

Abstract
Despite the success of immune checkpoint blockade therapy, few strategies sufficiently overcome immunosuppression within the tumor microenvironment (TME). Targeting regulatory T cells (Tregs) is challenging, because perturbing intratumoral Treg function must be specific enough to avoid systemic inflammatory side effects. Thus, no Treg-targeted agents have proven both safe and efficacious in patients with cancer. Neuropilin-1 (NRP1) is recognized for its role in supporting intratumoral Treg function while being dispensable for peripheral homeostasis. Nonetheless, little is known about the biology of human NRP1 Tregs and the signals that regulate NRP1 expression. Here, we report that NRP1 is preferentially expressed on intratumoral Tregs across six distinct cancer types compared to healthy donor peripheral blood [peripheral blood lymphocyte (PBL)] and site-matched, noncancer tissue. Furthermore, NRP1+ Treg prevalence is associated with reduced progression-free survival in head and neck cancer. Human NRP1+ Tregs have broad activation programs and elevated suppressive function. Unlike mouse Tregs, we demonstrate that NRP1 identifies a transient activation state of human Tregs driven by continuous T cell receptor (TCR) signaling through the mitogen-activated protein kinase pathway and interleukin-2 exposure. The prevalence of NRP1+ Tregs in patient PBL correlates with the intratumoral abundance of NRP1+ Tregs and may indicate higher disease burden. These findings support further clinical evaluation of NRP1 as a suitable therapeutic target to enhance antitumor immunity by inhibiting Treg function in the TME.
INTRODUCTION
On the basis of the success of immune checkpoint blockade (ICB) for patients with cancer, it is clear that reversal of CD8+ T cell exhaustion increases objective clinical responses (1). This paradigm has held true across numerous solid tumor types, from melanoma (Mel) to hepatocellular carcinoma, with varying response rates (2). However, even in the best-performing clinical indications, only 20 to 30% of patients achieve an objective response, highlighting that additional barriers to antitumor immunity exist beyond CD8+ T cell exhaustion. One key obstacle is overcoming checkpoint-independent immunosuppression within the tumor microenvironment (TME).
Many immunoregulatory cells are actively recruited, differentiated, and stimulated at the tumor site, including regulatory T cells (Tregs), tumor-associated macrophages, and myeloid-derived suppressor cells (3–5). Although these immunosuppressive cell types are required for physiologic homeostasis, they can markedly impede productive antitumor immunity and are known to drive resistance to ICB (6). Among this group, Tregs are consistently associated with worse clinical outcomes in human solid tumors (7–16) and thus are a major impediment for advancing cancer immunotherapy.
Marked by the master transcription factor forkhead box protein P3 (FOXP3), Tregs maintain immune equilibrium by minimizing inflammatory damage to host tissues (17). The potency of Treg function is demonstrated in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, wherein mutations of the FOXP3 gene locus engender a deficit in Treg development, resulting in debilitating autoimmunity (18). In cancer, the anti-inflammatory function of Tregs is commandeered for the protection of the tumor. Activated Tregs are often more prevalent in human tumors compared to normal, site-matched tissue and secondary lymphoid organs (13, 19). Furthermore, Treg-specific depletion in mouse models restores antitumor immunity (20); however, because of the essential role Tregs play in immune homeostasis, tumor rejection coincides with peripheral autoimmunity (21). Therefore, future translational strategies must discriminate between homeostatic and tumor-specific Treg programs with the goal of identifying a therapeutic opportunity that minimizes toxicity.
The transmembrane co-receptor neuropilin-1 (NRP1; CD304) has been identified as a prime candidate to specifically target intratumoral Tregs (22). NRP1 can be expressed by multiple immune, vascular, neuronal, and endothelial cell types; however, it is a key regulator of intratumoral Treg function (22). In murine studies, the requirement of NRP1 expression for optimal Treg function is constrained to the TME. Specifically, Treg-restricted NRP1 deletion or pharmacologic blockade restores antitumor immunity in mice challenged with implantable tumors without generating any aberrant autoimmune side effects (21, 23). Furthermore, loss of NRP1 reverses the phenotype of intratumoral Tregs, causing them to produce interferon-γ, a phenomenon termed as “infectious fragility” (24). Upon binding semaphorin-4a (SEMA4A), NRP1-mediated recruitment of phosphatase and tensin homolog (PTEN) to the immunologic synapse restrains protein kinase B (also known as AKT) activity, thereby stabilizing Treg activation (21).
Despite evidence supporting the importance of NRP1 in murine cancer models, the extent to which NRP1 shapes Treg function in human cancer remains underexplored. Whereas a high proportion of mouse resting or naïve Tregs express NRP1 (25–27), few Tregs in healthy (noncancer) human donors express NRP1 (28). Although increased NRP1 expression has been noted on Tregs in draining lymph nodes and tumors of patients (23, 24, 29–31), the degree of expression is lower than that observed in mouse studies. Nonetheless, initial reports are consistent with observations from mouse models regarding NRP1+ Treg phenotype, function, and potential clinical impact (23, 24, 31). With a current phase 1b clinical trial underway for an anti-NRP1 monoclonal antibody targeting the SEMA4A binding domain (NCT03565445), the need to more rigorously understand how findings from mouse studies translate to human disease is paramount. No study to date has characterized intratumoral and peripheral Tregs for NRP1 expression in more than two solid tumors nor has any report systematically addressed which signals within the TME drive NRP1 expression on Tregs in human tumors.
In this study, we evaluated whether NRP1 modulates Treg function in human cancer. We hypothesized that NRP1+ Tregs would be elevated in patients with cancer; that NRP1 expression on Tregs would be indicative of a highly activated phenotype, driven by signals enriched in the TME; and that a higher frequency of NRP1+ Tregs would correlate with poor prognosis. We analyzed 375 fresh patient samples from six distinct cancer types in conjunction with 85 fresh samples from relevant healthy tissue controls, paired with in vitro suppression assays, mechanistic interrogation of NRP1 drivers, and correlative clinical analysis of our prospectively accrued patient cohort. Overall, our findings support the idea that antagonizing NRP1-dependent Treg function to enhance antitumor immunity may augment standard-of-care immunotherapy.
RESULTS
NRP1+ Tregs are restricted to the TME in patients with solid malignancies
With comprehensive characterization of NRP1 expression in human cancer lacking, we prospectively collected 375 fresh blood and tumor samples from six cancer types with a minimum of 12 individuals per sample group (fig. S1A and table S1). We also collected 85 control samples including healthy donor (HD) peripheral blood [peripheral blood lymphocyte (PBL)] and site-matched, noncancer specimens from the tonsil, ovary, and lung (fig. S1, B and C, and table S2). This approach enabled comparisons between peripheral and intratumoral Tregs across multiple tumor types and noncancer tissues.
As previously reported (24), the total NRP1 protein expression (herein denoted as NRP1TOT) was determined by fixing and permeabilizing cells and staining with an antibody specific for the intracellular tail of NRP1. In addition, we incorporated a noncompeting cell surface NRP1-directed antibody to quantify the fraction of NRP1+ Tregs that could interact with ligands (herein denoted as NRP1SURF). Unexpectedly, the discordant expression of NRP1TOT versus NRP1SURF was observed in patient samples (Fig. 1A), revealing that Tregs (FOXP3+, CD25hi, CD4+ T cells) from patients with cancer contained intracellular stores of NRP1 protein (Fig. 1, A and B). Furthermore, 21.6% of patients with cancer had a higher fraction Tregs expressing NRP1SURF in their peripheral blood than any HD we measured, and this rose to 52.4% when looking at tumor samples (Fig. 1C and fig. S1C). NRP1 is only abundantly detected on HD peripheral Tregs after in vitro stimulation and was not associated with donor age (Fig. 1A and fig. S1D) (28).
Fig. 1. The proportion of NRP1+ regulatory T cells in solid tumors is negatively correlated with disease outcomes.
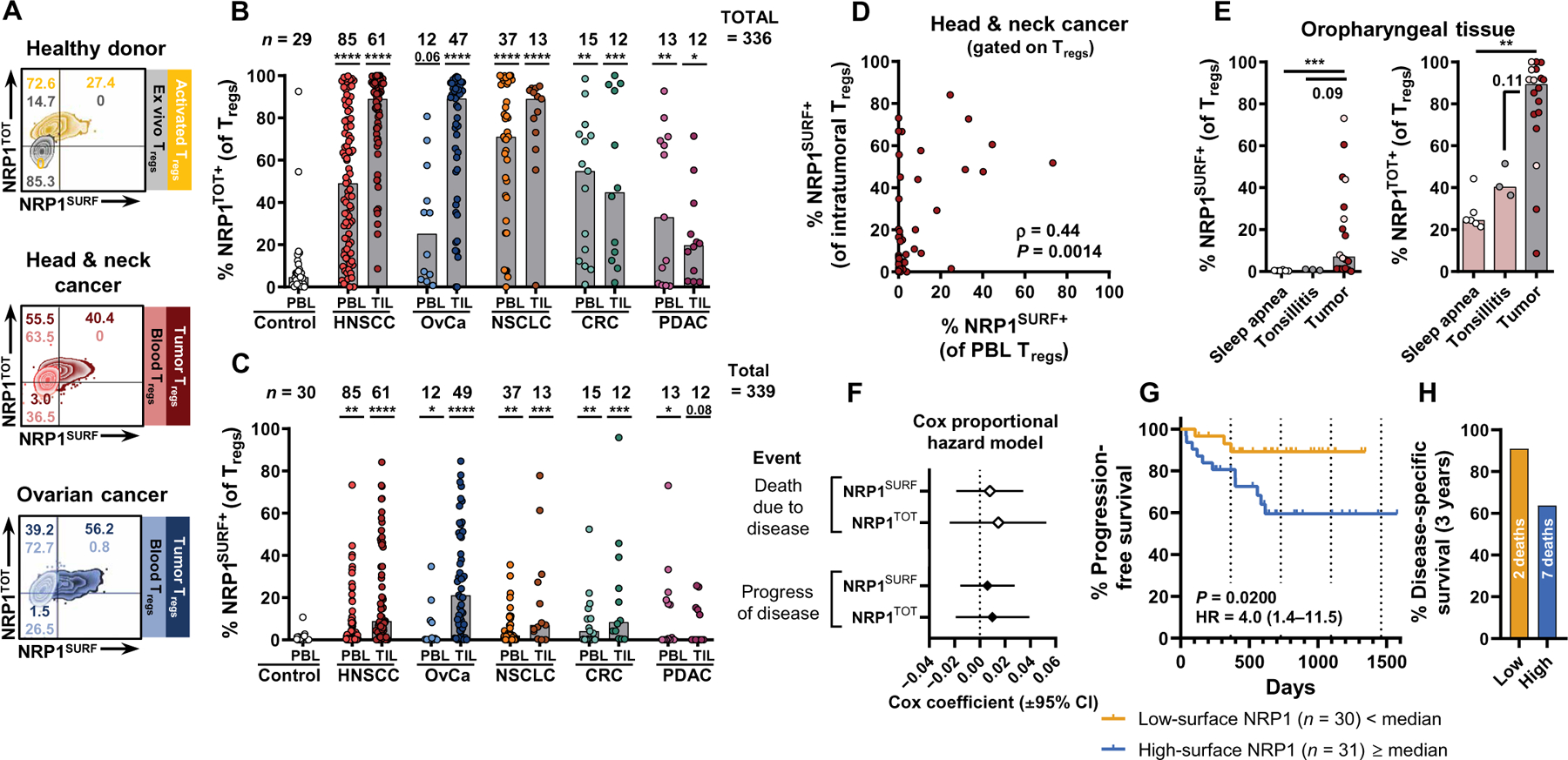
(A) Representative flow cytometry staining of NRP1 is shown for healthy donor (HD) blood and for blood and tumor samples from patients with head and neck squamous cell carcinoma (HNSCC) or ovarian cancer (OvCa). For HD samples, in vitro–activated Treg NRP1 expression profiles are overlaid in purple. For samples from patients with cancer, both blood and tumor Treg NRP1 expression profiles are overlaid. Total NRP1 (NRP1TOT) staining used a distinct antibody clone specific for the intracellular tail of NRP1 during intracellular staining. (B and C) Tabulation of NRP1 expression [(B) (NRP1TOT) and (C) (NRP1SURF)] on Tregs across five cohorts of treatment-naïve patients with solid tumors. Melanoma was excluded from this table because not all patients were treatment naïve (data are available in fig. S3). Bars indicate median of expression. Data were analyzed by a nonparametric Kruskal-Wallis test with multiple comparisons to control PBL [controlled for a false discovery rate (FDR) of 5% and reported adjusted P values as q values). *q < 0.05, **q < 0.01, ***q < 0.001, and ****q < 0.0001. NSCLC, non–small cell lung cancer; CRC, colorectal cancer; PDAC, pancreatic ductal adenocarcinoma. (D) Expression of NRP1SURF on Tregs from patients with HNSCC is shown matched between tumor biopsy and peripheral blood (n = 50). Spearman’s nonparametric correlation analysis results are reported. (E) Expression of NRP1 on Tregs is diminished in site-matched nontumor tissue specimens in the oropharynx (oral cavity, larynx, and hypopharynx excluded; n = 6 sleep apnea, n = 6 tonsilitis, and n = 1 tumor). Oropharyngeal tumors are a subset of HNSCC tumors. Tonsil tumors are marked in pink. Bars indicate the median of expression. Data were analyzed using a nonparametric Kruskal-Wallis test with multiple comparisons (FDR of 5%). **q < 0.01 and ***q < 0.001. (F) Univariate Cox proportional hazard model was used to evaluate NRP1SURF and NRP1TOT as continuous predictors of clinical outcome. Cox coefficients are reported with a 95% confidence interval (CI). (G) Intratumoral NRP1 expression was divided on the basis of the median of expression of NRP1SURF for HNSCC. Progression-free survival (PFS) was compared between the groups. A log-rank (Mantel-Cox) test was used to compare survival curves. HR, hazard ratio. (H) Disease-specific survival for the same groups as in (G) is shown after 3 years of follow-up. See fig. S4A for the full disease-specific survival curve.
