

Abstract
Genes embed their evolutionary history in the form of various alleles. Presence–absence variants (PAVs) are extreme cases of such alleles, where a gene present in one haplotype does not exist in another. Because PAVs may result from either birth or death of a gene, PAV genes and their alternative alleles, if available, can represent a basis for rapid intraspecific gene evolution. Using long-read sequencing technologies, this study traced the possible evolution of PAV genes in the PD1074 and CB4856 C. elegans strains as well as their alternative alleles in 14 other wild strains. We updated the CB4856 genome by filling 18 gaps and identified 46 genes and 7,460 isoforms from both strains not annotated previously. We verified 328 PAV genes, out of which 46 were C. elegans-specific. Among these possible newly born genes, 12 had alternative alleles in other wild strains; in particular, the alternative alleles of three genes showed signatures of active transposons. Alternative alleles of three other genes showed another type of signature reflected in accumulation of small insertions or deletions. Research on gene evolution using both species-specific PAV genes and their alternative alleles may provide new insights into the process of gene evolution.
INTRODUCTION
Genetic variation shapes gene sequences from their birth to their death. Gene duplication and divergence are the most representative examples through which one functional gene is duplicated in multiple copies via segmental or whole-genome duplication so that the copied genes acquire divergent sequences and new functions (1–4). A new gene can also be born in a non-genic region through a process in which non-genic transcripts acquire open reading frames and functions, which is called de novo gene birth (4–12). Conversely, genes may disappear as genetic variation disrupts their functions or deletes the entire gene sequences, resulting in gene death (4). These concepts have advanced our understanding of how genes are newly born or vanished, but details of these events remain elusive in a short time scale at the intraspecific level.
Mutations are clearly the main cause of gene birth and death. Single-nucleotide variants (SNVs) or small indels in functional genes may cause a pseudogenisation process that destroys their gene function (13). Large mutations, which result in structural variants, may lead to the disappearance of entire genes (14–20). De novo gene birth by mutation has been thoroughly studied in various organisms such as C. elegans, fruit fly, rice and yeast (10,12,21). Inter- or intra-species gene/pseudogene comparison served to confirm that some newly generated mutations allow pseudogenes to acquire new open reading frames, leading to new gene formation. However, most previous studies have focused on comparing alleles with similar gene/pseudogene structures, rather than those with very different structures caused by large mutations, possibly because of technical limitations.
A type of genetic variants, presence–absence variants (PAVs), is represented by genes present in some genomes but absent in others within the same species (14). PAV genes might be fast-evolving genes, as they are able to reflect gene birth or death (22). In addition, PAV genes may contribute to adaptation to changing environments, including pathogen infection, antitumor-agent synthesis or disease resistance in plants as well as immunity in animals (15–19). Recently, another case of gene evolution was also analysed using PAVs in several molluscs (20). Thus, PAVs may provide evidence of how genes evolve, but some of their characteristics make it difficult to precisely identify PAVs. PAVs typically do not contain conserved or essential genes as they should not have a disruptive loss-of-function mutant phenotype, thus homology-based gene prediction may fail to detect such orphan genes. High-quality genome assembly and evidence-based gene annotation methods can be employed, but stereotypical sequencing techniques are based on short-read sequencing technologies that sometimes result in fragmented genomes and do not cover full-length transcripts. Advances in long-read sequencing technologies resolve these limitations and allow detecting PAVs at the population level (23–25).
Caenorhabditis elegans is a model organism suitable for identifying PAVs at the population level. It has a small genome of 102 Mb, and hundreds of wild strains are isolated and cryopreserved (26–28). In a previous study, genes absent in 12 wild strains, including CB4856, but present in the reference strain were discovered; however, genes present specifically in these strains could not be identified at the sequence level because the authors used a hybridisation-based method (29). Long-read sequencing-based high-quality genomic resources are resolving these technical limitations, thus providing the opportunity to better understand PAVs in C. elegans populations. For instance, a clonal selection-descendant strain of the N2 reference strain, PD1074, was established to eliminate mutations accumulated in N2 and its high-quality genome containing only two gaps was completed (28). Moreover, the genome of CB4856—a genetically divergent strain from the reference strain—was completed at the pseudo-chromosome level, and high-quality genomes of 14 wild strains were also assembled (30,31). These high-quality genome assemblies further clear the way to determine which genes represent PAV genes in C. elegans and identify their alternative alleles, which could lead to clarifying the processes of possible gene birth or death.
Here, we analysed C. elegans-specific PAV genes and their alternative alleles in order to infer possible scenarios of gene evolution within a given species. We identified previously non-annotated genes and transcripts from both PD1074 and CB4856 genomes using long-read RNA sequencing. We analysed the possible gene birth time points of PAV genes between PD1074 and CB4856 by characterising whether they are Caenorhabditis lineage-specific or C. elegans-specific and showed that C. elegans-specific PAV genes and their alternative alleles identified from 14 C. elegans wild strains manifest snapshots of gene evolution such as complex small insertions and deletions (indels), active transposon signatures and gene duplication and divergence signatures. In summary, the analysis of alternative alleles of the PAV genes within a given species could be a useful tool to understand clues of gene evolution occurring within short time periods.
MATERIALS AND METHODS
C. elegans strains and maintenance
C. elegans worms belonging to two strains, PD1074 and CB4856, were cultured under standard culture conditions (32,33).
Genomic DNA extraction and Oxford Nanopore Technologies sequencing
Mixed-stage worms of the CB4856 strain grown at 20°C were harvested and washed 5 times with M9 buffer. Worms were lysed in Cell Lysis Solution from The Gentra Puregene® Cell and Tissue Kit (Qiagen) with 0.1 mg/mL proteinase K and 1% β-mercaptoethanol at 55°C for 2 h. We purified DNA three times using phenol/chloroform extraction and ethanol precipitation coupled with phase-lock gel to minimise DNA shearing. DNA dissolved in TE buffer was treated with 10 μg/mL RNase for 2 h after the first extraction and precipitation step. DNA was treated with the Oxford Nanopore Technologies (ONT) SQK-LSK109 library preparation kit and the DNA library was sequenced using FLO-MIN106.
Total RNA extraction and Pacific Biosciences sequencing
To obtain as many different transcripts as possible, we combined RNA samples from worms grown at three different temperatures. First, mixed-stage worms of both PD1074 and CB4856 strains were grown at 15°C, 20°C or 25°C. Each sample was separately harvested with M9 buffer. Worms were incubated in M9 with rotation for 30 min to remove bacteria in the gut and washed three times with M9. Worms collected in a volume of about 200 μL were treated with 2 mL of TRIzol solution and subsequently disrupted by six freeze-thaw cycles. RNA was extracted with chloroform/isopropanol precipitation. Each RNA sample was quantified and samples collected at three different temperatures of each strain were pooled in equal amounts. Macrogen (South Korea, https://www.macrogen.com/en/main) conducted RNA library preparation and Iso-Seq using the Pacific Biosciences (PacBio) Sequel System.
Gap filling in the CB4856 genome
We conducted basecalling of long-read DNA sequencing reads and trimming of adapter sequences using Guppy basecaller (version 3.4.1) with the default setting. We extracted reads > 20 kb and aligned them to the CB4856 genome (Supplementary Table S1) (30) using Minimap2 (version 2.17; minimap2 -ax map-ont) (34). The selected reads were then filtered based on their primary alignments using Samtools (version 1.9; samtools view -f 0 × 10 -q 2 or samtools view -q 2) (35). The filtered reads were aligned to the CB4856 genome again using Minimap2 and gap-filling reads were confirmed by visualising their alignments using NUCmer and mummerplot from MUMmer package (version 4.0.0 beta2) (36) and Gnuplot (version 5.0, patch level 3). We collected the reads that aligned to both flanking contigs of gaps and manually replaced reference sequences with their corresponding gap-filling read sequences (Supplementary Table S3).
