

Abstract
Permanently charged and ionizable organic compounds (IOC) are a large and diverse group of compounds belonging to many contaminant classes, including pharmaceuticals, pesticides, industrial chemicals, and natural toxins. Sorption and mobility of IOCs are distinctively different from those of neutral compounds. Due to electrostatic interactions with natural sorbents, existing concepts for describing neutral organic contaminant sorption, and by extension mobility, are inadequate for IOC. Predictive models developed for neutral compounds are based on octanol–water partitioning of compounds (Kow) and organic-carbon content of soil/sediment, which is used to normalize sorption measurements (KOC). We revisit those concepts and their translation to IOC (Dow and DOC) and discuss compound and soil properties determining sorption of IOC under water saturated conditions. Highlighting possible complementary and/or alternative approaches to better assess IOC mobility, we discuss implications on their regulation and risk assessment. The development of better models for IOC mobility needs consistent and reliable sorption measurements at well-defined chemical conditions in natural porewater, better IOC-, as well as sorbent characterization. Such models should be complemented by monitoring data from the natural environment. The state of knowledge presented here may guide urgently needed future investigations in this field for researchers, engineers, and regulators.
Keywords: ionizable organic compound, anion, cation, zwitterion, sorption model, environmental risk assessment, contaminant fate
Short abstract
We argue for the development of new concepts to describe mobility of charged organic compounds and present starting points.
For regulators, engineers, and researchers, the mobility of contaminants is crucial for assessing their potential to contaminate groundwater and surface waters. The mobility of an organic compound is generally inversely related to its tendency to sorb. Widely used approaches to assess sorption were developed for neutral compounds but are inadequate to describe the complex behavior of permanently charged and ionizable organic compounds (IOC). Common examples of IOC are weak acids and bases that have a pH-dependent fraction of species with a negative or positive charge, respectively, due to (de)protonation. Some compounds are permanently charged (ionic) under environmental conditions and/or exist in a zwitterionic form with both positive and negative charges in the same structure. Numerous contaminants of concern are IOC, including many pharmaceuticals, pesticides, industrial chemicals, such as dyes and polymer building blocks, as well as most per- and polyfluoroalkyl substances, and natural toxins.
Compounds that are (partially) charged at environmental pH make up more than half of all substances recently categorized as priority “persistent, mobile, and toxic” (PMT) or “very persistent, very mobile” (vPvM) substances. These compounds pose a threat to clean and safe drinking water if emitted in substantial volumes, due to their high mobility, persistence, and limited removability from water.1−3 In addition, approximately 48% of all compounds registered under Europe’s REACH regulation are (partially) charged at environmentally relevant pH (4–9).4 A recent screening for persistent, mobile (PM), and vPvM compounds in surface water underlines the importance of IOC, as 85% of the identified compounds were expected to be charged at environmental pH.5 What distinguishes IOC from well-studied neutral organic compounds is that their sorption behavior, and consequently, their mobility in the environment, depends, often dramatically, on the local pH, water hardness, and mineral composition of soils or sediments. Therein, IOC sorption, but also bioaccumulation,6 and ecotoxicity7 strongly differ between uncharged neutral, negatively charged, positively charged, and zwitterionic species.
Sorption affinity can be expressed as the solid–water equilibrium distribution coefficient Kd, which is the ratio of chemical concentration in the solid phase (Cs, μg/kg) to that in the aqueous phase (Caq, μg/L) at equilibrium:
| 1 |
For neutral organic compounds, it has been established since the 1980s that soil/sediment organic matter (SOM) is the key sorptive phase (sorbent).8,9 To ease comparison of sorption data between different sorbents, it is common practice to normalize measured Kd values to the fraction of organic carbon in soil or sediment (fOC), resulting in KOC values (L/kgOC), that allow for a more generalizable quantification of organic compound sorption:10
| 2 |
While the KOC for a given compound is not a universal constant and can vary with the structure and composition of SOM, variation of KOC in common soil and sediment organic matter is typically within a factor of 2,11 or in the worst case an order of magnitude for neutral organic chemicals.12 However, KOC can increase by several orders of magnitude if the SOM includes highly condensed aromatic fractions of pyrogenic material (“black carbon”).13 Nevertheless, KOC is commonly used to assess contaminant mobility in regulatory frameworks such as the European Biocide regulation,14 and the Food and Agriculture Organization of the United Nations guideline on soil contamination.15 As experimental Kd and KOC values are not always available, octanol–water partitioning based approaches are commonly used to estimate these parameters for screening purposes.
Here, we maintain that the widely used octanol–water partitioning- and KOC-based approaches are not well applicable for assessing sorption and mobility of IOC. We discuss compound and soil properties driving sorption of IOC, highlight limitations of current models, and discuss possible complementary and/or alternative approaches to better assess IOC mobility for researchers, engineers, and regulators.
Octanol–Water Partitioning
Following pioneering work by Karickhoff et al. in 19799 for sediments, quantitative relationships between the KOC and the octanol–water partition coefficient (Kow) obtained in independent experiments have been widely applied in sorption and mobility assessments of neutral hydrophobic compounds:16
| 3 |
where Kow is the ratio of concentrations in the (water-saturated) octanol and (octanol-saturated) water at equilibrium, and a and b are regression parameters. The application of Kow as a proxy for KOC to assess organic compound sorption assumes that partitioning into the bulk SOM phase is the predominant sorption process, and that octanol is a good surrogate for SOM, which as we will discuss later, for IOC it is not.
