

ABSTRACT
Infections caused by the Gram-positive bacterium Staphylococcus aureus remain a significant health threat globally. The production of bicomponent pore-forming leukocidins plays an important role in S. aureus pathogenesis. Transcriptionally, these toxins are primarily regulated by the Sae and Agr regulatory systems. However, the posttranslational regulation of these toxins is largely unexplored. In particular, one of the leukocidins, LukAB, has been shown to be both secreted into the extracellular milieu and associated with the bacterial cell envelope. Here, we report that a major cell wall hydrolase, autolysin (Atl), controls the sorting of LukAB from the cell envelope to the extracellular milieu, an effect independent of transcriptional regulation. By influencing the sorting of LukAB, Atl modulates S. aureus cytotoxicity toward primary human neutrophils. Mechanistically, we found that the reduction in peptidoglycan cleavage and increased LukAB secretion in the atl mutant can be reversed through the supplementation of exogenous mutanolysin. Altogether, our study revealed that the cell wall hydrolase activity of Atl and the cleavage of peptidoglycan play an important role in controlling the sorting of S. aureus toxins during secretion.
KEYWORDS: LukAB, MRSA, Staphylococcus aureus, autolysin, cell envelope, cell wall, neutrophils, pore-forming toxins, protein secretion
INTRODUCTION
The Gram-positive bacterium Staphylococcus aureus is commonly found as a colonizer on the skin and mucosae of human host without causing disease (1, 2). Upon gaining access to deeper tissues, S. aureus can cause a wide array of diseases in humans, including skin and soft tissue infections, sepsis, endocarditis, pneumonia, and osteomyelitis (3, 4). The treatment of S. aureus infections is complicated by the emergence of antibiotic resistance. In fact, methicillin-resistant S. aureus (MRSA) is considered a serious threat by the U.S. Centers for Disease Control and Prevention (5).
The current epidemic MRSA lineage in the United States, USA300, is dominant in both community- and hospital-associated MRSA infections (6–9). The versatile lifestyle of USA300 is facilitated by a large repertoire of virulence factors (10). The cytotoxins produced by USA300 play a key role in pathogenesis by forming pores on the targeted host cells, leading to cell death (11). The bicomponent leukocidins are a family of cytotoxins that form hetero-octameric β-barrel pores comprising four pairs of two subunits, designated as slow (S) and fast (F) components based on their chromatography elution profile (12, 13). A single USA300 S. aureus strain can produce up to five leukocidins. These are Panton-Valentine leukocidin (LukSF-PV or PVL), γ-hemolysins HlgAB and HlgCB, leukocidin ED (LukED), and leukocidin AB (LukAB, also known as LukGH) (14). The sequence identity among PVL, HlgAB, HlgCB, and LukED is 60 to 80% within each S-subunit and F-subunit family, while LukAB only shares 30 to 40% sequence identity with the other leukocidins (15). Among these toxins, LukAB has been shown to be the major contributor in killing human phagocytes in tissue-culture infection models with primary human cells (16–20). LukAB targets human cells by recognizing CD11b and HVCN1 as receptors, which dictates its human specificity due to the dissimilarity of these receptors between humans and rodents (21, 22).
While leukocidins are secreted toxins, LukAB is found both secreted and associated with the bacterial cell (17, 18, 23). The sorting of LukAB follows a multistep process controlled by the cell envelope, resulting in differential deposition of the toxin on the bacterial cell or into the extracellular milieu, dependent on growth conditions (23). The cell envelope of S. aureus is composed of the cell membrane, the peptidoglycan cell wall, membrane-anchored lipoteichoic acid (LTA), and cell wall-anchored wall teichoic acid (WTA) (24). The cell membrane is composed of five major types of glycerolipids, phosphatidylglycerol (PG), diacylglycerol (DAG), lysyl-PG (LPG), diglucosyl-DAG (Glc2-DAG), and cardiolipin (25, 26). Previously, LPG and Glc2-DAG were shown to be important for sorting LukAB from the bacterial cell envelope to the extracellular milieu (23).
The process of cell wall synthesis and turnover in S. aureus is controlled by a series of enzymes. We previously identified autolysin (Atl), the major cell wall hydrolase in S. aureus, as an enzyme involved in the LukAB sorting process (23). Atl is produced as a preproprotein of ∼137 kDa that is composed of a signal peptide for general secretion (Sec)-dependent membrane translocation, a propeptide of unknown function, and two catalytically active domains, amidase (AM) and glucosaminidase (GL) (27). The AM domain is an N-acetylmuramyl-l-alanine amidase that cleaves between MurNAc in the peptidoglycan backbone and l-alanine in the stem peptide (28, 29). The GL domain is an exo-β-N-acetylglucosaminidase that cuts the glycan backbone, resulting in GlcNAc-MurNAc disaccharide units (30). An S. aureus atl mutant is characterized by defects in autolysis activity, daughter cell separation, and attachment to polymer surfaces (31–33).
Here, we characterized the role of Atl in the secretion of LukAB and determined the impact of Atl on the cytotoxicity of S. aureus toward primary human phagocytes. We show that the atl mutant exhibits increased cytotoxicity toward primary human polymorphonuclear leukocytes (PMNs), which is the result of increased secretion of LukAB. The regulation of LukAB secretion by Atl is independent of transcription, but it is attributed to the differential sorting of LukAB. We show that the increased secretion of LukAB in the atl mutant can be chemically complemented by the addition of mutanolysin, an enzyme that cleaves peptidoglycan and disaggregates the atl-deficient cells. These results indicate that the cell wall hydrolase activity of Atl controls the postmembrane sorting of a critical virulence factor in USA300 S. aureus.
RESULTS
Increased protein secretion in the atl mutant.
Through a genetic screen using a transposon insertion library of USA300 S. aureus, the Nebraska transposon mutant library (34), we noticed that the atl transposon mutant produced more LukAB in the culture supernatant than the parental USA300 strain (23). To confirm the results of the screen, we constructed a mutant where the full atl open reading frame (ORF) was deleted in USA300, and we examined the secretion of LukAB in this mutant by immunoblotting. Indeed, the atl mutant showed ∼50% increase in the level of LukA, a proxy for LukAB, in the culture supernatant compared to wild type (WT) (Fig. 1A). A mutant strain lacking all leukocidins and hla (Δtoxins) was included as a negative control (Fig. 1A). We took advantage of a previously reported plasmid expression system for complementation studies (33). The production of full-length Atl in the atl mutant background restored LukAB secretion to the WT level (Fig. 1B). However, production of the AM or GL domain alone failed to complement the LukAB secretion phenotype (Fig. 1B).
FIG 1.