Iso-Seq data processing
We used the IsoSeq package (version 3.3.0) to process Iso-Seq data (https://github.com/PacificBiosciences/IsoSeq_SA3nUP/). First we generated circular consensus sequences (CCS) using CCS (version 4.2.0; ccs –min-rq 0.8 –min-passes 1) and removed library-prep-primers, poly(A) tails and artificial concatemers using lima (version 1.11.0; lima –isoseq –dump-clips –peek-guess) and IsoSeq (version 3.3.0; isoseq3 refine –require-polya). After clustering and polishing full-length reads using IsoSeq (version 3.3.0; isoseq3 cluster –verbose –use-qvs), we aligned high-quality full-length non-concatemer reads to the PD1074 genome (WS274) or the CB4856 genome using Minimap2 (version 2.17; minimap2 -ax splice -uf –secondary = no -C5, -O6,24 -B4) (Supplementary Table S1) (34). Finally, we extracted 5′ non-degraded isoforms using cDNA_Cupcake ToFU scripts (version 12.0.0, collapse_isoforms_by_sam.py –dun-merge-5-shorter and filter_away_subset.py; https://github.com/Magdoll/cDNA_Cupcake) (37,38).
Transferring gene annotations from the PD1074 genome to the CB4856 genome using LiftOver
To annotate genes of CB4856, we conducted a chain alignment process between the CB4856 and PD1074 genomes using the same-species liftover construction method (http://genomewiki.ucsc.edu/index.php/Same_species_lift_over_construction) from UCSC (39). Referring to the method of Yoshimura et al. (28), targetChunkSize and queryChunkSize were modified from 10,000,000 to 22,000,000 and a typographical error was corrected from ‘B = 'basename [ file]’ to ‘B = 'basename [ file]'’. The PD1074 gene annotation contained genes lifted from the reference N2 gene annotation and genes predicted based on the PD1074 genome. We only transferred PD1074 gene annotations (WS274) that originated from the N2 genome as they were better validated (liftOver -gff).
Annotation of isoforms obtained by Iso-Seq
We annotated 5′ non-degraded high-quality full-length isoforms using SQANTI3 (version 1.1; sqanti3_qc.py –aligner_choice = minimap2 –fl_count) (37). We used the PD1074 genome and its N2-origin gene annotations as well as the CB4856 genome and transferred gene annotations as references for the SQANTI3 analysis. Then, we categorised the annotated genes as follows: known genes with only known isoforms, known genes with newly detected isoforms, newly detected gene candidates and fusion or non-curated genes.
Validating newly detected genes and their protein identity
We built BLAST databases of the PD1074 genome, CB4856 genome, N2 transcriptome (WS274; mRNA, ncRNA, pseudogenic and transposon transcripts), PD1074 transcripts (WS274) and our PD1074 long-read transcripts (Supplementary Table S1). After identifying and masking out low complexity parts of the sequences using DustMasker (version 1.0.0; dustmasker -infmt fasta -parse_seqids -outfmt maskinfo_asn1_bin), we built BLAST databases using Makeblastdb (version 2.7.1+; makeblastdb -input_type fasta -dbtype nucl -parse_seqids). We then filtered out newly detected gene candidates from the SQANTI3 results by searching their CCS read sequences in all databases stated above (BLASTn version 2.7.1+; default setting with e-value < 0.001). Finally, the validated gene sequences were searched using the NCBI BLASTp database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins) and WormBase BLAST/BLAT tool (https://wormbase.org/tools/blast_blat) to determine their protein identity and using Batch CD-search (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) to identify any conserved domains (40).
Identifying PAVs and determining their characteristics
Genomic sequences of the PD1074 genes annotated by SQANTI3 and non-transferred genes by LiftOver of the PD1074 gene annotations, were analysed in the CB4856 genome database using BLASTn (version 2.7.1+; blastn for sequences > 50 bp or blastn -task blastn-short for sequences ≤ 50 bp) to identify PD1074-specific genes. CB4856-specific genes were identified by searching genomic sequences of the CB4856 SQANTI3 results in the PD1074 genome database. We considered PAVs as all genes from each strain that did not exhibit any significant identity in BLAST search results.
We searched these PAVs in the genomes of other 14 wild strains (DL238, ECA36, ECA396, JU310, JU1400, JU2526, EG4725, JU2600, MY2147, NIC2, NIC526, QX1794, MY2693 and XZ1516), whose resource information is compiled in Supplementary Table S1 (31). We first indexed each genome database for the 14 wild strains using Makeblastdb (version 2.7.1+; makeblastdb -input_type fasta -dbtype nucl-parse_seqids). After performing a BLAST search (BLASTn version 2.7.1+; default setting with e-value < 0.001) with the genomic sequences of PAV genes, we classified our PAVs status in each strain according to their coverage in BLAST results for the corresponding strain as follows: no significant identity—absence, significant identity with coverage ≥ 90%—presence, in-between—alternative. Raw data are summarised in Supplementary Tables S12 and S13.
These strain-specific protein-coding genes were also analysed against the NCBI protein database and the Batch CD-search database to identify any similarity to known protein or domain sequences. We used only genes with e-value < 0.001 and categorised them as genes that have significant identity with genes of C. elegans, Caenorhabditis, other organisms and none of them. For PD1074-specific genes, we also tested whether they have known RNAi phenotypes using the SimpleMine tool in WormBase (https://wormbase.org//tools/mine/simplemine.cgi) (41). The phenotypes were confirmed through literature search (42–48).
Genetic relatedness
A VCF file containing 963,027 biallelic SNVs from a previous study (31) was filtered for 16 wild C. elegans strains and converted to the PHYLIP format. The distance matrix and pseudo-rooted (XZ1516) neighbour-joining tree were created from this PHYLIP file using dist.ml and the NJ function using the phangorn (version 2.5.5) R package. The tree was visualised using the ggtree (version 1.16.6) R package.
Identifying alternative alleles in wild strains
Alternative alleles present in wild strains were identified by searching for presence alleles of our PAV genes in the genome assemblies of the 14 wild strains (31). The presence allele sequences included UTR regions. Because the searched sequences were partial in the 14 genome assemblies, we defined the alternative alleles as the searched sequences and their flanking sequences extended to the length of unsearched regions in the corresponding presence alleles. We obtained the coverage of each alternative allele by aligning their sequences to the N2 genome (WS279) using the BLAST/BLAT tool of WormBase (https://wormbase.org/tools/blast_blat) (49).
Identifying genes that are coding in one strain, but non-coding in the other strain
To identify pairs of non-coding transcripts and their coding counterparts in PD1074 and CB4856, we used non-coding transcripts validated by our long-read RNA sequencing data. These non-coding transcript sequences of PD1074 and CB4856 were searched in genomic sequences of each other, by using BLASTn (version 2.7.1+; e-value < 0.001, coverage ≥ 90%, and identity ≥ 95%). We only used subset of these non-coding and coding gene pairs by filtering out non-coding genes that had any coding transcripts in the same strain and coding genes that were not covered by its non-coding counterparts (coverage ≥ 50%). These final coding and non-coding gene pairs were used to determine whether or not the non-coding transcripts had start and stop codons and whether any part of the coding sequence was different from its counterpart non-coding sequence in terms of SNPs, structural variants, and splicing variants.