Since the neutral, charged, and (if relevant) zwitterionic species of IOC partition differently into octanol, in these cases Kow is replaced with an operational partitioning ratio called Dow. Dow is the concentration ratio of the sum of all species in octanol (C0) to the sum of all species in water (Cw) at equilibrium and at a given pH and ionic composition:
| 4 |
Generally, partition of a charged compound into octanol requires partition of an accompanying counterion to maintain electroneutrality in solution. Therefore, the extent to which a charged species partitions from water into octanol depends on the concentration and type of available counterions in the aqueous phase.17,18 If it is assumed that partitioning of the charged species is negligible compared to the neutral species, the calculation of Dow is simplified to19
| 5 |
| 6 |
However, if the charged species do interact with soil constituents as explored in the next sections, the approach is inadequate to estimate sorption and mobility of IOC. Moreover, eqs 5 and 6 cannot be used for permanently charged compounds such as quaternary ammonium cations, where Kow (neutral) does not exist. Additionally, hydrophobic domains in other parts of the IOC, charge delocalization over many atoms in the IOC (e.g., dinoseb, pentachlorophenoxide17), as well as hydrophobic organic counterions, can facilitate partitioning of an IOC into octanol as net-neutral ion pairs. Lastly, surfactant-like IOCs with a hydrophobic tail (e.g., many per- and polyfluoroalkyl substances) can form emulsions at high concentrations, which could affect their partitioning between organic matrices (octanol/SOM). Models to estimate Dow are generally not capable of adequately accounting for these factors, resulting in erroneous Dow estimates. This is especially true for cations and zwitterions, where models such as the Estimation Programs Interface (EPI) Suite20 do not yield meaningful estimates. For example, the EPI Suite by default assigns very low Dow values (log Dow = −6) to compounds with quaternary nitrogen structures, but ignores the ionized moiety in other compounds and treats them as if they were neutral.21 Even more importantly, as we will explore in the next sections, no matter how Dow is determined, Dow is not suitable for modeling IOC sorption when the charged species substantially affects sorption.
Octanol–Water Partitioning Is Not Suitable for Describing IOC mobility
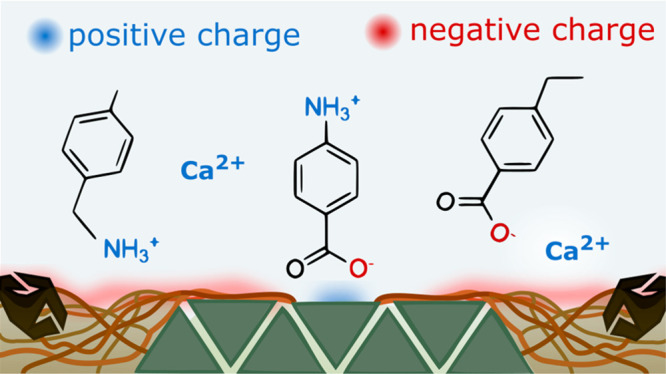
The application of Kow as a proxy for KOC to assess organic compound sorption and mobility assumes that partitioning into the SOM phase is the only/dominant sorption process. Models based on Kow or Dow do not consider that increasing pH results in increasing negative charge density in soil,22 as explained later. This negative charge repels organic anions and attracts organic cations, which Dow cannot reflect, as shown in Figure 1.
Figure 1.

Mobility of IOC in soils and sediments depends not only on hydrophobicity, but is additionally affected by the surface charge of soil constituents, pore water chemistry, and IOC speciation. PZC = sorbent point of zero charge; above this pH overall surface charge is negative, Dow = water-chemistry dependent octanol–water partitioning coefficient, pKa = IOC dissociation constant. Black solid lines and colored dashed lines represent hydrophobicity and mobility, respectively. The colored ranges represent the influence of counterion concentration.
Weak bases which form cations at pH < pKa of the corresponding acid, experience electrostatic repulsion at very low pH, which increases their mobility, followed by a minimal mobility due to electrostatic attraction toward negatively charged mineral and SOM moieties with increasing pH, and finally an intermediate mobility at pH ≫ pKa, where the neutral species is predominant.23 In contrast, for weak acids, Dow would be high and mobility would be correspondingly low at pH ≪ pKa, where the compound exists predominantly in the neutral form. As the pH transitions through the pKa, Dow is expected to decrease and mobility to increase as the compound is converted to the anionic form which is repulsed by negatively charged soil moieties.
The type and concentration of naturally occurring (counter)ions can modulate IOC sorption and mobility, as illustrated by the dashed lines in Figure 1. Importantly, the (counter)ion-dependent change in Dow does not cover the changes on the sorbent side brought about by the presence of counterions. For example, for cations, Dow increases with higher salinity because of the increased concentration of counterions that aid formation of ion pairs.17,18 However, in real soils or sediments the higher concentration of cations would compete for sorption sites and thus actually decrease sorption of cationic compounds.24,25 By contrast, (counter)ions could increase sorption for anionic compounds by decreasing electrostatic repulsion from negatively charged moieties.
Octanol is not a Suitable Surrogate for SOM
The free energy of sorption (ΔGsorp), which is linearly related to the logarithm of Kd, can be expressed as the sum of the contributions from net driving forces for removal of the solute from water and placing it in association with the solid. These driving forces include: van der Waals forces of dispersion and induction (ΔGvdW); polar forces including dipole–dipole, charge-dipole, and hydrogen (H)-bonding (ΔGpolar); Coulomb interactions between full charges (ΔGcoul), and the hydrophobic effect (ΔGhyd). The hydrophobic effect, also referred to as cavity formation energy, results from the sum of forces that limit the solubility of molecules in water. Its underlying cause is the disruption of the cohesive energy of water due to the greater ordering of water molecules and the lower number of water–water H-bonds in the hydration shell of the nonpolar moiety compared to the bulk water phase.26−28
Octanol is regarded an acceptable surrogate for SOM with respect to ΔGhyd and ΔGvdW. Thus, as shown in Figure 2, the best estimations of KOC from Kow exist for neutral, nonpolar molecules, where on average the log KOC is slightly smaller than log Kow.9,29 Octanol is less suitable with respect to ΔGpolar because octanol engages only in dipolar and ordinary (weak) H-bonding interactions of its aliphatic – OH group and misses many other polar interactions between sorbates and SOM. This is the reason why KOC-Kow correlations are somewhat poorer for polar compared to apolar compounds.29 For IOC, where ΔGcoul is relevant, the pH-dependent “DOC“ has become a common parameter used instead of KOC.3,19 For IOC, octanol is even less suitable as a surrogate for SOM with respect to ΔGcoul because, unlike SOM, octanol contains no charged groups. Consequently, for Dow-derived DOC estimations of IOC, errors substantially increase further and become meaningless. As shown in Figure 2, available Dow values can be several orders of magnitude smaller than experimentally measured DOC values for IOC, due to both ΔGcoul not being accounted for by Dow and the pH dependence being substantially more sensitive for Dow than DOC.
