

Abstract
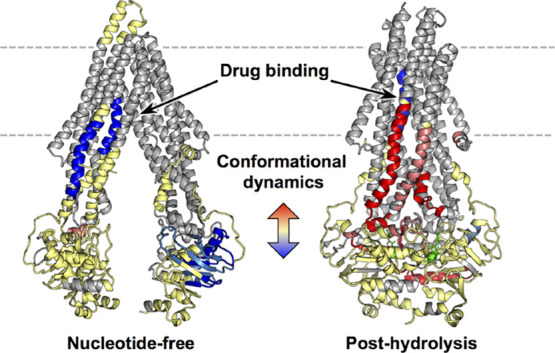
The ABC efflux pump P-glycoprotein (P-gp) transports a wide variety of drugs and is inhibited by others. Some drugs stimulate ATP hydrolysis at the nucleotide binding domains (NBDs) and are transported, others uncouple ATP hydrolysis and transport, and others inhibit ATP hydrolysis. The molecular basis for the different behavior of these drugs is not well understood despite the availability of several structural models of P-gp complexes with ligands bound. Hypothetically, ligands differentially alter the conformational dynamics of peptide segments that mediate the coupling between the drug binding sites and the NBDs. Here, we explore by hydrogen-deuterium exchange mass spectrometry the dynamic consequences of a classic substrate and inhibitor, vinblastine and zosuquidar, binding to mouse P-gp (mdr1a) in lipid nanodiscs. The dynamics of P-gp in nucleotide-free, pre-hydrolysis, and post-hydrolysis states in the presence of each drug reveal distinct mechanisms of ATPase stimulation and implications for transport. For both drugs, there are common regions affected in a similar manner, suggesting that particular networks are the key to stimulating ATP hydrolysis. However, drug binding effects diverge in the post-hydrolysis state, particularly in the intracellular helices (ICHs 3 and 4) and neighboring transmembrane helices. The local dynamics and conformational equilibria in this region are critical for the coupling of drug binding and ATP hydrolysis and are differentially modulated in the catalytic cycle.
Introduction
The ATP-binding cassette efflux transporter ABCB1 (P-glycoprotein; “P-gp”) exports from cells a remarkable range of structurally diverse drugs, nutraceuticals, and toxins, and it plays a critical role in drug–drug interactions and drug resistance.1−4 As a result of the central role that P-gp plays in drug response, its structure–function–dynamic relationships have been studied intensively. The P-gp fold includes two pseudo symmetric halves with 12 transmembrane helices (TMHs) arranged in two transmembrane domains (TMDs) that communicate with two nucleotide binding domains (NBDs) on the cytosolic side of the membrane. In the absence of ATP, the NBDs are predominantly separated and sample a range of distance distributions while the drug binding site is “inward facing” (the “IF” state) as demonstrated by numerous crystallographic and cryo-electron microscopy structures.5−13 Upon ATP binding but prior to ATP hydrolysis (the “pre-hydrolysis” state), the NBDs sample a dimerized state in order for hydrolysis to occur. Although the literature does not unambiguously demonstrate that the NBDs fully dimerize when nucleotide is bound to both, cryo-EM, molecular dynamics (MD) simulations, and in vivo fluorescence resonance energy transfer (FRET) suggest that a greater fraction of P-gp molecules adopt a conformation with closer contact between NBDs.14−16 This pre-hydrolysis state has been modeled by addition of non-hydrolyzable ATP analogs, addition of ATP to catalytically inactive mutants, and addition of ATP in the absence of Mg2+. Upon ATP hydrolysis at presumably one NBD (the “post-hydrolysis” state), there are further changes in the TMDs that allow egress of a substrate to the extracellular matrix. The post-hydrolysis state has historically been modeled by “vanadate trapping”, in which ATP is hydrolyzed and the released phosphate is replaced by tightly bound vanadate. As with other ATPases, the vanadate trapped state is widely regarded as a model for the hydrolytic transition state of the ATPase activity although we refer to it here as the “post-hydrolytic” state.17−19 Transport substrates or inhibitors bound in the TMDs activate, decrease, or have no effect on the ATPase activity in the NBDs that are 30–50 Å away. Cryo-EM and crystallographic studies have revealed structural details about some of these macroconformational states,5,11−13 but the mechanisms by which drugs or inhibitors differentially transmit information from their binding sites to the NBDs remain enigmatic. Ligand-dependent dynamics are a likely source of the functional differences of substrates vs inhibitors.
In fact, EPR-based DEER studies,20,21 LRET,22 atomic force microscopy (AFM),23,24 and H/D exchange mass spectrometry (HDX-MS)25−27 have demonstrated the highly dynamic nature of P-gp, which is critical for its catalytic cycle that requires large-scale conformational changes and for its substrate promiscuity. MD simulations have also been critical for supporting and driving mechanistic studies of conformational changes in P-gp.15,28−31 Collectively, the studies that monitor conformational dynamics indicate that each of these macroconformations is an ensemble with additional conformational heterogeneity, which could play a role in long-range communication between the drug-binding sites in the TMDs and the NBDs. In addition, the conformational plasticity of P-gp is hypothetically a determinant of the complex differences in functional effects of various ligands, wherein some transport substrates activate or inhibit ATP hydrolysis in a concentration-dependent manner, while others are inhibitors of both transport and ATP hydrolysis, and some activate ATP hydrolysis without being transported. The dynamic networks that control coupling of transport and ATP hydrolysis are not established.
The recently published structural models and dynamic experiments suggest that distinct communication networks between the TMHs and NBDs are differentially engaged by substrates vs inhibitors. Although substrates occupy a large “binding cavity” near the outer membrane leaflet lined by TMHs 1, 5, 6, 7, 10, 11, and 12, potent inhibitors bind in the cavity and “spill” across a smaller opening known as the “vestibule” and into the access channel near the inner leaflet.13 The access channel is hypothesized to provide a route of access for substrates entering from the inner membrane leaflet to the binding cavity, and the vestibule may provide a gating function between them. The inhibitor zosuquidar is particularly interesting and unique because it occupies the binding cavity and part of the vestibule without protruding into the access channel. Zosuquidar is not a substrate and is, in fact, a potent inhibitor of transport of other ligands but it also differentially stimulates or inhibits ATP hydrolysis, depending on specific mutations or a lipid environment.12,13,21,32,33 These differential effects are hypothetically due to the ability of zosuquidar to interact with different TMHs or occupy different cavities under different conditions. Here, we exploit zosuquidar and vinblastine with H/D exchange mass spectrometry (HDX-MS) to probe the conformational networks between the vestibule and the NBDs that are hypothetically distinct from those between the binding cavity and the NBDs. We specifically compare zosuquidar with vinblastine under identical conditions that yield differential effects on ATPase activity in a single defined lipid environment, to identify ligand-dependent, lipid-independent, conformational dynamics that are linked to function. We have previously described changes in HDX of wild-type murine P-gp in the IF and post-hydrolysis vanadate-trapped state in the absence of substrates or inhibitors.25 Others have reported HDX results for the nucleotide bound, pre-hydrolysis state in the absence of a substrate with the active site mutants (ExxxQExxxQ).26 However, no HDX results have been reported that map changes in dynamics caused by substrates or inhibitors.