RESULTS
Updating the CB4856 genome with ONT long reads
Because precise identification of PAVs partly depends on genome quality, we first updated the previously published CB4856 genome (30) by filling gaps between contigs with ONT long-read DNA sequencing data. We obtained 57-fold coverage of DNA sequences with a read length N50 of 4,284 bp and a total read length of 5.9 Gb (Supplementary Table S2 and Supplementary Figure S1A). To maximise the benefits of long-read sequencing, we used only long reads with over 20-kb read length and aligned the resulting 12,982 reads to the CB4856 genome (Supplementary Table S2 and Supplementary Figure S1B). We found 27 reads able to connect two flanking contigs around the gaps and filled 18 gaps with these reads (Supplementary Table S3 and Supplementary Figure S2). We updated the previous CB4856 genome composed of 76 contigs with a 103-Mb genome made of 54 contigs (Table 1). We used this updated genome for further analyses.
Table 1.
Statistics of the Kim genome and the Lee genome for the CB4856 strain.
| CB4856 genome (PacBio CLR + Illumina reads) | CB4856 genome + ONT reads | |
|---|---|---|
| (Kim genome (30)) | (Lee genome, this study) | |
| Number of bases (bp) | 102,914,785 | 102,934,386 |
| Contigs | 76 | 58 |
| Gaps | 69 | 51 |
| N50 contig size (bp) | 2,786,967 | 3,615,580 |
| Maximum contig size (bp) | 9,650,681 | 9,650,681 |
| Minimum contig size (bp) | 13,928 | 13,928 |
Discovery of previously non-annotated genes in PD1074
After filling nucleotide gaps, we processed the long-read RNA sequencing data obtained by a PacBio Iso-Seq platform to identify genes that are not annotated previously, but can be detected by using full-length transcripts. First, we generated 14 million raw reads of PD1074 (total 30 Gb, N50 2.7 kb) and processed these reads to obtain 8,630 high-quality, full-length and unique transcripts characterised by non-degraded 5′ end, poly(A) tail and polished sequences (Figure 1A, Supplementary Table S2 and Supplementary Figure S1C). These high-quality transcripts corresponded to 6,218 genes (Figure 1A) out of which 4,045 contained only known isoforms, but 1,916 genes contained non-annotated isoforms of previously known genes (Supplementary Table S4). A total 3,021 isoforms were newly detected, while 177 genes did not match any known gene annotation and were therefore categorised as newly detected gene candidates (Supplementary Table S4). These several thousands of newly detected isoforms support the hypothesis that our long-read RNA sequencing data were suitable for detecting non-annotated transcripts.
Figure 1.
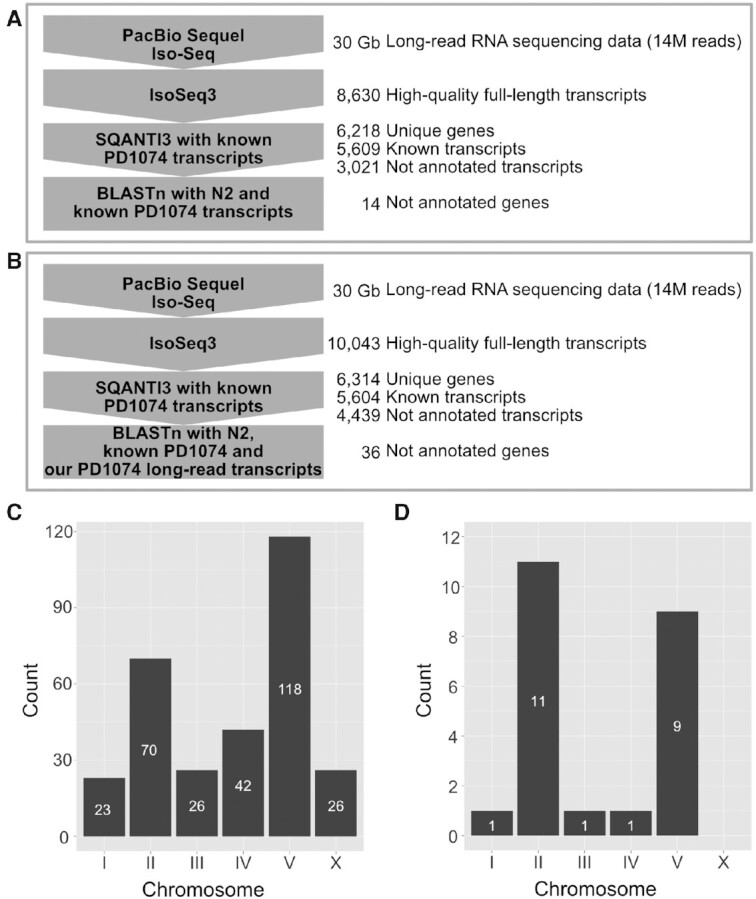
Schematic overview of data analysis to identify previously non-annotated genes and chromosomal distribution of strain-specific genes. (A and B) Computational workflows for finding previously non-annotated genes of (A) PD1074 and (B) CB4856 using long-read RNA sequencing. PacBio Iso-Seq data were processed using IsoSeq3 to produce high-quality full-length transcripts, SQANTI3 to extract newly detected gene candidates by comparing the transcripts with known PD1074 transcripts and BLASTn to verify the candidates by searching for them in either (A) the N2 and PD1074 known gene databases or (B) the N2 and PD1074 known gene databases supplemented with our long-read PD1074 transcripts database. (C and D) Chromosomal distribution of (C) PD1074- and (D) CB4856-specific genes.
Among the newly detected transcripts, 42 corresponded to genes belonging to the ribosomal RNA cluster located on the right end of chromosome I (Supplementary Figure S3). Their full-length transcripts were longer than those of previously annotated genes (50), suggesting that our long-read data properly annotated true gene sequences of previously fragmented genes.
We further validated the 177 newly detected gene candidates comprising 182 full-length transcripts to narrow down the list of true undetected genes which do not contain any known sequence. We searched for them across the known transcript sequences of PD1074 and its ancestral strain, N2, using BLASTn (Figure 1A). We found that 10 non-coding and 2 coding genes did not match any sequence in BLASTn search (Figure 1A and Supplementary Table S5).
To characterise the protein identities of the two coding genes, we searched for their protein sequences in the NCBI protein database and found that the first candidate encoded by PB.PD.4262 was similar to hypothetical proteins of other Caenorhabditis species (Supplementary Table S5). However, the second candidate encoded by PB.PD.6031 had no significant similarity with any protein sequence within the database (Supplementary Table S5). This observation implies that PB.PD.6031 may have been newly born in C. elegans.
Almost all PD1074 genes have homology in the CB4856 genome
We compared genes between PD1074 and CB4856 by transferring well-annotated gene information from PD1074 to the CB4856 genome (39). Out of 19,954 protein-coding and 26,290 non-coding PD1074 genes, 18,071 (90.6%) protein-coding and 24,992 (95.1%) non-coding genes were transferred to the CB4856 genome (Supplementary Table S6). Only 0.2% of the transferred genes were found on different chromosomes and 68.5% of these transcripts were transferred from the PD1074 chromosome V to the CB4856 chromosome II (Supplementary Table S7), which is consistent with previously reported data on this translocated region (30). In total, 6.9% of the PD1074 genes were not transferred and 33% of these transcripts were located on the PD1074 chromosome V (Supplementary Tables S6 and S7), possibly resulting from small rearrangements in chromosomes V between PD1074 and CB4856 (30,31).
