

Abstract

Chronic wasting disease (CWD) is a prion disease of cervids (deer, elk, moose, etc.). It spreads readily from CWD-contaminated environments and among wild cervids. As of 2022, North American CWD has been found in 29 states, four Canadian provinces and South Korea. The Scandinavian form of CWD originated independently. Prions propagate their pathology by inducing a natively expressed prion protein (PrPC) to adopt the prion conformation (PrPSc). PrPC and PrPSc differ solely in their conformation. Like other prion diseases, transmissible CWD prions can arise spontaneously. The CWD prions can respond to selection pressures resulting in the emergence of new strain phenotypes. Annually, 11.5 million Americans hunt and harvest nearly 6 million deer, indicating that CWD is a potential threat to an important American food source. No tested CWD strain has been shown to be zoonotic. However, this may not be true for emerging strains. Should a zoonotic CWD strain emerge, it could adversely impact the hunting economy and game meat consumers.
Introduction
Approximately 11.5 million Americans hunt deer annually.1 They support a 30 billion (USD)/year industry that results in an annual harvest of nearly 6 million cervids. This harvest yields more than 250 000 tons of meat per year, accounting for 2.5% of the meat consumed in the United States.2 Unlike meat procured from commercial sources, meat harvested from wild game is not inspected by regulatory agencies. It is presumed that hunters can determine if the animal is fit for consumption by inspection.3 By contrast, commercially available meat from farmed cervids is inspected prior to sale. The 21st century saw the dramatic expansion of a disease among wild cervids that may significantly impact food consumed by millions of Americans.
Chronic wasting disease (CWD) is a transmissible spongiform encephalopathy (TSE) of cervids (e.g., elk, deer, and moose).4 CWD is caused by a pathological protein called a prion.5 CWD-infected animals appear to be healthy throughout most of the disease course, even though they are afflicted with a prion disease that will eventually kill them. It is difficult to identify a CWD-infected animal solely by observation, as clinical signs are often subtle and only apparent for a comparatively short time as the disease course approaches its fatal conclusion. Unlike some of the other prion diseases, CWD prions are distributed throughout an infected cervid’s tissues. This means that CWD prions are present in the meat consumed by a hunter and a hunter’s family, friends, and neighbors. North American cervids are large animals; an adult moose can weigh 1500 pounds, an adult elk more than 1100 pounds, and an adult deer 200 or more pounds (http://www.montana4h.org/documents/resources/outdoor_educatoin/Big%20Game%20Booklet%20-%202016.pdf). This abundance of meat suggests it is likely to be widely distributed among people in or associated with the hunting community. Thus, this formerly obscure disease of wild and farmed cervids can have a substantial impact on American consumers. The potential impact of CWD extends far beyond the hunting community and the cervid farming industry (Figure 1).
Figure 1.
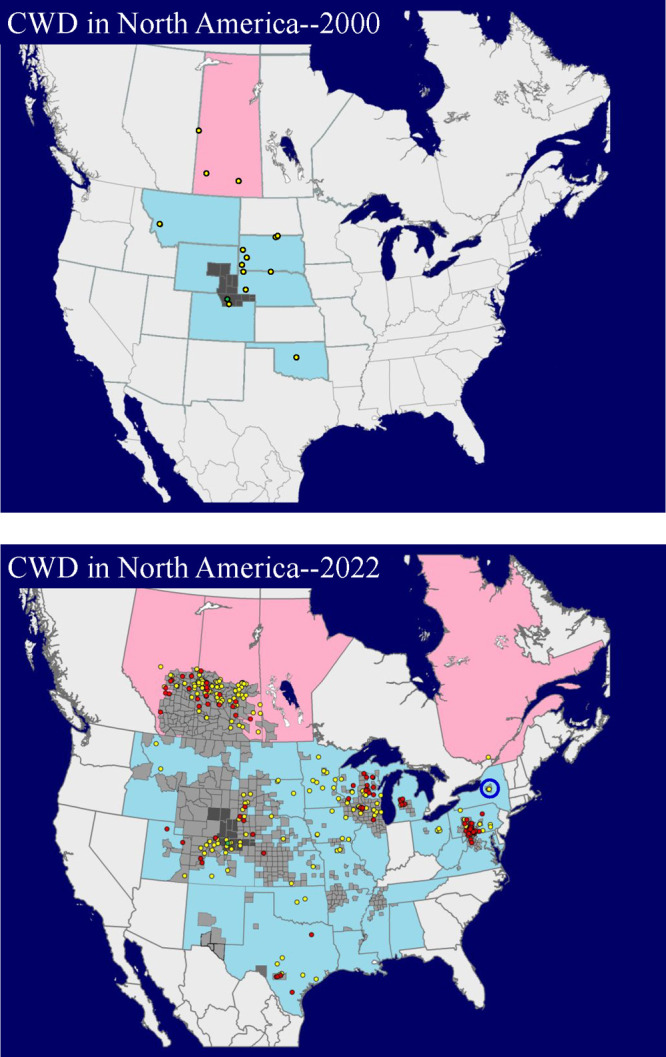
Comparison of CWD distribution in North America in 2000 and February of 2022. In 2000, CWD was a comparatively rare disease, restricted to five states and one Canadian province. By 2022, North American CWD is widely distributed, found in twenty-nine states and four Canadian provinces. Twenty-eight states and three Canadian provinces have detected CWD in wild cervids. In the United States, county-wide CWD incidence can exceed 25%. Depopulated CWD-infected cervid farms are indicated as yellow dots. Red dots indicate CWD-infected cervid farms that have yet to be depopulated. The single green dot indicates the location where CWD was first detected. The dark gray regions indicate the known distribution of CWD in free ranging cervids prior to 2000. The light gray regions indicate counties where CWD has been diagnosed in free ranging cervid populations. The blue circle indicates the only known area in which CWD has been eradicated from a wild population. Data (February 2022) courtesy of Bryan J. Richards and the United States Geological Survey (USGS).
Nature of the Prion Contagion
The neologism prion (proteinaceous infectious particle) was constructed to capture the concept of a protein that transmits heritable and infectious information (Figure 2).5,6 Subsequently, researchers discovered that mammals expressed a gene encoding a natively expressed prion protein (PrPC) with the same primary structure (linear amino acid sequence). Even though prions (PrPSc) and the natively expressed prion protein (PrPC) possess the same primary structure, they have dramatically different properties. Prions are infectious and difficult to inactivate by conventional means. They can survive autoclaving; cooking does not inactivate them. Researchers estimate that an oral infectious dose for deer is between 100 and 300 ng of brain material from a CWD-infected deer.7 CWD in wild cervids represents a novel potential threat to the food supply.
Figure 2.

Chemical nature of heritable information transfer in organisms. DNA and RNA transmit the information through covalent bonds. In contrast, a prion transmits heritable information through its conformation and not through covalent bonds. The existence of prion strains (Prion1–Prionn) implies that prions can adapt in response to natural selection pressures.
The natively expressed prion protein (PrPC) is encoded in a single gene. Although PrPC is required for prion pathology, it is not pathogenic. The human form of the gene encoding human PrPC is referred to as PRNP; in other mammals, it is referred to as Prnp. Even though the sequence of PrPC is highly conserved among mammals, Prnp-ablated transgenic mice and goats with a natural functional equivalence of Prnp ablation appear normal.8−10Prnp-ablated animals are resistant to infection by prions.8,9,11 Despite significant efforts to elucidate its role, the function of PrPC remains uncertain.12
Prions (PrPSc) replicate within a host by inducing the natively expressed prion protein (PrPC) to adopt the PrPSc conformation, yielding more PrPSc to continue the process.5 The covalent structures of PrPC and PrPSc have been carefully analyzed by mass spectrometry and amino acid sequencing.13 These studies revealed that PrPC and PrPSc possess identical primary structures. Additionally, they have identical covalent post translational modifications: a single disulfide bond, a glycosylphosphatidylinositol (GPI) anchor, and two sites of variable glycosylation.14 Similar glycoform variations are observed in the PrPC and PrPSc conformations.15−17 Oxidation of methionine was proposed to be a covalent prion signature, but this was subsequently shown to be incorrect.18−20 PrPC and PrPSc are conformers, as the only demonstrated difference between the two is conformational.
The primary and secondary structures of the monomeric PrPC protein are well established (Figures 3 and 4); the structure of multimeric PrPSc is less certain. NMR studies of bacterially derived recombinant PrP (rPrP) show its secondary structure is composed of random coil, α-helix and a very small amount of β-sheet (Figure 4).21,22 These same secondary structures are also seen in NMR-based analysis of native bovine PrPC, whose GPI anchor has been cleaved, but whose variable asparagine-linked glycosylation remains intact.23 PrPC is a monomer. Monomeric PrPC has no resistance to proteinase K (PK) digestion.
Figure 3.

Cartoon of deer PrPC. The three post translational modifications, a disulfide bond, glycosylphosphatidylinositol (GPI) anchor, and variable asparagine-linked glycosylation found on asparagine 184 or 200 are shown. The relevant secondary structures are indicated and based on an X-ray structure (PDB identifier: 4YXK).34 The asparagine-linked glycosylation is based on that observed in hamsters as is the glycosylation shown for the GPI-anchor.35,36 The inset shows a Western blot of brain-derived elk or white-tailed deer PrPC. The location of the di-, mono-, and unglycosylated glycoforms are indicated. The blot was probed with the GE8 mAb (https://www.ed.ac.uk/roslin/facilities-resources/tse-resource-centre/reagents/monoclonal-antibodies/ge8).
Figure 4.

Pymol image of the X-ray structure of recombinant deer PrP (PrPC conformation). The location of the N-terminus, C-terminus, disulfide bond, the two sites where asparagine-linked glycosylation would occur, α-helices, and β-sheets are indicated. The image is based on an X-ray structure whose coordinates have been deposited into the Protein Data Bank (4YXK).34 The structure image comprises amino acids 123–231.
The structure of PrPSc has been analyzed by Fourier transform infrared spectroscopy and hydrogen–deuterium exchange mass spectrometry.24,25 Analysis of this data revealed that PrPSc’s secondary structure is mostly β-sheet, with some unstructured elements, and no α-helical regions. In contrast to PrPC, PrPSc is a multimer, whose quaternary structure is required to stabilize its tertiary structure (conformation).26,27 Multimeric PrPSc has significant resistance to PK digestion.5
There are two competing β-sheet-based models for the tertiary structure of PrPSc, the four-rung-β-solenoid structure (4RβS) and the parallel in-register intermolecular β-sheet (PIRIBS) structure (Figures 5 and 6).28−30 4RβS is a computational model that incorporates steric constraints (e.g., glycosylation, GPI anchor, amino acid side chain size and charge, intact disulfide bond), spectral, and other experimental evidence.28,31 The model is based on pectate lyase E from Erwinia chrysanthemi.32 The PIRIBS structure is based on a cryo-EM analysis of the 263K strain of hamster-adapted scrapie30 and from a solid-state NMR analysis of a recombinant prion.29 Although these structures seem to be incompatible, a computational model suggests that each can be used, via a deformed template process, to generate the other.33
Figure 5.

Pymol images of a 4-rung-β-solenoid (4RβS) computational model for mouse PrPSc.28 The edge view (A) and the axial view (B) of the 4RβS are shown. The secondary structure is shown in cartoon form. The surface is indicated by mesh. Asparagine residues are shown as spheres (carbon [black], hydrogen [white], nitrogen [red], and oxygen [blue]). The close packing of the amino acid side chains is shown. This indicates that subtle changes of register in one portion of the molecule can lead to unfavored interactions in other portions of the molecule. Such unfavorable interactions may destabilize the entire structure.
Figure 6.

Pymol image of a parallel in-register intermolecular β-sheet (PIRIBS) structure of the 263K strain of hamster-adapted scrapie (PrPSc).30 The secondary β-sheet structure is shown in a cartoon format. The surface of the structure is designated with mesh. Asparagine residues are shown as spheres (carbon [black], hydrogen [white], nitrogen [red], and oxygen [blue]). The close packing of the amino acid side chains is shown. This indicates that a change of register can lead to unfavored interactions within the molecule. The image is based on the cryo-electron microscopy (cryo-EM) coordinate data deposited in the PDB (7LNA; https://www.rcsb.org/structure/7LNA).
Prion propagation is dependent upon the ability of PrPSc to induce the primary structure of the host’s PrPC to adopt a prion (PrPSc) conformation.5 The extent to which the primary sequence of a host’s PrPC is incompatible with PrPSc replication is referred to as the species barrier,37 even though this barrier to prion replication can exist within a species.38 For example, sheep expressing a PrPC polymorphism with alanine, arginine, and arginine at positions 136, 154, and 171 (A136R154R171), respectively, are resistant to infection with classical sheep scrapie, even though the progenitor prion and potential substrate are both derived from sheep PrPC.39−42 While the A136R154R171 sheep are resistant to classical scrapie, they are readily infected with the Nor98 or atypical strain of scrapie.43,44 In A136R154R171 sheep infected with atypical scrapie, the scrapie prions are composed entirely of refolded A136R154R171 sheep PrPC. Thus, the resistance to prion infection can be overcome if a different prion strain or conformation, which is compatible with the refolded host’s PrPC, infects that host.
Cervid PrPC possesses polymorphisms that influence prion propagation.45 Unlike sheep, there are no known cervid PrPC polymorphisms that completely protect cervids from CWD. Cervid PrPC polymorphisms are distributed throughout the PrPC protein.46 The polymorphisms associated with lower rates of prion propagation include histidine at position 95 (H95), serine at position 96 (S96), glycine at position 116 (G116), leucine at position 132 (L132), phenylalanine at position 225 (F225), and lysine at position 226 (K226).47−53 These polymorphisms extend the incubation period and reduce susceptibility to prion infection.54,55 Other polymorphisms at position 226 (glutamine [Q226] and glutamic acid [E226]) influence the distribution of prions in the bodies of CWD infected animals.56 Monitoring prion diseases requires the detection of prions and discrimination among prion polymorphisms and strains.46
A given mammalian host can be infected by more than one prion strain.57 Each prion strain propagates its distinct conformation and consequent pathogenic phenotype. These phenotypic differences include longer or shorter incubation periods, tissue tropism, and the ability to infect other species. Classical sheep scrapie readily propagates in sheep by using sheep PrPC as a substrate, but it does not infect humans (zoonotic). Bovine spongiform encephalopathy (BSE) induces bovine PrPC to refold into the BSE conformation. BSE is also zoonotic.58 BSE can induce sheep PrPC to adopt the BSE conformation.59,60 Even though the resulting PrPSc is composed entirely of refolded sheep PrPC, it possesses the zoonotic BSE conformation.61 A recently described tragic laboratory accident confirmed that sheep passaged BSE is zoonotic.62 The infectivity of a prion does not reside in the primary structure of the refolded PrPC comprising it; instead, it resides solely in the conformation or tertiary structure of the refolded PrPC.5
Even though a prion’s pathogenic information resides solely in a conformation (tertiary structure) stabilized by multimerization (quaternary structure) and not in the primary sequence of a nucleic acid, a prion’s conformation can respond to natural selection pressures. The ability of a strain to propagate is determined by the steric constraints imposed by refolding the disulfide bond, GPI anchor, asparagine-linked glycosylation, and amino acids of PrPC to conform with the β-sheets of the 4RβS or PIRIBS structure. Each alternate conformation propagates its distinct phenotype, which means that a conformation which can more readily amplify under a given selection pressure will predominate over those less readily propagating conformations. In principle, a prion conformation can evolve in response to natural selection pressures.
