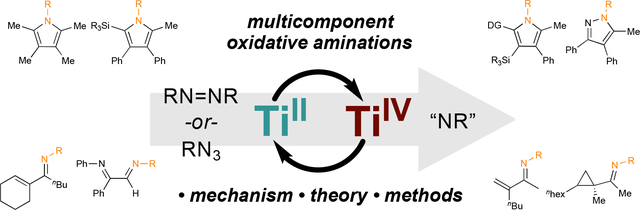
The key reactive species in Ti-catalyzed oxidative amination reactions is a Ti imido (Ti≡NR), which can be generated from either aryl diazenes (RN=NR), or organic azides (RN3). These Ti imidos can then undergo [2+2] cycloadditions with alkynes, resulting in intermediates that can be coupled to an array of other unsaturated functional groups including alkynes, alkenes, nitriles, and nitrosos. This basic reactivity pattern has been extended into a broad range of catalytic and stoichiometric oxidative multicomponent coupling reactions of alkynes and other reactive small molecules, leading to multicomponent syntheses of various heterocycles and aminated building blocks.
For example, catalytic oxidative coupling of Ti imidos with two different alkynes leads to pyrroles, while stoichiometric oxidative coupling with alkynes and nitriles leads to pyrazoles. These heterocycle syntheses often yield substitution patterns that are complementary to classical condensation routes, and provide access to new electron-rich, highly substituted heteroaromatic scaffolds. Further, catalytic oxidative alkyne carboamination reactions can be accomplished via reaction of Ti imidos with alkynes and alkenes, yielding α,β-unsaturated imine or cyclopropylimine building blocks. New catalytic and toichiometric oxidative amination methods such as alkyne α-diimination, isocyanide imination, and ring-opening oxidative amination of strained alkenes are continuously emerging as a result of better mechanistic understanding of Ti redox catalysis.
Ultimately, these Ti-catalyzed and -mediated oxidative amination methods demonstrate the importance of examining often-overlooked elements like the early transition metals through the lens of modern catalysis: rather than a lack of utility, these elements frequently have undiscovered potential for new transformations with orthogonal or complementary selectivity to their late transition metal counterparts.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
INTRODUCTION
Nitrogen-based functional groups are ubiquitous in bioactive molecules.5,6 Recently, powerful methods have evolved around new ways of constructing C-N bonds, including alkene hydroamination,7,8 aziridination,9 oxidative C-H amination,10 and multicomponent cycloadditions.11 To a large extent, this impressive development has been the domain of late transition metals (LTMs), raising the question of the role that early transition metals (ETMs) such as Ti can have in engendering new amination reactions.
ETMs have the potential to access different structures (e.g. metal-ligand multiple bonds) and elementary reaction steps than LTMs, potentially enabling new, orthogonal bond-forming strategies.12 ETMs are also earth abundant: the crustal concentration of Ti is greater than all other transition metals combined, Fe excluded.13 There is significant opportunity to reinvestigate this often-overlooked part of the transition series through the lens of modern catalysis.14 In fact, catalysis with TiIII-based reagents has experienced a renaissance owing to increased interest in/understanding of selective radical-based strategies.15–18
While there are myriad examples of Ti-catalyzed15 or stoichiometric19 reduction reactions, Ti-catalyzed oxidation reactions involving metal-centered 2-electron redox are rare, owing to the difficulty in reduction of TiIV to TiII.20 Nonetheless, developments in redox noninnocent ligands21 over the past 20 years have made ETM redox catalysis an attractive target.
This Account will explore recent developments in Ti-catalyzed and -mediated oxidative amination reactions, using mechanistic insight to explain fundamental principles of catalysis and motivate selective reaction development. These oxidative aminations are built off of the (1) reactivity of Ti≡NR imido complexes, which undergo [2+2] cycloaddition16 with alkynes; and (2) facile bimolecular cleavage of N=N bonds in aryl diazenes by TiII. Based on this chemistry, diverse multicomponent reactions have been developed that yield important heterocycles and aminated building blocks (Figure 1).
Figure 1.

Summary of oxidative amination reactions catalyzed or mediated by Ti imido (Ti≡NR) complexes.
CATALYTIC [2+2+1] SYNTHESIS OF PYRROLES
In 2016, we reported the formal [2+2+1] synthesis of pyrroles (1) from alkynes and azobenzene, catalyzed by py3TiCl2(NPh) (Figure 2A).1 This reaction was significant as the first example of catalytic oxidative C-N bond formation with Ti. Tethered diynes (1a, 1b) and simple alkynes (1c) undergo reactions with equal efficiency (Figure 2B). Unsymmetric internal alkynes (1e, 1f) yield regioisomeric mixtures, where the product distribution is often statistical. Terminal alkynes (1g, 1h) also yield regioisomeric mixtures but have a strong preference for formation of 2,4-disubstituted product 1. Qualitatively, the effect of alkyne structure on reaction rate is terminal (1g, 1h) > polarized (1f) > internal dialkyl (1c) > internal diaryl (1d). In the case of highly reactive terminal and polarized alkynes, an excess of aryl diazene is sometimes needed to suppress unwanted alkyne cyclotrimerization.
Figure 2.

Initial demonstration of TiII/TiIV-catalyzed synthesis of pyrroles along with representative examples. Regioisomeric ratios (1 : 1’ : 1”) in parentheses.
This catalytic [2+2+1] pyrrole synthesis has formed the underpinnings for a large array of oxidative nitrene transfer reactions that have been developed in our labs, and mechanistic studies have been a key driver in their development. As a foundation for later work, details of each step of the mechanism will be discussed first along with historical context and computational studies, before returning to new reaction discovery.
[2+2+1] pyrrole synthesis is an extension of ETM imido-catalyzed alkyne hydroamination.8 In Ti-catalyzed alkyne hydroamination, [2+2] Ti≡NR (IM1) + alkyne cycloaddition (TS1) leads to an azatitanacyclobutene (IM2), which undergoes protonolysis by a primary amine to liberate enamine (imine) 3 (Figure 3B). Inspiration for [2+2+1] pyrrole synthesis was borne out of a study of (ONO)TiBn2 (2) catalyzed 2-butyne hydroamination (Figure 3A).23 Reactions with excess 2-butyne catalyzed by 2 resulted in trace amounts of pyrrole 1i and C6Me6 4. We speculated that these sideproducts were generated through 2nd alkyne insertion into IM2, leading to an azatitanacyclohexadiene (IM3) that underwent reductive elimination to form 1i and a TiII species which cyclotrimerized remaining 2-butyne to 4. This hypothesis led to the discovery of simple Ti complexes (e.g. py3TiCl2(NPh)) and oxidants (aryl diazenes,1 later organic azides24) capable of closing the catalytic cycle for pyrrole production.
Figure 3.

Earlier studies of 2-butyne hydroamination led to the observation of minor byproducts resulting from 2nd alkyne insertion into IM2.
[2+2+1] Mechanism – Overview.
A summary of the mechanism of Ti-catalyzed formal [2+2+1] pyrrole synthesis2 is presented in Figure 4, along with rate laws derived from variable time normalization analysis25 (VTNA). Computational studies2,26 indicate that the active species has 1 coordinated pyridine (IM4), although there may be complex/changing speciation resulting from reversible pyridine coordination to various intermediates along the cycle.
Figure 4.