Discovery of previously non-annotated genes in CB4856
We additionally generated long-read RNA sequencing data for CB4856 and reported previously non-annotated transcript information. We processed 14 million raw reads (total 30 Gb, N50 2.6 kb) and obtained 10,043 high-quality, full-length and unique transcripts using PacBio Iso-Seq methods (Figure 1B, Supplementary Table S2 and Supplementary Figure S1D). These processed transcripts belonged to 6,314 genes (Figure 1B and Supplementary Table S4); among them, 3,296 genes contained only known isoforms, 2,369 genes contained non-annotated isoforms, and 471 genes were categorised as newly detected gene candidates (Supplementary Table S4).
These newly detected gene candidates of CB4856 were further validated using BLAST searches against N2 and PD1074 known transcripts and our PD1074 long-read transcripts, as some of them might exist in the PD1074 genome even if not transferred. Among the 471 candidates, we determined that 26 protein-coding and 8 non-coding genes, including 54 transcripts, were true newly detected genes as they had no significant matches to any known transcript (Supplementary Table S8). Most of these newly detected protein-coding genes were similar to other proteins of either C. elegans or other Caenorhabditis species, but PB.CB.1096, PB.CB.2931 and PB.CB.3084 were found to encode proteins never reported previously because their protein sequences had no significant similarity with any protein sequence in the NCBI database (Supplementary Table S8).
PAVs were mainly located in hyper-divergent genomic regions
On the basis of these updated transcript data, we identified PAVs differing between PD1074 and CB4856 by comparing PD1074 transcripts to the CB4856 genome to obtain PD1074-specific genes and vice versa for discovering CB4856-specific genes. Considering PD1074, we found that 117 protein-coding and 188 non-coding genes, containing 331 transcripts, were PD1074-specific genes and of them, 239 PAVs were not found in the previous report that used a hybridisation method (Supplementary Tables S9 and S11) (29). We also analysed whether these PAV genes are located in genetically hyper-divergent regions in C. elegans (31). These regions exhibit mega base-level haplotypes identified by SNVs of 609 wild strains and occupy approximately 20% of the C. elegans genome. We found that 67% of PD1074-specific genes (204 out of 305) were located in hyper-divergent regions (Supplementary Table S9), suggesting that hyper-divergent regions are variable not only at SNV level but also at PAV level. Additionally, 96 genes were found on the right arm of chromosome V, further supporting that this location can be susceptible to rapid changes both between and within species (Figure 1C and Supplementary Table S9) (30,31,50). For CB4856, we found 21 protein-coding and two non-coding CB4856-specific genes (Supplementary Tables S10 and S11), most being localised on chromosomes II and V (Figure 1D and Supplementary Table S10).
We analysed potential functions of these PAV genes by comparison with known RNAi phenotypes (41). Because C. elegans RNAi phenotypes have been studied in the N2 background but not in the CB4856 background, we had an opportunity to investigate only PD1074-specific genes. Among the 117 PD1074-specific protein-coding genes, 11 were reported to have various RNAi phenotypes such as embryonic lethality, reduced brood size, growth variants, oocyte development defects, accumulated cell corpses, protein aggregation variants and hypersensitivity to cadmium and Bacillus thuringiensis toxins (Supplementary Table S9) (42–48). Although cadmium hypersensitivity was examined in CB4856 as well, this strain showed a similar hypersensitivity response to that of N2 (51).
Most PAVs were common in C. elegans wild strains but not conserved in other nematodes
In the succeeding experiments, we investigated whether the discovered PAVs were prevalent in natural populations by searching for our PAV sequences in other high-quality genome assemblies among the reported 14 C. elegans wild strains (31). We confirmed that 211 (64.3%) of our PAVs exhibited presence–absence patterns in other wild strains (Figures 2A-B and Supplementary Tables S12 and S13). Specifically, all the CB4856-specific genes derived solely from full-length transcript data had almost identical sequences (>90% of identity) in ≥ 1 wild strains (4.9 strains on average) (Figure 2B and Supplementary Table S13). These results suggest the possibility that the discovered CB4856-specific genes were not artefacts that emerged specifically from our CB4856 genome assembly, but the genuine genetic variants shared among natural C. elegans populations. Conversely, among the total 305 PD1074-specific genes, 208 had almost identical sequences in ≥ 1 wild strains (8.4 strains on average), but the remaining 97 genes did not match to any wild strain (Figure 2A and Supplementary Table S12). Whether these 97 genes were born during PD1074 domestication or if they are present in other wild strains not used in our study needs to be verified further. Interestingly, 58 PD1074-specific genes and 9 CB4856-specific genes exhibited ≥ 10% genomic difference in ≥ 1 wild strains, as compared to our PAV sequences, suggesting that these genomic regions are still rapidly changing (Figures 2A-B and Supplementary Tables S12 and S13). We also analysed the genetic distance of the 14 wild strains, but there was no notable concordance between genetic distance and PAV patterns (Figures 2A-B, Supplementary Figure S4). We further tested whether these PAV genes have similar patterns with selective sweep in C. elegans. However, only 23% of PD1074-specific genes (70 out of 305) were located in selectively swept regions (46.2% of genomic regions); the PAV patterns of only 0.7% (2 out of 305) were the same as their selective sweep patterns, suggesting that these PAV patterns did not result from selective sweep in C. elegans.
Figure 2.

Homology searches for PAV genes to identify alternative alleles and C. elegans-specific protein-coding genes. (A and B) Presence–absence patterns of (A) PD1074-specific and (B) CB4856-specific genes in 14 wild strains. The y-axis represents (A) 306 PD1074-specific and (B) 23 CB4856-specific genes and the x-axis represents wild strains. Red or blue fields indicate if a gene is present or absent in the corresponding strain, respectively. White fields indicate that an allele of a wild strain has substantially different sequences from alleles present in either PD1074 (A) or CB4856 (B) but still possesses > 10% homology sequences. Schematic illustrations of a neighbour-joining tree among the wild strains (A and B) generated from segregating sites. NIC2 is the one most closely related to N2 and PD1074, and DL238 is the one most closely related to CB4856 (red arrows). (C and D) Protein homology search results for (C) PD1074- and (D) CB4856-specific protein-coding genes. Each number means that the discovered genes have similarity with proteins belonging to C. elegans, other Caenorhabditis species or other organisms.
Thereafter, we examined whether PD1074- and CB4856-specific genes were conserved in other species at the protein level by comparison with the NCBI protein database. Among 117 PD1074- and 21 CB4856-specific protein-coding genes, protein sequences of 75 PD1074- and 16 CB4856-specific genes exhibited at least partial similarity to proteins of other Caenorhabditis species and, among these genes, 24 PD1074- and 7 CB4856-specific genes matched to the corresponding protein sequences of other nematodes (Figures 2C-D and Supplementary Tables S14 and S15). Specifically, 4 PD1074- and 1 CB4856-specific genes were only conserved in C. inopinata, the close relative species of C. elegans, but not in other Caenorhabditis species (Supplementary Tables S14 and S15), suggesting that these genes have emerged from a common ancestor of C. elegans and C. inopinata. Intriguingly, proteins encoded by the remaining 41 PD1074- and 5 CB4856-specific genes had no significant similarity with any protein sequence in the database, except some C. elegans proteins and protein domains, suggesting that they are newly born genes in C. elegans (Figures 2C-D and Supplementary Tables S9, S10, S14 and S15).