The means by which prion conformations can adapt to selection pressures may occur by several mechanisms. To propagate, a prion conformation needs to be compatible with the steric constraints imposed by the primary structure and the post-translational modifications (disulfide bond, asparagine-linked glycosylation, and GPI anchor) of a host’s PrPC. In addition, the tertiary structure needs to be compatible with multimerization of the conformers (vide supra). If a prion conformation is incompatible with the primary structure and post translational modifications of a given PrPC, then it may not propagate efficiently. The deformed template-based model posits that a prion can be initiated via an imperfect or deformed template.63 The resulting prion can propagate more efficiently than its deformed template progenitor and, as a result, predominate. Another model posits that a prion may be composed of an ensemble of conformations. In this ensemble, one conformation predominates, but each retains the ability propagate its own conformation.64,65 If a new host is exposed to the ensemble, then it is possible that a minor conformational component may propagate more efficiently and predominate, resulting in a new infection. Alternately, if a conformation can propagate using more than one PrPC polymorphism, then it can propagate in hosts possessing those polymorphisms.64,65 Like nucleic acid-based self-replicating agents (cells, viruses, etc.), some prion strains may propagate more efficiently in response to natural selection pressures. Unlike nucleic acid-based agents, prions adapt to selection pressures by conformational changes instead of changes in nucleic acid sequences (mutations).
The simplest example of a conformation responding to selection pressures occurs with the emergence of a sporadic prion disease. A sporadic prion disease occurs when a prion conformation independently emerges with the ability to replicate by refolding the host’s PrPC. While a sporadic prion can amplify in a host, it usually is not transmitted from a living host to a new host. After the host dies, however, its body will decompose and release prions into the environment where they can infect a new host.
A conformation that facilitates oral transmissibility, confers a significant advantage to that prion conformation. For example, bovine spongiform encephalopathy (BSE) is orally transmissible to domestic cattle through the consumption of BSE-contaminated feed (meat and bone meal). If the prion conformation can replicate in peripheral tissues, then it has the potential to be shed and be horizontally transmitted to another host. This occurs with classical scrapie in sheep and CWD in cervids. In domestic cattle, BSE prions do not replicate well in peripheral tissues, so they are not horizontally transmissible among domestic cattle. Thus, the ability to replicate in peripheral tissues imparts a selective advantage to that prion conformation by allowing it to be more easily spread to new hosts.
Selective advantages occur when a prion conformation gains the ability to refold different PrPC polymorphisms or greater stability in the environment. A conformation that can replicate in a host expressing a different PrPC polymorphism will have a selective advantage by expanding the host range of that prion conformation. Similarly, a prion conformation that can replicate in new species will be able to further expand its host range. A prion conformation that is more stable in the environment, will remain viable in the environment longer. This will afford it more opportunities to transmit to a new host. In these ways a prion’s conformation can adapt to propagate more efficiently among hosts with different PrPC polymorphisms or persist in the environment for a longer period of time.
Atypical scrapie provides insight into a prion’s ability to mutate its conformation in response to natural selection pressures. Atypical scrapie is a sporadic sheep prion disease, characterized by an unusual distribution in the brain, limited peripheral distribution, and a geographically diverse, but infrequent occurrence with no horizontal transmission. Unlike classical scrapie, atypical scrapie can propagate using sheep ARR (alanine at position 136, arginine at position 154, and arginine at position 171) PrPC, which is resistant to infection by classical scrapie.44 Additionally, atypical scrapie propagates well in sheep expressing other PrPC polymorphisms.44 When atypical scrapie is transmitted to sheep, it can convert to the CH1641 strain of classical scrapie.66 Unlike atypical scrapie, the CH1641 strain of classical scrapie is horizontally transmissible among sheep, a clear selective advantage. Other researchers have shown that, under experimental conditions, atypical scrapie is the progenitor of BSE.67,68 These results indicate that the conformation of a prion can adapt to spread by horizontal transmission among members of a species and to new species.
The unintentional spread of BSE to other species provides insight into how the conformation of a prion can facilitate changes in transmission to other species. In domestic cattle, BSE is readily transmitted by the consumption of BSE-contaminated feed.69,70 Even though BSE is orally transmissible, it has limited peripheral prion distribution and, consequently, no horizontal transmission. Sheep are susceptible to oral transmission of BSE and, unlike domestic cattle, the resulting prions are spread readily throughout the lymphatic system of infected animals.71 During the BSE crisis in the UK, BSE was transmitted orally to a variety of captive and companion animals. In captive kudu, BSE replicates in peripheral tissues72 and is horizontally transmitted in kudu, unlike most other BSE-infected animals. In humans, variant CJD (vCJD; human form of the BSE conformation) is present in the peripheral tissues and has been iatrogenically transmitted through blood transfusions and factor VIII concentrate (vide infra). The BSE conformation has properties that facilitate its oral transmission to a variety of species and in a few of those, it can amplify in peripheral tissues and, as a result, become horizontally transmissible.
Analysis of PrPSc usually requires its denaturation, which inactivates it but also results in the loss of the prion’s characteristic conformation.5,73 The most common means of identifying the prion conformation is to digest a sample with proteinase K (PK), which leaves covalent changes that are detectable even after denaturation. PrPC is completely digested by PK, while PK digestion of PrPSc results in the cleavage and digestion of exposed amino acids to yield a characteristic PK-resistant peptide referred to as PrP27-30 because of its characteristic migration on a denaturing SDS-PAGE gel. Other prions have different PK-resistant cores, which are covalent differences that can be used to distinguish among these prion strains after denaturation. PK digestion results in characteristic covalent modifications that are retained after the distinct prion conformation is lost.
A prion is defined as an infectious agent that causes a distinct transmissible spongiform encephalopathy.5 Demonstrating infectivity requires the inoculation of experimental animals with the prion and then observing the characteristic clinical signs, incubation period, and distinct pathology. Inoculation of experimental animals provides insight into the transmissibility of prions into other species. These experiments can be performed on relevant animals, such as domestic cattle, sheep, elk, deer, etc. Such experiments are necessarily limited by the size of the experimental animals, the extended incubation times, and the consequent costs associated with their housing, feeding, and handling. Prnp ablated mice have been engineered to express transgenic PrPC from other species, such as deer, elk, domestic cattle, sheep, humans, etc.74 The use of transgenic mice has revolutionized the study of prion disease.
Human Prion Diseases
The three principle forms of human prion disease are among the most closely studied prion diseases.75 In the United States, the incidence of human prion disease is between 1 to 1.5 patients per 1 million of the population (https://www.cdc.gov/prions/cjd/index.html). Humans can be afflicted by Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker (GSS) disease, and fatal familial insomnia (FFI).76 A fourth entirely sporadic and very rare (<2% of prion diseases) human prion disease, variably protease-sensitive prionopathy (VPSPr), has recently been described.77 Some forms of CJD are familial (fCJD), as are all cases of FFI and GSS.78,79 Familial forms of FFI, GSS, and fCJD are all autosomal dominant.78,79 A sporadic form of FFI, sporadic familial insomnia (SFI), has also been identified.80 CJD is transmissible and has been iatrogenically transmitted (iCJD).81 Human prion diseases are known to be sporadic, hereditary, and transmissible.75,82 85% of CJD cases are sporadic, with no discernible cause. CJD is also an inherited disease in approximately 5–15% of cases. fCJD is the only known disease to be both heritable and transmissible. Sporadic CJD, like the familial, iatrogenic, or acquired forms, is also transmissible. Such information can inform our understanding of CWD.
Bovine spongiform encephalopathy (BSE)83 or “mad cow” disease is the only known zoonotic prion disease.58 It was transmitted through the consumption of BSE-contaminated food. The human form of BSE is referred to as variant Creutzfeldt–Jakob disease (vCJD).84 With the exception of two heterozygous (M/V at position 129) patients,85,86 all symptomatic vCJD patients express PrPC that is homozygous for methionine at position 129.87 The disease has afflicted 260 people worldwide (Creutzfeldt–Jakob Disease Fact Sheet|National Institute of Neurological Disorders and Stroke (nih.gov). Unlike CJD or fCJD, vCJD prions are widely distributed in the patient’s peripheral tissues.88 vCJD has been transmitted through blood transfusions and it has infected patients that are homozygous (M/M) or heterozygous (M/V) at position 129.89,90 Evidence of infection in methionine/valine heterozygous presymptomatic patients has been observed. vCJD was detected through a CJD surveillance program established in the UK. Since then, many countries have established analogous CJD surveillance programs to detect the emergence of novel forms of CJD.91
The study of the human prion disease Kuru informs our current understanding of TSEs.92 Hadlow pointed out the similarity of sheep scrapie and kuru, which provided the linkage between an obscure human disease and an older disease of sheep, which greatly enhanced our understanding of TSEs.93,94 Kuru was largely restricted to members of the Fore language group in the highlands of New Guinea. Kuru is the Fore word for “to shake” or “tremble” from a fever or cold. The disease is transmitted through mortuary feasts in which participants practice endocannibalism of their deceased relatives. Kuru was extensively studied by Dr. David Carleton Gajdusek, other scientific researchers, and anthropologists. The last of these mortuary feasts is believed to have occurred in the late 1950s. The incubation period of Kuru is long, and the last known Kuru patient died in 2005.95 Kuru is now a historical disease.
Although Kuru is now an extinct disease, its impact on the genetic makeup of the Fore people remains. For cultural and not biological reasons, Kuru disproportionately infected adult women and children of either sex, but largely spared adult males.92 The disease altered the ratio of adult males to females to 3:1 in some villages and 2:1 for the South Fore group.92 Since 1957, Kuru is thought to have killed 2500 people in a population of approximately 12 000.96 A retrospective analysis showed that the most vulnerable population with the shortest incubation period consisted of people who were homozygous for methionine at position 129 of PrPC; those that were homozygous for valine at the same position had longer incubation periods.97 Those that were heterozygous (methionine and valine) at position 129 of PrPC were the most resistant and had the longest incubation periods.97 When researchers compared the PRNP genes of the Fore population born before and after the cessation of the endocannibalism, they noted strong selection pressure for the heterozygous (M/V) polymorphisms at position 129 of PrPC and then a return to a more even distribution of PrPC polymorphisms at position 129, respectively.98 A detailed survey of the PRNP genes found in the areas of high Kuru incidence showed the presence of a novel polymorphism where glycine was replaced with valine at position 127 (G127V) in PrPC. The G127V polymorphism is believed to impart resistance to Kuru infection.99 Experiments with transgenic mice expressing human PrPC with valine at position 127 show that these mice are resistant to kuru, CJD, and vCJD.100
The study of human TSEs reveals the complexities of the origin of prion diseases, their transmission and their influence on the genetics of an afflicted group of people. Human prion diseases can be sporadic, familial, and transmitted. Most CJD cases are sporadic. vCJD, the human form of BSE, shows the existence of at least one zoonotic prion conformation. BSE prions were transmitted to vCJD patients by the consumption of properly cooked but BSE-contaminated foods that were derived from inspected animals. The study of Kuru revealed that a sporadic CJD case can become a transmissible prion disease if those prions are consumed. The selection pressure of the Kuru epidemic altered the distribution of PRNP among the Kuru affected Fore population. These observations can provide insight into what may occur in cervid populations as CWD becomes even more common.
CWD in North America
The term chronic wasting disease (CWD) was originally used to describe the clinical signs of a disease associated with captive cervids in the late 1960s. In 1978, CWD was shown to be a TSE (prion disease) of captive mule deer.4 By 1981, CWD was found in captive Rocky Mountain elk (Cervus elaphus nelsoni).101 In 1985, it was found in wild mule deer (Odocoileus hemionus hemionus) and by 1990 in free-ranging white-tailed deer (Odocoileus virginianus).102 The first case of CWD in a wild moose (Alces alces shirasi) was discovered in a hunter-killed moose in 2005.103 In 2012, CWD was identified in a farmed red deer (Cervus elaphus).104
The expansion of CWD in North America in the 21st century is illustrated in Figure 1 and is caused by the ready transmission of the disease. Some of the expansion is undoubtedly due to relocation of infected, but asymptomatic, animals as has occurred in the Toronto Zoo and in South Korea (vide infra). CWD is the only known prion disease that is transmissible among wild animals through their natural behaviors.4 Researchers have shown that CWD was transmitted to mule deer cohoused with CWD-infected animals, housed in pens formerly occupied by infected animals, or housed in pens containing the decomposed carcass (after 1.8 years) of a CWD-infected mule deer.105 Even after thorough cleaning and being left fallow for a year, a CWD-contaminated environment remained infectious.106 CWD prions avidly bind to soils and yet remain infectious.107,108 Urine, feces, blood, or saliva from CWD-infected cervids have been used to infect transgenic cervidized mice.109−111 White-tailed deer have been experimentally infected by the transfusion of blood from CWD-infected white-tailed deer.112 At the turn of the millennium CWD was a comparatively rare disease largely restricted to Colorado and Wyoming. Twenty-one years later, CWD has been found in wild cervid populations in 28 states, three Canadian provinces, Norway, Sweden, and Finland (Figures 1 and 7).
Figure 7.
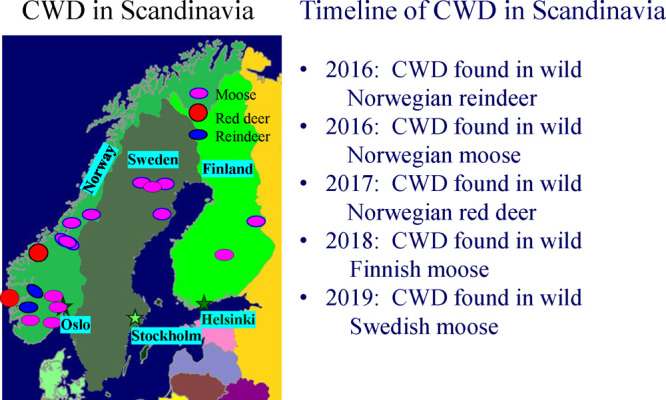
Geographical locations of CWD incidence in Scandinavia and a relevant timeline. CWD has been found in wild cervids in Norway, Sweden, and Finland. The capitals of the three countries (Oslo, Stockholm, and Helsinki, respectively) are indicated. The European Union surveyed wild and farmed cervids prior to 2016 and did not detect the presence of CWD in any of the tested animals (Chronic wasting disease (CWD)—information in English—Hjorteviltportalen).
CWD was introduced from the United States to Canada and, then, was exported from Canada to South Korea. A retrospective analysis found that a Colorado-born mule deer in the Toronto Zoo died of CWD in 1976.113 In 1996, CWD was found in a farmed elk (Cervus elaphus nelsoni) in the Canadian province of Saskatchewan.114 A farmed CWD-infected white-tailed deer was found in the Canadian province of Alberta.114 Subsequently CWD was found in wild mule deer in Saskatchewan.114 On October 14, 2021, CWD was found in a Manitoban wild mule deer (https://news.gov.mb.ca/news/index.html?item=52700). CWD was imported to South Korea through asymptomatic CWD-infected Canadian elk (Cervus elaphus nelsoni).115 The disease was subsequently transmitted to South Korean born elk (Cervus elaphus nelsoni). Later it was transmitted to farmed South Korean red deer (Cervus elaphus), sika deer (Cervus nippon), and crosses of these species.115,116 Importation of CWD-infected cervids has led to outbreaks in the past.
The incubation period of CWD depends on the species of the host cervid.117 The CWD incubation period for wild mule deer is estimated to be 2–2.5 years. Wild elk are estimated to have an incubation period of 2–5 years. The incubation period is also strongly influenced by the host’s Prnp genotype (vide infra). The length of the incubation period directly influences the survival of an infected animal and the spread of the disease.