Mechanism of py3TiCl2(NPh)-catalyzed [2+2+1] pyrrole synthesis. Computed free energies (M06/6–311G(d,p)/SMD, PhCF3, 115 °C) for R = Me. Reaction energy span reads from IM4-Dimer to IM8.
First, TiIV imido IM4 undergoes [2+2] cycloaddition with an alkyne to form azatitanacyclobutene IM5. Next, a second alkyne inserts into IM5 to form azatitanacyclohexadiene IM6 in the rate-determining step. Reductive elimination from IM6 next yields a TiII intermediate, either free or as “masked” TiII backdonating to the bound pyrrole (IM7). IM7 can then either unproductively trimerize alkyne or be trapped by azobenzene to form TiII η2-azobenzene adduct IM8, which disproportionates into a TiIV imido (IM4 and IM4-Dimer) to close the cycle. Reoxidation of IM7 is facile, and examples of Ti imido synthesis by TiII azobenzene disproportionation/oxidation go back several decades.27,28 Disproportionation is likely bimetallic although the exact mechanism (homodimerization of IM8 as drawn, or reaction29 of IM8 with free TiII) remains unclear. Disproportionation is irreversible (no diazene crossover has been observed), likely owing to the formation of strong Ti≡NR bonds. Half order dependence on [Ti] indicates an equilibrium between the precatalyst, IM4, and IM4-Dimer, consistent with kinetic analyses of Ti-catalyzed alkyne hydroamination30,31 where Ti imidos are well-known to dimerize.8
Second Alkyne Insertion.
Despite many studies of [2+2] cycloaddition of Ti imidos, there are only two examples of alkyne insertion into group 4 azametallacyclobutenes like IM5 (Figure 5). First, Walsh and Bergman32 reported that reaction of Zr bis(amide) 5 with excess MeCCMe resulted in [2+2] cycloaddition and 2nd alkyne insertion to 6. Mountford33 isolated 8 from reaction of 7 with excess arylacetylene. Similarly, Odom reported that Mo and W bis(imido)s undergo double reaction with cyclooctyne to form azametallacycloehexadienes which liberate pyrrole upon heating.34
Figure 5.

Examples of group 4 imidos that undergo reaction with 2 equivalents of alkyne.
Insertions of other unsaturated species (e.g. nitriles,35 aldehydes,36 and imines.37) into azatitanacyclobutenes have also been reported. Notably, Odom has reported a versatile class of catalytic iminoamination reactions via isocyanide insertion.38,39 Cloke reported a stoichiometric [2+2+1] synthesis of 1,2,4-azadiphosphole via double phosphaalkyne reaction with py3TiCl2(NtBu).40
In [2+2+1] pyrrole synthesis, a second alkyne inserts into IM5 since (unlike in hydroamination) there is no amine for protonolysis. The 2nd alkyne insertion is more kinetically challenging than [2+2] cycloaddition (or protonolysis), owing to sterically-encumbered 6-coordinate TS3. For reactions of dialkylacetylenes catalyzed by py3TiCl2(NPh), kinetic (2nd order [EtCCEt]) and computational analysis (Figure 4, TS3 ΔG‡ = 29.6 kcal/mol vs. TS2 ΔG‡ = 22.0 kcal/mol; MeCCMe) indicate 2nd insertion is rate-determining.
C-N Reductive Elimination.
Ti organometallics are more likely to undergo H-abstraction41 or radical reaction42 than σ-bond reductive elimination. Thus, it is surprising that IM6 undergoes facile C-N reductive elimination (Figure 4, TS4).
Since reductive elimination occurs after rate-limiting TS3, this step has only been examined computationally. Comparative free energy profiles for reductive elimination from two major computational studies2,26 are presented in Figure 6. Although significant free energy differences arise between these models (resulting from different levels of theory and computational approaches: gas-phase geometry optimizations followed by single-point calculations26 vs. initial geometry optimization in solvent2) it is clear that C-N reductive elimination is a low barrier process (TS4 ΔG‡ = 20.1 or 10.6 kcal/mol). There is debate on whether the reoxidation of IM7 occurs through associative interchange with PhNNPh (TS5 ΔG‡ = 17.5 or 14.9 kcal/mol) or through pyrrole dissociation to free/solvated TiII (IM10 ΔG = 32.1 or 9.9 kcal/mol). Given the abundance of potential π-acceptors in these reactions (alkyne, azobenzene, solvent), an associative interchange mechanism with some stabilizing ligand is certainly plausible.
Figure 6.

Computational free energies of reductive elimination and pyrrole displacement/reoxidation by PhNNPh. Black lines (M06/6–311G(d,p)/SMD, PhCF3, 115 °C) from ref 20. Red lines (gas-phase B3LYP/6–311G(d,p) followed by SPE at M06-L/6–311++G(d,p)/PCM, PhCF3, 25 °C) from ref 22. Solid lines follow associative exchange of pyrrole by PhNNPh (TS5); dotted lines follow dissociative exchange (IM10).
Why is reductive elimination so facile? First, backbonding of the resultant low valent TiII species into the pyrrole product π* orbitals stabilizes IM7. This backbonding can be seen in the elongation (~1.45 Å vs ~1.38 Å in N-Ph pyrrole43) of the C-N bonds of the calculated structure of IM7 (Figure 7A), as well as in NICS and NBO analysis. Synergistic backbonding with π-acceptor products provides a thermodynamic rationale for why reductive elimination is favorable, similar to how redox noninnocent ligands cooperatively attenuate metal electron density. Formation of 2-electron π-backbonded species may also help attenuate competitive radical TiIII processes; indeed, significant work on the reactivity of π-backbonded “masked” TiII fragments has been reported.28,44,45
Figure 7.

(A) Calculated bond lengths (Å) and IBOs (B and C) for reductive elimination from IM6 (left) through TS4 (middle) to IM7 (right). Figure adapted from ref 2.
The second factor contributing to facile reductive elimination is that C-N coupling proceeds through a π electrocyclic mechanism rather than Ti-N/Ti-C σ-bond coupling. This hypothesis emerged from intrinsic bond orbital (IBO) analysis46 of reductive elimination (Figure 7, B and C), where one can observe the π-to-σ transformation seen in electrocyclic reactions: the N-Ti π bond in IM6 (blue) rotates and attacks C4 to form the new C-N bond, while the π bonds on the metallacycle (orange and green) shift to localize on C1, C2, and C3. Concurrent with C-N bond formation, the Ti-C and Ti-N σ-bonding orbitals rotate perpendicular to the forming pyrrole plane and form a π-backbond with Ti. The overall transformation47 is similar to an aza-Nazarov cyclization.48,49
Electrocyclic reductive elimination kinetically enables much of the Ti redox catalysis herein; further, it may play an important role in reactions across the periodic table. For example, pericyclic redox mechanisms have been demonstrated in asymmetric Tsuji allylic alkylation,50 PIII/PV reductive transposition,51 and lanthanide-promoted pyrimidine formation.52
Studies into well-behaved examples of electrocyclic reductive elimination will be important for better understanding the requirements for these transformations. Fortier recently reported a reversible thiophene C-S oxidative addition/reductive elimination sequence from arene-masked TiII complex 9 (Figure 8).53 DFT calculations of oxidative addition show Ti backdonation into the thiophene π* orbital prior to bond torsion and C-S bond cleavage, similar to the reverse C-N bond formation (Figure 7). Photoexcitation of 10 results in a ligand-to-metal charge transfer, which triggers electrocyclic ring-closure and regeneration of thiophene and 9. Unlike pyrrole/py3TiCl2(NPh) catalysis where reductive elimination is thermodynamically favored, thiophene oxidative addition is favored with 9. This difference could be a result of ligand effects (electron-rich and bulky in 9, electron-deficient in py3TiCl2(NPh)), or a reflection of the aromatic stabilization of pyrrole relative to thiophene.
Figure 8.

Reversible oxidative addition/reductive elimination of thiophene to a masked TiII complex.
CATALYST DEVELOPMENT
Discovery of Catalysts.
Given the initial substoichiometric reactions with 2 (Figure 3) and the relationship of the [2+2+1] reaction with alkyne hydroamination, we tested many highly active hydroamination catalysts for [2+2+1] reactivity. Disappointingly, most complexes exhibited little or no catalytic activity, although several stoichiometrically generate pyrrole and cyclotrimerize alkynes.54
Instead, simple electron-deficient Ti halides55 remain the most effective catalysts (Figure 9A), with more electron-deficient56 13 catalyzing the formation of 1j the fastest compared to 12 and 11 (this trend holds across most alkynes, although 13 competitively trimerizes terminal alkynes). The rationale for this activity is that rate-determining alkyne binding and insertion into TS3 (Figure 4) relies on dative donation of the alkyne π-bond to Ti, which will be stronger for electron-deficient complexes. Further, TS3 is sterically encumbered (forming IM6 from IM4 requires 3 fac-binding sites), so lower-coordinate complexes insert more readily—for example, removal of excess pyridine (14 vs 11) significantly increases reaction rate, indicating strong pyridine inhibition.
Figure 9.

