

Abstract
The deadliest complication of infection by Plasmodium parasites, cerebral malaria, accounts for the majority of malarial fatalities. Although our understanding of the cellular and molecular mechanisms underlying the pathology remains incomplete, recent studies support the contribution of systemic and neuroinflammation as the cause of cerebral edema and blood-brain barrier (BBB) dysfunction. All Plasmodium species encode an orthologue of the innate cytokine, Macrophage Migration Inhibitory Factor (MIF), which functions in mammalian biology to regulate innate responses. Plasmodium MIF (PMIF) similarly signals through the host MIF receptor CD74, leading to an enhanced inflammatory response. We investigated the PMIF-CD74 interaction in the onset of experimental cerebral malaria (ECM) and liver stage Plasmodium development by using a combination of CD74 deficient (Cd74−/−) hosts and PMIF deficient parasites. Cd74−/− mice were found to be protected from ECM and the protection was associated with the inability of brain microvessels to present parasite antigen to sequestered and pathogenic Plasmodium-specific CD8+ T cells. Infection of WT hosts with PMIF-deficient sporozoites or infection of Cd74−/− hosts with WT sporozoites impacted the survival of infected hepatocytes and subsequently reduced blood-stage associated inflammation, contributing to protection from ECM. We recapitulated these finding with a novel pharmacologic PMIF-selective antagonist that reduced PMIF/CD74 signaling and fully protected mice from ECM. These findings reveal a conserved mechanism for Plasmodium usurpation of host CD74 signaling and suggest a tractable approach for new pharmacologic intervention.
Introduction
Malaria caused by parasites of the genus Plasmodium is the most deadly parasitic disease, causing approximately half a million deaths annually (1). Plasmodium sporozoites enter the skin through the bite of infected Anopheles mosquitoes and transit through the bloodstream to invade the liver. A single infected hepatocyte produces tens of thousands of erythrocyte-infectious merozoites and initiates the erythrocytic cycle of infection. The subsequent erythrocytic stage of infection produces the disease’s clinical manifestations (2), including the most severe complication of P. falciparum infection: cerebral malaria leading to impaired consciousness, seizures, coma, and subsequent mortality (3). The experimental cerebral malaria (ECM) animal model by infection of susceptible C57BL/6J mice with Plasmodium berghei ANKA (PbA) reproduces many of the neurological signs and pathologic changes of human cerebral malaria (4). ECM is triggered by parasitized erythrocytes in the cerebral microvasculature leading to the production of inflammatory molecules such as IFN-γ, granzyme B, and perforin, and is associated with the recruitment and accumulation of effector CD8+ T cells in the CNS (5, 6).
Both host and parasite factors contribute to the pre- and erythrocytic stages of infection and severe malaria development. Plasmodium parasites express intricate strategies to evade immune detection and destruction. It is noteworthy that all Plasmodium species analyzed genetically encode an orthologue of the mammalian cytokine macrophage migration inhibitory factor (MIF) (7, 8). MIF sustains activation responses by promoting innate cell survival, which occurs by signaling through its cognate receptor CD74, leading to sustained ERK1/2 activation and reducing cellular p53 activity (9, 10, 11). Mammalian MIF also engage the non-cognate receptors CXCR2 and CXCR4 (12) Plasmodium MIF (PMIF) is highly conserved in all known Plasmodium genomes; for instance, only a single amino acid distinguishes murine Plasmodium berghei from human P. falciparum PMIF (7, 8). Recent evidence has implicated PMIF in the growth and development of liver-stage parasites (13, 14), and PMIF binds with high affinity to the host receptor CD74 (15, 16), which has been independently identified as a susceptibility factor for murine Plasmodium infection (17).
In the present study, we show that PbA infected Cd74−/− mice are resistant to ECM. Cerebral malaria onset further relies on the contribution of endothelial cell CD74, which is upregulated in the brains of infected mice in the presence of PMIF to promote parasite antigen presentation to brain-sequestered Plasmodium-specific CD8+ T cells. Mice infected with PbAmif- parasites were only resistant to ECM when infected with sporozoites, reinforcing the idea that liver-stage Plasmodium infection is critical for ECM development (18). In agreement with prior studies (13, 14), our data also support a central role for PMIF in Plasmodium liver infection. PMIF activates the hepatocellular host MIF receptor CD74 to inhibit the apoptosis of infected hepatocytes, thus promoting Plasmodium development and replication and leading to systemic and neuroinflamation. These findings were recapitulated by pharmacologic inhibition of the PMIF/CD74 interaction with a novel, PMIF-selective small molecule antagonist (19, 20) that reduced the survival of infected hepatocytes, decreased liver-stage parasite burden and fully protected mice from acute cerebral malaria.
Materials and Methods
Mice
Female WT or Cd74−/− C57BL/6J mice between 6–10 weeks of age were purchased from The Jackson Laboratory and used for the study. Cd8−/−Cd74−/− mice were obtained by crossing Cd8−/− with Cd74−/− mice. Swiss Webster mice were obtained from The Jackson Laboratory. All animals were maintained in a specific pathogen-free facility at Yale Animal Resource Center. All animal procedures followed federal guidelines and were approved by the Yale University Animal Care and Use Committee, approval number 2017-10929.
Parasites and infection
PbAWT (MR4), PbAmif- (Leiden Malaria Group) (15), or PbAWT-GFP-luciferase (MR4) parasites were cycled between Swiss Webster mice and Anopheles stephensi mosquitoes. For erythrocytic infection, cryopreserved stocks of infected red blood cells (iRBCs) were injected (106 iRBCs/mouse), and blood parasitemia was monitored by Giemsa-stained blood smears and flow cytometry (14). For the pre-erythrocytic stage infection, salivary gland sporozoites were extracted from infected mosquitoes on day 19 post-blood meal infection. WT or Cd74−/− C57BL/6J mice were infected by i.v. tail injection of 2000 PbAWT, PbAmif- or PbAWT-GFP-luciferase sporozoites, and blood patency was monitored beginning day 3 by blood smear and flow cytometry. Liver parasite burden was monitored at 48 h after infection using an IVIS imaging system (Caliper) or quantitative PCR (14).
For adoptive transfer, splenocytes were isolated six days after infection of WT or Cd74−/− mice infected with 106 PbAWT iRBC, CD8 T cells were purified with anti-CD8 (Ly-2, Miltenyi Biotech) according to the manufacturer’s protocol. 1×107 cells were transferred, i.v. into recipient C57BL6/J Cd8−/− or Cd8−/−Cd74−/− mice and mice infected three days after with 106 PbAWT iRBC.