Here, we demonstrate with HDX that zosuquidar and vinblastine differentially modulate the dynamics of structural elements far from their binding sites. The ligand-dependent differences in HDX at various stages of the catalytic cycle provide new detail about communication pathways between drug binding sites and the NBDs. Major differences emerge between the “uncoupler” zosuquidar vs inhibitory concentrations of vinblastine in the post-hydrolysis states, but they are modest to minimal in earlier stages of the catalytic cycle. In addition, the results reveal that conformational heterogeneity is prevalent throughout the P-gp catalytic cycle even in the presence of substrates or inhibitors.
Materials and Methods
Protein Purification and Nanodisc Incorporation
Hexa-histidine tagged MSP1D1 was expressed in E. coli and purified as previously described,20 and the his-tag was cleaved. C-terminally his-tagged mouse (MDR1A, UniProtKB P21447) codon-optimized P-gp (N-glycosylation sites mutated, N83Q/N87Q/N90Q34) was expressed in Pichia pastoris. P-gp pellets were lysed twice using a French-Press at 20,000 psi. The lysate was then centrifuged in three stages: 3500, 13,000, and 44,000 rpm. The pellet was discarded in the first two stages. The pelleted microsomes from the 44,000 rpm spin were solubilized in 1% n-dodecyl-β-d-maltoside (DDM) by passage through 18, 20, 22, and 25 gauge needles once each. Solubilized microsomes were loaded onto a His-60 nickel column followed by washing with 20 mM imidazole buffer also containing 50 mM Tris, 50 mM NaCl, 30% glycerol, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 0.1% DDM, pH 8.0. Imidazole buffer (300 mM) was used to elute the protein, with an initial 10% step to remove other species followed by 65% for P-gp elution. Pure fractions were concentrated and buffer exchanged using Amicon spin concentrators before passing through a DEAE column, to which the majority of impurities bind. The flow-through was concentrated using Amicon spin concentrators and quantified by gel-densitometry using BSA as a standard and stored at −80 °C in 20% glycerol, 10 mM Tris pH 8.0, 1 mM TCEP, and 0.1% DDM.
Nanodisc reactions were prepared with 1:10:70 P-gp:MSP1D1:DMPC molar ratios, with final concentrations of P-gp of 1–2 μM and 7–14 ng/mL. DDM was added to solubilize lipid at a 1:6 lipid:DDM molar ratio. Reactions were made in 20 mM Tris, 100 mM NaCl, 1 mM TCEP, pH 7.4 buffer, and residual 4% glycerol from addition of P-gp. Reactions were nutated at room temperature (24 °C) for 1 h. To initiate nanodisc formation, reaction mixtures were transferred to 0.25 g/mL Amberlite XAD-2 beads for 30 min with gentle mixing, followed by another 0.25 g/mL addition for 15 min, all at room temperature, with a final ratio of 14–28 ng of P-gp to 1 g of beads. P-gp nanodiscs were captured and eluted by the P-gp his-tag and further purified by SEC on a Superdex200 column at room temperature.
ATP Hydrolysis Assays
P-gp protein (2 μg) (14 pmol) in nanodiscs was used in duplicate samples per assay; averages and standard deviations are shown in Figure 1 for two separate P-gp and nanodisc preparations. Samples were prepared in ATPase buffer (50 mM Tris, 150 mM NH4Cl, 5 mM MgSO4, 5 mM dithiothreitol [DTT], pH 7.4). For the drug series, 2.5 mM ATP was used in all samples. A final concentration of 1% DMSO was maintained in all samples. Samples were incubated at 37 °C for 10 min prior to ATP addition. After adding ATP, samples were incubated at 37 °C for 1 h. Reactions were quenched by addition of SDS to a final concentration of 5% (v/v). Measurement of free phosphate was performed as described by Chifflet et al.,35 measuring absorbance at 850 nm.
Figure 1.

ATPase activity of P-gp nanodiscs in the presence of the two drugs. Average and standard deviation of duplicate samples for two separate preparations of P-gp nanodisc from two separate P-gp preparations are shown. Samples included 2.5 mM ATP. The ATP hydrolysis rate is shown as fold stimulation of basal (at 2.5 mM) activity, which averaged 38 nmol Pi/min/mg P-gp. The presence of vinblastine stimulates ATP hydrolysis at low concentrations but inhibits hydrolysis at higher concentrations. Zosuquidar stimulates ATP hydrolysis at all concentrations probed but is not completely soluble above 100 μM. Arrows indicate drug concentrations used for HDX-MS (8 or 80 μM vinblastine and 40 μM zosuquidar). Fits to the classical two-binding site (stimulatory/inhibitory) equation are for visualization only as standard errors of fits are too large for interpretation.
Hydrogen Deuterium Exchange Mass Spectrometry
Samples were split into two separate datasets, prepared from different P-gp preparations, and sample preparation and injection were performed at separate times. Dataset 1 consists of apo, AMPPNP, ATP/VO4, zosuquidar, zosuquidar and AMPPNP, zosuquidar and ATP/VO4, and vinblastine (80 μM) and AMPPNP. Dataset 2 consists of apo, ATP/VO4, vinblastine (8 and 80 μM), and vinblastine with ATP/VO4 (8 and 80 μM vinblastine). Within each dataset, all samples were made from the same P-gp nanodisc preparation, on the same day, and injected in the same run. Changes in deuterium uptake were calculated within each dataset so that delta deuterium uptake (but not absolute uptake) can be compared between datasets. Exchange samples were made in duplicate. No more than one sample for each condition was lost due to instrumentation errors; these are apparent in Supporting Information Figure S6 where standard deviation is lacking for one condition/time point. The PPPI peptide standard was included in all samples. All samples contained a final 1% DMSO for direct comparison to drug-containing samples.