NRP1 expression (NRP1TOT and NRP1SURF) was robust across multiple tumor types, with NRP1SURF being preferentially expressed by intratumoral Tregs compared to PBL Tregs except in pancreatic ductal adenocarcinoma (PDAC), which had a low median expression in both PBL and tumor-infiltrating lymphocyte (TIL) (Fig. 1, B and C; treatment-naïve tumor cohorts). NRP1 expression was correlated between intratumoral and peripheral Tregs in patients with head and neck squamous cell cancer (HNSCC) (NRP1SURF, Fig. 1D; NRP1TOT, fig. S2A). Compared to HD PBL, patients with cancer had significantly more NRP1TOT+ Tregs in their peripheral blood and tumor (P ≤ 0.05 in 9 of 10 cohorts; Fig. 1B). To determine whether NRP1 expression was simply an indicator of tissue residence or trafficking, we analyzed noncancer, site-matched specimens of the oropharynx (matched with a subset of HNSCC). We found that both NRP1SURF and NRP1TOT are enriched in the TME (Fig. 1E). Similar findings were observed in the lung tissue comparing normal tissue (both smoker and nonsmoker) and chronic obstructive pulmonary disease (COPD) tissue with non–small cell lung cancer (NSCLC) specimens (fig. S2B). Moreover, Tregs in benign ovarian lesions had little to no NRP1 expression compared to those in malignant epithelial ovarian cancer (OvCa; fig. S2C). Together, these findings from three distinct tissue/tumor pairs suggest that NRP1SURF expression among Tregs is unique to the TME in these tissues. NRP1SURF expression was enriched on Tregs compared to CD4+ effector T cells (CD4+ Teff) and CD8+ T cells in HNSCC tumors, although also correlated between T cell subsets (fig. S2D). In contrast, NRP1TOT expression was comparable between T cell subsets (fig. S2E). Analogous observations were made in OvCa tumors and HNSCC PBL (fig. S2, F to I). This suggests that Tregs may be more responsive to cues within the TME that drive NRP1SURF.
The distribution of NRP1+ Tregs (NRP1TOT and NRP1SUR) within each tumor type was distinct (fig. S3). HNSCC, OvCa, and NSCLC had the highest median values for NRP1SURF and NRP1TOT (fig. S3B), and in HNSCC and OvCa, NRP1SURFand NRP1TOT were correlated with each other. Patients with these cancer types had a high proportion of NRP1+ Tregs compared to colorectal cancer (CRC), PDAC, and Mel (defined by the median NRP1SURF and NRP1TOT values aggregated across tumor types; see Materials and Methods; fig. S3, A and B, quadrants III and IV). The relative enrichment of individuals in quadrant I for CRC, PDAC, and Mel suggests that Tregs may be exposed to fewer stimuli within these TME that regulate NRP1 expression. Microsatellite stable (MSS) CRC and PDAC are poorly immunogenic tumors, compared to the others in our cohort (2, 32, 33). We hypothesized that this contributes to the similarity of NRP1SURF and NRP1TOT abundance between PBL and TIL samples of these cancers and overall low abundance of NRP1SURF+ Tregs (Fig. 1, B and C). Furthermore, the Mel cohort in our study was distinct from the others, because a high proportion of individuals were pretreated, many with ICB (fig. S3, C and D), whereas all other cancer cohorts were treatment naïve at the time of biopsy. Lack of response to prior therapy may indicate that these individuals had particularly immune-suppressed TMEs, which may concomitantly affect NRP1 expression. Together, NRP1 can be expressed by Tregs in many cancer types, and signals in the TME may be responsible for driving NRP1 expression.
Prevalence of intratumoral NRP1SURF+ Tregs is correlated with decreased survival in HNSCC
Because NRP1SURF+ Tregs are enriched in solid tumors, we hypothesized that the proportion of intratumoral Tregs expressing NRP1SURF would be negatively associated with the patient outcome. We evaluated this hypothesis in HNSCC and OvCa, as we prospectively accrued the most tumor specimens in these cohorts (fig. S1A). We analyzed NRP1 in both tumors with respect to progression-free survival (PFS) and other clinical parameters. In HNSCC, we found that the proportion of NRP1+ Tregs was not a continuous predictor of outcome (Fig. 1F); however, when we grouped individuals above or below the median proportion of NRP1SURF+ Tregs, we found that individuals in the high group had significantly reduced PFS compared to those with fewer NRP1SURF+ Tregs (P = 0.0200; Fig. 1G and fig. S4), with a lower rate of disease-specific survival at 3 years (Fig. 1H). Conversely, NRP1TOT expression was not associated with a clinical difference, neither as a continuous predictor nor with the cutoff as the median (Fig. 1F and fig. S4B). We did not observe a strong association between NRP1SURF and PFS in OvCa (fig. S5). Nonetheless, continued follow-up may reveal a difference in PFS between these groups in the OvCa cohort.
Although other clinical parameters were also associated with PFS (fig. S6A), most did not display a relationship with NRP1 expression (fig. S6B), suggesting that our observations are not strongly confounded by other clinical characteristics. Nonetheless, of the nine patients with HNSCC subsequently treated with immunotherapy, six fell into the above median NRP1SURF expression group (66%), with five experiencing a progression of disease. If we remove these individuals from our analysis to mitigate potential confounding, then the P value calculated for these data no longer falls under a significance level of 0.05 [P = 0.099; hazard ratio (HR) of 3.53; fig. S6C], likely due to decreased statistical power from fewer samples and events. Because it is unlikely that immunotherapy deleteriously affected subsequent outcome (34), it is more likely that that intratumoral Treg NRP1 expression typified a subset of patients whose disease was unable to be controlled with other treatment modalities. Thus, the association does not undermine our findings but rather supports the claim that the proportion of NRP1+ Tregs is prospectively associated with disease severity, possibly through increased Treg-mediated immunosuppression in the TME.
Intratumoral NRP1SURF+ Tregs exhibit a multifaceted suppressive program
On the basis of the mechanistic analysis of NRP1+ Tregs in mouse models of cancer (21, 24), we hypothesized that NRP1 expression would be associated with increased Treg activation, stability, and survival in human tumors. Other research has reported an enrichment of FOXP3, human leukocyte antigen (HLA)–DR, and glucocorticoid-induced tumor necrosis factor receptor (TNFR)–related protein (GITR) expression for NRP1+ CD4+ T cells in lymph nodes from patients with cancer (31). However, we aimed to evaluate a more complete Treg-specific activation signature combined with the suppressive function of intratumoral Tregs in vitro. We first focused on comparing the differential protein expression between the NRP1SURF+ and NRP1SURF− subsets because only cell surface NRP1 would interact with ligands to influence Treg function in the TME (fig. S7A). The proportion of activated Tregs in HNSCC and OvCa was increased within the NRP1SURF+ subpopulation, as revealed by expression of early activation marker CD69 (Fig. 2, A and B), as well as by T cell immunoreceptor with immunoglobulin (Ig) and immunoreceptor tyrosine-based inhibitory motif domains (TIGIT) and inducible T cell costimulator (ICOS) expression (ICOS only for OvCa; fig. S7, B and C) (35–37). We also observed a higher proportion of NRP1SURF+ Tregs expressing the chemokine receptor CCR8 (CD198) (Fig. 2, A and B) (38). In addition to canonical markers of activation and tumor residence, HNSCC NRP1SURF+ Tregs were enriched for multiple TNFR superfamily members (TNFRSFs), including GITR, 4–1BB (Fig. 2, C and D), OX40, and TNFR2 (fig. S7, D and E). Although the proportion of Tregs expressing NRP1SURF and TNFRSFs is comparable between HNSCC and OvCa, we only observed a correlation between NRP1 and TNFRSFs in Tregs from HNSCC tumors (Fig. 2, C and D, and fig. S7, F to I).
Fig. 2. NRP1 is coexpressed with numerous activation markers on intratumoral Tregs.
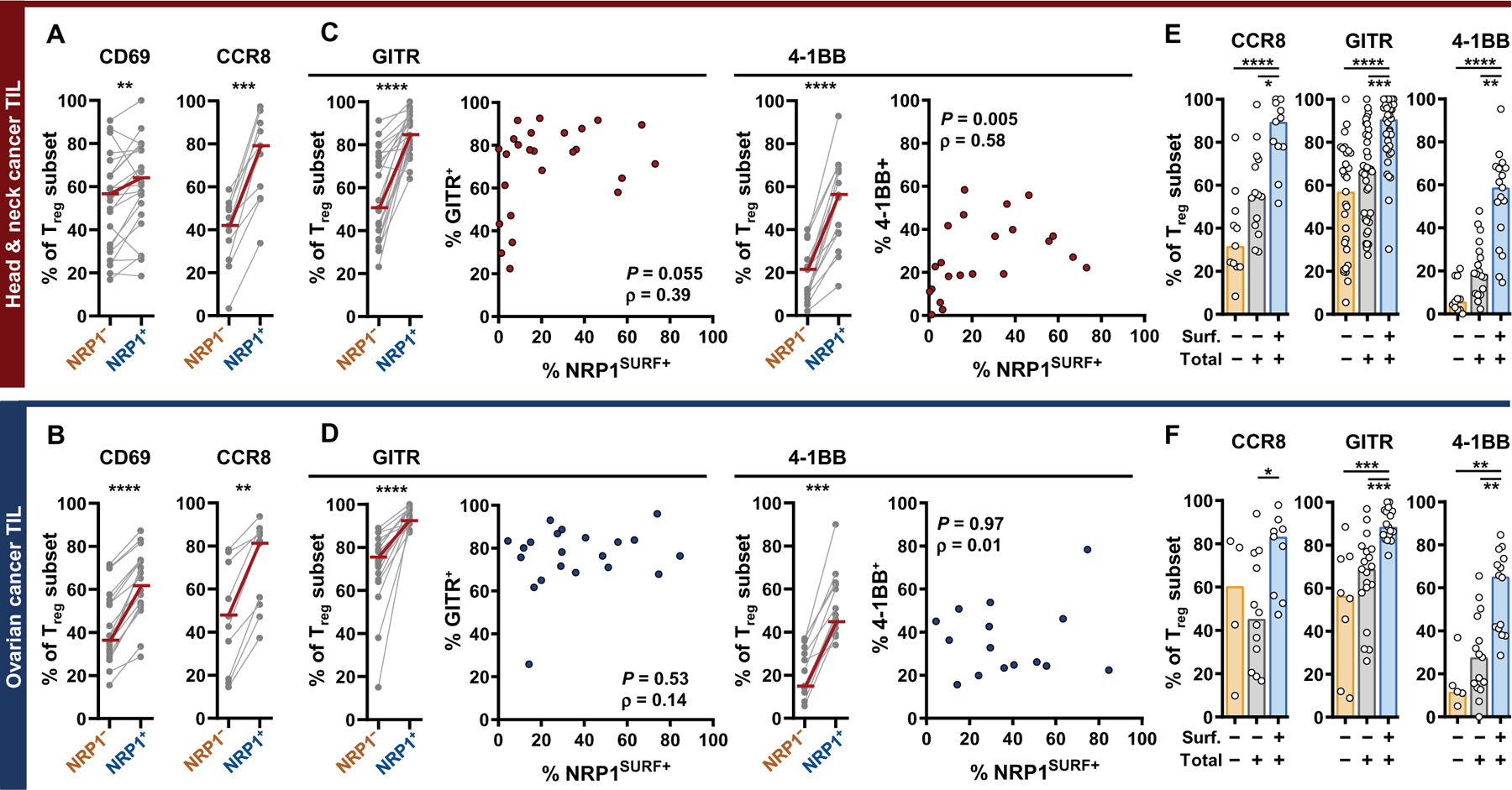
(A and B) NRP1SURF+ is indicated as NRP1+. Intratumoral NRP1+ Tregs are enriched for an activation phenotype as demonstrated by CD69 and CCR8 expression. Expression is reported as a percentage of respective Treg subpopulations. Median values are indicated by overlaid red bars. The number of samples is between 11 and 22 depending on the marker. Statistical comparison was done by paired, nonparametric t test (Wilcoxon). **P < 0.01, ***P < 0.001, and ****P < 0.0001. (C and D) Intratumoral NRP1+ Tregs consistently express high concentrations of TNF receptor superfamily (TNFRSF) members, GITR and 4–1BB. Median values are indicated by overlaid red bars. The number of samples is between 14 and 22 depending on the marker in the column graphs. Statistical comparison was done by paired, nonparametric t test (Wilcoxon). ***P < 0.001 and ****P < 0.0001. Overall nonparametric (Spearman’s) correlation of intratumoral surface NRP1+ Tregs with either GITR+ or 4–1BB+ as indicated is shown on the right side (the number of samples is between 15 and 25). (E and F) Quantification of Tregs that are completely NRP1 negative versus those that express the protein but do not have it on the cell surface (Total+, Surface−) or that express protein and have it on the cell surface (Total+, Surface+) is shown. The number of samples in each subgroup is between 4 and 34 depending on the marker and subgroup. Differences between the three subpopulations were assessed by nonparametric Kruskal-Wallis test with comparisons between all three groups controlled for with a 5% FDR. Bars indicate the median of expression. *q < 0.05, **q < 0.01, ***q < 0.001, and ****q < 0.0001.
