

ABSTRACT
Persistent infections generally involve a complex balance between protective immunity and immunopathology. We used a murine model to investigate the role of inflammatory monocytes in immunity and host defense against persistent salmonellosis. Mice exhibit increased susceptibility to persistent infection when inflammatory monocytes cannot be recruited into tissues or when they are depleted at specific stages of persistent infection. Inflammatory monocytes contribute to the pathology of persistent salmonellosis and cluster with other cells in pathogen-containing granulomas. Depletion of inflammatory monocytes during the chronic phase of persistent salmonellosis causes regression of already established granulomas with resultant pathogen growth and spread in tissues. Thus, inflammatory monocytes promote granuloma-mediated control of persistent salmonellosis and may be key to uncovering new therapies for granulomatous diseases.
KEYWORDS: CCR2+ cells, inflammatory monocyte, Salmonella, chronic, granuloma, host response, infection, pathogenesis, persistent
INTRODUCTION
Bacterial pathogens that cause persistent or chronic infections pose a significant public health problem. Examples of bacterial pathogens that can cause persistent or chronic infections in humans include Mycobacterium tuberculosis, Helicobacter pylori, Borrelia burgdorferi, Brucella abortus, and Salmonella enterica (1–5). Persistent infections generally involve a complex balance between protective immunity and immunopathology. New insights into mechanisms that control this balance are needed to better combat persistent infections.
Host-adapted Salmonella enterica serovar Typhi (STy), the causative agent of typhoid fever in humans, is responsible for 21.6 million illnesses annually (6–8). A significant percentage (1 to 6%) of typhoid patients become chronic carriers of STy (2, 9, 10). Individuals who are chronically infected with STy are often asymptomatic. However, they transmit disease via fecal shedding of the bacteria. Thus, chronically infected individuals are a critical reservoir of STy (1, 9).
Histologically, persistent infections with STy have been associated with granulomas in peripheral tissues and bone marrow, as well as localization of the bacteria in the gallbladder (9, 11–16). These granulomas are composed primarily of macrophages (histiocytes) mixed with lymphocytes and fewer plasma cells and neutrophils. They are sites of chronic inflammation referred to as typhoid nodules, presumably triggered by persistent STy. The presence of Salmonella enterica in tissues is a serious medical problem, especially in areas of the world where comorbidities such as malaria, human immunodeficiency virus infection, and malnutrition impair the immune system, leading to higher incidences of relapse and acute and recurrent infection (9, 17–26).
As part of our studies aimed at elucidating mechanisms by which bacterial pathogens avoid immune clearance, we began to investigate the role of inflammatory monocytes (IM; Ly6Chi Ly6G− CD11b+ cells) in immunity and host defense (27–30). IM are innate immune cells that originate from progenitors in bone marrow (BM). During infection, IM are recruited from BM into tissues. Once there, IM can exert direct antimicrobial effects, transport bacteria to draining lymph nodes, differentiate into macrophages and dendritic cells, contribute to T cell priming, and orchestrate functions of other cell types (31–34). IM and other subsets of monocytes are an important component of the host response to Salmonella enterica (27, 29, 35–40). However, little is known about their role in the pathogenesis of persistent salmonellosis.
Here, we show that, in a murine model of persistent salmonellosis, IM cluster with other cells in pathogen-containing granulomas and, furthermore, that depletion of IM during the chronic phase of persistent infection causes regression of already established granulomas with resultant pathogen growth and spread in tissues. These findings establish that IM play a key role in granuloma-mediated control of persistent salmonellosis.
RESULTS
NRAMP1+ mice exhibit increased susceptibility to STm infection when IM cannot be recruited from BM into tissues due to Ccr2 or Ccl2 deficiency.
The lack of a convenient, tractable mammalian model of STy infection has been a significant limitation in gaining new insights into the chronic carrier state (41, 42). However, infection of 129 × 1/SvJ mice with Salmonella enterica serovar Typhimurium (STm) recapitulates a number of hallmarks that characterize the pathology of persistent STy infection in humans and has served as a useful model to elucidate mechanisms of intestinal persistence and chronic carriage in tissues (42–44). We previously utilized this model and showed that IM accumulate in tissues of mice infected with STm (29).
To define the role of IM in immunity and host defense against persistent STm infection, we used strains of genetically modified mice available in the C57BL/6J strain background. In contrast to 129 × 1/SvJ mice, which are naturally resistant to STm infection, C57BL/6J mice are highly susceptible to STm infection. Susceptibility to STm is associated with a single point mutation in the Slc11a1/Nramp1 gene, resulting in a glycine-to-aspartic acid substitution at position 169 of the primary sequence of SLC11A1/NRAMP1 (45). This divalent cation transporter restricts the replication of a number of different intracellular pathogens within phagocytes by limiting essential metal availability (45). C57BL/6J mice carry two copies of the Slc11a1/Nramp1D169 allele and, therefore, are phenotypically SLC11A1/NRAMP1−.
To limit the number of genetic crosses necessary to be able to use genetically modified mouse strains available in the C57BL/6J strain background, C57BL/6J mice were crossed with C57BL/6J Slc11a1/Nramp1G169 mice (46). These transgenic mice in the C57BL/6J strain background are homozygous for the dominant Slc11a1/Nramp1G169 allele and have been used previously to study aspects of persistent STm infection (46–50). The resulting F1 hybrid mice, which are phenotypically SLC11A1/NRAMP1+ and referred to here as NRAMP1+ mice, were inoculated with phosphate-buffered saline (PBS) or STm suspended in PBS. Consistent with our published findings (29), IM accumulated over a period of at least 60 days in spleens (Fig. 1A and B; Fig. S1A) and livers (Fig. S1B and C) of infected NRAMP1+ mice. Thus, infection of NRAMP1+ mice with STm serves as a useful model to investigate the role of IM in the pathogenesis of persistent salmonellosis.
FIG 1.
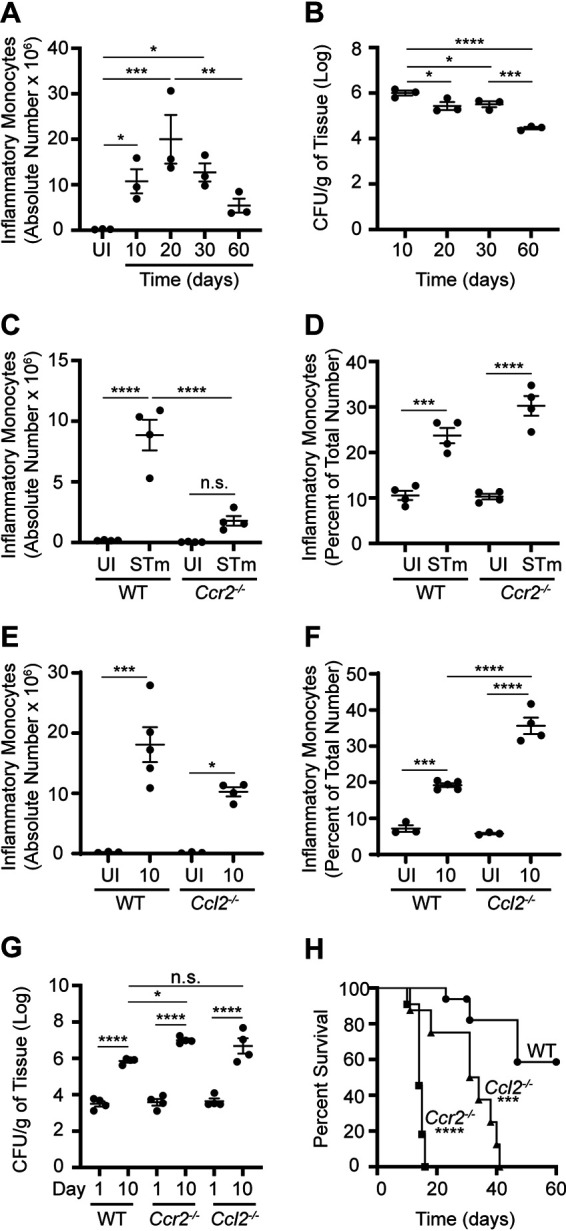
NRAMP1+ mice exhibit increased susceptibility to STm infection when IM cannot be recruited from BM into tissues due to Ccr2 or Ccl2 deficiency. (A) Absolute numbers of IM in spleens from NRAMP1+ mice (n = 3 per group) inoculated with PBS or STm. Spleens were harvested at different times (up to day 60) after inoculation. (B) Corresponding numbers of STm CFU recovered per gram of spleen tissue. (C) Absolute numbers of IM in spleens from NRAMP1+ (WT) and Ccr2−/− NRAMP1+ (Ccr2−/−) mice (4 per group) inoculated with PBS or STm. Spleens were harvested on day 10 after inoculation. (D) Corresponding percentages of IM in BM. (E) Absolute numbers of IM in spleens from NRAMP1+ (WT) and Ccl2−/− NRAMP1+ (Ccl2−/−) mice (3 to 5 per group) inoculated with PBS or STm. Spleens were harvested on day 10 after inoculation. (F) Corresponding percentages of IM in BM. (G) Numbers of STm CFU recovered per gram of spleen tissue from NRAMP1+ (WT), Ccr2−/− NRAMP1+ (Ccr2−/−), and Ccl2−/− NRAMP1+ (Ccl2−/−) mice (4 per group) inoculated with STm. Spleens were harvested on day 1 or day 10 after inoculation. (H) Survival of NRAMP1+ (WT), Ccr2−/− NRAMP1+ (Ccr2−/−), and Ccl2−/− NRAMP1+ (Ccl2−/−) mice (8 to 16 per group) inoculated with STm. Data are means with standard errors of the means (SEM) and individual data points (A to G) or survival in each group (H) and were analyzed by use of one-way analysis of variance (ANOVA) with Fisher’s least significant difference (LSD) (A and B) or Sidak’s (C to G) post hoc test or by use of a log rank test (H). ****, P < 0.0001; ***, P < 001; **, P < 0.01; *, P < 0.05; n.s., not significant. Each dot in the figures represents one mouse. Data are from a single experiment that is representative of two independent experiments (A to G) or are cumulative from two independent experiments (H). Also see Fig. S1.