Figure 2.
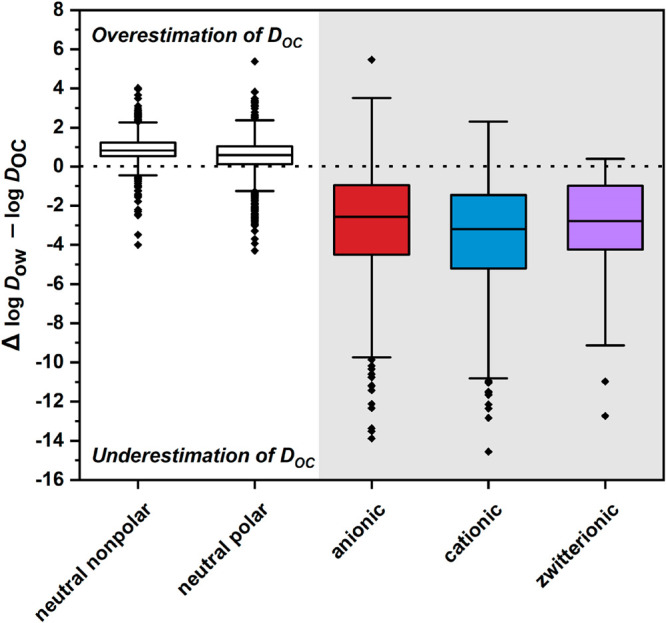
Differences (Δ) comparing lowest available Dow in the pH range 4−919 with measured DOC. D = K for neutral compounds. The dotted line at Δ = 0 indicates the point where Dow = DOC. Charged species are highlighted in color. The extent of the boxplot relates to the uncertainty associated with predicting sorption from Kow/Dow for a given compound. The middle line in the box corresponds to the median, the box to the 25% quantiles and the whiskers to the 1.5-fold interquartile range. Dow being extremely lower than experimental DOC is substantially influenced by the larger pH dependence of Dow over this pH range, and Coulombic interactions with SOM not being considered in Dow. All boxplots are based on data presented in more detail by Arp et al.,30 which compiled experimental KOC, Kow, and pKa data from the eChemPortal,31 and additional sources.29,32 Sample size: neutral nonpolar (n = 703), neutral polar (n = 1066), anionic (n = 488), cationic (n = 607), zwitterionic (n = 71).
IOC can Partake in a Variety of Interactions in Soil not Represented by Octanol
There are a number of sorption mechanisms of IOC in soil/sediment that are not captured at all by octanol-based models. Partitioning of organic compounds into octanol, whether they are ionized or not, is generally linear with solute concentration. While (ab)sorption of most neutral compounds into “soft” amorphous SOM phases is also close to linear, the same is not true for minerals and “hard” crystalline SOM phases (e.g., coal, black carbon), which can show moderate to strong nonlinearity of (ad)sorption with solute concentration.10 Here, the Kd generally decreases with increasing concentration, because adsorption sites are occupied preferentially in the order of the energy gain they enable, which varies. Deviation from linearity is more pronounced for organic anions and cations relative to neutral molecules, showing L- or H-type isotherms and additional sorbent-specific effects (e.g., for black carbon).33,34
As Illustrated in Figure 3, a number of interactions that do not occur for neutral compounds can occur for charged species (panels highlighted in gray in Figure 3). None of the following interactions are possible with octanol: Nonspecific electrostatic attraction or repulsion by charged moieties can direct the sorption of charged species (d,f in Figure 3), which can be described by the Donnan potential.35 Specific interactions of charged species with individual sorption sites widely differ among IOC, but often involve interactions between charged functional groups or aromatic structures in the IOC.36,37 The degree of aromatic condensation of SOM and black carbon can play an important role in the sorption of aromatic and heterocyclic compounds, which can interact via several types of π-electron donor–acceptor interactions (b,g, h in Figure 3).38−41 Weak acids and bases are capable of forming very strong, “charge-assisted” H-bonds (CAHB, c* in Figure 3) when acidic sites on SOM and black carbon have similar pKa values as the IOC.42 The degree of hydration can also affect sorption site accessibility by crowding out solute molecules,43 or by disrupting SOM–SOM contact points within the solid phase.44,45
Figure 3.

Key drivers and interactions for sorption of different groups of organic compounds under acidic conditions (top row) and alkaline conditions (bottom row). Compound groups with representative examples from left to right: neutral nonpolar compounds, neutral polar compounds, anionic compounds, cationic compounds, and zwitterionic compounds. Panels with charged species are highlighted in gray. Possible drivers and interactions: a = hydrophobic effect, b = π–π electron donor–acceptor interaction, c = H-bond, c* = charge assisted H-bond, d = electrostatic repulsion, e = cation bridging, f = electrostatic attraction, g = anion – π bond, h = cation – π bond.