Because NRP1SURF+ Tregs are also NRP1TOT+ (Fig. 1A), we queried whether NRP1TOT expression contributed to the above observations by further subdividing Tregs into three groups based on NRP1 expression: NRP1−, NRP1TOT+SURF−, and NRP1TOT+SURF+ (referred to as NRPSURF+ elsewhere; fig. S8A). NRP1TOT+SURF− Tregs (indicated by gray bars) had an intermediate expression of several markers that we investigated (Fig. 2, E and F, and fig. S8); however, their expression was significantly lower than that of NRP1TOT+SURF+ Tregs (P < 0.05 for all markers shown, indicated by blue bars) and not readily distinguishable from NRP1− Tregs.
In addition, intratumoral NRP1SURF+ Tregs expressed 20% more FOXP3 protein and 30 to 40% more CD25 protein compared to NRP1SURF− Tregs residing in the same TME (fig. S9A). This indicates that NRP1SURF+ Tregs are more stable, because both FOXP3 and CD25 are hallmarks of Treg stability (39). Similarly, NRP1SURF+ Tregs also expressed 15% more Bcl2 protein, a critical antiapoptotic regulator, and a higher proportion of NRP1SURF+ Tregs expressed Ki67, marking recent or ongoing entry into the cell cycle (fig. S9, B and C). These findings were consistent in HNSCC and OvCa tumors. We found that programmed death 1 (PD-1) and Helios were only significantly coexpressed with NRP1 in OvCa (P < 0.001; fig. S9, D and E). Collectively, these findings demonstrate general consistency of the NRP1SURF+ Treg phenotype across two distinct tumor types, albeit with minor differences.
Given these data, we next hypothesized that intratumoral NRP1SURF+ Tregs would be more suppressive than matched NRP1SURF− Tregs. We observed increased suppression by NRP1SURF+ Tregs from either HNSCC or OvCa tumors compared to NRP1SURF− Tregs (P = 0.01164 for HNSCC; Fig. 3, A and B) (40). When performed in the presence of an NRP1-specific blocking antibody, the functional advantage of OvCa TIL NRP1SURF+ Tregs is restrained (P = 0.05075; Fig. 3B). The antibody used was specifically designed to bind the SEMA4A binding domain of NRP1 (domain b1; patent US20190119389A1), which blocks the key interaction shown to maintain Treg stability (21). Nonetheless, this remains an unexpected finding as NRP1SURF+ Tregs are enriched for expression of numerous function- and stability-associated proteins. Furthermore, we observed that the proportion of NRP1SURF+ Tregs in OvCa tumors was correlated with the prevalence of CD45RA− CD8+ T cells, which are enriched for antigen-experienced cells (Fig. 3, C and D, and fig. S9, F and G) (41). This trend was not observed in HNSCC (Fig. 3C).
Fig. 3. NRP1 is associated with increased suppressive function.
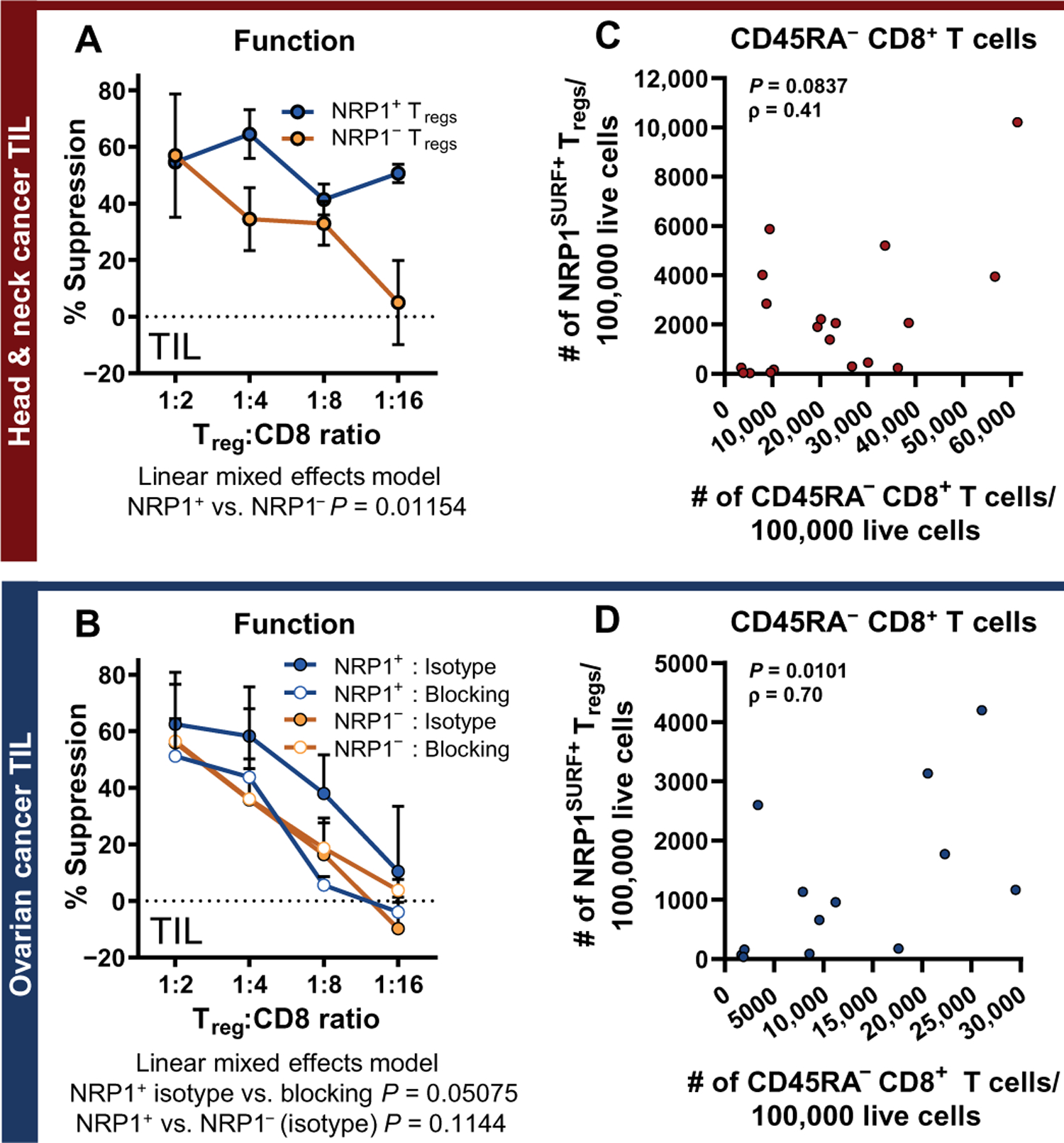
(A and B) Microsuppression assay comparing NRP1+ and NRP1− Tregs isolated from the same tumors are shown. Data are presented as means + SEM. Data are pooled from multiple experiments [three experiments for (A); four experiments for the NRP1SURF− group in (B) and two experiments for the NRP1SURF+ group in (B)]. Significance was analyzed by comparing the linear mixed effects model for the dataset to a null model via likelihood ratio test. A targeted NRP1 antagonist (10 μg/ml) was compared to the isotype control and reduced the suppressive function of OvCa intratumoral NRP1+ Tregs (P = 0.05075). (C and D) Nonparametric (Spearman’s) correlation is shown for the intratumoral count of NRP1SURF+ Tregs with the intratumoral count of CD45RA− CD8+ T cells (both normalized to 100,000 live cells) in samples of HNSCC (n = 19) and OvCa (n = 13), respectively.
T cell activation signals in the TME drive surface expression of NRP1 on Tregs
Because NRP1SURF expression is critical for a suppressive program in intratumoral Tregs in human cancer, we next investigated what signals drive and maintain its expression. Our prior observation that Tregs residing in noncancer site–matched tissues have lower NRP1SURF expression than tumors at the same site suggests that unique signals in the TME may govern NRP1SURF expression (Fig. 1E and fig. S2, B and C). Given the importance of T cell receptor (TCR) stimulation in Treg function and stability and the difference in neoantigen load in tumors versus normal tissues, we hypothesized that T cell activation initiates NRP1 expression in the TME. In vitro TCR stimulation of patient PBL Treg with interleukin-2 (IL-2) resulted in marked NRP1SURF up-regulation after 6 days but minimal change after 3 days (Fig. 4A). Increased NRP1TOT expression was also observed by flow cytometry with similar kinetics (fig. S10A). Furthermore, NRP1SURF up-regulation is a late activation event compared to other activation-associated receptors, such as GITR, or inhibitory receptors, such as PD-1 and TIGIT, which are expressed after 3 days of stimulation (fig. S10B).
Fig. 4. T cell activation signals in the TME drive Treg NRP1 expression.
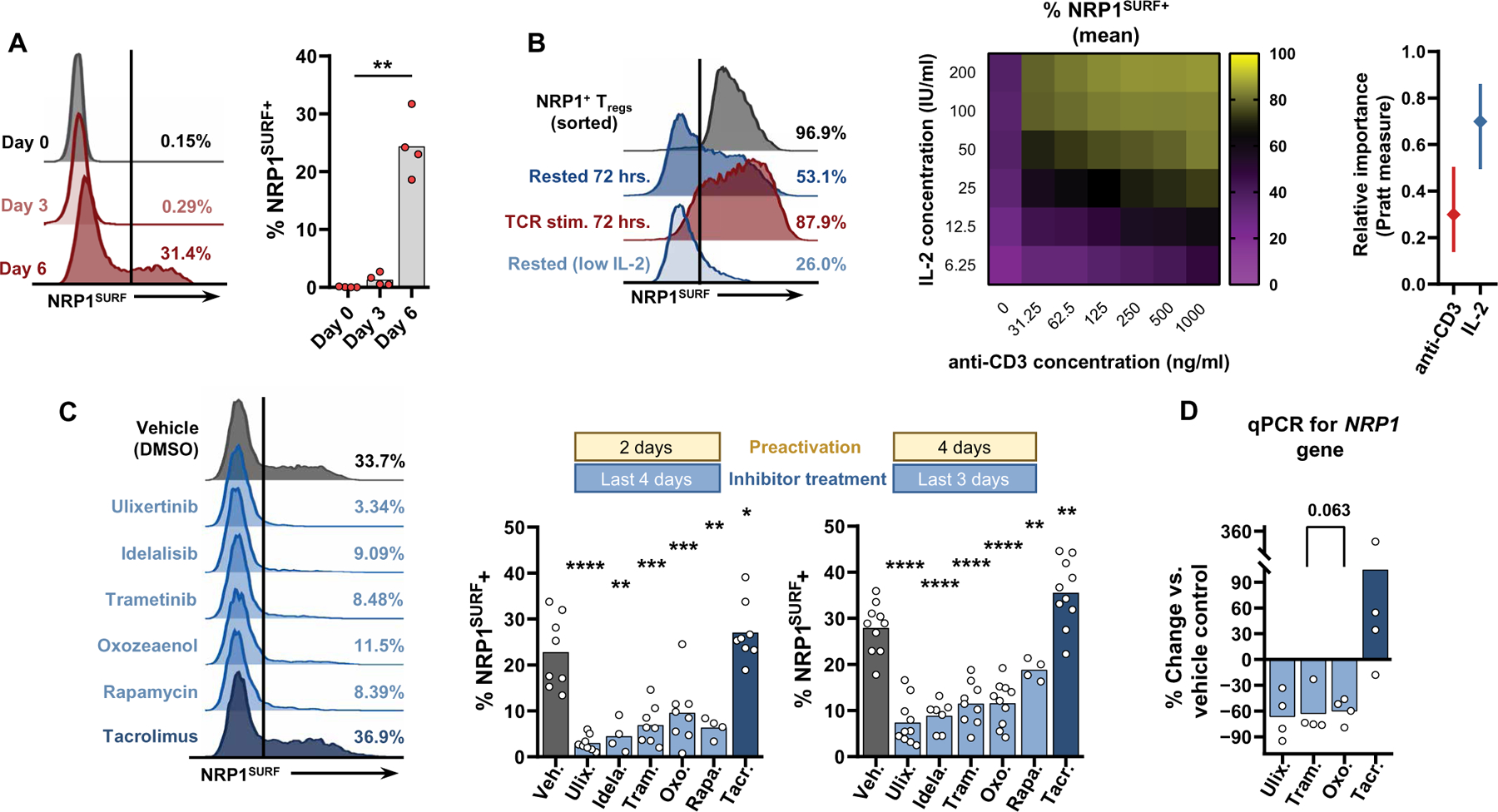
(A) In vitro stimulation of HNSCC peripheral Tregs with plated anti-CD3 (0.5 μg/ml), soluble anti-CD28 (1 μg/ml), and IL-2 (200 IU/ml) induce NRP1SURF expression after 6 days of stimulation. Stimulation duration was tested by one-way ANOVA. Data are from one experiment because all samples were thawed and processed simultaneously. Bars indicate the mean of expression. N = 4. **P < 0.01. (B) HD lymphocytes were processed, and Tregs were immediately selected and stimulated, as described above. After a 6-day in vitro stimulation, NRP1SURF+ Tregs were isolated by fluorescence-activated cell sorting and cultured at the indicated conditions for 72 hours. Heatmap displays the mean values from four independent experiments. The relative importance of IL-2 or anti-CD3 was calculated using a multiparameter linear regression analysis with bootstrapping (1000 replicates) to generate 95% CIs. The Pratt measure is the product of the regression coefficient and the zero-order correlation for each predictor. (C) HD Tregs were stimulated in vitro as described above. After either 2 or 4 days, the cells were redistributed to 20,000 per well where they were restimulated (as previously) in the presence of the indicated inhibitors. The secondary stimulation lasted for 4 or 3 days, respectively, before analysis. NRP1SURF expression data across 4 to 10 independent experiments are tabulated. Results were compared using a mixed effects model, with an FDR of 5% and corrected for differences in sphericity with the Geisser-Greenhouse correction. Thus, treatment effects were evaluated, paired to the subject’s vehicle control expression. Bars indicate the mean of expression. P values adjusted for multiple comparisons are reported as q values; *q < 0.05, **q < 0.01, ***q < 0.001, and ****q < 0.0001. (D) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) for NRP1 gene expression within in vitro inhibitor-treated Tregs is shown. Values are reported as the percent change versus the vehicle-treated (DMSO) condition. The paired CT values of the inhibitor conditions versus the sample vehicle control were compared by a nonparametric one-way ANOVA with 5% FDR for the four independent experiments.