Halide and pyridine effects on the rate of [2+2+1] pyrrole formation. Bottom: in situ catalysts from air-stable TiCl4(THF)2 provide a convenient entrypoint into catalysis.
ETM complexes are often unfairly disregarded as impractical owing to their purported air sensitivity. However, Ti halide imido catalysts can easily be made in situ from the air-stable precursor TiCl4(THF)2 through reduction by Zn powder (Figure 9B).57 Here, single-electron reduction to TiIII is enough to generate a Ti imido through disproportionation with azobenzene. This in situ protocol can be used to carry out virtually all of the reactions reported in this Account on the benchtop, typically with only mild impacts on yield.
Regioselective Catalyst Design.
A limitation of initial [2+2+1] pyrrole syntheses was that reactions with unsymmetrical alkynes (e.g. 1e-1h, Figure 2B) were not regioselective. Thus, regiocontrol was an initial goal of catalyst development. Early challenges in rational design led us to attempt higher-throughput reaction screens from in situ reaction of [TiCl2(NPh)]n58 with L donors, forming “LnTiCl2(NPh)” (Figure 10A). From these in situ studies and follow-ups with pre-formed “LnTiCl2(NPh),” it appeared that substituted pyridine ligands could have subtle effects on the regioselectivity of phenylpropyne homocoupling—for example, 2,6-lutidine (17) was moderately selective (~2:1 against other regioisomers) for 2,5-Me2-1,3,4-Ph3-1H-pyrrole 1f” (Figure 10B).
Figure 10.

Design of regioselective catalysts for [2+2+1] pyrrole synthesis from phenylpropyne.
Using a new computational/statistical prediction tool, iterative supervised principal component analysis (ISPCA),59 led to the prediction (and experimental confirmation) that complexes of sterically encumbered electron-rich pyridines (18) would be highly selective for 1f”. This selectivity was rationalized as a result of (1) steric control of 2nd alkyne insertion (ΔΔG‡ (TS3-TS3”) = 3.4–3.8 kcal/mol) imparted by the pyridine ligand; and (2) strongly donating pyridines disfavoring the dissociation equilibria to non-ligated (nonselective) species (Figure 10C).
Other Catalysts.
Tsurugi and Mashima found that V-based precatalysts, in combination with PhN(SiMe3)2 19 (used to make M=NPh in situ) are competent for [2+2+1] coupling (Figure 11A).60 Other metals (NbCl5, TaCl5, MoCl5, WCl6) in combination with 19 also yielded pyrrole with lower yields. These observations indicate that catalytic oxidative amination manifolds may be broadly accessible with ETMs.61
Figure 11.

V-catalyzed [2+2+1] pyrrole synthesis from alkynes and azobenzene.
The VCl3(THF)3/PhN(SiMe3)2 scope (Figure 11B) is similar to that catalyzed by [py2TiCl2(NPh)]2, although unsymmetrical alkynes (1f) had different selectivities (notably, no formation of 1f’). Additionally, there were several mechanistic differences (Figure 11C): first, the active species is a bis(imido) IM12.62 Second, the rate law is 2nd order in [VCl3(THF)3] and 1st order in [5-decyne] and [azobenzene], indicating that bimetallic reoxidation of IM11 (VIII) to IM12 (VV) is kinetically relevant and that IM11 may be the resting state, consistent with the higher oxidation potential of VIII compared to TiII.
MULTICOMPONENT REACTIONS WITH ALKYNES
A key observation from mechanistic studies of Ti-catalyzed pyrrole synthesis2 was that alkyne engagement occurred in 2 distinct steps, opening the door for potential multicomponent reactions through selective reactivity at each step. Accordingly, significant progress has been made in multicomponent oxidative aminofunctionalization of alkynes, where the azatitanacyclobutene [2+2] cycloadduct can be intercepted by a variety of other substrates.
[2+2+1] Alkyne Heterocouplings: Functionalized Pyrrole Synthesis.
In an effort to further exert regiocontrol over [2+2+1] pyrrole synthesis, we envisioned that heterocoupling of two stereoelectronically differentiated alkynes may lead to better regioselectivity, assuming the chemoselectivity of alkyne coupling could be controlled.
We had earlier found that 1-phenyl-2-trimethylsilylacetylene (20a) was unreactive in [2+2+1] pyrrole synthesis, as it is incapable of facile [2+2] cycloaddition with py3TiCl2(NPh). Thus, TMS-substituted alkynes were targeted as selective 2nd insertion partners in a [2+2+1] heterocoupling manifold—and found to be remarkably efficient and selective alkynes for cross-selective pyrrole formation (Figure 12A).3 This reaction was demonstrated with many TMS-substituted alkynes, including aryl (e.g. 21a, 21b), conjugated (21c), alkyl (21d), coupled to PhCCMe and other internal alkynes (e.g. 21e-g) (Figure 12B). This modularity allows for direct synthesis of regioisomeric pyrroles such as 21b and 21f. Additional selective heterocouplings have subsequently been reported with boryl and stannyl alkynes, yielding 2-boryl- or 2-stannyl-substituted pyrroles.63
Figure 12.

Chemoselective [2+2+1] alkyne heterocouplings with TMS-substituted alkynes. Ln = pyCl2.
Regiocontrol for both the TMS-substituted alkyne and the partner alkyne is a function of electronic demands. In [2+2] cycloaddition, TS2/IM5 better stabilize building ∂+ on the C-CH3 rather than on C-Ph in TS2’/IM5’ (ΔΔG‡ = −1.5 kcal/mol, ΔG = −4.7 kcal/mol) (Figure 12C). For 2nd alkyne insertion, the silyl group can hyperconjugate to stabilize partial positive charge buildup64 on Cβ during TS6, and the resultant Ti-Cα-SiMe3 bond is stronger than the alternative Ti-Cα-R bond. Similar effects have been observed in other group 4 insertions65,66 and have been used to affect regioselective reductive couplings.67
The chemoselectivity of 2nd alkyne insertion is driven by alkyne coordination. Here, electron-rich alkynes such as TMS-protected alkynes will bind more strongly to TiIV, since there is no possibility for backdonation from TiIV. A bonus effect is that TMS-alkyne reactions are significantly faster (t ≤ 1.5 h), and run at lower temperatures (≤ 90 °C) compared to homocoupling1 of alkynes. These advantages are also an effect of the electron-rich TMS-protected alkyne: since 2nd insertion is rate-limiting, facile alkyne coordination likely leads to faster insertion.
During investigation of the alkyne heterocoupling scope, 2-pyridyl pyrrole 21h’ was found to be the major product from 20h, rather than the expected 3-substituted 21h. This is a result of pyridine coordination (Figure 13, TS6’), which directs the TMS-alkyne insertion to occur with opposite regioselectivity. We next developed methods to exploit this directing effect, demonstrating directed insertion with 9 functional groups.68 The directing effect can be tuned by changing the catalyst: while 2-py-substituted 20h results in high 21h’ selectivity with [py2TiCl2(NPh)]2, the weaker Lewis base o-OMe 20i is completely unselective. However, moving to the more Lewis acidic catalyst (THF)3TiI2(NPh) results in highly 2-selective coupling of 20i owing to the stronger Lewis acid-base interaction. On the other hand, more Lewis basic 20h results in no catalysis with (THF)3TiI2(NPh) due to inhibition by pyridine coordination.
Figure 13.

Directing group effects can invert regioselectivity in [2+2+1] alkyne heterocoupling reactions. Structure of 21 shown in Figure 12.
Installation of silyl, boryl, and stannyl groups onto the pyrrole provides a platform for post-functionalizations such as electrophilic aromatic substitutions (22) or cross-coupling reactions (25) (Figure 14A/B). 21 and 24 are hydrolytically unstable and must be further transformed in situ, while stable stannyl pyrroles can be isolated. These methods can be telescoped into multistep syntheses, as demonstrated in a formal synthesis of Lamellarin R3,69 (26) and the synthesis of a 1,5-biaryl pyrrole EP1 receptor antagonist68,70 27 (Figure 14C).
Figure 14.

