

Abstract
While somatic mutations in colorectal cancer (CRC) are well characterized, little is known about the accumulation of cancer mutations in the normal colon prior to cancer. Here we have developed and applied an ultra-sensitive, single-molecule mutational test based on CRISPR-DS technology, which enables mutation detection at extremely low frequency (<0.001) in normal colon from patients with and without CRC. This testing platform revealed that normal colon from patients with and without CRC carry mutations in common colorectal cancer genes, but these mutations are more abundant in patients with cancer. Oncogenic KRAS mutations were observed in the normal colon of about one third of patients with CRC but in none of the patients without CRC. Patients with CRC also carried more TP53 mutations than patients without cancer, and these mutations were more pathogenic and formed larger clones, especially in patients with early onset CRC. Most mutations in the normal colon were different from the driver mutations in tumors, suggesting that the occurrence of independent clones with pathogenic KRAS and TP53 mutations is a common event in the colon of individuals who develop CRC. These results indicate that somatic evolution contributes to clonal expansions in the normal colon and that this process is enhanced in individuals with cancer, particularly in those with early onset CRC.
Keywords: somatic mutation, clonal expansions, cancer evolution, early onset colorectal cancer, cancer risk prediction, early cancer detection, precancer, carcinogenic fields, ultra-deep sequencing, duplex sequencing, single molecule sequencing, aging
INTRODUCTION
Colorectal cancers, like many other solid tumors, form through a process of somatic evolution that takes decades (1). This fact and our current understanding of the molecular pathogenesis of colorectal adenomas and adenocarcinoma imply that clones primed with cancer mutations exist within histologically normal tissue for years prior to cancer diagnosis. Providing convincing evidence of this proposed phenomenon is of critical importance for early cancer detection, risk prediction, and prevention (2). However, the robust identification of these mutant clones has been challenging due to the lack of methods able to reliably detect small subsets of mutant cells within morphologically normal tissue.
Two approaches to identify somatic mutations in normal tissue consist of either reducing the sample size to very small or microscopic regions in which mutant clones represent a sizable proportion of cells (3), or of expanding single-cells in vitro to larger clones amenable for next-generation sequencing (NGS) (4). These approaches have been applied to the characterization of somatic mutations in normal tissues including skin (5,6), esophagus (7,8), endometrium (9,10), bladder (11,12), colon (13,14), and others (15,16). Many of these studies have revealed an unexpected abundance of cancer driver mutations in histologically normal tissue (17,18) and are rapidly expanding our understanding of the role of somatic evolution in human aging (19,20). However, little is known about how this process differs in individuals with and without cancer, and whether this knowledge can be harnessed to improve cancer prediction and prevention. A main challenge relies on the fact that these approaches require the analysis of a large number of samples per individual and are very labor-intensive, precluding large cohort studies and translational applications.
An alternative approach to detect low frequency somatic mutations within normal tissue consists of performing ultra-deep sequencing using high-accuracy NGS methods such as duplex sequencing (DS) (21). DS employs double-stranded molecular tags, which enable error correction by consensus sequence independently in each DNA strand, effectively decreasing the error rate of sequencing from 10−3 to <10−7 (22). Because each duplex read corresponds to an original DNA molecule, this method enables the detection of single mutant DNA molecules among thousands of non-mutant genomes, thus providing extreme resolution to identify mutant cells in normal tissue by analyzing a single biopsy. The trade-off for this high-throughput, high-sensitive approach for mutation detection is that it requires large sequencing capabilities and therefore is mostly suitable for small target regions. We have used DS to perform ultra-deep sequencing of TP53 in normal gynecological tissues across the human lifespan, revealing a progressive enrichment of TP53 pathogenic mutations with older age (23). We have also demonstrated the presence of cancer driver TP53 mutations in peritoneal fluid (24), uterine lavage (23), and Pap test DNA (25) of women with and without ovarian cancer. Women with cancer tended to have higher TP53 mutation burden (24,25), suggesting increased TP53 somatic evolution in association with cancer progression.
Colorectal cancer is the second most common cause of cancer death in the USA and its incidence has been increasing in individuals under 50 years of age (26). The need for better biomarkers for CRC prediction, coupled with easy access to normal tissue via colonoscopy, makes this cancer type especially suitable to study the potential of clonal expansion detection in histologically normal tissue to identify early cancer progression. Our goal was to investigate whether mutations in common CRC genes (TP53, KRAS, PIK3CA and BRAF) could be detected by ultra-deep sequencing (>1,000x) in single, histologically normal colon biopsies, and to determine whether they were more frequent in individuals with CRC than in those who are cancer-free. These genes were selected because, together with APC, they constitute the 5 most frequently mutated genes in CRC. However, in contrast to APC, they accumulate mutations in localized hotspot regions, thus providing excellent targets for the development of ultra-sensitive sequencing tests for early cancer detection. We used a version of DS called CRISPR-DS (27), which employs CRISPR-based target enrichment to increase library preparation efficiency for small target panels. We demonstrate the feasibility of our approach to detect low frequency cancer driver mutations in normal colon, and the potential clinical value of these mutations as CRC risk markers. Our results suggest that this approach could be useful for CRC risk prediction in younger patients, who constitute a rapidly increasing subset of the population at risk (28).
MATERIALS AND METHODS
Subjects and samples
This study included normal colon mucosa samples (n=47) collected at the University of Washington Medical Center and affiliated practice sites (Seattle, WA, USA) from 24 patients without colorectal adenocarcinoma (CRC) undergoing colonoscopic screening or surveillance and from 23 patients with a newly diagnosed primary invasive colorectal adenocarcinoma undergoing surgical resection (Fig. 1A). Clinico-pathological characteristics of patients are provided in Supplementary Table S1 and Supplementary Methods. The two groups of patients were matched by age and polyp formation and were enriched with young individuals to explore differences in somatic mutations in early vs late onset CRC. We analyzed normal left colon epithelium in individuals without CRC and normal epithelium mostly from left colon and distant from tumor (>10cm) in individuals with CRC. Only one patient had neoadjuvant therapy. In all but one case, MSI was determined by mismatch repair defect based on routine clinical immunohistochemistry of proteins MLH1, MSH2, MSH6, and PMS2 in tumor Formalin-Fixed Paraffin-Embedded (FFPE) tissue sections. Two cases were MSI positive (Supplementary Table S1). None of the patients had hereditary cancer syndrome as determined by clinical and family history and MSI testing with reflex genetic testing for hereditary cancer genes using a variety of multi-gene panels in the two MSI positive cases. FFPE tumor blocks from patients with CRC were histologically examined and microdissected in 19 cases with sufficient tumor content. DNA extraction and library preparation from tumor DNA (Supplementary Methods) was performed after all normal tissue was analyzed to avoid cross-contamination. Patients provided written informed consent for study enrollment and sample collection. The study was conducted in accordance with recognized ethical guidelines, which include but are not restricted to U.S. Common Rule, Belmont Report, Declaration of Helsinki, and Nuremberg Code, and following protocols approved by Institutional Review Board committees at the University of Washington and the Fred Hutchinson Cancer Research Center. DNA from colorectal cancer cell lines HCT116, HT29 and SW480 was used for method validation (Supplementary Methods).
Figure 1. CRISPR-DS enables ultra-sensitive detection of cancer gene mutations in normal colon samples.