Hepatocyte infection, apoptosis induction, Annexin V assay, and quantification of p53 by western blotting
For apoptosis assessment, 1×105 HepG2 cells (ATCC) were seeded in complete EMEM medium (ATCC) (10% FBS (Atlanta Biologicals), 1% streptomycin/penicillin (Thermo-Fisher), and infected with 2 ×103 PbAWT or PbAmif- sporozoites. 48 h after infection, cells were treated with 1 mM of SNP (Sodium Nitroprusside, Sigma) for 4 h or left untreated as a control. For PMIF pharmacologic inhibition, cells were treated with 26k (10 nM, 100 nM) or with an equivalent concentration of vehicle (0.1% DMSO) (Sigma) before infection. Cells then were detached with Acutase (MP Bio), and cell suspensions split for Western blot or Annexin V analysis. For Annexin V analyses, cells were stained with Pacific Blue-Annexin V and 7AAD (7-aminoactinomycin D) (Biolegend) before running in an LSRII cytometer (BD Biosciences). For quantification of p53 by western blotting, PbAWT-infected HepG2 hepatocytes were detached with Accutase (MP Biolabs) and pelleted. Western blots were performed by lysing cell pellets in RIPA buffer (ThermoFisher) according to standard protocols and using antibodies directed against p53-Ser15 (clone 1C12) or total p53 (pAb) (Cell Signaling Technology). For quantification, density signals were normalized to an anti-β-actin Ab (LICOR) and developed with anti-mouse or anti-rabbit Abs conjugated with HRP (LI-COR Biosciences). Membranes were visualized using an Odyssey-Fc imaging system (LI-COR Biosciences). Each western blot panel was developed from the same membrane that was re-probed after stripping.
Quantification of liver-stage PbAWT and PbAmif- infection, and Cd74 knockdown
HepG2 liver cells infected with PbAWT or PbAmif- sporozoites were lysed at 24 h or 48 h after infection, cellular proteins transferred to PVDF membranes (Millipore), and analyzed by western blotting using anti-CSP (MRA-100) and anti-MSP-1 (MRA-667) antibodies obtained from MR4 ATCC (Manassas, VA). β-actin was used as a loading control. For treatment with siRNA, hepatocytes were transfected with 15 pmol of siRNA (Ambion) targeting CD74 mRNA (3 target sequences in exon 2) or siCtrl (scrambled unrelated sequence) as a negative control. siRNA was complexed with Lipofectamine RNAimax reagent (ThermoFisher) and added to the cells for 24 h; cells then were infected with PbAWT sporozoites.
Murine PbA infection and 26k treatment
C57BL/6J mice received three i.p. injections of 26k (4-(3-methoxy-5-methylphenoxy)-2-(4-methoxyphenyl)-6-methylpyridine) (19), synthesized by CheminPharma LLC (Branford, Connecticut), at 80 mg/kg after dissolution in PEG400 (Sigma-Aldrich) in a sonicating water bath. HP-P-cyclodextrin (Sigma-Aldrich) was added to prepare a 4 mg/ml solution. Control mice received vehicle alone. Immediately after the first i.p. injection, mice were infected i.v. with 2×103 PbA-luc-GFP sporozoites; the second and third injections of 26k were given 24 h and 48 h later, and always after measurement of PbA liver burden. The PbA liver burden was quantified 48 h after infection by luminescence emission after luciferin injection (Perkin Elmer) using an IVIS apparatus (Caliper). Livers were excised 48 h after infection, the total RNA extracted and purified with Trizol (Life Technologies), and parasites quantified by RT-qPCR using primers for Pb18s.
Brain microvessel cross‐presentation assay
WT, Cd74−/−, Cd8−/− or Cd8−/−Cd74−/− C57BL/6J mice were infected by intravenous injection of sporozoites or by intraperitoneal injection of PbA infected red blood cells. Infected mice were sacrificed when the signs of ECM (head deviation, ataxia) were manifested. Control naïve mice were sacrificed contemporaneously with the experimental group. The technique for isolating brain microvessels and quantification of cross-presentation of the parasite-derived GAP50 epitope by LR-BSL8.4a reporter T cells (21) was performed according to the published protocol (22). Quantification of β-galactosidase activity by activated LR-BSL8.4a reporter cells was performed by using a luminescence β-galactosidase assay (ThermoFisher) and the resultant signal quantified with a microplate reader (Tecan).
Isolation of Brain-infiltrating lymphocytes for flow cytometry.
Naïve or infected WT and Cd74−/− C57BL/6J mice were sacrificed and perfused intra-cardially with 20 ml 1x DPBS (‘ ‘Dulbecco’s Phosphate Buffer Saline). Brains were minced in RPMI, digested with 10 μg/ml of DNase I (Sigma) and 0.5 mg/ml for 30 min at 37°C, and homogenized with a pestle then filtered with a 70 μm cell-strainer (BD Falcon). The suspension then was centrifuged at 200xg for 5min, and the pellet re-suspended in 90% Percoll (GE Healthcare) and overlaid with a 70%, 50% and 30% Percoll gradient. After centrifugation at 500xg for 10 min, the cell interphase was collected treated with RBC lysis buffer, washed once, and re-suspended in complete RPMI medium (containing 10% FBS (Atlanta Biologicals) and 1% penicillin-streptomycin (Thermo Fisher)). 100 μl of brain cell suspensions were stimulated with 1 μM of peptide (SQLLNAKYL) in the presence of 10 μg/ml Brefeldin A. After 5 h incubation at 37°C, the cells were centrifuged and washed once with 100 μl DPBS + 5% FBS. The cells then were re-suspended and incubated with FITC-labelled SQLLNAKYL-H-2b tetramer for 15 min on ice before staining with anti-CD8-PE Cy7 (clone 53–6.7, Biolegend), anti-CD4-PerCP Cy 5.5 (clone RM4, Biolegend), and anti-CD11a antibodies in the presence of FCR-block for 20 min on ice. Cells were washed, pelleted, and permeabilized by re-suspension in 100 μl Fix/Perm buffer on ice for 15 min. Cells then were washed once with 1x Perm Wash buffer (BD Bioscience) and stained with anti-IFN-γ-APC-Cy7 (clone XMG1.2, Biolegend) and GrmzB-Pe-Cy7 (clone NGZB, eBioescience) in 1x Perm Wash buffer (BD Bioesciences) for 20 min on ice. Finally, cells were washed, centrifuged once, and re-suspended in 200 μl PBS+5% FBS for flow cytometry. The data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed using the FlowJo software (version 10).