Alternative alleles of PAV genes may provide evidence of new gene formation
We assumed that the discovered C. elegans-specific PAV genes were recently born through de novo gene birth and, if this was correct, other alleles in the C. elegans wild strains would have signatures of rapid gene evolution, because alleles without a specific gene variant (‘absence’) should have gained coding capacities to become ‘presence’ alleles. We analysed whether these C. elegans-specific 46 PAV genes have alternative alleles, besides presence and absence alleles, among previously published high-quality genome assemblies of 14 C. elegans wild strains. We found that 34 PAV genes exhibited only either presence or absence alleles in all 14 genomes, but the remaining 12 PAV genes had alternative alleles partially aligned to the corresponding presence alleles (Figures 3A-C, Supplementary Table S16 and Supplementary Figure S5).
Figure 3.

Transposon-mediated allele formation signatures of alternative alleles in two PAV genes. (A, B and C) Schematic allele structure presentation of alternative alleles of (A) C40D2.4, (B) Y113G7A.12 and (C) PB.CB.5378.1. (D) Jumping out of the transposon HELICOP1 may produce different alleles of C40D2.4. HELICOP1 transposon (grey block) is located next to the presence allele (left black vertical bars in the central black line) but far from other alternative alleles in DL238 and ECA36. The alternative alleles have partial sequences of C40D2.4 and F59H6.15 or F59H6.9. One of the three alleles of JU2526 contains inverted partial sequences of C40D2.4 and inverted flanking sequences of F59H6.15. These sequences, belonging to two different regions, are connected by another transposon, CELE14B, instead of HELICOP1. (E) In the two alternative alleles of Y113G7A.12, 3′ UTR sequences flank the sequences of the CELE46A transposon and a repeat region. (F) An alternative allele of PB.CB.5378.1 in ECA396 contains partial original gene sequences and Tc3 family transposon, which do not reside near the original gene in CB4856.
Because these alternative alleles may reflect distinguishable events of gene birth or pseudogenisation, we analysed the corresponding presence and alternative alleles in detail. First, the alternative alleles of six genes showed high similarity with other annotated genes (>31.5% coverage, >80% identity) and these annotated genes also showed high similarity with the corresponding presence alleles (>46.7% coverage, >86% identity), implying that these genes may have been generated through gene duplication and divergence. Among the other six genes, alternative alleles of Y46C8AL.11, Y43F8B.22 and PB.CB.5376 were characterised by several small fragments (approximately 50 bp). These small fragments exhibited no similarity with presence alleles but possessed high similarity to some other sequences in the N2 genome (Supplementary Figure S5 and Supplementary Table S16). We could not find any trace of either duplication or exon shuffling for these alternative alleles, which suggests that their corresponding genes had evolved through extensive indels by unknown mechanisms.
Alternative alleles of the remaining three genes, C40D2.4, Y113G7A.12 and PB.CB.5378.1, exhibited characteristic signatures of transposon action. C40D2.4 had five different alternative alleles and its presence allele was found next to the sequences of the HELICOP1 transposon. No sequence of this transposon flanked the alternative alleles and the sequences of three of them were composed of partial sequences of C40D2.4 and its closely located genes (F59H6.15 or F59H6.9) or non-genic regions possibly derived from transposon jumping out (Figure 3C). Moreover, an alternative allele of JU2526 was found to contain partial sequences of another transposon, CELE14B, between inverted partial C40D2.4 sequences and inverted flanking F59H6.15 sequences (Figure 3C). The presence allele of Y113G7A.12 was found to contain transposon sequences included in the TIR23T5A_CE family. In the two alternative alleles of Y113G7A.12, a transposon sequence was missing and only a partial 3′ UTR region of Y113G7A.12 remained. These partial sequences were also fragmented into two parts and partial sequences of CELE46A transposon and repeat sequences of the N2 genome were found located between the two parts of the 3′ UTR region, presenting an evidence for active transposons. PB.CB.5378.1 also had two different alternative alleles. Its presence allele had no transposon sequences; however, one alternative allele contained 1.2-kb transposon sequences of the Tc3 family, and the other alternative allele contained 80-bp transposon sequences of the CELE46B family (Figure 3D). These results suggest that active transposons have affected either birth or pseudogenisation of these three genes.
Non-coding genes and their coding counterparts revealed another mechanism of gene birth and death
We hypothesised that some genes may be coding ones in one strain, but non-coding in the other strain, as coding genes can be pseudogenised into non-coding genes and non-coding genes can be newly born to become coding genes. We first collected valid non-coding transcripts in PD1074 or CB4856 in our long-read RNA sequencing data, and searched their coding gene counterparts in the other strain (Supplementary Table S17). We found that a total 14 PD1074 and 4 CB4856 non-coding genes did not have any coding transcript in the corresponding strain, but had coding genes in the other strain. Four out of the 18 genes were previously classified as nematode-specific peptide gene. Among the 18 genes, 11 PD1074 and 2 CB4856 non-coding genes contained mismatches or indels in exons of their coding gene counterparts. In addition, 3 PD1074 and 1 CB4856 non-coding genes exhibited an additional feature of gene birth or death, that is, loss of start codon.
DISCUSSION
How genes are born and die is an important question in evolutionary genetics, but the answer remains elusive, because this process requires a long time to be fully elucidated. Here, we tried to glimpse into how genes evolved by using genetic resources of C. elegans wild strains and long-read sequencing technologies to finely resolve their variations. Specifically, we used 46 species-specific PAV genes and alternative alleles of some PAV genes to identify rapid gene evolution snapshots of gene birth and death because these genes might have been newly born in C. elegans and not fixed to presence alleles. Of these genes, 34 did not show any homology at either protein or domain level with other species; therefore, they may not be genes formed by gene duplication, segmental duplication or whole-genome duplication events, in addition to insertion events of the active transposon (52–54). Unfortunately, we could not find any evidence to understand how these 34 genes have evolved. However, we found that the remaining 12 C. elegans-specific protein-coding PAV genes have alternative alleles in other wild isolates. Six out of these genes had similarities to other annotated genes, suggesting that these genes have been generated by gene duplications and changed by mutations. The reason why gene duplication is a major mechanism to produce C. elegans-specific genes should be further addressed as it might happen by chance or from the fact that segmental duplication can produce many genes in a single replication.
On the contrary, alternative alleles of the other six genes did not exhibit gene duplication signatures, suggesting that they represent early forms generated through de novo gene birth. Three genes, C40D2.4, Y113G7A.12 and PB.CB.5378.1, exhibited either transposon-mediated de novo gene birth or pseudogenisation, similarly to new genes that have gained transcription factor functions through transposon insertion, as previously reported in a mammalian study (52). However, while this study has shown that active transposons must be inserted into the existing genes to become DNA-binding motifs, our study shows that the potential for gene birth or death can be achieved through active transposable elements (Figures 4A-B). Transposon insertion into a genomic region may add new sequences to existing non-genic sequences, generating a new gene (Figure 4A). In another case, transposon jumping out from a genomic region can lead to a fusion between its surrounding sequences, creating a new gene (Figure 4B). The other three genes, which may not result from active transposons, exhibited another gene evolution process that accumulated small indels. Although the exact sequential process remains elusive, alternative alleles containing small indels may reveal a de novo gene birth process from non-genic, absence alleles into genic, presence alleles (or a pseudogenisation process, vice versa) (Figure 4C).
Figure 4.