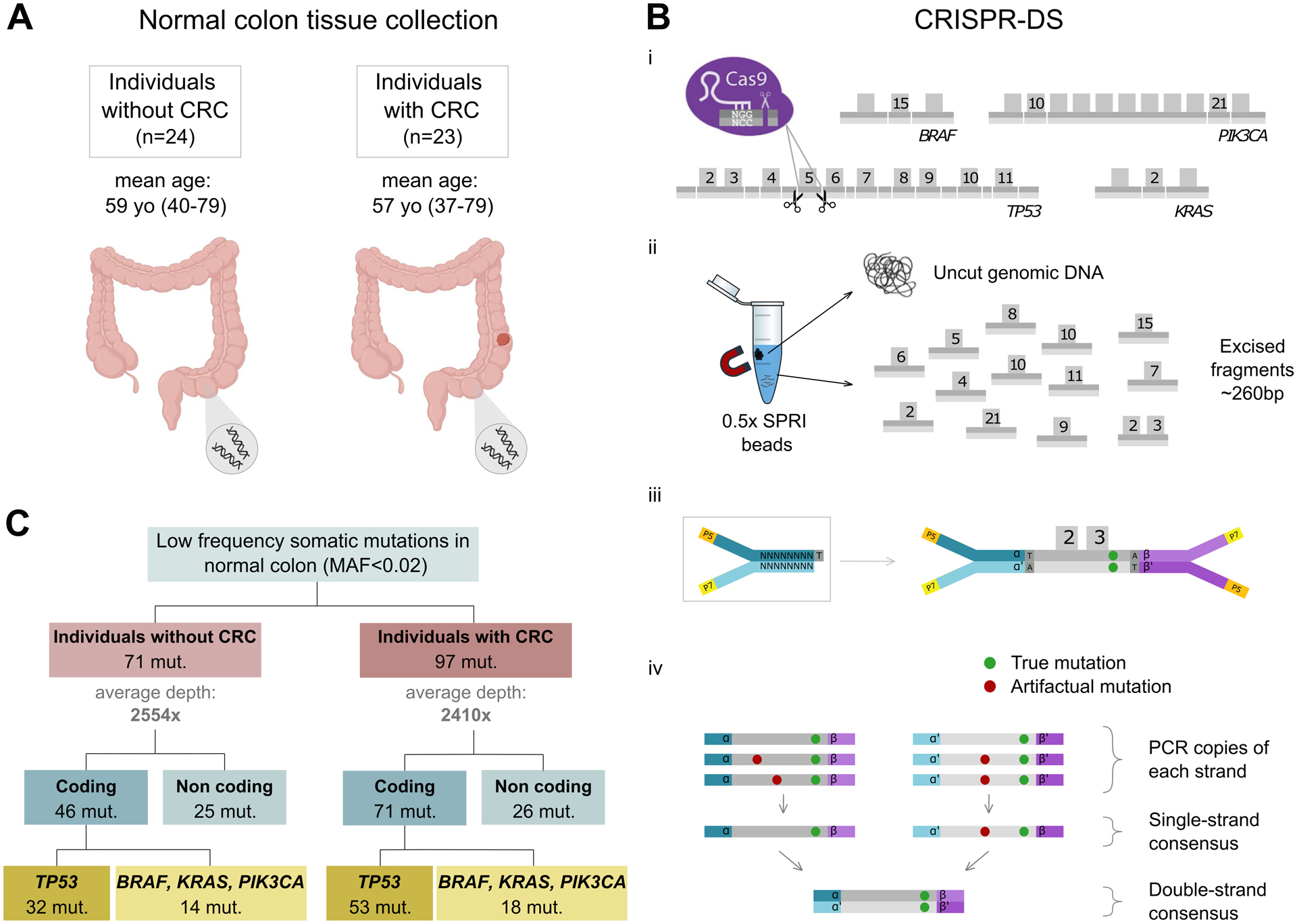
A. Normal colon biopsies were procured from individuals with and without CRC. B. CRISPR-DS. (i) CRISPR-Cas9 guides were designed to target common oncogenic mutation regions in BRAF, PIK3CA and KRAS as well as the full coding region of TP53 in fragments of ~260bp. (ii) Size selection with SPRI beads was used to enrich for excised fragments. (iii) Excised fragments were ligated to adapters with random double-stranded molecular tags. (iv) The generation of single-strand and double-strand consensus sequences from reads sharing the same molecular tags enables the elimination of artifactual mutations. Adapted from Nachmanson et al (ref. 27). C. Low frequency somatic mutations in cancer genes are identified in normal colon from patients with and without CRC.
CRISPR guide design
CRISPR-DS employs CRISPR-Cas9 digestion of target regions followed by size selection of excised fragments as a method for efficient target enrichment prior to library preparation (27) (Fig. 1B). We used Benchling (RRID:SCR_013955) to design guide RNAs (gRNAs) to excise the coding regions of the TP53 gene and the hotspot mutation codons of BRAF, KRAS and PIK3CA genes into fragments of ~250–280bp. Then we used the CRISPOR web tool (RRID:SCR_015935) (29) to select the best candidates, which included 24 gRNAs (Supplementary Table S2) that excised the target region into 13 fragments with a total panel size of 3461bp. The panel comprised 1953 coding bp and 1508 non-coding bp from intronic regions flanking the excised exons.
CRISPR-DS
Genomic DNA from normal colon tissues and CRC cell lines was processed for CRISPR-DS as previously described with minor modifications (27) (Supplementary Methods). Hybridization capture was performed with 120bp biotinylated xGen Lockdown probes (Integrated DNA Technology, Coralville, IA, USA) designed to target the selected regions of TP53, BRAF, KRAS and PIK3CA (Supplementary Table S3). Libraries were sequenced using 150 PE reads on a MiSeq Illumina platform on site or HiSeq at Genewiz (South Plainfield, NJ), allocating ~2 million reads per sample. Sequencing reads were analyzed as previously described (27) using pipeline v1.1.4 from https://github.com/Kennedy-Lab-UW/Duplex-Seq-Pipeline. Mutant Allele Frequency (MAF) was calculated for each mutation as the number of mutated duplex reads divided by the duplex depth at the given position.
Calculation of mutation frequency
For each sample, the overall duplex depth was calculated as the total number of duplex nucleotides sequenced divided by the size of the panel. On average, for each sample we sequenced 8.6M duplex nucleotides corresponding to a duplex depth of 2,484x (minimum 1,268x; maximum 4,306x) (Supplementary Table S4). To correct for the variability in sequencing depth across samples, comparisons were made based on mutation frequencies, which were calculated as the number of mutations in a given region (e.g., coding, non-coding, TP53 coding) divided by the total number of duplex nucleotides sequenced in that region. Coding included nucleotides in coding exons plus 2bp boundary nucleotides to capture splice site mutations, and non-coding included all the remaining nucleotides in the target regions. Similarly, mutation frequencies were calculated for specific types of mutations (e.g., drivers) by dividing the number of mutations in the category of interest by the total number of duplex nucleotides sequenced in the target region. Mutation counts and corresponding mutation frequencies for each sample are indicated in Supplementary Table S4.
Mutational analysis
Coding mutations were extracted from MAF files and were further annotated by mutation type (missense, nonsense, splice, indel and synonymous), mutation spectrum (C>A, C>G, C>T, T>A, T>C and T>G), localization in CpG dinucleotides, and driver mutations (Supplementary Methods). The list of annotated coding mutations for oncogenes and TP53 are presented in Supplementary Tables S5 and S6, respectively. Large intestine carcinoma variants from COSMIC (v.95) derived from whole genome screens and filtered for the target regions in the study were used to determine the mutation spectrum (6 possible nucleotide substitutions) in cancer samples (n= 2,768 mutations). COSMIC data derived from whole genome screens for the genes of interest was also used to determine the distribution of CRC mutations within protein domains.
Statistical analysis
Correlations were tested with Spearman’s rank test. Comparison of mutation frequency means across groups of individuals was performed by Mann-Whitney U test. Associations between categorical variables were tested with Fisher’s Exact Test. All tests were two-sided at an alpha level (type 1 error rate) of 0.05. The predictive model was estimated with the glmnet R package (30), with parameters for Lasso logistic regression. The penalization parameter was selected to restrict the model to 5 covariates. Predictive accuracy was calculated with the area under the ROC curve and its 95% confidence intervals as implemented in the pROC R package (31). Statistical analyses were performed with SPSS version 25 (RRID:SCR_002865) and R version 3.6.3.
Software avalability
Software is available at https://github.com/Kennedy-Lab-UW/Duplex-Seq-Pipeline.
Data availability Statement
Sequencing data from this study have been submitted to the NCBI BioProject database (RRID:SCR_004801, https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA767868.
RESULTS
CRISPR-DS enables ultra-sensitive detection of mutations in normal colon and CRC cell lines
We performed ultra-deep sequencing of normal colonic epithelium of 24 individuals without CRC and 23 individuals with CRC (Fig. 1A) using CRISPR-DS (Fig. 1B), a method that incorporates CRISPR-Cas9 target excision to eliminate problems associated with DNA sonication (32) and to increase sequencing efficiency by target enrichment with size selection prior to library preparation (27). We adapted CRISPR-DS to excise the exons that carry common oncogenic mutations in BRAF, KRAS and PIK3CA and the full coding region of TP53. Duplex adapters containing 8bp random molecular tags were used to uniquely label each DNA molecule to enable double-strand error correction as previously described (21,27) (Fig. 1B).