Patient Samples and PMIF-CD74 binding assay
Sera from a previously characterized Zambian cohort of P. falciparum-infected patients were used in the study (16, 23). The interaction between PMIF and CD74 was analyzed as previously described (20, 24). Briefly, 96-well plate were coated with 26 ng/ml of recombinant CD74 ectodomain (aa 114–243) in PBS and incubated overnight at 4°C. After washing with PBS/0.1% Tween-20, the plate was blocked with Superblock reagent (Pierce) for 2 hours. Biotinylated recombinant PMIF (5ng/ml) was incubated for 45 min, with human serum (diluted 1:1000) from a previously described repository of healthy donors or subjects with uncomplicated or complicated malaria (16). After washing and incubating with Streptavidin-HRP (Roche), the peroxidase substrate is 3,3’−5,5’-tetramethylbenzidine (TMB, Roche) was added, and after 20 min incubation, the reaction was stopped with 1N H2SO4/HCl. The results were expressed as the percentage of binding in the presence versus the absence of serum.
Statistical analysis
All statistical analysis was performed as described before (15), using Software Prism v.6.0, (GraphPad). Statistical significance was indicated at p values of less than 0.05, 0.01, or 0.001. All data were expressed as a mean ± SD of at least two independent experiments. Mouse survival times were analyzed by the Mantel-Cox log-rank test. All other data were first tested for Gaussian distribution of values using a D’Agostino-Pearson normality test. The statistical significance of differences was assessed using the Kruskal-Wallis or Mann–Whitney U test for non-parametric data distribution and ANOVA or Student’s t-test for parametric data.
Ethics Approval
All animal procedures followed federal guidelines and were approved by the Yale University Animal Care and Use Committee, approval number 2017-10929. De-identified human sera collected from a prior study were used for in vitro ELISA studies (Yale HIC #0804003730).
Data Availability
All study data are included in the article and SI Appendix.
Results
CD74 is overexpressed in the brain of PbA infected mice and contributes to ECM development.
We recently demonstrated that PMIF exerts its inflammatory effects by signaling through the host receptor CD74 (16). To examine the potential role of CD74 in the pathogenesis of ECM, we measured the expression of CD74 in PbA-infected mouse brains during ECM and observed an increase in Cd74 mRNA expression compared with uninfected mice (Fig. 1A). We next challenged WT and Cd74−/− mice with PbAWT iRBCs and assessed ECM development. While 100% of the WT mice exhibited neurological symptoms within 7–8 days after infection, PbA-infected Cd74−/− mice were fully protected from ECM and succumbed to hyperparasitemia only 30 days after infection (Fig. 1B, C and S1A). Moreover, we found no significant differences in parasitemia between PbAWT-infected WT or Cd74−/− mice during the asymptomatic blood-stage, suggesting that Cd74 deficiency does not affect parasite replication in the erythrocyte (Fig. S1B). The same results were observed in Cd74−/− mice infected with PbAmif- parasites, whic develop normally and achieve similar parasitemia in BALB/c or C57BL/6J mice (14–16) (Fig. S1C, D, and E). The protection of Cd74−/− mice was associated with the downregulation of IFN-γ, perforin, and granzyme B expression in the brains of Cd74−/− versus WT mice but without an appreciable difference in the quantity of brain sequestered parasites, suggesting that CD74 is necessary for the neuroinflammatory response to Plasmodium infection (Fig. 1D and S1F).
Fig. 1. CD74 is overexpressed in the brain of PbA infected mice and contributes to ECM development.
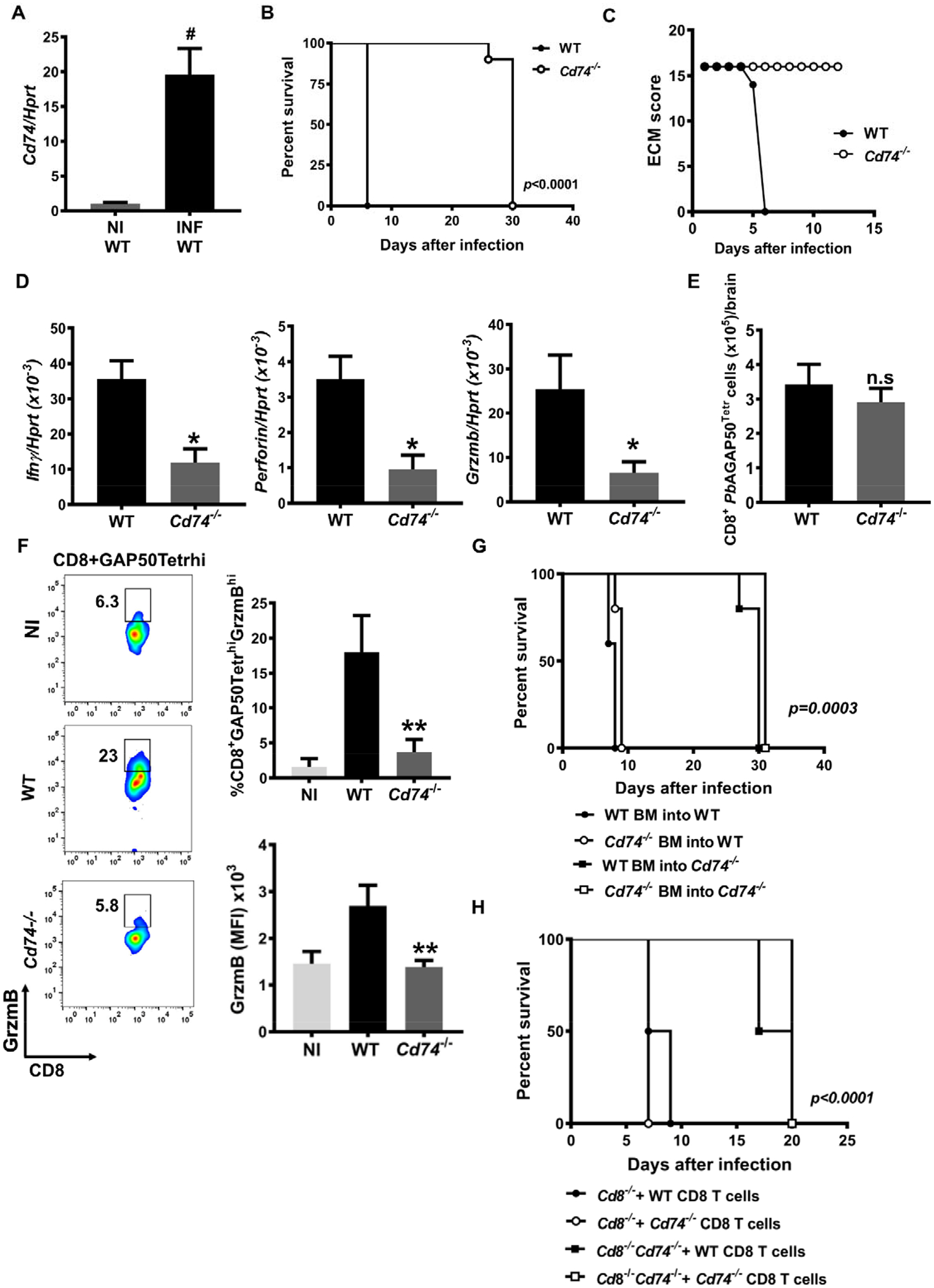
A, Cd74 mRNA expression measured by qPCR in the brains of wild type (WT) C57BL6/J mice that were either non-infected (NI WT) or infected with 106 PbA iRBC (INF WT). Results are shown as mean ± SD from two independent experiments with 6 animals per group and experiment: #p<0.0001 by Mann-Whitney test. CD74 deficient (Cd74−/−) and WT C57BL6/J mice were infected i.p with 106 PbA iRBC and the B, Kaplan–Meier survival plots for WT and Cd74−/− mice following infection with PbA and C, ECM malaria score were assessed. Data are from two pooled independent experiments with 10 animals per group; p < 0.0001 by log-rank (Mantel Cox) test. Bars represent the ± SD of n=10 WT and n=10 