Heteroatom-substituted pyrroles provide convenient handles for further functionalization and can be used in synthesis.
Ultimately, Ti-catalyzed [2+2+1] protocols are extremely modular/flexible strategies for the synthesis of highly substituted and/or electron-rich N-aryl pyrroles. They are complementary to condensation (Paal-Knorr, Lewis acid-catalyzed, etc.) or cycloisomerization strategies, each of which typically perform best for electron-deficient pyrroles (e.g. acyl-substituted) and often have specific regiochemical limitations.71 A current limitation of Ti-catalyzed protocols is the inability to access N-protected or N-H pyrroles arising from several factors: (1) incompatibility of the Ti complexes with oxygenated functional groups (Ts, CO2R, etc.); (2) thermally-triggered radical decomposition of R-NN-R (R ≠ Ar) diazenes; and (3) substitution reactivity of Ti-X with R3SiN3 that generates Ti-N3 complexes. However, with intentional catalyst and methods development it should be possible to overcome these challenges in the future.
Alkyne + Alkene Coupling: Oxidative Alkyne Carboamination.
Unlike alkynes, most alkenes do not readily undergo [2+2] cycloaddition with Ti imidos. Thus, alkenes can also potentially be incorporated into multicomponent oxidative amination reactions through selective 2nd insertion of the alkene. Reactions of tethered enynes (28) catalyzed by [py2TiCl2(NPh)]2 resulted in the formation of either α,β-unsaturated imines (29) or cyclopropylimines (30), the products of oxidative alkyne carboamination (Figure 15A).72 Both products are formed from azatitanacyclohexadiene IM15 (Figure 15C) which then undergoes either (1) pericyclic Cα-Cɣ reductive coupling (TS7) to form IM16 (then 30) or (2) β-H elimination to IM17, followed by reductive elimination to form 29. C-N reductive elimination to 2,3-dihydropyrroles is not observed, presumably because Cα-Cβ saturation in IM15 prevents electrocyclic ring closure.
Figure 15.

Ti-catalyzed oxidative carboamination of alkynes with alkenes and azobenzene, along with mechanism showing divergent pathways to 29 and 30. Regioisomeric ratios (29:30) reported in parentheses. Purple β-H in 28 tracked throughout for clarity.
Product selectivity (29:30) is very sensitive to the starting enyne structure (Figure 15B). For example, propylene-linked 28a is selective for α,β-unsaturated imine 29a (85:15), while butylene-linked 28d is unselective (49:51). β-deuterated 28b is less selective than 28a (53:47) as the kinetic isotope effect makes β-D elimination less favorable. β-Me substituted 28e, which cannot undergo β-H elimination, yields exclusively 30e. Internal tethered enynes 28c and 28f yield exclusively α,β-unsaturated 29c and 29f.
The mechanism for IM15 formation is similar to previous examples.2 However, the mechanism for β-H elimination and reductive elimination from IM17 reveals interesting new reactivity. Computational analysis by Wang73 indicates that reductive elimination from IM17 likely occurs via reinsertion of Ti-H into either Cα (IM18) followed by associative interchange with azobenzene to liberate 29 and regenerate IM4.
Evidence for a Ti-H reinsertion mechanism can be seen in intermolecular reactions (Figure 16A). Oxidative carboamination of 3-hexyne or 2-butyne with allylanisole (31) and azobenzene results in the formation of unconjugated β,ɣ-unsaturated imine 32. Since 14 is no longer bicyclic and thus has more rotational degrees of freedom, Ti-H insertion into the remote Cɣ position can occur, leading to IM18’ (Figure 16B). Unfortunately, product selectivity in Ti-catalyzed oxidative carboamination remains under subtle substrate control and the specific control elements remain unclear—for example, switching from 2-butyne to 3-hexyne completely inverts the selectivity (15:85 to 71:29) in reactions with 31.
Figure 16.

Intermolecular carboamination leads to β,ɣ-unsaturated imine 32 instead of α,β-unsaturated 29 resulting from differential Ti-H reinsertion in IM17. PMP = para-methoxyl-phenyl.
Related redox-neutral group 4-catalyzed alkyne carboaminations were reported by Bergman74 and Mindiola75 (Figure 17). In these examples, insertion of diaryl aldimines (33) into Ti/Zr azametallacyclobutenes (IM19) led to diazametallacyclohexenes (IM20), which undergo retro-[4+2] sequences to liberate α,β-unsaturated imines 34 and regenerate the Ti/Zr imido. Although limited to aryl aldimines and alkynes, further investigation of this reaction class is warranted given increased interest in carboamination catalysis across the periodic table.76–78
Figure 17.

Examples of group 4 imido-catalyzed alkyne carboamination via a redox-neutral mechanism.
Alkyne + Nitrile Coupling: Pyrazole Synthesis.
Pyrazoles are pharmaceutically important heterocycles that are commonly made via Knorr-like condensation of hydrazines onto 1,3-diketones, or via 1,3-dipolar cycloaddition of hydrazones with alkynes. Regioselectivity issues in these reactions, along with potential safety concerns around hydrazines, provide a compelling rationale to develop alternative synthetic routes.
Nitriles were explored as potential 2nd insertion partners in an effort to extend Ti redox catalysis to pyrazole synthesis. Livinghouse previously demonstrated intermolecular capture of an azatitanacyclobutene (generated by intramolecular [2+2] cycloaddition of 37) by isobutyronitrile, leading to diazatitanacyclohexadiene IM21 (Figure 18A).35 Thus, the basic coupling framework for [2+2+1] pyrazole synthesis was already in place, although this reaction would require N-N reductive elimination from the diazatitanacycle. Examples of organometallic N-N bond-forming methods are extremely limited;79,80 however, since C-N reductive elimination in the related pyrrole synthesis occurs via electrocyclization, we hypothesized that a similar mechanism could allow access to this unusual N-N coupling.
Figure 18.

A: precedent for stoichiometric isobutyronitrile insertion into [2+2] cycloadducts. B: multicomponent pyrazole synthesis via oxidation-induced N-N coupling.
Unfortunately, attempts at Ti-imido-catalyzed [2+2+1] pyrazole synthesis have mostly failed. Diazatitanacycle IM22 (Figure 18B) is too stable to undergo facile reductive elimination; compared to IM6, the introduction of a 2nd strong Ti-N bond in IM22 results in significant stabilization.
However, the stability of diazatitanacycle IM22 meant that they could be synthesized in situ (or isolated) from alkynes, nitriles, and imidos and subjected to further reactivity. Although IM22 is TiIV, we hypothesized that ligand-based oxidation may promote electrocyclic N-N bond formation. Several oxidants provide facile N-N coupling of IM22, with 2 equiv. TEMPO performing best. This protocol was then developed into a 1-pot, 2-step multicomponent cyclization/oxidation sequence for the synthesis of a diverse array of pyrazoles (39) (Figure 18B).4
Pyrazole formation requires 2 oxidation equivalents for full conversion, although N-N coupling could occur through three oxidation states: the “default” oxidation state IM22, 1-electron oxidized IM23, or 2-electron oxidized IM24 (Figure 19A) Previously, we had shown that TiII synthons could ring-open 2H-azirines,81 and thus hypothesized that ring-opening of 2-imino-2H-azirine 40 (isomer of pyrazole 41) by TiII, TiIII, or TiIV would lead to diazatitanacycles with oxidation states analogous to IM22, IM23, or IM24, respectively, such that we could probe the oxidation state coupling question. Reaction of 40 with Cp2Ti(BTMSA) led to stable IM22, reaction with TiCl4 led to full conversion to 40, and reaction with TiCl3(THF)3 led to a 50:50 mixture of 40 and IM22 (Figure 19B).4 These stoichiometric reactions indicate that N-N coupling occurs via IM24, which can be accessed either through direct 2-electron oxidation or via redox disproportionation of IM23 (likely the case for TEMPO).
Figure 19.