We first demonstrated the reproducibility, sensitivity, and specificity of the assay by deep sequencing DNA from 3 common CRC cell lines with driver mutations in the selected target genes (HCT116, SW480 and HT29) (Supplementary Methods). All the expected driver mutations were identified in addition to multiple low frequency (<0.1) mutations in HCT116 and several low frequency mutations in SW480, in agreement with the known levels of single nucleotide variants in these cell lines (33) (Supplementary Fig. S1A). An independent technical replicate experiment identified all the expected driver mutations, all the mutations with Mutant Allele Frequency (MAF) as low as 0.001, and a subset of the very rare mutations below 0.001, despite the decreased likelihood of resampling a rare event (Supplementary Fig. S1A). Remarkably, in HT29, which is the most genetically stable of the cell lines analyzed, no mutations except for the two clonal driver mutations were identified in the two replicates, demonstrating the high specificity of the assay. Based on this data, we estimated the error rate of the assay to be <6×10−8, which is comparable to other DS estimates (22,34). To further demonstrate the sensitivity and accuracy of the assay in an independent experiment, we spiked DNA from HT29 into DNA from HCT116 at 3 different ratios (1:10, 1:20, 1:100). The two driver HT29 mutations were observed at the expected frequencies in the 3 mixes even when present at low level (0.01 and 0.003) (Supplementary Fig. S1B).
We then used CRISPR-DS to sequence the normal colon of individuals with and without CRC. While the mean duplex depth across samples was variable, all samples reached a minimum of 1,000x duplex depth and the average depth for both groups of patients was similar (Fig. 1C). Overall, CRISPR-DS yielded a total of 404M duplex nucleotides, with 227M in coding regions and 177M in non-coding regions. A total of 168 mutations were identified: 117 coding and 51 non-coding (Fig. 1C). All these mutations had low MAF (<0.02). To correct for the fact that more mutations might be identified in samples with more sequenced nucleotides, sample comparisons were made based on mutation frequencies. Mutation counts and corresponding mutation frequencies for each sample are shown in Supplementary Table S4.
Individuals with CRC carry a higher frequency of coding mutations than individuals without cancer in a non-age dependent manner
We first compared coding and non-coding mutation frequencies in the normal colon of patients with and without CRC. Patients with CRC had a significantly higher coding mutation frequency in normal colon than patients without cancer (Mann-Whitney U test p=0.006, Fig. 2A) even when separating the patients by early and late-stage CRC (Supplementary Fig. S2). Non-coding mutation frequency, however, was similar in patients with and without CRC (Fig. 2A). Moreover, in patients with CRC, but not in patients cancer-free, the frequency of coding mutations was significantly higher than the frequency of non-coding mutations (Mann-Whitney U test p=0.001, Supplementary Fig. S3). Interestingly, the non-coding mutation frequency significantly increased with age (Spearman’s correlation p=0.024), but this trend was not observed for the coding mutation frequency (Fig. 2B). In addition, higher non-coding mutation frequency correlated with advanced epigenetic age in the normal colon as measured by the Horvath clock, the PhenoAge clock, and the EpiTOC clock, which are well-established measurements of epigenetic aging (35,36) (Fig. 2C). Coding mutations did not associate with lower or higher epigenetic age determined by these clocks. Altogether, these results indicate that while intronic, non-functional mutations accumulate with chronological and biological aging in the normal colon, coding mutations in cancer driver genes exceed the age-related background level, especially in patients that develop CRC, suggesting that functional mutations are selected and clonally expanded in the normal colon of these patients.
Figure 2. Normal colon of patients with CRC has higher, not age-related, coding mutation frequency and a mutation spectrum similar to cancers.
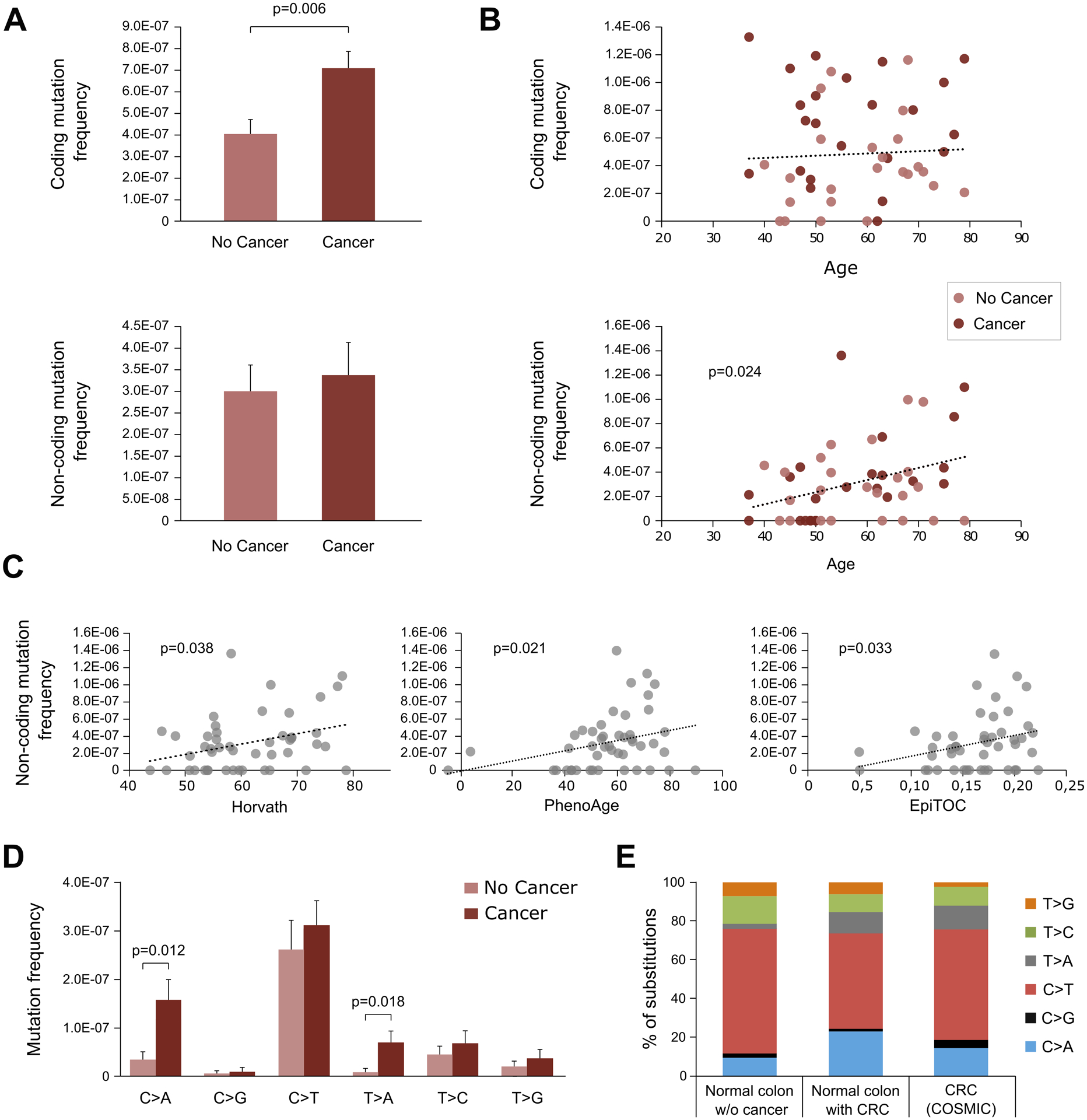
A. Coding and non-coding mutation frequency in normal colon from individuals with and without cancer. Mutation frequency is calculated as the number of mutations divided by the total number of duplex nucleotides sequenced in the coding or non-coding target regions, respectively. P-value corresponds to Mann-Whitney U test. Error bars represent standard error of the mean. B. Coding and non-coding mutation frequency and its correlation with age. P-value corresponds to Spearman’s correlation. C. Non-coding mutation frequency correlation with Horvath, PhenoAge and EpiTOC epigenetic clocks. P-values correspond to Spearman’s correlation. D. Frequency of coding mutation by substitution type compared between normal colon of individuals with and without cancer. P-values correspond to Mann-Whitney U test. Error bars represent standard error of the mean. E. Mutation spectrum compared between coding nucleotide substitutions from normal colon of individuals without CRC (n=42), with CRC (n=65), and CRC COSMIC data for whole genome screens restricted to the study targets (n=2,768). Only significant p-values are displayed.