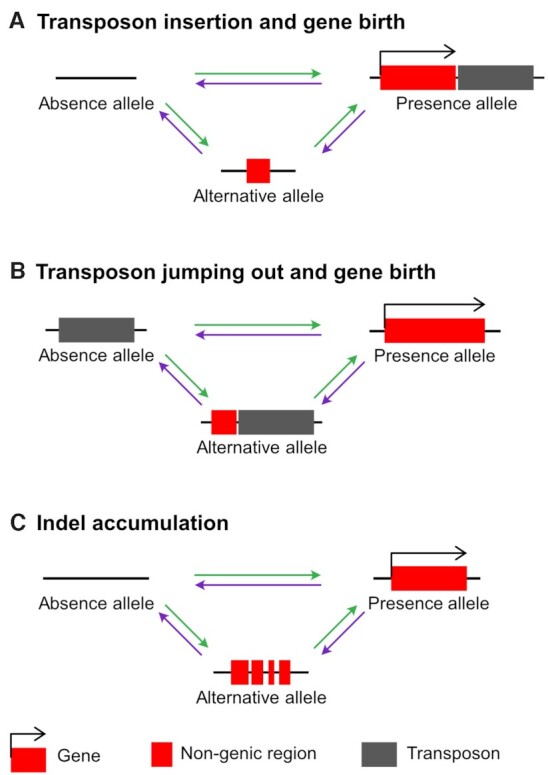
Models of PAV gene evolution. (A) A new gene formation or gene loss process by transposon insertion. (B) A new gene formation or gene loss process by transposon jumping out. (C) New gene formation or pseudogenisation by small indels. Changes in nucleotide sequences caused by small indels lead to birth or death of a gene. Red boxes represent genic or pseudogenised genic regions. Black right-angle arrows indicate coding potential of the region. Grey boxes show transposons. Horizontal black lines represent genomic regions. Green arrows represent gene birth processes and purple arrows represent gene loss processes.
Transposons belonging to the TIR23T5A_CE and the TC3 families found in the presence allele of Y113G7A.12 and an alternative allele of PB.CB.5378.1 are DNA transposons known to be able to transfer to a new part of a genome through the cut-and-paste mechanism (55,56). The HELICOP1 transposon, located next to C40D2.4 in PD1074, is a helitron, which replicates elsewhere in the genome through the rolling circle mechanism (57). Helitrons have also been reported to move to new locations through the cut-and-paste mechanism in maize (58). These cut-and-paste processes cause double-strand breaks in areas from where the transposon has been pulled out and must be repaired by either homologous recombination or non-homologous end-joining (59). In particular, non-homologous end-joining may cause small indels, increase the mutation rate of surrounding sequences and/or insert substantially large sequences (60–62). This process may probably contribute to the generation of genes and alternative alleles that we found.
Among PD1074-specific PAV genes, 38.7% were found mainly concentrated on chromosome V, demonstrating that this chromosome is a hotspot for rapid evolution. Our results are highly consistent with those reported previously, implying that chromosomes II and V of N2 contained most genes deleted in 12 wild strains (29) and that chromosomes II and V of C. briggsae contained a lower number of orthologous genes between C. elegans and C. briggsae genomes (50). In addition, chromosome V of C. elegans has much more hyper-divergent regions than those of other chromosomes (31). Moreover, these characteristics are enriched in the chromosome arm, consistent with previous findings showing that many core genes are located in the chromosome centre and new genes mainly located on the arm (50,63). This suggests that higher density of transposable elements, higher crossover rates or possibly intensive rearrangements in the chromosome arms may have contributed to gene evolution (30,50,64).
Some of the discovered PAV genes could be important for adaptation to different environmental conditions, as they are known to exhibit RNAi phenotypes for important traits such as embryonic lethality and reduced brood size, but their absence alleles still exist in different genetic backgrounds (42–48). Although these results suggest that PAV-related phenotypes can be detected in the CB4856 strain, we could not verify their phenotypes owing to the absence of a genome-wide RNAi or a mutant study on the CB4856 genetic background. One exception was the cadmium-hypersensitive phenotype. However, a phenotypic difference in cadmium hypersensitivity was not observed between N2 and CB4856 (51), implying that the PAV gene may have unknown epistatic or complement genetic relationships. If we consider the advantage of powerful reverse genetics tools developed for C. elegans, we would be able to define whether PAV-related phenotypic variations exist, and if so, we might be able to find the answer on how PAVs affect such phenotypic variations in different genetic architectures.
Interestingly, we identified new various isoforms using long-read RNA sequencing, even though gene annotation of C. elegans has been updated for over several decades. Among the reported PD1074 long-read transcripts, 35% unique transcripts were categorised as previously non-annotated isoforms, suggesting that many more isoforms are present in C. elegans, not detected with conventional approaches. In humans, it is predicted that genes have dozens to hundreds of isoforms per gene on average (65,66); therefore, if we succeed to obtain many more full-length transcripts in a simple organism such as C. elegans, we would be probably able to identify almost all transcripts, which would allow for a further understanding and defining what a gene may represent at the whole species level. In addition, long-read sequencing technologies are being rapidly developed, leading to the first complete human genome (67), allowing for highly accurate-long reads (approximately 99.9% accuracy) (68) and ultra-long reads (approximately 1 Mb of read length) (69). Soon, it will serve to update current long-read-based genome assemblies (70) and provide the complete genomes of any species, further improving the precise annotation of undetected isoforms. Although it is still difficult to grasp whether alternatively spliced isoforms are functional, previous studies showed that the expression of different isoforms may depend on environmental changes, which are known to affect survival and evolution in various organisms (71,72). Thus, it is expected that we can get a deeper insight into this simple organism by elucidating under which condition various isoforms are expressed and what function they have.
In summary, in this study we presented possible processes that may exist between gene birth and death by examining presence, absence and alternative alleles of C. elegans-specific genes. Gene duplication and divergence was found to be a key mechanism for the formation of new genes, but alternative alleles accumulating small indels were found in some genes, while other genes evolved via active transposons. Although the present results are still insufficient to fully understand the de novo gene birth process, future development of high-quality genomes and genetic models of more diverse wild strains and close relative species of C. elegans will enable a precise interpretation of both gene birth and pseudogenisation processes at much higher resolution. It will deepen our understanding of how different organisms acquire and discard genes and how they diverge into different species.
DATA AVAILABILITY
Our genome assemblies and raw PacBio reads were submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA764925. Scripts for all analysis are available from GitHub (https://github.com/JLee1962/PAVs-in-C.elegans).
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: Conceptualisation, J.K. and B.Y.L.; Methodology, B.Y.L. and J.K.; Formal Analysis, B.Y.L.; Investigation, B.Y.L.; Writing-Original Draft, J.K. and B.Y.L.; Writing-Review & Editing, J.K., B.Y.L. and J.L.; Funding Acquisition, J.L.; Supervision, J.L. and J.K. Thanks to Chuna Kim for providing the initial idea of this study, and to Daehan Lee for providing the neighbour-joining tree. Caenorhabditis Genetics Center provided the PD1074 and CB4856 strains.
Contributor Information
Bo Yun Lee, Research Institute of Basic Sciences, Seoul National University, Seoul 08826, Korea; Institute of Molecular Biology and Genetics, Seoul National University, Seoul 08826, Korea.
Jun Kim, Research Institute of Basic Sciences, Seoul National University, Seoul 08826, Korea; Department of Biological Sciences, Seoul National University, Gwanak-ro 1, Gwanak-gu, Seoul 08826, Korea.