CC chemokine receptor 2 (CCR2) plays a critical role in the emigration of Ly6Chi monocytes from BM, with CCR2 expression being a hallmark of IM (51). To evaluate the role of CCR2 in persistent STm infection, Ccr2−/− mice in the C57BL/6J strain background were crossed with NRAMP1+ mice to ultimately generate Ccr2−/− NRAMP1+ offspring. These mice were inoculated with PBS or STm. NRAMP1+ mice inoculated with PBS or STm were used as wild-type (WT) controls. On day 10 after inoculation, IM had accumulated in spleens (Fig. 1C) and livers (Fig. S1D) of infected WT but not Ccr2−/− mice. Consistent with the notion that emigration of IM from BM is dependent on Ccr2 (51), it appeared as if IM had accumulated in BM of infected Ccr2−/− mice to a greater degree than in WT mice (Fig. 1D). Similar results were obtained when we generated and used Ccl2−/− NRAMP1+ mice (Fig. 1E and F; Fig. S1E), which lack CC chemokine ligand 2 (CCL2), a known ligand for CCR2 (52, 53). Thus, Ccr2 and Ccl2 are required for STm-induced recruitment of IM from BM into tissues.
To evaluate the roles of Ccr2 and Ccl2 in host susceptibility to persistent STm infection, Ccr2−/− and Ccl2−/− NRAMP1+ mice were again inoculated with STm. NRAMP1+ mice inoculated with STm were used as WT controls. On day 1 and day 10 after inoculation, organ burden assays were performed to determine bacterial loads. On day 1 after inoculation, similar numbers of STm CFU were recovered from spleens (Fig. 1G) and livers (Fig. S1F) of infected WT, Ccr2−/−, and Ccl2−/− mice. On day 10 after inoculation, however, increased numbers of STm CFU were recovered from spleens (Fig. 1G) and livers (Fig. S1F) of infected Ccr2−/− and Ccl2−/− mice relative to WT mice, with data from spleens of infected Ccl2−/− mice trending. In parallel experiments, survival of STm-infected WT, Ccr2−/−, and Ccl2−/− mice was monitored over time. Ccr2−/− and Ccl2−/− mice succumbed to STm infection faster than WT mice, with Ccr2−/− mice succumbing fastest (Fig. 1H). Thus, Ccr2−/− and Ccl2−/− NRAMP1+ mice exhibit increased susceptibility to STm infection, with Ccr2−/− NRAMP1+ mice being most susceptible.
Collectively, these results suggest that NRAMP1+ mice exhibit increased susceptibility to STm infection when IM cannot be recruited from BM into tissues.
Transient depletion of IM renders NRAMP1+ mice more susceptible to STm infection.
To define the role of IM at specific stages of persistent STm infection, NRAMP1+ mice were crossed with CCR2-DTR mice (54), which are transgenic mice in the C57BL/6J strain background that express simian diphtheria toxin receptor (DTR) and cyan fluorescent protein (CFP) driven by the mouse Ccr2 promoter. The resulting CCR2-DTR NRAMP1+ offspring were inoculated with STm. NRAMP1+ littermates inoculated with STm were used as WT controls. On day 21 after inoculation, PBS, or diphtheria toxin (DT) suspended in PBS, was administered to the mice, the effect of which was evaluated 3 days later by use of flow cytometry and organ burden assay. Administration of DT had caused IM to be depleted in spleens of infected CCR2-DTR but not WT mice (Fig. 2A). Furthermore, increased numbers of STm CFU were recovered from spleens of infected CCR2-DTR mice treated with DT relative to CCR2-DTR mice treated with PBS and WT mice treated with DT or PBS (Fig. 2B). Similar results were obtained in liver (Fig. S2A and B). Notably, decreased numbers of IM had accumulated in spleens of infected CCR2-DTR mice treated with PBS relative to WT mice treated with PBS (Fig. 2A). At present, the biological relevance of this effect is unclear. Consistent with the notion that expression of Ccr2 is not entirely specific for IM, administration of DT also caused depletion of other, minor subsets of CCR2+ cells, including cells that likely differentiated from IM (Fig. S2C and D). DT-mediated depletion of CCR2+ cells was transient, as numbers of IM present in spleens of infected CCR2-DTR NRAMP1+ mice returned to near normal by day 5 after DT treatment (Fig. 2C). Despite the reemergence of IM, however, the numbers of STm CFU recovered from spleens of infected CCR2-DTR NRAMP1+ mice were higher on day 5 after DT treatment than on day 1 or day 3 after DT treatment (Fig. 2D).
FIG 2.
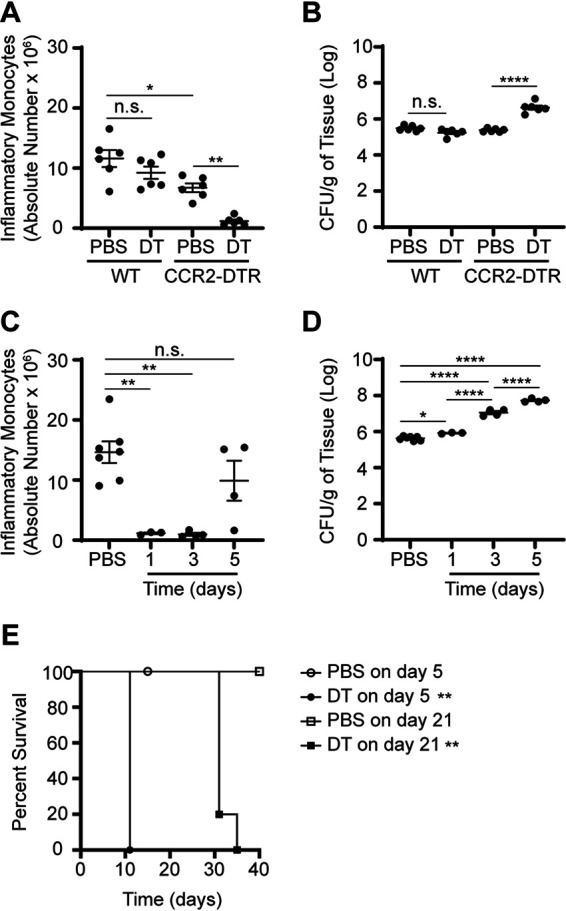
Transient depletion of IM renders NRAMP1+ mice more susceptible to STm infection. (A) Absolute numbers of IM in spleens from NRAMP1+ (WT) and CCR2-DTR NRAMP1+ (CCR2-DTR) mice (6 per group) treated with PBS or DT on day 21 after inoculation with STm. Spleens were harvested at on day 3 after treatment. (B) Corresponding numbers of STm CFU recovered per gram of spleen tissue. (C) Absolute numbers of IM in spleens from CCR2-DTR NRAMP1+ mice (4 to 8 per group) treated with PBS or DT on day 21 after inoculation with STm. Spleens were harvested on day 1, day 3, or day 5 after treatment. (D) Corresponding numbers of STm CFU recovered per gram of spleen tissue. (E) Survival of CCR2-DTR NRAMP1+ mice (4 or 5 per group) treated with PBS or DT on day 5 or day 21 after inoculation with STm. Data are means with SEM and individual data points (A to D) or survival in each group (E) and were analyzed by use of one-way ANOVA with Sidak’s post hoc test (A to D) or by use of a log rank test (E). ****, P < 0.0001; **, P < 0.01; *, P < 0.05; n.s., not significant. Each dot in the figures represents one mouse. Data are from a single experiment that is representative of two independent experiments (A to D) or are cumulative from two independent experiments (E). Also see Fig. S2.