To further explore the nature of the coding mutations present in the normal colon of patients with and without cancer, we classified them by mutational spectrum (C>A, C>G, C>T, T>A, T>C, T>G). While all types of mutations were more frequent in patients with cancer, two types were significantly overrepresented: C>A (Mann-Whitney U test p=0.012) and T>A (Mann-Whitney U test p=0.018) (Fig. 2D). These two types of mutations were also enriched in CRC based on data from the Catalogue of Somatic Mutations in Cancer (COSMIC) (37) restricted to the study targets (Fig. 2E), indicating higher resemblance to CRC mutations in normal colon of individuals with CRC.
KRAS and TP53 driver mutations are abundant in the colon of patients with CRC
We then explored the distribution of coding mutations by gene (Fig. 3A–D). For BRAF, PIK3CA and KRAS, we deep sequenced the exons with CRC mutation hotspots according to the COSMIC database. We found several mutations in BRAF and PIK3CA, but none of the BRAF mutations (0/7) corresponded to the canonical hotspot V600E mutation and only two of the PIK3CA mutations (2/12) corresponded to the 3 most common PIK3CA mutations in CRC (E545K, H1047R and E542K, which account for >40% of CRC PIK3CA mutations according to COSMIC (37)) (Fig. 3A–B and Supplementary Table S5). In contrast, 7 out of 13 KRAS mutations identified (54%) corresponded to oncogenic hotspot mutations in codons 12 or 13 (Fig. 3C). Remarkably, these 7 oncogenic KRAS mutations were all identified in normal colon from individuals with CRC. Overall, 30% of patients with CRC carried a KRAS hotspot mutation in normal colon compared to none of the patients without CRC (Fig. 3E, Fisher’s Exact Test p=0.004).
Figure 3. Normal colon carries mutations in common CRC genes, but these mutations are more abundant and pathogenic in patients with CRC.
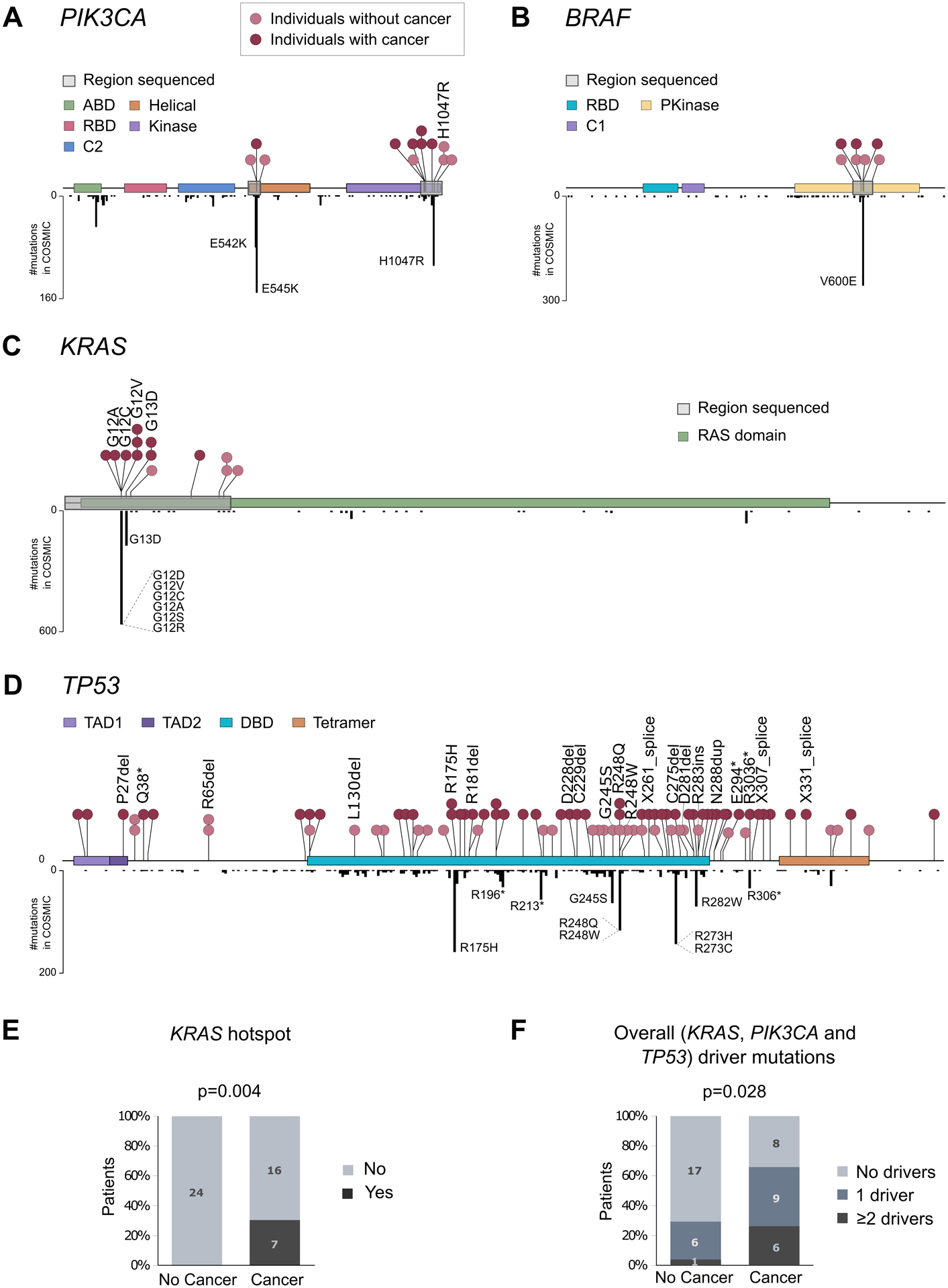
A-C. Distribution of mutations in PIK3CA, BRAF and KRAS in normal colon (above gene diagram) and in CRC samples from COSMIC database (below gene diagram). Normal colon mutations are color-coded by individuals with or without CRC and mutations corresponding to cancer hotspots are indicated. D. Distribution of mutations across TP53 in normal colon (above gene diagram) and in CRC samples from COSMIC database (below gene diagram). E. Percentage of patients with and without CRC that carry KRAS hotspot mutations in normal colon. F. Percentage of patients with and without CRC that carry one or more different cancer driver mutations in PIK3CA, KRAS, or TP53 in their normal colon. P-values correspond to Fisher’s Exact Test. ABD: adapter-binding domain; RBD: Ras-binding domain; Pkinase: protein tyrosine kinase domain; TAD: transactivation domain; DBD: DNA-binding domain; Tetramer: tetramerization domain.
Given the tumor suppressor role of TP53, we deep sequenced all its coding exons. We identified a total of 85 coding mutations, which mostly clustered in the DNA binding domain of the protein and coinciding with areas of high density of CRC mutations in COSMIC (Fig. 3D and Supplementary Table S6). This clustering suggests that TP53 mutations in normal colon are not random but follow patterns of selection similar to those operative in CRC. About one in 10 substitutions identified (9.4%) corresponded to the top ten most common TP53 substitutions in CRC (hotspots), which account for about half of CRC TP53 mutations reported in COSMIC. In addition, 17.7% of TP53 mutations were high impact mutations (indels, nonsense, or splice), which severely affect protein function. In total, more than a quarter of TP53 mutations identified in normal colon (27.1%) were either hotspots or high impact mutations. These mutations are likely to confer a selective advantage to the cells that carry them and were considered TP53 driver mutations for the purpose of mutation classification in the study. TP53 driver mutations were identified in the normal colon of 25% of patients without CRC and 52.2% of patients with CRC.