2H-azirine ring-opening with various oxidation states of Ti reveals that N-N coupling occurs through a 2e-oxidized species IM24, although 1-electron oxidized IM23 can also disproportionate into IM24.
Alkyne Diimination.
Since IM22 underwent electrocyclic N-N coupling upon oxidation, we next aimed to explore the potential for IM22 to undergo other cycloaddition reactions. We envisioned that IM22 could serve as an electron-rich diene in [4+2] cycloaddition reactions with polar dienophiles.82 Reactions of C-nitrosos (43) with preformed diazatitanacycle 42 were explored, resulting in rapid, near-quantitative formation of α-diimines 44, along with expulsion of p-tolunitrile (Figure 20A). Interestingly, the yields of α-diimines are consistent irrespective of C-nitroso substitution—perhaps a reflection of the strong driving force of Ti-O formation.
Figure 20.

Development of stoichiometric Ti- and nitrile-mediated alkyne α-diimination.
Based on these results, a 2-step, 1-pot oxidative alkyne diimination method was developed (Figure 20B).82 As in the multicomponent pyrazole syntheses, the α-diimine yields are determined by the efficiency of forming diazatitanacycle IM25 and use of excess MeCN as the nitrile component significantly improves metallacycle formation. This alkyne diimination provides facile and modular access to fully unsymmetric α-diimines (45), which are often impossible to make via stepwise condensation (which is reversible). Regioisomeric series of α-diimines can be easily constructed by swapping substituents on the various components, for example in 45a-45c (Figure 20C).
Computational analysis indicates that this reaction proceeds via [4+2] cycloaddition of the C-nitroso across IM25 to form IM26, followed by a retro-[4+2] cycloaddition that expels nitrile and produces IM27 (Figure 20B). From IM27, N-O bond cleavage via α-N elimination (IM28) generates a diimine-coordinated Ti oxo species which liberates 45.
This two-step reaction sequence is required for productive α-diimine formation because C-nitrosos undergo direct [2+2] cycloaddition with Ti≡NR to produce Ti=O and RN=NR. Thus, the nitrile component serves as a promoter in the reaction, first forming the key intermediate IM25 and then being expelled prior to product formation.
OTHER OXIDATIVE AMINATION REACTIONS
Although group 4-catalyzed oxidative amination reactions have primarily been explored with alkynes, other types of reactive functional groups are also capable of undergoing catalytic oxidation. There is a wealth of examples of stoichiometric reactions of unsaturated functional groups with group 4 metal imido complexes, which serve as the inspiration for development of new catalytic protocols.
Ring-Opening Oxidative Amination of Methylenecyclopropanes.
Strained exocyclic double bonds, like those in methylenecyclopropanes (MCPs), undergo [2+2] cycloadditions with Ti imidos. Eisen demonstrated ring-opening hydroamination83,84 of 2-aryl MCPs (46), suggesting that ring strain confers “sp-like” character onto the alkene (Figure 21A) and promotes cycloaddition. After [2+2] cycloaddition, β-C elimination and aminolysis generates 47.
Figure 21.

Hydroamination and oxidative amination of methylene cyclopropanes (MCPs). Isolated yields. PMP = para-methoxyl-phenyl.
Ti-catalyzed ring-opening oxidative aminations of MCPs (48) with PhNNPh are also possible (Figure 21B), yielding unusual α-methylene imines (49) or cyclic α,β-unsaturated imines (50).85 These reactions follow a similar mechanism to MCP hydroamination, however here IM30 undergoes β-H elimination to IM31 (Figure 21C). From IM31, hydride re-insertion (similar in catalytic alkyne carboamination, Figure 16B) results in formation of the backbonded α,β-unsaturated imine intermediate IM32, which can undergo associative exchange with azobenzene to close the catalytic cycle. The regioselectivity of ring-opening is opposite that observed in Eisen’s hydroamination83,84 and is a substituent effect: aryl MCPs undergo scission between Caryl and C=CH2, while alkyl MCPs undergo scission between Cunsubst and C=CH2.
Nitrene Isocyanation and Carbonylation.
In a seminal example from Heyduk, redox-noninnocent86 Zr complex 51 catalyzed transfer of nitrenes from alkyl azides to isocyanides, forming unsymmetrical carbodiimides (52) (Figure 22A).87 Isocyanide insertion into group 4 metal imidos is well-known, but the resultant η2-carbodiimides are often unreactive due to strong backbonding. With 51, catalytic turnover can be achieved because the NNN ligand in IM35 can accept the pair of electrons that were previously backbonding into the η2-carbodiimide and render the carbodiimide labile. Similarly, Wolczanski reported catalytic nitrene carbonylation using a Ti complex of a redox noninnocent diamide, diimine ligand.88
Figure 22.