To further define the effect of CCR2+ cell depletion on host susceptibility to persistent salmonellosis, CCR2-DTR NRAMP1+ mice were again inoculated with STm. On day 5 or day 21 after inoculation, PBS or DT was administered to the mice, the effect of which was evaluated by monitoring survival of the mice over time. No deaths were observed among the PBS-treated control mice (Fig. 2E). In contrast, all the DT-treated mice died, regardless of when DT was administered, with mice treated on day 5 after inoculation dying faster than mice treated on day 21 after inoculation (Fig. 2E). Importantly, no deaths were observed among PBS- or DT-treated mice left uninfected (Fig. S2E). Remarkably, when DT was administered to the mice on day 170 after inoculation, all the DT-treated mice died, with no deaths observed among the PBS-treated control mice (Fig. S2F), suggesting that CCR2+ cells are also involved in immunity and host defense during later phases of the infection.
Collectively, these results indicate that transient depletion of IM (i.e., CCR2+ cells) renders NRAMP1+ mice more susceptible to STm infection.
IM contribute to the pathology of persistent STm infection and cluster with other cells in STm-containing granulomas.
To further define the role of IM in the pathogenesis of persistent salmonellosis, we felt it was of fundamental importance to examine methodically and in detail the pathology of persistent STm infection and to visualize the spatial distribution of CCR2+ cells in tissues. Therefore, NRAMP1+ mice were crossed with CCR2-GFP mice (54), which are transgenic mice in the C57BL/6J strain background that express green fluorescent protein (GFP) driven by the mouse Ccr2 promoter. The resulting CCR2-GFP NRAMP1+ offspring were inoculated with PBS or STm. On day 21 after inoculation, tissues were harvested and analyzed by blind, qualitative histopathology and flow cytometry.
Hematoxylin and eosin staining revealed that livers of infected mice contained myriad inflammatory foci, scattered multifocally throughout the hepatic parenchyma with a random distribution (Fig. 3A). Sites of inflammation were variable in size, ranging from approximately 100 to 1,000 μm, and contained mixed inflammatory infiltrates composed of macrophages, with fewer neutrophils, lymphocytes, and plasma cells. Smaller foci were composed exclusively of neutrophils (Fig. 3B) and were interpreted to be sites of recent, acute bacterial colonization subsequent to ongoing or recurring bacteremia. Larger foci contained a mixed population of inflammatory cells (Fig. 3C) and were interpreted to be sites of chronic infection. The larger, chronic lesions contained abundant macrophages and exhibited typical features of mature granuloma development with epithelioid macrophages and peripheral lymphocyte recruitment (Fig. 3D). In addition, livers of infected mice also contained frequent multifocal aggregates of hematopoietic precursor cells that were interpreted to be sites of extramedullary hematopoiesis (EMH) (Fig. 3E). Furthermore, mononuclear inflammatory cell infiltrates were also observed surrounding blood vessels and within portal tracts (Fig. S3A). This feature of nonsuppurative perivascular and periportal inflammation was interpreted as a response to systemic inflammation (i.e., reactive hepatitis) and contributed only minimally to the overall hepatic inflammation (Fig. S3A). Occasional liver sections contained solitary or coalescing areas of coagulative necrosis, which were interpreted to be areas of ischemia due to endotoxin-mediated vascular injury. Staining for F4/80 revealed that Kupffer cell hyperplasia and hypertrophy was abundant and represented a significant reactive inflammatory feature of this model (Fig. S3B). Staining for CD31 revealed an expansion of hepatic sinusoids to accommodate the marked proliferation of Kupffer cells, while no evidence for STm-induced endothelial proliferation or reduplication of hepatic arterioles was observed (Fig. S3C). Staining for cytokeratin-7 revealed no evidence of significant STm-induced biliary hyperplasia (Fig. S3D). Masson’s trichome staining revealed that hepatic fibrosis is not a major feature of this infection model on day 21 after inoculation (Fig. S3E).
FIG 3.

IM contribute to the pathology of persistent STm infection and cluster with other cells in STm-containing granulomas. (A) Representative image of hematoxylin- and eosin-stained liver sections from CCR2-GFP NRAMP1+ mice (4 per group) inoculated with PBS (left) or STm (right). Magnification, ×20. Bar, 50 μm. (B to E) Representative image of hematoxylin- and eosin-stained liver sections from CCR2-GFP NRAMP1+ mice (4 per group) inoculated with STm. Magnification, ×40. Bar, 20 μm. (B) Small acute inflammatory nodule composed predominantly of neutrophils. (C) Small subacute inflammatory nodule composed of a mixture of neutrophils and mononuclear cells with emergence of epithelioid macrophages. (D) Larger inflammatory nodule composed of a mixed population of inflammatory cells with abundant macrophages and fewer neutrophils and lymphocytes. (E) Focus of hypercellularity composed of hematopoietic precursor cells consistent with EMH. (F) Representative image of multicolor immunohistochemical staining for GFP (blue) and STm CSA-1 (brown) on liver sections from CCR2-GFP NRAMP1+ mice (4 per group) inoculated with STm. (Left) Magnification, ×20. Bar, 50 μm. (Right) Magnification, ×40. Bar, 20 μm. The arrow points to the brown color used to visualize STm. (G) Representative image of multicolor immunohistochemical staining for GFP (blue) and Ly6G (brown) on liver sections from CCR2-GFP NRAMP1+ mice (4 per group) inoculated with STm. Magnification, ×20. Bar, 50 μm. (H) Representative image of multicolor immunohistochemical staining for GFP (blue) and CD4 (brown) on liver sections from CCR2-GFP NRAMP1+ mice (4 per group) inoculated with STm. Magnification, ×20. Bar, 50 μm. Data are from a single experiment that is representative of two independent experiments. Also see Fig. S3.
As for the spatial distribution of CCR2+ cells (e.g., IM) and STm in liver, staining for both GFP (blue) and STm common structural antigen 1 (CSA-1) (brown) revealed that most CCR2+ cells had clustered with other cells in inflammatory foci at sites of tissue infection that were interpreted as being STm-containing granulomas (Fig. 3F). Notably, the granulomas contained scant STm organisms (i.e., they were paucibacillary), and only few CCR2+ cells were scattered throughout the hepatic sinusoids. No CCR2+ cells were observed in EMH lesions (Fig. S3F) or in livers from mice inoculated with PBS (Fig. S3G). Consistent with the notion that expression of Ccr2 is not entirely specific for IM, parallel flow-cytometric analysis revealed that most, but not all, CCR2+ cells are IM or cells that likely differentiated from IM (Fig. S3H to J).
As for the spatial distribution of cells other than CCR2+ cells in liver, staining for GFP (blue) and Ly6G (brown) or CD4 (brown) revealed that both Ly6G+ cells (e.g., neutrophils) and CD4+ cells (e.g., Th1 cells) were among the cells that had congregated with CCR2+ cells in STm-containing granulomas (Fig. 3G and H).