The prevalence of CWD among wild cervids is related to an animal’s gender.118 Wild mule deer males have a CWD incidence that is 2.4-fold greater than wild females.118 The reindeer herd in the Nordfjella region of Norway contained more females (1278) than males (1081). Among these reindeer, the number of CWD positive males (13) was disproportionately greater than that for females (6).119 Adult Wisconsin male deer are disproportionately infected with CWD compared to females or yearlings of either gender (Figure 8). These results suggest that, because of behavioral differences, CWD generally infects more wild male animals than female ones, even though both genders are equally susceptible to CWD.4 Behavior-based differences in susceptibility to CWD can, thus, be added to the factors that influence the spread of CWD.
Figure 8.

CWD prevalence by age and gender of tested cervids in Iowa county, Wisconsin over time. CWD prevalence is higher for adult males than adult females or yearlings of either gender. Data from Wisconsin Department of Natural Resources (DNR).
CWD in South Korea
The first reported incidence of CWD found outside of North America occurred in South Korea.115 The animals were located in a South Korean cervid farm. The infected elk were identified in 2001, but were imported from Canada in 1997.115 Subsequently, more elk were found to be infected with CWD.120 All of those elk were infected with the same strain of CWD.121 Further investigation revealed that Canadian CWD was subsequently transmitted to farmed sika deer (Cervus nippon), red deer (Cervus elaphus), and cross-bred animals.116,122 The CWD outbreak in South Korea was restricted to farmed cervids and originated from asymptomatic but CWD-infected imported Canadian elk.
The South Korean experience reveals the extent to which imported CWD-infected animals can spread CWD to other cervid species. The tissues from the farmed South Korean CWD-infected sika deer were used to orally dose other captive sika deer.122 All of the orally dosed sika deer succumbed to CWD.122 Red deer have also been experimentally infected with CWD.123 A farmed Minnesota red deer was naturally infected with CWD.104 The first case of a wild CWD-infected red deer was found in Norway (vide infra).124 The Norwegian red deer was infected with a strain of CWD that was different from that infecting the South Korean or Minnesotan red deer. The South Korean experience provides insight into how the unintentional importation of CWD can lead to its natural spread among different cervid species.
CWD in Scandinavia
The recent discovery of CWD in Norway, Sweden, and Finland (Figure 7) is perplexing. The white-tailed deer currently inhabiting Finland are descended from animals imported from Minnesota in 1934 and 1948 (https://www.rcinet.ca/eye-on-the-arctic/2018/07/09/finland-deer-white-history-wildlife-minnesota-hunting/). Finnish authorities have surveyed their white-tailed deer from 2003 to 2015 and found no cases of CWD among the 643 animals tested. Other than those animals, Scandinavia has not imported cervids from North America.125
Scandinavian CWD appears to have emerged independently of North American CWD. The first cases of CWD were found in wild Norwegian reindeer (Rangifer tarandus tarandus)125 and moose (Alces alces).126 Wild Norwegian red deer (Cervus elaphus) were found to be infected with CWD in 2017 and 2021 (https://www.vetinst.no/en/news/the-veterinary-institute-has-confirmed-cwd-in-the-red-deer-from-etne-in-norway).124 In 2018, a CWD-infected moose was identified in Finland (https://www.ruokavirasto.fi/en/farmers/animal-husbandry/animal-health-and-diseases/animal-diseases/wildlife/chronic-wasting-disease-cwd-in-cervids/). In March 2019, the first Swedish case of CWD was found in a moose.127 Unlike CWD in South Korea, the Scandinavian strains of CWD have an independent origin and are not derived from North American CWD strains.126,128,129
Norwegian reindeer from Nordfjella zone 1 were infected with a “classical” form of CWD, where detectable CWD prions were found in lymph nodes only or in the lymph nodes and in the brain.119,125 This distribution is consistent with a natural transmission of CWD where prions are presumed to be ingested. After ingestion, they amplify in peripheral tissues and are eventually moved to the brain where they amplify and inevitably cause the death of the infected animal. The first CWD positive Nordfjella zone 1 reindeer died of CWD.125 In this index case, CWD prions were widely distributed throughout the animal’s body and brain. Most of the other CWD positive reindeer from Nordfjella zone 1 only showed prions in their lymph nodes and not in their brains. The overall incidence of the disease in the reindeer herd was approximately 0.8%; males were disproportionately infected with CWD.119 The distribution of CWD prions in the tissues found in reindeer and the gender ratio of infected reindeer were consistent with a naturally acquired and not spontaneous origin.
In contrast to the CWD found in Norwegian reindeer, the CWD strains infecting Scandinavian moose and red deer were found to be atypical (https://www.vetinst.no/en/news/the-veterinary-institute-has-confirmed-cwd-in-the-red-deer-from-etne-in-norway). Atypical prion diseases typically afflict animals that are older and are not thought to be acquired. An atypical prion disease implies a sporadic rather than acquired origin. Furthermore, atypical prion diseases are generally not thought to be naturally transmissible (horizontal or vertical) to other animals.130
In Sweden, there are approximately 400 000–500 000 moose. Approximately 100 000 animals are harvested each year. As a result of this hunting pressure, bull moose rarely live longer than 5 years. Moose cows are under less hunting pressure so they can live 10–20 years. Three of the four CWD positive Swedish moose were at least 16 years old (https://www.sva.se/en/animals/wildlife/map-of-chronic-wasting-disease-cwd/).127 The other was a hunter harvested 10-year-old animal. The two CWD positive Finnish moose were 15 and 18 years old (https://www.ruokavirasto.fi/en/farmers/animal-husbandry/animal-health-and-diseases/animal-diseases/news/tse-found-in-a-moose-killed-in-laukaa/). The first three Norwegian moose identified in 2018 as being infected with CWD were 13 or 14 years old.126 In 2019, two moose, 12 and 20 years old, tested CWD-positive, and in 2021, a 13-year-old Norwegian moose was identified with CWD. As noted previously, atypical or sporadic prion disease is more often found in older animals and the prion distribution is very different from that observed in classical or naturally transmitted prion diseases.
The distribution of prions found in Scandinavian moose and red deer were markedly different than those found in North American animals. The Norwegian moose (Alces alces) presented detectable amounts of CWD prions in the brain but not in the peripheral tissues.126 This was also true for the CWD positive wild hunter harvested Norwegian red deer (Cervus elaphus) found in 2017.124 Three Swedish moose also showed prions in their brains, but not in their peripheral tissues.127 The CWD infecting the Finnish moose is not thought to have been acquired from another animal (https://www.ruokavirasto.fi/en/farmers/animal-husbandry/animal-health-and-diseases/animal-diseases/news/tse-found-in-a-moose-killed-in-laukaa/). The first hunter harvested moose (Jackson County, Colorado) showed CWD prions in peripheral and brain tissues, which is consistent with an acquired disease.103 The CWD-infected captive red deer found in Minnesota also presented with CWD prions in peripheral and brain tissues.104 Unlike Scandinavian moose and red deer, North American moose and red deer were infected with classical CWD.
The Norwegian red deer, moose, and Swedish and Finnish moose were all infected with atypical CWD. Bioassay-based analysis of three Norwegian moose showed that each of them was infected with its own distinct strain of CWD.128 The Norwegian red deer was also infected with its own distinct sporadic strain of CWD.124 That prions were detected in the brains but not in peripheral tissues of Scandinavian moose and red deer is consistent with a sporadic origin for the CWD found in the 15 Scandinavian moose and two red deer. These numbers suggest that sporadic CWD has an incidence of more than 1 per 100 000 moose. Even though the CWD in Norwegian moose and red deer is atypical or sporadic, it is transmissible.128
The presence of atypical CWD in Scandinavia shows that spontaneous cases of CWD continue to occur in Scandinavian moose and red deer. The prions from the atypical CWD-infected moose and red deer have been experimentally transmitted to bank voles by intracranial inoculation with high efficiency.128 Thus, although atypical CWD may not be horizontally transmissible by living animals, their prions remain infectious. When wild animals die, their bodies are almost always scavenged and left to decompose. This will inevitably lead to a localized prion-contaminated environment. Classical CWD was experimentally transmitted to naïve mule deer after the animals were cohoused in a paddock with a decomposed (1.8 years of decomposition) carcass from a CWD-infected mule deer.105 This is not surprising since a single infectious oral dose of classical CWD is between 100 and 300 ng of CWD-infected brain tissue.7 In this way, an atypical prion disease originating in a Scandinavian moose or red deer could be transmitted to another cervid via an environment contaminated with atypical prions. If such a rare event occurred, then it is possible that the ingested atypical CWD prions might be more widely distributed in peripheral tissues as has been observed in experimental sheep orally dosed with atypical scrapie.131 One of the atypical strains of Norwegian CWD appears to adopt the North American CWD phenotype after experimental inoculation into transgenic cervidized mice.132 The Scandinavian experience suggests conversion of sporadic CWD to transmissible CWD may not be an extremely rare event.
The herd of CWD-infected wild Norwegian reindeer was restricted to an isolated location, which suggests a practical means of controlling the spread of CWD. If the entire herd could be depopulated, and the land left fallow for an interval (5 years), then the reintroduction of uninfected animals would demonstrate a means of eradicating CWD. Norway started the process by successfully depopulating the entire reindeer herd in the Nordfjella region.133 It remains to be seen if the reintroduced healthy animals become infected. However, in the meantime, CWD has been found in a reindeer outside of the Nordfjella region (https://www.vetinst.no/en/news/chronic-wasting-disease-cwd-identified-in-a-wild-reindeer-at-hardanger-plateau). Unfortunately, this means that Norway’s attempt to eradicate CWD in its wild reindeer population failed.
CWD Transmission to Other Species
Researchers have attempted experimental transmission of CWD prions to other animals to assess their vulnerability to CWD. The most common means of experimental transmission is by intracranial (ic) inoculation. The purpose of this approach is to assess the theoretical possibility of transmission. While ic transmission of prions is not a natural means of transmission, it does provide an indication of the potential for interspecies transmission of those prions. This potential for interspecies transmission is assessed by the number of inoculated animals that develop disease (attack rate) and the time before the animals become sick (incubation period). A low attack rate with a long incubation period indicates that there is a significant barrier to the interspecies transmission of CWD. A higher attack rate and a shorter incubation period suggests a less significant species barrier to CWD transmission exists. Unfortunately, these results are only true for the strain used in each experiment. If a different strain of CWD is used, the results may be different, even though both are composed of isosequential refolded PrPC. These experiments provide an assessment of the threat of CWD to agriculturally important animals.
CWD has been transmitted to domestic cattle, sheep, and other mammals.134−139 When domestic cattle are inoculated (ic) with CWD from mule deer, they succumb to disease, but the attack rate is low and the incubation period long.134,135 The first passage of elk derived CWD in domestic cattle also had a low attack rate.139 In contrast, the attack rate for the first passage white-tailed deer derived CWD to domestic cattle was higher.136 The difference in attack rate for the different deer CWD inocula may be due to differences in the titer of CWD in the inocula or the use of different CWD strains. Upon repassage of cattle passaged mule deer CWD, the attack rate was 100% and the incubation period decreased compared to the first passage.140 Cattle were not infected by oral dosing with CWD141 and cattle that grazed for seven years on pastures that were probably contaminated with CWD remained free of prion disease.142 CWD has been transmitted to sheep, but the attack rate is low and transmission seems to depend upon the polymorphism in the host sheep’s expressed PrPC.137 One attempt to transmit (ic) CWD to potential scavengers (raccoons) was unsuccessful.143 Another attempt was successful but with a low attack rate, suggesting that CWD is only inefficiently transmitted to raccoons.138 Other potential scavengers (ferrets) were infected by ic inoculation, but not by oral dosing with CWD.144 The ferret passaged CWD was transmissible to hamsters, unlike unpassaged CWD, suggesting ferret passaged CWD can infect different hosts than cervid passaged CWD.145 Goats are susceptible to infection from ic inoculated CWD.146 Oral transmission to these animals failed or was not done. These experimental results suggest that CWD is not readily transmissible to other agriculturally important or noncervid scavenger species.
Cervids Are Susceptible to Other Prion Diseases
Researchers have successfully transmitted other prion diseases to cervids. BSE has been transmitted (ic) to red deer with a 100% attack rate.147 Red deer orally dosed with a large inoculum of BSE succumbed to disease, but the attack rate was low and the incubation period extended.148 The tissue distribution of prions in the bodies of red deer experimentally infected with BSE was more restricted than in the bodies of red deer infected with CWD.149 In addition, digestion with PK and analysis by Western blot showed that the prions from CWD-infected and BSE-infected red deer were readily distinguishable.149 Both farmed and wild red deer have been naturally infected with prions, but they resemble CWD and not BSE.104,116,124 Inoculation of red deer passaged BSE into cervidized transgenic mice showed that the inoculum had a shorter incubation period, suggesting that, upon repassage, the BSE conformation could more efficiently propagate using cervid PrPC as a substrate.150 During the BSE crisis in the United Kingdom, red deer were fed proprietary supplements that may have been contaminated with BSE and yet no BSE was observed in farmed red deer. These experiments show that the BSE conformation can propagate using cervid PrPC as a substrate. They also show that the transmission of BSE to cervids is likely to be inefficient.
Sheep scrapie has been transmitted to cervids. When sheep scrapie is transmitted (ic) to elk the attack rate is less than 100% and dependent upon the elk’s PrPC polymorphism.151−153 Cervidized (elk) transgenic mice are susceptible to scrapie infection and infection by sheep passaged BSE.154 White-tailed deer are susceptible to scrapie by ic inoculation.155 This suggests the theoretical possibility of scrapie transmission to cervids. Other researchers have posited that CWD may have originated from scrapie. This seems unlikely as sheep and cervids coexist in many parts of the world, but CWD spread among wild cervid populations was, until its recent (2016) discovery in Scandinavia, restricted to North America.
Zoonotic Potential of CWD
CWD has been transmitted to primates, squirrel monkeys (Simia sciurea) and cynomolgus macaques (Macaca fascicularis). Squirrel monkeys (Simia sciurea) are New World monkeys. They have been experimentally infected after either ic inoculation or oral dosing with CWD prions.156,157 Analogous experiments were performed on Old World cynomolgus macaques (Macaca fascicularis).157,158 In one set of experiments, cynomolgus macaques remained disease-free after either ic or oral inoculation with CWD prions.157,158 In an analogous experiment using a different CWD inoculum, the cynomolgus macaques showed subtle signs of disease after an extended incubation period (http://www.cste2.org/Webinars/files/CWD_Slides_FINAL.pdf and https://www.cdc.gov/prions/cwd/transmission.html). These contradictory results may have been the result of some experimental artifact, or they may have arisen from the inadvertent use of different CWD strains.
Currently there is no evidence that North American CWD has been transmitted to people. Epidemiological studies have not associated Creutzfeldt–Jakob disease incidence with CWD prevalence.159,160 A previous report associating CWD consumption with a cluster of CJD cases was shown to be the result of a misdiagnosis, and there was no association between the occurrence of CJD and the consumption of cervid meat potentially contaminated with CWD prions.161 When North American CWD prions are inoculated (ic) into transgenic mice engineered to express human PrPC (humanized mice), the mice are not infected.162,163 Recent experiments have shown that CWD prions from Norwegian moose and reindeer are not transmissible to humanized mice.164 Other known human prions or zoonotic prions cause disease in humanized mice.165 This suggests that the North American and Norwegian CWD prions used in these experiments will not propagate with human PrPC and, therefore, are unlikely to be zoonotic. The limitation of these studies is that they only apply to the CWD conformations that were used and not the current or future CWD strains.
Unfortunately, CWD is not a static disease. It is a disease that can arise spontaneously (vide supra). In addition, CWD prions, like all prions, have the capacity to respond to natural selection pressures. Furthermore, CWD can impart negative selection pressures on the common and more vulnerable PrPC polymorphisms. In the future, the common PrPC genotypes are unlikely to be same as those currently common. As has been seen experimentally, those new genotypes may propagate prion strains with different properties. What those properties will be are completely unknown. Thus, their potential for zoonosis is also unknown.