A remaining question was whether NRP1SURF marks a stable lineage of human Tregs or a transient activation state. After stimulating HD PBL Tregs for 6 days to drive NRP1SURF expression, NRP1SURF+ Tregs were isolated by fluorescence-activated cell sorting (FACS) and cultured in the presence of IL-2 without continued stimulation (Fig. 4B). After 72 hours of rest, about half of the Tregs down-regulated NRP1SURF. However, if stimulation was maintained after purification, then nearly all cells retained NRP1SURF.
We also found that, when the concentration of IL-2 during rest was decreased 32-fold (notated as “low IL-2”), the proportion of NRP1SURF+ Tregs was further diminished (45% average reduction; Fig. 4B). A full titration of both TCR stimulation (via plated anti-CD3 density) and IL-2 further confirmed that both signals participate in sustaining NRP1SURF expression after initial activation (Fig. 4B and fig. S10, C to G). A clear dose-response was observed for decreasing IL-2 concentrations across all anti-CD3 concentrations; however, Treg expression of NRP1SURF was highly sensitive to a low concentration of anti-CD3 when sufficient IL-2 was present (fig. S10D). Multiparameter linear regression suggests that IL-2 concentration is twice as important as TCR stimulation for maintaining NRP1SURF expression on Tregs (Fig. 4B). Similarly, FOXP3 expression was heavily dependent on IL-2 concentration when minimal anti-CD3 was present (fig. S10G). In contrast, the persistence of inhibitory receptors PD-1 and TIGIT was more dependent on anti-CD3 density within the range tested (fig. S10E). We also observed that ex vivo intratumoral NRP1SURF+ Tregs expressed higher phosphorylated signal transducer and activator of transcription 5 (pSTAT5) and phosphorylated extracellular signal–related kinase 1/2 [pERK1/2; also known as mitogen-activated protein kinase 3 (MAPK3)] relative to NRP1SURF− Tregs (fig. S10, H and I). This supports the notion that both signals contribute to NRP1 expression in cancer. Furthermore, pSTAT5 expression was correlated with CD25 expression in these intratumoral Tregs (fig. S10H).
Next, we evaluated various pathways of the TCR signaling cascade to determine whether they influenced NRP1SURF expression on stimulated Tregs. We used a panel of well-characterized small-molecule inhibitors that target various TCR signaling kinases. Three of the most potent inhibitors of NRP1SURF expression were ulixertinib (BVD-523), trametinib, and 5Z-7-oxozeaenol, which inhibit ERK1/2, MAPK2 kinases 1/2 (MEK1/2; also known as MAP2K1/2), and ERK2, respectively (Fig. 4C). 5Z-7-Oxozeaenol also has inhibitory function against transforming growth factor–β–activated kinase 1 (TAK1; also known as MAP3K7). These agents restricted NRP1SURF up-regulation by 60 to 80% compared to the vehicle control group [dimethyl sulfoxide (DMSO)], providing strong evidence that the MAPK pathway was participating in NRP1SURF up-regulation. Our observation was further supported by the use of other redundant MAPK pathway targeting agents that also decreased NRP1SURF expression (fig. S11A). Reverse transcription quantitative polymerase chain reaction (RT-qPCR) after inhibitor treatment suggests that a large proportion of the regulation of NRP1 was at the level of gene transcription because the observed decrease in NRP1 gene transcript of 60 to 70% largely matches what was observed by flow (R = 0.88, P < 0.001; Fig. 4D and fig. S11B). Similarly, no consistent changes in the FOXP3 gene were observed (fig. S11C).
Idelalisib, a phosphoinositide 3-kinase (PI3K)–δ inhibitor, and rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, also reduced NRP1SURF expression; however, these pathways are known to be broadly important for Treg identity, survival, and function (42–46). Thus, they may affect a broader activation program in Tregs, as we saw greater reductions in Ki67, GITR, and 4–1BB with these agents, particularly idelalisib (fig. S11, A and D). Because these kinases also participate in the regulation and function of AKT, it is possible that their effect on NRP1 expression is partially due to AKT-mediated FOXP3 dysregulation and Treg instability (42). Conversely, treatment with tacrolimus, a clinically applied calcineurin inhibitor, generated a modest increase in NRP1SURF protein and gene expression. We did not see an effect on NRP1 regulation with inhibitors targeting p38 MAPK, cyclic adenosine monophosphate response element–binding protein (CREB), or ribosomal s6 kinases 1/2/3 (RSK1/2/3) (SB202190, 666–15, and LJH685, respectively; fig. S11, A and D). We observed comparable effects on NRP1SURF expression with all inhibitors tested with a reduced treatment duration (Fig. 4C and fig. S11D), providing evidence that the MAPK/ERK pathway participates in NRP1SURF regulation on Tregs, whereas nuclear factor of activated T cells is in opposition.
Circulating NRP1SURF+ Tregs may serve as a prognostic marker of disease burden
We aimed to elucidate whether the proportion of NRP1+ Tregs in PBL could be a useful indicator of patients who may benefit most from NRP1-directed immunotherapy. Unlike the current standard-of-care immunotherapy targets, such as PD-1 and cytotoxic T lymphocyte–associated protein 4, NRP1 is elevated and detectable in patient PBL and correlated with intratumoral expression (Fig. 1, B and D). Specifically, patients with OvCa had a significant enrichment of NRP1SURF+ PBL Tregs, in contrast to patients with benign ovarian lesions (P < 0.01; fig. S12A). Thus, systemic NRP1 expression on Tregs may have clinical utility.
We found that NRP1SURF+ peripheral Tregs in HNSCC are enriched for the expression of activation markers, including ICOS and GITR (Fig. 5A), compared to matched peripheral NRP1SURF− Tregs. Furthermore, in HNSCC PBL, NRP1TOT+ Tregs and NRP1TOT+SURF+ Tregs have a more activated phenotype compared to NRP1− Tregs (Fig. 5A and fig. S12, B and C). Using HD PBL Treg NRP1 expression to define a meaningful cutoff (fig. S12D), we grouped patients with HNSCC by their peripheral expression of NRP1 and found that those with a high proportion of NRP1SURF+ Tregs trended toward decreased PFS and decreased DSS at 3 years (PFS, P = 0.0939, HR = 2.3; Fig. 5B and fig. S12E). We next determined whether a high prevalence of NRP1SURF+ Tregs in both blood and tumor compartments resulted in worse outcome than one alone. We previously observed that the NRP1SURF expression was correlated between TIL and PBL in HNSCC (Fig. 1D) and found that our patient grouping based on PBL NRP1SURF+ Treg prevalence was significantly associated with TIL prevalence (P < 0.001; Fig. 5C). Almost all patients with HNSCC (12 of 13) with a high proportion of peripheral NRP1SURF+ Tregs also had NRP1SURF expression above the median intratumoral value (Fig. 5C), reinforcing the point that PBL NRP1SURF expression reflects tumor expression for a subset of patients. The rate of recurrence, median tumor burden, and median lines of therapy for disease control was higher in those individuals who had a high proportion of NRP1SURF+ Tregs in both tumor and blood samples compared to high abundance of NRP1SURF+ Tregs in the tumor alone (Fig. 5, C and D). Compared to all other patients, those with a high prevalence of NRP1SURF+ Tregs in both PBL and TIL had worse PFS (P = 0.0152; Fig. 5E and fig. S12F). The P value calculated for the comparison of PFS between all three groups from Fig. 5D did not fall below the significance level of 0.05 due to the decreased power from the fewer patients per group (log-rank test between all three groups yields P = 0.0593; Forest plot of risk for each group is also shown; fig. S12, G and H). Like in the tumor, NRP1TOT expression in the PBL did not discriminate between groups with differing clinical outcome (fig. S12, I and J). Assessment of NRP1SURF as a blood marker may identify a subset of patients with poor prognosis that could be ideal candidates from NRP1-directed immunotherapy (fig. S13).
Fig. 5. Circulating NRP1SURF+ Tregs may serve as a prognostic indicator.
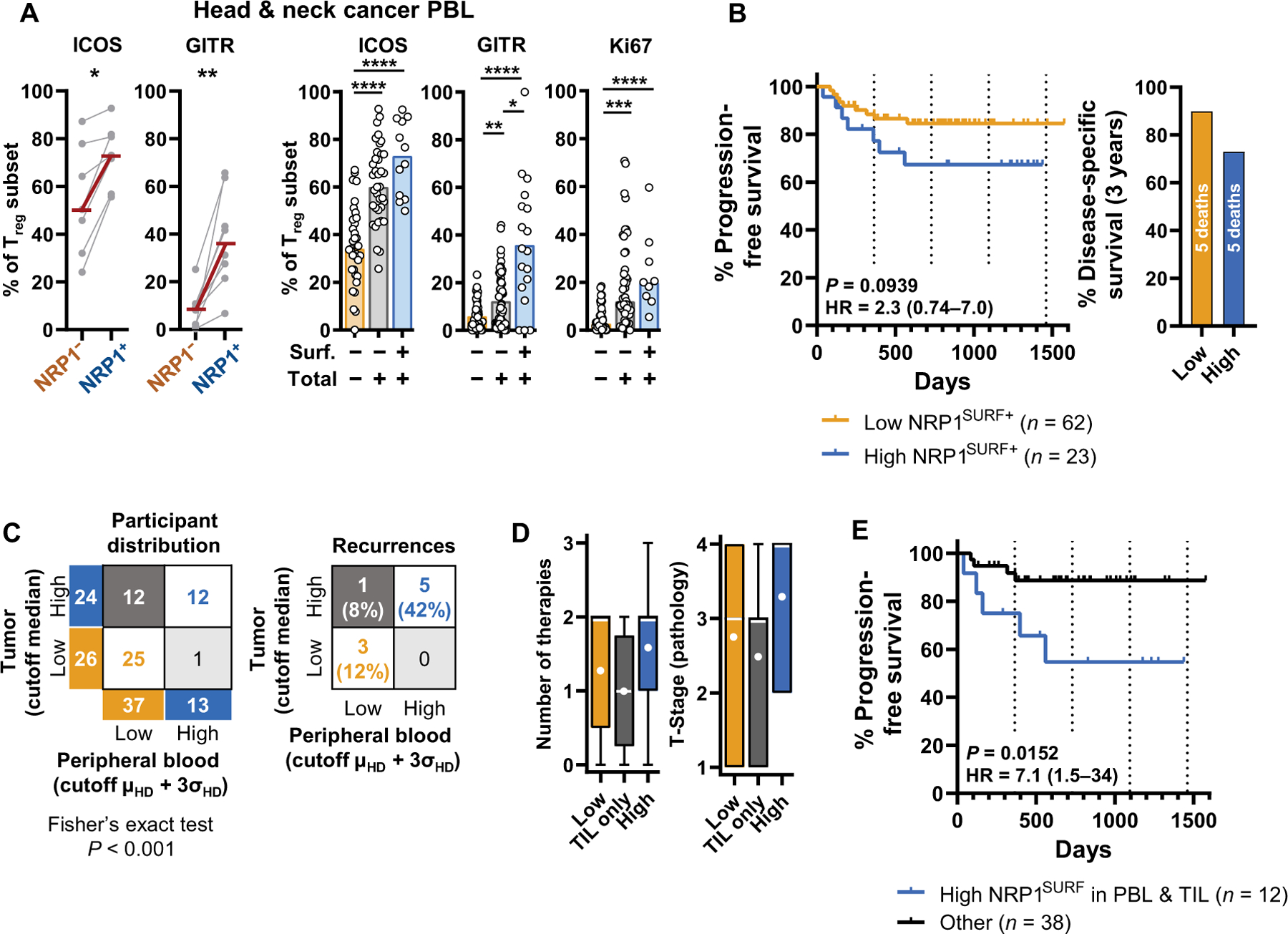
(A) Expression of ICOS, GITR, and Ki67 in peripheral blood NRP1SURF+ Tregs isolated from patients with HNSCC is shown. Paired nonparametric t test (Wilcoxon; n = 7 and 8, respectively) and nonparametric Kruskal-Wallis test (the number of samples ranges from 9 to 63 depending on the marker and subgroup) were used. Bars indicate the median of expression. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (B) Kaplan-Meier curve for individuals grouped by Treg NRP1SURF in PBL as a predictor of PFS is shown. Curve comparison was conducted using the log-rank Mantel-Cox test. P value with the HR ± 95% CI is reported. Disease-specific survival for the same groups after 3 years of follow-up is shown on the right. (C) Concordance of assignment based on peripheral or intratumoral NRP1SURF expression is tabulated according to the groups previously described [HNSCC TIL cohort median for intratumoral Tregs; HD mean (μHD) plus 3 SDs (σHD) for PBL Tregs] and tested by Fisher’s exact test. Relative recurrence rates of corresponding groups are reported to the right. (D) Box plots show the number of lines of therapy (range 0 to 3) and pathology-defined T-stage; Tumor-stage (range, 1 to 4) for the three groups defined in (C): high NRP1SURF in both tumor and blood (blue; right), tumor alone (gray; middle), and low NRP1SURF (orange; left). These measures may correlate with disease burden. The white line indicates the median; the white dot indicates the mean. (E) A Kaplan-Meier curve for PFS is shown comparing high in both the TIL and PBL (blue) versus all other patients (black). Curve comparison was conducted using a log-rank Mantel-Cox test.