Collectively, these results describe the liver pathology of persistent STm infection of mice, which is like that observed in people with chronic typhoid (12, 14–16, 55–57), and establish that CCR2+ cells (e.g., IM) cluster with other cells (e.g., neutrophils, Th1 cells) in STm-containing granulomas.
Transient depletion of IM impairs granuloma-mediated control of persistent STm infection.
To further define the role of IM in immunity and host defense against persistent salmonellosis, CCR2-DTR NRAMP1+ mice were again inoculated with PBS or STm. On day 21 after inoculation, DT was administered to the mice, the effect of which was evaluated over time by use of multicolor immunohistochemistry. Administration of PBS was used as a control. Staining for CFP (blue) and STm (brown) revealed that, by day 1 after DT treatment, CCR2+ cells (e.g., IM) had been depleted from STm-containing granulomas in liver (Fig. 4A to C; Fig. S4A to C). Consistent with the notion that DT-mediated depletion of IM is transient (Fig. 2C), increased staining for CFP was observed by day 5 after DT treatment (Fig. 4A to C; Fig. S4A to C). The spatial distribution of the blue color used to visualize CCR2+ cells revealed that while most CCR2+ cells colocalized with infectious foci in what were taken to be altered STm-containing granulomas, many CCR2+ cells were scattered haphazardly throughout the tissues at this time point (Fig. 4A and B). Remarkably, the intensity of the brown color used to visualize STm had seemingly increased by day 3 after DT treatment and had increased significantly by day 5 after DT treatment (Fig. 4A, B, and D; Fig. S4A, B, and D). Furthermore, the spatial distribution of the brown color used to visualize STm suggested that the bacteria had spread throughout the tissue (Fig. 4A and B, Fig. S4A and B). Staining for CFP (blue) and Ly6G (brown) or CD4 (brown) revealed that DT treatment not only had caused depletion of CCR2+ cells but also had led to a marked loss of both Ly6G+ and CD4+ cells associated with the granulomas (Fig. S4E and S4F). Given that Ly6G+ cells and most CD4+ cells (assuming CD4 T cells) do not express CCR2 (Fig. S3J), these results suggest that DT treatment causes direct depletion of CCR2+ cells and indirect depletion of Ly6G+ and CD4+ cells, with the effect of DT treatment on CCR2+ cells being the precipitating effect that then leads to the marked loss of Ly6G+ and CD4+ cells.
FIG 4.

Transient depletion of IM impairs granuloma-mediated control of persistent STm infection. (A and B) Representative images of multicolor immunohistochemical staining for CFP (blue) and STm CSA-1 (brown) on liver sections from CCR2-DTR NRAMP1+ mice (2 to 11 per group) treated with PBS or DT on day 21 after inoculation with STm. Magnifications, ×10 (A) and ×40 (B). Livers were harvested on day 1, day 3, or day 5 after treatment. Arrows point to the brown color used to visualize STm. (C and D) Corresponding quantitative analysis of total areas of strong positivity for the blue color used to visualize IM (C) and the brown color used to visualize STm (D). Quantification is reported as percent of positive tissues for each stain. Data are means with SEM and individual data points and were analyzed by use of one-way ANOVA with Sidak’s post hoc test (C and D). ****, P < 0.0001; **, P < 0.01; *, P < 0.05. Data are from a single experiment that is representative of two independent experiments. Each dot in the figures represents one mouse (C and D). Also see Fig. S4.
Collectively, these results indicate that transient depletion of IM (i.e., CCR2+ cells) impairs granuloma-mediated control of persistent STm infection and suggest that once granuloma-mediated control of persistent STm infection is lost, it cannot easily be regained.
DISCUSSION
We report that NRAMP1+ mice exhibit increased susceptibility to STm infection when IM (i.e., CCR2+ cells) cannot be recruited from BM into tissues or when they are depleted at specific stages of persistent STm infection. Furthermore, we report that CCR2+ cells (e.g., IM) cluster with other cells (e.g., neutrophils and Th1 cells) in STm-containing granulomas and that their depletion during the chronic phase of persistent STm infection causes regression of already established granulomas with resultant pathogen growth and spread in tissues. These findings indicate that IM (i.e., CCR2+ cells) promote granuloma-mediated control of persistent STm infection.
Our comprehensive histopathological analysis of chronic hepatic lesions associated with persistent STm infection of NRAMP1+ mice indicates that this type of systemic salmonellosis is characterized by the formation of granulomas and the induction of EMH, Kupffer cell hyperplasia, and various reactive inflammatory changes. These findings are consistent with the notion that persistent infections usually involve a complex balance between protective immunity and immunopathology.
Effective control and clearance of intracellular bacterial pathogens generally require a type 1 immune response that involves the recruitment of pathogen-specific T cells, particularly Th1 cells. These cells coordinate and amplify the host response to the pathogen through classical activation of macrophages, heightening their microbicidal functions. In several clinically important infections, however, the bacteria are sufficiently resistant to the microbicidal effects of classically activated (M1) macrophages such that they are incompletely eliminated by them. Some can even set up long-term residence in the macrophages and incapacitate them. This gives rise to a low level of infection (paucibacillary) that requires an ongoing Th1 response to limit pathogen growth and spread. In this circumstance, the chronic coordination between Th1 cells and macrophages underlies the formation of granulomas (58–62). Whether granulomatous responses to persistent infections are harmful to the host is unknown (59).
Granulomas are the pathological hallmark of persistent infections caused by intracellular bacterial pathogens (59). Our findings indicate that the granulomatous response to persistent STm infection is at its core protective and that the benefits of the response far outweigh the costs associated with it. We postulate that IM promote the formation of granulomas and that, to some extent, constant replenishment of IM may be needed to maintain granulomas and sequester STm within discrete sites in tissues, impeding their growth and systemic spread. As the granulomatous response to STm evolves, it is possible that the heterogeneity among the CCR2+ cells changes and that CCR2+ cells other than IM (e.g., IM-derived cells, minor subsets of CD4 and CD8 T cells) could also be involved in granuloma-mediated control during later phases of the infection.
Granulomas that form during persistent STm infection contain both M1 and alternatively activated (M2) macrophages but are T cell sparse (43, 63, 64). Within these granulomas, most STm reside in M1 macrophages (63). However, persistence of STm in tissues is associated with residence in M2 macrophages (65–67). Recent studies have shown that STm employs the effector SteE to direct macrophage polarization toward an M2 state and establish a metabolic environment conducive to long-term persistence, a process that antagonizes tumor necrosis factor alpha (TNF-α)-mediated pathogen restriction (64, 68, 69). Interestingly, neutralization of TNF-α during the chronic phase of persistent salmonellosis results in the regression of already established granulomas and reactivation of STm growth and spread in tissues (64, 70, 71). Our finding that depletion of IM during the chronic phase of persistent salmonellosis similarly results in the regression of already established granulomas with resultant growth and spread of STm in tissues suggests that these cells may promote granuloma-mediated disease containment by producing TNF-α.
In summary, our work establishes that IM (i.e., CCR2+ cells) promote granuloma-mediated control of persistent STm infection, providing new insights into a host-pathogen standoff that is a stalemate. Future studies on how STm exploits host-protective granulomas for long-term persistence, expansion, and eventual dissemination will undoubtedly provide new insights into the role of IM in immunity and host defense and may be key to uncovering new therapies for a multitude of granulomatous diseases.
MATERIALS AND METHODS
Experimental model and subject details. (i) Mouse strains.
This study used C57BL/6J mice (The Jackson Laboratory, stock no. 000664; RRID, IMSR_JAX:000664), C57BL/6J Slc11a1/Nramp1G169 mice (46), B6.129S4-Ccr2tm1Ifc/J (Ccr2−/−) mice (The Jackson Laboratory, stock no. 004999; RRID, IMSR_JAX:004999), B6.129S4-Ccl2tm1Rol/J (Ccl2−/−) mice (The Jackson Laboratory, stock no. 004434; RRID, IMSR_JAX:004434), CCR2-GFP mice (54), and CCR2-DTR mice (54). Small breeding colonies of these strains of mice were maintained in the specific-pathogen-free animal facility at Stony Brook University. Mouse genotyping was performed by use of established protocols. Studies used both male and female mice aged 8 to 20 weeks, sex and age matched by group. All mice were housed in the Division of Laboratory Animal Resources at Stony Brook University. The animal facility at Stony Brook University is accredited by AAALAC and licensed by the USDA and the Department of Health of the State of New York.