Junho Lee, Research Institute of Basic Sciences, Seoul National University, Seoul 08826, Korea; Institute of Molecular Biology and Genetics, Seoul National University, Seoul 08826, Korea; Department of Biological Sciences, Seoul National University, Gwanak-ro 1, Gwanak-gu, Seoul 08826, Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
The Samsung Science and Technology Foundation under Project Number SSTF-BA1501-52; National Research Foundation of Korea grant funded by the Korean government (MEST) [2019R1A6A1A10073437 to J.K.].
Conflict of interest statement. None declared
REFERENCES
- 1. Jacob F. Evolution and tinkering. Science. 1977; 196:1161–1166. [DOI] [PubMed] [Google Scholar]
- 2. Dennis M.Y., Eichler E.E.. Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev. 2016; 41:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marlétaz F., Firbas P.N., Maeso I., Tena J.J., Bogdanovic O., Perry M., Wyatt C.D.R., de la Calle-Mustienes E., Bertrand S., Burguera D.et al.. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature. 2018; 564:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Oss S.B., Carvunis A.R.. De novo gene birth. PLos Genet. 2019; 15:e1008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Begun D.J., Lindfors H.A., Kern A.D., Jones C.D.. Evidence for de novo evolution of testis-expressed genes in the drosophila yakuba/Drosophila erecta clade. Genetics. 2007; 176:1131–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levine M.T., Jones C.D., Kern A.D., Lindfors H.A., Begun D.J.. Novel genes derived from noncoding DNA in drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc. Natl. Acad. Sci. USA. 2006; 103:9935–9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Begun D.J., Lindfors H.A., Thompson M.E., Holloway A.K.. Recently evolved genes identified from drosophila yakuba and d. erecta accessory gland expressed sequence tags. Genetics. 2006; 172:1675–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carvunis A.R., Rolland T., Wapinski I., Calderwood M.A., Yildirim M.A., Simonis N., Charloteaux B., Hidalgo C.A., Barbette J., Santhanam B.et al.. Proto-genes and de novo gene birth. Nature. 2012; 487:370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang W., Gao Y., Long M., Shen B.. Origination and evolution of orphan genes and de novo genes in the genome of caenorhabditis elegans. Sci China Life Sci. 2019; 62:579–593. [DOI] [PubMed] [Google Scholar]
- 10. Zhang L., Ren Y., Yang T., Li G., Chen J., Gschwend A.R., Yu Y., Hou G., Zi J., Zhou R.et al.. Rapid evolution of protein diversity by de novo origination in oryza. Nat. Ecol. Evol. 2019; 3:679–690. [DOI] [PubMed] [Google Scholar]
- 11. Vakirlis N., Hebert A.S., Opulente D.A., Achaz G., Hittinger C.T., Fischer G., Coon J.J., Lafontaine I.. A molecular portrait of de novo genes in yeasts. Mol. Biol. Evol. 2018; 35:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao L., Saelao P., Jones C.D., Begun D.J.. Origin and spread of de novo genes in drosophila melanogaster populations. Science. 2014; 343:769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart M.K., Clark N.L., Merrihew G., Galloway E.M., Thomas J.H.. High genetic diversity in the chemoreceptor superfamily of caenorhabditis elegans. Genetics. 2005; 169:1985–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trowsdale J., Barten R., Haude A., Stewart C.A., Beck S., Wilson M.J.. The genomic context of natural killer receptor extended gene families. Immunol. Rev. 2001; 181:20–38. [DOI] [PubMed] [Google Scholar]
- 15. Winzer T., Gazda V., He Z., Kaminski F., Kern M., Larson T.R., Li Y., Meade F., Teodor R., Vaistij F.E.et al.. A papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science. 2012; 336:1704–1708. [DOI] [PubMed] [Google Scholar]
- 16. Gabur I., Chawla H.S., Lopisso D.T., von Tiedemann A., Snowdon R.J., Obermeier C.. Gene presence-absence variation associates with quantitative verticillium longisporum disease resistance in brassica napus. Sci. Rep. 2020; 10:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang L., Lv Y., Li T., Zhao H., Zhang T.. Identification and characterization of presence/absence variation in maize genotype Mo17. Genes Genom. 2015; 37:503–515. [Google Scholar]
- 18. Rosa R.D., Alonso P., Santini A., Vergnes A., Bachere E.. High polymorphism in big defensin gene expression reveals presence-absence gene variability (PAV) in the oyster crassostrea gigas. Dev. Comp. Immunol. 2015; 49:231–238. [DOI] [PubMed] [Google Scholar]
- 19. Shen J., Araki H., Chen L., Chen J.Q., Tian D.. Unique evolutionary mechanism in R-genes under the presence/absence polymorphism in arabidopsis thaliana. Genetics. 2006; 172:1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calcino A.D., Kenny N.J., Gerdol M.. Single individual structural variant detection uncovers widespread hemizygosity in molluscs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2021; 376:20200153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu T.C., Leu J.Y., Lin W.C.. A comprehensive analysis of transcript-supported de novo genes in saccharomyces sensu stricto yeasts. Mol. Biol. Evol. 2017; 34:2823–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takahashi-Kariyazono S., Sakai K., Terai Y.. Presence-absence polymorphisms of single-copy genes in the stony coral acropora digitifera. BMC Genomics. 2020; 21:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gao L., Gonda I., Sun H., Ma Q., Bao K., Tieman D.M., Burzynski-Chang E.A., Fish T.L., Stromberg K.A., Sacks G.L.et al.. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019; 51:1044–1051. [DOI] [PubMed] [Google Scholar]
- 24. Liu Y., Du H., Li P., Shen Y., Peng H., Liu S., Zhou G.A., Zhang H., Liu Z., Shi M.et al.. Pan-Genome of wild and cultivated soybeans. Cell. 2020; 182:162–176. [DOI] [PubMed] [Google Scholar]
- 25. Li C., Xiang X., Huang Y., Zhou Y., An D., Dong J., Zhao C., Liu H., Li Y., Wang Q.et al.. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nat. Commun. 2020; 11:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cook D.E., Zdraljevic S., Roberts J.P., Andersen E.C.. CeNDR, the caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 2017; 45:D650–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crombie T.A., Zdraljevic S., Cook D.E., Tanny R.E., Brady S.C., Wang Y., Evans K.S., Hahnel S., Lee D., Rodriguez B.C.et al.. Deep sampling of hawaiian caenorhabditis elegans reveals high genetic diversity and admixture with global populations. Elife. 2019; 8:e50465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshimura J., Ichikawa K., Shoura M.J., Artiles K.L., Gabdank I., Wahba L., Smith C.L., Edgley M.L., Rougvie A.E., Fire A.Z.et al.. Recompleting the caenorhabditis elegans genome. Genome Res. 2019; 29:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maydan J.S., Lorch A., Edgley M.L., Flibotte S., Moerman D.G.. Copy number variation in the genomes of twelve natural isolates of caenorhabditis elegans. BMC Genomics. 2010; 11:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim C., Kim J., Kim S., Cook D.E., Evans K.S., Andersen E.C., Lee J.. Long-read sequencing reveals intra-species tolerance of substantial structural variations and new subtelomere formation in c. elegans. Genome Res. 2019; 29:1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee D., Zdraljevic S., Stevens L., Wang Y., Tanny R.E., Crombie T.A., Cook D.E., Webster A.K., Chirakar R., Baugh L.R.et al.. Balancing selection maintains hyper-divergent haplotypes in caenorhabditis elegans. Nat. Ecol. Evol. 2021; 5:794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brenner S. The genetics of caenorhabditis elegans. Genetics. 1974; 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sulston J., Hodgkin J.. Wood W.B. The Nematode Caenorhabditis elegans. 1988; NY: Cold Spring Harbor Laboratory Press; 587–606. [Google Scholar]