Cd74−/− mice and pooled from three independent experiments. D, Transcriptional expression of IFNγ, perforin and granzyme B was measured in brain tissue of PbA infected WT and Cd74−/− mice on day 7 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments: *p=0.0159 and *p=0.0317 by two-tailed Mann-Whitney test. Brain infiltrating lymphocytes from WT and Cd74−/− PbA infected mice were isolated 7 days after infection, and the number of E, pathogenic tetramer-labeled brain CD8+ T cells (CD8+GAP50Tetrahi), expressing F, the proinflammatory marker GrzmB (CD8+GAP50TetrahiGrzmBhi) measured by flow cytometry. Data are shown as mean ± SD of n=6 WT and n=6 Cd74−/− mice and pooled from two independent experiments; n.s.=non-significant; *p=0.0022 by two-tailed Mann-Whitney test. G, Kaplan-Meier survival of mixed BM chimeric mice after i.p infection with with 106 PbAWT iRBC. BM chimeras were generated by reconstitution of lethally irradiated WT or Cd74−/− mice with BM cells from WT mice or Cd74−/− mice. Data are form two pooled independent experiments with a total of 6 animals per group; p=0.0003 by log-rank (Mantel Cox) test. H, Kaplan-Meier survival plots for C57BL6/J mice Cd8−/− and Cd8−/−Cd74−/− receiving CD8+ T cells isolated from PbA infected WT or Cd74−/− and infected with i.p injection with 106 PbAWT iRBC. Data are form three pooled independent experiments with 6 animals per group; p<0.0001 by log-rank (Mantel Cox) test.
CD8+ T cells are essential for ECM development and contribute directly to human cerebral malaria (4, 25) and ECM pathology (22). Thus, we investigated if CD8+ T cells from Cd74−/− mice have an impaired response to PbA infection. We measured the amount of brain sequestered CD8+ T cells responding to PbA using a T cell receptor tetramer specific to the PbAGAP50 antigen (21). Notably, the amount of brain-sequestered PbAGAP50-specific CD8+ T cells was not significantly different between WT and Cd74−/− mice (Fig. 1E), indicating that in Cd74−/− mice CD8+ T cells can be primed in the spleen. Nevertheless, in Cd74−/− mice, CD8 T cell effector functions were strongly suppressed, as indicated by the reduced frequency of PbGAP50-specific CD8+ T cells expressing the ECM-associated inflammatory molecule Granzyme B (Fig. 1F). Our findings suggest that PbA responsive CD8+ T cells from Cd74−/− mice undergo priming in the spleen and trafficking to the brain during PbA blood-stage infection but do not express the inflammatory effector response associated with ECM. CD4+ T cells are necessary for the pathogenic role of PbA responsive CD8+ T cells and ECM development (26). Cd74−/− mice have a lower number of thymic and peripheral CD4+CD8− T cells, and the peripheral CD4+ T cells express TCR at a reduced level (27). Nevertheless, CD4+ T cells from Cd74−/− mice respond to mitogenic stimuli as well as WT CD4+ T cells (28). We therefore examined the origin of the resistance to ECM in bone marrow chimeric mice generated by injecting bone marrow from WT or Cd74−/− mice into lethally irradiated WT or Cd74−/− mice. Seven weeks after bone marrow engraftment, we infected mice with PbAWT iRBC and followed parasitemia, cell survival and ECM manifestations. Infected Cd74−/− chimeras transplanted with Cd74−/− or WT bone marrow were resistant to ECM and succumbed by hyperparasitemia at day 30. Only, suggesting that resistance due to CD74 deficiency in bone marrow does not impair the T cell responses against infection and ECM development (Fig. 1G, S1G).
We also examined if a dysfunctional cytotoxic response in Cd74 deficient CD8+ T cells reduced ECM symptoms. For this, we adoptively transferred CD8+ T cells from WT or Cd74−/− PbA-infected mice into naïve Cd8−/− or Cd8−/−Cd74−/− recipient mice and infected them with PbA-infected red blood cells (iRBCs) three days later. Recipient Cd8−/− mice that received WT or Cd74−/− CD8+ T cells from PbA-infected mice showed signs of ECM and succumbed by day 10, whereas recipient Cd8−/− Cd74−/− mice receiving WT or Cd74−/− CD8+ T cells from PbAWT infected mice did not show ECM symptoms and succumbed by 20 days after infection (Fig. 1H). Together, these data support the conclusion that CD8+ T cells from Cd74−/− mice are primed by PbA antigens but cannot induce ECM, supporting the role of brain-expressed CD74 in the development of ECM pathology.
Cross-presentation of Plasmodium antigen by brain endothelium is CD74 dependent.