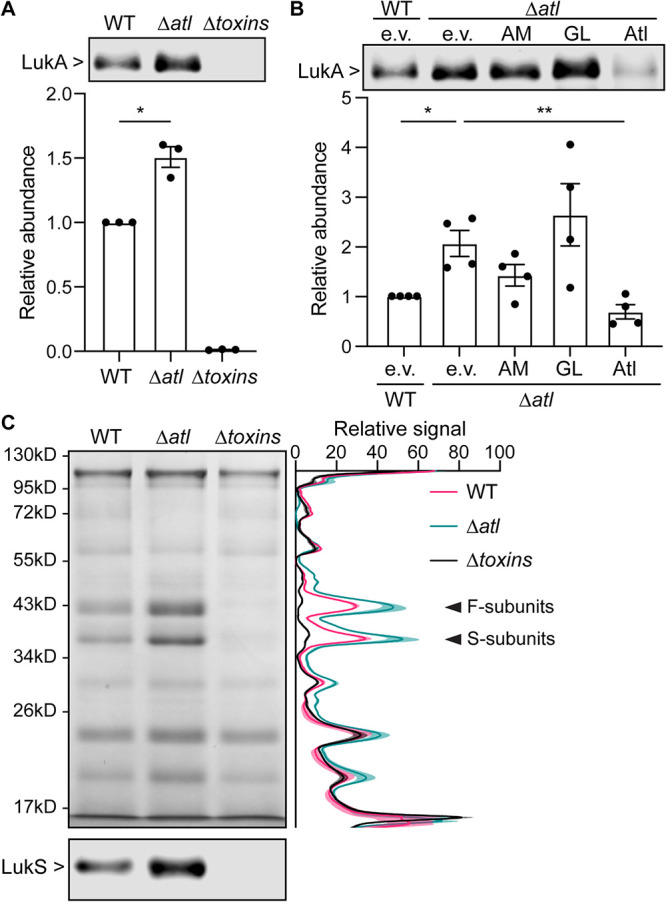
Atl influences the secretion of leukocidins. (A) Immunoblot of LukA in the culture supernatant of WT, the atl mutant, and a mutant strain depleted of all pore-forming toxins (Δtoxins). A representative immunoblot (top) and the mean ± SEM of the LukA signal from 3 independent experiments are shown (bottom). *, P ≤ 0.05 comparing WT and the atl mutant by a paired t test. (B) Immunoblot of LukA in the culture supernatant of WT containing empty vector (e.v.) and the atl mutant containing the indicated Atl domains or full-length Atl. A representative immunoblot (top) and the mean ± SEM of LukA signal from 4 independent experiments (bottom) are shown. *, P ≤ 0.05; **, P ≤ 0.01 by repeated-measures (RM) one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison tests. (C) Exoprotein profile as detected in an InstantBlue-stained SDS-PAGE (top) and immunoblot of leukocidin S subunits (bottom) of indicated strains. Signals of the InstantBlue-stained SDS-PAGE from 3 independent experiments were quantified and plotted on the right. Shaded area indicates SEM. The arrows point to the bands at the size of leukocidins. The immunoblot is a representative image of 3 independent experiments.
We next examined the role of Atl in protein secretion in USA300. The protein levels in the culture supernatant of early-stationary-phase bacteria were semiquantitatively analyzed by total protein staining. In general, the atl mutant secreted a greater amount of exoproteins than WT (Fig. 1C). The most significant difference was observed with the two bands corresponding to the S and F subunits of leukocidins (Fig. 1C, arrows). Because of the high sequence similarity between PVL, LukED, HlgAB, and HlgCB, we used a pan-leukocidin antibody to detect the S subunits of these leukocidins. The immunoblot showed that indeed the leukocidins were oversecreted in the atl mutant (Fig. 1C). Taken together, these results demonstrate that Atl negatively regulates the secretion of leukocidins.
Atl influences the virulence of USA300.
Leukocidins impair host immune responses by directly targeting and killing phagocytes (14, 20, 35, 36). We next sought to examine the virulence potential of the atl mutant using a tissue culture model of infection where primary human PMNs were infected with live USA300, and PMN lysis was measured by quantifying the release of the mammalian lactate dehydrogenase (LDH) enzyme. These studies revealed that the atl mutant lysed significantly more PMNs than WT USA300 (Fig. 2A). LukAB was responsible for the observed PMN killing by the atl mutant, as deletion of lukAB in the WT or atl mutant resulted in significantly reduced cell death (Fig. 2A).
FIG 2.
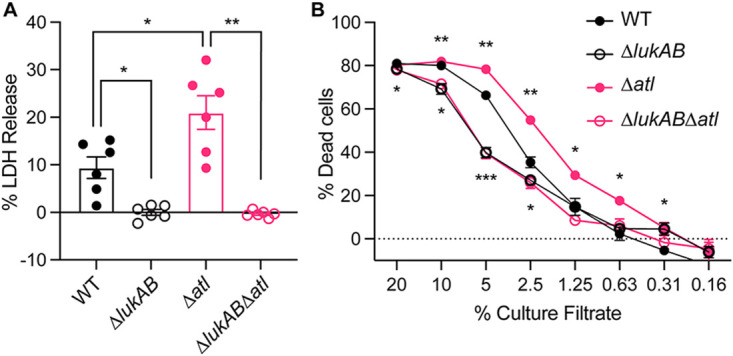
The killing of PMNs by WT and isogenic mutant strains. (A) Infection of PMNs with indicated strains for 2 h with an MOI of 10. PMN lysis was measured by LDH release. Data show mean ± SEM of PMNs isolated from 6 independent blood donors. *, P ≤ 0.05; **, P ≤ 0.01 by RM one-way ANOVA with Tukey’s multiple-comparison tests. (B) Intoxication of PMNs with cell-free supernatants from indicated strains. The PMN viability was measured by CellTiter. Data show mean ± SEM of PMNs isolated from 6 independent blood donors. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 compared to WT by RM one-way ANOVA with Dunnett’s multiple-comparison tests.
To examine if the increased LukAB secretion observed in an atl mutant contributes to the enhanced cytotoxicity of the atl mutant, we incubated primary human PMNs with different concentrations of bacteria-free culture supernatant from USA300 and then evaluated PMN viability by measuring cellular metabolic activity. Consistent with the infection results, the culture supernatant of the atl mutant was more cytotoxic than the WT (Fig. 2B). When lukAB was deleted from the atl mutant, this strain showed the same cytotoxicity as the lukAB isogenic mutant of WT (Fig. 2B), indicating that secreted LukAB is responsible for the increased virulence of atl in this model.
Atl does not affect the transcription of leukocidin genes.
Next, we investigated how the atl mutation leads to increased secretion of leukocidins. We first examined whether the atl mutation has any transcriptional effect on the leukocidins by measuring the activities of leukocidin promoters in the WT strain and the atl isogenic mutant. We chose to evaluate the promoters of lukAB and pvl, as these two toxins are abundantly produced and their promoter activities are comparable under in vitro culture conditions (37, 38). We measured promoter activities at both the exponential (3 h) and early stationary phase (4.5 h), as the transcriptional regulation may occur prior to the protein secretion. At both time points, the promoter activities of lukAB and pvl remained the same in the atl mutant as the WT (Fig. 3A and B).
FIG 3.