Prions Can Evolve and May Alter the Genetics of Cervid Populations
The negative impact of CWD on wild mule deer populations was revealed by studying those populations in areas with high CWD incidences (Figure 1; dark gray areas where CWD was present prior to 2000). CWD-infected animals weaned fewer fawns than did uninfected animals.166 CWD-infected deer were more susceptible to predation by mountain lions.167,168 CWD-infected deer were more likely to be killed by automobiles.169 The average additional life expectancy of CWD infected mule deer was estimated to be 1.6 years.168 In contrast, the same measure for uninfected animals is 5.2 years.168 Among naturally infected cervids, males are infected at much higher rates than females (Figure 8).168 This occurs despite the observation that there is no intrinsic difference in infectivity among captive animals.4 CWD has caused mule deer population declines in Wyoming.170 A similar decline was noted in Colorado.168 These observations suggest that CWD infection places a negative natural selection pressure on cervids, which may alter the population genetics of cervids. Thus, currently rare PrP polymorphisms are likely to become more common, if they offer the survival advantage of a longer CWD incubation period or a greater resistance to CWD infection. These observations suggest that CWD will change the relative abundance of PrP polymorphisms in cervid populations and will perturb population ratios of males to females.
Among cervids, certain genotypes confer resistance to infection.50,171 When white-tailed deer expressing different PrPC polymorphisms (Q95G96/Q95G96 [homozygous for Q at position 95 and G at position 96], Q95G96/Q95S96, Q95S96/Q95S96, H95G96/Q95G96, or H95G96/Q95S96) were orally dosed with equal amounts of wild type CWD (Q95G96/Q95G96), the wild type animals succumbed much faster than did the other genotypes.54,55 Analogous experiments with elk expressing a polymorphism at position 132 (M or L), showed that animals with the L polymorphism survived for a longer period after oral dosing compared to M homozygous animals.172 Wild white-tailed deer expressing polymorphisms (S at position 96) are underrepresented among wild deer naturally infected with CWD.47,49 Furthermore, wild white-tailed deer expressing an S polymorphism at position 96 show a greater than 4-fold reduction in CWD prevalence than do wild type deer (G96/G96) and when infected with CWD, show a 48% greater additional life expectancy compared to homozygous wild type CWD-infected white-tailed deer.173 Among farmed white-tailed deer, those expressing rare polymorphisms (H95, S96, A116, or K226) are less likely to be infected with CWD compared to those expressing wild type PrPC (Q95G96/Q95G96).50 Additionally, among those animals who were CWD-infected and expressing rare polymorphisms, CWD was noticeably less progressed than those animals expressing wild type PrPC.50 In captive and wild elk, animals with an L polymorphism at position 132 are underrepresented among CWD-infected animals.48 In mule deer, the presence of the F polymorphism at position 225 is underrepresented in wild mule deer infected with CWD.52 These differences in susceptibility to prion disease may be driving changes in the distribution of Prnp polymorphisms in wild deer.173,174
Careful study of North American and Scandinavian CWD isolates has revealed the presence of 11 (or more) different CWD strains.128,144,175−178 North American elk also propagate at least two CWD strains.179−181 White-tailed deer propagate the CWD strains Wisc-1 and H95+.182 Two additional strains are described in heterozygous white-tailed deer expressing the A and G polymorphisms at position 116 of PrPC.175 Norwegian scientists have identified six distinct strains of Norwegian CWD.124,128 These observations indicate that both North American and Scandinavian cervids are infected with distinct CWD prion strains.
When prions from the most common polymorphism (wild type) present in white-tailed deer are used to experimentally infect white-tailed deer with different polymorphisms, the resulting prions have different properties.182 As noted previously, while-tailed deer with rare genotypes show an extended incubation period before they succumb to the disease compared to wild type controls.54,55 Furthermore, the distribution of CWD prions in the bodies of those white-tailed deer with some rare Prnp genotypes show less peripheral distribution than is observed in analogously infected wild type white-tailed deer.183 This suggests that animals expressing these rare PrPC polymorphisms are more resistant to CWD infection and, if infected, may transmit CWD less efficiently. The prions from some of the orally dosed white-tailed deer expressing the rarer polymorphisms could be transmitted to wild type mice, while others could not.184 Elk, homozygous for leucine at position 132, show a longer incubation period after being experimentally infected with CWD compared to methionine homozygous or methionine/leucine heterozygous elk.172 These animals are also susceptible to sheep scrapie.185 Thus, wild type prions can be transmitted to animals with rarer PrPC polymorphisms, but the resulting prions may have different transmission properties; they may infect previously invulnerable species.184 In addition, some of the rare genotypes are vulnerable to prions from other species.185
CWD prions can evolve. Scandinavian CWD appears to represent an independent emergence of CWD. In Norway, cervids propagate at least six different CWD conformations.124,128,129 In North America, mule deer CWD has altered the distribution of cervid Prnp genotypes in some areas.186 In principle, infecting deer that have rare genotypes with CWD may alter the strain properties of the progenitor prions. The modes of CWD transmission, transmission among wild animals through their natural behaviors and from CWD-contaminated environments, are beyond human control. Thus, CWD represents an unintentional experiment in prion evolution. CWD strain properties may be altered when they infect cervids with currently rare PrPC genotypes. The new strains may be capable of infecting species that the progenitor strains cannot. Transmission of wild type CWD prions to rarer genotypes extends the incubation period of the disease, allowing infected animals to survive longer.54,55 This may result in prion shedding for an extended period relative to wild type infections. On the other hand, these animals have also been shown to have lower levels of prions in their peripheral tissues, suggesting that fewer prions will be shed. Experimental evidence shows that a cervid PrPC polymorphism (serine at position 96) may impede wild type CWD prion propagation enough to prevent subsequent transmission in the wild.187 Predicting the future of CWD can, therefore, be problematic. This is especially true considering that red deer have been shown to propagate the zoonotic BSE conformation,147−149 even though no cervid has been naturally infected with BSE. Equally problematic is presuming that the CWD of the future will resemble the CWD of the past.
CWD Detection Methods
In the United States, there are two regulatory diagnostic tests for prions (https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/nvap/NVAP-Reference-Guide/Control-and-Eradication/Chronic-Wasting-Disease). Immunohistochemistry (IHC) is the “gold” standard for prion diagnostics among regulators. Regulatory tissues (retropharyngeal lymph nodes and obex) are formaldehyde fixed and then embedded in paraffin. Thin slices of the paraffin embedded tissue are placed on glass slides, the paraffin removed, and fixed tissue stained with dyes or prepared for antibody-based staining. IHC shows the presence of prions and their consequent pathology. IHC can be used to identify some CWD prion strains. The other diagnostic test relies on Western blot or ELISA-based analysis. Prions can be selectively isolated (e.g., ultracentrifugation, phosphotungstic acid, proprietary resins) and then detected by Western blot or ELISA. Alternately, the tissue can be digested with proteinase K (PK), which completely digests the endogenous PrPC, but leaves a partially digested PrPSc core (PrP27-30), which can be detected by monoclonal antibody-based ELISA or Western blot. These methods can be used to distinguish among some of the known CWD prion strains but cannot necessarily be used to discriminate among all of them.
Mass spectrometry has been used to directly detect CWD prions.18,46 The most sensitive means of directly detecting prions is a multiple reaction monitoring-based (MRM) mass spectrometry method.188,189 It has been used to detect CWD prions at lower levels than are detectable by ELISA-based methods.18 In addition, it has been used to discriminate among CWD prion strains expressed in heterozygous white-tailed deer.46 In principle, a mass spectrometry-based analysis of covalently modified prions that was used to distinguish among strains of hamster-adapted scrapie could be adapted to distinguish among CWD prion strains.190
In vitro and in vivo prion amplification methods have been used to detect CWD prions and distinguish among prion strains. The most reliable means of amplifying prions is to inoculate a susceptible animal, monitor the disease course, and then characterize the resulting pathology. The animals can include cervids, transgenic mice expressing cervid PrPC (cervidized mice), or other experimental animals, such as bank voles (a universal prion acceptor)191 or transgenic mice expressing bank vole PrPC.74 Animal inoculation yields authentic prions and is used to distinguish among CWD strains.128 Unfortunately, in vivo amplification requires the use of experimental animals, involves the expenditure of considerable resources, and is very time intensive.
Protein misfolding cyclic amplification (PMCA) and real-time-quaking-induced conversion (RT-QuIC) are in vitro methods of amplification. RT-QuIC and PMCA amplify some but not all human prion strains.192 PMCA and related techniques require a prion seed, PrPC from brain homogenates, and sonication to amplify the infectious prion conformation.193 Unfortunately, PMCA and related amplification approaches require animals (mostly transgenic mice) as a PrPC source. PMCA has been used to detect CWD prions in blood and other tissues.194−196 RT-QuIC is a newer in vitro technique that uses bacteria-derived recombinant PrP (rPrP) as a substrate for a prion template-mediated amplification of a misfolded (noninfectious) form of rPrP.197 RT-QuIC has been used to detect CWD in feces, blood, and ear punches in CWD-infected North American cervids.198−200 RT-QuIC has been used to detect Norwegian CWD prions.201 The misfolded rPrP is not infectious, so it is not a true prion amplification method. Although RT-QuIC is very sensitive, it can take 87 h to complete.197 It also requires rPrP, which because of its limited commercial availability is produced by the laboratories performing the test, and therefore, lacks the standardized uniformity that is characteristic of commercial kits. RT-QuIC is becoming more widely used and may become an approved diagnostic technique in the future.
Hunters’ Use of Testing
The Centers for Disease Control and Prevention (CDC) does not recommend consumption of CWD-infected animals (https://www.cdc.gov/prions/cwd/index.html). Ample experimental evidence exists to indicate that the known CWD strains are almost certainly not zoonotic. In addition, there is no epidemiological evidence to suggest that CWD is zoonotic.159,160 Nonetheless, the amount of CWD in a red deer is considerable, estimated to be 83 000 infectious units (one mouse bioassay ic ID50).202 Unfortunately, approximately 10 000 ID50s are found in tissues that a hunter would consume.202 These estimates suggest that a significant amount of infectivity (∼13%) is retained in tissues likely to be consumed by a hunter and could be problematic if a zoonotic strain of CWD emerged. Currently, consumption of CWD-infected tissue, while not recommended, appears to carry a low risk.
Reliable methods of detecting CWD prions in hunter harvested cervids are currently available. Testing of hunter harvested cervids show that CWD prevalence tends to increase over time. New York remains the only state to successfully eradicate CWD in a local wild cervid population (https://www.dec.ny.gov/docs/wildlife_pdf/cwdbooklet2019.pdf). Outside of New York, once a wild cervid population is found to be infected with “classical” CWD, the incidence of CWD increases over time in that population. This trend can be seen in the documented increase in CWD prevalence in Wisconsin’s wild cervids (Figure 9).
Figure 9.

Change in CWD incidence over time in Wisconsin by county. The incidence is defined as percentage of CWD-infected animals among those tested. Thirteen counties are indicated: Adams (A), Columbia (Co), Crawford (Cr), Dane (D), Grant (Gt), Green (Gn), Iowa (I), Jefferson (J), Lafayette (L), Richland (Ri), Rock (Ro), Sauk (S), and Walworth (W). The graph shows that once CWD is identified in a wild population, its incidence tends to increase over time. The county-wide incidence can exceed 25%. Local (within county) incidence may be greater than the county-wide incidence. The geographic locations of the counties are shown in the inset map. (https://dnr.wisconsin.gov/topic/wildlifehabitat/cwd.html).
Even though CWD has been known in Wisconsin for 18 years, the number of hunter-harvested deer tested for CWD is consistently less than 7% of the state-wide harvested animals (Figure 10; https://dnr.wisconsin.gov/topic/wildlifehabitat/cwd.html). More than 75% of the CWD positive deer identified in Wisconsin are found in just four counties (Dane, Iowa, Richland, and Sauk). While the greatest proportion of hunter-harvested deer testing positive for CWD is found these four counties, the proportion of animals tested for CWD in these counties remains less than 25% of the cervids harvested there (Figure 11). This is important because even though the deer are harvested in these four counties, the hunters harvesting those deer come from all 50 states to hunt them (Figure 12). Although testing is available and the known CWD incidence is high, most hunters appear to choose not to test their animals.
Figure 10.
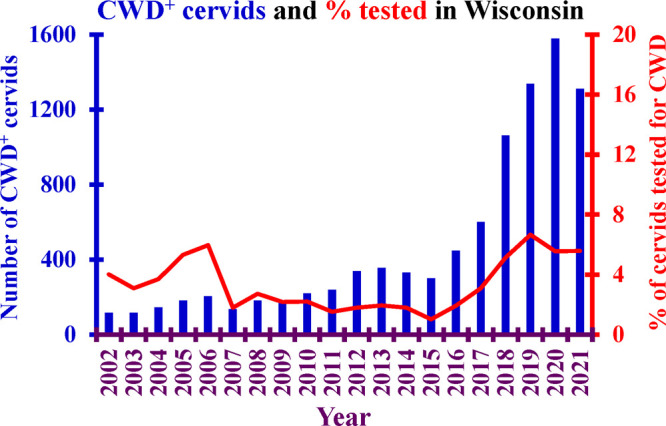
Graph of the number of CWD-infected (CWD+) cervids (blue bar graph) and the percentage of hunter harvested animals tested for CWD (red line) in the state of Wisconsin by year. This double axis graph covers the years 2002–2021. The statewide percentage of hunter-harvested cervids tested for CWD (red line) has never exceeded 7%. Data from Wisconsin DNR.
Figure 11.

Graph of the number of CWD-infected (CWD+) cervids (blue bar graph) and the percentage of hunter harvested cervids tested for CWD (red line graph) in the Wisconsin counties of Dane, Iowa, Richland, and Sauk by year. This double-axis graph covers the years 2002–2021. The percentage of hunter-harvested cervids tested for CWD has fallen from a 40% high in 2006 to a 23% rate in 2021. Data from Wisconsin DNR.
Figure 12.

Graph showing the home ZIP codes of hunters who harvested deer (n > 32 000; 2016–2017) in four Wisconsin counties (Dane, Iowa, Richland, and Sauk). This graph shows that deer harvested in regions with high incidences of CWD may not remain there. Image by Bryan J. Richards, United States Geological Survey (USGS; https://www.usgs.gov/media/images/home-zip-codes-hunters-harvesting-deer-4-wi-counties-2016-17). Data from Wisconsin DNR.
Unfortunately, the CWD of today is unlikely to be the CWD of the future. As its incidence continues to increase, the effect it has on selection pressures will likely perturb the current population ratios of cervid Prnp as Kuru did with the PRNP distribution in the Fore people (vide supra). The experience in Scandinavia suggests that new CWD strains emerge sporadically and may become transmissible. This suggests an uncertain and unpredictable future.
Conclusions
CWD represents a distinct food safety challenge. Even though wild cervid meat provides between 2% and 3% of the meat calories consumed in the United States, it is not inspected prior to consumption. It is presumed that a hunter can visually inspect an animal and determine if that animal appears to be healthy. Such observations are unreliable, however, since CWD-infected wild cervids may appear to be healthy even when they are infected with CWD. Thorough cooking is a means of inactivating conventional pathogens, but cooking will not inactivate prions. The CDC recommends against consumption of CWD-infected tissues. This means that animals need to be tested prior to consumption. Currently, the time it takes for a hunter to be informed of the results of a CWD test can take as long as 3–4 weeks (https://www.outdooralabama.com/cwd-sampling). In many locations, CWD testing is voluntary. Resolving these issues requires improving testing protocols and improving hunter outreach and education programs. These issues represent a unique food safety challenge.