DISCUSSION
The consistent enrichment of NRP1+ Tregs across six tumor types and the comparable NRP1-associated phenotypes in HNSCC and OvCa suggest that NRP1 may play a key role for Tregs in multiple human cancers. Furthermore, in some indications, the prevalence of NRP1SURF+ Tregs was associated with worse patient outcomes and advanced disease. Our study deepens what is known about NRP1 biology on human Tregs by tabulating NRP1 expression in the tumor and blood of across a broad population of patients with cancer and providing comprehensive examination of the suppressive program of NRP1-expressing Tregs. In this way, our work lays a foundation for future clinical evaluation of this putative modulator of Treg function and stability.
The profile of Treg NRP1 expression across human tissue samples paired with our mechanistic in vitro analysis of human NRP1 regulation highlights the advantages of this marker for clinical development. A key concern with Treg-directed immunotherapy is the off-tumor toxicity due to a broad inhibition of Treg function or depletion, especially at sites of environmental interface such as the gastrointestinal tract. Although NRP1 identifies thymic-derived Tregs in mice (47), human Tregs do not express high NRP1SURF in the absence of activation or in noncancer tissues. Because NRP1SURF expression is a late marker of human Treg cell activation and is only expressed by a subset of intratumoral Tregs, NRP1 antagonism may preferentially affect the most suppressive Tregs in the TME whose presence is also positively correlated with CD45RA− CD8+ T cell infiltration. This strategy could prove sufficient to permit effective antitumor immunity without compromising systemic immune homeostasis.
Many groups have set out to find intratumoral Treg functional markers that could offer utility as a therapeutic target or biomarker (38, 48). These studies did not identify NRP1 as a key target; however, this is likely due to the approach taken. Whereas these studies took an unbiased, transcriptomic view to identify intratumoral-enriched, Treg-specific molecules, we focused on NRP1 due to our prior studies in murine cancer models (21, 24). Although NRP1 expression on Tregs is unique to the TME in our study, the degree of expression is highly variable between patients. With a limited study cohort, NRP1 may not have been expressed consistently enough to warrant comparison to other molecules. Furthermore, NRP1 is not fully Treg specific but rather can be expressed at lower abundance on other T cell subsets (49). NRP1 has also been shown to be expressed highly on myeloid cells, some tumor cells, and tumor-associated vasculature (50–52). In addition, although NRP1SURF was not a continuous predictor of outcome, grouping individuals by high and low NRP1SURF+ Treg prevalence yielded insight. Interpreted with a biological perspective, in view of the highly skewed distribution of NRP1 expression, this observation may indicate that a threshold of NRP1SURF+ Tregs must be achieved to exert sufficient suppressive impact on the TME.
Our finding that TCR signals and IL-2 exposure both contribute to NRP1SURF regulation may explain why there is broad interpatient heterogeneity of Treg NRP1 expression. This also may explain the difference in the proportion of NRP1+ Tregs between different cancers, such as HNSCC versus PDAC. In general, we observed a higher proportion of NRP1SURF+ Tregs in more immunogenic cancers, whereas PDAC and CRC (all of which were confirmed MSS) had a lower median proportion of NRP1SURF+ Tregs. Differences in driver mutations, tumor mutational burden, and HLA expression between these tumors likely alter T cell–mediated tumor surveillance by modulating tumor antigen prevalence and consequently affect NRP1SURF+ expression on Tregs. Similarly, the minimal NRP1 expression observed in noncancer, site-matched tissues could be because of the paucity of neoantigens to stimulate T cell responses. For some individuals, NRP1SURF up-regulation on Tregs may be a regulatory mechanism to attenuate productive immunity, because the same signals that drive CD8+ T cell function also drove Treg NRP1SURF expression. We observe a correlation between antigen-experienced CD45RA− CD8+ T cells and NRP1SURF+ Tregs in tumors. Given the dependence on TCR activation, it remains to be seen whether NRP1SURF+ Tregs in human tumors comprise a clonal, tumor-reactive population whose TCRs have high affinity for tumor-associated antigens. Treg TCR affinity for altered self-neoepitopes in cancer cells is likely higher than for normal self-antigens because Treg generation requires escaping negative thymic selection. Thus, NRP1SURF may identify Tregs with tumor-specific TCRs, which are likely the most potent suppressors in the TME.
Our observation that NRP1SURF relies more on IL-2 concentration for maintained expression, akin to FOXP3, which is known to be a direct target of IL-2–mediated STAT5 activation, is the first indication in human Tregs that STAT5 participates in NRP1 regulation. This may be achieved either by directly interacting with the NRP1 gene locus, which has already been demonstrated in human cancer cells (53), or by indirect mechanisms through FOXP3. As IL-2 alone does not drive NRP1 expression, STAT5 signaling may contribute to NRP1 expression by enforcing chromatin accessibility of the NRP1 gene locus (54). Given the dependence of FOXP3 expression on IL-2 and STAT5, we cannot determine in our system whether NRP1 expression was lost in low IL-2 conditions directly from reduced pSTAT5 or from the instability of Treg identity via decreased FOXP3. The dependence on IL-2 concentration to maintain NRP1SURF expression may explain why Tregs preferentially express NRP1SURF in the TME. Tregs have a competitive advantage to consume IL-2 because of their constitutive CD25 expression. This presumably enables a higher fraction of them to retain NRP1 expression compared to other intratumoral T cell subsets. We observed increased pSTAT5 and CD25 expression in NRP1SURF+ Tregs.
The mechanism by which NRP1 expression is regulated on Tregs is a key finding because of the stark difference between NRP1 on mouse and human Tregs. Our findings clearly implicate the MAPK/ERK and PI3K/AKT/mTOR pathways in contributing to human Treg NRP1 regulation. Both sets of pathways can have broad impacts on T cell survival and activation (42, 43, 46, 55), and there are numerous downstream molecules that may also participate. Although our data exclude p38, CREB, and RSK, alternative potential targets include JunB, Atf2, Elk1, cFOS, c-Myc, Erythroblast Transformation Specific family (ETS), and others (55–57). Furthermore, the delayed kinetics of NRP1 up-regulation relative to other markers suggests that NRP1 may not participate in tuning initial Treg cell activation programs but rather reinforces the activated state under chronic stimulation. Future studies to elucidate the regulatory circuitry may reveal therapeutic opportunities to disrupt intratumoral Treg function. Immuno-monitoring in ongoing clinical trials using the inhibitors tested could provide further insight as well. In particular, ulixertinib and trametinib are under evaluation for MAPK-mutant or B-Rapidly Accelerated Fibrosarcoma (BRAF)-mutant advanced solid tumors, respectively (58, 59), whereas idelalisib is effective for treating numerous hematological malignancies including relapsed chronic lymphocytic leukemia in combination with rituximab (60).
Our observation of the concordant expression of NRP1 with numerous TNFRSFs suggests that these proteins may be regulated by overlapping mechanisms. This is supported by the fact that the same inhibitors that perturbed NRP1 expression also had modest effects on GITR and 4–1BB expression. In addition, GITR and 4–1BB persistence after stimulation had dual dependency on TCR and IL-2 similar to NRP1SURF. The role of TNFRSFs on intratumoral Tregs remains controversial, with numerous studies in mouse models suggesting that TNFRSF agonism destabilizes Tregs (61–63); however, this is likely at least in part due to Treg depletion in these models because Tregs express the highest abundance of these proteins (61, 64). Similar to NRP1, mouse Tregs constitutively express TNFRSFs, whereas human Tregs only express them upon activation (except for TNFR2). In the context of human cancer, recent work demonstrated that intratumoral 4–1BB+ Tregs are highly suppressive in human tumors and associated with poor outcomes (65, 66). Our study supports the notion that TNFRSF+ Tregs are highly suppressive, because their expression is enriched in the NRP1SURF+ compartment. Nonetheless, we postulate that NRP1 may be more central to intratumoral Treg suppressive capacity as NRP1 blockade mitigated the NRP1-associated suppressive advantage despite high TNFRSF expression.
Although we did not observe an association between NRP1TOT expression on Tregs and outcome, we hypothesize that it accelerates the up-regulation of NRP1SURF on Tregs with T cell activation. We observed not only higher NRP1TOT in samples from patients with cancer but also a trend toward increased expression in inflamed, noncancer tissue. In addition, whereas NRP1TOT+SURF− had statistically distinct phenotypes from NRP1SURF+ Tregs in the TME, they tended to be more similar in PBL. This may indicate that these Tregs retain a portion of the NRP1-associated phenotype after tumor egress despite NRP1SURF down-regulation. Further work is needed to determine whether this phenomenon contributes to disease.
Informed development and evaluation of NRP1 antagonists may provide therapeutic benefit for cancer. These drugs would have a broad applicability across cancers with low risk for toxicity based on our preclinical observations. However, note that Treg expression of NRP1 did vary between the cohorts we examined, and thus, careful consideration of disease indices is warranted to maximize therapeutic opportunity. NRP1 has additional functions in the TME that may prove additive to its effect on Treg suppressive function (22). Within the immune compartment, NRP1 has been implicated in mediating Treg recruitment to the TME in a vascular endothelial growth factor–dependent manner (67), supporting tolerogenic myeloid cell functions (68–70), and restricting CD8+ T cell antitumor memory (49). NRP1 also directly supports tumor cell growth, metastatic potential, and associated neoangiogenesis (52, 71, 72). Moreover, a major goal in immuno-oncology is the identification of reliable prognostic biomarkers to inform therapeutic decision-making. Accessibility of tissue for follow-up is a limitation of current immunohistochemical approaches. In this light, NRP1 is a unique target because NRP1SURF can be readily detected on peripheral Tregs in a subset of individuals, who may have the greatest need for aggressive treatment options. One may also reason that these individuals would be ideal candidates for an NRP1-targetting therapeutic, although this remains to be demonstrated. As a predictive marker, detection of NRP1SURF+ Tregs in patient PBL has strong specificity, but strategies to overcome its modest sensitivity would greatly increase the utility of this readout. Our report is timely given that an anti-NRP1 monoclonal antibody designed to inhibit Treg function by binding the SEMA4A binding domain of NRP1 is under clinical assessment in combination with nivolumab for patients with advanced solid tumors (NCT03565445). Furthermore, the NRP1 antagonist that we tested is a preclinical version of the same antibody clone. Our results support the biological rationale of this trial and suggest that monitoring Treg responses after treatment may reveal the dominant mechanisms of action for this agent in patients.