Samples were prepared in 20 mM Tris, 100 mM NaCl, 1 mM TCEP, 5 mM MgSO4, pH 7.4 buffer at a 10× concentration of 5 μM. Concentrations (1×) of drugs (40 μM zosuquidar and 8 or 80 μM vinblastine) and nucleotide (1 mM AMPPNP or 1 mM ATP/250 μM VO4) were maintained in the P-gp nanodisc stocks and in the deuteration buffer. Samples were pre-incubated with drug and nucleotide for 1 h prior to deuteration at room temperature (24 °C). Samples were diluted 10-fold in an equivalent deuterated buffer (95%) for a final deuterium content of 86%. Deuteration reactions proceeded for 1 min, 8 min, 1 h, and 8 h at room temperature (24 °C). Undeuterated samples were similarly prepared in water-based buffer. Reactions were quenched by 1:1 addition to quench buffer (0.6% formic acid [FA], 1 mM TCEP, 0.2% DDM) on ice for a final pH of ∼2.55. Samples were immediately transferred to filter spin-tubes on ice with 60 mg of pepsin in quench buffer and vortexed. Samples were incubated on ice for 4 min, at which point 3 mg zirconium oxide beads (slurry in quench) were added and vortexed. Samples were vortexed after another 30 s and centrifuged after another 30 s to retain the zirconium oxide beads on the filter. Passed-through samples were transferred to tubes on ice and flash frozen in liquid nitrogen.
Samples were stored in liquid nitrogen and removed for 5 min on ice and 1.5 min at room temperature just prior to injection onto a mobile in-house LC system connected to a Waters Synapt-G2. The LC system was maintained at 1 °C, and peptides were trapped on a Vanguard BEH Shield RP18 1.7 μm trap column (2.1 × 5 mm; Waters), flowing loading buffer (2% acetonitrile and 0.1% trifluoroacetic acid) at 200 μL/min for 5 min, after which they were separated over a Hypersil 1.9 μm C18 column (1 × 50 mm; Thermo Scientific) using an acetonitrile gradient at 40 μL/min for 10 min; most peptides were eluted before 45% acetonitrile. The aqueous buffer contained 2% acetonitrile, 0.1% FA, and 0.025% trifluoroacetic acid, and the organic buffer contained 0.1% FA in acetonitrile. The C18-separated peptides were then separated by ion mobility and measured by Q-TOF. The StepWave ion guide settings were set to minimize nonuniform deuterium loss during desolvation.36 The trap and resolving columns were washed extensively to reduce sample carryover.37,38
Peptides were identified primarily using MSE and the ProteinLynx Global SERVER software with a minimum overall score of 6.0. Weaker intensity and lower scoring peptides were also confirmed using tandem MS on a Thermo LTQ-Orbitrap and Protein Prospector software. Deuterium uptake was determined for all peptides using HX-Express v239 due to the abundance of bimodal spectra. Bimodal deconvolution was used for any broad isotopic distribution, but quantification of EX1 kinetics was only considered for well-deconvoluted cases (evaluated visually and by statistical metrics in the software). In deuterium uptake plots, the weighted average of deuterium uptake in each state is presented. Deuterium uptake for each peptide was normalized to the number of amides in the peptide, omitting the first two residues. Percent deuteration is based on the theoretical maximum deuterium uptake of 86%. For determining significantly affected peptides, a 95% confidence interval based on the average of standard deviations across all peptides per sample was used as in previous cases of small changes in uptake.27,40,41 Peptides with two or more significantly different timepoints and a sum percent deuteration greater than ±3 were determined as affected unless trends were inconsistent for highly overlapping peptides or different charge states.
Results
Changes in ATP Hydrolysis upon Drug Binding
The known P-gp substrate vinblastine stimulates ATP hydrolysis at low concentrations but can inhibit hydrolysis at higher concentrations depending on conditions.21,42 This behavior requires multiple binding of vinblastine. In our HDX analysis of the effects of low and high concentrations of vinblastine binding to P-gp NDs, in either a nucleotide-free state or post-hydrolysis state, we observed no significant dynamic changes at low drug concentrations (Figure S7). The absence of an effect at low concentrations is difficult to interpret because the HDX will include a contribution from drug-free P-gp, P-gp with one vinblastine bound, and P-gp with two vinblastines bound. Therefore, our HDX analysis focuses on the effects of a high vinblastine concentration at which ATPase inhibition is observed in our P-gp ND system (Figure 1). Importantly, this concentration of vinblastine allows for comparison of P-gp states where ATPase function is inhibited, with two vinblastines bound, vs states that have two zosuquidars bound and are stimulated. Specifically for our system, zosuquidar stimulates ATP hydrolysis at all concentrations including those expected to have multiple zosuquidars bound, as observed by others.21 Zosuquidar exhibits variable effects, likely depending on the specific P-gp construct and the lipid/detergent environment.12,13,21,33 In our MSP1D1 nanodisc system with DMPC lipids and codon-optimized, glycan-free, murine P-gp,34 zosuquidar is consistently an activator of ATPase activity up to the highest concentrations attainable, above which zosuquidar is insoluble. A similar increase in ATPase activity was observed in cys-less mouse P-gp in MSP1D1E3 nanodiscs composed of POPC and E. coli polar lipids.21 As the presence of the detergent was observed to switch zosuquidar effects from inhibitory (in native membranes) to stimulatory (in detergent micelles),33 we quantified the residual DDM detergent present in our P-gp ND preparations (Figure S1). We detected approximately one molecule of DDM per 500 P-gp NDs or even less DDM, indicating that residual DDM does not contribute to zosuquidar behavior in our system. Additionally, to confirm our assay conditions in a more native membrane context, we performed the same drug series analysis on murine P-gp in HEK293 vesicles (Figure S2). Vinblastine displays comparable stimulatory and inhibitory behavior as in nanodiscs. Zosuquidar causes ATPase inhibition at low concentrations as reported by Shukla et al.,33 but we observe modest recovery of activity at higher concentrations not probed in that study.