Acknowledgments
The author would like to acknowledge the assistance of Melissa Erickson-Beltran in preparing this manuscript.
Glossary
Abbreviations Used
- BSE
bovine spongiform encephalopathy
- CDC
Centers for Disease Control and Prevention
- CJD
Creutzfeldt–Jakob disease
- DNR
Department of Natural Resources
- GSS
Gerstmann–Sträussler–Scheinker
- CWD
chronic wasting disease
- FFI
fatal familial insomnia
- GPI
glycosylphosphatidylinositol
- mAb
monoclonal antibody
- PMCA
protein misfolding cyclic amplification
- PRNP
gene encoding human prion protein
- Prnp
gene encoding mammalian prion protein
- PrP
prion protein
- PrPC
natively expressed and noninfectious conformer of the prion protein
- PrPSc
infectious conformer of the prion protein
- rPrP
recombinant PrP
- RT-QuIC
real-time quaking-induced conversion
- TSE
transmissible spongiform encephalopathy
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
The author declares no competing financial interest.
References
- Arnett E. B.; Southwick R. Economic and social benefits of hunting in North America. Int. J. Environ. Sci. 2015, 72, 734–745. 10.1080/00207233.2015.1033944. [DOI] [Google Scholar]
- Johnson J. L.; Zamzow B. K.; Taylor N. T.; Moran M. D. Reported U.S. wild game consumption and greenhouse gas emissions savings. Hum. Dimens. Wildl. 2021, 26, 65–75. 10.1080/10871209.2020.1799266. [DOI] [Google Scholar]
- Hedman H. D.; Varga C.; Duquette J.; Novakofski J.; Mateus-Pinilla N. E. Food Safety Considerations Related to the Consumption and Handling of Game Meat in North America. Vet. Sci. 2020, 7, 188. 10.3390/vetsci7040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. S.; Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. 10.7589/0090-3558-16.1.89. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. Prions. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13363–13383. 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S. B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Denkers N. D.; Hoover C. E.; Davenport K. A.; Henderson D. M.; McNulty E. E.; Nalls A. V.; Mathiason C. K.; Hoover E. A. Very low oral exposure to prions of brain or saliva origin can transmit chronic wasting disease. PLoS One 2020, 15, e0237410. 10.1371/journal.pone.0237410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H.; Aguzzi A.; Sailer A.; Greiner R. A.; Autenried P.; Aguet M.; Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B.; Groth D.; Serban A.; Koehler R.; Foster D.; Torchia M.; Burton D.; Yang S. L.; DeArmond S. J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 10608–10612. 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benestad S. L.; Austbo L.; Tranulis M. A.; Espenes A.; Olsaker I. Healthy goats naturally devoid of prion protein. Vet. Res. 2012, 43, 87. 10.1186/1297-9716-43-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen Ø.; Espenes A.; Reiten M. R.; Vuong T. T.; Malachin G.; Tran L.; Andréoletti O.; Olsaker I.; Benestad S. L.; Tranulis M. A.; Ersdal C. Goats naturally devoid of PrP(C) are resistant to scrapie. Vet. Res. 2020, 51, 1. 10.1186/s13567-019-0731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill A. C.; Castle A. R. The cellular and pathologic prion protein. Handb. Clin. Neurol. 2018, 153, 21–44. 10.1016/B978-0-444-63945-5.00002-7. [DOI] [PubMed] [Google Scholar]
- Stahl N.; Baldwin M. A.; Teplow D. B.; Hood L.; Gibson B. W.; Burlingame A. L.; Prusiner S. B. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry 1993, 32, 1991–2002. 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- Stahl N.; Baldwin M.; Teplow D. B.; Hood L. E.; Beavis R.; Chait B.; Gibson B. W.; Burlingame A. L.; Prusiner S. B.. Cataloging post-translational modifications of the scrapie prion protein by mass spectrometry. In Prion Diseases of Humans and Animals, 1 st ed.; Prusiner S. B., Collinge J., Powell J., Anderton B., Eds.; Ellis Horwood: New York, 1992; pp 361–379. [Google Scholar]
- Rudd P. M.; Wormald M. R.; Wing D. R.; Prusiner S. B.; Dwek R. A. Prion glycoprotein: structure, dynamics, and roles for the sugars. Biochemistry 2001, 40, 3759–3766. 10.1021/bi002625f. [DOI] [PubMed] [Google Scholar]
- Endo T.; Groth D.; Prusiner S. B.; Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989, 28, 8380–8388. 10.1021/bi00447a017. [DOI] [PubMed] [Google Scholar]
- Stimson E.; Hope J.; Chong A.; Burlingame A. L. Site-specific characterization of the N-linked glycans of murine prion protein by high-performance liquid chromatography/electrospray mass spectrometry and exoglycosidase digestions. Biochemistry 1999, 38, 4885–4895. 10.1021/bi982330q. [DOI] [PubMed] [Google Scholar]
- Silva C. J.; Dynin I.; Erickson M. L.; Requena J. R.; Balachandran A.; Hui C.; Onisko B. C.; Carter J. M. Oxidation of methionine 216 in sheep and elk prion protein is highly dependent upon the amino acid at position 218 but is not important for prion propagation. Biochemistry 2013, 52, 2139–2147. 10.1021/bi3016795. [DOI] [PubMed] [Google Scholar]
- Silva C. J.; Onisko B. C.; Dynin I.; Erickson M. L.; Vensel W. H.; Requena J. R.; Antaki E. M.; Carter J. M. Assessing the role of oxidized methionine at position 213 in the formation of prions in hamsters. Biochemistry 2010, 49, 1854–1861. 10.1021/bi901850n. [DOI] [PubMed] [Google Scholar]
- Canello T.; Engelstein R.; Moshel O.; Xanthopoulos K.; Juanes M. E.; Langeveld J.; Sklaviadis T.; Gasset M.; Gabizon R. Methionine sulfoxides on PrPSc: a prion-specific covalent signature. Biochemistry 2008, 47, 8866–8873. 10.1021/bi800801f. [DOI] [PubMed] [Google Scholar]
- Hornemann S.; Korth C.; Oesch B.; Riek R.; Wider G.; Wuthrich K.; Glockshuber R. Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett. 1997, 413, 277–281. 10.1016/S0014-5793(97)00921-6. [DOI] [PubMed] [Google Scholar]
- Riek R.; Hornemann S.; Wider G.; Billeter M.; Glockshuber R.; Wuthrich K. NMR structure of the mouse prion protein domain PrP(121–231). Nature 1996, 382, 180–182. 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- Hornemann S.; Schorn C.; Wuthrich K. NMR structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep. 2004, 5, 1159–1164. 10.1038/sj.embor.7400297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnovas V.; Baron G. S.; Offerdahl D. K.; Raymond G. J.; Caughey B.; Surewicz W. K. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. 10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron G. S.; Hughson A. G.; Raymond G. J.; Offerdahl D. K.; Barton K. A.; Raymond L. D.; Dorward D. W.; Caughey B. Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 2011, 50, 4479–4490. 10.1021/bi2003907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger-Kawahara C. G.; Kempner E.; Groth D.; Gabizon R.; Prusiner S. B. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology 1988, 164, 537–541. 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- Alper T.; Haig D. A.; Clarke M. C. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966, 22, 278–284. 10.1016/0006-291X(66)90478-5. [DOI] [PubMed] [Google Scholar]
- Spagnolli G.; Rigoli M.; Orioli S.; Sevillano A. M.; Faccioli P.; Wille H.; Biasini E.; Requena J. R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. 10.1371/journal.ppat.1007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Pastor M.; Codeseira Y. B.; Spagnolli G.; Eraña H.; Fernández L. C.; Martin D.; Bravo S.; López-Lorenzo N.; Iglesias A.; López-Moreno R.; Sabaté R.; Veiga S.; Rezaei H.; Biasini E.; Sánchez-Pedregal V. M.; Castilla J.; Requena J. R. Solid state NMR reveals a parallel in register architecture for an infectious recombinant prion. bioRxiv 2021, 10.1101/2021.07.20.453078v1. [DOI] [Google Scholar]
- Kraus A.; Hoyt F.; Schwartz C. L.; Hansen B.; Artikis E.; Hughson A. G.; Raymond G. J.; Race B.; Baron G. S.; Caughey B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551. 10.1016/j.molcel.2021.08.011. [DOI] [PubMed] [Google Scholar]
- Wille H.; Requena J. R. The Structure of PrP(Sc) Prions. Pathogens 2018, 7, 20. 10.3390/pathogens7010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietzke S. E.; Scavetta R. D.; Yoder M. D.; Jurnak F. The Refined Three-Dimensional Structure of Pectate Lyase E from Erwinia chrysanthemi at 2.2 A Resolution. Plant Physiol. 1996, 111, 73–92. 10.1104/pp.111.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolli G.; Rigoli M.; Novi Inverardi G.; Codeseira Y. B.; Biasini E.; Requena J. R. Modeling PrP(Sc) Generation Through Deformed Templating. Front. Bioeng. Biotechnol. 2020, 8, 590501. 10.3389/fbioe.2020.590501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baral P. K.; Swayampakula M.; Aguzzi A.; James M. N. X-ray structural and molecular dynamical studies of the globular domains of cow, deer, elk and Syrian hamster prion proteins. J. Struct. Biol. 2015, 192, 37–47. 10.1016/j.jsb.2015.08.014. [DOI] [PubMed] [Google Scholar]
- Stahl N.; Baldwin M. A.; Hecker R.; Pan K. M.; Burlingame A. L.; Prusiner S. B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 1992, 31, 5043–5053. 10.1021/bi00136a600. [DOI] [PubMed] [Google Scholar]
- Rudd P. M.; Endo T.; Colominas C.; Groth D.; Wheeler S. F.; Harvey D. J.; Wormald M. R.; Serban H.; Prusiner S. B.; Kobata A.; Dwek R. A. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 13044–13049. 10.1073/pnas.96.23.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattison I. H.Experiments with Scrapie with Special Reference to the Nature of the Agent and the Pathology of the Disease. In Slow, Latent and Temperate Virus Infections (NINDB Monograph 2), Gajdusek C. J., Gibbs C. J., Alpers M. P., Eds.; U.S. Government Printing Office: Washington D. C., 1965; pp 249–257. [Google Scholar]
- Hunter N.; Hope J.; McConnell I.; Dickinson A. G. Linkage of the scrapie-associated fibril protein (PrP) gene and Sinc using congenic mice and restriction fragment length polymorphism analysis. J. Gen. Virol. 1987, 68, 2711–2716. 10.1099/0022-1317-68-10-2711. [DOI] [PubMed] [Google Scholar]
- Goldmann W.; Hunter N.; Foster J. D.; Salbaum J. M.; Beyreuther K.; Hope J. Two alleles of a neural protein gene linked to scrapie in sheep. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 2476–2480. 10.1073/pnas.87.7.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belt P. B.; Muileman I. H.; Schreuder B. E.; Bos-de Ruijter J.; Gielkens A. L.; Smits M. A. Identification of five allelic variants of the sheep PrP gene and their association with natural scrapie. J. Gen. Virol. 1995, 76, 509–517. 10.1099/0022-1317-76-3-509. [DOI] [PubMed] [Google Scholar]
- Hunter N.; Goldmann W.; Foster J. D.; Cairns D.; Smith G. Natural scrapie and PrP genotype: case-control studies in British sheep. Vet. Rec. 1997, 141, 137–140. 10.1136/vr.141.6.137. [DOI] [PubMed] [Google Scholar]
- Tongue S. C.; Pfeiffer D. U.; Warner R.; Elliott H.; Del Rio Vilas V. Estimation of the relative risk of developing clinical scrapie: the role of prion protein (PrP) genotype and selection bias. Vet. Rec. 2006, 158, 43–50. 10.1136/vr.158.2.43. [DOI] [PubMed] [Google Scholar]
- Cassmann E. D.; Mammadova N.; Moore S. J.; Benestad S.; Greenlee J. J. Transmission of the atypical/Nor98 scrapie agent to Suffolk sheep with VRQ/ARQ, ARQ/ARQ, and ARQ/ARR genotypes. PLoS One 2021, 16, e0246503. 10.1371/journal.pone.0246503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Dur A.; Beringue V.; Andreoletti O.; Reine F.; Lai T. L.; Baron T.; Bratberg B.; Vilotte J. L.; Sarradin P.; Benestad S. L.; Laude H. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 16031–16036. 10.1073/pnas.0502296102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arifin M. I.; Hannaoui S.; Chang S. C.; Thapa S.; Schatzl H. M.; Gilch S. Cervid Prion Protein Polymorphisms: Role in Chronic Wasting Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 2271. 10.3390/ijms22052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva C. J.; Erickson-Beltran M. L.; Duque Velasquez C.; Aiken J. M.; McKenzie D. A General Mass Spectrometry-Based Method of Quantitating Prion Polymorphisms from Heterozygous Chronic Wasting Disease-Infected Cervids. Anal. Chem. 2020, 92, 1276–1284. 10.1021/acs.analchem.9b04449. [DOI] [PubMed] [Google Scholar]
- O’Rourke K. I.; Spraker T. R.; Hamburg L. K.; Besser T. E.; Brayton K. A.; Knowles D. P. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J. Gen. Virol. 2004, 85, 1339–1346. 10.1099/vir.0.79785-0. [DOI] [PubMed] [Google Scholar]
- O’Rourke K. I.; Besser T. E.; Miller M. W.; Cline T. F.; Spraker T. R.; Jenny A. L.; Wild M. A.; Zebarth G. L.; Williams E. S. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 1999, 80, 2765–2679. 10.1099/0022-1317-80-10-2765. [DOI] [PubMed] [Google Scholar]
- Johnson C.; Johnson J.; Vanderloo J. P.; Keane D.; Aiken J. M.; McKenzie D. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J. Gen. Virol. 2006, 87, 2109–2114. 10.1099/vir.0.81615-0. [DOI] [PubMed] [Google Scholar]
- Haley N. J.; Merrett K.; Buros Stein A.; Simpson D.; Carlson A.; Mitchell G.; Staskevicius A.; Nichols T.; Lehmkuhl A. D.; Thomsen B. V. Estimating relative CWD susceptibility and disease progression in farmed white-tailed deer with rare PRNP alleles. PLoS One 2019, 14, e0224342. 10.1371/journal.pone.0224342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane D. P.; Barr D. J.; Bochsler P. N.; Hall S. M.; Gidlewski T.; O’Rourke K. I.; Spraker T. R.; Samuel M. D. Chronic wasting disease in a Wisconsin white-tailed deer farm. J. Vet. Diagn. Invest. 2008, 20, 698–703. 10.1177/104063870802000534. [DOI] [PubMed] [Google Scholar]
- Jewell J. E.; Conner M. M.; Wolfe L. L.; Miller M. W.; Williams E. S. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J. Gen. Virol. 2005, 86, 2127–2134. 10.1099/vir.0.81077-0. [DOI] [PubMed] [Google Scholar]
- Fox K. A.; Jewell J. E.; Williams E. S.; Miller M. W. Patterns of PrPCWD accumulation during the course of chronic wasting disease infection in orally inoculated mule deer (Odocoileus hemionus). J. Gen. Virol. 2006, 87, 3451–3461. 10.1099/vir.0.81999-0. [DOI] [PubMed] [Google Scholar]