SOM is not Always the Predominant Sorbent of IOC
Sorption models based on KOC/DOC are conceptually not sufficient for capturing the full range of factors influencing IOC mobility in many soils and sediments. Such sorbents are complex mixtures of minerals, SOM, black carbon, colloids, and pore water containing dissolved organic matter (DOM) and dissolved inorganic ions, including anions such as Cl–, NO3–, H2PO4–/HPO42–, SO42–, and HCO3–/CO32–, as well as cations such as Na+, K+, Ca2+ and Mg2+. IOC sorption to surfaces and nanometer-size pores of minerals and black carbon can be affected by all these substances.36,46,47
Most soil constituents, including SOM, black carbon, phyllosilicate minerals, and Mn oxides, exhibit an overall negative surface charge at pH of 4–9.22 The negative charge predominating on soil/sediment surfaces derives mainly from oxygen-containing functional groups that dissociate with increasing pH (e.g., carboxyl-, and hydroxyl groups). These functional groups determine the solid’s capacity to bind cations via cation exchange interactions, which can be quantified as the cation exchange capacity (CEC) at a given pH. SOM, black carbon, and clay minerals are especially high in CEC and are thus crucial to the mobility of cations.48 Sorption of organic cations to clay minerals depends on surface charge distribution, as well as type of exchangeable cations.49 Some organic anions (e.g., carboxylates, sulfonates) can also undergo surface bonding on mineral surfaces by ligand exchange with the underlying metal ions.33
On the other hand, only ∼7% of the numerous minerals in global soils have surfaces that are net positively charged at ambient pH, most importantly Fe-oxides and Al-oxides.22 Anion exchange can occur in the presence of these positively charged minerals. However, anion exchange capacity (AEC) is usually much smaller than CEC. As DOM and many types of colloids in the porewater are composed of negatively charged polyelectrolytes of different molecular sizes, which can compete with IOC anions for positively charged sites that are accessible to them. Thus, whatever AEC is inherent to soils or sediments is reduced by adsorption of DOM and/or aggregation with negatively charged minerals.
Implications for Regulation and Risk Assessment
Regulatory criteria for contaminant mobility in soil are critically important to protect surface water, groundwater, and drinking water.50 The emphasis of mobility for risk assessment has recently been reinforced in the European Commissioǹs “Chemicals Strategy for Sustainability towards a Toxic Free Environment”, which states that mobility should be included in a wide number of activities related to chemical regulation, in order to reduce exposure to hazardous substances via groundwater, drinking water and other pristine water bodies.51
In 1989 Gustafson52 combined soil half-lives and KOC values to estimate pesticide leachability. Today these two parameters are still used, as substances that degrade easily or sorb strongly are less prone to percolate to groundwater or pass bank filtration. For instance, the European regulatory framework for bioicides uses a KOC of 500 L/kgOC and soil half-life of 21 days as threshold values for groundwater risk assessment.14KOC and DOC threshold values for the mobility criteria for PMT and vPvM substances to be adopted by the European Classification, Labeling and Packaging (CLP) and REACH regulations are currently under discussion, and are expected to be finalized in 2022.53 As of now, the thresholds being investigated by the European Commission are a log DOC < 3 within a pH range of 4–9 to be considered mobile, and substances with a log DOC < 2 to be considered very mobile.19,54 Revised European chemical regulations that include PMT/vPvM substances could potentially mandate experimental DOC assessments of all persistent substances in Europe, which is a key market for the chemical industry.
Currently, experimental Kd values, which would reflect the variety of possible soil (mineral) compositions and water–chemical conditions in the environment, are not widely available. Thus, estimated DOC or Dow values could be used as screening parameter to prioritize substances for experimental determination. As discussed previously, errors in the Dow-to-DOC correlations for IOC can be substantial and are more pronounced for modeled than for experimental Dow data.19 This renders the use of Dow for risk assessment problematic. However, this does not invalidate the role of Kow as a screening parameter for neutral nonpolar and neutral polar compounds, or arguably very large Dow to screen for nonmobility of IOCs (considering Dow are generally < DOC). Dow is, however, not capable of substituting experimentally determined sorption parameters for IOC. For local mobility assessments, DOC or even soil-specific Kd values need to be measured, due to substantial uncertainties in Dow extrapolations. To aid the comparison of such values, soil mapping could be helpful, using databases from soil sciences and regulators.55,56 Still, local measurements are not always possible, and even if they were, they are impractical for inclusion in generalized chemical regulation.
Moving Forward
In addition to simple relationships between sorption and Kow/Dow, more sophisticated quantitative structure–property relationships (QSPR) exist to estimate the sorption of neutral compounds to a vast number of sorbents.57 The appeal of these approaches is their capacity to yield mechanistic insights into sorption in dependence of compound properties (e.g., polarizability, H-bonding abilities). QSPR approaches based on such descriptors for charged species have been proposed.58,59 However, as the behavior of charged compounds strongly depends not only on pH, but also on the ionic composition in water, determining generalizable descriptors is not always straightforward. In addition, most QSPR approaches are developed for pure solvents or sorbents and fall short of describing complex mixtures of SOM, minerals, and black carbon which contain varying sorption sites and exhibit different CEC.
Mobility and sorption of IOC are more complex and variable than that of neutral compounds, as a larger number of factors can modulate their behavior. Most key interactions for charged compounds are not driven by hydrophobicity but rather by IOC speciation and sorbent surface charge, as well as the amount and composition of other ions in solution. Because of the complexity of IOC mobility, the emergence of a single and generalizable best-for-all parameter as alternative to experimentally determined KOC/DOC values is unlikely. It is important to deduce from the discussion above, that for IOC, experimentally determined Kd for soil should not simply be converted to DOC since multiple soil components contribute to overall IOC sorption and mobility. Until better approaches are developed, experimentally determined Kd, and by extension KOC/DOC, values for diverse soil or sediment types are the only available parameters for initial sorption and mobility assessments for chemical regulation.