Distinct Macrostates of P-gp Pre-Hydrolysis and Post-Hydrolysis
To understand the potential impact of bound drug at each stage of the catalytic cycle, we probed changes in deuterium uptake by HDX-MS of drug-free, vinblastine-bound (inhibitory concentration of 80 μM), and zosuquidar-bound P-gp NDs in nucleotide-free, AMPPNP bound (pre-hydrolysis), and ATP/VO4-trapped (post-hydrolysis) states. Mg2+ was present in all samples. It was first necessary to examine changes in dynamics that accompany conversion from the IF macroconformation to the pre-hydrolysis and post-hydrolysis states in the absence of the drug or inhibitor. We depict in the figures the nucleotide-bound pre-hydrolysis state mimicked by the AMPPNP bound P-gp as being “closed” in order to distinguish it from the apo state. We emphasize that, although the available data suggest that the population shifts toward the closed form in the nucleotide-bound state relative to the apo state, we do not know what fraction of P-gp is in the NBD dimerized state. We previously reported changes between nucleotide-free and post-hydrolysis states25 in P-gp NDs and detergent/lipid micelles, and more recently, Kopcho et al. described HDX changes in these states as well as a pre-hydrolysis state mimicked by ATP bound to a catalytically inactive form of P-gp in detergent.26 We obtained extensive coverage of the NBDs, ICH3, ICH4, the extracellular loop, and the more solvent-accessible portions of the TMHs (see Figure S3 for the coverage map and Table S1 for experiment statistics). Throughout the following discussion, we refer to “increased protection” as a decrease in the rate of H/D exchange and “less protection” or “deprotection” as an increase in the isotope exchange. P-gp displays a broad range of dynamic behaviors that are highly consistent between different preparations (Figures S4 and S5). In addition, we observe widespread EX1 exchange kinetics as we reported previously, and these processes are referred to in the last section.
Our analysis comprehensively shows predominantly increased protection from H/D exchange in many individual peptides throughout the protein in both nucleotide bound states compared to the apo state, consistent with less dynamic, more ordered, local conformers (Figure 2 and Figures S6 and S7). However, the post-hydrolysis state induces greater protection in NBD1, ICH3, and ICH4 than the pre-hydrolysis state. This is generally consistent with the Kopcho et al. findings with the catalytically inactive mutant of P-gp in detergent as opposed to our studies with wild-type P-gp in NDs.26 Most changes with the post-hydrolytic state occur throughout NBD1 and increased protection of NBS2 motifs (Walker A and A-loop) in NBD2, but not specific elements of NBS1, indicating nonsymmetric effects upon hydrolysis to the NBSs. This suggests the possibility of different hydrolysis or nucleotide binding parameters at the two NBSs or the possibility that the post-hydrolysis state has released nucleotide from NBS1. The nucleotide-free, AMPPNP-bound, and ATP/VO4-trapped macrostates are distinct by HDX-MS with large changes in overall deuterium uptake and changes in EX1 kinetics, which are discussed below. These changes are larger and more widespread in the drug-bound states than the changes observed in the corresponding drug-free states for each of the three macroconformations (IF, pre-hydrolysis, and post-hydrolysis, next sections). Based on HDX, the conversion between macroconformations is clearly driven by nucleotide binding and hydrolysis rather than drug binding, consistent with conformational distributions observed by other experimental approaches.21,22 The HDX changes observed in the presence of drug vs the absence of the drug do not preclude occupation of the pre- and post-hydrolysis states we have defined but do alter the macrostates in complex ways.
Figure 2.

Large dynamic changes in pre- and post-hydrolysis states define macro states of the P-gp catalytic cycle. (A) Sequence plots of the changes in HDX across time points for the pre-hydrolysis (AMPPNP) and post-hydrolysis (ATP/VO4) states relative to the nucleotide-free state (top) and changes between post- and pre-hydrolysis (bottom). (B) Peptides showing significant HDX changes plotted on the ATP-bound structure (PDB ID: 6C0V(16)). (C) Top-down view from membrane into cytoplasm of NBDs and ICHs. See Figures S6 and S7 for individual peptide plots and sequence plots for each time point.
Zosuquidar Alters TMD Arrangement in the Post-Hydrolysis State
Although it is not clear if a nucleotide-free state is long-lived for P-gp in vivo, this state is the most tractable, and structural changes induced by drug binding could indicate mechanisms for changes in nucleotide affinity. The modest effects we observe on the KM of ATP hydrolysis with each drug suggest that there could be differences in nucleotide affinity. In the nucleotide-free state, zosuquidar binding induces protection in a localized part of NBD2 including the A-loop and Walker B motifs, TMH6, and TMH11 (Figure 3). A slight decrease in protection is observed in ICH4 (peptide 900–906) but without prominent effects on the flanking helices that are typically observed as well.
Figure 3.

Zosuquidar binding to P-gp alters HDX-MS in different catalytic states. (A) Peptides that show significant HDX changes for zosuquidar binding to nucleotide-free, pre-hydrolysis (AMPPNP), and post-hydrolysis (ATP/VO4) states of P-gp relative to the drug-free states of the catalytic cycle, plotted on apo and ATP-bound structures (PDB IDs: 5KPI and 6C0V(10,16)). (B) Top-down view from membrane into cytoplasm of NBDs and ICHs. (C) Sequence plot of the sum of changes in HDX across time points. See Figures S6 and S7 for individual peptide plots and sequence plots for each time point.
Relative to a drug-free pre-hydrolysis state, the zosuquidar-bound pre-hydrolysis state exhibits increased protection in NBD2. The affected regions are some of those impacted in the nucleotide-free zosuquidar-bound state but to a lesser extent (still includes the Walker B motif), with the largest effects still observed in the 1092–1109 region. NBD1 shows slight decreases in protection in regions that include the Walker B motif of NBS1. As in the nucleotide-free, zosuquidar-bound state, TMH6 is still protected, consistent with zosuquidar binding in the hydrophobic cavity.
Remarkably, zosuquidar affects the post-hydrolysis state more intensely than other states of the catalytic cycle. There is a clear and localized effect on the post-hydrolysis state that is unique to zosuquidar. Both ICH3, ICH4, and helices 10 and 11 are substantially less protected with zosuquidar bound to the post-hydrolysis state than in the absence of zosuquidar. The NBDs show some effects with zosuquidar bound to the post-hydrolysis state, mainly decreased protection for peptides neighboring ICH4 that include the Q-loop and D-loop of NBD1. These results imply that the presence of zosuquidar greatly alters the TMH arrangement and/or stability in the post-hydrolysis (presumably outward-facing) state and that subtle effects are translated down to the NBDs. The changes observed in the NBDs are not comparable to the large changes seen between different nucleotide-bound states. This is novel structural evidence of a possible mechanism of uncoupling of transport (specifically TMD behavior) from ATP hydrolysis.