Our current study has several limitations that are also opportunities for future research. First, interpretation of our clinical analysis is constrained by the number of individuals accrued and median follow-up time. Because disease control was robust in our HNSCC cohort, the paucity of progression events over the follow-up duration limits the effect size. Similarly, we did not have power to determine an unbiased cut point and rather selected the median NRP1SURF value. Additional larger-scale cohorts are necessary to support our exploratory findings. Second, our study is unable to discern which aspects of the NRP1-associated Treg phenotype are causally determined by NRP1 expression and signaling. Although NRP1-mediated signaling is expected to be largely conserved between mice and humans, because there is >95% homology in the NRP1 cytoplasmic domain, these experiments have not been undertaken in human Tregs to date. Similarly, our study did not identify specific transcription factors that induce or suppress NRP1 expression; however, these questions warrant further study. Third, another shortcoming was the limited number of biological replicates with which we were able to perform Treg functional assays. Nonetheless, note that our efforts represent a step beyond what has previously been undertaken for NRP1+ Treg suppression assays because we isolated NRP1SURF+ Tregs from primary tumors and used a blocking antibody that is currently under clinical evaluation. In contrast, prior studies isolated Tregs from patient lymph nodes, had low NRP1+ purity, or did not fractionate the Treg population by NRP1 expression or did not use multiple biological replicates (23, 31).
In conclusion, the efficacy of single-agent immunotherapy is limited by checkpoint-independent mechanisms of immune suppression, such as immunosuppressive cell populations including Tregs. Given the absence of effective agents for diminishing Treg function in cancer, investigating targets such as NRP1 is of great importance. Our study suggests that NRP1 may be a prominent feature of intratumoral Treg phenotypes in some patients with cancer or in particular cancer types, raising the possibility that NRP1 blockade may complement ICB by abrogating Treg-mediated counter-regulation of inflamed tumors.
MATERIALS AND METHODS
Study design
Our study first sought to accumulate sufficient samples from patients with cancer to determine whether NRP1 expression in these patients differed from healthy individuals. We also sought to determine whether NRP1 expression was associated with differential Treg phenotypes and functional capabilities in patients with cancer. Last, we sought to determine the mechanisms regulating NRP1 expression in human patients. Measurements of these parameters, including the readouts of out functional in vitro assay and inhibitor screen, were made using flow cytometry because it readily provides expression data at a per-cell level.
On the basis of prior data (24), our power analysis suggested that 10 patient samples per cohort would achieve a power of 0.90 at an alpha of 0.05 to detect differences in the prevalence of NRP1+ Tregs. We accumulated this number of samples for all patient cohorts. Our inclusion criteria were patients with primary cancer at the indicated sites with our exclusion criteria being prior treatment for the primary cancer of interest or existing recurrent cancer. We accumulated additional samples in the study time that met our inclusion and exclusion criteria to accommodate subsequent phenotyping and clinical analyses. All data were included and reported without the exclusion of outliers. Similar power analyses for our phenotyping analysis and driving experiments suggested that 16 and 4 samples per group would be sufficient to achieve a power of 0.80 at an alpha of 0.05.
All tumor tissues were acquired under the University of Pittsburgh Cancer Institute Institutional Review Board (IRB)–approved protocols (HNSCC: 99–069; Mel: 96–099; OvCa: PRO15100457; lung: STUDY19060269; CRC: PRO19070174; PDAC: STUDY20040316), with written informed consent obtained from each patient in conjunction with the University of Pittsburgh Cancer Institute HNSCC, Melanoma, and Lung Cancer Specialized Programs of Research Excellence (SPOREs). Noncancer tissue specimens for tonsil, ovary, and lung samples were collected on the same IRB-approved protocols as the respective tumor samples.
Tumor samples were obtained at the time of surgery from consecutive immunotherapy-naïve patients undergoing surgical resection for treatment and collected into RPMI 1640 with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Peripheral blood (PBL) was drawn by venipuncture into tubes with EDTA anticoagulant. There were no restrictions on cancer subtype, smoking status, age, or race. Control donor PBL was collected through an approved material transfer agreement protocol with Vitalant. Mel tumor specimens were obtained from viably cryopreserved, banked samples and thawed immediately before analysis. This was the only cohort with treatment history before tumor biopsy (or peripheral blood) collection. For all samples, we were blinded to patient information and clinical characteristics through sample processing, data acquisition, and data analysis. An honest broker was designated for each patient cohort, per IRB guidelines, and maintained access to the protected patient information.
For our outcome studies, samples from our patient cohorts were analyzed at the time of surgical resection and followed prospectively for outcome. We requested deidentified clinical characteristics for HNSCC and OvCa cohorts from the designated honest broker after the completion of flow cytometric analysis. The summary of the patients evaluated can be found in table S1. Our intention was to conduct an exploratory investigation of the correlation between NRP1 expression and clinical outcome. All individuals with HNSCC had locally advanced disease at the time of surgical resection and had not received treatment for their disease. Patients with OvCa also had not received treatment for their disease before surgical resection.
We chose to perform an outcome-related analysis in our HNSCC cohort because it had the largest sample size. NRP1 expression in the intratumoral Treg compartment was first assessed as a continuous predictor of outcome using the Cox proportional hazards test. We validated that the proportional hazard assumption was met in this dataset. Although NRP1 was not a continuous predictor, we reasoned that the highly skewed biological distribution of NRP1 expression warranted investigation of NRP1 as a categorical predictor. We selected the median value of NRP1+ Treg prevalence as the cut point between “high” and “low” NRP1 expression groups for both NRP1SURF and NRP1TOT. We evaluated the resultant Kaplan-Meier curves using the log-rank test. Lost to follow-up and death from noncancer causes were censored events in both the progression-free and disease-specific survival analyses. Competing risk event analysis was performed but did not reveal any substantial insights. We evaluated the sensitivity of our cut point by also examining the curves when the cohort was split into tertiles on the basis of NRP1SURF expression.
Subsequently, we tested the potential for our findings to be confounded by other clinical characteristics. To address this, we performed iterative, univariate Cox proportional hazard tests for each covariate in relation to disease progression and reported the P value from the Wald test (likelihood ratio test results were also considered). For those parameters for which the P value was less than 0.05 for either the Wald or likelihood ratio test, we evaluated whether there was an association between that characteristic and NRP1+ Treg prevalence. We did not find that NRP1 expression was substantially enriched with any of the covariates, suggesting that our observations for NRP1 as a potential discrete predictor of response are not meaningfully affected by an association with another factor.
The same steps as above were performed for our OvCa tumor dataset and for our HNSCC peripheral blood dataset. For the latter, instead of using the median, the cut point was defined as the HD PBL mean plus 3 SDs so that the cut point was set to more than 99.85% of the HD distribution, assuming a normal distribution. This would thus serve as an unbiased way to discriminate those individuals whose PBL Tregs had substantially increased NRP1SURF+ Treg prevalence compared to HDs. We also tested the cutoff points of 1 and 2 SDs above the mean, which only added three and zero individuals to the NRP1SURF+-high group, respectively, and did not alter our findings. All other analyses were the same.
Tissue processing
PBLs were isolated from the whole blood (both from HDs and patients with cancer) by density gradient centrifugation in Ficoll-Paque (GE Healthcare) for 20 min at 400g with the brake off. Residual red blood cells were lysed with BD Pharm Lyse. Subsequent centrifugation cycles at 140g were used to purify the cell suspension. Peripheral blood mononuclear cells (PBMCs) were rested at 37°C before later use (from 0 to 24 hours).
Tissue specimens (both tumor and noncancerous tissue) were subjected to a tissue-specific processing protocol that included antibiotic wash, mechanical digestion, and enzymatic digestion (for select tissue sites). Tissues from mucosal sites (head and neck and lung) were incubated with antibiotic-supplemented media (1% penicillin, 1% streptomycin, and 1% amphotericin B) for 30 min before processing. After removing the transportation media, tissues were weighed and mechanically digested using scalpels (achieving tissue pieces of 1 to 3 mm in diameter). For ovarian, lung, pancreatic, and colorectal tissues, an enzymatic digestion using Liberase DL (50 μg/ml) (Roche), a combination of type I/II collagenase, was performed for 15 min at 37°C in 5 ml of serum-free media. Enzymatic digestion was stopped by washing the tissue with complete RPMI 1640 (supplemented with 10% FBS, nonessential amino acids, 2 mM l-glutamine, 1% penicillin/streptomycin, and 1 mM sodium pyruvate). Tissue pieces were transferred to a 100-μm filter and further mechanically digested. Tissues were repeatedly washed with media to ensure complete collection of infiltrating cells. Cell suspensions were centrifuged at 400g for 5 min and treated with BD Pharm Lyse to remove any residual red blood cells. Tissue-infiltrating cells were used immediately after processing, resting for no longer than 4 hours at 37°C. If experiments were not conducted on the same day as the surgery, then undigested tissue specimens were incubated at 4°C overnight (no more than 16 hours).
Flow cytometry
Cells were stained across multiple panels with the antibodies listed below. Staining buffer was composed of phosphate-buffered saline (PBS) with 0.1% azide, 10 mM Hepes, and 2% FBS. Staining for surface antigens was performed first for 25 min at 4°C (all antibody dilutions 1:100), followed by viability staining using a 1:4000 solution of fixable dye [eFluor (eF) 780, Invitrogen eBioscience] for 15 min at 4°C. Cells for analysis were fixed and permeabilized using the FOXP3 transcription factor staining buffer set (eBiosience) overnight. The next day, intracellular staining was conducted at room temperature for 25 min. Cells were washed and stored in staining buffer before analysis. Samples were acquired using a BD LSRFortessa II flow cytometer, and data were analyzed using FlowJo V10. Analysis of Treg phenotypes and subpopulation phenotypes was not performed whether the cell count was less than 50 recorded cells within the analysis gate. This required recording between 5 × 105 and 1 × 107 events depending on sample viability and cell population prevalence.
Antibodies and reagents included the following: CD304 (NRP1)–phycoerythrin (PE) clone 12C2 (BioLegend), CD304 (NRP1; intracellular)–Alexa Fluor (AF) 647 clone EPR3113 (used at 1:300 dilution; Abcam), CellTrace Violet (CTV) (Invitrogen), Fixable Viability Dye eF780 (Invitrogen eBioscience), 4–1BB–peridinin-chlorophyll-protein (PerCP)–eF710 clone 4B4 (eBioscience), Bcl2-AF488 clone 100 (BioLegend), CCR8–Brilliant Violet (BV) 421 clone 433H (BD Biosciences), CD3-AF700 clone SP34–2 (BD Bioscience), CD4-PerCP/Cy5.5 clone RPA-T4 (BioLegend), CD4–Brilliant Ultraviolet 395 clone RPA-T4 (BD Biosciences), CD8-AF700 clone RPA-T8 (eBioscience), CD8-BV421 clone RPA-T8 (BD Biosciences), CD8-BV711 clone RPA-T8 (BioLegend), CD14-BV785 clone M5E2 (BioLegend), CD19-PE/Cy7 clone HIB19 (BioLegend), CD25-BV650 clone BC96 (BioLegend), CD45-BV510 clone H130 (BD Biosciences), CD45RA-BV510 clone H1100 (BioLegend), CD69-BV785 clone FN50 (BioLegend), CD120b (TNFR2)–PE/Cy7 clone 3G7A02 (BioLegend), CD127-allophycocyanin clone A019D5 (BioLegend), FOXP3-eF450 clone PCH101 (eBioscience), GITR-PE/Cy7 clone 108–17 (BioLegend), GITR-AF488 clone 108–17 (BioLegend), Helios-PE/Dazzle clone 22F6 (BioLegend), ICOS-BV785 clone C398.4A (BioLegend), Ki67-BV711 clone Ki-67 (BioLegend), OX40-BV510 clone Ber-ACT35 (BioLegend), PD-1–PE/Cy7 clone eBioJ105 (eBioscience), pSTAT5-PerCP-Cy5.5 clone 47/Stat5 (pY694) (BD Bioscience), pERK1/2-PE/Cy7 clone 6B8B69 (BioLegend), TIGIT-PE/Dazzle clone A15153G (BioLegend), and SEMA4A-PerCP/eF710 clone 5E3 (eBioscience).
Treg isolation
Tregs were isolated from PBMC or tissue cell suspensions either by bead selection (STEMCELL) or FACS. FACS was performed by staining with CD4 (clone RPA-T4), CD25 (clone BC96), and CD127 (clone A019D5) in staining buffer (as described above), where Tregs were identified as CD4+, CD25HI, and CD127DIM. NRP1SURF+ Tregs were isolated by further discrimination of NRP1 expression (clone 12C2). FACS was performed using either Beckman Coulter MoFlo Astrios or Sony MA-900 machine operated by the University of Pittsburgh Medical Center (UPMC) Hillman Cancer Center Cytometry Facility. If Tregs were being isolated from cryopreserved specimens, then lymphocytes were thawed and rested overnight (12 to 16 hours) in IL-2 (R&D Systems; 200 IU/ml) before Treg selection and stimulation.
Microsuppression assays and NRP1 blockade
HD PBMCs were isolated from whole blood. Naïve CD8+ T cells were isolated by bead selection (STEMCELL), whereas antigen-presenting cells (APCs) were collected by FACS (viable, SSCHI, and SEMA4A+). Tregs were collected as described above. The isolated naïve CD8+ T cells were labeled with 5 μM CTV dye per the manufacturer’s instructions to track their proliferation (Invitrogen).