- 34. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marcais G., Delcher A.L., Phillippy A.M., Coston R., Salzberg S.L., Zimin A.. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 2018; 14:e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tardaguila M., de la Fuente L., Marti C., Pereira C., Pardo-Palacios F.J., Del Risco H., Ferrell M., Mellado M., Macchietto M., Verheggen K.et al.. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res. 2018; 28:396–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gordon S.P., Tseng E., Salamov A., Zhang J., Meng X., Zhao Z., Kang D., Underwood J., Grigoriev I.V., Figueroa M.et al.. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One. 2015; 10:e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M.et al.. The UCSC genome browser database: 2021 update. Nucleic Acids Res. 2021; 49:D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C.J., Lu S., Chitsaz F., Derbyshire M.K., Geer R.C., Gonzales N.R.et al.. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017; 45:D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harris T.W., Arnaboldi V., Cain S., Chan J., Chen W.J., Cho J., Davis P., Gao S., Grove C.A., Kishore R.et al.. WormBase: a modern model organism information resource. Nucleic Acids Res. 2020; 48:D762–D767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernandez A.G., Gunsalus K.C., Huang J., Chuang L.S., Ying N., Liang H.L., Tang C., Schetter A.J., Zegar C., Rual J.F.et al.. New genes with roles in the c. elegans embryo revealed using RNAi of ovary-enriched ORFeome clones. Genome Res. 2005; 15:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rual J.F., Ceron J., Koreth J., Hao T., Nicot A.S., Hirozane-Kishikawa T., Vandenhaute J., Orkin S.H., Hill D.E., van den Heuvel S.et al.. Toward improving caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004; 14:2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sakaki K., Yoshina S., Shen X., Han J., DeSantis M.R., Xiong M., Mitani S., Kaufman R.J.. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc. Natl. Acad. Sci. USA. 2012; 109:8079–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cui Y., McBride S.J., Boyd W.A., Alper S., Freedman J.H.. Toxicogenomic analysis of caenorhabditis elegans reveals novel genes and pathways involved in the resistance to cadmium toxicity. Genome Biol. 2007; 8:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Green R.A., Kao H.L., Audhya A., Arur S., Mayers J.R., Fridolfsson H.N., Schulman M., Schloissnig S., Niessen S., Laband K.et al.. A high-resolution c. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell. 2011; 145:470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zullig S., Neukomm L.J., Jovanovic M., Charette S.J., Lyssenko N.N., Halleck M.S., Reutelingsperger C.P., Schlegel R.A., Hengartner M.O.. Aminophospholipid translocase TAT-1 promotes phosphatidylserine exposure during c. elegans apoptosis. Curr. Biol. 2007; 17:994–999. [DOI] [PubMed] [Google Scholar]
- 48. Kao C.Y., Los F.C., Huffman D.L., Wachi S., Kloft N., Husmann M., Karabrahimi V., Schwartz J.L., Bellier A., Ha C.et al.. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011; 7:e1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stein L.D., Bao Z., Blasiar D., Blumenthal T., Brent M.R., Chen N., Chinwalla A., Clarke L., Clee C., Coghlan A.et al.. The genome sequence of caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 2003; 1:E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Evans K.S., Brady S.C., Bloom J.S., Tanny R.E., Cook D.E., Giuliani S.E., Hippleheuser S.W., Zamanian M., Andersen E.C.. Shared genomic regions underlie natural variation in diverse toxin responses. Genetics. 2018; 210:1509–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cosby R.L., Judd J., Zhang R., Zhong A., Garry N., Pritham E.J., Feschotte C.. Recurrent evolution of vertebrate transcription factors by transposase capture. Science. 2021; 371:eabc6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crow K.D., Wagner G.P.. What is the role of genome duplication in the evolution of complexity and diversity. Mol. Biol. Evol. 2006; 23:887–892. [DOI] [PubMed] [Google Scholar]
- 54. Meyer A., Schartl M.. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol. 1999; 11:699–704. [DOI] [PubMed] [Google Scholar]
- 55. C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998; 282:2012–2018. [DOI] [PubMed] [Google Scholar]
- 56. van Luenen H.G., Colloms S.D., Plasterk R.H.. The mechanism of transposition of Tc3 in c. elegans. Cell. 1994; 79:293–301. [DOI] [PubMed] [Google Scholar]
- 57. Kapitonov V.V., Jurka J.. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007; 23:521–529. [DOI] [PubMed] [Google Scholar]
- 58. Li Y., Dooner H.K.. Excision of helitron transposons in maize. Genetics. 2009; 182:399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Krasileva K.V. The role of transposable elements and DNA damage repair mechanisms in gene duplications and gene fusions in plant genomes. Curr. Opin. Plant Biol. 2019; 48:18–25. [DOI] [PubMed] [Google Scholar]
- 60. Wicker T., Yu Y., Haberer G., Mayer K.F., Marri P.R., Rounsley S., Chen M., Zuccolo A., Panaud O., Wing R.A.et al.. DNA transposon activity is associated with increased mutation rates in genes of rice and other grasses. Nat. Commun. 2016; 7:12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gorbunova V., Levy A.A.. Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucleic Acids Res. 1997; 25:4650–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim C., Sung S., Kim J., Lee J.. Repair and reconstruction of telomeric and subtelomeric regions and genesis of new telomeres: implications for chromosome evolution. Bioessays. 2020; 42:e1900177. [DOI] [PubMed] [Google Scholar]
- 63. Prabh N., Roeseler W., Witte H., Eberhardt G., Sommer R.J., Rodelsperger C.. Deep taxon sampling reveals the evolutionary dynamics of novel gene families in pristionchus nematodes. Genome Res. 2018; 28:1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Woodruff G.C., Teterina A.A.. Degradation of the repetitive genomic landscape in a close relative of caenorhabditis elegans. Mol. Biol. Evol. 2020; 37:2549–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sedlazeck F.J., Lee H., Darby C.A., Schatz M.C.. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet. 2018; 19:329–346. [DOI] [PubMed] [Google Scholar]
- 66. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S.et al.. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A.et al.. The complete sequence of a human genome. Science. 2022; 376:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D.et al.. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019; 37:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jain M., Koren S., Miga K.H., Quick J., Rand A.C., Sasani T.A., Tyson J.R., Beggs A.D., Dilthey A.T., Fiddes I.T.et al.. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018; 36:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim E., Kim J., Kim C., Lee J.. Long-read sequencing and de novo genome assemblies reveal complex chromosome end structures caused by telomere dysfunction at the single nucleotide level. Nucleic Acids Res. 2021; 49:3338–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Trevisan G.L., Oliveira E.H., Peres N.T., Cruz A.H., Martinez-Rossi N.M., Rossi A.. Transcription of aspergillus nidulans pacC is modulated by alternative RNA splicing of palB. FEBS Lett. 2011; 585:3442–3445. [DOI] [PubMed] [Google Scholar]
- 72. Nilsen T.W., Graveley B.R.. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010; 463:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Our genome assemblies and raw PacBio reads were submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA764925. Scripts for all analysis are available from GitHub (https://github.com/JLee1962/PAVs-in-C.elegans).