Vinblastine at Inhibitory Concentrations Alters NBD Stability in the Post-Hydrolysis State
Vinblastine at a high, inhibitory concentration (80 μM) binding to P-gp at different catalytic states induces many different changes compared to those observed for zosuquidar. Low, stimulatory concentrations of vinblastine resulted in no significant HDX changes in either a nucleotide-free post-hydrolysis state (Figure S7). However, the lower, 8 μM concentration of vinblastine would likely not be sufficient to saturate P-gp NDs in the HDX samples. For inhibitory vinblastine binding (presumably two molecules), decreased protection is observed for ICH4 in the nucleotide-free state as well as nearby regions of NBD1, including the Walker B motif (Figure 4). Similar to zosuquidar, the A-loop in NBS2 is slightly more protected in the presence of the higher concentration of vinblastine. Also similar to zosuquidar, there are minimal changes in the pre-hydrolytic state when vinblastine is bound, with only slight protection observed in the 1092–1109 region of NBD2, but none of the covered NBS motifs are affected. It should be noted that the experiment probing the pre-hydrolysis state with vinblastine was performed separately (see Materials and Methods) from vinblastine in the nucleotide-free and post-hydrolysis states; therefore, we are conservative with comparisons between the pre-hydrolytic state and the other two catalytic states. The post-hydrolytic state with two vinblastines bound shows widespread protection throughout the NBDs, including many of the NBS motifs (Figure 4B). Interestingly, ICH4 is less protected, while ICH3 is more protected. The differences in the post-hydrolytic states in the presence of saturating vinblastine vs zosuquidar clearly identify possible distinct mechanisms of action.
Figure 4.

An inhibitory concentration of vinblastine binding to P-gp alters HDX-MS in different catalytic states. (A) Peptides that show significant HDX changes for vinblastine binding to nucleotide-free, pre-hydrolysis (AMPPNP), and post-hydrolysis (ATP/VO4) states of P-gp relative to the drug-free states of the catalytic cycle, plotted on apo and ATP-bound structures (PDB IDs: 5KPI and 6C0V). (B) Top-down view from membrane into cytoplasm of NBDs and ICHs. (C) Sequence plot of the sum of changes in HDX across time points. See Figures S6 and S7 for individual peptide plots and sequence plots for each time point.
Bound Drugs Alter Slow Conformational Exchange of ICH4
As previously reported, P-gp in lipid NDs and detergent/lipid micelles exhibits EX1 kinetics, or conformational exchange, across a broad range of timescales.25,27 This behavior is observed via bimodal isotopic distributions in the mass spectrum for a given peptide. Specifically, one can observe two slowly exchanging states for a region if the two conformations differ in their degree of solvent exposure or hydrogen bonding, and therefore deuterium uptake. The rate of conformational exchange must be much slower than the rate of HDX in order to observe EX1 kinetics. These results demonstrate the complexity and heterogeneity of the P-gp conformational ensemble even within one macrostate of the protein. For peptides that exhibit EX1 behavior with well-deconvoluted isotopic distributions, we demonstrated that in the ATP/VO4 trapped state, the rates of slow conformational exchange throughout the protein were greatly decreased. In our comparison of P-gp dynamics between lipid environments (P-gp in DMPC NDs with or without cholesterol), the majority of affected peptides showed bimodal isotopic distributions.27 We find with drug binding that most of the peptides that are more strongly affected also show bimodal distributions, consistent with a role for conformational heterogeneity in drug binding.
One of the most affected regions across many conditions probed here that also shows well-resolved bimodal peptides is ICH4 and the flanking helices 10 and 11. Under all conditions, this region (example peptide 901–915, Figure 5) experiences slow exchange between protected and very unprotected conformations with a half-life on the order of ∼20 min, although the motion is complex and does not fit a single exponential. The rate of this exchange in the absence of the drug is greatly reduced in the pre-hydrolysis state and even more so in the post-hydrolysis state, resulting in large increases in overall protection. In the presence of the drug, however, some states exhibit decreased protection due to an increased rate of this slow conformational exchange. For example, bound zosuquidar does not affect ICH4 in the nucleotide-free or pre-hydrolysis states but greatly increases conformational exchange in the post-hydrolysis state, bringing the exchange kinetics closer to that of the nucleotide-free state. Inhibitory levels of vinblastine also increase the rate of slow exchange of ICH4, but this is most strongly observed in the nucleotide-free state, with a more modest change in protection observed in the post-hydrolysis state. Together, these results demonstrate that the rates of interconversion between local conformations in ICH4 and adjacent TMHs 10 and 11 are differentially affected by the binding of vinblastine at inhibitory concentrations and zosuquidar. These ligands have a true “dynamic” effect on the rates of conformational exchange that is communicated from the drug binding sites to ICH4.
Figure 5.

Drug binding impacts slow conformational exchange at different catalytic stages. (A) Mass spectra of peptide 901–915 encompassing ICH4 across several conditions and time points. The relative proportions of the two conformer populations change among conditions. (B) Summary plots of proportion of the protected population vs time (blue species in panel A); vinblastine and apo are from a separate dataset. (C) Position of peptide 901–915 in an open state (PDB ID: 5KPI).
Discussion
There are ample biological and biochemical studies on the functional consequences of various substrates and inhibitors interacting with P-gp. In principle, the design of drugs that target P-gp functionality, especially inhibitors to be used in conjunction with chemotherapy, could be informed by a structural understanding of how molecules interact with P-gp. However, the mechanisms by which P-gp responds to different molecules (substrates, inhibitors, couplers, uncouplers, etc.) remain elusive for several reasons: [1] P-gp demonstrates a high level of conformational plasticity even within each macro-conformational state of the catalytic cycle. Canonical structural techniques that can be applied to P-gp (crystallography and cryo-EM) are ideal for capturing high-resolution structures for a predominant conformation but not for describing a range of conformations and their proportions.5−13,43 Each drug might differentially interact with different conformations within the ligand free ensemble (conformational selection) and alter the landscape of each state of the catalytic cycle (induced fit). Differential effects on these macrostates have been observed with other structural techniques20−22 (DEER and LRET) that quantify distance distributions under a number of conditions. [2] While these results provide the essential framework for characterizing P-gp-drug interactions, these static structural “snapshots” do not describe the dynamic effects that bound drugs have on P-gp as it traverses the complex conformational landscape of its catalytic cycle. Our HDX-MS study reveals possible conformational changes due to drug binding in different catalytic states and identifies regions that show drug-dependent changes in conformational exchange.