For cations, where electrostatic attraction to negatively charged surfaces often drives sorption, CEC normalized Kd values (KCEC) have been proposed as a complementary approach to the use of KOC.46 Sorption of organic cations to specific soil components (standardized SOM and Illite clay), have been compared to sorption to natural soils.46 This comparison found that sorption to the clay fraction had a negligible contribution to the Kd for an OC-enriched soil, whereas for a clayish soil the SOM sorption strongly underestimated the Kd, which could be largely accounted for by including the Illite clay sorption affinity. For organocations, mobility estimates for a suite of soil types could thus be based on simple experimental measurements (in this example fOC, CECsoil, KSOM, Kclay). In another study, maximum sorption capacity of black carbon for the dicationic herbicide paraquat was proportional to the square of the CEC of the black carbon, suggesting that the dication associated in a bidentate fashion with appropriately spaced negative sites on the sorbent.43 Thus, measurements of IOC sorption to pure soil constituents (SOM, clay minerals, black carbon) at specific pH and ionic strength conditions may offer a solid base for improved IOC mobility estimates. Although no one single “standard” SOM exists, a growing sorption data set on IOC has become available for Pahokee peat,60,61 and many processes such as influence of ionic strength, hardness, and pH dependency are relatively constant for other SOM types.60
In future approaches, DOC could be complemented by pH-dependent KCEC for cations, and extended to pH- and ionic-strength-dependent sorption measurements of key scenarios (e.g., a soil with a low OC content, a high CEC, and a low ionic strength would likely show large discrepancies between DOC and KCEC). A similar approach could also be developed for anions. To close the gap between regulation and science, researchers may develop compound-group specific “realistic worst-case” scenarios that could be applied in risk assessment. For example, considering interactions in Figure 3, anions could be investigated at very high pH and low ionic strength, where electrostatic repulsion increases mobility and the available cations for charge shielding and cation bridging are minimized. By contrast, the mobility of cations could be measured at low pH and high ionic strength, where soils are partially positively charged, CEC is lowered, and inorganic cations can compete for sorption sites. A more detailed categorization of IOC would need to be developed for such an approach to account for complex molecules with multiple functional groups, as well as physical accessibility to sorption sites resulting from differences in sorbate conformation, sorbent geometry, and chemical structure (e.g., aromaticity).
Neural-network-based models combining compound and sorbent parameters could yield improved estimations for IOC sorption,47 and combined with sensitivity analysis may be a good starting point to identify key parameters for further model development. Ideally, in future approaches, molecular and geometrical properties of IOC will specify which interactions a given species can undergo and allow for categorization and prioritization of compound classes. This categorization could then result in a set of descriptors and/or probe compounds tailored to the compound class of interest. Based on these compound groups, tailored predictive models based on consistent sets of experimental data could be developed. These data should include sorption coefficients to a number of well characterized soil constituents (SOM, black carbon, clay minerals) as well as soils and sediments with varying compositions using high throughput experimental systems, as can be run using soil column chromatography approaches.62 Such approaches could also account for additional factors affecting IOC sorption, such as DOM and other compounds competing for sorption sites, as well as temperature, which can also alter IOC and sorbent functional group speciation.63 Field monitoring of potential contaminants under saturated conditions would be a valuable complementary approach to measuring sorption under well-defined conditions. Recent developments in analytical chemistry make it possible to measure a very wide range of IOC in environmental samples.5,64 These measurements may aid future model developments and allocation of IOC to substance classes with different environmental behavior.
Predictive approaches will continue to be necessary at least for preliminary assessment and screening purposes. To develop better models for IOC mobility, it is crucial to create consistent and reliable data sets with (i) well documented and correctly determined molecular properties including pKa and Dow, (ii) well documented sorbent properties including organic carbon and black carbon content, mineral composition as well as pH dependent CEC, (iii) sorption data measured under different well-defined chemical conditions in water and soil (pH and ionic composition) under saturated conditions, and (iv) the consideration of additional complex interactions such as the air–water interface under unsaturated conditions which are important for a number of compound such as per- and polyfluoroalkyl substances.65 Predictive models aiming to improve risk assessment should integrate findings from monitoring studies for model calibration and validation, which can help to identify conceptual shortcomings and to expand the scope of a given model on a relevance and need basis.
Acknowledgments
M.S. acknowledges funding by the RECETOX research infrastructure (the Czech Ministry of Education, Youth and Sports: LM2018121), the CETOCOEN PLUS project (CZ.02.1.01/0.0/0.0/15_003/0000469), and the CETOCOEN EXCELLENCE Teaming 2 project supported by the Czech ministry of Education, Youth and Sports (No CZ.02.1.01/0.0/0.0/17_043/0009632).
Biography

Gabriel Sigmund is a Junior Research Group Leader at the Centre for Microbiology and Environmental Systems Science, University of Vienna. Gabriel’s ongoing research focuses on (i) effects of pyrogenic carbon in postfire landscapes on organic matter cycling, (ii) biochar and wood-based activated carbon for sustainable contaminant remediation, and (ii) mobility of ionic and ionizable organic contaminants, which is critical for assessing the emerging contaminant class of persistent mobile and toxic (PMT) substances, as discussed in this feature.
The authors declare no competing financial interest.
References
- Jin B.; Huang C.; Yu Y.; Zhang G.; Arp H. P. H. The Need to Adopt an International PMT Strategy to Protect Drinking Water Resources. Environ. Sci. Technol. 2020, 54 (19), 11651–11653. 10.1021/acs.est.0c04281. [DOI] [PubMed] [Google Scholar]
- Hale S. E.; Arp H. P. H.; Schliebner I.; Neumann M. What’s in a Name: Persistent, Mobile, and Toxic (PMT) and Very Persistent and Very Mobile (VPvM) Substances. Environ. Sci. Technol. 2020, 54 (23), 14790–14792. 10.1021/acs.est.0c05257. [DOI] [PubMed] [Google Scholar]
- Reemtsma T.; Berger U.; Arp H. P. H.; Gallard H.; Knepper T. P.; Neumann M.; Quintana J. B.; Voogt P. de. Mind the Gap: Persistent and Mobile Organic Compounds—Water Contaminants That Slip Through. Environ. Sci. Technol. 2016, 50 (19), 10308–10315. 10.1021/acs.est.6b03338. [DOI] [PubMed] [Google Scholar]
- Arp H. P. H.; Hale S. E.. REACH: Improvement of Guidance and Methods for the Identification and Assessment of PMT/VPvM Substances, 2019.