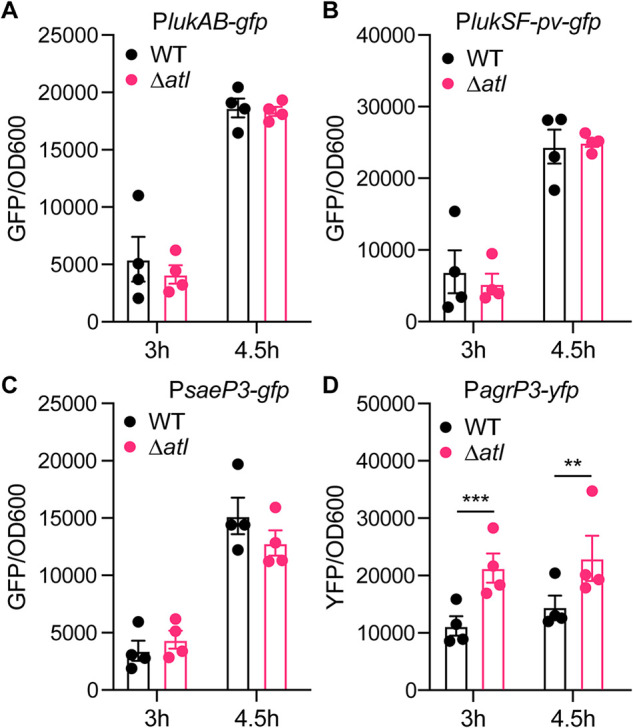
Promoter activities of leukocidins and regulators. Activities of the lukAB (A), lukSF-pv (pvl) (B), saeP3 (C), and agrP3 (D) promoters in the WT and atl mutant at the exponential (3 h) and early-stationary (4.5 h) phases. The indicated promoters were fused to gfp or yfp genes. The promoter activities were measured as fluorescence and normalized by OD600. Data show mean ± SEM from 4 independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.00 by two-way ANOVA with Sidak multiple-comparison test.
In addition to the leukocidins, we also observed an increased secretion of other exoproteins in the atl mutant (Fig. 1C). To further dissect the effect of Atl on the production of exoproteins, we measured the promoter activities of two master regulators of exoprotein gene expression, sae and agr (39, 40). The promoter activity of sae remained the same in the WT and atl mutant (Fig. 3C). However, the promoter activity of agr was increased in the atl mutant (Fig. 3D), potentially due to the increased local concentration of autoinducing peptides in the unseparated atl cell clusters. The increased agr activity may lead to greater production of Sae-independent exoproteins such as PSMs (41), which could also promote the release of cytoplasmic proteins (42). In conclusion, the observed increased secretion of leukocidins in the atl mutant is not the result of increased transcription of the leukocidin genes.
Atl controls the sorting of LukAB.
While the other leukocidins are primarily secreted into the extracellular milieu, LukAB is found both secreted and associated with the bacterial cell envelope (17, 18, 23). The secretion of leukocidins is thought to start with translocating across the cell membrane through the Sec pathway (43, 44). After being released from the cell membrane, LukAB can be retained in the cell envelope. The distribution of LukAB in different compartments of the cell envelope and its secretion into the culture supernatant are dependent on the bacterial growth phase (Fig. 4A) and the presence of LPG and Glc2-DAG (23). To evaluate if Atl also controls the sorting of LukAB, we examined the presence of LukAB associated with the bacterial cell versus in the culture supernatant at different growth phases (Fig. 4B). Whenever LukAB was detectable in the culture supernatant, more LukAB was found in the atl mutant than the WT. On the other hand, the levels of LukAB associated with the bacterial cell were significantly reduced in the atl mutant compared to WT (Fig. 4B). The reduction of the bacterial cell-associated LukAB was complemented by producing full-length Atl (Fig. 4C). Thus, Atl negatively regulates LukAB sorting from the cell envelope to the extracellular milieu.
FIG 4.

Sorting of LukAB in the atl mutant. (A) Diagram of LukAB localization with different compartments of the cell envelope. LukAB is secreted into the culture supernatant at the exponential and early-stationary phases but is absent in the supernatant at the late-stationary phase. LukAB can be found associated with the bacterial cells in all growth phases. The bacterial cell can be further separated into the membrane-proximal and surface-exposed compartments. Most LukAB is found in the membrane-proximal compartment at the exponential phase but in the surface-exposed compartment at the late-stationary phase. CM, cell membrane. CW, cell wall. (B) Representative immunoblots of LukA in the cultures from different growth times (top) and the quantification of the LukA signal in the culture supernatant or associated with the bacterial cell from the immunoblot results (bottom). The signal was normalized to 50 ng purified recombinant LukAB on each membrane. Data show mean ± SEM from 3 independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 by two-way ANOVA with Sidak multiple-comparison test. (C) Immunoblot of LukA associated with the bacterial cell in WT containing empty vector (e.v.), the atl mutant containing empty vector (e.v.), or plasmid-expressed full-length Atl. Figure shows a representative immunoblot from 3 independent experiments. (D) Representative immunoblots (top) and quantifications of the LukA signal (bottom) in the membrane-proximal or surface-exposed compartments associated with the bacterial cell. Relative abundance (R.A.) of LukA signal was normalized to 50 ng purified recombinant LukAB on each membrane. Data show mean ± SEM from 3 independent experiments. P values between WT and Δatl determined by two-tailed paired t tests as follows: membrane proximal, 0.04; surface exposed, 0.12.
The bacteria-associated LukAB can be further separated into the membrane-proximal and surface-exposed compartments (Fig. 4A). After membrane translocation, LukAB first accumulates in the membrane-proximal compartment before being sorted to the cell surface and extracellular milieu (23). We found that in the atl mutant, LukAB levels were reduced in both bacteria-associated compartments compared to WT (Fig. 4D), suggesting that the atl mutant is more efficient in releasing LukAB from the cell envelope into the extracellular milieu.
The LukAB sorting is dependent on the cell wall hydrolase activity.
Atl is a prominent cell wall hydrolase that controls cell wall turnover (30–32). As a result of insufficient cleavage of peptidoglycan, the atl mutant cells are connected by peptidoglycan threads, resulting in significant cell clusters (Fig. 5A) (30–32). Mutanolysin, a muramidase produced by Streptomyces globisporus, has been shown to be able to digest S. aureus peptidoglycan threads on the cell surface without causing cell lysis (31). Indeed, by culturing the atl mutant with mutanolysin, the cells were efficiently separated (Fig. 5A). Consistent with this, mutanolysin significantly reduced the size of cell clusters in the atl mutant, although the median cluster size of the mutanolysin-treated atl mutant remained slightly higher than the WT (Fig. 5B).
FIG 5.