The results reveal potential mechanisms of differential communication between drug-binding sites in the TMHs and the NBDs, for transport substrates vs inhibitors (Figure 6). Comparison of HDX in the drug-bound nucleotide-free states indicates that there are only minor, localized changes in dynamics. The HDX properties of the NBDs do not change dramatically upon addition of vinblastine or zosuquidar, although both drugs induce modest increases in the rates of HDX in different NBDs. Vinblastine increases exchange in the Walker B motif of NBS1, whereas zosuquidar largely decreases exchange in NBD2. Interestingly, both ligands decrease exchange in the A-loop of NBS2, a region implicated in determining catalytic efficiency based on DEER in the post-hydrolysis state.21 Together, these effects are consistent with the macroscopic conformation remaining in the IF state. For both ligands, the effects on dynamics in the absence of nucleotide are modest and not widespread. These results are consistent with various types of structural studies. The crystal and cryo-EM structures of P-gp bound to the drug in the absence of nucleotide show only slight changes in secondary/tertiary structure; for example, a comparison of P-gp with taxol or zosuquidar bound shows that a slight shift near the binding site translates down to minor shifting of the NBDs with respect to one another.12 LRET and DEER distance distributions in P-gp NDs show variable effects on the NBD-NBD distance. LRET suggests verapamil alone causes a shift toward the closed form but DEER, and native mass spectrometry,44 suggests no substantial shifts in this conformational distribution (larger fractions open/closed) with drug binding to a nucleotide-free state. Based on DEER, P-gp populates a new conformation upon addition of zosuquidar (but not vinblastine at low, stimulatory concentrations), in which the NBDs and intracellular region of the TMDs are closer (more closed state) but not achieving a nucleotide-bound-like state. Under our conditions, this state might not be highly populated or produces similar HDX behavior to the conformations already observed.
Figure 6.

Summary of HDX-MS changes throughout catalytic cycle in drug-free, substrate-bound (high concentration of vinblastine), and inhibitor-bound states. Only parts of the protein with substantial HDX coverage are labeled: NBDs, ICH3, ICH4, and connecting TMHs are represented with generalized HDX protection indicated by color (red = deprotected and blue = protected).
In the pre-hydrolysis state modeled by the addition of AMPPNP, the effects of either vinblastine or zosuquidar on the NBDs remain modest and relatively indistinct. Compared to the drug-free pre-hydrolysis state, the pre-hydrolysis state with vinblastine or zosuquidar exhibits a very slight decrease in exchange in NBD2 in the region of 1092–1109. That is, the NBD-dimerized pre-hydrolysis states in the presence of vinblastine or zosuquidar differ from the canonical open, IF states but not greatly from each other. However, the LRET of Mg2+-ATP-bound P-gp with verapamil increases the fraction of NBDs in a “closed”, shorter-distance state.22 Upon progressing to the post-hydrolysis state, however, the vinblastine and zosuquidar complexes populate distinctly different conformational ensembles with different dynamic signatures. Specifically, zosuquidar does not greatly affect the NBDs in the post-hydrolysis macroconformation but does alter the state of the TMHs, while vinblastine induces widespread, modest effects throughout the protein, especially the NBDs.
Previous cryo-EM studies of antibody-bound P-gp in lipid nanodiscs containing brain lipids indicated that when two zosuquidar molecules are bound, they mainly occupy the substrate binding cavity, but one protrudes into the “vestibule” created by TMHs 5, 7, 9, and 12.19 In contrast, with vincristine bound (highly similar to vinblastine), the molecule is fully encompassed in the binding cavity. Comparison of this nucleotide-free state with the pre-hydrolysis state observed in an ATP-bound structure shows the movement of TMH9 conflicts with inhibitors bound in the vestibule. This indicates a mechanism by which inhibitors block transport. Although we do not have evidence of transport inhibition in the same lipid context used in our HDX study, numerous studies demonstrate zosuquidar behaving as a transport inhibitor in different cell and liposome contexts.32,45,46 A recent MD study also demonstrates a broader range of distances between the NBDs and TMDs and increased lipid access events in the presence of the inhibitor tariquidar.28 Our results for zosuquidar in the post-hydrolysis state are consistent with these findings. Zosuquidar in the post-hydrolysis state decreases protection for TMH9 and neighboring ICH3, as well as ICH4 and neighboring TMHs 10 and 11. Therefore, the canonical OF state is not achieved, defining a clear mechanism of transport inhibition. The minimal effects on the NBDs suggest that ATP is hydrolyzed in a manner comparable to a drug-free cycle, therefore producing “uncoupling” of transport and ATP hydrolysis. Conversely, the widespread effects throughout the NBDs in the post-hydrolysis state with vinblastine bound suggest a mechanism for ATPase inhibition. Increased protection throughout the NBDs suggests an alteration in the communication network for hydrolysis; this post-hydrolysis state does not resemble the pre-hydrolysis state. Although it is possible that the second hydrolysis step could be hindered, hydrolysis might be more or less “symmetric” at the NBSs, or hydrolyzed nucleotide and/or the NBDs have tighter affinities, reducing the frequency with which P-gp can complete a hydrolysis cycle.
The observed effects due to either vinblastine or zosuquidar binding to P-gp-NDs are somewhat different from those we observed when cholesterol is included in the NDs.27 This suggests that our previously reported effects from cholesterol are predominantly due to environmental changes in the ND as opposed to cholesterol in the binding pocket. The predominant effects with cholesterol are increased protection throughout NBD2 and ICH3 through a shift in the equilibrium distribution of conformations within the apo macrostate; modest increases in exchange are observed in NBD1/ICH4. In contrast, with either drug binding to the nucleotide-free state, only minor to modest effects are observed but they do either decrease exchange in NBD2 or increase exchange in NBD1/ICH4. Cholesterol does not induce effects in HDX that are very similar to vinblastine binding, but there are several peptides protected in NBD2 by both cholesterol and zosuquidar. Therefore, a possible trend in dynamics with ATP hydrolysis is preserved (cholesterol increases ATPase activity). Overall, this suggests possible common mechanisms of ATPase stimulation caused by various drugs bound to P-gp or lipid components in the membrane, but drugs and lipids may produce their effects by interacting at different remote sites. For example, cholesterol is observed to be crowded around the TMDs in nanodiscs,12 vinblastine is likely bound high in the binding pocket,47,48 and zosuquidar extends down into the vestibule.13
Our findings emphasize the need to use techniques that can capture small changes in conformational heterogeneity, approaches that quantify dynamic motions, and mimics for multiple states of the catalytic cycle. In the context of numerous other structural mechanistic studies of P-gp, HDX-MS provides crucial insight into subtle dynamic consequences when various ligands bind. Comprehensive, collaborative efforts in the field with distinct approaches will allow us to better characterize drug binding sites and how they change through the catalytic cycle. Given the dynamic interplay of lipids with P-gp portals to the drug binding pocket, it is also important to characterize regulation of P-gp conformations/function by lipid composition.
Acknowledgments
We thank Miklos Guttman for the use of a home-built HDX-LC system. We thank Dale Whittington for helping us develop the DDM quantitation assay.