Examples of catalytic carbodiimide formation via isocyanide imination redox noninnocent Zr complexes (A) and Ti halide complexes (B).
We hypothesized isocyanide imination with simple Ti imidos using azobenzene or organoazides as the nitrene source may be possible (Figure 22B), since azobenzene is a strong π-acceptor and could serve the same role as Heyduk’s/Wolczanski’s redox-noninnocent ligands. A small screen of Ti imido halides showed that Cl, Br, and I derivatives (11–14, from Figure 9) were competent catalysts for imination of tBuNC with PhNNPh,89 with Br complex 53 deemed the best balance between reactivity and catalyst cost. Reactions proceeded effectively with tBuNC yielding unsymmetric 54, while 2,6-Me2-PhNC and CyNC were poorly reactive owing to product inhibition. Catalysis with these latter substrates could be accomplished by switching to a bulkier nitrene source such as AdN3. DFT calculations support an azobenzene-bound mechanism, wherein the carbodiimide product release (IM38) is triggered by electron transfer from the η2-carbodiimide to azobenzene.
CONCLUSION
In summary, this Account discusses the surprisingly versatile 2-electron redox chemistry of Ti imido complexes, driven by substrates and products that can π-backbond effectively at key states during catalysis. Fundamental mechanistic studies of Ti redox catalysis in the context of [2+2+1] pyrrole synthesis have led to new strategies for the synthesis of important 5-membered aromatic heterocycles, as well as other aminated products.
Looking forward, there still remains significant opportunity to exploit the fundamental reactivity of early transition metal complexes for redox catalysis—which, importantly, will likely have orthogonal selectivity and functional group tolerance when compared to late transition metal-catalyzed methods. A significant ongoing challenge will be in uncovering new strategies for catalytic turnover for cases where π-backbonding does not provide enough thermodynamic stabilization for metal reduction, or in cases where pericyclic reactivity may not be possible. Here, drawing inspiration from modern catalytic methods (electrochemistry, photoredox catalysis, etc.) that have not been employed frequently with early transition metals will be critical for opening new doors in catalytic oxidative reactions.
KEY REFERENCES.
Gilbert, Z. W.; Hue, R. J.; Tonks, I. A. Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nature Chemistry 2016, 8, 63–68. We report the first example catalytic oxidative nitrene transfer with Ti, leading to the formation of pyrroles.1
Davis-Gilbert, Z. W.; Wen, X.; Goodpaster, J. D.; Tonks, I. A. Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2+2+1] Pyrrole Synthesis. J. Am. Chem. Soc. 2018, 140, 7267–7281. A detailed combined experimental and theoretical study of the mechanism of Ti-catalyzed oxidative amination, revealing a new electrocyclic mechanism for C-N reductive elimination and kinetic and mechanistic details regarding the overall catalytic cycle.2
Chiu, H.-C.; Tonks, I. A. Trialkylsilyl-Protected Alkynes as Selective Cross Coupling Partners in Ti-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem. Int. Ed. 2018, 57, 6090–6094. The first example of selective multicomponent reactions of Ti imidos and alkynes with 3rd components, in this case yielding highly regioselective reactions for the formation of silyl pyrroles. These pyrroles were then used as precursors to the natural product Lamellarin R.3
Pearce, A. J.; Harkins, R. P.; Reiner, B. R.; Wotal, A. C.; Dunscomb, R. J.; Tonks, I. A. Multicomponent Pyrazole Synthesis from Alkynes, Nitriles, and Titanium Imido Complexes via Oxidatively-Induced N-N Bond Coupling. J. Am. Chem. Soc. 2020, 142, 4390. Demonstration of a useful multicomponent pyrazole synthesis through N-N coupling chemistry, taking advantage of our understanding of selective insertion into azatitanacycles and electrocyclic reductive elimination reactions.4
ACKNOWLEDGMENT
To all of the students, postdocs, and collaborators in the group over the past eight years: thank you for all of your hard work, creativity, perseverance, and teamwork. The success of all of the projects reported herein is a reflection of your collective strengths. This work has been generously supported by the University of Minnesota, the NIH (R35GM119457), and the Alfred P. Sloan Foundation.
Funding Sources
No competing financial interests have been declared.
Biographical Information
Ian A Tonks is the Lloyd H. Reyerson Professor in the Department of Chemistry at the University of Minnesota – Twin Cities, and Associate Editor for the ACS journal Organometallics. He received his B.A. from Columbia University, followed by his Ph.D. from the California Institute of Technology, and postdoctoral studies at the University of Wisconsin – Madison. His research program is focused on earth abundant catalysis and developing new strategies for CO2 utilization in polymer synthesis.
REFERENCES
- 1.Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nature Chemistry 2016, 8, 63–68. [DOI] [PubMed] [Google Scholar]
- 2.Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2+2+1] Pyrrole Synthesis. J. Am. Chem. Soc. 2018, 140, 7267–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu H-C; Tonks IA Trialkylsilyl-Protected Alkynes as Selective Cross Coupling Partners in Ti-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem. Int. Ed. 2018, 57, 6090–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearce AJ; Harkins RP; Reiner BR; Wotal AC; Dunscomb RJ; Tonks IA Multicomponent Pyrazole Synthesis from Alkynes, Nitriles, and Titanium Imido Complexes via Oxidatively-Induced N-N Bond Coupling. J. Am. Chem. Soc. 2020, 142, 4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns and Frequency of Nitrogen Heterocycles among US FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274 [DOI] [PubMed] [Google Scholar]
- 6.Ertl P; Altmann E; McKenna JM The Most Common Functional Groups in Bioactive Molecules and How Their Popularity Has Evolved over Time. J. Med. Chem. 2020, 63, 8408–8418. [DOI] [PubMed] [Google Scholar]
- 7.Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- 8.Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. [DOI] [PubMed] [Google Scholar]
- 9.Degennaro L; Trinchera P; Luisi R Recent Advances in the Stereoselective Synthesis of Aziridines. Chem. Rev. 2014, 114, 7881–7929. [DOI] [PubMed] [Google Scholar]
- 10.Park Y; Kim Y; Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- 11.Roglans A; Pla-Quintana A; Solà M Mechanistic Studies of Transition-Metal-Catalyzed [2+2+2] Cycloaddition Reactions. Chem. Rev. 2021, 121, 1894–1979. [DOI] [PubMed] [Google Scholar]
- 12.Nugent WA; Mayer JM Metal-Ligand Multiple Bonds: The Chemistry of Transition Metal Complexes Containing Oxo, Nitrido, Imido, Alkylidene, or Alkylidyne Ligands. (Wiley, 1988). [Google Scholar]
- 13.Hunt AJ; Farmer TJ; Clark JH in Element Recovery and Sustainability (ed. Hunt AJ) 1–8 (Royal Society of Chemistry, 2013). [Google Scholar]
- 14.Schafer L; Hill M; Tonks IA Organometallic Complexes of Electrophilic Elements for Selective Synthesis. Organometallics 2018, 37, 4311–4312. [Google Scholar]
- 15.McCallum T; Wu X; Lin S Recent Advances in Titanium Radical Redox Catalysis. J. Org. Chem. 2019, 84, 14369–14380. [DOI] [PubMed] [Google Scholar]
- 16.Rodríguez MC; García IR; Maecker RNR; Morales LP; Oltra JE; Martínez AR Cp2TiCl: An Ideal Reagent for Green Chemistry? Org. Process Res. Dev. 2017, 21, 911–923. [Google Scholar]
- 17.Yao C; Dahmen T; Gansäuer A; Norton J Anti-Markovnikov Alcohols via Epoxide Hydrogenation through Cooperative Catalysis. Science 2019, 364, 764–767. [DOI] [PubMed] [Google Scholar]
- 18.Gansäuer A From Enantioselective to Regiodivergent Epoxide Opening and Radical Arylation – Useful or Just Interesting? Synlett 2021, 32, 447–456. [Google Scholar]
- 19.Negishi E Controlled Carbometallation as a New Tool for Carbon-Carbon Bond Formation and its Application to Cyclization. Acc. Chem. Res. 1987, 20, 65–72. [Google Scholar]