(ii) Ethics statement.
Studies that used live mice were performed in accordance with relevant institutional and national guidelines and regulations. All experiments involving mice were approved by the Stony Brook University Institutional Animal Care and Use Committee (protocol number 2012-2075-11.12.21-R2-MI) and conformed to the relevant regulatory standards. Euthanasia of mice with carbon dioxide was conducted in accordance with the American Veterinary Medical Association Guidelines for Euthanasia of Animals (2013 Report of the AVMA Panel of Euthanasia). The Stony Brook University Animal Care and Use program operates in accordance with the U.S. Department of Agriculture Animal Welfare Act (1966), regulation (CFR, 2009), and policies; the Health Research Extension Act (1985); the Public Health Service Policy on Humane Care and Use of Laboratory Animals (2002); the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011), the New York State Law (Chapter II: Administrative Rules and Regulations, Chapter II, Part 55 State Sanitary Code 16), and other applicable federal, state, and local laws, regulations, policies, and guidelines.
(iii) Microbe strain and growth conditions.
This study used STm strain IR715 (72), a spontaneous nalidixic acid-resistant derivative of STm strain 14028. With the use of standard microbiological techniques, bacteria were grown aerobically overnight at 37°C in 3 mL of lysogeny broth or on lysogeny agar supplemented with 50 μg/mL of nalidixic acid (Alfa Aesar catalog no. J63550-14; CAS no. 3374-05-8).
Method details. (i) Mouse infections and treatments.
Naive mice were injected intravenously with 0.1 mL of PBS or 5 × 103 CFU of STm suspended in PBS. Tenfold serial dilutions of the inoculum were plated on lysogeny agar to confirm the inoculum titer. At various times after injection (up to day 60), tissues were harvested and processed for downstream analysis, and organ burden assays were performed to enumerate STm CFU. Where noted, mice were injected intraperitoneally with 0.1 mL of PBS or DT (List Biological Laboratories catalog no. 150) (20 ng/g of body weight) suspended in PBS. Death was an endpoint for some of the studies. Euthanasia of mice was performed by inhalation of carbon dioxide followed by cervical dislocation, a method that is consistent with the recommendations of the AVMA Panel on Euthanasia.
(ii) Flow-cytometric analysis.
Spleens were collected postmortem, immersed in RP-10 medium (RPMI 1640 medium supplemented with 10% fetal bovine serum, 0.2 M l-Gln, 0.1 M HEPES, 50 μM 2-mercaptoethanol), and passed through 70-μm nylon filters to obtain single-cell suspensions. Resulting samples were suspended in ACK lysing buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA [pH 7.4]), incubated for 3 min at room temperature to lyse red blood cells, washed twice in PBS, suspended in fluorescence-activated cell sorting (FACS) buffer (1% bovine serum albumin [BSA] in PBS), and passed through 70-μm nylon filters prior to cell staining.
Livers were collected postmortem and cut into small pieces that were then transferred into gentleMACS C tubes containing 10 mL of dissociation buffer (3 mM CaCl2, 5% BSA, and 3 U/mL collagenase type 2 [Worthington Biochemical Corporation catalog no. LS004176] in PBS). Resulting samples were twice subjected to the gentleMACS Dissociator program Liver_02 and incubated for 45 min at 37°C, with agitation. Samples were then twice subjected to the gentleMACS Dissociator program Heart_01 and filtered through 70-μm nylon filters to obtain single-cell suspensions. Cells were resuspended in 8 mL of a 44% Percoll solution (GE Healthcare catalog no. 17-5445-02) (100% Percoll solution is 9 parts Percoll and 1 part 10× PBS) that was then overlaid onto 5 mL of a 67% Percoll solution and centrifuged (1,600 rpm) for 20 min at room temperature, with the brake off. Cells at the interface between the 44% and 67% Percoll solutions were collected, added to 10 mL of RP-10 medium, washed, and resuspended in FACS buffer prior to staining.
Cells were stained in the presence of Fc block (purified anti-CD16/32 antibody; BioLegend catalog no. 101302; RRID, AB_312801) with peridinin chlorophyll protein (PerCP)-conjugated anti-CD11b antibody (BioLegend catalog no. 101230; RRID, AB_2129374), allophycocyanin (APC)-conjugated anti-Ly6C antibody (BioLegend catalog no. 128016; RRID, AB_1732076), phycoerythrin (PE)-conjugated anti-Ly6G antibody (BioLegend catalog no. 127608; RRID, AB_1186099), BV510-conjugated anti-CD11b antibody (BioLegend catalog no. 101263; RRID, AB_2629529), BV711-conjugated anti-CD4 antibody (BioLegend catalog no. 100549; RRID, AB_11219396), PerCP-conjugated anti-NK1.1 antibody (BioLegend catalog no. 108726; RRID, AB_2132707), PE-Cy7-conjugated anti-CD11c antibody (BioLegend catalog no. 117317; RRID, AB_493569), BV650-conjugated anti-F4/80 antibody (BioLegend catalog no. 123149; RRID, AB_2564589), BV421-conjugated anti-NK1.1 antibody (BioLegend catalog no. 108732; RRID, AB_2562218), BV785-conjugated anti-CD3 antibody (BioLegend catalog no. 100355; RRID, AB_2565969), BV711-conjugated anti-B220 (CD45R) antibody (BD Biosciences catalog no. 563892; RRID, AB_2738470), BV785-conjugated anti-Ly6G antibody (BioLegend catalog no. 127645; RRID, AB_2566317), PacBlue-conjugated anti-CD11c antibody (BioLegend catalog no. 117322; RRID, AB_755988), PerCP-conjugated anti-CD3 antibody (BioLegend catalog no. 100326; RRID, AB_893317), APC-Cy7-conjugated anti-CD8b antibody (BioLegend catalog no. 126619; RRID, AB_2563950), PE-Cy7-conjugated anti-I-Ab antibody (BioLegend catalog no. 116420; RRID, AB_10575296), PE-conjugated anti-CD301 antibody (BioLegend catalog no. 145704; RRID, AB_2561961), Alexa Fluor 700 dye (Life Technologies catalog no. A-20010), or Zombie Violet dye (BioLegend catalog no. 423114), or combinations thereof. Cells were then fixed with fixation buffer (BioLegend catalog no. 420801) and subjected to flow-cytometric analysis.
Data were acquired on a BD FACScan flow cytometer with the Cytek digital extra parameter upgrade (DxP8) and FlowJo Collectors’ Edition software or a Cytek Aurora (4 laser; 16V-14B-10YG-8R) spectral flow cytometer and were analyzed with FlowJo, LLC, software (https://www.flowjo.com).
(iii) Histopathological analysis.
Livers were collected postmortem, fixed in 10% buffered formalin for 4 h at room temperature and washed extensively in PBS before being embedded in paraffin. Embedded liver tissues were routinely sectioned (5 μm per section) and stained in the van der Velden laboratory or by HistoWiz, Inc. Histochemical staining with hematoxylin and eosin was used for observation of cell nuclei, cytoplasm, and tissue architecture. Histochemical staining with Masson’s trichrome was used for detailed observation of type I collagen and analysis of fibrillar collagen deposition. Immunohistochemical staining with anti-CD31 antibody (Cell Signaling Technology catalog no. 77699; RRID, AB_2722705) (1:100) was used for identification and analysis of vascular proliferations. Immunohistochemical staining with anti-cytokeratin-7 antibody (Abcam catalog no. ab181598) (1:8,000) was used for identification and analysis of biliary ductular reactions. Immunohistochemical staining with anti-F4/80 antibody (Invitrogen catalog no. 14-4801-82; RRID, AB_467558) (1:200) was used for analysis of Kupffer cell proliferation and macrophage recruitment to inflammatory foci.