- Johnson C. J.; Herbst A.; Duque-Velasquez C.; Vanderloo J. P.; Bochsler P.; Chappell R.; McKenzie D. Prion protein polymorphisms affect chronic wasting disease progression. PLoS One 2011, 6, e17450. 10.1371/journal.pone.0017450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. W.; Wolfe L. L.; Sirochman T. M.; Sirochman M. A.; Jewell J. E.; Williams E. S. Survival patterns in white-tailed and mule deer after oral inoculation with a standardized, conspecific prion dose. J. Wildl. Dis. 2012, 48, 526–529. 10.7589/0090-3558-48.2.526. [DOI] [PubMed] [Google Scholar]
- Bian J.; Christiansen J. R.; Moreno J. A.; Kane S. J.; Khaychuk V.; Gallegos J.; Kim S.; Telling G. C. Primary structural differences at residue 226 of deer and elk PrP dictate selection of distinct CWD prion strains in gene-targeted mice. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 12478–12487. 10.1073/pnas.1903947116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M.; Baiardi S.; Parchi P. Understanding Prion Strains: Evidence from Studies of the Disease Forms Affecting Humans. Viruses 2019, 11, 309. 10.3390/v11040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce M. E.; Will R. G.; Ironside J. W.; McConnell I.; Drummond D.; Suttie A.; McCardle L.; Chree A.; Hope J.; Birkett C.; Cousens S.; Fraser H.; Bostock C. J. Transmissions to mice indicate that ’new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- Foster J. D.; Hope J.; McConnell I.; Bruce M.; Fraser H. Transmission of bovine spongiform encephalopathy to sheep, goats, and mice. Ann. N.Y. Acad. Sci. 1994, 724, 300–303. 10.1111/j.1749-6632.1994.tb38920.x. [DOI] [PubMed] [Google Scholar]
- Foster J. D.; Hope J.; Fraser H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet. Rec. 1993, 133, 339–341. 10.1136/vr.133.14.339. [DOI] [PubMed] [Google Scholar]
- Bruce M.; Chree A.; McConnell I.; Foster J.; Pearson G.; Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos. Trans. R. Soc. London B Biol. Sci. 1994, 343, 405–411. 10.1098/rstb.1994.0036. [DOI] [PubMed] [Google Scholar]
- Brandel J. P.; Vlaicu M. B.; Culeux A.; Belondrade M.; Bougard D.; Grznarova K.; Denouel A.; Plu I.; Bouaziz-Amar E.; Seilhean D.; Levasseur M.; Haik S. Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure. N. Engl. J. Med. 2020, 383, 83–85. 10.1056/NEJMc2000687. [DOI] [PubMed] [Google Scholar]
- Makarava N.; Baskakov I. V. The evolution of transmissible prions: the role of deformed templating. PLoS Pathog. 2013, 9, e1003759. 10.1371/journal.ppat.1003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J.; Clarke A. R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- Aguzzi A.; Heikenwalder M.; Polymenidou M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- Simmons M. M.; Moore S. J.; Lockey R.; Chaplin M. J.; Konold T.; Vickery C.; Spiropoulos J. Phenotype shift from atypical scrapie to CH1641 following experimental transmission in sheep. PLoS One 2015, 10, e0117063. 10.1371/journal.pone.0117063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huor A.; Espinosa J. C.; Vidal E.; Cassard H.; Douet J. Y.; Lugan S.; Aron N.; Marin-Moreno A.; Lorenzo P.; Aguilar-Calvo P.; Badiola J.; Bolea R.; Pumarola M.; Benestad S. L.; Orge L.; Thackray A. M.; Bujdoso R.; Torres J. M.; Andreoletti O. The emergence of classical BSE from atypical/Nor98 scrapie. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 26853–26862. 10.1073/pnas.1915737116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín B.; Otero A.; Lugan S.; Espinosa J. C.; Marín-Moreno A.; Vidal E.; Hedman C.; Romero A.; Pumarola M.; Badiola J. J.; Torres J. M.; Andréoletti O.; Bolea R. Classical BSE prions emerge from asymptomatic pigs challenged with atypical/Nor98 scrapie. Sci. Rep. 2021, 11, 17428. 10.1038/s41598-021-96818-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konold T.; Arnold M. E.; Austin A. R.; Cawthraw S.; Hawkins S. A.; Stack M. J.; Simmons M. M.; Sayers A. R.; Dawson M.; Wilesmith J. W.; Wells G. A. Bovine spongiform encephalopathy: the effect of oral exposure dose on attack rate and incubation period in cattle—An update. BMC Res. Notes 2012, 5, 674. 10.1186/1756-0500-5-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells G. A. H.; Konold T.; Arnold M. E.; Austin A. R.; Hawkins S. A. C.; Stack M.; Simmons M. M.; Lee Y. H.; Gavier-Widen D.; Dawson M.; Wilesmith J. W. Bovine spongiform encephalopathy: the effect of oral exposure dose on attack rate and incubation period in cattle. J. Gen. Virol. 2007, 88, 1363–1373. 10.1099/vir.0.82421-0. [DOI] [PubMed] [Google Scholar]
- McGovern G.; Martin S.; Jeffrey M.; Dexter G.; Hawkins S. A.; Bellworthy S. J.; Thurston L.; Algar L.; Gonzalez L. Minimum Effective Dose of Cattle and Sheep BSE for Oral Sheep Infection. PLoS One 2016, 11, e0151440 10.1371/journal.pone.0151440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham A. A.; Kirkwood J. K.; Dawson M.; Spencer Y. I.; Green R. B.; Wells G. A. Bovine spongiform encephalopathy infectivity in greater kudu (Tragelaphus strepsiceros). Emerg. Infect. Dis. 2004, 10, 1044–1049. 10.3201/eid1006.030615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S. B.; Groth D.; Serban A.; Stahl N.; Gabizon R. Attempts to restore scrapie prion infectivity after exposure to protein denaturants. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2793–2797. 10.1073/pnas.90.7.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diack A. B.; Alibhai J. D.; Manson J. C. Gene Targeted Transgenic Mouse Models in Prion Research. Prog. Mol. Biol. Transl. Sci. 2017, 150, 157–179. 10.1016/bs.pmbts.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Asher D. M.; Gregori L. Human transmissible spongiform encephalopathies: Historic view. Handb. Clin. Neurol. 2018, 153, 1–17. 10.1016/B978-0-444-63945-5.00001-5. [DOI] [PubMed] [Google Scholar]
- Ironside J. W.; Ritchie D. L.; Head M. W. Prion diseases. Handb. Clin. Neurol. 2018, 145, 393–403. 10.1016/B978-0-12-802395-2.00028-6. [DOI] [PubMed] [Google Scholar]
- Notari S.; Appleby B. S.; Gambetti P. Variably protease-sensitive prionopathy. Handb. Clin. Neurol. 2018, 153, 175–190. 10.1016/B978-0-444-63945-5.00010-6. [DOI] [PubMed] [Google Scholar]
- Takada L. T.; Kim M. O.; Metcalf S.; Gala I. I.; Geschwind M. D. Prion disease. Handb. Clin. Neurol. 2018, 148, 441–464. 10.1016/B978-0-444-64076-5.00029-6. [DOI] [PubMed] [Google Scholar]
- Ladogana A.; Kovacs G. G. Genetic Creutzfeldt–Jakob disease. Handb. Clin. Neurol. 2018, 153, 219–242. 10.1016/B978-0-444-63945-5.00013-1. [DOI] [PubMed] [Google Scholar]
- Cracco L.; Appleby B. S.; Gambetti P. Fatal familial insomnia and sporadic fatal insomnia. Handb. Clin. Neurol. 2018, 153, 271–299. 10.1016/B978-0-444-63945-5.00015-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Kitamoto T.; Mizusawa H. Iatrogenic Creutzfeldt–Jakob disease. Handb. Clin. Neurol. 2018, 153, 207–218. 10.1016/B978-0-444-63945-5.00012-X. [DOI] [PubMed] [Google Scholar]
- Zerr I.; Parchi P. Sporadic Creutzfeldt–Jakob disease. Handb. Clin. Neurol. 2018, 153, 155–174. 10.1016/B978-0-444-63945-5.00009-X. [DOI] [PubMed] [Google Scholar]
- Harman J. L.; Silva C. J. Bovine spongiform encephalopathy. J. Am. Vet. Med. Assoc. 2009, 234, 59–72. 10.2460/javma.234.1.59. [DOI] [PubMed] [Google Scholar]
- Will R. G.; Ironside J. W.; Zeidler M.; Cousens S. N.; Estibeiro K.; Alperovitch A.; Poser S.; Pocchiari M.; Hofman A.; Smith P. G. A new variant of Creutzfeldt–Jakob disease in the UK. Lancet 1996, 347, 921–925. 10.1016/S0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- Mok T.; Jaunmuktane Z.; Joiner S.; Campbell T.; Morgan C.; Wakerley B.; Golestani F.; Rudge P.; Mead S.; Jager H. R.; Wadsworth J. D.; Brandner S.; Collinge J. Variant Creutzfeldt–Jakob Disease in a Patient with Heterozygosity at PRNP Codon 129. N. Engl. J. Med. 2017, 376, 292–294. 10.1056/NEJMc1610003. [DOI] [PubMed] [Google Scholar]
- Kaski D.; Mead S.; Hyare H.; Cooper S.; Jampana R.; Overell J.; Knight R.; Collinge J.; Rudge P. Variant CJD in an individual heterozygous for PRNP codon 129. Lancet 2009, 374, 2128. 10.1016/S0140-6736(09)61568-3. [DOI] [PubMed] [Google Scholar]
- Mead S.; Poulter M.; Uphill J.; Beck J.; Whitfield J.; Webb T. E.; Campbell T.; Adamson G.; Deriziotis P.; Tabrizi S. J.; Hummerich H.; Verzilli C.; Alpers M. P.; Whittaker J. C.; Collinge J. Genetic risk factors for variant Creutzfeldt–Jakob disease: A genome-wide association study. Lancet Neurol. 2009, 8, 57–66. 10.1016/S1474-4422(08)70265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandel J. P.; Knight R. Variant Creutzfeldt–Jakob disease. Handb. Clin. Neurol. 2018, 153, 191–205. 10.1016/B978-0-444-63945-5.00011-8. [DOI] [PubMed] [Google Scholar]
- Llewelyn C. A.; Hewitt P. E.; Knight R. S.; Amar K.; Cousens S.; Mackenzie J.; Will R. G. Possible transmission of variant Creutzfeldt–Jakob disease by blood transfusion. Lancet 2004, 363, 417–421. 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- Bishop M. T.; Diack A. B.; Ritchie D. L.; Ironside J. W.; Will R. G.; Manson J. C. Prion infectivity in the spleen of a PRNP heterozygous individual with subclinical variant Creutzfeldt–Jakob disease. Brain 2013, 136, 1139–1145. 10.1093/brain/awt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson N.; Brandel J. P.; Green A.; Hermann P.; Ladogana A.; Lindsay T.; Mackenzie J.; Pocchiari M.; Smith C.; Zerr I.; Pal S. The importance of ongoing international surveillance for Creutzfeldt–Jakob disease. Nat. Rev. Neurol. 2021, 17, 362–379. 10.1038/s41582-021-00488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberski P. P.; Gajos A.; Sikorska B.; Lindenbaum S. Kuru, the First Human Prion Disease. Viruses 2019, 11, 232. 10.3390/v11030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlow W. J. Kuru likened to scrapie: the story remembered. Philos. Trans. R. Soc. London B Biol. Sci. 2008, 363, 3644. 10.1098/rstb.2008.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlow W. J. Scrapie and kuru. Lancet 1959, 274, 289–290. 10.1016/S0140-6736(59)92081-1. [DOI] [Google Scholar]
- Alpers M. P. Review. The epidemiology of kuru: Monitoring the epidemic from its peak to its end. Philos. Trans. R. Soc. London B Biol. Sci. 2008, 363, 3707–3713. 10.1098/rstb.2008.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S.; Brown P.; Cervenakova L.; Garruto R. M.; Alpers M. P.; Gajdusek D. C.; Goldfarb L. G. Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J. Infect. Dis. 2001, 183, 192–196. 10.1086/317935. [DOI] [PubMed] [Google Scholar]
- Cervenakova L.; Goldfarb L. G.; Garruto R.; Lee H. S.; Gajdusek D. C.; Brown P. Phenotype-genotype studies in kuru: implications for new variant Creutzfeldt–Jakob disease. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 13239–13241. 10.1073/pnas.95.22.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead S.; Stumpf M. P.; Whitfield J.; Beck J. A.; Poulter M.; Campbell T.; Uphill J. B.; Goldstein D.; Alpers M.; Fisher E. M.; Collinge J. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003, 300, 640–643. 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- Mead S.; Whitfield J.; Poulter M.; Shah P.; Uphill J.; Campbell T.; Al-Dujaily H.; Hummerich H.; Beck J.; Mein C. A.; Verzilli C.; Whittaker J.; Alpers M. P.; Collinge J. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. 10.1056/NEJMoa0809716. [DOI] [PubMed] [Google Scholar]
- Asante E. A.; Smidak M.; Grimshaw A.; Houghton R.; Tomlinson A.; Jeelani A.; Jakubcova T.; Hamdan S.; Richard-Londt A.; Linehan J. M.; Brandner S.; Alpers M.; Whitfield J.; Mead S.; Wadsworth J. D.; Collinge J. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. 10.1038/nature14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. S.; Young S. Spongiform encephalopathy of Rocky Mountain elk. J. Wildl. Dis. 1982, 18, 465–471. 10.7589/0090-3558-18.4.465. [DOI] [PubMed] [Google Scholar]
- Spraker T. R.; Miller M. W.; Williams E. S.; Getzy D. M.; Adrian W. J.; Schoonveld G. G.; Spowart R. A.; O’Rourke K. I.; Miller J. M.; Merz P. A. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J. Wildl. Dis. 1997, 33, 1–6. 10.7589/0090-3558-33.1.1. [DOI] [PubMed] [Google Scholar]
- Baeten L. A.; Powers B. E.; Jewell J. E.; Spraker T. R.; Miller M. W. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). J. Wildl. Dis. 2007, 43, 309–314. 10.7589/0090-3558-43.2.309. [DOI] [PubMed] [Google Scholar]
- Schwabenlander M. D.; Culhane M. R.; Hall S. M.; Goyal S. M.; Anderson P. L.; Carstensen M.; Wells S. J.; Slade W. B.; Armien A. G. A case of chronic wasting disease in a captive red deer (Cervus elaphus). J. Vet. Diagn. Invest. 2013, 25, 573–576. 10.1177/1040638713499914. [DOI] [PubMed] [Google Scholar]
- Miller M. W.; Williams E. S.; Hobbs N. T.; Wolfe L. L. Environmental sources of prion transmission in mule deer. Emerg. Infect. Dis. 2004, 10, 1003–1006. 10.3201/eid1006.040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. W.; Williams E. S. Prion disease: Horizontal prion transmission in mule deer. Nature 2003, 425, 35–36. 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- Kuznetsova A.; McKenzie D.; Cullingham C.; Aiken J. M. Long-Term Incubation PrP(CWD) with Soils Affects Prion Recovery but Not Infectivity. Pathogens 2020, 9, 311. 10.3390/pathogens9040311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. J.; Phillips K. E.; Schramm P. T.; McKenzie D.; Aiken J. M.; Pedersen J. A. Prions adhere to soil minerals and remain infectious. PLoS Pathog. 2006, 2, e32. 10.1371/journal.ppat.0020032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley N. J.; Seelig D. M.; Zabel M. D.; Telling G. C.; Hoover E. A. Detection of CWD prions in urine and saliva of deer by transgenic mouse bioassay. PLoS One 2009, 4, e4848. 10.1371/journal.pone.0004848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiason C. K.; Powers J. G.; Dahmes S. J.; Osborn D. A.; Miller K. V.; Warren R. J.; Mason G. L.; Hays S. A.; Hayes-Klug J.; Seelig D. M.; Wild M. A.; Wolfe L. L.; Spraker T. R.; Miller M. W.; Sigurdson C. J.; Telling G. C.; Hoover E. A. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 2006, 314, 133–136. 10.1126/science.1132661. [DOI] [PubMed] [Google Scholar]