Brain vascular endothelium becomes activated during malaria infection with the ability to process and cross-present Plasmodium antigens (21), thereby contributing to the T cell effector response and inflammation that underlies ECM (22). In addition to the role of CD74 as the cognate MIF receptor (10), it functions intracellularly as the MHC class II invariant chain (29) and has been implicated in an MHC class I cross-presentation pathway for cytolytic T lymphocytes (CTL) (30). We hypothesized that CD74 expressed by activated brain endothelium may cross-present PbA antigens to prime infiltrating CD8+ T cells. Accordingly, we assessed the ability of PbA antigen-pulsed, brain-derived endothelium to activate T cells by employing the LR-BSL8.4a reporter T cell line that expresses LacZ in response to the PbA-GAP50 epitope (6). Cd74−/− brain-derived endothelial cells were less able to activate LR-BSL8.4a T cells in the presence of PbA antigens when compared to WT brain-derived endothelial cells (Fig. 2A). We confirmed these results by isolating brain microvessels from PbA-infected WT and Cd74−/− mice at the time of ECM development and incubating them with LR-BSL8.4a reporter T cells for measurement of LacZ expression. Microvessels from PbA-infected WT mice showed a greater ability to cross-present PbA antigens than microvessels from Cd74−/− mice (Fig. 2B), which may be related to the ability of CD74 to facilitate endolysosomal MHC I cross presentation (30) We next assessed cross-presentation of PbA antigens ex vivo by using brain microvessels from Cd8−/− or Cd8−/−Cd74−/− mice infected with PbA after adoptive transfer with CD8+ T cells from WT or Cd74−/− mice. The microvessels from recipient Cd8−/− mice receiving WT or Cd74−/− CD8+ T cells from PbA infected mice had a greater ability to cross-present PbA antigens (Fig. 2C). Together, these results indicate that CD74 expression by brain endothelial cells contributes to cross-presentation of PbA antigens to CD8+ T cells and to ECM development.
Fig. 2. Cross-presentation of Plasmodium antigen by brain endothelium is Cd74 dependent.
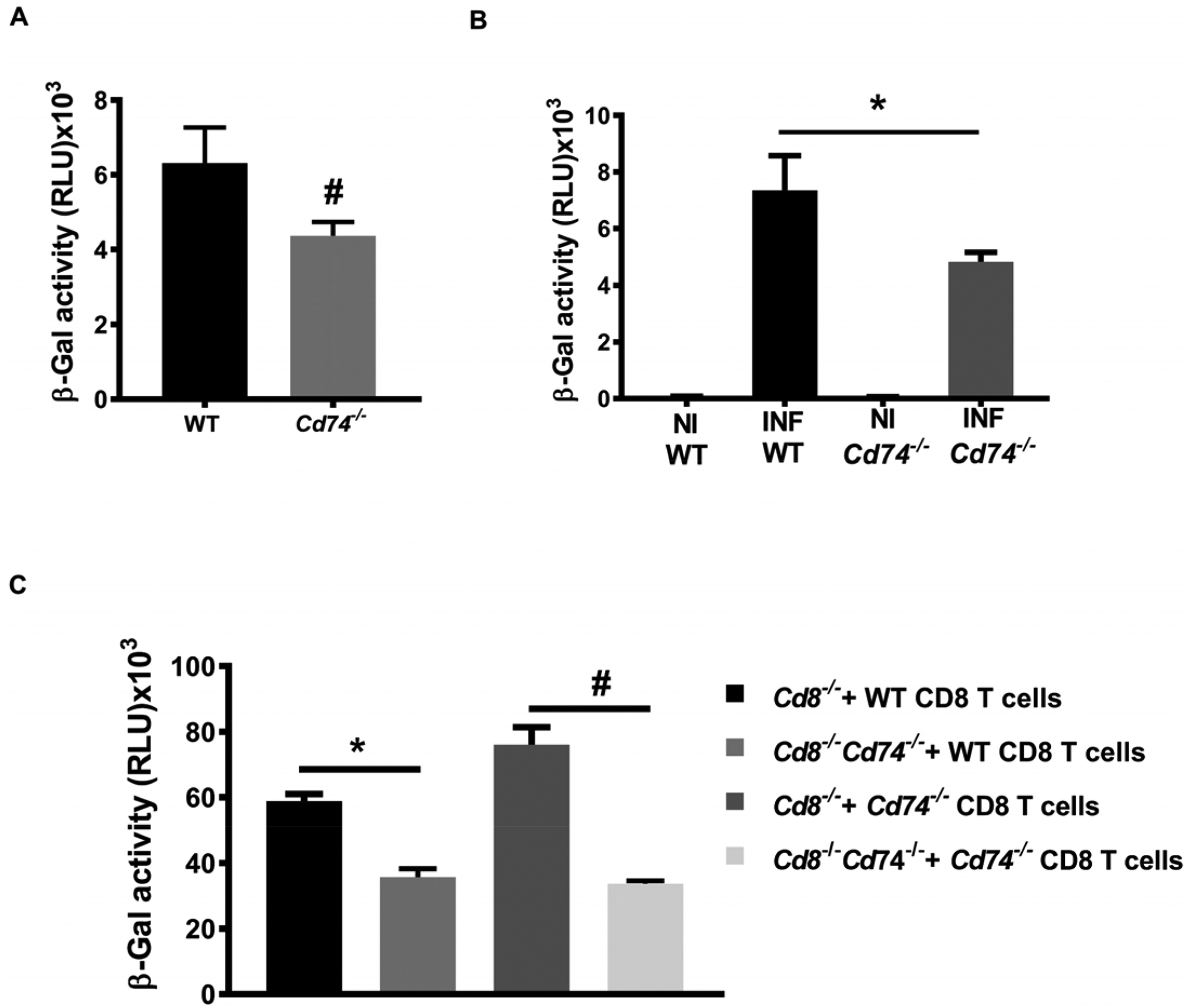
Cross-presentation of PbAGAP50 by brain endothelial cells. A, BEC isolated from WT and Cd74−/− mice were stimulated with 10 ng/ml IFNγ for 24 h, and then incubated for additional 24h with PbA mature stage iRBCS before co-culture with LR-BSL8.4a reporter cells overnight prior to β-galactosidase activity assessment. Data are shown as mean ± SD of three independent biological replicates performed in triplicate; #p<0.0001 by Mann-Whitney test. B, Brain microvessel cross-presentation of PbAGAP50 from naïve and PbA infected WT and Cd74−/− mice. Mice were infected with 106 PbA, iRBC and brain microvessels were isolated when WT-infected mice exhibited neurological signs at 7 days after infection and co-incubated with LR-BSL8.4a reporter cells for 24 h and then assessed for β-galactosidase activity. Data are shown as mean ± SD of n=6 mice per group and pooled from two independent biological replicates; **p=0.0021 by Mann-Whitney test. C, Brain microvessel cross-presentation of PbAGAP50 from Cd8−/− and Cd8−/−Cd74−/− receiving CD8+ T cells isolated from PbA infected WT or Cd74−/− mice three days before the infection with 106 PbAWT iRBC. Brain microvessels were isolated when the first infected mice exhibited neurological signs at 7 days after infection and co-incubated with LR-BSL8.4a reporter cells for 24 h and then assessed for β-galactosidase activity. Data are shown as mean ± SD of n=5 mice per group and pooled from two independent biological replicates; #p=0.0017 and **p<0.0001 by Kruskal-Wallis test.