Mutanolysin complements the phenotype of the atl mutant. (A) Light microscopy of WT and Δatl cells at the early stationary phase (4.5 h), treated with or without mutanolysin. The cell wall was stained with BODIPY FL vancomycin. Scale bar, 1 μm. (B) Area of the connected cells on a two-dimensional projection of WT and Δatl, treated with or without mutanolysin. Each dot is a cluster of cells. The line indicates the median. **, P ≤ 0.01; ****, P < 0.0001 by ordinary one-way ANOVA with Tukey’s multiple-comparison test. (C) Autolysis activity of WT and Δatl in 0.05% Triton X-100 in PBS, supplemented with or without mutanolysin. Data show mean ± SEM from 3 independent experiments. (D) Surface positive charge of WT and Δatl measured by cytochrome c binding to the cell surface. Data show mean ± SEM from 3 independent experiments. **, P ≤ 0.01 by RM one-way ANOVA with Tukey’s multiple-comparison test. (E) Representative immunoblots (top) and quantification (bottom) of LukA in the culture supernatant or bacterial cell lysate of WT and Δatl treated with or without mutanolysin. Bars show mean ± SEM from n = 4 (supernatant) or n = 7 (bacterial cell) independent experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001 by RM one-way ANOVA with Tukey’s multiple-comparison test.
The autolysis activity of USA300 depends on the presence of cell wall hydrolases. In the atl mutant, the Triton X-100-induced autolysis is disrupted (33). However, supplementation with mutanolysin restored the autolysis activity of the atl mutant (Fig. 5C). Last, we observed that the changes in peptidoglycan and cell clustering resulted in a reduction in the surface charge of the atl mutant, a property that was also reversed by mutanolysin (Fig. 5D). These data established that mutanolysin serves as an exogenous source of cell wall hydrolase that can restore the defects in cell wall turnover exhibited by the atl mutant.
To examine if the restoration of cell wall hydrolysis is sufficient to control LukAB sorting in the atl mutant, we examined the localization of LukAB in USA300 strains cultured with or without mutanolysin. We observed that when the atl mutant was treated with mutanolysin, the secretion and bacterial cell association of LukAB returned to WT levels (Fig. 5E). Thus, the multistep sorting of LukAB depends on proper cleavage of peptidoglycan.
DISCUSSION
In this study, we examined the contribution of a prominent cell wall hydrolase, Atl, to the sorting of LukAB. We show that in the atl mutant, increased amounts of LukAB, as well as other leukocidins, were released into the extracellular milieu. This altered toxin secretion is independent of transcriptional regulation. Instead, our data establish that Atl influences the protein sorting process, as less LukAB was found associated with the cell envelope. Using mutanolysin to complement the phenotype exhibited by the atl mutant, our data indicate that the cell wall hydrolase activity of Atl is the key regulator for sorting LukAB from the cell envelope to the extracellular milieu. As a result of enhanced sorting, deletion of atl in USA300 results in increased cytotoxicity toward primary human PMNs. Taken together, by studying the role of Atl in sorting LukAB, our study revealed that cell wall hydrolase activity is not only crucial for cell wall turnover but also for the sorting of toxins across the cell wall in S. aureus.
The expression of Atl is tightly regulated by multiple master regulators, such as negative regulators MgrA (45), ArlRS (46), LytSR (47), and SarA (48), as well as the positive regulators Agr (48) and CidAB (49). Posttranscriptionally, Atl is suppressed by the presence of WTA (50, 51). The complex regulatory system of Atl allows S. aureus to respond to different environmental conditions and adjust the rate of cell wall degradation and separation of dividing cells. In addition, the current study demonstrates that the release of virulence factors is also controlled by Atl.
We envision that the activity of Atl creates a depot for LukAB to be associated with the cell envelope (Fig. 6). Spatially, the primary structure of the cell wall of Gram-positive bacteria, peptidoglycan, is a single macromolecule with pores ranging from 6 nm in diameter at the internal surface to 39 nm in diameter at the external surface (52). The elasticity of the cell wall enables bacteria to resist high osmotic pressure, but the plasticity of the cell wall is what allows for cell wall turnover mediated by cell wall synthases and hydrolases in order for bacteria to grow (53). During plastic deformation, depots may be created in the cell envelope for proteins to reside. Meanwhile, active hydrolysis is more likely to generate reactive residues that allow noncovalent interactions such as hydrophobic or electrostatic interactions between LukAB and peptidoglycan fragments. Nevertheless, it is also possible that the Atl activity influences the level of other cell envelope structures that interact with LukAB in the cell envelope.
FIG 6.

The sorting of LukAB is dependent on Atl cell wall hydrolase activity. Model of how Atl may influence the sorting of LukAB. In WT, the activity of Atl creates depots for LukAB in the cell envelope by breaking down the peptidoglycan strands. In the atl mutant, such depots are scarce, and more LukAB is therefore sorted into the extracellular milieu. Created with BioRender.com.
Previously, Atl has been reported to modulate the expression and secretion of other proteins. In the atl mutant, multiple cell wall hydrolases are transcriptionally upregulated and are more abundant in the extracellular milieu, potentially compensating for the loss of atl (54). Atl is important for the excretion of a number of cytoplasmic proteins, while the transcription of corresponding genes remains unchanged (54). These proteins lack a signal peptide that allows them to be secreted through canonical pathways. Cell lysis, cell envelope modulation by Atl, and the generation of extracellular vesicles are thought to be the major routes for the release of these cytoplasmic proteins (55). Here, we established the role of Atl in coordinating the secretion of toxins in USA300 and showed that the absence of Atl leads to enhanced secretion of other exoproteins, including leukocidins. Interestingly, we observed an increased agr promoter activity but not the promoters of sae or leukocidins in the atl mutant. The transcription of lukAB and lukSF-PV is directly regulated by SaeR, while both sae and leukocidins are regulated by agr through RNAIII and Rot (56–59). The unsynchronized promoter activities may suggest additional regulatory factors involved in toxin expression.
The finding that mutanolysin reverts the phenotypes of the atl mutant suggests that the cleavage of peptidoglycan, but not Atl itself, influences the sorting of toxins. A previous study has reported that another cell wall hydrolase, SagB, is also important for the secretion of a subset of proteins, particular adhesins and superantigen-like proteins in S. aureus strain Newman (60). Although Atl contains both AM and GL domains, the AM domain of Atl contributed greater to the murein hydrolase activity (33), which is in agreement with our data that the AM domain showed a partial complementation of LukAB sorting (Fig. 1B). Instead, SagB is the major GL that determines the length of glycan chains in S. aureus (60, 61). It is yet unclear whether SagB influences the sorting of leukocidins in USA300. The accumulation of unprocessed peptidoglycan in the atl mutant versus the elongated glycan chains without cross-peptides in the sagB mutant may alter distinct aspects of the cell envelope and thus likely differentially contribute to the sorting of selective groups of exoproteins. Studying the contribution of different cell wall hydrolases to the sorting of exoproteins could provide a new avenue to understanding how Gram-positive bacteria transport proteins across the cell wall.
MATERIALS AND METHODS
Bacterial growth and culture conditions.