To investigate whether normal colon samples carried concurrently more than one cancer driver mutation, we plotted all the mutations identified for each gene in each patient (Fig. 4). In patients without CRC, we only observed one sample with multiple cancer driver mutations (2 in TP53 and 1 in PIK3CA). However, in patients with CRC we identified 6 samples with multiple cancer driver mutations, including 4 samples with driver mutations in KRAS and TP53, and 2 samples with multiple driver mutations in TP53. Overall, patients with CRC were more likely to carry one or more cancer driver mutations in normal colon than patients without CRC (Fisher’s Exact Test p=0.028, Fig. 3F). The common detection of single and multiple cancer driver mutations in the normal colon of patients with CRC suggests a prevalent process of selection and clonal expansion in these patients.
Figure 4. Mutations in normal colon of patients with CRC are often different from mutations in synchronous tumors and, in early onset CRC patients, driver mutations are frequently observed forming large clones.
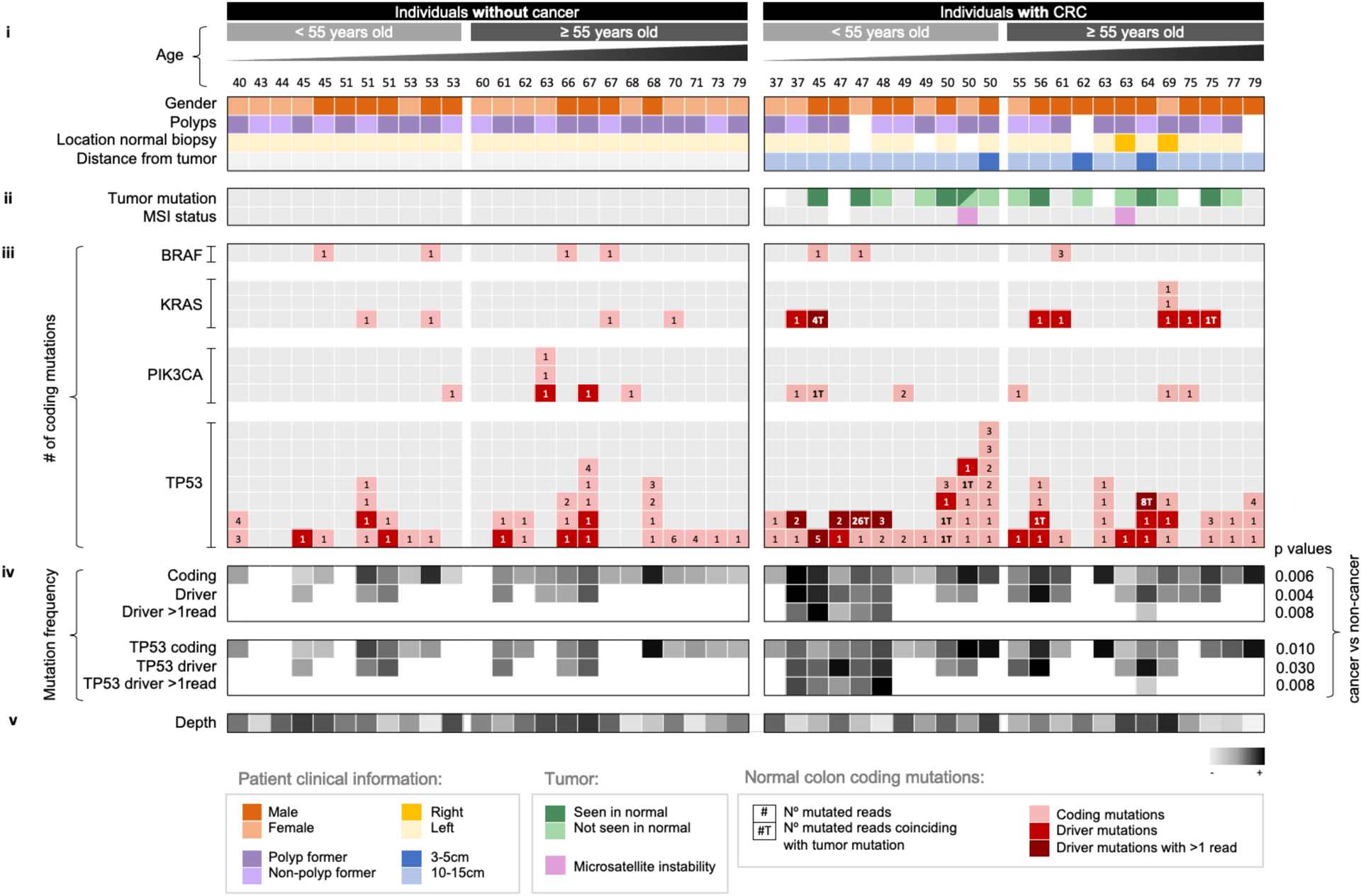
Each column corresponds to a patient. Patients are grouped by cancer status and sorted by ascending age. Panels of data indicate (i) clinical information, (ii) presence of tumor mutation in normal tissue and MSI status, (iii) normal colon mutation counts for each gene, (iv) normal colon mutation frequency, and (v) depth. In (i) and (ii), white squares indicate that the information is not available and grey squares indicate negative. Tumor mutation was negative for four cases that did not show any mutation in the 4 tested genes. Normal colon mutations for each gene (iii) are indicated with squares that contain the number of mutated reads color coded for mutations that are coding, drivers, and drivers with more than one (>1) mutated duplex read. ‘T’ next to the number indicates that the mutation was observed in the synchronous tumor. Driver mutations were conservatively defined as oncogenic hotspots and TP53 hotspots, nonsense, splice, and indel mutations. Greyscale heatmaps (iv) show mutation frequency values based on mutations that are coding, driver, and driver with >1 duplex read for all genes and TP53 only. P-values correspond to Mann-Whitney U test comparison of the mean frequency between individuals with and without CRC. Depth (v) indicates average duplex depth for all positions sequenced.
Of note, the MAF of the mutations identified, including driver mutations, was very low (<0.004 except for one mutation at 0.01) (Supplementary Fig. S4, Table S5, and Table S6), which makes them unidentifiable by standard sequencing methods (38). CRISPR-DS, however, enables accurate detection of very low frequency mutations, providing an ultra-high resolution view of the landscape of common CRC mutations in normal colon. The higher representation of cancer driver mutations in the normal colon of individuals with CRC indicates an excess of mutant clones in these patients whose detection by ultra-sensitive sequencing could be valuable for CRC risk prediction.
Most mutations in normal colon of patients with CRC differ from mutations identified in the cancers of the same patients
We then investigated whether the mutations observed in the normal colon of individuals with CRC coincided with those detected in the synchronous tumor, indicating possible clonal origin. We sequenced the same 4 gene regions in tumor DNA from 19 patients with available tumor tissue and catalogued all non-synonymous mutations and indel mutations with MAF>0.1 (Supplementary Methods and Supplementary Table S7). In 4 tumors, no such mutations were identified, likely because they were driven by other non-sequenced genes. In 7 out of 15 tumors, at least one non-synonymous TP53, KRAS or PIK3CA mutation was identified in the tumor as well as the normal tissue (Fig. 4 and Supplementary Table S7). However, in all cases, the normal tissue also carried additional cancer gene mutations not detected in the tumor. In addition, in 8 cases, tumor TP53, KRAS and PIK3CA mutations could not be identified in the normal tissue, which nevertheless carried other mutations in these genes. Overall, out of 44 non-synonymous TP53, KRAS, BRAF, or PIK3CA mutations identified in the normal colon of 15 individuals with sequenced tumor data, only 9 (20.5%) coincided with a synchronous tumor mutation, indicating that most mutant clones observed in normal colon are not precursors of the clone that eventually progressed to CRC. These results suggest that multiple independent mutant clones might be abundant in normal colonic mucosa of individuals at risk of CRC, which increases the chance of eventually giving rise to a tumor.
Of note, two of the patients with CRC had microsatellite instability (MSI) (Fig. 4), but we did not observe any differences in the normal colon mutation profiles of these patients compared to the rest of the patients in the study. While mutations in individuals without CRC were less abundant, in these patients we identified positive associations between the frequency of TP53 coding mutations and polyps formation (Mann-Whitney U test p=0.021) (Supplementary Fig. S5). Patients with polyps have an increased risk of developing CRC (39). Thus, these results are consistent with TP53 clonal expansions playing a role in the risk of progression to CRC.