Glossary
Abbreviations
- P-gp
P-glycoprotein
- HDX-MS
hydrogen deuterium exchange mass spectrometry
- TMD
transmembrane domain
- TMH
transmembrane helix
- ICH
intracellular helix
- NBD
nucleotide binding domain
- ND
nanodisc
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- MSP
membrane scaffold protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00056.
Quantitation of the residual DDM detergent in nanodiscs, ATPase activity of P-gp in HEK239 vesicles, HDX-MS coverage map, table of HDX-MS parameters, absolute HDX across P-gp sequence for the apo state in both datasets, HDX individual peptide plots, and Delta HDX plots for all timepoints and states (PDF)
Accession Codes
MDR1A: UniProtKB P21447.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by NIH R01GM121603 to W.M.A. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Sugawara I.; Kataoka I.; Morishita Y.; Hamada H.; Tsuruo T.; Itoyama S.; Mori S. Tissue Distribution of P-Glycoprotein Encoded by a Multidrug-Resistant Gene as Revealed by a Monoclonal Antibody, mrk 16. Cancer Res. 1988, 48, 1926–1929. [PubMed] [Google Scholar]
- Cordon-Cardo C.; O’Brien J. P.; Boccia J.; Casals D.; Bertino J. R.; Melamed M. R. Expression of the multidrug resistance gene product (P-Glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem. 1990, 38, 1277–1287. 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- Clarke R.; Leonessa F.; Trock B. Multidrug resistance/P-glycoprotein and breast cancer: Review and meta-analysis. Semin. Oncol. 2005, 32, S9–S15. 10.1053/j.seminoncol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Khan M.; Maryam A.; Mehmood T.; Zhang Y.; Ma T. Enhancing activity of anticancer drugs in multidrug resistant tumors by modulating p-glycoprotein through dietary nutraceuticals. Asian Pac. J. Cancer Prev. 2015, 16, 6831–6839. 10.7314/APJCP.2015.16.16.6831. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. B.; Szewczyk P.; Grimard V.; Lee C. W.; Martinez L.; Doshi R.; Caya A.; Villaluz M.; Pardon E.; Cregger C.; Swartz D. J.; Falson P. G.; Urbatsch I. L.; Govaerts C.; Steyaert J.; Chang G. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13386–13391. 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk P.; Tao H.; McGrath A. P.; Villaluz M.; Rees S. D.; Lee S. C.; Doshi R.; Urbatsch I. L.; Zhang Q.; Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 732–741. 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A.; Lee S. C.; Tao H.; Speir J. A.; Chang G.; Urbatsch I. L.; Potter C. S.; Carragher B.; Zhang Q. Distinct Conformational Spectrum of Homologous Multidrug ABC Transporters. Structure 2015, 23, 450–460. 10.1016/j.str.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank G. A.; Shukla S.; Rao P.; Borgnia M. J.; Bartesaghi A.; Merk A.; Mobin A.; Esser L.; Earl L. A.; Gottesman M. M.; Xia D.; Ambudkar S. V.; Subramaniam S. Cryo-EM analysis of the conformational landscape of human P-glycoprotein (ABCB1) during its catalytic cycle. Mol. Pharmacol. 2016, 90, 35–41. 10.1124/mol.116.104190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L.; Zhou F.; Pluchino K. M.; Shiloach J.; Ma J.; Tang W. K.; Gutierrez C.; Zhang A.; Shukla S.; Madigan J. P.; Zhou T.; Kwong P. D.; Ambudkar S. V.; Gottesman M. M.; Xia D. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. 10.1074/jbc.M116.755884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Küng R.; Kowal J.; McLeod R. A.; Tremp N.; Broude E. V.; Roninson I. B.; Stahlberg H.; Locher K. P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. 2018, 115, E1973–E1982. 10.1073/pnas.1717044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Kowal J.; Broude E.; Roninson I.; Locher K. P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. 10.1126/science.aav7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosol K.; Romane K.; Irobalieva R. N.; Alam A.; Kowal J.; Fujita N.; Locher K. P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 26245–26253. 10.1073/pnas.2010264117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iram S. H.; Gruber S. J.; Raguimova O. N.; Thomas D. D.; Robia S. L. ATP-binding cassette transporter structure changes detected by intramolecular fluorescence energy transfer for high-throughput screenings. Mol. Pharmacol. 2015, 88, 84–94. 10.1124/mol.114.096792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L.; Aller S. G. Equilibrated atomic models of outward-facing P-glycoprotein and effect of ATP binding on structural dynamics. Sci. Rep. 2015, 5, 7880. 10.1038/srep07880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. 10.1126/science.aar7389. [DOI] [PubMed] [Google Scholar]
- Urbatsch I. L.; Sankaran B.; Weber J.; Senior A. E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995, 270, 19383–19390. 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- Dey S.; Ramachandra M.; Pastan I.; Gottesman M. M.; Ambudkar S. V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10594–10599. 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbatsch I. L.; Tyndall G. A.; Tombline G.; Senior A. E. P-glycoprotein catalytic mechanism: Studies of the ADP-vanadate inhibited state. J. Biol. Chem. 2003, 278, 23171–23179. 10.1074/jbc.M301957200. [DOI] [PubMed] [Google Scholar]
- Verhalen B.; Dastvan R.; Thangapandian S.; Peskova Y.; Koteiche H. A.; Nakamoto R. K.; Tajkhorshid E.; McHaourab H. S. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 2017, 543, 738–741. 10.1038/nature21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastvan R.; Mishra S.; Peskova Y. B.; Nakamoto R. K.; Mchaourab H. S. Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 2019, 364, 689–692. 10.1126/science.aav9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi M. E.; Mok L.; Swartz D. J.; Singh A.; Fendley G. A.; Urbatsch I. L.; Altenberg G. A. Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis. J. Biol. Chem. 2017, 292, 20412–20424. 10.1074/jbc.M117.814186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigdel K. P.; Wilt L. A.; Marsh B. P.; Roberts A. G.; King G. M. The conformation and dynamics of P-glycoprotein in a lipid bilayer investigated by atomic force microscopy. Biochem. Pharmacol. 2018, 156, 302–311. 10.1016/j.bcp.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. H.; Sigdel K. P.; Schaefer K. G.; Mensah G. A. K.; King G. M.; Roberts A. G. The effects of anthracycline drugs on the conformational distribution of mouse P-glycoprotein explains their transport rate differences. Biochem. Pharmacol. 2020, 174, 113813 10.1016/j.bcp.2020.113813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiarong Li M.; Guttman M.; Atkins W. M. Conformational dynamics of P-glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale. J. Biol. Chem. 2018, 293, 6297–6307. 10.1074/jbc.RA118.002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopcho N.; Chang G.; Komives E. A. Dynamics of ABC Transporter P-glycoprotein in Three Conformational States. Sci. Rep. 2019, 9, 15092. 10.1038/s41598-019-50578-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouser A. F.; Alam Y. H.; Atkins W. M. Cholesterol Asymmetrically Modulates the Conformational Ensemble of the Nucleotide-Binding Domains of P-Glycoprotein in Lipid Nanodiscs. Biochemistry 2021, 60, 85–94. 10.1021/acs.biochem.0c00824. [DOI] [PubMed] [Google Scholar]