S. aureus strains, plasmids, and primers used in this study are listed in Table 1. S. aureus USA300 strain AH-LAC (62) was used in all experiments as the wild-type (WT) strain. Escherichia coli IM08B (63) was used for cloning and was grown in Luria-Bertani broth with 100 μg/mL ampicillin or 20 μg/mL chloramphenicol when appropriate. S. aureus was routinely grown at 37°C on tryptic soy agar (TSA). For plasmid selection, erythromycin was added at 5 μg/mL, and chloramphenicol was added at 10 μg/mL in the overnight culture or 5 μg/mL in the subculture. When appropriate, mutanolysin was supplemented at 10 U/mL in the subculture.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer identifier | Name | Description | Reference or source |
|---|---|---|---|
| Strains | |||
| VJT15.77 | AH-LAC (WT) | ErmS USA300 parent strain | 62 |
| VJT23.52 | ΔlukAB | AH-LAC with a clean deletion of the lukAB operon | 18 |
| VJT58.79 | Δtoxins | AH-LAC mutant strain lacking all bicomponent leukocidins and α-toxin | 71 |
| VJT80.33 | Δatl | AH-LAC carrying the tetM cassette in replacement of the atl gene | This study |
| VJT80.97 | ΔlukAB Δatl | AH-LAC ΔlukAB carrying the tetM cassette in replacement of the atl gene | This study |
| Plasmids | |||
| pOS1-PlukAB-gfp | lukAB promoter controlling expression of a superfolder GFP | This lab | |
| pOS1-Ppvl-gfp | lukSF-pv promoter controlling expression of a superfolder GFP | This lab | |
| pOS1-Psae-gfp | sae P1 promoter controlling expression of a superfolder GFP | 72 | |
| pDB59 PagrP3-yfp | rnaIII promoter controlling expression of YFP | 73 | |
| pJB128 (e.v.) | Insertless complement plasmid control (e.v.) | 33 | |
| pJB111 (AM) | Complement plasmid expressing the amidase domain of Atl (AM) | 33 | |
| pJB135 (GL) | Complement plasmid expressing the glucosaminidase domain of Atl (GL) | 33 | |
| pJB141 (Atl) | Complement plasmid expressing the full-length Atl (Atl) | 33 | |
| Primers | |||
| VJT1836 | tetM.F | GATTGTAAAATAACAAATATTGGTACATG | This study |
| VJT1837 | tetM.R | CAAAAGGTATCAATGAAGCAAGAAATATTG | This study |
| VJT3023 | atl-up.F | CTCTAGAACTAGTGGATCCCCCGGGattagctataaagatgattcacaac | This study |
| VJT3024 | atl-up.R | TACCAATATTTGTTATTTTACAATCtctatttattactcctaacatttattaattattac | This study |
| VJT3025 | atl-down.F | TTTCTTGCTTCATTGATACCTTTTGgcaacatgaacataggatcaaaag | This study |
| VJT3026 | atl-down.R | GCTGGGTACCGGGCCCCCCCTCGAGaataatctctctcttttaatgaagtc | This study |
S. aureus cultures were grown in 5 mL of medium in 15-mL tubes with shaking at a 45° angle at 37°C. S. aureus was grown overnight in tryptic soy broth (TSB), and a 1:100 dilution of overnight cultures was subcultured into TSB. Unless otherwise specified, S. aureus was grown to early stationary phase (4.5 h of subculture) and was collected and normalized by optical density at 600 nm (OD600) for further experimental analysis.
Generation of the atl mutant.
The Δatl mutant was generated by replacing the atl locus with tetM gene encoding tetracycline resistance using the pIMAY* allelic exchange system (64). The primers are listed in Table 1. The Δatl mutation was transduced into ΔlukAB background by phage 80α to generate ΔlukAB Δatl. To complement the phenotype of Δatl, plasmids producing full-length Atl, the AM domain, or the GL domain (33) were electroporated into the Δatl strain.
Fractionation of bacterial culture.
The culture supernatant was prepared by trichloroacetic acid (TCA) precipitation. In brief, normalized bacterial cultures were pelleted by centrifugation at 4,000 rpm for 10 min. The culture supernatant was filtered through a 0.2-μm filter and precipitated at 4°C overnight with 10% (vol/vol) TCA. The precipitated proteins were washed with 100% ethanol, pelleted, air-dried, and solubilized with 8 M urea for 30 min at room temperature. The solution was mixed with 2× SDS sample buffer and boiled for 10 min.
To prepare the bacterial cell lysate, 1 mL of the normalized bacterial culture was washed with phosphate-buffered saline (PBS) and lysed with 100 μg/mL lysostaphin (Ambi Products LLC), 40 U/mL DNase, 40 μg/mL RNase A, and 1× Halt protease inhibitor (Thermo Fisher) in lysis buffer (10 mM MgCl2, 1 mM CaCl2 in 50 mM Tris, pH 7.5) for 30 min at 37°C. The lysate was mixed with 4× SDS sample buffer and boiled for 10 min.
To separate surface-exposed and membrane-proximal compartments, the OD600-normalized and washed bacterial pellet was incubated with 1× protease inhibitor in PBS for 10 min at 37°C, followed by incubation with 1% (wt/vol) SDS and 1× protease inhibitor in PBS for 30 min at room temperature. After centrifugation at 13,000 rpm for 2 min, the supernatant was collected as the surface-exposed fraction. The resulting pellet, namely, the membrane-proximal fraction, was washed three times with PBS containing 1× protease inhibitor and lysed as described above. Both fractions were mixed with 4× SDS sample buffer and boiled for 10 min.
Coomassie staining and immunoblotting.
In 12% SDS-PAGE gels, equal volumes of protein samples prepared from OD600-normalized bacterial culture were loaded into each well, and proteins were separated by SDS-PAGE. To examine the exoprotein profile, the gels were stained with InstantBlue Coomassie protein stain (Expedeon). Quantification of the exoprotein profiles was performed using the Fiji distribution of the ImageJ software (65, 66). For each lane, a line with the same width of the lane was drawn vertically, and the profile of the line was recorded. The starting point of each line was normalized to the peak signal of the top band. The baseline was set to be the minimum value of each lane.
For immunoblotting, proteins were transferred onto a nitrocellulose membrane. The membrane was blocked with 5% milk, probed with rabbit anti-LukA (1:5,000) (16) or rabbit anti-LukE (for detecting the S subunit of other leukocidins) (1:5,000) (67) as the primary antibodies, and incubated with Alexa Fluor 680-conjugated goat anti-rabbit IgG (Invitrogen; 1:25,000) as a secondary antibody. Due to high sequence similarity, the anti-LukE antibody is able to detect the respective S subunits of HlgAB, HlgCB, LukED, and PVL. Images were acquired with the Odyssey CLx imaging system (Li-Cor Biosciences). Quantification of protein signals was performed using the Western analysis with the Image Studio software (Li-Cor Biosciences). Protein signals were normalized to WT or a purified recombinant protein control on each membrane.
Infection assays.
Primary human neutrophils (polymorphonuclear neutrophils [PMNs]) were isolated from buffy coats from anonymous, consenting healthy donors (New York Blood Center) as described previously (68).