Where noted, liver sections were subjected to multicolor enzymatic immunohistochemical staining. Specifically, embedded liver tissues were sectioned (5 μm per section), deparaffinized in xylene, and rehydrated by immersing samples in a series of ethanol solutions of decreasing concentrations. Heat-based antigen retrieval was performed using citrate-based antigen-unmasking solution (Vector Laboratories catalog no. H-3300; RRID, AB_2336226), with use of a microwave. Slides were allowed to cool for at least 30 min at room temperature before being rinsed in tap water. Using a hydrophobic pen, a boundary was drawn around the tissue sections, which were then incubated for 10 min at room temperature with 3% hydrogen peroxide buffer. Slides were then rinsed in tap water followed by 1× TBST buffer (20 mM Tris, 150 mM NaCl, 0.1% Tween 20).
To visualize STm, slides were incubated overnight at 4°C with 2.5% horse serum (Vector Laboratories catalog no. S-2012; RRID, AB_2336618), incubated with an avidin/biotin blocking kit (Vector Laboratories catalog no. SP-2001; RRID, AB_2336231) according to the manufacturer’s instructions, and incubated overnight at 4°C with anti-STm CSA-1 primary antibody (KPL BacTrace catalog no. 5310-0322; RRID, AB_2890923), diluted 1:1,000 in 1× Tris-buffered saline-Tween (TBST) buffer. Slides were then incubated for 30 min at room temperature with ready-to-use biotinylated horse anti-goat secondary antibody (Vector Laboratories catalog no. BP-9500; RRID, AB_2336123) according to the manufacturer’s instructions, incubated for 30 min at room temperature with Vectastain Elite ABC-HRP reagent (Vector Laboratories catalog no. PK-7100; RRID, AB_2336827), incubated for 1 min at room temperature with 3,3′-diaminobenzidine-containing horseradish peroxidase substrate solution (Dako catalog no. K3467) to develop the desired brown stain intensity, and rinsed in tap water.
To visualize CD4+ cells (e.g., Th1 cells), slides were incubated for 1h at room temperature with 2.5% horse serum, incubated with an avidin/biotin blocking kit according to the manufacturer’s instructions, and incubated overnight at 4°C with anti-CD4 primary antibody (Abcam catalog no. 183685; RRID, AB_2686917), diluted 1:500 in 1× TBST buffer. Slides were then incubated for 30 min at room temperature with ready-to-use biotinylated horse anti-rabbit secondary antibody (Vector Laboratories catalog no. BP-1100) according to the manufacturer’s instructions, incubated for 30 min at room temperature with Vectastain Elite ABC-HRP reagent, incubated for 5 min at room temperature with 3,3′-diaminobenzidine-containing horseradish peroxidase substrate solution to develop the desired brown stain intensity, and rinsed in tap water.
To visualize Ly6G+ cells (e.g., neutrophils), slides were incubated for 1 h at room temperature with 2.5% rabbit serum (Vector Laboratories catalog no. S-5000; RRID, AB_2336619), incubated with an avidin/biotin blocking kit according to the manufacturer’s instructions, and incubated overnight at 4°C with anti-Ly6G primary antibody (BioLegend catalog no. 127601; RRID, AB_1089179), diluted 1:500 in 1× TBST buffer. Slides were then incubated for 30 min at room temperature with ready-to-use biotinylated mouse adsorbed rabbit anti-rat secondary antibody (Vector Laboratories catalog no. BA-4001; RRID, AB_10015300) according to the manufacturer’s instructions, incubated for 30 min at room temperature with Vectastain Elite ABC-HRP reagent, incubated for 5 min at room temperature with 3,3′-diaminobenzidine-containing horseradish peroxidase substrate solution to develop the desired brown stain intensity, and rinsed in tap water.
To visualize CCR2+ cells (e.g., IM), slides were incubated for 3 to 4 h at 4°C with 5% BSA blocking solution, incubated with an avidin/biotin blocking kit according to the manufacturer’s instructions, and incubated overnight at 4°C with biotinylated anti-GFP antibody (GeneTex catalog no. GTX26658; RRID, AB_371422; detects both GFP and CFP), diluted 1:500 in 1× TBST buffer. Slides were then incubated for 30 min at room temperature with Vectastain Elite ABC-AP reagent (Vector Laboratories catalog no. AK-5000; RRID, AB_2336792), incubated for 30 min at room temperature with 5-bromo-4-chloro-3-indolyl phosphate- and nitro blue tetrazolium-containing alkaline phosphatase substrate solution (Vector Laboratories catalog no. SK-5400; RRID, AB_2336236) to develop the desired blue stain intensity, and rinsed in tap water.
Stained tissue sections were dehydrated by immersing samples in a series of ethanol solutions of increasing concentrations to xylene and mounted using a nonaqueous mounting medium, after which slides were digitized by HistoWiz, Inc. Blind, qualitative histopathological analysis of digitized slides was performed by a board-certified veterinary anatomic pathologist using the latest version of QuPath software (https://qupath.github.io).
Quantification and statistical analysis.
Quantification of multicolor immunohistochemical stains was performed by HistoWiz, Inc. Specifically, stained liver sections were analyzed using the latest version of Indica Labs HALO software (https://www.indicalab.com/halo). Digitized images were imported into the HALO database, tissue areas were detected using an automated tissue detection classifier, and detected regions were manually corrected to exclude folds, edge staining effects, and artifacts. Image analysis was performed using the Indica Labs Area Quantification v 2.1.3 algorithm. Color characteristics of the brown color used to visualize STm and the blue color used to visualize IM were identified from positive samples, and thresholds were set corresponding to low-, medium-, and high-intensity staining for GFP and STm within tissues. Results, including the total areas of strong positivity for each stain analyzed across the whole slide, as well as the average optical densities for each stain within the positively identified areas, were exported on an image-by-image basis for use in postprocessing statistical analysis. Representative regions of interest were also exported.
The significance of the results described in this study was determined by use of a number of different statistical analyses performed with Prism 6 software (GraphPad Software, Inc.; https://www.graphpad.com). All of the details regarding the statistical analysis of the data can be found in the figure legends. P values of less than 0.05 were considered statistically significant.
This study did not generate any new unique reagents, datasets, or codes.
ACKNOWLEDGMENTS
We thank Ferric Fang (University of Washington) for sharing C57BL/6J Slc11a1/Nramp1G169 mice, Eric Pamer (University of Chicago) for sharing CCR2-GFP and CCR2-DTR mice, Juei-Suei Chen for help with mouse breeding and colony maintenance, James Bliska (Dartmouth) for helpful discussions, Jorge Benach (Stony Brook University) for encouraging us to investigate the role of IM in immunity and host defense, and Brian Sheridan (Stony Brook University) for providing feedback on the manuscript. Public Health Service grants AI101221, AI140002, and AI153280 awarded by the National Institutes of Health to A.W.M.V.D.V. supported this research.
Footnotes
Supplemental material is available online only.
Contributor Information
Adrianus W. M. van der Velden, Email: a.vandervelden@stonybrook.edu.