A serial twofold dilution of Tregs was prepared across five rows of a round-bottom 96-well plate (Costar), where the Treg count was 1000 cells in the first well. Two thousand APCs, 2000 CTV-labeled naïve CD8+ T cells, and anti-CD3 (0.5 μg/ml) (eBioscience, clone OKT3) were added to all wells. Thus, the Treg:CD8+ T cell ratio extends from 1:2 to 1:64. A no-proliferation control composed of labeled CD8+ T cells alone was included, as was a maximum proliferation control composed of only naïve CD8+ T cells and APCs with anti-CD3 (0.5 μg/ml). The cells were cocultured at 37°C for 5 days with 5% CO2, after which they were harvested and stained for analysis by flow cytometry. Experiments were always run with technical duplicates. For NRP1 blockade experiments, NRP1-directed antibody (10 μg/ml) (PTZ21722, Potenza Therapeutics) or an IgG2a isotype control antibody was added to the coculture. Cell preparation, counts, and culture conditions remained unchanged otherwise.
Data were analyzed for significance by comparing a linear mixed effect model for the dataset to a null model using the likelihood ratio test. The lmer function was used in RStudio to generate the mixed effects model–treating treatment (isotype or NRP1 blockade), Treg phenotype (NRP1+ or NRP1−), and the CD8:Treg ratio as multilevel factors and patient ID as a random effect. Thus, the model accounts for differences across the dataset on a per-patient basis and is not biased by examination at a single ratio or data point.
T cell culture and stimulation
Isolated primary T cells were cultured in complete RPMI 1640 (cRPMI 1640; described above) at 37°C. For stimulation experiments, agonistic anti-CD3 (eBioscience, clone OKT3) was adhered to the plate well in PBS (0.5 μg/ml; incubated for 1 hour at 37°C), after which cells were cultured in cRPMI 1640 supplemented by anti-CD28 (1.0 μg/ml) (eBioscience, clone CD28.2) and recombinant human IL-2 (200 IU/ml) (PeproTech) at 37°C. In addition, as indicated, concentrations of anti-CD3 or IL-2 were serially diluted across a 96-well plate before culturing isolated Tregs. When cultured in round-bottom 96-well plates, Tregs were plated at a concentration of 1 × 105 cells/ml in 200 μl of media.
RT-qPCR for NRP1 expression
Cells from HNSCC PBL were isolated by FACS (APCs) or bead selection (Tregs) as described above. Activated Tregs were stimulated 4 to 5 days before isolation. Between 30,000 and 1 × 106 cells were collected per sample. Cell pellets were frozen at −80°C until all samples were accrued. For the RT-qPCR after inhibitor treatment, samples were collected from in vitro–treated cultures at day 7 and the following procedure was started immediately. Samples were lysed and RNA was harvested (QIAGEN RNeasy kit) per the manufacturer’s instructions. After reverse transcription, NRP1 gene amplification was executed with the primers listed below and the SYBR Green system (Thermo Fisher Scientific) with 100 nM primers and diluted cDNA templates. Samples were run for 40 cycles at 95°C for 20 s and 60°C for 60 s. All samples were run in triplicate. Human RPLP0 was used as a housekeeping expression control. Quantification was performed by the ∆∆Ct method. NRP1, AAGGTTTCTCAGCAAACTACAGTG (forward) and GGGAAGAAGCTGTGATCTGGTC (reverse); FOXP3, GAAACAGCACATTCCCAGAGTTC (forward) and ATGGCCCAGCGGATGAG (reverse); and RPLP0, TAAACCCTGCGTGGCAATC (forward) and TTGTCTGCTCCCACAATGAAA (reverse).
Inhibition of T cell activation signals
Isolated primary Tregs were stimulated as described above for either 2 or 4 days. After the prestimulation, cells were divided into anti-CD3–precoated, round-bottom, 96-well plates at a concentration of 1 × 105 cells/ml in 100 μl of cRPMI 1640. One hundred microliters of 2× solution of the indicated inhibitors was added to the cells. This solution was prepared by diluting the stock in DMSO to be 200× the working concentration and subsequently diluted 100-fold in cRPMI 1640. Thus, the concentration of DMSO in all conditions (including vehicle control) was 0.5% (v/v). Cells remained in stimulation for an additional 4 or 3 days, respectively (according to initial stimulation) at 37°C. Cell phenotypes were recorded by flow cytometry after the initial stimulation to ensure viability. At the assay end point, multiplexed flow cytometry was used to characterize the cell phenotypes. Reagents used are as follows (with used concentration): ulixertinib [2.5 μM; also known as BVD-523, Chemical Abstracts Service (CAS) no. 869886–67-9), trametinib (10 nM; also known as GSK1120212, CAS no. 871700–17-3], 5Z-7-oxozeaenol (400 nM; CAS no. 253863–19-3), TPCA-1 (90 nM, CAS no. 507475–17-4), idelalisib (12.5 μM; CAS no. 870281–82-6), rapamycin (0.5 nM; CAS no. 53123–88-9), binimetinib (60 nM; also known as MEK162, CAS no. 606143–89-9), FR 180204 (2.55 μM; CAS no. 865362–74-9), PD0325901 (1.65 nM; CAS no. 391210–10-9), SB202190 (500 nM; CAS no. 152121–30-7), 666–15 (405 nM; CAS no. 1433286–70-4), LJH685 (30 nM; CAS no. 1627710–50-2), and tacrolimus (1.5 μM; also known as FK506, CAS no. 104987–11-3).
Statistical analysis
Raw, individual-level data for any experiment, where n < 20, are presented in data file S1. Analysis conducted using GraphPad Prism v8.0.0 included unpaired Student’s t test (fig. S1D), nonparametric Kolmogorov-Smirnov t test (fig. S2C), paired nonparametric t test (Wilcoxon) (Fig. 2, A to D; Fig. 5A; fig. S7, B to E; fig. S9, C to E; fig. S10, H and I; and fig. S12B), one-sample two-tailed t test (fig. S9, A and B), nonparametric Kruskal-Wallis test (Fig. 1, B, C, and E; Fig. 2, E and F; Fig. 4C; Fig. 5A; fig. S2, B and D to I; fig. S3D; fig. S4, A and B; fig. S5, B and C; fig. S6, B and C; fig. S8, B and C; fig. S11, C and D; and fig. S12, A, B, E, and H), mixed effects model (using GraphPad Prism; Fig. 4C and fig. S11, A and B), one-way analysis of variance (ANOVA) (Fig. 4A and fig. S10, A and B), repeated measures one-way ANOVA (fig. S11, C and D), nonparametric correlation analysis (Spearman rho) (Fig. 1D; Fig. 2, C and D; Fig. 3, C and D; fig. S2, A and D to I; fig. S3, A and B; fig. S7, H and I; fig. S9G; fig. S10H; and fig. S11C), and Kaplan-Meier curve generation with log-rank tests (Fig. 1G; Fig. 5, B and E; fig. S4; fig. S5, B and C; fig. S6C; and fig. S12, E, F, and H). RStudio v1.1.423 was used for monovariate Cox proportional hazard models (Fig. 1F and figs. S5A and S6A), linear mixed effects model (using RStudio; Fig. 3, A and B), multiparameter linear regression (Fig. 4B and fig. S10, E to G), and Fisher’s exact test (Fig. 5C and fig. S12G). P values or q values (adjusted for multiple comparisons) with a two-sided type I error of less than 5% were considered significant. In all appropriate instances, P values were corrected for multiple comparisons using a false discovery rate of 5% and were noted in figure legends as q values. Monovariate Cox proportional hazard regression models were used to relate NRP1 expression profiles to outcomes. Cell frequency and expression profile correlations were assessed by Pearson’s correlation coefficient. Graphs show individual samples with the mean or median represented by a bar (as appropriate), with the number of individual experiments listed in the legend.
Supplementary Material
Acknowledgments:
We thank everyone in the Vignali Laboratory and Bruno Laboratory for all constructive comments and advice during this project; Y. Gong (University of Pittsburgh) for providing the signaling inhibitor compounds and helpful discussion; M. Meyer, P. Dascani, and B. Janjic from the UPMC Hillman Cancer Center Cytometry Facility for cell sorting; C. Sander, E. Rush, J. Ward, L. Dzubinski, S. Hostein, A. Gaither Davis, A. Laslavic, J. Hetrick, and C. Reeder for assistance with consenting patients and for providing deidentified clinical information; and L. Wang and N. Siripong of the University of Pittsburgh Clinical and Translational Science Institute for guidance with the clinical analysis. The NRP1 antibody (PTZ21722; previously developed by Potenza Therapeutics) is a property of Astellas Pharma Inc. and can be requested from them directly.
Funding:
This work was supported by the NIH [R01 CA203689 and P01 AI108545 (to D.A.A.V.); P50 CA121973-09 (to J.M.K.) and Career Development Award to T.C.B.; P50 CA159981 (to R.E.), Career Development Award to T.C.B., and Biospecimen CORE (to F.M.), with matching funds from the Hillman Family Foundation; Early Detection Research Network U01-CA152753 (to R.E.S.); P50 CA097190 (to R.L.F. and D.A.A.V.; project 3 Co-I); and F31 CA243168 (to C.A.C.)], the Foundation for Women’s Cancer (to T.C.B.), the Rivkin Center for Ovarian Cancer (to T.C.B.), Sponsored Research Agreements (SRAs) from Potenza Therapeutics and Astellas Pharma (to D.A.A.V.), a Hillman Postdoctoral Fellowship for Innovative Cancer Research (to A.R.C.), biostatistics consultation supported by grant number UL1-TR-001857 (University of Pittsburgh Clinical and Translational Science Institute), Sampson Endowed Chair in Thoracic Surgical Oncology at University of Pittsburgh (to A.P.), and this project used the UPMC Hillman Cancer Center and Tissue and Research Pathology/Pitt Biospecimen Core shared resource, which is supported in part by award P30CA047904.
Footnotes
Competing interests: D.A.A.V. declares competing financial interests and has submitted patents covering Nrp1 that are licensed or pending and is entitled to a share in net income generated from licensing of these patent rights for commercial development (therapies based on control of regulatory T cell stability and function via a neuropilin-1:semaphorin axis; application no. 16/438,696). DAAV: Cofounder and stock holder—Novasenta, Tizona, and Trishula; stock holder—Oncorus, Werewolf, and Apeximmune; patents licensed and royalties—Astellas and Bristol Myers Squibb; scientific advisory board member—Tizona, Werewolf, F-Star, Bicara, and Apeximmune; consultant—Astellas, Bristol Myers Squibb, Almirall, Incyte, and G1 Therapeutics; research funding—Bristol Myers Squibb, Astellas, and Novasenta. R.L.F. consults for or is on the scientific advisory board for Aduro Biotech Inc., AstraZeneca/MedImmune, Bristol-Myers Squibb, EMD Serono, GlaxoSmithKline, Iovance Biotherapeutics Inc., MacroGenics Inc., Merck, Nanobiotix, Numab Therapeutics AG, Oncorus Inc., Ono Pharmaceutical Co. Ltd., PPD (Benitec, Immunicum), Regeneron Pharmaceuticals Inc., Novasenta, and Torque Therapeutics Inc. R.L.F. receives research funding from AstraZeneca/MedImmune, Bristol Myers Squibb, Tesaro, and Novasenta and has stock from Novasenta. T.C.B. consults for or is on the scientific advisory board for Bespoke Biotherapeutics, iTeos Therapeutics, and Walking Fish Therapeutics. T.C.B. receives research support from Alkermes Inc. and Pfizer. R.E.S. receives research support from Freenome and Immunovia Inc. All other authors declare that they have no competing interests. J.L. is a stockholder in Johnson and Johnson, Intuitive Surgical, and Medtronics. J.L. is an inventor and patent holder for a method outlining a rapid, multiplex RT-PCR protocol (PCR method; patent # WO2002070751A1).