- Neuwald I.; Muschket M.; Zahn D.; Berger U.; Seiwert B.; Meier T.; Kuckelkorn J.; Strobel C.; Knepper T. P.; Reemtsma T. Filling the Knowledge Gap: A Suspect Screening Study for 1310 Potentially Persistent and Mobile Chemicals with SFC- and HILIC-HRMS in Two German River Systems. Water Res. 2021, 204, 117645. 10.1016/j.watres.2021.117645. [DOI] [PubMed] [Google Scholar]
- Armitage J. M.; Erickson R. J.; Luckenbach T.; Ng C. A.; Prosser R. S.; Arnot J. A.; Schirmer K.; Nichols J. W. Assessing the Bioaccumulation Potential of Ionizable Organic Compounds: Current Knowledge and Research Priorities. Environ. Toxicol. Chem. 2017, 36 (4), 882–897. 10.1002/etc.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher B. I.; Abagyan R.; Embry M.; Klüver N.; Redman A. D.; Zarfl C.; Parkerton T. F. Recommendations for Improving Methods and Models for Aquatic Hazard Assessment of Ionizable Organic Chemicals. Environ. Toxicol. Chem. 2020, 39 (2), 269–286. 10.1002/etc.4602. [DOI] [PubMed] [Google Scholar]
- Chiou C. T.; Peters L. J.; Freed V. H. A Physical Concept of Soil-Water Equilibria for Nonionic Organic Compounds. Science 1979, 206 (4420), 831–832. 10.1126/science.206.4420.831. [DOI] [PubMed] [Google Scholar]
- Karickhoff S. W.; Brown D. S.; Scott T. A. Sorption of Hydrophobic Pollutants on Natural Sediments. Water Res. 1979, 13 (3), 241–248. 10.1016/0043-1354(79)90201-X. [DOI] [Google Scholar]
- Hamaker J.; Thompson J.. Adsorption. In Goring GAI, Hamaker JW (eds) Organic Chemicals in the Soil Environment, Vol 1. Dekker; New York, 1972; pp 49–143. [Google Scholar]
- Kile D. E.; Chiou C. T.; Zhou H..; Li H..; Xu O.. Partition of Nonpolar Organic Pollutants from Water to Soil and Sediment Organic Matters. Environ. Sci. Technol. 1995, 29 (5), 1401–1406. 10.1021/es00005a037. [DOI] [PubMed] [Google Scholar]
- Niederer C.; Schwarzenbach R. P.; Goss K. U. Elucidating Differences in the Sorption Properties of 10 Humic and Fulvic Acids for Polar and Nonpolar Organic Chemicals. Environ. Sci. Technol. 2007, 41 (19), 6711–6717. 10.1021/es0709932. [DOI] [PubMed] [Google Scholar]
- Cornelissen G.; Gustafsson O.; Bucheli T.; Jonker M.; Koelmans A.; van Noort P. Extensive Sorption of Organic Compounds to Black Carbon, Coal, and Kergen in Sediments and Soils: Mechanisms and Consequences for Distribution, Bioaccumulation, and Biodegradation. Environ. Sci. Technol. 2005, 39 (18), 6881–6895. 10.1021/es050191b. [DOI] [PubMed] [Google Scholar]
- ECHA, E. C. A . Guidance on Biocides Legislation: Vol. IV Environment, Assessment & Evaluation (Parts B+C), 2017. [Google Scholar]
- FAO . Assessing Soil Contamination A Reference Manual; Food and Agriculture Organization - Information Division, 2000. [Google Scholar]
- Doucette W. J. Quantitative Structure-Activity Relationships for Predicting Soil-Sediment Sorption Coefficients for Organic Chemicals. Environ. Toxicol. Chem. 2003, 22 (8), 1771–1788. 10.1897/01-362. [DOI] [PubMed] [Google Scholar]
- Jafvert C. T.; Westall J. C.; Grieder E.; Schwarzenbach R. P. Distribution of Hydrophobic Ionogenic Organic Compounds between Octanol and Water: Organic Acids. Environ. Sci. Technol. 1990, 24 (12), 1795–1803. 10.1021/es00082a002. [DOI] [Google Scholar]
- Westall J. C.; Leuenberger C.; Schwarzenbach R. P. Influence of PH and Ionic Strength on the Aqueous-Nonaqueous Distribution of Chlorinated Phenols. Environ. Sci. Technol. 1985, 19 (2), 193–198. 10.1021/es00132a014. [DOI] [Google Scholar]
- Neumann M.; Schliebner I.. Protecting the Sources of Our Drinking Water: The Criteria for Identifying Persistent, Mobile and Toxic (PMT) Substances and Very Persistent and Very Mobile (VPvM) Substances under EU Regulation REACH (EC) No 1907/2006; Umweltbundesamt, 2019. [Google Scholar]
- USEPA . Estimation Programs Interface SuiteTM for Microsoft® Windows, v 4.11; United States Environmental Protection Agency: Washington, DC, 2012. [Google Scholar]
- Mannhold R.; Poda G. I.; Ostermann C.; Tetko I. V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on More than 96,000 Compounds. J. Pharm. Sci. 2009, 98 (3), 861–893. 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- Kleber M.; Bourg I. C.; Coward E. K.; Hansel C. M.; Myneni S. C. B.; Nunan N. Dynamic Interactions at the Mineral–Organic Matter Interface. Nature Reviews Earth & Environment 2021, 2, 1–20. 10.1038/s43017-021-00162-y. [DOI] [Google Scholar]
- Sibley S. D.; Pedersen J. A. Interaction of the Macrolide Antimicrobial Clarithromycin with Dissolved Humic Acid. Environ. Sci. Technol. 2008, 42 (2), 422–428. 10.1021/es071467d. [DOI] [PubMed] [Google Scholar]
- Droge S.; Goss K.-U. Effect of Sodium and Calcium Cations on the Ion-Exchange Affinity of Organic Cations for Soil Organic Matter. Environ. Sci. Technol. 2012, 46 (11), 5894–5901. 10.1021/es204449r. [DOI] [PubMed] [Google Scholar]