PMIF contributes to the development of ECM by promoting PbA liver stage development.
Precedent studies have shown that PbA parasites genetically deficient in PMIF (PbAmif-) develop normally in their mosquito hosts and during blood-stage infection (15). To examine the contribution of PMIF to the development of ECM, we infected C57BL6/J mice with wild-type PbA (PbAWT) or PbAmif- iRBC. There was no difference in ECM manifestations and all the mice succumbed by day seven after infection (Fig. 3A) (14, 16). Recent studies have shown that liver-stage Plasmodium infection is critical for ECM development (18). We and others have demonstrated that PMIF is necessary for efficient Plasmodium liver-stage development of the parasite and that its absence impairs blood-stage patency (13, 14). Accordingly, mice infected with PbAmif- sporozoites did not exhibit ECM signs and survived until day 25 when compared with mice infected with PbAWT sporozoites; the later mice exhibited neurological symptoms followed by mortality 8–9 days after infection (Fig. 3B). We measured the expression of CD74 in the brains of infected mice during ECM and observed an increase in Cd74 mRNA expression in PbAWT infected mice when compared with PbAmif- infected mice (Fig. 3C). The expression of the inflammatory molecules IFN-γ, perforin, and granzyme B in the brains of mice infected with PbAWT sporozoites also was higher than in mice infected with PbAmif- sporozoites (Fig. 3D).
Fig. 3. PMIF contributes to the development of ECM.
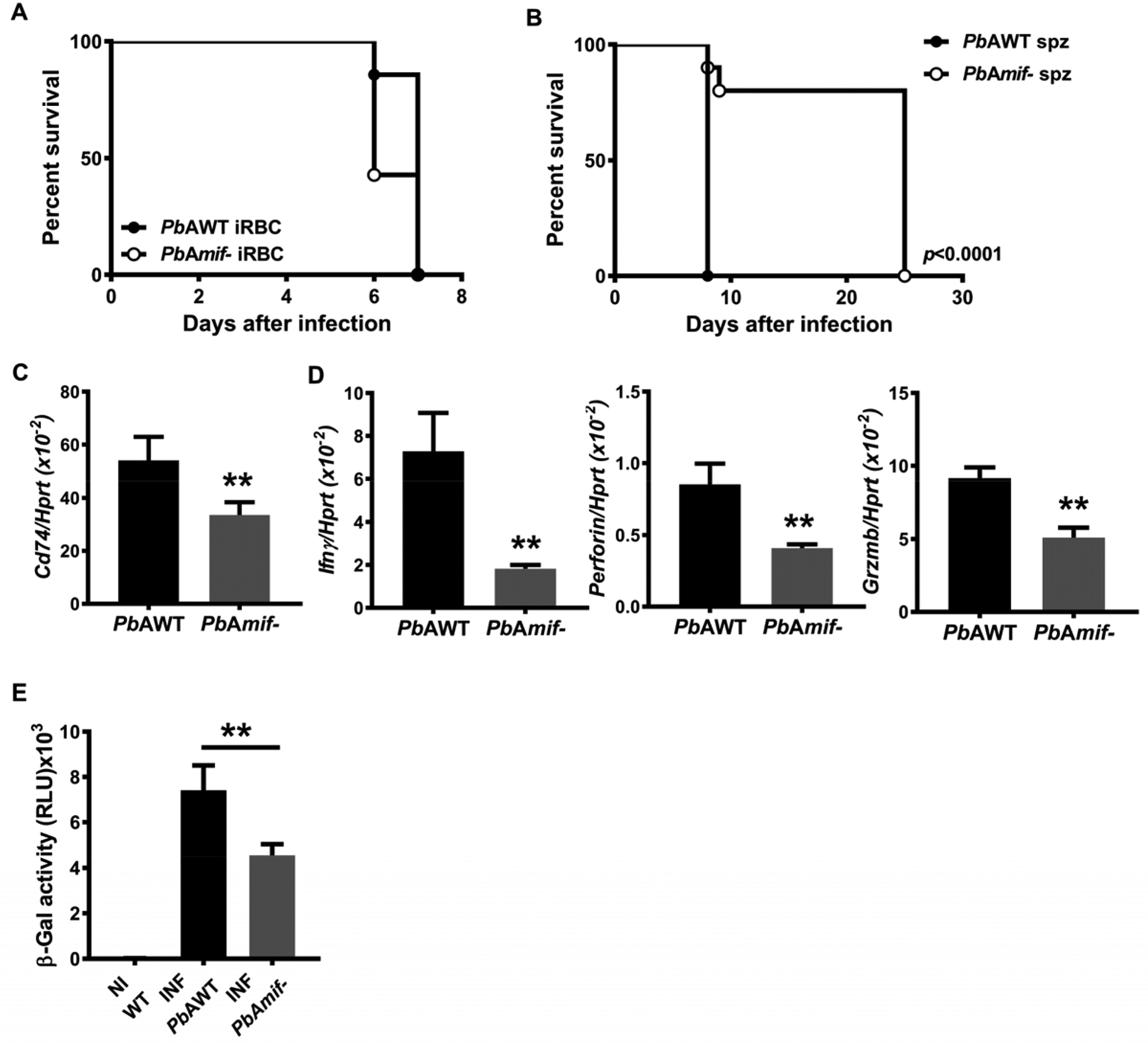
A, Kaplan-Meier survival plots for C57BL6/J mice infected with i.p with 106 PbAWT or PbAmif- iRBC. Data are form two pooled independent experiments with a total of 6 animals per group. n.s: non-significant by log-rank (Mantel Cox) test. B, Kaplan–Meier survival plots for C57BL6/J mice infected with 2×103 PbAWT or PbAmif- sporozoites. Data are from three pooled independent experiments with a total of 6 animals per group; p< 0.0001 by log-rank (Mantel Cox) test. WT C57BL6/J mice were infected i.v with 2×103 PbAWT or PbAmif- sporozoites. C, CD74 transcript expression in the brain of infected mice was measured by q-PCR at day 8 after infection. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; **p=0.0022 by Mann-Whitney test. D, Transcriptional expression of IFNγ, Perforin and Granzyme B was measured in brain tissue of PbAWT or PbAmif- infected WT mice on day 8 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; **p=0.0022 for Perforin and IFNγ and **p=0.0317 for Granzyme B, by two-tailed Mann-Whitney test. E, Brain microvessel cross-presentation of PbAGAP50 from naïve (NI) and PbAWT or PbAmif− infected WT mice. Mice were infected with PbAWT or PbAmif- sporozoites and brain microvessels were isolated when PbAWT-infected mice exhibited neurological signs at 7 days after infection. Microvessels then were co-incubated with LR-BSL8.4a reporter cells for 24 h and then assessed for β-galactosidase activity. Data are shown as mean ± SD of n=6 mice per group and pooled from two independent biological replicates; **p=0.0021 by Mann-Whitney test.