- 20.Beaumier EP; Pearce AJ; See XY; Tonks IA Modern Applications of Low Valent Early Transition Metals in Synthesis and Catalysis. Nat. Rev. Chem. 2019, 3, 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyaskovskyy V; de Bruin B Redox Non-Innocent Ligands: Versatile New Tools to Control Catalytic Reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar]
- 22.Kawakita K; Kakiuchi Y; Tsurugi H; Mashima K; Parker B; Arnold J; Tonks IA Reactivity of Terminal Imido Complexes of Group 4–6 Metals: Stoichiometric and Catalytic Reactions Involving Cycloaddition with Unsaturated Organic Molecules. Coord. Chem. Rev. 2020, 407, 213118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tonks IA; Meier JC; Bercaw JE Alkyne Hydroamination and Trimerization with Titanium Bis(phenolate)pyridine Complexes: Evidence for Low-Valent Titanium Intermediates and Synthesis of an Ethylene Adduct of Titanium(II). Organometallics 2013, 32, 3451–3457. [Google Scholar]
- 24.Pearce AJ; See XY; Tonks IA Oxidative Nitrene Transfer from Azides to Alkynes via Ti(II)/Ti(IV) Redox Catalysis: Formal [2+2+1] Synthesis of Pyrroles. Chem. Commun. 2018, 54, 6891–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burés J Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem. Int. Ed. 2016, 55, 16084–16087. [DOI] [PubMed] [Google Scholar]
- 26.Guo J; Deng X; Song C; Lu Y; Qu S; Dang Y; Wang Z-X Differences Between the Elimination of Early and Late Transition Metals: DFT Mechanistic Insights into the Titanium-Catalyzed Synthesis of Pyrroles from Alkynes and Diazenes. Chem. Sci. 2017, 8, 2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duchateau R; Williams AJ; Gambarotta S; Chiang MY Carbon-Carbon Double-Bond Formation in the Intermolecular Acetonitrile Reductive Coupling Promoted by a Mononuclear Titanium(II) Compound. Preparation and Characterization of Two Titanium(IV) Imido Derivatives. Inorg. Chem. 1991, 30, 4863–4866. [Google Scholar]
- 28.Hill JE; Profilet RD; Fanwick PE; Rothwell IP Synthesis, Structure, and Reactivity of Aryloxo(imido)titanium Complexes. Angew. Chem. Int. Ed. 1990, 29, 664–665. [Google Scholar]
- 29.Gray SD; Thorman JL; Adamian VA; Kadish KM; Woo LK Synthesis, Electrochemistry, and Imido Transfer Reactions of (TTP)Ti(η2-PhN=NPh). Inorg. Chem. 1998, 37, 1–4. [DOI] [PubMed] [Google Scholar]
- 30.Pohlki F; Doye S The Mechanism of the [Cp2TiMe2]-Catalyzed Intermolecular Hydroamination of Alkynes. Angew. Chem. Int. Ed. 2001, 40, 2305–2308. [DOI] [PubMed] [Google Scholar]
- 31.Straub BF; Bergman RG The Mechanism of Hydroamination of Allenes, Alkynes, and Alkenes Catalyzed by Cyclopentadienyl-Imido Complexes: A Density Functional Study. Angew. Chem. Int. Ed. 2001, 40, 4632–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walsh PJ The Synthesis and Chemistry of Organozirconocene Complexes Containing Zirconium-Nitrogen Double and Single Bonds. Ph.D. Thesis, University of California – Berkeley, Berkeley, CA, 1990. [Google Scholar]
- 33.Vujkovic N; Ward BD; Maisse-François A; Wadepohl H; Mountford P; Gade LH Imido-Alkyne Coupling in Titanium Complexes: New Insights into the Alkyne Hydroamination Reaction. Organometallics 2007, 26, 5522–5534. [Google Scholar]
- 34.Lokare KS; Ciszewski JT; Odom AL Group-6 Imido Activation by a Ring-Strained Alkyne. Organometallics 2004, 23, 5386–5388. [Google Scholar]
- 35.McGrane PL; Jensen M; Livinghouse T Intramolecular [2+2] cycloadditions of Group IV metal-imido complexes. Applications to the synthesis of dihydropyrrole and tetrahydropyridine derivatives. J. Am. Chem. Soc. 1992, 114, 5459–5460. [Google Scholar]
- 36.Hanna TA; Baranger AM; Bergman RG Reactivity of Zirconocene Azametallacyclobutenes: Insertion of Aldehydes, Carbon Monoxide, and Formation of a,b-Unsaturated Imines. Formation and Trapping of [Cp2Zr=O] in a [4+2] Retrocycloaddition. J. Org. Chem. 1996, 61, 4532–4541. [DOI] [PubMed] [Google Scholar]
- 37.Ruck RT; Bergman RG Reactions of Imines with Azazirconacyclobutenes and Generation of Electron-Deficient Imidozirconocene Complexes. Organometallics 2004, 23, 2231–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odom AL; McDaniel TJ Titanium-Catalyzed Multicomponent Couplings: Efficient One-Pot Syntheses of Nitrogen Heterocycles. Acc. Chem. Res., 2015, 48, 2822–2833. [DOI] [PubMed] [Google Scholar]
- 39.Cao C; Shi Y; Odom AL A Titanium-Catalyzed 3-Component Coupling to Generate α,β-Unsaturated β-Iminoamines. J. Am. Chem. Soc 2003, 125, 2880–2881. [DOI] [PubMed] [Google Scholar]
- 40.Cloke FGN; Hitchcock PB; Nixon JF; Wilson DJ; Tabellion F; Fischbeck U; Preuss F; Regitz M Synthetic, structural and theoretical studies on new aromatic 1,2,4-azadiphosphole ring systems: crystal and molecular structure of P2C2But2NPh. Chem. Commun. 1999, 2363–2364. [Google Scholar]
- 41.Negishi E; Takahashi T Patterns of Stoichiometric and Catalytic Reactions of Organozirconium and Related Complexes of Synthetic Interest. Acc. Chem. Res. 1994, 27, 124–130. [Google Scholar]
- 42.Siebeneicher H; Doye S Dimethyltitanocene Cp2TiMe2: A Useful Reagent for C–C and C–N Bond Formation J. Prakt. Chem. 2000, 342, 102–106. [Google Scholar]
- 43.Meindl K; Henn J; Kocher N; Leusser D; Zachariasse KA; Sheldrick GM; Koritsansky T; Stalke D Experimental Charge Density Studies of Disordered N-Phenylpyrrole and N-(4-Fluorophenyl)pyrrole. J. Phys. Chem. A 2009, 113, 9684–9691. [DOI] [PubMed] [Google Scholar]
- 44.Thorn MG; Hill JE; Waratuke SA; Johnson ES; Fanwick PE; Rothwell IP J. Am. Chem. Soc. 1997, 119, 8630–8641. [Google Scholar]
- 45.Rosenthal U Update for Reactions of Group 4 Metallocene Bis(trimethylsilyl)acetylene Complexes: A Never-Ending Story? Organometallics 2020, 39, 4403–4414. [Google Scholar]
- 46.Knizia G; Klein JEMN Electron flow in reaction mechanisms --- revealed from first principles Angew. Chem. Int. Ed. 2015, 54, 5518–5522. [DOI] [PubMed] [Google Scholar]
- 47.This mechanism may be involved in other pyrrole syn-theses. See: Matsui K; Shibuya M; Yamamoto Y Synthesis of pyrroles via ruthenium-catalyzed nitrogen-transfer [2 + 2+1] cycloaddition of α,ω-diynes using sulfoximines as nitrene surrogates. Commun. Chem. 2018, 1, 21.Haut F-L; Feichtinger NJ; Plangger I; Wein LA; Müller M; Streit T-N; Wurst K; Podewitz M; Magauer T Synthesis of Pyrroles via Consecutive 6π-Electrocyclization/Ring Contraction of Sulfilimines. J. Am. Chem. Soc. 2021. 143, 9002–9008.
- 48.Vinogradov MG; Turova OV; Zlotin SG Nazarov reaction: current trends and recent advances in the synthesis of natural compounds and their analogs. Org. Biomol. Chem. 2017, 15, 8245–8269. [DOI] [PubMed] [Google Scholar]
- 49.Dieker J; Frölich R; Würthwein E-U Substituted 3-Hydroxypyrroles from 1-Azapenta-1,4-dien-3-ones: The Aza-Nazarov Reaction – Synthesis and Quantum Chemical Calculations. Eur. J. Org. Chem. 2006, 5339–5356. [Google Scholar]
- 50.Cusumano AQ; Goddard III WA; Stoltz BM The Transition Metal Catalyzed [π2s + π2s + σ2s + σ2s] Pericyclic Reaction: Woodward–Hoffmann Rules, Aromaticity, and Electron Flow. J. Am. Chem. Soc. 2020, 142, 19033–19039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reichl KD; Dunn NL; Fastuca NJ; Radosevich AT Biphilic Organophosphorus Catalysis: Regioselective Reductive Transposition of Allylic Bromides via PIII/PV Redox Cycling. J. Am. Chem. Soc. 2015, 137, 5292–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lv Z-J; Chai Z; Zhu M; Wei J; Zhang W-X Selective Coupling of Lanthanide Metallacyclopropenes and Nitriles via Azametallacyclopentadiene and η2-Pyrimidine Metallacycle. J. Am. Chem. Soc. 2021, 143, 9151–9161. [DOI] [PubMed] [Google Scholar]
- 53.Gómez-Torres A; Aguilar-Calderón JR; Saucedo C; Jordan A; Metta-Magaña A; Pinter B; Fortier S Reversible oxidative-addition and reductive elimination of thiophene from a titanium complex and its thermally-induced hydrodesulphurization chemistry. Chem. Commun. 2020, 56, 1545–1548. [DOI] [PubMed] [Google Scholar]