- Kapoor K.; Pant S.; Tajkhorshid E. Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein. Chem. Sci. 2021, 12, 6293–6306. 10.1039/D0SC06288J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L.; Aller S. G. Allosteric Role of Substrate Occupancy Toward the Alignment of P-glycoprotein Nucleotide Binding Domains. Sci. Rep. 2018, 8, 14643. 10.1038/s41598-018-32815-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonito C. A.; Ferreira R. J.; Ferreira M. J. U.; Gillet J. P.; Cordeiro M. N. D. S.; dos Santos D. J. V. A. Theoretical insights on helix repacking as the origin of P-glycoprotein promiscuity. Sci. Rep. 2020, 10, 9823. 10.1038/s41598-020-66587-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen P. C.; Verhalen B.; Wilkens S.; Mchaourab H. S.; Tajkhorshid E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 2013, 288, 19211–19220. 10.1074/jbc.M113.450114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chufan E. E.; Kapoor K.; Ambudkar S. V. Drug-protein hydrogen bonds govern the inhibition of the ATP hydrolysis of the multidrug transporter P-glycoprotein. Biochem. Pharmacol. 2016, 101, 40–53. 10.1016/j.bcp.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S.; Abel B.; Chufan E. E.; Ambudkar S. V. Effects of a detergent micelle environment on P-glycoprotein (ABCB1)-ligand interactions. J. Biol. Chem. 2017, 292, 7066–7076. 10.1074/jbc.M116.771634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Swartz D. J.; Protasevich I. I.; Brouillette C. G.; Harrell P. M.; Hildebrandt E.; Gasser B.; Mattanovich D.; Ward A.; Chang G.; Urbatsch I. L. A gene optimization strategy that enhances production of fully functional P-Glycoprotein in Pichia pastoris. PLoS One 2011, 6, e22577 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chifflet S.; Torriglia A.; Chiesa R.; Tolosa S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal. Biochem. 1988, 168, 1–4. 10.1016/0003-2697(88)90002-4. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Wales T. E.; Whittington D.; Engen J. R.; Brown J. M.; Lee K. K. Tuning a High Transmission Ion Guide to Prevent Gas-Phase Proton Exchange during H/D Exchange MS Analysis. J. Am. Soc. Mass Spectrom. 2016, 27, 662–668. 10.1007/s13361-015-1330-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J.; Rand K. D.; Beuning P. J.; Engen J. R. False EX1 signatures caused by sample carryover during HX MS analyses. Int. J. Mass Spectrom. 2011, 302, 19–25. 10.1016/j.ijms.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R.; Manikwar P.; Hickey J. M.; Arora J.; Middaugh C. R.; Volkin D. B.; Weis D. D. Minimizing carry-over in an online pepsin digestion system used for the H/D exchange mass spectrometric analysis of an IgG1 monoclonal antibody. J. Am. Soc. Mass Spectrom. 2012, 23, 2140–2148. 10.1007/s13361-012-0485-9. [DOI] [PubMed] [Google Scholar]
- Guttman M.; Weis D. D.; Engen J. R.; Lee K. K. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J. Am. Soc. Mass Spectrom. 2013, 24, 1906–1912. 10.1007/s13361-013-0727-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller I. R.; Slivacka M.; Nielsen A. K.; Rasmussen S. G. F.; Gether U.; Loland C. J.; Rand K. D. Conformational dynamics of the human serotonin transporter during substrate and drug binding. Nat. Commun. 2019, 10, 1687. 10.1038/s41467-019-09675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paço L.; Zarate-Perez F.; Clouser A. F.; Atkins W. M.; Hackett J. C. Dynamics and Mechanism of Binding of Androstenedione to Membrane-Associated Aromatase. Biochemistry 2020, 59, 2999–3009. 10.1021/acs.biochem.0c00460. [DOI] [PubMed] [Google Scholar]
- Li M. J.; Nath A.; Atkins W. M. Differential Coupling of Binding, ATP Hydrolysis, and Transport of Fluorescent Probes with P-Glycoprotein in Lipid Nanodiscs. Biochemistry 2017, 56, 2506–2517. 10.1021/acs.biochem.6b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonghin N.; Collins R. F.; Barbieri A.; Shafi T.; Siebert A.; Ford R. C. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct. Biol. 2018, 18, 1. 10.1186/s12900-018-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J.; Wang S. C.; Politis A.; Reading E.; Ma J.; Biggin P. C.; Zhou M.; Tao H.; Zhang Q.; Chang G.; Morgner N.; Robinson C. V. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9704–9709. 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling J. J.; Shepard R. L.; Cao J.; Law K. L.; Norman B. H.; Kroin J. S.; Ehlhardt W. J.; Baughman T. M.; Winter M. A.; Bell M. G.; Shih C.; Gruber J.; Elmquist W. F.; Dantzig A. H. Pharmacological characterization of LY335979: A potent cyclopropyldibenzosuberane modulator of P-glycoprotein. Adv. Enzyme Regul. 1997, 37, 335–347. 10.1016/S0065-2571(96)00021-0. [DOI] [PubMed] [Google Scholar]
- Melchior D. L.; Sharom F. J.; Evers R.; Wright G. E.; Chu J. W. K.; Wright S. E.; Chu X.; Yabut J. Determining P-glycoprotein-drug interactions: Evaluation of reconstituted P-glycoprotein in a liposomal system and LLC-MDR1 polarized cell monolayers. J. Pharmacol. Toxicol. Methods 2012, 65, 64–74. 10.1016/j.vascn.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggemann E. P.; Currier S. J.; Gottesman M. M.; Pastan I. Characterization of the azidopine and vinblastine binding site of P- glycoprotein. J. Biol. Chem. 1992, 267, 21020–21026. 10.1016/S0021-9258(19)36791-2. [DOI] [PubMed] [Google Scholar]
- Martin C.; Higgins C. F.; Callaghan R. The vinblastine binding site adopts high- and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry 2001, 40, 15733–15742. 10.1021/bi011211z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.