The systemic expression of the inflammatory cytokines IL-12 and IFN-γ is significantly higher in WT mice infected with PbAWT than in mice infected with PbAmif- (14, 16) (Fig. S2A). Precedent studies have shown that while host MIF circulates in high concentrations in Plasmodium-infected patients or mice, the genetic absence of MIF in infected mice does not influence the circulating levels of TNF-α or IFN-γ (31). We confirmed the inflammatory role of PMIF during PbA infection by infecting Mif−/− mice with PbAWT or PbAmif- parasites and measuring the circulating levels of IFNγ and IL-12. The levels of IL-12 and IFNγ were significantly higher in Mif−/− mice infected with PbAWT parasites than in mice infected with PbAmif-, supporting the important contribution of PMIF to inflammation during infection (Fig. S2B).
To evaluate the contribution of the PMIF/CD74 interaction to Plasmodium antigen cross-presentation and ECM development, we infected WT mice with PbAWT or PbAmif- sporozoites isolated from brain microvessels at the time of ECM onset and incubated them with LR-BSL8.4a reporter T cells. We observed that microvessels from PbAWT infected mice exhibited a greater ability to cross-present PbA antigens than microvessels from PbAmif- infected mice (Fig. 3E). Together, these data confirm the role of PMIF in liver-stage Plasmodium infection and the subsequent promotion of systemic inflammation and neuroinflammation during blood-stage infection and ECM development.
PMIF promotes Plasmodium-infected hepatocyte survival and p53 inhibition through host CD74.
Prior studies of Plasmodium-infected liver cells have implicated important pro-survival roles for hepatocyte growth factor signaling (32, 33) and the tumor suppressor p53 (34). Mammalian and parasite MIF molecules promote monocyte survival by decreasing p53 phosphorylation at Ser15, thereby countering cellular stress and programmed cell death (9, 11). Our results suggest that PMIF regulates PbA liver-stage development and promotes the development of ECM. We observed that intracellular parasite content was reduced in HepG2 cells infected with PbAmif- sporozoites compared with PbAWT sporozoites (Fig. 4A). We used circumsporozoite (CSP) and merozoite surface protein-1 (MSP-1) as indicators of parasite maturation (35). While CSP was expressed in similar levels (Fig. S3A), there was reduced expression of MSP-1 in the HepG2 cells infected with PbAmif- parasites, suggesting that PMIF is not necessary for hepatocyte infection but may have a permissive role in pre-erythrocytic parasite development (Fig S3B). To assess the mechanistic role of PMIF in liver-stage parasite development, we examined its effect on the survival of infected liver cells by treating infected HepG2 cells with the nitric oxide (NO) donor sodium nitroprusside (SNP) to induce p53 accumulation and apoptosis. We found that HepG2 cells infected with PbAmif- sporozoites were significantly more susceptible to NO-induced apoptosis than cells cultured with PbAWT parasites, despite a reduced infection level compared with PbAWT sporozoites (Fig. 4B). The protection from apoptosis observed in PbAWT infected HepG2 cells was associated with decreased phospho-p53Ser15 and intracellular p53 content compared with PbAmif- infected cells (Fig. 4C). Induction of apoptosis in PbAmif- versus PbAWT infected cells also was associated with increased Akt phosphorylation (Fig. S3C). We confirmed these in vitro findings by infecting mice with PbAWT or PbAmif- sporozoites. The livers of PbAmif- infected mice showed an 80% reduction in parasite burden compared with the livers of PbAWT infected mice, and this was associated with an attendant decrease in expression of the host pro-survival gene Bcl-2 and an increase in the expression of the pro-apoptotic gene Bad (Fig. S3D).
Fig. 4. PMIF promotes Plasmodium-infected hepatocyte survival and p53 inhibition through host CD74.