- 54.See XY; Beaumier EP; Davis-Gilbert ZW; Dunn PL; Pearce AJ; Wheeler TA; Tonks IA Generation of TiII Alkyne Trimerization Catalysts in the Absence of Strong Metal Reductants. Organometallics 2017, 36, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ironically, we were initially using these complexes as synthons to make more complex Ti imidos. See:Adams N; Bigmore HR; Blundell TL; Boyd CL; Dubberley SR; Sealey AJ; Cowley AR; Skinner MEG; Mountford P New Titanium Imido Synthons: Syntheses and Supramolecular Structures. Inorg. Chem. 2005, 44, 2882–2894.
- 56.DiFranco SA; Maciulis NA; Staples RJ; Batrice RJ; Odom AL Evaluation of Donor and Steric Properties of Anionic Ligands on High Valent Transition Metals. Inorg. Chem. 2012, 51, 1187–1200. [DOI] [PubMed] [Google Scholar]
- 57.Davis-Gilbert ZW; Kawakita K; Blechschmidt DC; Tsurugi H; Mashima K; Tonks IA In Situ Catalyst Generation and Benchtop-Compatible Entrypoints for Ti-Catalyzed Nitrene Transfer Reactions. Organometallics 2018, 37, 4439–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pennington DA; Horton PN; Hursthouse MB; Bochmann M; Lancaster SJ Synthesis and catalytic activity of dinuclear imido titanium complexes: the molecular structure of [Ti(NPh)Cl(μ-Cl)(THF)2]2. Polyhedron 2005, 24, 151–156. [Google Scholar]
- 59.See XY; Wen X; Wheeler TA; Klein CK; Goodpaster JD; Reiner BR; Tonks IA Iterative Principal Component Analysis-Driven Ligand Design for Regioselective Ti-Catalyzed [2+2+1] Pyrrole Synthesis. ACS Catalysis 2020, 10, 13504–13517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kawakita K; Beaumier EP; Kakiuichi Y; Tsurugi H; Tonks IA; Mashima K Bis(imido)vanadium(V)-Catalyzed [2+2+1] Coupling of Alkynes and Azobenzenes Giving Multi-Substituted Pyrroles. J. Am. Chem. Soc. 2019, 141, 4194–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang W; Nakajima K; Nishibayashi Y Synthesis of 1,2,4-azadiphosphole derivatives based on vanadium-catalyzed [2+2+1] cycloaddition reactions of azobenzenes with phosphaalkynes. RSC Adv. 2020, 10, 12730–12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Group 5 imido complexes often require a 2nd strong π donor to be activated toward cycloaddition. See: de With J; Horton AD Orpen AG Unusual [2 + 2] cycloaddition adducts of an imidovanadium complex with alkynes and ethene: conversion to .eta.3–1-azaallyl and ethenyl complexes. Organometallics 1993, 12, 1493–1496.
- 63.Cheng Y; Klein CK; Tonks IA Synthesis of Pentasubstituted 2-Aryl Pyrroles from Boryl and Stannyl Alkynes via One-Pot Sequential Ti-Catalyzed [2+2+1] Pyrrole Synthesis/Cross Coupling Reactions. Chem. Sci. 2020, 11, 10236–10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dallaire C; Brook MA The .beta.-effect with vinyl cations: kinetic study of the protiodemetalation of silyl-, germyl-, and stannylalkynes. Organometallics 1993, 12, 2332–2338. [Google Scholar]
- 65.Lefeber C; Ohff A; Tillack A; Baumann W; Kempe R; Burlakov VV; Rosenthal U Representation and regioselective reactions of the phosphine-free zirconocene-alkyne complex Cp2Zr(THF)(tBuC2 SiMe 3). J. Organomet. Chem. 1995, 501, 189–194. [Google Scholar]
- 66.List AK; Koo Kwangmo, Reingold AL; Hillhouse GL Preparation of trimethylsilyl alkyne complexes of bis(pentamethylcyclopentadienyl)zirconium, (η5-C5Me5)2Zr(RC≡CSiMe3), and their regioselective reactions with nitrous oxide. Inorg. Chim. Acta. 1998, 270, 399–404. [Google Scholar]
- 67.Micalizio GC; Mizoguchi H The Development of Alkoxide-Directed Metallacycle-Mediated Annulative Cross-Coupling Chemistry. Isr. J. Chem. 2017, 57, 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiu H-C; See XY; Tonks IA Dative Directing Group Effects in Ti-Catalyzed [2+2+1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catal. 2019, 9, 216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Imbri D; Tauber J; Opatz T Synthetic Approaches to the Lamellarins—A Comprehensive Review. Mar. Drugs 2014, 12, 6142–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hall A; Atkinson S; Brown SH; Chessell IP; Chowdhury A; Clayton NM; Coleman T; Giblin GMP; Gleave RJ; Hammond B; Healy MP; Johnson MR; Michel AD; Naylor A; Novelli R; Spalding DJ; Tang SP Structure−Activity Relationships of 1,5-Biaryl Pyrroles as EP1 Receptor Antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3657–3662. [DOI] [PubMed] [Google Scholar]
- 71.Gulevich AV; Dudnik AS; Chernyak N; Gevorgyan V Transition Metal-Mediated Synthesis of Monocyclic Aromatic Heterocycles. Chem. Rev. 2013, 113, 3084–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davis-Gilbert ZW; Yao LJ; Tonks IA Ti-catalyzed Multicomponent Oxidative Carboamination of Alkynes with Alkenes and Diazenes. J. Am. Chem. Soc. 2016. 138, 14570–14573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo J; Lu Y; Zhao R; Liu Z; Menberu W; Wang Z-X Strong Preference of the Redox-Neutral Mechanism over the Redox Mechanism for the TiIV Catalysis Involved in the Carboamination of Alkyne with Alkene and Diazene. Chem. Eur. J. 2018, 24, 7010–7025. [DOI] [PubMed] [Google Scholar]
- 74.Ruck RT; Zuckerman RL; Krska SW; Bergman RG Carboamination: Additions of Imine C=N Bonds Across Alkynes Catalyzed by Imidozirconium Complexes. Angew. Chem. Int. Ed. 2004, 43, 5372–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basuli F; Aneetha H; Huffman JC; Mindiola DJ A Fluorobenzene Adduct of Ti(IV), and Catalytic Carboamination to Prepare α,β-Unsaturated Imines and Triaryl-Substituted Quinolines. J. Am. Chem. Soc. 2005, 127, 17992–17993. [DOI] [PubMed] [Google Scholar]
- 76.Piou T; Rovis T Rhodium-catalysed syn-carboamination of alkenes via a transient directing group. Nature 2015, 527, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation, J. Am. Chem. Soc. 2017, 139, 11261–11270. [DOI] [PubMed] [Google Scholar]
- 78.Gockel SN; Buchanan TL; Hull KL Cu-Catalyzed Three-Component Carboamination of Alkenes. J. Am. Chem. Soc. 2018, 140, 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neumann JJ; Suri M; Glorius F Efficient Synthesis of Pyrazoles: Oxidative C-C/N-N Bond-Formation Cascade. Angew. Chem., Int. Ed. 2010, 49, 7790–7794. [DOI] [PubMed] [Google Scholar]
- 80.Diccianni JB; Hu C; Diao T N-N Bond Forming Reductive Elimination via a Mixed-Valent Nickel(II)-Nickel(III) Intermediate. Angew. Chem., Int. Ed. 2016, 55, 7534–7538. [DOI] [PubMed] [Google Scholar]
- 81.Desnoyer AN; See XY; Tonks IA Diverse Reactivity of Diazatitanacyclohexenes: Coupling Reactions of 2H-Azirines Mediated by Titanium(II). Organometallics 2018, 37, 4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frye CW; Egger DT; Kounalis E; Pearce AJ; Cheng Y; Tonks IA α-Diimine Synthesis via Titanium-Mediated Multicomponent Diimination of Alkynes with C-Nitrosos. ChemRxiv 2021, DOI: 10.33774/chemrxiv-2021-dmdqr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smolensky E; Kapon M; Eisen MS Catalytic Intermolecular Hydroamination of Methylenecyclopropanes. Organometallics 2005, 24, 5495–5498. [Google Scholar]
- 84.Smolensky E; Kapon M; Eisen MS Intermolecular Hydroamination of Methylenecyclopropane Catalyzed by Group IV Metal Complexes. Organometallics 2007, 26, 4510–4527. [Google Scholar]
- 85.Beaumier EP; Ott AA; Wen X; Davis-Gilbert ZW; Wheeler TA; Topczewski JJ; Goodpaster JD; Tonks IA Ring-Opening Oxidative Amination of Methylenecyclopropanes with Diazenes via TiII/TiIV Redox Catalysis. Chem. Sci. 2020, 11, 7204–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Munhá RF; Zarkesh RA; Heyduk AF Group transfer reactions of d0 transition metal complexes: redox-active ligands provide a mechanism for expanded reactivity. Dalton Trans. 2013, 42, 3751–3766. [DOI] [PubMed] [Google Scholar]
- 87.Nguyen AI; Zarkesh RA; Lacy DC; Thorson MK; Heyduk AF Catalytic nitrene transfer by a zirconium(iv) redox-active ligand complex. Chem. Sci. 2011, 2, 166–169. [Google Scholar]
- 88.Heins SP; Wolczanski PT; Cundari TR; MacMillan SN Redox non-innocence permits catalytic nitrene carbonylation by (dadi)Ti=NAd (Ad = adamantyl). Chem. Sci. 2017, 8, 3410–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beaumier EP; McGreal M; Pancoast AR; Wilson RH; Moore JT; Graziano BJ; Goodpaster JD; Tonks IA Carbodiimide Synthesis via Ti-Catalyzed Nitrene Transfer from Diazenes to Isocyanides. ACS Catal. 2019, 9, 11753–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]