Clones with cancer driver mutations are larger in patients with early CRC
Next, we asked whether the mutations identified in the normal colon of individuals with CRC were not only more abundant but also corresponded to larger clones. As duplex depth indicates the number of haploid genomes sequenced, the number of duplex reads containing a given mutation is proportional to the relative size of the clone carrying the mutation. We observed that patients with CRC not only had more driver mutations, but driver mutations were often detected in multiple reads, indicating they were in larger clones (Fig. 4). The two largest clones observed corresponded to mutations also identified in the synchronous tumors. Overall, the frequency of coding, driver, and driver in more than one read mutations (for all genes and for TP53 only) was statistically significantly higher in the normal colon of individuals with cancer compared to those without cancer (Fig. 4). However, the differences in the frequency of large mutant clones were driven by patients with early CRC, since large clones were especially common in individuals that developed CRC younger than age 50. Almost half of the younger patients (5/11) had large mutant clones in their normal mucosa compared to only one in 12 older patients with CRC. None of the patients without CRC had large mutant clones in their normal mucosa. These results suggest differences in the factors mediating clonal expansion and possibility progression to CRC in young and old individuals.
TP53 mutations in normal colon are more commonly pathogenic in individuals with CRC and more closely resemble mutations reported in CRC
TP53 mutations were further assessed using Seshat, a web service tool that provides functional data specific for TP53 variants, including frequency in the UMD database and predicted pathogenicity (40). We classified the mutations according to their location in the protein DNA-binding domain, their frequency in cancer, and their pathogenicity (See Supplementary Methods). Patients with CRC had higher frequency of TP53 mutations located in the DNA binding domain, common in cancer, and predicted to be pathogenic, than individuals without CRC (Fig. 5A).
Figure 5. TP53 mutations identified in normal colon are more pathogenic in individuals with CRC than in cancer-free individuals and more closely resemble TP53 mutations identified in CRC.
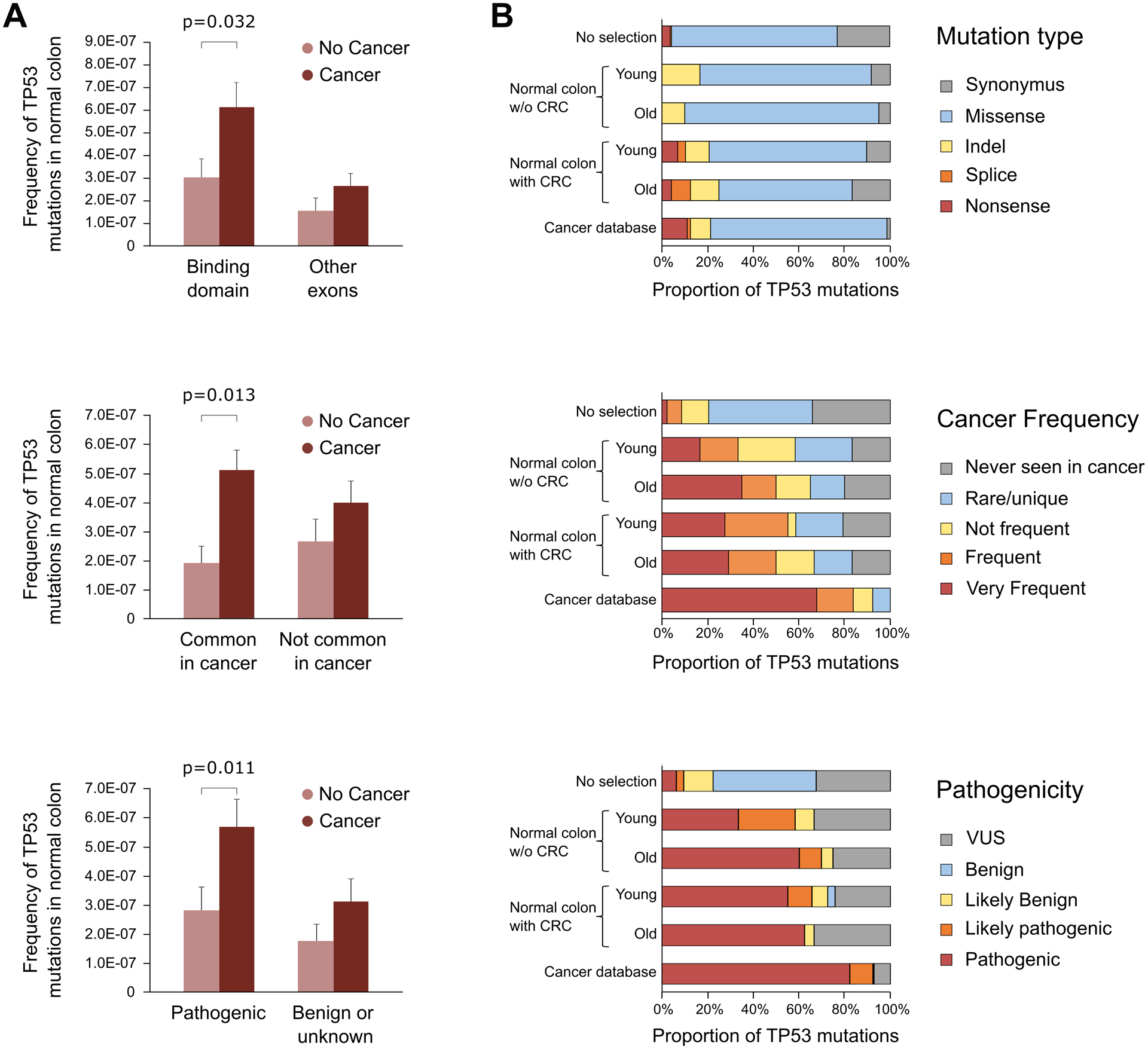
A. TP53 mutation frequency of individuals with and without CRC was compared based on mutations localized in the binding domain, mutations common in CRC, and mutations predicted to be pathogenic. Data was extracted from Seshat (ref. 38). Only significant p-values of Mann-Whitney U test are displayed. Error bars represent standard error of the mean. B. Distribution of TP53 mutations by mutation type, cancer frequency, and pathogenicity in normal colon of young (<55 years old) and old (≥55 years old) individuals without and with CRC compared to all possible TP53 mutations in the coding region (no selection, n= 3,546) and TP53 mutations reported in CRC in the UMD cancer database (n=17,681). Number of TP53 mutations in each group: young without CRC n=12; old without CRC n=20; young with CRC n=29; old with CRC n=24.
We then investigated the type, frequency, and pathogenicity of TP53 mutations observed in normal colon compared with mutations reported in colon carcinomas in the UMD database (2021, n=17,681) and with all the possible TP53 coding mutations in the theoretical absence of selection (n=3,546) (Fig. 5B). Mutations identified in normal colon samples were predominantly missense, similar to mutations reported in CRC or in TP53 in the absence of selection. However, only the normal colon of patients with CRC carried nonsense and splicing mutations, which are considered highly damaging, in similar proportions to what is observed in the cancer database. In normal colon, the distribution of mutations frequent in cancer and pathogenic clearly differed from the expected pattern under no selection and strongly resembled the pattern observed in cancers, especially in normal colon from older individuals and those with CRC (Fig. 5B). These results suggest a common process of positive selection of TP53 mutant clones in normal colon and CRC, which appears to be enhanced with aging and in those patients that develop CRC.
Integrative mutational analysis and proof-of-principle studies for the development of a CRC predictor
Our results provide support for the concept of creating a CRC risk predictor based on the normal colonic mucosa mutational profile. An essential step towards that goal is to construct a predictive model summarizing and harmonizing the mutational analysis results. Due to multicollinearity and to avoid overfitting, we used regularized logistic regression with Lasso penalty estimated to determine the 5 variables that were the best predictors (Supplementary Table S8). All quantitative variables with prior demonstrated significance in univariate analyses were included in the model as well as their interaction with age in order to determine potential differential effects between young and old individuals. The variables with the largest effects were the frequency of driver mutations (OR=2.16) and the presence of hotspots in KRAS (OR=1.86). Additional information was gained when considering the frequency of TP53 coding mutations, TP53 mutations common in cancer, and the interaction between driver mutations with > 1 supporting read and age (ORs of 1.26, 1.066, and 1.26, respectively). This later interaction indicates that the risk of CRC increases with increased frequency of larger clones (represented by mutations identified in more than 1 read) but only in younger individuals. The predicted accuracy of the model was good, with AUC = 0.69 95% CI: 0.53–0.85 after 5-fold cross-validation. While this preliminary analysis included a small number of cases and requires validation in larger studies, it demonstrates the potential of this approach for the development of a CRC predictor based on the mutational analysis of biopsies collected from histologically normal mucosa.