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Pauken KE, Wherry EJ, Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yarchoan M, Hopkins A, Jaffee EM, Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med 377, 2500–2501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Togashi Y, Shitara K, Nishikawa H, Regulatory T cells in cancer immunosuppression— Implications for anticancer therapy. Nat. Rev. Clin. Oncol 16, 356–371 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, Vonderheide RH, Pittet MJ, Jain RK, Zou W, Howcroft TK, Woodhouse EC, Weinberg RA, Krummel MF, Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI, Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol 166, 678–689 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S, Mechanisms of resistance to immune checkpoint blockade: Why does checkpoint inhibitor immunotherapy not work for all patients? Am. Soc. Clin. Oncol. Educ. Book 39, 147–164 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Liyanage UK, Moore TT, Joo H-G, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC, Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol 169, 2756–2761 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH, Regulatory CD4+CD25+ T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res 61, 4766–4772 (2001). [PubMed] [Google Scholar]
- 9.Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B, Increase of regulatory T cells in the peripheral blood of cancer patients. Clin. Cancer Res 9, 606–612 (2003). [PubMed] [Google Scholar]
- 10.Karube K, Ohshima K, Tsuchiya T, Yamaguchi T, Kawano R, Suzumiya J, Utsunomiya A, Harada M, Kikuchi M, Expression of FoxP3, a key molecule in CD4+CD25+ regulatory T cells, in adult T-cell leukaemia/lymphoma cells. Br. J. Haematol 126, 81–84 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Gray CP, Arosio P, Hersey P, Association of increased levels of heavy-chain ferritin with increased CD4+ CD25+ regulatory T-cell levels in patients with melanoma. Clin. Cancer Res 9, 2551–2559 (2003). [PubMed] [Google Scholar]
- 12.Ihara F, Sakurai D, Horinaka A, Makita Y, Fujikawa A, Sakurai T, Yamasaki K, Kunii N, Motohashi S, Nakayama T, Okamoto Y, CD45RA−Foxp3high regulatory T cells have a negative impact on the clinical outcome of head and neck squamous cell carcinoma. Cancer Immunol. Immunother 66, 1275–1285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W, Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Shang B, Liu Y, S.-j. Jiang, Y. Liu, Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep 5, 15179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boucek J, Mrkvan T, Chovanec M, Kuchar M, Betka J, Boucek V, Hladikova M, Betka J, Eckschlager T, Rihova B, Regulatory T cells and their prognostic value for patients with squamous cell carcinoma of the head and neck. J. Cell. Mol. Med 14, 426–433 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun W, Li W-J, Wu C-Y, Zhong H, Wen W-P, CD45RA-Foxp3high but not CD45RA+Foxp3low suppressive T regulatory cells increased in the peripheral circulation of patients with head and neck squamous cell carcinoma and correlated with tumor progression. J. Exp. Clin. Cancer Res 33, 35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vignali DAA, Collison LW, Workman CJ, How regulatory T cells work. Nat. Rev. Immunol 8, 523–532 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Bras S, Geha RS, IPEX and the role of Foxp3 in the development and function of human Tregs. J. Clin. Invest 116, 1473–1475 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akimova T, Zhang T, Negorev D, Singhal S, Stadanlick J, Rao A, Annunziata M, Levine MH, Beier UH, Diamond JM, Christie JD, Albelda SM, Eruslanov EB, Hancock WW, Human lung tumor FOXP3+ Tregs upregulate four “Treg-locking” transcription factors. JCI Insight 2, e94075 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Kostareli E, Suffner J, Garbi N, Hämmerling GJ, Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur. J. Immunol 40, 3325–3335 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Delgoffe GM, Woo S-R, Turnis ME, Gravano DM, Guy C, Overacre AE, Bettini ML, Vogel P, Finkelstein D, Bonnevier J, Workman CJ, Vignali DAA, Stability and function of regulatory T cells is maintained by a neuropilin-1–semaphorin-4a axis. Nature 501, 252–256 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chuckran CA, Liu C, Bruno TC, Workman CJ, Vignali DA, Neuropilin-1: A checkpoint target with unique implications for cancer immunology and immunotherapy. J. Immunother. Cancer 8, e000967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung K, Kim J-A, Kim Y-J, Lee H-W, Kim C-H, Haam S, Kim Y-S, A neuropilin-1 antagonist exerts anti-tumor immunity by inhibiting the suppressive function of intratumoral regulatory T cells. Cancer Immunol. Res 8, 46–56 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, Horne W, Moskovitz JM, Kolls JK, Sander C, Shuai Y, Normolle DP, Kirkwood JM, Ferris RL, Delgoffe GM, Bruno TC, Workman CJ, Vignali DAA, Interferon-γ drives Treg fragility to promote anti-tumor immunity. Cell 169, 1130–1141.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, Ruminski P, Weiss D, Von Schack D, Bluestone JA, Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med 209, 1713–1722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, Yang Y, Floess S, Huehn J, Oh S, Li MO, Niec RE, Rudensky AY, Dustin ML, Littman DR, Lafaille JJ, Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med 209, 1723–1742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh K, Hjort M, Thorvaldson L, Sandler S, Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naïve mice. Sci. Rep 5, 7767 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio M-T, Intyre EM, Lepelletier Y, Hermine O, Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur. J. Immunol 39, 1466–1471 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Battaglia A, Buzzonetti A, Baranello C, Ferrandina G, Martinelli E, Fanfani F, Scambia G, Fattorossi A, Metastatic tumour cells favour the generation of a tolerogenic milieu in tumour draining lymph node in patients with early cervical cancer. Cancer Immunol. Immunother 58, 1363–1373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battaglia A, Buzzonetti A, Martinelli E, Fanelli M, Petrillo M, Ferrandina G, Scambia G, Fattorossi A, Selective changes in the immune profile of tumor-draining lymph nodes after different neoadjuvant chemoradiation regimens for locally advanced cervical cancer. Int. J. Radiat. Oncol. Biol. Phys 76, 1546–1553 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Battaglia A, Buzzonetti A, Monego G, Peri L, Ferrandina G, Fanfani F, Scambia G, Fattorossi A, Neuropilin-1 expression identifies a subset of regulatory T cells in human lymph nodes that is modulated by preoperative chemoradiation therapy in cervical cancer. Immunology 123, 129–138 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, Martinelli E, Ciardiello F, Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev 76, 22–32 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Johnson BA III, Yarchoan M, Lee V, Laheru DA, Jaffee EM, Strategies for increasing pancreatic tumor immunogenicity. Clin. Cancer Res 23, 1656–1669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferris RL, Blumenschein G Jr., Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Iglesias Docampo LC, Haddad R, Rordorf T, Kiyota N, Tahara M, Monga M, Lynch M, Geese WJ, Kopit J, Shaw JW, Gillison ML, Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med 375, 1856–1867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, Sharpe AH, Quintana FJ, Mathis D, Benoist C, Hafler DA, Kuchroo VK, Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fourcade J, Sun Z, Chauvin J-M, Ka M, Davar D, Pagliano O, Wang H, Saada S, Menna C, Amin R, Sander C, Kirkwood JM, Korman AJ, Zarour HM, CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight 3, e121157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D-Y, Xiong X-Z, ICOS+ Tregs: A functional subset of Tregs in immune diseases. Front. Immunol 11, 2104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva Ekaterina EV, Dmitriy DMC, Rudensky Alexander AY, Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C, Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Collison LW, Vignali DAA, In vitro Treg suppression assays. Methods Mol. Biol 707, 21–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akbar AN, Terry L, Timms A, Beverley PC, Janossy G, Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells. J. Immunol 140, 2171–2178 (1988). [PubMed] [Google Scholar]
- 42.Pompura SL, Dominguez-Villar M, The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol 103, 1065–1076 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Chapman NM, Zeng H, Nguyen T-LM, Wang Y, Vogel P, Dhungana Y, Liu X, Neale G, Locasale JW, Chi H, mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat. Commun 9, 2095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ali K, Soond DR, Piñeiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B, Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510, 407–411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahmad S, Abu-Eid R, Shrimali R, Webb M, Verma V, Doroodchi A, Berrong Z, Samara R, Rodriguez PC, Mkrtichyan M, Khleif SN, Differential PI3Kδ signaling in CD4+ T-cell subsets enables selective targeting of T regulatory cells to enhance cancer immunotherapy. Cancer Res 77, 1892–1904 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Chellappa S, Kushekhar K, Munthe LA, Tjønnfjord GE, Aandahl EM, Okkenhaug K, Taskén K, The PI3K p110δ isoform inhibitor idelalisib preferentially inhibits human regulatory T cell function. J. Immunol 202, 1397–1405 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, von Boehmer H, Buer J, Hansen W, Neuropilin-1: A surface marker of regulatory T cells. Eur. J. Immunol 34, 623–630 (2004). [DOI] [PubMed] [Google Scholar]
- 48.De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJP, Provasi E, Sarnicola ML, Panzeri I, Moro M, Crosti M, Mazzara S, Vaira V, Bosari S, Palleschi A, Santambrogio L, Bovo G, Zucchini N, Totis M, Gianotti L, Cesana G, Perego RA, Maroni N, Ceretti AP, Opocher E, De Francesco R, Geginat J, Stunnenberg HG, Abrignani S, Pagani M, Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45, 1135–1147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C, Somasundaram A, Manne S, Gocher AM, Szymczak-Workman AL, Vignali KM, Scott EN, Normolle DP, John Wherry E, Lipson EJ, Ferris RL, Bruno TC, Workman CJ, Vignali DAA, Neuropilin-1 is a T cell memory checkpoint limiting long-term antitumor immunity. Nat. Immunol 21, 1010–1021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roy S, Bag AK, Singh RK, Talmadge JE, Batra SK, Datta K, Multifaceted role of neuropilins in the immune system: Potential targets for immunotherapy. Front. Immunol 8, 1228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M, Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92, 735–745 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Niland S, Eble JA, Neuropilins in the context of tumor vasculature. Int. J. Mol. Sci 20, 639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi F, Shang L, Pan B-Q, Wang X-M, Jiang Y-Y, Hao J-J, Zhang Y, Cai Y, Xu X, Zhan Q-M, Wang M-R, Calreticulin promotes migration and invasion of esophageal cancer cells by upregulating Neuropilin-1 expression via STAT5A. Clin. Cancer Res 20, 6153–6162 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Rawlings JS, Gatzka M, Thomas PG, Ihle JN, Chromatin condensation via the condensin II complex is required for peripheral T-cell quiescence. EMBO J 30, 263–276 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rincón M, Flavell RA, Davis RJ, Signal transduction by MAP kinases in T lymphocytes. Oncogene 20, 2490–2497 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Chang J-H, Hu H, Sun S-C, Survival and maintenance of regulatory T cells require the kinase TAK1. Cell. Mol. Immunol 12, 572–579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA, Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 17, 1263–1293 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, Patel MR, Shapiro GI, Mier JW, Tolcher AW, Wang-Gillam A, Sznol M, Flaherty K, Buchbinder E, Carvajal RD, Varghese AM, Lacouture ME, Ribas A, Patel SP, De Crescenzo GA, Emery CM, Groover AL, Saha S, Varterasian M, Welsch DJ, Hyman DM, Li BT, First-in-class ERK1/2 inhibitor Ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov 8, 184–195 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Subbiah V, Lassen U, Élez E, Italiano A, Curigliano G, Javle M, de Braud F, Prager GW, Greil R, Stein A, Fasolo A, Schellens JHM, Wen PY, Viele K, Boran AD, Gasal E, Burgess P, Ilankumaran P, Wainberg ZA, Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 21, 1234–1243 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O’Brien SM, Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med 370, 997–1007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schoenhals JE, Cushman TR, Barsoumian HB, Li A, Cadena AP, Niknam S, Younes AI, da Silva Caetano M, Cortez MA, Welsh JW, Anti-glucocorticoid-induced tumor necrosis factor–related protein (GITR) therapy overcomes radiation-induced Treg immunosuppression and drives abscopal effects. Front. Immunol 9, 2170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schaer DA, Budhu S, Liu C, Bryson C, Malandro N, Cohen A, Zhong H, Yang X, Houghton AN, Merghoub T, Wolchok JD, GITR pathway activation abrogates tumor immune suppression through loss of regulatory T-cell lineage stability. Cancer Immunol. Res 1, 320–331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Piconese S, Valzasina B, Colombo MP, OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med 205, 825–839 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buchan SL, Dou L, Remer M, Booth SG, Dunn SN, Lai C, Semmrich M, Teige I, Mårtensson L, Penfold CA, Chan HTC, Willoughby JE, Mockridge CI, Dahal LN, Cleary KLS, James S, Rogel A, Kannisto P, Jernetz M, Williams EL, Healy E, Verbeek JS, Johnson PWM, Frendéus B, Cragg MS, Glennie MJ, Gray JC, Al-Shamkhani A, Beers SA, Antibodies to costimulatory receptor 4–1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 49, 958–970.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Freeman ZT, Nirschl TR, Hovelson DH, Johnston RJ, Engelhardt JJ, Selby MJ, Kochel CM, Lan RY, Zhai J, Ghasemzadeh A, Gupta A, Skaist AM, Wheelan SJ, Jiang H, Pearson AT, Snyder LA, Korman AJ, Tomlins SA, Yegnasubramanian S, Drake CG, A conserved intratumoral regulatory T cell signature identifies 4–1BB as a pan-cancer target. J. Clin. Invest 130, 1405–1416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toker A, Nguyen LT, Stone SC, Yang SYC, Katz SR, Shaw PA, Clarke BA, Ghazarian D, Al-Habeeb A, Easson A, Leong WL, McCready DR, Reedijk M, Guidos CJ, Pugh TJ, Bernardini MQ, Ohashi PS, Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin. Cancer Res 24, 5685–5696 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Hansen W, Hutzler M, Abel S, Alter C, Stockmann C, Kliche S, Albert J, Sparwasser T, Sakaguchi S, Westendorf AM, Schadendorf D, Buer J, Helfrich I, Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med 209, 2001–2016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M, Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24, 695–709 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Miyauchi JT, Caponegro MD, Chen D, Choi MK, Li M, Tsirka SE, Deletion of Neuropilin 1 from microglia or bone marrow–derived macrophages slows glioma progression. Cancer Res 78, 685–694 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wallerius M, Wallmann T, Bartish M, Östling J, Mezheyeuski A, Tobin NP, Nygren E, Pangigadde P, Pellegrini P, Squadrito ML, Pontén F, Hartman J, Bergh J, De Milito A, De Palma M, Östman A, Andersson J, Rolny C, Guidance molecule SEMA3A restricts tumor growth by differentially regulating the proliferation of tumor-associated macrophages. Cancer Res 76, 3166–3178 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Rizzolio S, Tamagnone L, Multifaceted role of neuropilins in cancer. Curr. Med. Chem 18, 3563–3575 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Jia H, Cheng L, Tickner M, Bagherzadeh A, Selwood D, Zachary I, Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br. J. Cancer 102, 541–552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.