The PMN infection assays were carried out at the concentration of 2 × 105 cells per well in RPMI without phenol red (Gibco) supplemented with 0.1% human serum albumin (HSA) and 10 mM HEPES in tissue culture-treated 96-well flat-bottom plates. The PMNs were infected with S. aureus at a multiplicity of infection (MOI) of 10 and incubated at 37°C in 5% CO2 for 2 h. Following infection, cells were pelleted by centrifugation at 1,500 rpm for 5 min, and lactate dehydrogenase (LDH) release was measured using the CytoTox-One homogeneous membrane integrity assay (Promega). In brief, 25 μL of culture supernatant was mixed with 25 μL of LDH reagent and incubated for 15 min at room temperature. Fluorescence was measured using a PerkinElmer 2103 EnVision multilabel plate reader (excitation, 555 nm; emission, 590 nm) and normalized to wells containing cells without S. aureus (0% cell lysis) and cells with 0.05% Triton X-100 (100% cell lysis).
Cytotoxicity assays.
Bacteria were subcultured for 4 h, normalized to an OD600 of ∼1.2, and pelleted by centrifuging at 4,000 rpm for 10 min. The supernatant was filtered through a 0.22-μm filter. PMNs were seeded at 2 × 105 cells per well in RPMI without phenol red (Gibco) supplemented with 10% fetal bovine serum (FBS) and incubated with different concentrations of culture filtrates at 37°C and 5% CO2. After 1 h, 10 μL of CellTiter 96 AQueous One solution (Promega) was added to each well, and the plate was incubated for another 1.5 h at 37°C and 5% CO2. The absorbance at 490 nm was measured using a PerkinElmer 2103 EnVision multilabel plate reader.
Promoter reporter assays.
The promoter activities were measured using individual gene promoters fused to a superfolder green fluorescent protein (GFP) (PlukAB, PlukSF-pv, Psae) or yellow fluorescent protein (YFP) (Pagr). Bacteria were washed and diluted 1:2 with PBS. The GFP or YFP fluorescence and OD600 in 200 μL of the suspension were measured using a PerkinElmer EnVision 2103 multilabel reader. The GFP/YFP signal was normalized by the OD600 readings.
Cytochrome c binding assay.
The positive surface charge was determined based on bacterial ability to repulse cationic protein cytochrome c as described previously (69, 70). Early-stationary-phase bacteria were adjusted to an OD600 of ∼1.2. Aliquots of 2 mL bacteria were washed twice with sodium acetate buffer (20 mM, pH 4.6). The pellet was resuspended in 0.5 mL 0.25 mg/mL cytochrome c in sodium acetate buffer and incubated with shaking for 15 min at 37°C. The bacteria were centrifuged at 13,000 rpm for 2 min, the supernatant was aliquoted, and the absorbance at 410 nm was measured using a PerkinElmer 2103 EnVision multilabel plate reader. The measurement was normalized to 0.25 mg/mL cytochrome c as the percentage of unbound cytochrome c, and then normalized to WT.
Microscopy of cell clustering.
To visualize the cell shape of WT and Δatl by microscopy, washed bacterial cells were settled on a clean coverslip for 15 min and fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. The coverslip was washed three times and stained in the dark with 1 μg/mL BODIPY FL vancomycin (Invitrogen) in PBS for 10 min. The coverslip was washed three times, mounted with Fluoromount-G, and air-dried overnight in the dark.
Slides were imaged using Plan-Apochromat 63×/1.4 oil DIC M27 Elyra objective on a Zeiss 880 laser scanning confocal microscope with Airyscan. The BODIPY FL channel was imaged using 488-nm excitation and 495- to 550-nm emission filters. Z-stacks of fluorescent channels were collected at 0.17-μm steps to cover the depth of the bacterial cell clusters. Three-dimensional Airyscan processing was performed for raw images using the Zen software (Zeiss) with automatic strength. A single slice of brightfield was captured as a reference for bacteria position. Three random fields were collected for each condition in each experiment, and two independent experiments were performed.
Images were processed in the Fiji distribution of the ImageJ software (65, 66). Stack images were Z-projected by maximum intensity. The cluster areas were identified as fluorescent signal over 2,000 and including all holes. Each separate cluster was saved as a region of interest (ROI) and measured for area.
Autolysis assay.
The autolysis assay was adapted from a previous study (50). In brief, overnight culture of WT and Δatl was washed and diluted 1:4 into PBS plus 0.05% Triton X-100 with or without 10 U/mL mutanolysin. The bacteria were grown at 37°C under shaking condition, and the OD600 was measured every 15 min using a Bioscreen C.
ACKNOWLEDGMENTS
We thank members of the Torres laboratory for discussions and Ashley L. DuMont for insightful comments on the manuscript. We thank Yan Deng and Michael Cammer (Microscopy Laboratory, New York University Grossman School of Medicine) for assistance in performing the microscopy experiments. We thank Jeffrey L. Bose (University of Kansas Medical Center) for kindly providing the plasmids to express full-length and the single domains of Atl. The pIMAY* plasmid was a gift from Angelika Gründling (Addgene; plasmid no. 121441).
This work was supported in part by the NIH National Institute of Allergy and Infectious Diseases award numbers AI099394 and AI105129 to V.J.T. The NYU Langone’s Microscopy Laboratory is partially supported by the Laura and Isaac Perlmutter Cancer Center Support Grant P30CA016087 from the NIH National Cancer Institute. V.J.T. is a Burroughs Wellcome Fund Investigator in the pathogenesis of infectious diseases.
X.Z. and V.J.T. designed the study. X.Z. performed experiments with help from S.X.M. and A.S.J. X.Z. and V.J.T. wrote the manuscript, and all authors commented on the manuscript.
V.J.T. has consulted for Janssen Research & Development, LLC, and has received honoraria from Genentech and Medimmune. V.J.T. is also an inventor on patents and patent applications filed by New York University, which are currently under commercial license to Janssen Biotech Inc. Janssen Biotech Inc. provides research funding and other payments associated with a licensing agreement.
Footnotes
This article is a direct contribution from Victor J. Torres, a member of the Infection and Immunity Editorial Board, who arranged for and secured reviews by Lindsey Shaw, University of South Florida, and Alexander Horswill, University of Colorado School of Medicine and the Denver VA Healthcare System.
Contributor Information
Victor J. Torres, Email: Victor.Torres@nyulangone.org.