DISCUSSION
In this study we used deep sequencing with CRISPR-DS to perform high resolution single-molecule characterization of common colorectal cancer mutations in normal colon of individuals with and without CRC. We found that patients with CRC carried abundant oncogenic KRAS and TP53 driver mutations in normal colonic epithelium distant from the primary tumor and that these clones were larger in patients with early onset CRC. In addition, most of the mutations identified in normal colon were different from the mutations in cancers, suggesting the presence of multiple, independent mutant clones in association with CRC progression. These results expand our understanding of somatic evolution in the colon, offer insights about different mechanisms of carcinogenesis in early vs late onset CRC, and raise the possibility of using normal colon biopsies for CRC risk assessment.
Prior studies have demonstrated an age associated increase of somatic mutations in normal colon (13,14,41) in agreement with our findings for non-coding mutations. By focusing on cancer driver mutations and leveraging ultra-deep, single-molecule sequencing, our study expands these initial findings and demonstrates that, in addition to the age-related increase of somatic mutations, the normal colon of individuals with CRC frequently carries clones with oncogenic KRAS and pathogenic TP53 mutations. In some cases, we identified the same mutation in the normal biopsy and the tumor, which could be explained by contamination at sample collection, a mutation in development (42), or a very large epithelial clonal expansion (at least 10cm long) from which the cancer evolved. Large clonal expansions have been previously described in the colon of patients with ulcerative colitis (43–45), who are prone to CRC, and exemplify the concept of carcinogenic fields (also known as field effect or field cancerization) by which the normal cell population is replaced by a cancer-primed cell population that is morphologically normal but carries some of the phenotypes required for malignancy (46). In most cases in our study, however, driver mutations in normal colon did not coincide with the driver mutation in the tumor, suggesting the presence of fields composed by multiple precancerous clonal expansions as opposed to a single large clonal patch.
While some of the driver mutations in normal colon could be derived from clonal hematopoiesis, we previously demonstrated that the contribution of leukocyte mutations to other tissues appears to be relatively uncommon in middle-aged individuals (23). Nevertheless, future larger studies dedicated to characterizing the extension and composition of precancerous fields in CRC would benefit from analyzing matched peripheral blood to better characterize this issue. Additionally, careful consideration of the histological composition of samples is critical to avoid bias due to highly variable epithelial content.
Lee-Six et al recently demonstrated that by the sixth decade of life around 1% of normal colon crypts carry a clonal driver mutation, but these mutations occur in genes rarely mutated in CRC and similarly in patients with and without CRC (14). The authors suggested that these mutations likely contribute to crypt colonization whereas mutations in TP53 and KRAS might enable subsequent preneoplastic transformation. Our results support this hypothesis by demonstrating that oncogenic KRAS mutations and pathogenic TP53 mutations are infrequent in normal colon of patients without CRC but abundant in individuals that progressed to CRC. TP53 mutations are also frequent in patients with polyps, who are at risk of CRC progression, suggesting a very early connection to cancer risk. KRAS mutations have been implicated in the lateral expansion of mutant crypts (47,48) providing a plausible mechanism for the generation of large patches of mutant cells in normal colon. In addition, phylogenetic reconstruction of CRC evolution has revealed that TP53 and KRAS mutations are very early events, which take place after APC mutations decades prior to the development of CRC (1). Our results expand these findings by revealing that TP53 and KRAS mutations can be identified even in histologically normal epithelium of patients with CRC suggesting that multiple mutant clones accumulate in these individuals through life, one of which eventually evolves into cancer.
In this study, we purposely included individuals with early onset CRC to investigate the nature of clonal expansions in the normal colon when cancer develops at younger age. The incidence of early onset CRC has increased substantially in the last 2 decades for unclear reasons (28). We discovered that almost one half of younger individuals with CRC (5/11) harbor a large clone containing a TP53 driver mutation suggesting that the development of CRC in a large proportion of young individuals might be related to large expansions of TP53 mutant clones in normal colon. Interestingly, early-onset CRC has been reported to have a high prevalence of TP53 mutations and whole genome doubling (49), which suggests, in concordance to our data, that there might be different carcinogenic processes in early onset vs late onset CRC, the former involving frequent TP53 loss in early stages of carcinogenesis.
A limitation of our study is that the gene panel was small and therefore, we cannot extract information about mutational signatures, which are helpful to elucidate mutation etiology. In addition, APC was not included in the panel due to its large size and lack of mutational hotspots, but its absence limits the interpretation of clonal relationships. The goal of this study, however, was to determine whether ultra-sensitive sequencing of normal colon could have clinical value to assess CRC risk and, towards that goal, smaller panels are preferred to facilitate clinical applicability. We have demonstrated that a panel based solely on TP53 and KRAS might already have value for CRC risk assessment. A smaller panel enables to dedicate sequencing resources to achieve higher depth and/or screen more biopsies, providing more accurate estimates of clone size and abundance. This approach is well aligned with recently proposed efforts to develop panels of hotspot cancer driver mutations as biomarkers of cancer risk using error corrected NGS (50). Another limitation of the study is that normal colon prior to the development of CRC was not tested. Moving forward, that assessment will be critical to demonstrate predictive value. The easy accessibility of the colon and established colonoscopic surveillance make it possible to obtain biopsies prior to CRC progression offering an excellent scenario for cancer risk prediction as well as a unique opportunity to study clonal evolution as a function of aging and exposures.
In summary, we have demonstrated that normal colon from patients with and without CRC carry mutations in common colorectal cancer genes, but these mutations are more abundant in patients with cancer. In addition, individuals with cancer carry more mutations that are canonical cancer drivers, especially in KRAS and TP53, and these mutations tend to be found in larger clones. Our results support the notion that somatic evolution contributes to clonal expansions in the normal colon and that this process is enhanced in individuals with cancer and, most significantly, in those with early onset CRC. These findings open the possibility for the development of a CRC predictor based on ultra-deep analysis of mutations in normal colonic biopsies.
Supplementary Material
SIGNIFICANCE.
This work suggests prevalent somatic evolution in the normal colon of patients with colorectal cancer, highlighting the potential of employing ultra-sensitive gene sequencing to predict disease risk.
ACKNOWLEDGMENTS
We thank Dr. Scott Kennedy for manuscript revision and members of the Risques and Kennedy lab at the University of Washington, members of the Grady lab at the Fred Hutchinson Cancer Research Center and members of the Peinado lab at the Institut Germans Trias i Pujol for technical assistance. We are grateful to the ColoCare and GiCare Studies for providing patient samples and we deeply thank all the patients who donated samples, without which this research would not have been possible.
Financial Support:
This work was supported in part by i-PFIs (IFI16/00012) and M-AES (MV19/00042) research fellowships from Instituto de Salud Carlos III to JM; Agencia Estatal de Investigación (RTI2018-094009-B-100) to MAP; R21CA240885 and startup funds from the Department of Laboratory Medicine and Pathology to RAR; R50CA233042 to MY; P01CA077852, P30CA042014, P30CA15704, R01189184, R01CA194663, R01CA207371, R01CA220004, U01CA206110, U54CA143862, Listwin Family Foundation, Cottrell Family Fund, R.A.C.E. Charities, Seattle Translational Tumor Research, Rodger C. Haggitt Endowed Chair, and W.H. Geiger Family Foundation to WMG; and Agency for Management of University and Research Grants (AGAUR) of the Catalan Government grant 2017SGR723 to VM.