- Droge S. T. J.; Goss K.-U. Sorption of Organic Cations to Phyllosilicate Clay Minerals: CEC-Normalization, Salt Dependency, and the Role of Electrostatic and Hydrophobic Effects. Environ. Sci. Technol. 2013, 47 (24), 14224–14232. 10.1021/es403187w. [DOI] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437 (7059), 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Lazaridis T. Solvent Size vs Cohesive Energy as the Origin of Hydrophobicity. Acc. Chem. Res. 2001, 34 (12), 931–937. 10.1021/ar010058y. [DOI] [PubMed] [Google Scholar]
- Southall N. T.; Dill K. A.; Haymet A. D. J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106 (3), 521–533. 10.1021/jp015514e. [DOI] [Google Scholar]
- Bronner G.; Goss K. U. Predicting Sorption of Pesticides and Other Multifunctional Organic Chemicals to Soil Organic Carbon. Environ. Sci. Technol. 2011, 45 (4), 1313–1319. 10.1021/es102553y. [DOI] [PubMed] [Google Scholar]
- Arp H. P. H.; Hale S. E.. On the Persistence and Mobility of Organic Contaminants Detected in Freshwater Resources. in prep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OECD . eChemPortal : The Global Portal to Information on Chemical Substances https://www.echemportal.org/echemportal/ (accessed 2022/01/05).
- Arp H. P. H.; Brown T. N.; Berger U.; Hale S. E. Ranking REACH Registered Neutral, Ionizable and Ionic Organic Chemicals Based on Their Aquatic Persistency and Mobility. Environmental Science: Processes and Impacts 2017, 19 (7), 939–955. 10.1039/C7EM00158D. [DOI] [PubMed] [Google Scholar]
- McBride M. B.Environmental Chemistry of Soils; Oxford University Press, 1994. [Google Scholar]
- Xiao F.; Pignatello J. J. Effect of Adsorption Nonlinearity on the Ph-Adsorption Profile of Ionizable Organic Compounds. Langmuir 2014, 30 (8), 1994–2001. 10.1021/la403859u. [DOI] [PubMed] [Google Scholar]
- Donnan F. G. Theory of Membrane Equilibria and Membrane Potentials in the Presence of Non-Dialysing Electrolytes. A Contribution to Physical-Chemical Physiology. J. Membr. Sci. 1995, 100 (1), 45–55. 10.1016/0376-7388(94)00297-C. [DOI] [Google Scholar]
- Schönsee C. D.; Wettstein F. E.; Bucheli T. D. Phytotoxin Sorption to Clay Minerals. Environmental Sciences Europe 2021, 33 (1), 36. 10.1186/s12302-021-00469-z. [DOI] [Google Scholar]
- MacKay A. A.; Vasudevan D. Polyfunctional Ionogenic Compound Sorption: Challenges and New Approaches To Advance Predictive Models. Environ. Sci. Technol. 2012, 46 (17), 9209–9223. 10.1021/es301036t. [DOI] [PubMed] [Google Scholar]
- Kah M.; Sigmund G.; Xiao F.; Hofmann T. Sorption of Ionizable and Ionic Organic Compounds to Biochar, Activated Carbon and Other Carbonaceous Materials. Water Res. 2017, 124, 673–692. 10.1016/j.watres.2017.07.070. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Hyun S.; Pignatello J. J.; Lee L. S. Evidence for Pi-Pi Electron Donor-Acceptor Interactions between Pi-Donor Aromatic Compounds and Pi-Acceptor Sites in Soil Organic Matter through PH Effects on Sorption. Environ. Sci. Technol. 2004, 38, 4361–4368. 10.1021/es035379e. [DOI] [PubMed] [Google Scholar]
- Xiao F.; Pignatello J. J. Π+-π Interactions between (Hetero)Aromatic Amine Cations and the Graphitic Surfaces of Pyrogenic Carbonaceous Materials. Environ. Sci. Technol. 2015, 49 (2), 906–914. 10.1021/es5043029. [DOI] [PubMed] [Google Scholar]
- Xiao F.; Pignatello J. J. Interactions of Triazine Herbicides with Biochar: Steric and Electronic Effects. Water Res. 2015, 80, 179–188. 10.1016/j.watres.2015.04.040. [DOI] [PubMed] [Google Scholar]
- Li X.; Pignatello J. J.; Wang Y.; Xing B. New Insight into Adsorption Mechanism of Ionizable Compounds on Carbon Nanotubes. Environ. Sci. Technol. 2013, 47 (15), 8334–8341. 10.1021/es4011042. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Duan P.; Schmidt-Rohr K.; Pignatello J. J. Physicochemical Changes in Biomass Chars by Thermal Oxidation or Ambient Weathering and Their Impacts on Sorption of a Hydrophobic and a Cationic Compound. Environ. Sci. Technol. 2021, 55 (19), 13072–13081. 10.1021/acs.est.1c04748. [DOI] [PubMed] [Google Scholar]
- Graber E. R.; Borisover M. D. Hydration-Facilitated Sorption of Specifically Interacting Organic Compounds by Model Soil Organic Matter. Environ. Sci. Technol. 1998, 32 (2), 258–263. 10.1021/es9705957. [DOI] [Google Scholar]
- Graber E. R.; Borisover M. Exploring Organic Compound Interactions with Organic Matter: The Thermodynamic Cycle Approach. Colloids Surf., A 2005, 265 (1), 11–22. 10.1016/j.colsurfa.2005.02.039. [DOI] [Google Scholar]
- Droge S. T. J.; Goss K.-U. Development and Evaluation of a New Sorption Model for Organic Cations in Soil: Contributions from Organic Matter and Clay Minerals. Environ. Sci. Technol. 2013, 47 (24), 14233–14241. 10.1021/es4031886. [DOI] [PubMed] [Google Scholar]