Andreas J. Bäumler, University of California, Davis
REFERENCES
- 1.Williams RE. 1963. Healthy carriage of Staphylococcus aureus: its prevalence and importance. Bacteriol Rev 27:56–71. 10.1128/br.27.1.56-71.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 3.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 4.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG, Jr.. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2019. Antibiotic resistance threats in the United States, 2019. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 6.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alam MT, Read TD, Petit RA, III, Boyle-Vavra S, Miller LG, Eells SJ, Daum RS, David MZ. 2015. Transmission and microevolution of USA300 MRSA in U.S. households: evidence from whole-genome sequencing. mBio 6:e00054. 10.1128/mBio.00054-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talan DA, Krishnadasan A, Gorwitz RJ, Fosheim GE, Limbago B, Albrecht V, Moran GJ, The EMERGEncy ID Net Study Group . 2011. Comparison of Staphylococcus aureus from skin and soft-tissue infections in US emergency department patients, 2004 and 2008. Clin Infect Dis 53:144–149. 10.1093/cid/cir308. [DOI] [PubMed] [Google Scholar]
- 9.Souli M, Ruffin F, Choi SH, Park LP, Gao S, Lent NC, Sharma-Kuinkel BK, Thaden JT, Maskarinec SA, Wanda L, Hill-Rorie J, Warren B, Hansen B, Fowler VG. 2019. Changing characteristics of Staphylococcus aureus bacteremia: results from a 21-year, prospective, longitudinal study. Clin Infect Dis 69:1868–1877. 10.1093/cid/ciz112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thammavongsa V, Kim HK, Missiakas D, Schneewind O. 2015. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543. 10.1038/nrmicro3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otto M. 2014. Staphylococcus aureus toxins. Curr Opin Microbiol 17:32–37. 10.1016/j.mib.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodin AM. 1959. Fractionation of a leucocidin from Staphylococcus aureus. Biochem J 73:225–237. 10.1042/bj0730225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodin AM. 1960. Purification of the two components of leucocidin from Staphylococcus aureus. Biochem J 75:158–165. 10.1042/bj0750158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonzo F, III, Torres VJ. 2014. The bicomponent pore-forming leucocidins of Staphylococcus aureus. Microbiol Mol Biol Rev 78:199–230. 10.1128/MMBR.00055-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DuMont AL, Torres VJ. 2014. Cell targeting by the Staphylococcus aureus pore-forming toxins: it's not just about lipids. Trends Microbiol 22:21–27. 10.1016/j.tim.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, Torres VJ. 2011. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79:814–825. 10.1111/j.1365-2958.2010.07490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ventura CL, Malachowa N, Hammer CH, Nardone GA, Robinson MA, Kobayashi SD, DeLeo FR. 2010. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5:e11634. 10.1371/journal.pone.0011634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DuMont AL, Yoong P, Surewaard BG, Benson MA, Nijland R, van Strijp JA, Torres VJ. 2013. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infect Immun 81:1830–1841. 10.1128/IAI.00095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melehani JH, James DB, DuMont AL, Torres VJ, Duncan JA. 2015. Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog 11:e1004970. 10.1371/journal.ppat.1004970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berends ETM, Zheng X, Zwack EE, Menager MM, Cammer M, Shopsin B, Torres VJ. 2019. Staphylococcus aureus impairs the function of and kills human dendritic cells via the LukAB toxin. mBio 10:e01918-18. 10.1128/mBio.01918-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DuMont AL, Yoong P, Day CJ, Alonzo F, III, McDonald WH, Jennings MP, Torres VJ. 2013. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci USA 110:10794–10799. 10.1073/pnas.1305121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perelman SS, James DBA, Boguslawski KM, Nelson CW, Ilmain JK, Zwack EE, Prescott RA, Mohamed A, Tam K, Chan R, Narechania A, Pawline MB, Vozhilla N, Moustafa AM, Kim SY, Dittmann M, Ekiert DC, Bhabha G, Shopsin B, Planet PJ, Koralov SB, Torres VJ. 2021. Genetic variation of staphylococcal LukAB toxin determines receptor tropism. Nat Microbiol 6:731–745. 10.1038/s41564-021-00890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng X, Marsman G, Lacey KA, Chapman JR, Goosmann C, Ueberheide BM, Torres VJ. 2021. The cell envelope of Staphylococcus aureus selectively controls the sorting of virulence factors. Nat Commun 12:6193. 10.1038/s41467-021-26517-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koch HU, Haas R, Fischer W. 1984. The role of lipoteichoic acid biosynthesis in membrane lipid metabolism of growing Staphylococcus aureus. Eur J Biochem 138:357–363. 10.1111/j.1432-1033.1984.tb07923.x. [DOI] [PubMed] [Google Scholar]
- 26.Fischer W. 1994. Lipoteichoic acid and lipids in the membrane of Staphylococcus aureus. Med Microbiol Immunol 183:61–76. 10.1007/BF00277157. [DOI] [PubMed] [Google Scholar]
- 27.Oshida T, Sugai M, Komatsuzawa H, Hong YM, Suginaka H, Tomasz A. 1995. A Staphylococcus aureus autolysin that has an N-acetylmuramoyl-L-alanine amidase domain and an endo-beta-N-acetylglucosaminidase domain: cloning, sequence analysis, and characterization. Proc Natl Acad Sci USA 92:285–289. 10.1073/pnas.92.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zoll S, Patzold B, Schlag M, Gotz F, Kalbacher H, Stehle T. 2010. Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog 6:e1000807. 10.1371/journal.ppat.1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buttner FM, Zoll S, Nega M, Gotz F, Stehle T. 2014. Structure-function analysis of Staphylococcus aureus amidase reveals the determinants of peptidoglycan recognition and cleavage. J Biol Chem 289:11083–11094. 10.1074/jbc.M114.557306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nega M, Tribelli PM, Hipp K, Stahl M, Gotz F. 2020. New insights in the coordinated amidase and glucosaminidase activity of the major autolysin (Atl) in Staphylococcus aureus. Commun Biol 3:695. 10.1038/s42003-020-01405-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi J, Komatsuzawa H, Yamada S, Nishida T, Labischinski H, Fujiwara T, Ohara M, Yamagishi J, Sugai M. 2002. Molecular characterization of an atl null mutant of Staphylococcus aureus. Microbiol Immunol 46:601–612. 10.1111/j.1348-0421.2002.tb02741.x. [DOI] [PubMed] [Google Scholar]
- 32.Biswas R, Voggu L, Simon UK, Hentschel P, Thumm G, Gotz F. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259:260–268. 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 33.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spaan AN, van Strijp JAG, Torres VJ. 2017. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15:435–447. 10.1038/nrmicro.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tam K, Lacey KA, Devlin JC, Coffre M, Sommerfield A, Chan R, O'Malley A, Koralov SB, Loke P, Torres VJ. 2020. Targeting leukocidin-mediated immune evasion protects mice from Staphylococcus aureus bacteremia. J Exp Med 217:e20190541. 10.1084/jem.20190541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balasubramanian D, Ohneck EA, Chapman J, Weiss A, Kim MK, Reyes-Robles T, Zhong J, Shaw LN, Lun DS, Ueberheide B, Shopsin B, Torres VJ. 2016. Staphylococcus aureus coordinates leukocidin expression and pathogenesis by sensing metabolic fluxes via RpiRc. mBio 7:e00818-16. 10.1128/mBio.00818-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chapman JR, Balasubramanian D, Tam K, Askenazi M, Copin R, Shopsin B, Torres VJ, Ueberheide BM. 2017. Using quantitative spectrometry to understand the influence of genetics and nutritional perturbations on the virulence potential of Staphylococcus aureus. Mol Cell Proteomics 16:S15–S28. 10.1074/mcp.O116.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novick RP, Geisinger E. 2008. Quorum sensing in staphylococci. Annu Rev Genet 42:541–564. 10.1146/annurev.genet.42.110807.091640. [DOI] [PubMed] [Google Scholar]