Footnotes
Conflict of interest: RAR is a consultant and equity holder at TwinStrand Biosciences and an equity holder at NanoString Technologies. RAR is named inventor on patents owned by the University of Washington and licensed to TwinStrand Biosciences. RAR’s laboratory has received research support from TwinStrand Biosciences. MAP is cofounder and equity holder at Aniling. MAP’s laboratory has received research funding from Celgene. VM is equity holder of Aniling. WMG is an Advisory Board member for SEngine, Guardant Health, and Freenome. He is a consultant for Diacarta. He receives non financial support from Tempus and Lucid Technologies.
REFERENCES
- 1.Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature 2020;578:122–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fittall MW, Van Loo P. Translating insights into tumor evolution to clinical practice: promises and challenges. Genome medicine 2019;11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis P, Moore L, Sanders MA, Butler TM, Brunner SF, Lee-Six H, et al. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat Protoc 2021;16:841–71 [DOI] [PubMed] [Google Scholar]
- 4.Jager M, Blokzijl F, Sasselli V, Boymans S, Janssen R, Besselink N, et al. Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat Protoc 2018;13:59–78 [DOI] [PubMed] [Google Scholar]
- 5.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015;348:880–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang J, Fewings E, Chang D, Zeng H, Liu S, Jorapur A, et al. The genomic landscapes of individual melanocytes from human skin. Nature 2020;586:600–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science 2018;362:911–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 2019;565:312–7 [DOI] [PubMed] [Google Scholar]
- 9.Suda K, Nakaoka H, Yoshihara K, Ishiguro T, Tamura R, Mori Y, et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep 2018;24:1777–89 [DOI] [PubMed] [Google Scholar]
- 10.Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, et al. The mutational landscape of normal human endometrial epithelium. Nature 2020;580:640–6 [DOI] [PubMed] [Google Scholar]
- 11.Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020;370:75–82 [DOI] [PubMed] [Google Scholar]
- 12.Li R, Du Y, Chen Z, Xu D, Lin T, Jin S, et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 2020;370:82–9 [DOI] [PubMed] [Google Scholar]
- 13.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 2016;538:260–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019;574:532–7 [DOI] [PubMed] [Google Scholar]
- 15.Li R, Di L, Li J, Fan W, Liu Y, Guo W, et al. A body map of somatic mutagenesis in morphologically normal human tissues. Nature 2021;597:398–403 [DOI] [PubMed] [Google Scholar]
- 16.Moore L, Cagan A, Coorens THH, Neville MDC, Sanghvi R, Sanders MA, et al. The mutational landscape of human somatic and germline cells. Nature 2021;597:381–6 [DOI] [PubMed] [Google Scholar]
- 17.Fiala C, Diamandis EP. Mutations in normal tissues-some diagnostic and clinical implications. BMC Med 2020;18:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy SR, Zhang Y, Risques RA. Cancer-Associated Mutations but No Cancer: Insights into the Early Steps of Carcinogenesis and Implications for Early Cancer Detection. Trends Cancer 2019;5:531–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Risques RA, Kennedy SR. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet 2018;14:e1007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijg J, Dong X. Pathogenic Mechanisms of Somatic Mutation and Genome Mosaicism in Aging. Cell 2020;182:12–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A 2012;109:14508–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat Protoc 2014;9:2586–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salk JJ, Loubet-Senear K, Maritschnegg E, Valentine CC, Williams LN, Higgins JE, et al. Ultra-Sensitive TP53 Sequencing for Cancer Detection Reveals Progressive Clonal Selection in Normal Tissue over a Century of Human Lifespan. Cell Rep 2019;28:132–44 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krimmel JD, Schmitt MW, Harrell MI, Agnew KJ, Kennedy SR, Emond MJ, et al. Ultra-deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc Natl Acad Sci U S A 2016;113:6005–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krimmel-Morrison JD, Ghezelayagh TS, Lian S, Zhang Y, Fredrickson J, Nachmanson D, et al. Characterization of TP53 mutations in Pap test DNA of women with and without serous ovarian carcinoma. Gynecologic oncology 2020;156:407–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel RL, Fedewa SA, Anderson WF, Miller KD, Ma J, Rosenberg PS, et al. Colorectal Cancer Incidence Patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nachmanson D, Lian S, Schmidt EK, Hipp MJ, Baker KT, Zhang Y, et al. Targeted genome fragmentation with CRISPR/Cas9 enables fast and efficient enrichment of small genomic regions and ultra-accurate sequencing with low DNA input (CRISPR-DS). Genome Res 2018;28:1589–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoffel EM, Murphy CC. Epidemiology and Mechanisms of the Increasing Incidence of Colon and Rectal Cancers in Young Adults. Gastroenterology 2020;158:341–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 2018;46:W242–W5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 2010;33:1–22 [PMC free article] [PubMed] [Google Scholar]
- 31.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics 2011;12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park G, Park JK, Shin SH, Jeon HJ, Kim NKD, Kim YJ, et al. Characterization of background noise in capture-based targeted sequencing data. Genome biology 2017;18:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal Cancer Cell Lines Are Representative Models of the Main Molecular Subtypes of Primary Cancer. Cancer Research 2014;74:3238–47 [DOI] [PubMed] [Google Scholar]
- 34.Valentine CC, Young RR, Fielden MR, Kulkarni R, Williams LN, Li T, et al. Direct quantification of in vivo mutagenesis and carcinogenesis using duplex sequencing. Proceedings of the National Academy of Sciences 2020;117:33414–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 2018;19:371–84 [DOI] [PubMed] [Google Scholar]
- 36.Wang T, Maden SK, Luebeck GE, Li CI, Newcomb PA, Ulrich CM, et al. Dysfunctional epigenetic aging of the normal colon and colorectal cancer risk. Clin Epigenetics 2020;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 2019;47:D941–D7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet 2018;19:269–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Click B, Pinsky PF, Hickey T, Doroudi M, Schoen RE. Association of Colonoscopy Adenoma Findings With Long-term Colorectal Cancer Incidence. JAMA 2018;319:2021–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tikkanen T, Leroy B, Fournier JL, Risques RA, Malcikova J, Soussi T. Seshat: A Web service for accurate annotation, validation, and analysis of TP53 variants generated by conventional and next-generation sequencing. Human mutation 2018;39:925–33 [DOI] [PubMed] [Google Scholar]
- 41.Abascal F, Harvey LMR, Mitchell E, Lawson ARJ, Lensing SV, Ellis P, et al. Somatic mutation landscapes at single-molecule resolution. Nature 2021;593:405–10 [DOI] [PubMed] [Google Scholar]
- 42.Pareja F, Ptashkin RN, Brown DN, Derakhshan F, Selenica P, Da Silva EM, et al. Cancer Causative Mutations Occurring in Early Embryogenesis. Cancer discovery 2021:candisc.1110.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker KT, Salk JJ, Brentnall TA, Risques RA. Precancer in ulcerative colitis: the role of the field effect and its clinical implications. Carcinogenesis 2018;39:11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi CR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol 2017;14:218–29 [DOI] [PubMed] [Google Scholar]
- 45.Salk JJ, Salipante SJ, Risques RA, Crispin DA, Li L, Bronner MP, et al. Clonal expansions in ulcerative colitis identify patients with neoplasia. Proc Natl Acad Sci U S A 2009;106:20871–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer 2018;18:19–32 [DOI] [PubMed] [Google Scholar]
- 47.Nicholson AM, Olpe C, Hoyle A, Thorsen AS, Rus T, Colombe M, et al. Fixation and Spread of Somatic Mutations in Adult Human Colonic Epithelium. Cell stem cell 2018;22:909–18 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olpe C, Khamis D, Chukanova M, Skoufou-Papoutsaki N, Kemp R, Marks K, et al. A Diffusion-like Process Accommodates New Crypts During Clonal Expansion in Human Colonic Epithelium. Gastroenterology 2021;161:548–59 e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim JE, Choi J, Sung CO, Hong YS, Kim SY, Lee H, et al. High prevalence of TP53 loss and whole-genome doubling in early-onset colorectal cancer. Exp Mol Med 2021;53:446–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris KL, Myers MB, McKim KL, Elespuru RK, Parsons BL. Rationale and Roadmap for Developing Panels of Hotspot Cancer Driver Gene Mutations as Biomarkers of Cancer Risk. Environ Mol Mutagen 2020;61:152–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data from this study have been submitted to the NCBI BioProject database (RRID:SCR_004801, https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA767868.