- Sigmund G.; Gharasoo M.; Hüffer T.; Hofmann T. Deep Learning Neural Network Approach for Predicting the Sorption of Ionizable and Polar Organic Pollutants to a Wide Range of Carbonaceous Materials. Environ. Sci. Technol. 2020, 54 (7), 4583–4591. 10.1021/acs.est.9b06287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfitt R. L.; Giltrap D. J.; Whitton J. S. Contribution of Organic Matter and Clay Minerals to the Cation Exchange Capacity of Soils. null 1995, 26 (9–10), 1343–1355. 10.1080/00103629509369376. [DOI] [Google Scholar]
- Haderlein S. B.; Weissmahr K. W.; Schwarzenbach R. P. Specific Adsorption of Nitroaromatic Explosives and Pesticides to Clay Minerals. Environ. Sci. Technol. 1996, 30 (2), 612–622. 10.1021/es9503701. [DOI] [Google Scholar]
- European Commission . Setting out the Data Requirements for Plant Protection Products, in Accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council Concerning the Placing of Plant Protection Products on the Market, 2013. [Google Scholar]
- European Comission . Communication from the Commission to the European Parliament, The Council, The European Economic and Social Committee and the Committee of the Regions Chemicals Strategy for Sustainability Towards a Toxic-Free Environment COM/2020/667 Final, 2020. [Google Scholar]
- Gustafson D. I. Groundwater Ubiquity Score: A Simple Method for Assessing Pesticide Leachability. Environ. Toxicol. Chem. 1989, 8 (4), 339–357. 10.1002/etc.5620080411. [DOI] [Google Scholar]
- Chemicals legislation – revision of REACH Regulation to help achieve a toxic-free environment https://ec.europa.eu/info/law/better-regulation/have-your-say/initiatives/12959-Chemicals-legislation-revision-of-REACH-Regulation-to-help-achieve-a-toxic-free-environment_en (accessed 2021/11/05).
- European Commission . European Commission Ad Hoc Meeting of CARACAL PBT/VPvB/PMT/VPvM Criteria 30 September 2021. Topic: Discussion on PMT/VPvM Possible Criteria in CLP. Ad-Hoc CA/03/2021. 9 Pp. Brussels. 2021; European Commission, 2021. [Google Scholar]
- Soil Survey Staff, Natural Conservation Service, United States Department of Agriculture . Web Soil Survey https://websoilsurvey.nrcs.usda.gov/.
- Joint Research Centre - European Soil Data Centre (ESDAC) . European Soil Database & Soil Properties https://esdac.jrc.ec.europa.eu/resource-type/european-soil-database-soil-properties.
- Endo S.; Goss K. U. Applications of Polyparameter Linear Free Energy Relationships in Environmental Chemistry. Environ. Sci. Technol. 2014, 48 (21), 12477–12491. 10.1021/es503369t. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; William E.; Acree J. The Transfer of Neutral Molecules, Ions and Ionic Species from Water to Wet Octanol. Phys. Chem. Chem. Phys. 2010, 12 (40), 13182–13188. 10.1039/c0cp00695e. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Acree W. E. Descriptors for Ions and Ion-Pairs for Use in Linear Free Energy Relationships. Journal of Chromatography A 2016, 1430, 2–14. 10.1016/j.chroma.2015.07.023. [DOI] [PubMed] [Google Scholar]
- Droge S. T. J.Sorption of Polar and Ionogenic Organic Chemicals. In Bioavailability of Organic Chemicals in Soil and Sediment; Ortega-Calvo J. J.; Parsons J. R., Eds.; The Handbook of Environmental Chemistry; Springer International Publishing: Cham, 2020; pp 43–80. 10.1007/698_2020_517. [DOI] [Google Scholar]
- Schönsee C. D.; Wettstein F. E.; Bucheli T. D. Disentangling Mechanisms in Natural Toxin Sorption to Soil Organic Carbon. Environ. Sci. Technol. 2021, 55 (8), 4762–4771. 10.1021/acs.est.0c06634. [DOI] [PubMed] [Google Scholar]
- Bi E.; Schmidt T. C.; Haderlein S. B. Practical Issues Relating to Soil Column Chromatography for Sorption Parameter Determination. Chemosphere 2010, 80 (7), 787–793. 10.1016/j.chemosphere.2010.05.006. [DOI] [PubMed] [Google Scholar]
- Aumeier B. M.; Augustin A.; Thönes M.; Sablotny J.; Wintgens T.; Wessling M. Linking the Effect of Temperature on Adsorption from Aqueous Solution with Solute Dissociation. Journal of Hazardous Materials 2022, 429, 128291. 10.1016/j.jhazmat.2022.128291. [DOI] [PubMed] [Google Scholar]
- Schulze S.; Zahn D.; Montes R.; Rodil R.; Quintana J. B.; Knepper T. P.; Reemtsma T.; Berger U. Occurrence of Emerging Persistent and Mobile Organic Contaminants in European Water Samples. Water Res. 2019, 153, 80–90. 10.1016/j.watres.2019.01.008. [DOI] [PubMed] [Google Scholar]
- Brusseau M. L.; Guo B. Air-Water Interfacial Areas Relevant for Transport of per and Poly-Fluoroalkyl Substances. Water Res. 2021, 207, 117785. 10.1016/j.watres.2021.117785. [DOI] [PMC free article] [PubMed] [Google Scholar]