- Tamguney G.; Miller M. W.; Wolfe L. L.; Sirochman T. M.; Glidden D. V.; Palmer C.; Lemus A.; DeArmond S. J.; Prusiner S. B. Asymptomatic deer excrete infectious prions in faeces. Nature 2009, 461, 529–532. 10.1038/nature08289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammadova N.; Cassmann E.; Greenlee J. J. Successful transmission of the chronic wasting disease (CWD) agent to white-tailed deer by intravenous blood transfusion. Res. Vet. Sci. 2020, 133, 304–306. 10.1016/j.rvsc.2020.10.009. [DOI] [PubMed] [Google Scholar]
- Dubé C.; Mehren K. G.; Barker I. K.; Peart B. L.; Balachandran A. Retrospective investigation of chronic wasting disease of cervids at the Toronto Zoo, 1973–2003. Can. Vet. J. 2006, 47, 1185–1193. [PMC free article] [PubMed] [Google Scholar]
- Kahn S.; Dubé C.; Bates L.; Balachandran A. Chronic wasting disease in Canada: Part 1. Can. Vet. J. 2004, 45, 397–404. [PMC free article] [PubMed] [Google Scholar]
- Sohn H. J.; Kim J. H.; Choi K. S.; Nah J. J.; Joo Y. S.; Jean Y. H.; Ahn S. W.; Kim O. K.; Kim D. Y.; Balachandran A. A case of chronic wasting disease in an elk imported to Korea from Canada. J. Vet. Med. Sci. 2002, 64, 855–858. 10.1292/jvms.64.855. [DOI] [PubMed] [Google Scholar]
- Sohn H. J.; Roh I. S.; Kim H. J.; Suh T.-Y.; Park K. J.; Park H. C.; Kim B. WS-03: Epidemiology of chronic wasting disease in Korea. Prion 2016, 10, S15–S21. 10.1080/19336896.2016.1163048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. S.; Miller M. W. Chronic wasting disease in deer and elk in North America. Rev. Sci. Technol. 2002, 21, 305–316. 10.20506/rst.21.2.1340. [DOI] [PubMed] [Google Scholar]
- Miller M. W.; Conner M. M. Epidemiology of chronic wasting disease in free-ranging mule deer: spatial, temporal, and demographic influences on observed prevalence patterns. J. Wildl. Dis. 2005, 41, 275–290. 10.7589/0090-3558-41.2.275. [DOI] [PubMed] [Google Scholar]
- Mysterud A.; Madslien K.; Viljugrein H.; Vikøren T.; Andersen R.; Güere M. E.; Benestad S. L.; Hopp P.; Strand O.; Ytrehus B.; Røed K. H.; Rolandsen C. M.; Våge J. The demographic pattern of infection with chronic wasting disease in reindeer at an early epidemic stage. Ecosphere 2019, 10, e02931. 10.1002/ecs2.2931. [DOI] [Google Scholar]
- Kim T. Y.; Shon H. J.; Joo Y. S.; Mun U. K.; Kang K. S.; Lee Y. S. Additional cases of Chronic Wasting Disease in imported deer in Korea. J. Vet. Med. Sci. 2005, 67, 753–759. 10.1292/jvms.67.753. [DOI] [PubMed] [Google Scholar]
- Lee Y. H.; Sohn H. J.; Kim M. J.; Kim H. J.; Lee W. Y.; Yun E. I.; Tark D. S.; Cho I. S.; Balachandran A. Strain characterization of the Korean CWD cases in 2001 and 2004. J. Vet. Med. Sci. 2013, 75, 95–98. 10.1292/jvms.12-0077. [DOI] [PubMed] [Google Scholar]
- Sohn H. J.; Mitchell G.; Lee Y. H.; Kim H. J.; Park K. J.; Staskevicus A.; Walther I.; Soutyrine A.; Balachandran A. Experimental oral transmission of chronic wasting disease to sika deer (Cervus nippon). Prion 2020, 14, 271–277. 10.1080/19336896.2020.1857038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran A.; Harrington N. P.; Algire J.; Soutyrine A.; Spraker T. R.; Jeffrey M.; Gonzalez L.; O’Rourke K. I. Experimental oral transmission of chronic wasting disease to red deer (Cervus elaphus elaphus): Early detection and late stage distribution of protease-resistant prion protein. Can. Vet. J. 2010, 51, 169–178. [PMC free article] [PubMed] [Google Scholar]
- Vikøren T.; Våge J.; Madslien K. I.; Røed K. H.; Rolandsen C. M.; Tran L.; Hopp P.; Veiberg V.; Heum M.; Moldal T.; das Neves C. G.; Handeland K.; Ytrehus B.; Kolbjørnsen O.; Wisløff H.; Terland R.; Saure B.; Dessen K. M.; Svendsen S. G.; Nordvik B. S.; Benestad S. L. First Detection of Chronic Wasting Disease in a Wild Red Deer (Cervus elaphus) in Europe. J. Wildl. Dis. 2019, 55, 970–972. 10.7589/2018-10-262. [DOI] [PubMed] [Google Scholar]
- Benestad S. L.; Mitchell G.; Simmons M.; Ytrehus B.; Vikøren T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47, 88. 10.1186/s13567-016-0375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirisinu L.; Tran L.; Chiappini B.; Vanni I.; Di Bari M. A.; Vaccari G.; Vikøren T.; Madslien K. I.; Våge J.; Spraker T.; Mitchell G.; Balachandran A.; Baron T.; Casalone C.; Rolandsen C. M.; Røed K. H.; Agrimi U.; Nonno R.; Benestad S. L. Novel Type of Chronic Wasting Disease Detected in Moose (Alces alces), Norway. Emerg. Infect. Dis. 2018, 24, 2210–2218. 10.3201/eid2412.180702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ågren E. O.; Sörén K.; Gavier-Widén D.; Benestad S. L.; Tran L.; Wall K.; Averhed G.; Doose N.; Våge J.; Nöremark M. First Detection of Chronic Wasting Disease in Moose (Alces alces) in Sweden. J. Wildl. Dis. 2021, 57, 461–463. 10.7589/JWD-D-20-00141. [DOI] [PubMed] [Google Scholar]
- Nonno R.; Di Bari M. A.; Pirisinu L.; D’Agostino C.; Vanni I.; Chiappini B.; Marcon S.; Riccardi G.; Tran L.; Vikoren T.; Vage J.; Madslien K.; Mitchell G.; Telling G. C.; Benestad S. L.; Agrimi U. Studies in bank voles reveal strain differences between chronic wasting disease prions from Norway and North America. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 31417–31426. 10.1073/pnas.2013237117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritzkow S.; Gorski D.; Ramirez F.; Telling G. C.; Benestad S. L.; Soto C. North American and Norwegian Chronic Wasting Disease Prions Exhibit Different Potential for Interspecies Transmission and Zoonotic Risk. J. Infect. Dis. 2022, 225, 542–551. 10.1093/infdis/jiab385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous Regulation (EC) No 999/2001 of the European Parliament and of the Council of 22 May 2001 laying down rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies. Off. J. Eur. Communities 2001, L147, 1–40. [Google Scholar]
- Simmons M. M.; Moore S. J.; Konold T.; Thurston L.; Terry L. A.; Thorne L.; Lockey R.; Vickery C.; Hawkins S. A.; Chaplin M. J.; Spiropoulos J. Experimental oral transmission of atypical scrapie to sheep. Emerg. Infect. Dis. 2011, 17, 848–854. 10.3201/eid1705.101654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J.; Kim S.; Kane S. J.; Crowell J.; Sun J. L.; Christiansen J.; Saijo E.; Moreno J. A.; DiLisio J.; Burnett E.; Pritzkow S.; Gorski D.; Soto C.; Kreeger T. J.; Balachandran A.; Mitchell G.; Miller M. W.; Nonno R.; Vikoren T.; Vage J.; Madslien K.; Tran L.; Vuong T. T.; Benestad S. L.; Telling G. C. Adaptive selection of a prion strain conformer corresponding to established North American CWD during propagation of novel emergent Norwegian strains in mice expressing elk or deer prion protein. PLoS Pathog. 2021, 17, e1009748. 10.1371/journal.ppat.1009748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysterud A.; Rolandsen C. M. A reindeer cull to prevent chronic wasting disease in Europe. Nat. Ecol. Evol. 2018, 2, 1343–1345. 10.1038/s41559-018-0616-1. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Cutlip R. C.; Miller J. M.; Williams E. S.; Stack M. J.; Miller M. W.; O’Rourke K. I.; Chaplin M. J. Preliminary findings on the experimental transmission of chronic wasting disease agent of mule deer to cattle. J. Vet. Diagn. Invest. 2001, 13, 91–96. 10.1177/104063870101300121. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Kunkle R. A.; Cutlip R. C.; Miller J. M.; O’Rourke K. I.; Williams E. S.; Miller M. W.; Stack M. J.; Chaplin M. J.; Richt J. A. Experimental transmission of chronic wasting disease agent from mule deer to cattle by the intracerebral route. J. Vet. Diagn. Invest. 2005, 17, 276–281. 10.1177/104063870501700313. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Miller J. M.; Kunkle R. A.; Hall S. M.; Richt J. A. Susceptibility of cattle to first-passage intracerebral inoculation with chronic wasting disease agent from white-tailed deer. Vet. Pathol. 2007, 44, 487–493. 10.1354/vp.44-4-487. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Kunkle R. A.; Cutlip R. C.; Miller J. M.; Williams E. S.; Richt J. A. Transmission of chronic wasting disease of mule deer to Suffolk sheep following intracerebral inoculation. J. Vet. Diagn. Invest. 2006, 18, 558–565. 10.1177/104063870601800606. [DOI] [PubMed] [Google Scholar]
- Moore S. J.; Smith J. D.; Richt J. A.; Greenlee J. J. Raccoons accumulate PrP(Sc) after intracranial inoculation of the agents of chronic wasting disease or transmissible mink encephalopathy but not atypical scrapie. J. Vet. Diagn. Invest. 2019, 31, 200–209. 10.1177/1040638718825290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee J. J.; Nicholson E. M.; Smith J. D.; Kunkle R. A.; Hamir A. N. Susceptibility of cattle to the agent of chronic wasting disease from elk after intracranial inoculation. J. Vet. Diagn. Invest. 2012, 24, 1087–1093. 10.1177/1040638712461249. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Kunkle R. A.; Miller J. M.; Greenlee J. J.; Richt J. A. Experimental second passage of chronic wasting disease (CWD(mule deer)) agent to cattle. J. Comp. Pathol. 2006, 134, 63–69. 10.1016/j.jcpa.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Williams E. S. Chronic wasting disease. Vet. Pathol. 2005, 42, 530–549. 10.1354/vp.42-5-530. [DOI] [PubMed] [Google Scholar]
- Gould D. H.; Voss J. L.; Miller M. W.; Bachand A. M.; Cummings B. A.; Frank A. A. Survey of cattle in northeast Colorado for evidence of chronic wasting disease: geographical and high-risk targeted sample. J. Vet. Diagn. Invest. 2003, 15, 274–277. 10.1177/104063870301500309. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Kunkle R. A.; Miller J. M.; Cutlip R. C.; Richt J. A.; Kehrli M. E. Jr.; Williams E. S. Age-related lesions in laboratory-confined raccoons (Procyon lotor) inoculated with the agent of chronic wasting disease of mule deer. J. Vet. Diagn. Invest. 2007, 19, 680–686. 10.1177/104063870701900610. [DOI] [PubMed] [Google Scholar]
- Sigurdson C. J.; Mathiason C. K.; Perrott M. R.; Eliason G. A.; Spraker T. R.; Glatzel M.; Manco G.; Bartz J. C.; Miller M. W.; Hoover E. A. Experimental chronic wasting disease (CWD) in the ferret. J. Comp. Pathol. 2008, 138, 189–196. 10.1016/j.jcpa.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Bartz J. C.; Marsh R. F.; McKenzie D. I.; Aiken J. M. The host range of chronic wasting disease is altered on passage in ferrets. Virology 1998, 251, 297–301. 10.1006/viro.1998.9427. [DOI] [PubMed] [Google Scholar]
- Williams E. S.; Young S. Spongiform encephalopathies in Cervidae. Rev. Sci. Technol. 1992, 11, 551–567. 10.20506/rst.11.2.611. [DOI] [PubMed] [Google Scholar]
- Dagleish M. P.; Martin S.; Steele P.; Finlayson J.; Siso S.; Hamilton S.; Chianini F.; Reid H. W.; Gonzalez L.; Jeffrey M. Experimental transmission of bovine spongiform encephalopathy to European red deer (Cervus elaphus elaphus). BMC Vet. Res. 2008, 4, 17. 10.1186/1746-6148-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagleish M. P.; Martin S.; Steele P.; Finlayson J.; Eaton S. L.; Siso S.; Stewart P.; Fernandez-Borges N.; Hamilton S.; Pang Y.; Chianini F.; Reid H. W.; Goldmann W.; Gonzalez L.; Castilla J.; Jeffrey M. Susceptibility of European red deer (Cervus elaphus elaphus) to alimentary challenge with bovine spongiform encephalopathy. PLoS One 2015, 10, e0116094. 10.1371/journal.pone.0116094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.; Jeffrey M.; Gonzalez L.; Siso S.; Reid H. W.; Steele P.; Dagleish M. P.; Stack M. J.; Chaplin M. J.; Balachandran A. Immunohistochemical and biochemical characteristics of BSE and CWD in experimentally infected European red deer (Cervus elaphus elaphus). BMC Vet. Res. 2009, 5, 26. 10.1186/1746-6148-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery C. M.; Lockey R.; Holder T. M.; Thorne L.; Beck K. E.; Wilson C.; Denyer M.; Sheehan J.; Marsh S.; Webb P. R.; Dexter I.; Norman A.; Popescu E.; Schneider A.; Holden P.; Griffiths P. C.; Plater J. M.; Dagleish M. P.; Martin S.; Telling G. C.; Simmons M. M.; Spiropoulos J. Assessing the susceptibility of transgenic mice overexpressing deer prion protein to bovine spongiform encephalopathy. J. Virol. 2014, 88, 1830–1833. 10.1128/JVI.02762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamir A. N.; Miller J. M.; Cutlip R. C.; Stack M. J.; Chaplin M. J.; Jenny A. L. Preliminary observations on the experimental transmission of scrapie to elk (Cervus elaphus nelsoni) by intracerebral inoculation. Vet. Pathol. 2003, 40, 81–85. 10.1354/vp.40-1-81. [DOI] [PubMed] [Google Scholar]
- Hamir A. N.; Miller J. M.; Cutlip R. C.; Kunkle R. A.; Jenny A. L.; Stack M. J.; Chaplin M. J.; Richt J. A. Transmission of sheep scrapie to elk (Cervus elaphus nelsoni) by intracerebral inoculation: final outcome of the experiment. J. Vet. Diagn. Invest. 2004, 16, 316–321. 10.1177/104063870401600410. [DOI] [PubMed] [Google Scholar]