- 40.Liu Q, Yeo WS, Bae T. 2016. The SaeRS two-component system of Staphylococcus aureus. Genes (Basel) 7:81. 10.3390/genes7100081. [DOI] [Google Scholar]
- 41.Peschel A, Otto M. 2013. Phenol-soluble modulins and staphylococcal infection. Nat Rev Microbiol 11:667–673. 10.1038/nrmicro3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebner P, Luqman A, Reichert S, Hauf K, Popella P, Forchhammer K, Otto M, Gotz F. 2017. Non-classical protein excretion is boosted by PSMalpha-induced cell leakage. Cell Rep 20:1278–1286. 10.1016/j.celrep.2017.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ravipaty S, Reilly JP. 2010. Comprehensive characterization of methicillin-resistant Staphylococcus aureus subsp. aureus COL secretome by two-dimensional liquid chromatography and mass spectrometry. Mol Cell Proteomics 9:1898–1919. 10.1074/mcp.M900494-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schallenberger MA, Niessen S, Shao C, Fowler BJ, Romesberg FE. 2012. Type I signal peptidase and protein secretion in Staphylococcus aureus. J Bacteriol 194:2677–2686. 10.1128/JB.00064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ingavale S, van Wamel W, Luong TT, Lee CY, Cheung AL. 2005. Rat/MgrA, a regulator of autolysis, is a regulator of virulence genes in Staphylococcus aureus. Infect Immun 73:1423–1431. 10.1128/IAI.73.3.1423-1431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Memmi G, Nair DR, Cheung A. 2012. Role of ArlRS in autolysis in methicillin-sensitive and methicillin-resistant Staphylococcus aureus strains. J Bacteriol 194:759–767. 10.1128/JB.06261-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groicher KH, Firek BA, Fujimoto DF, Bayles KW. 2000. The Staphylococcus aureus lrgAB operon modulates murein hydrolase activity and penicillin tolerance. J Bacteriol 182:1794–1801. 10.1128/JB.182.7.1794-1801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujimoto DF, Bayles KW. 1998. Opposing roles of the Staphylococcus aureus virulence regulators, Agr and Sar, in Triton X-100- and penicillin-induced autolysis. J Bacteriol 180:3724–3726. 10.1128/JB.180.14.3724-3726.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice KC, Firek BA, Nelson JB, Yang SJ, Patton TG, Bayles KW. 2003. The Staphylococcus aureus cidAB operon: evaluation of its role in regulation of murein hydrolase activity and penicillin tolerance. J Bacteriol 185:2635–2643. 10.1128/JB.185.8.2635-2643.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schlag M, Biswas R, Krismer B, Kohler T, Zoll S, Yu W, Schwarz H, Peschel A, Gotz F. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol Microbiol 75:864–873. 10.1111/j.1365-2958.2009.07007.x. [DOI] [PubMed] [Google Scholar]
- 51.Biswas R, Martinez RE, Gohring N, Schlag M, Josten M, Xia G, Hegler F, Gekeler C, Gleske AK, Gotz F, Sahl HG, Kappler A, Peschel A. 2012. Proton-binding capacity of Staphylococcus aureus wall teichoic acid and its role in controlling autolysin activity. PLoS One 7:e41415. 10.1371/journal.pone.0041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasquina-Lemonche L, Burns J, Turner RD, Kumar S, Tank R, Mullin N, Wilson JS, Chakrabarti B, Bullough PA, Foster SJ, Hobbs JK. 2020. The architecture of the Gram-positive bacterial cell wall. Nature 582:294–297. 10.1038/s41586-020-2236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amir A, Babaeipour F, McIntosh DB, Nelson DR, Jun S. 2014. Bending forces plastically deform growing bacterial cell walls. Proc Natl Acad Sci USA 111:5778–5783. 10.1073/pnas.1317497111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pasztor L, Ziebandt AK, Nega M, Schlag M, Haase S, Franz-Wachtel M, Madlung J, Nordheim A, Heinrichs DE, Gotz F. 2010. Staphylococcal major autolysin (Atl) is involved in excretion of cytoplasmic proteins. J Biol Chem 285:36794–36803. 10.1074/jbc.M110.167312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ebner P, Gotz F. 2019. Bacterial excretion of cytoplasmic proteins (ECP): occurrence, mechanism, and function. Trends Microbiol 27:176–187. 10.1016/j.tim.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 56.Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, Voyich JM. 2010. SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J Infect Dis 201:241–254. 10.1086/649570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, Gaspin C, Vandenesch F, Romby P. 2007. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21:1353–1366. 10.1101/gad.423507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Killikelly A, Benson MA, Ohneck EA, Sampson JM, Jakoncic J, Spurrier B, Torres VJ, Kong XP. 2015. Structure-based functional characterization of repressor of toxin (Rot), a central regulator of Staphylococcus aureus virulence. J Bacteriol 197:188–200. 10.1128/JB.02317-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li D, Cheung A. 2008. Repression of hla by rot is dependent on sae in Staphylococcus aureus. Infect Immun 76:1068–1075. 10.1128/IAI.01069-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan YG, Frankel MB, Missiakas D, Schneewind O. 2016. SagB glucosaminidase is a determinant of Staphylococcus aureus glycan chain length, antibiotic susceptibility, and protein secretion. J Bacteriol 198:1123–1136. 10.1128/JB.00983-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wheeler R, Turner RD, Bailey RG, Salamaga B, Mesnage S, Mohamad SA, Hayhurst EJ, Horsburgh M, Hobbs JK, Foster SJ. 2015. Bacterial cell enlargement requires control of cell wall stiffness mediated by peptidoglycan hydrolases. mBio 6:e00660. 10.1128/mBio.00660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boles BR, Thoendel M, Roth AJ, Horswill AR. 2010. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5:e10146. 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monk IR, Tree JJ, Howden BP, Stinear TP, Foster TJ. 2015. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6:e00308-15. 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuster CF, Howard SA, Gründling A. 2019. Use of the counter selectable marker PheS* for genome engineering in Staphylococcus aureus. Microbiology (Reading) 165:572–584. 10.1099/mic.0.000791. [DOI] [PubMed] [Google Scholar]
- 65.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alonzo F, III, Benson MA, Chen J, Novick RP, Shopsin B, Torres VJ. 2012. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol 83:423–435. 10.1111/j.1365-2958.2011.07942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reyes-Robles T, Lubkin A, Alonzo F, III, Lacy DB, Torres VJ. 2016. Exploiting dominant-negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep 17:780. 10.15252/embr.201670010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ernst CM, Slavetinsky CJ, Kuhn S, Hauser JN, Nega M, Mishra NN, Gekeler C, Bayer AS, Peschel A. 2018. Gain-of-function mutations in the phospholipid flippase MprF confer specific daptomycin resistance. mBio 9:e01659-18. 10.1128/mBio.01659-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F. 1999. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410. 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 71.Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, Torres VJ, Chiu IM. 2018. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 9:37. 10.1038/s41467-017-02448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benson MA, Lilo S, Nygaard T, Voyich JM, Torres VJ. 2012. Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J Bacteriol 194:4355–4365. 10.1128/JB.00706-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yarwood JM, Bartels DJ, Volper EM, Greenberg EP. 2004. Quorum sensing in Staphylococcus aureus biofilms. J Bacteriol 186:1838–1850. 10.1128/JB.186.6.1838-1850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]