Andreas J. Bäumler, University of California, Davis
REFERENCES
- 1.Monack DM. 2013. Helicobacter and salmonella persistent infection strategies. Cold Spring Harb Perspect Med 3:a010348. 10.1101/cshperspect.a010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monack DM, Mueller A, Falkow S. 2004. Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol 2:747–765. 10.1038/nrmicro955. [DOI] [PubMed] [Google Scholar]
- 3.Byndloss MX, Tsolis RM. 2016. Chronic bacterial pathogens: mechanisms of persistence. Microbiol Spectr 4:VMBG-0020-2015. 10.1128/microbiolspec.VMBF-0020-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nadelman RB, Hanincová K, Mukherjee P, Liveris D, Nowakowski J, McKenna D, Brisson D, Cooper D, Bittker S, Madison G, Holmgren D, Schwartz I, Wormser GP. 2012. Differentiation of reinfection from relapse in recurrent Lyme disease. N Engl J Med 367:1883–1890. 10.1056/NEJMoa1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloom BR, Atun R, Cohen T, Dye C, Fraser H, Gomez GB, Knight G, Murray M, Nardell E, Rubin E, Salomon J, Vassall A, Volchenkov G, White R, Wilson D, Yadav P. 2017. Tuberculosis. In Holmes KK, Bertozzi S, Bloom BR, Jha P (ed), Major infectious diseases, 3rd ed. World Bank, Washington, DC. [Google Scholar]
- 6.Crump JA, Luby SP, Mintz ED. 2004. The global burden of typhoid fever. Bull World Health Organ 82:346–353. [PMC free article] [PubMed] [Google Scholar]
- 7.Bhan MK, Bahl R, Bhatnagar S. 2005. Typhoid and paratyphoid fever. Lancet 366:749–762. 10.1016/S0140-6736(05)67181-4. [DOI] [PubMed] [Google Scholar]
- 8.Merican I. 1997. Typhoid fever: present and future. Med J Malaysia 52:299–308. [PubMed] [Google Scholar]
- 9.Gunn JS, Marshall JM, Baker S, Dongol S, Charles RC, Ryan ET. 2014. Salmonella chronic carriage: epidemiology, diagnosis, and gallbladder persistence. Trends Microbiol 22:648–655. 10.1016/j.tim.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine MM, Black RE, Lanata C. 1982. Precise estimation of the numbers of chronic carriers of Salmonella typhi in Santiago, Chile, an endemic area. J Infect Dis 146:724–726. 10.1093/infdis/146.6.724. [DOI] [PubMed] [Google Scholar]
- 11.Bharadwaj S, Anim JT, Ebrahim F, Aldahham A. 2009. Granulomatous inflammatory response in a case of typhoid fever. Med Princ Pract 18:239–241. 10.1159/000204357. [DOI] [PubMed] [Google Scholar]
- 12.Mert A, Tabak F, Ozaras R, Ozturk R, Aki H, Aktuglu Y. 2004. Typhoid fever as a rare cause of hepatic, splenic, and bone marrow granulomas. Intern Med 43:436–439. 10.2169/internalmedicine.43.436. [DOI] [PubMed] [Google Scholar]
- 13.Muniraj K, Padhi S, Phansalkar M, Sivakumar P, Varghese RG, Kanungo R. 2015. Bone marrow granuloma in typhoid fever: a morphological approach and literature review. Case Rep Infect Dis 2015:628028. 10.1155/2015/628028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narechania S, Duran M, Karivedu V, Gopalakrishna KV. 2015. A case of typhoid fever with hepatic granulomas and enteritis. Case Rep Pathol 2015:745461. 10.1155/2015/745461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pais P. 1984. A hepatitis like picture in typhoid fever. Br Med J (Clin Res Ed) 289:225–226. 10.1136/bmj.289.6439.225-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satti MB, Al-Freihi H, Ibrahim EM, Abu-Melha A, Al-Ghassab G, Al-Idrissi HY, Al-Sohaibani MO. 1990. Hepatic granuloma in Saudi Arabia: a clinicopathological study of 59 cases. Am J Gastroenterol 85:669–674. [PubMed] [Google Scholar]
- 17.Bronzan RN, Taylor TE, Mwenechanya J, Tembo M, Kayira K, Bwanaisa L, Njobvu A, Kondowe W, Chalira C, Walsh AL, Phiri A, Wilson LK, Molyneux ME, Graham SM. 2007. Bacteremia in Malawian children with severe malaria: prevalence, etiology, HIV coinfection, and outcome. J Infect Dis 195:895–904. 10.1086/511437. [DOI] [PubMed] [Google Scholar]
- 18.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. 2012. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379:2489–2499. 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gal-Mor O. 2018. Persistent infection and long-term carriage of typhoidal and nontyphoidal salmonellae. Clin Microbiol Rev 32:e00088-18. 10.1128/CMR.00088-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon MA. 2008. Salmonella infections in immunocompromised adults. J Infect 56:413–422. 10.1016/j.jinf.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Gordon MA. 2011. Invasive nontyphoidal Salmonella disease: epidemiology, pathogenesis and diagnosis. Curr Opin Infect Dis 24:484–489. 10.1097/QCO.0b013e32834a9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lokken KL, Walker GT, Tsolis RM. 2016. Disseminated infections with antibiotic-resistant non-typhoidal Salmonella strains: contributions of host and pathogen factors. Pathog Dis 74:ftw103. 10.1093/femspd/ftw103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nielsen MV, Sarpong N, Krumkamp R, Dekker D, Loag W, Amemasor S, Agyekum A, Marks F, Huenger F, Krefis AC, Hagen RM, Adu-Sarkodie Y, May J, Schwarz NG. 2012. Incidence and characteristics of bacteremia among children in rural Ghana. PLoS One 7:e44063. 10.1371/journal.pone.0044063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reddy EA, Shaw AV, Crump JA. 2010. Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect Dis 10:417–432. 10.1016/S1473-3099(10)70072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh AL, Phiri AJ, Graham SM, Molyneux EM, Molyneux ME. 2000. Bacteremia in febrile Malawian children: clinical and microbiologic features. Pediatr Infect Dis J 19:312–318. 10.1097/00006454-200004000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Ao TT, Feasey NA, Gordon MA, Keddy KH, Angulo FJ, Crump JA. 2015. Global burden of invasive nontyphoidal Salmonella disease, 2010. Emerg Infect Dis 21:941–949. 10.3201/eid2106.140999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaughlin PA, Bettke JA, Tam JW, Leeds J, Bliska JB, Butler BP, van der Velden AWM. 2019. Inflammatory monocytes provide a niche for Salmonella expansion in the lumen of the inflamed intestine. PLoS Pathog 15:e1007847. 10.1371/journal.ppat.1007847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen JW, Tam JW, Okan NA, Mena P, Furie MB, Thanassi DG, Benach JL, van der Velden AWM. 2012. Phenotypic, morphological, and functional heterogeneity of splenic immature myeloid cells in the host response to tularemia. Infect Immun 80:2371–2381. 10.1128/IAI.00365-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tam JW, Kullas AL, Mena P, Bliska JB, van der Velden AW. 2014. CD11b+ Ly6Chi Ly6G- immature myeloid cells recruited in response to Salmonella enterica serovar Typhimurium infection exhibit protective and immunosuppressive properties. Infect Immun 82:2606–2614. 10.1128/IAI.01590-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Khairallah C, Sheridan BS, van der Velden AWM, Bliska JB. 2018. CCR2(+) Inflammatory monocytes are recruited to Yersinia pseudotuberculosis pyogranulomas and dictate adaptive responses at the expense of innate immunity during oral infection. Infect Immun 86:e00782-17. 10.1128/IAI.00782-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lauvau G, Loke P, Hohl TM. 2015. Monocyte-mediated defense against bacteria, fungi, and parasites. Semin Immunol 27:397–409. 10.1016/j.smim.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rivera A, Siracusa MC, Yap GS, Gause WC. 2016. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol 17:356–363. 10.1038/ni.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, Lee SH, Mendez J, Paun A, Sacks DL, Peters NC. 2017. Divergent roles for Ly6C+CCR2+CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PLoS Pathog 13:e1006479. 10.1371/journal.ppat.1006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serbina NV, Jia T, Hohl TM, Pamer EG. 2008. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol 26:421–452. 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolowschiak T, Mueller AA, Pisan LJ, Feigelman R, Felmy B, Sellin ME, Namineni S, Nguyen BD, Wotzka SY, Heikenwalder M, von Mering C, Mueller C, Hardt W-D. 2016. IFN-gamma hinders recovery from mucosal inflammation during antibiotic therapy for Salmonella gut infection. Cell Host Microbe 20:238–249. 10.1016/j.chom.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 36.Griffin AJ, Li LX, Voedisch S, Pabst O, McSorley SJ. 2011. Dissemination of persistent intestinal bacteria via the mesenteric lymph nodes causes typhoid relapse. Infect Immun 79:1479–1488. 10.1128/IAI.01033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rydstrom A, Wick MJ. 2007. Monocyte recruitment, activation, and function in the gut-associated lymphoid tissue during oral Salmonella infection. J Immunol 178:5789–5801. 10.4049/jimmunol.178.9.5789. [DOI] [PubMed] [Google Scholar]