- Madsen-Bouterse S. A.; Schneider D. A.; Zhuang D.; Dassanayake R. P.; Balachandran A.; Mitchell G. B.; O’Rourke K. I. Primary transmission of chronic wasting disease versus scrapie prions from small ruminants to transgenic mice expressing ovine or cervid prion protein. J. Gen. Virol. 2016, 97, 2451–2460. 10.1099/jgv.0.000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney G.; Miller M. W.; Giles K.; Lemus A.; Glidden D. V.; DeArmond S. J.; Prusiner S. B. Transmission of scrapie and sheep-passaged bovine spongiform encephalopathy prions to transgenic mice expressing elk prion protein. J. Gen. Virol. 2009, 90, 1035–1047. 10.1099/vir.0.007500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee J. J.; Smith J. D.; Kunkle R. A. White-tailed deer are susceptible to the agent of sheep scrapie by intracerebral inoculation. Vet. Res. 2011, 42, 107. 10.1186/1297-9716-42-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh R. F.; Kincaid A. E.; Bessen R. A.; Bartz J. C. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus). J. Virol. 2005, 79, 13794–13796. 10.1128/JVI.79.21.13794-13796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race B.; Meade-White K. D.; Miller M. W.; Barbian K. D.; Rubenstein R.; LaFauci G.; Cervenakova L.; Favara C.; Gardner D.; Long D.; Parnell M.; Striebel J.; Priola S. A.; Ward A.; Williams E. S.; Race R.; Chesebro B. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg. Infect. Dis. 2009, 15, 1366–1376. 10.3201/eid1509.090253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Race B.; Williams K.; Orrú C. D.; Hughson A. G.; Lubke L.; Chesebro B. Lack of Transmission of Chronic Wasting Disease to Cynomolgus Macaques. J. Virol. 2018, 92, e00550-18. 10.1128/JVI.00550-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawhinney S.; Pape W. J.; Forster J. E.; Anderson C. A.; Bosque P.; Miller M. W. Human prion disease and relative risk associated with chronic wasting disease. Emerg. Infect. Dis. 2006, 12, 1527–1535. 10.3201/eid1210.060019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams J. Y.; Maddox R. A.; Harvey A. R.; Schonberger L. B.; Belay E. D. Travel history, hunting, and venison consumption related to prion disease exposure, 2006–2007 FoodNet Population Survey. J. Am. Diet Assoc. 2011, 111, 858–863. 10.1016/j.jada.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Davis J. P.; et al. Fatal degenerative neurologic illnesses in men who participated in wild game feasts—Wisconsin, 2002. MMWR Morb. Mortal. Wkly. Rep. 2003, 52, 125–127. [PubMed] [Google Scholar]
- Wilson R.; Plinston C.; Hunter N.; Casalone C.; Corona C.; Tagliavini F.; Suardi S.; Ruggerone M.; Moda F.; Graziano S.; Sbriccoli M.; Cardone F.; Pocchiari M.; Ingrosso L.; Baron T.; Richt J.; Andreoletti O.; Simmons M.; Lockey R.; Manson J. C.; Barron R. M. Chronic wasting disease and atypical forms of bovine spongiform encephalopathy and scrapie are not transmissible to mice expressing wild-type levels of human prion protein. J. Gen. Virol. 2012, 93, 1624–1629. 10.1099/vir.0.042507-0. [DOI] [PubMed] [Google Scholar]
- Sandberg M. K.; Al-Doujaily H.; Sigurdson C. J.; Glatzel M.; O’Malley C.; Powell C.; Asante E. A.; Linehan J. M.; Brandner S.; Wadsworth J. D.; Collinge J. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J. Gen. Virol. 2010, 91, 2651–2657. 10.1099/vir.0.024380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth J. D. F.; Joiner S.; Linehan J. M.; Jack K.; Al-Doujaily H.; Costa H.; Ingold T.; Taema M.; Zhang F.; Sandberg M. K.; Brandner S.; Tran L.; Vikøren T.; Våge J.; Madslien K.; Ytrehus B.; Benestad S. L.; Asante E. A.; Collinge J. Humanised transgenic mice are resistant to chronic wasting disease prions from Norwegian reindeer and moose. J. Infect. Dis. 2021, jiab033. 10.1093/infdis/jiab033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth J. D.; Asante E. A.; Collinge J. Review: contribution of transgenic models to understanding human prion disease. Neuropathol. Appl. Neurobiol. 2010, 36, 576–597. 10.1111/j.1365-2990.2010.01129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulberger J.; Hobbs N. T.; Swanson H. M.; Bishop C. J.; Miller M. W. Estimating chronic wasting disease effects on mule deer recruitment and population growth. J. Wildl. Dis. 2010, 46, 1086–1095. 10.7589/0090-3558-46.4.1086. [DOI] [PubMed] [Google Scholar]
- Krumm C. E.; Conner M. M.; Hobbs N. T.; Hunter D. O.; Miller M. W. Mountain lions prey selectively on prion-infected mule deer. Biol. Lett. 2010, 6, 209–211. 10.1098/rsbl.2009.0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. W.; Swanson H. M.; Wolfe L. L.; Quartarone F. G.; Huwer S. L.; Southwick C. H.; Lukacs P. M. Lions and prions and deer demise. PLoS One 2008, 3, e4019. 10.1371/journal.pone.0004019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm C. E.; Conner M. M.; Miller M. W. Relative vulnerability of chronic wasting disease infected mule deer to vehicle collisions. J. Wildl. Dis. 2005, 41, 503–511. 10.7589/0090-3558-41.3.503. [DOI] [PubMed] [Google Scholar]
- DeVivo M. T.; Edmunds D. R.; Kauffman M. J.; Schumaker B. A.; Binfet J.; Kreeger T. J.; Richards B. J.; Schatzl H. M.; Cornish T. E. Endemic chronic wasting disease causes mule deer population decline in Wyoming. PLoS One 2017, 12, e0186512. 10.1371/journal.pone.0186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S. J.; Samuel M. D.; O’Rourke K. I.; Johnson C. J. The role of genetics in chronic wasting disease of North American cervids. Prion 2012, 6, 153–162. 10.4161/pri.19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamir A. N.; Gidlewski T.; Spraker T. R.; Miller J. M.; Creekmore L.; Crocheck M.; Cline T.; O’Rourke K. I. Preliminary observations of genetic susceptibility of elk (Cervus elaphus nelsoni) to chronic wasting disease by experimental oral inoculation. J. Vet. Diagn. Invest. 2006, 18, 110–114. 10.1177/104063870601800118. [DOI] [PubMed] [Google Scholar]
- Robinson S. J.; Samuel M. D.; Johnson C. J.; Adams M.; McKenzie D. I. Emerging prion disease drives host selection in a wildlife population. Ecol. Appl. 2012, 22, 1050–1059. 10.1890/11-0907.1. [DOI] [PubMed] [Google Scholar]
- Miller W. L.; Walter W. D. Spatial heterogeneity of prion gene polymorphisms in an area recently infected by chronic wasting disease. Prion 2019, 13, 65–76. 10.1080/19336896.2019.1583042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannaoui S.; Triscott E.; Duque Velasquez C.; Chang S. C.; Arifin M. I.; Zemlyankina I.; Tang X.; Bollinger T.; Wille H.; McKenzie D.; Gilch S. New and distinct chronic wasting disease strains associated with cervid polymorphism at codon 116 of the Prnp gene. PLoS Pathog. 2021, 17, e1009795. 10.1371/journal.ppat.1009795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrott M. R.; Sigurdson C. J.; Mason G. L.; Hoover E. A. Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J. Gen. Virol. 2012, 93, 212–221. 10.1099/vir.0.035006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond G. J.; Raymond L. D.; Meade-White K. D.; Hughson A. G.; Favara C.; Gardner D.; Williams E. S.; Miller M. W.; Race R. E.; Caughey B. Transmission and adaptation of chronic wasting disease to hamsters and transgenic mice: evidence for strains. J. Virol. 2007, 81, 4305–4314. 10.1128/JVI.02474-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney G.; Giles K.; Bouzamondo-Bernstein E.; Bosque P. J.; Miller M. W.; Safar J.; DeArmond S. J.; Prusiner S. B. Transmission of elk and deer prions to transgenic mice. J. Virol. 2006, 80, 9104–9114. 10.1128/JVI.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J.; Tatum T.; Hwang S.; Vrentas C.; West Greenlee M. H.; Kong Q.; Nicholson E.; Greenlee J. Novel Strain of the Chronic Wasting Disease Agent Isolated From Experimentally Inoculated Elk With LL132 Prion Protein. Sci. Rep. 2020, 10, 3148. 10.1038/s41598-020-59819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers R. C.; Kang H. E.; Napier D.; Browning S.; Seward T.; Mathiason C.; Balachandran A.; McKenzie D.; Castilla J.; Soto C.; Jewell J.; Graham C.; Hoover E. A.; Telling G. C. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154–1158. 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols T. A.; Nicholson E. M.; Liu Y.; Tao W.; Spraker T. R.; Lavelle M.; Fischer J.; Kong Q.; VerCauteren K. C. Detection of two dissimilar chronic wasting disease isolates in two captive Rocky Mountain elk (Cervus canadensis) herds. Prion 2021, 15, 207–215. 10.1080/19336896.2021.1982333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque Velasquez C.; Kim C.; Herbst A.; Daude N.; Garza M. C.; Wille H.; Aiken J.; McKenzie D. Deer Prion Proteins Modulate the Emergence and Adaptation of Chronic Wasting Disease Strains. J. Virol. 2015, 89, 12362–12373. 10.1128/JVI.02010-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero A.; Duque Velásquez C.; Johnson C.; Herbst A.; Bolea R.; Badiola J. J.; Aiken J.; McKenzie D. Prion protein polymorphisms associated with reduced CWD susceptibility limit peripheral PrP(CWD) deposition in orally infected white-tailed deer. BMC Vet. Res. 2019, 15, 50. 10.1186/s12917-019-1794-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A.; Velasquez C. D.; Triscott E.; Aiken J. M.; McKenzie D. Chronic Wasting Disease Prion Strain Emergence and Host Range Expansion. Emerg. Infect. Dis. 2017, 23, 1598–1600. 10.3201/eid2309.161474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green K. M.; Browning S. R.; Seward T. S.; Jewell J. E.; Ross D. L.; Green M. A.; Williams E. S.; Hoover E. A.; Telling G. C. The elk PRNP codon 132 polymorphism controls cervid and scrapie prion propagation. J. Gen. Virol. 2008, 89, 598–608. 10.1099/vir.0.83168-0. [DOI] [PubMed] [Google Scholar]
- LaCava M. E. F.; Malmberg J. L.; Edwards W. H.; Johnson L. N. L.; Allen S. E.; Ernest H. B. Spatio-temporal analyses reveal infectious disease-driven selection in a free-ranging ungulate. R. Soc. open sci. 2021, 8, 210802. 10.1098/rsos.210802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero A.; Duque Velásquez C.; Aiken J.; McKenzie D. White-tailed deer S96 prion protein does not support stable in vitro propagation of most common CWD strains. Sci. Rep. 2021, 11, 11193. 10.1038/s41598-021-90606-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onisko B. C.; Silva C. J.; Dynin I.; Erickson M.; Vensel W. H.; Hnasko R.; Requena J. R.; Carter J. M. Sensitive, preclinical detection of prions in brain by nanospray liquid chromatography/tandem mass spectrometry. Rapid. Commun. Mass. Spectrom. 2007, 21, 4023–4026. 10.1002/rcm.3310. [DOI] [PubMed] [Google Scholar]
- Silva C. J.; Onisko B. C.; Dynin I. C.; Erickson-Beltran M.; Requena J. R. Time of Detection of Prions in the Brain by Nanoscale Liquid Chromatography Coupled to Tandem Mass Spectrometry Is Comparable to Animal Bioassay. J. Agric. Food Chem. 2021, 69, 2279–2286. 10.1021/acs.jafc.0c06241. [DOI] [PubMed] [Google Scholar]
- Silva C. J.; Erickson-Beltran M. L.; Dynin I. C. Covalent Surface Modification of Prions: A Mass Spectrometry-Based Means of Detecting Distinctive Structural Features of Prion Strains. Biochemistry 2016, 55, 894–902. 10.1021/acs.biochem.5b01068. [DOI] [PubMed] [Google Scholar]
- Watts J. C.; Giles K.; Patel S.; Oehler A.; DeArmond S. J.; Prusiner S. B. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014, 10, e1003990. 10.1371/journal.ppat.1003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandel J. P.; Culeux A.; Grznarova K.; Levavasseur E.; Lamy P.; Privat N.; Welaratne A.; Denouel A.; Laplanche J. L.; Haik S. Amplification techniques and diagnosis of prion diseases. Rev. Neurol. (Paris) 2019, 175, 458–463. 10.1016/j.neurol.2019.06.002. [DOI] [PubMed] [Google Scholar]
- Morales R.; Duran-Aniotz C.; Diaz-Espinoza R.; Camacho M. V.; Soto C. Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 2012, 7, 1397–1409. 10.1038/nprot.2012.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramm C.; Pritzkow S.; Lyon A.; Nichols T.; Morales R.; Soto C. Detection of Prions in Blood of Cervids at the Asymptomatic Stage of Chronic Wasting Disease. Sci. Rep. 2017, 7, 17241. 10.1038/s41598-017-17090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramm C.; Soto P.; Nichols T. A.; Morales R. Chronic wasting disease (CWD) prion detection in blood from pre-symptomatic white-tailed deer harboring PRNP polymorphic variants. Sci. Rep. 2020, 10, 19763. 10.1038/s41598-020-75681-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramm C.; Gomez-Gutierrez R.; Soto C.; Telling G.; Nichols T.; Morales R. In Vitro detection of Chronic Wasting Disease (CWD) prions in semen and reproductive tissues of white tailed deer bucks (Odocoileus virginianus). PLoS One 2019, 14, e0226560. 10.1371/journal.pone.0226560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz M.; Cramm M.; Llorens F.; Muller-Cramm D.; Collins S.; Atarashi R.; Satoh K.; Orru C. D.; Groveman B. R.; Zafar S.; Schulz-Schaeffer W. J.; Caughey B.; Zerr I. The real-time quaking-induced conversion assay for detection of human prion disease and study of other protein misfolding diseases. Nat. Protoc. 2016, 11, 2233–2242. 10.1038/nprot.2016.120. [DOI] [PubMed] [Google Scholar]
- Ferreira N. C.; Charco J. M.; Plagenz J.; Orru C. D.; Denkers N. D.; Metrick M. A. 2nd; Hughson A. G.; Griffin K. A.; Race B.; Hoover E. A.; Castilla J.; Nichols T. A.; Miller M. W.; Caughey B. Detection of chronic wasting disease in mule and white-tailed deer by RT-QuIC analysis of outer ear. Sci. Rep. 2021, 11, 7702. 10.1038/s41598-021-87295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S.; Greenlee J. J.; Nicholson E. M. Real-Time Quaking-Induced Conversion Detection of PrP(Sc) in Fecal Samples From Chronic Wasting Disease Infected White-Tailed Deer Using Bank Vole Substrate. Front. Vet. Sci. 2021, 8, 643754. 10.3389/fvets.2021.643754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder A. M.; Henderson D. M.; Nalls A. V.; Wilham J. M.; Caughey B. W.; Hoover E. A.; Kincaid A. E.; Bartz J. C.; Mathiason C. K. In vitro detection of prionemia in TSE-infected cervids and hamsters. PLoS One 2013, 8, e80203. 10.1371/journal.pone.0080203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bistaffa E.; Vuong T. T.; Cazzaniga F. A.; Tran L.; Salzano G.; Legname G.; Giaccone G.; Benestad S. L.; Moda F. Use of different RT-QuIC substrates for detecting CWD prions in the brain of Norwegian cervids. Sci. Rep. 2019, 9, 18595. 10.1038/s41598-019-55078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin C.; Henderson D.; Benestad S. L.; Simmons M.; Adkin A. Estimating the amount of Chronic Wasting Disease infectivity passing through abattoirs and field slaughter. Prev. Vet. Med. 2019, 166, 28–38. 10.1016/j.prevetmed.2019.02.016. [DOI] [PubMed] [Google Scholar]