- 38.Rydstrom A, Wick MJ. 2009. Monocyte and neutrophil recruitment during oral Salmonella infection is driven by MyD88-derived chemokines. Eur J Immunol 39:3019–3030. 10.1002/eji.200939483. [DOI] [PubMed] [Google Scholar]
- 39.Toapanta FR, Bernal PJ, Fresnay S, Darton TC, Jones C, Waddington CS, Blohmke CJ, Dougan G, Angus B, Levine MM, Pollard AJ, Sztein MB. 2015. Oral wild-type Salmonella Typhi challenge induces activation of circulating monocytes and dendritic cells in individuals who develop typhoid disease. PLoS Negl Trop Dis 9:e0003837. 10.1371/journal.pntd.0003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauler TJ, Starr T, Nagy TA, Sridhar S, Scott D, Winkler CW, Steele-Mortimer O, Detweiler CS, Peterson KE. 2017. Salmonella meningitis associated with monocyte infiltration in mice. Am J Pathol 187:187–199. 10.1016/j.ajpath.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song J, Wilhelm CL, Wangdi T, Maira-Litran T, Lee S-J, Raetz M, Sturge CR, Mirpuri J, Pei J, Grishin NV, McSorley SJ, Gewirtz AT, Bäumler AJ, Pier GB, Galán JE, Yarovinsky F. 2016. Absence of TLR11 in mice does not confer susceptibility to Salmonella Typhi. Cell 164:827–828. 10.1016/j.cell.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsolis RM, Xavier MN, Santos RL, Baumler AJ. 2011. How to become a top model: impact of animal experimentation on human Salmonella disease research. Infect Immun 79:1806–1814. 10.1128/IAI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monack DM, Bouley DM, Falkow S. 2004. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNgamma neutralization. J Exp Med 199:231–241. 10.1084/jem.20031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez-Escobedo G, La Perle KM, Gunn JS. 2013. Histopathological analysis of Salmonella chronic carriage in the mouse hepatopancreatobiliary system. PLoS One 8:e84058. 10.1371/journal.pone.0084058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forbes JR, Gros P. 2001. Divalent-metal transport by NRAMP proteins at the interface of host-pathogen interactions. Trends Microbiol 9:397–403. 10.1016/S0966-842X(01)02098-4. [DOI] [PubMed] [Google Scholar]
- 46.Govoni G, Vidal S, Gauthier S, Skamene E, Malo D, Gros P. 1996. The Bcg/Ity/Lsh locus: genetic transfer of resistance to infections in C57BL/6J mice transgenic for the Nramp1 Gly169 allele. Infect Immun 64:2923–2929. 10.1128/iai.64.8.2923-2929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown DE, Libby SJ, Moreland SM, McCoy MW, Brabb T, Stepanek A, Fang FC, Detweiler CS. 2013. Salmonella enterica causes more severe inflammatory disease in C57/BL6 Nramp1G169 mice than Sv129S6 mice. Vet Pathol 50:867–876. 10.1177/0300985813478213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.DelGiorno KE, Tam JW, Hall JC, Thotakura G, Crawford HC, van der Velden AW. 2014. Persistent salmonellosis causes pancreatitis in a murine model of infection. PLoS One 9:e92807. 10.1371/journal.pone.0092807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karlinsey JE, Maguire ME, Becker LA, Crouch ML, Fang FC. 2010. The phage shock protein PspA facilitates divalent metal transport and is required for virulence of Salmonella enterica sv. Typhimurium. Mol Microbiol 78:669–685. 10.1111/j.1365-2958.2010.07357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaharik ML, Cullen VL, Fung AM, Libby SJ, Kujat Choy SL, Coburn B, Kehres DG, Maguire ME, Fang FC, Finlay BB. 2004. The Salmonella enterica serovar typhimurium divalent cation transport systems MntH and SitABCD are essential for virulence in an Nramp1G169 murine typhoid model. Infect Immun 72:5522–5525. 10.1128/IAI.72.9.5522-5525.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serbina NV, Pamer EG. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7:311–317. 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 52.Tsou C-L, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. 2007. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117:902–909. 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. 2008. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol 180:6846–6853. 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. 2009. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 6:470–481. 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Husain EH. 2011. Fulminant hepatitis in typhoid fever. J Infect Public Health 4:154–156. 10.1016/j.jiph.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 56.Pramoolsinsap C, Viranuvatti V. 1998. Salmonella hepatitis. J Gastroenterol Hepatol 13:745–750. 10.1111/j.1440-1746.1998.tb00726.x. [DOI] [PubMed] [Google Scholar]
- 57.Ramachandran S, Godfrey JJ, Perera MV. 1974. Typhoid hepatitis. JAMA 230:236–240. 10.1001/jama.1974.03240020026016. [DOI] [PubMed] [Google Scholar]
- 58.Pagan AJ, Ramakrishnan L. 2015. Immunity and immunopathology in the tuberculous granuloma. Cold Spring Harb Perspect Med 5:a018499. 10.1101/cshperspect.a018499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pagan AJ, Ramakrishnan L. 2018. The formation and function of granulomas. Annu Rev Immunol 36:639–665. 10.1146/annurev-immunol-032712-100022. [DOI] [PubMed] [Google Scholar]
- 60.Petersen HJ, Smith AM. 2013. The role of the innate immune system in granulomatous disorders. Front Immunol 4:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ravesloot-Chavez MM, Van Dis E, Stanley SA. 2021. The innate immune response to Mycobacterium tuberculosis infection. Annu Rev Immunol 39:611–637. 10.1146/annurev-immunol-093019-010426. [DOI] [PubMed] [Google Scholar]
- 62.Thakur A, Mikkelsen H, Jungersen G. 2019. Intracellular pathogens: host immunity and microbial persistence strategies. J Immunol Res 2019:1356540. 10.1155/2019/1356540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goldberg MF, Roeske EK, Ward LN, Pengo T, Dileepan T, Kotov DI, Jenkins MK. 2018. Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity 49:1090–1102.E7. 10.1016/j.immuni.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pham THM, Brewer SM, Thurston T, Massis LM, Honeycutt J, Lugo K, Jacobson AR, Vilches-Moure JG, Hamblin M, Helaine S, Monack DM. 2020. Salmonella-driven polarization of granuloma macrophages antagonizes TNF-mediated pathogen restriction during persistent infection. Cell Host Microbe 27:54–67.E5. 10.1016/j.chom.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, Mukundan L, Chawla A, Monack DM. 2013. Salmonella require the fatty acid regulator PPARdelta for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 14:171–182. 10.1016/j.chom.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCoy MW, Moreland SM, Detweiler CS. 2012. Hemophagocytic macrophages in murine typhoid fever have an anti-inflammatory phenotype. Infect Immun 80:3642–3649. 10.1128/IAI.00656-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nix RN, Altschuler SE, Henson PM, Detweiler CS. 2007. Hemophagocytic macrophages harbor Salmonella enterica during persistent infection. PLoS Pathog 3:e193. 10.1371/journal.ppat.0030193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gibbs KD, Washington EJ, Jaslow SL, Bourgeois JS, Foster MW, Guo R, Brennan RG, Ko DC. 2020. The Salmonella secreted effector SarA/SteE mimics cytokine receptor signaling to activate STAT3. Cell Host Microbe 27:129–139.E4. 10.1016/j.chom.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Panagi I, Jennings E, Zeng J, Günster RA, Stones CD, Mak H, Jin E, Stapels DAC, Subari NZ, Pham THM, Brewer SM, Ong SYQ, Monack DM, Helaine S, Thurston TLM. 2020. Salmonella effector SteE converts the mammalian serine/threonine kinase GSK3 into a tyrosine kinase to direct macrophage polarization. Cell Host Microbe 27:41–53.E6. 10.1016/j.chom.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mastroeni P, Skepper JN, Hormaeche CE. 1995. Effect of anti-tumor necrosis factor alpha antibodies on histopathology of primary Salmonella infections. Infect Immun 63:3674–3682. 10.1128/iai.63.9.3674-3682.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mastroeni P, Villarreal-Ramos B, Hormaeche CE. 1993. Effect of late administration of anti-TNF alpha antibodies on a Salmonella infection in the mouse model. Microb Pathog 14:473–480. 10.1006/mpat.1993.1046. [DOI] [PubMed] [Google Scholar]
- 72.Stojiljkovic I, Baumler AJ, Heffron F. 1995. Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol 177:1357–1366. 10.1128/jb.177.5.1357-1366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4. Download iai.00070-22-s0001.pdf, PDF file, 6.6 MB (6.8MB, pdf)