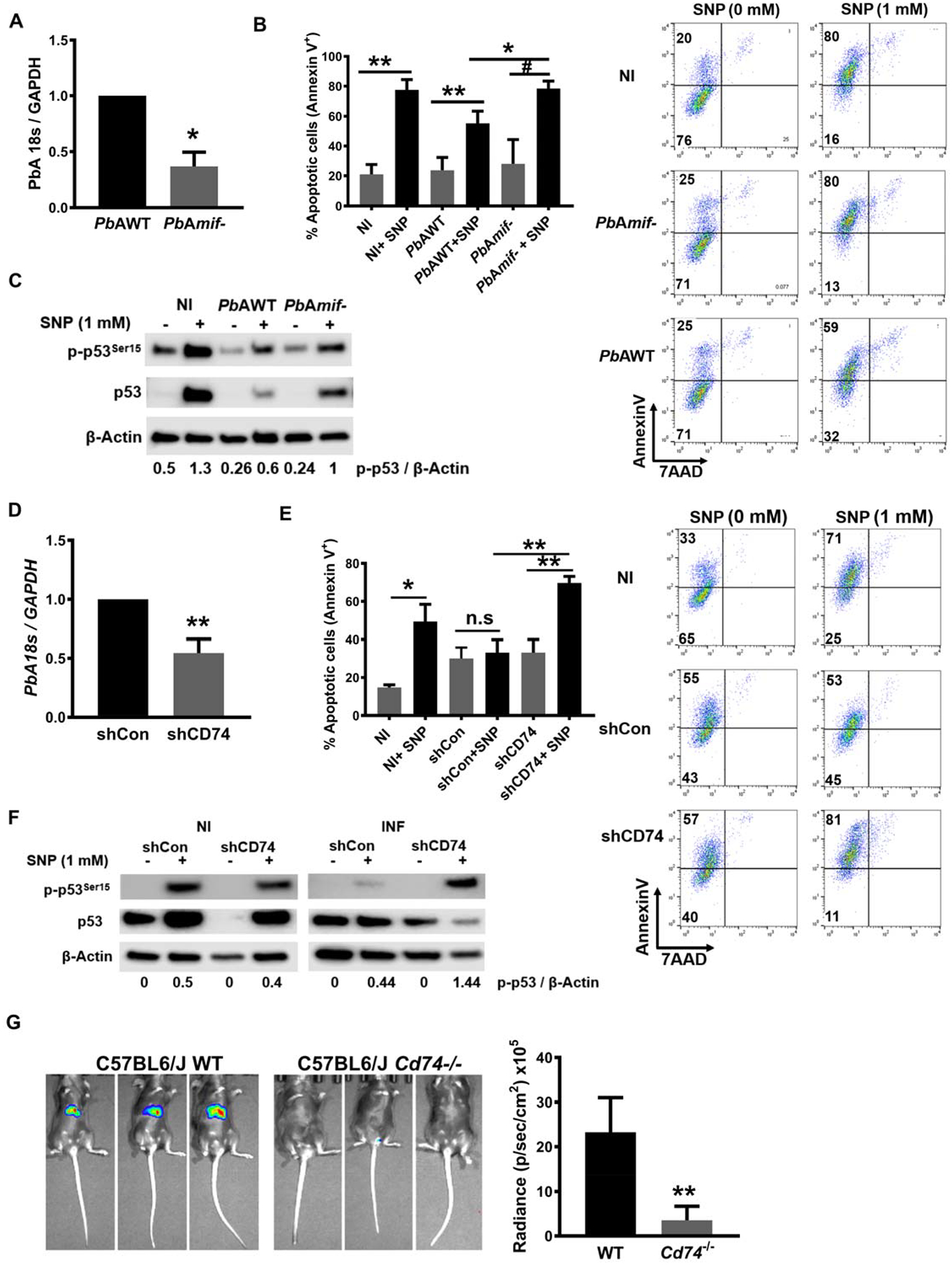
1×105 HepG2 cells were infected with 2×103 PbAWT or PbAmif- sporozoites. A, Parasite load was measured by quantitative PCR of PbA 18S rRNA relative to host GAPDH 48 h after infection. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; *p=0.0286 by Mann–Whitney test. 1×105 PbAWT or PbAmif- infected HepG2 cells, were cultured for 48 h and treated with 1 mM of the NO donor sodium nitroprusside (SNP) for 4 h to induce apoptosis. B, Percentage of apoptotic cells measured by AnnexinV and 7AAD (7-amino-actinomycin D) staining. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; *p=0.0133; **p=0.0011; Ѱp<0.0001; by Kruskal-Wallis test. C, Lysates of the same HepG2 cells in C were assessed for total p53 and p53Ser15 by Western blotting. NI: non-infected. Numerals represent the mean densitometric scanning ratios. Data are representative of two independent replicates experiments for the western blot analysis. 1×105 HepG2 cells were treated with 10 nM of shRNA directed at CD74 (shCD74) or a control shRNA (shCon), and infected 24 h later with 2×103 PbAWT sporozoites. D, Parasite load was measured by quantitative PCR of PbA 18S rRNA relative to host GAPDH. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; **p=0.002 by Mann–Whitney test. 1×105 shCon or shCd74 treated HepG2 cells, were infected with 2×103 PbAWT sporozoites and cultured for 48 h. shRNA cells were then treated with 1 mM of the NO donor SNP for 4 h to induce apoptosis E, Percentage of apoptotic (measured by AnnexinV and 7AAD staining) PbAWT infected HepG2 cells treated with shCon or shCD74. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; **p=0.0095 and n.s.=non-significant; by Kruskal-Wallis test. F, Lysates of the same hepatocytes as in E were assessed for total p53 and p53Ser15 by Western blotting with β-actin as loading control. NI: non-infected, INF: PbAWT infected. Numerals represent the mean densitometric scanning ratios. Data are representative of two independent replicate experiments for the western blot analysis. G, Wild type (WT) or Cd74−/− C57BL/6J mice were infected i.v. with 2×103 PbA-luciferase sporozoites and liver PbA-luc load quantified by luminescence at 48 h after infection. Bars represent the mean ± SD; **p=0.0022; by Mann-Whitney test.
We next confirmed the direct role of PMIF signaling through the host MIF receptor by studying sporozoite infection in HepG2 cells after the knockdown of CD74 (Fig. S3E). HepG2 cells treated with shCD74 to reduce CD74 expression had decreased parasite burden compared with treatment with a non-relevant shRNA (shCon) (Fig. 4D). As expected, PbAWT infected shCD74-treated cells were more susceptible to apoptosis than infected shCon-treated cells (Fig. 4E). Apoptosis induction also was associated with increased cellular p53Ser15 and p53 accumulation in the infected HepG2 cells with reduced CD74 expression (Fig. 4F). Infection of mice genetically deficient in CD74 (Cd74−/−) with PbAWT sporozoites revealed a significant reduction in the liver burden of Plasmodium parasites when compared to WT (Cd74+/+) mice (Fig. 4G), and this reduction was associated with a delay in blood-stage patency from 2 to 6 days post-infection (Fig. S3F). These results support the essential role of CD74 in mediating PMIF action and promoting Plasmodium pre-erythrocytic development leading to blood-stage infection.
Pharmacologic PMIF antagonism reduces PbA infection and protects against cerebral malaria.
Our experimental results support a central role in malaria infection for host CD74 and its activation by PMIF to promote the survival of infected hepatocytes, leading to inflammatory blood-stage infection and subsequent ECM pathophysiology. Anti-PMIF antibodies have been reported in malaria patients (36), and we assessed the ability of malaria-infected sera to interfere with PMIF binding to CD74 using an established ELISA employing the recombinant CD74 ectodomain (20, 24). Such sera inhibited PMIF binding to CD74 compared to sera from uninfected healthy controls (Fig. 5A). Moreover, sera from patients with clinically uncomplicated malaria were more effective in reducing PMIF/CD74 interaction than sera from those with complicated malaria (e.g., severe anemia, cerebral malaria (16)), suggesting that a more effective anti-PMIF serologic response may be associated with reduced inflammatory sequelae during infection.
Fig. 5. Inhibition of PMIF/CD74 axis is associated with protection from severe malaria.
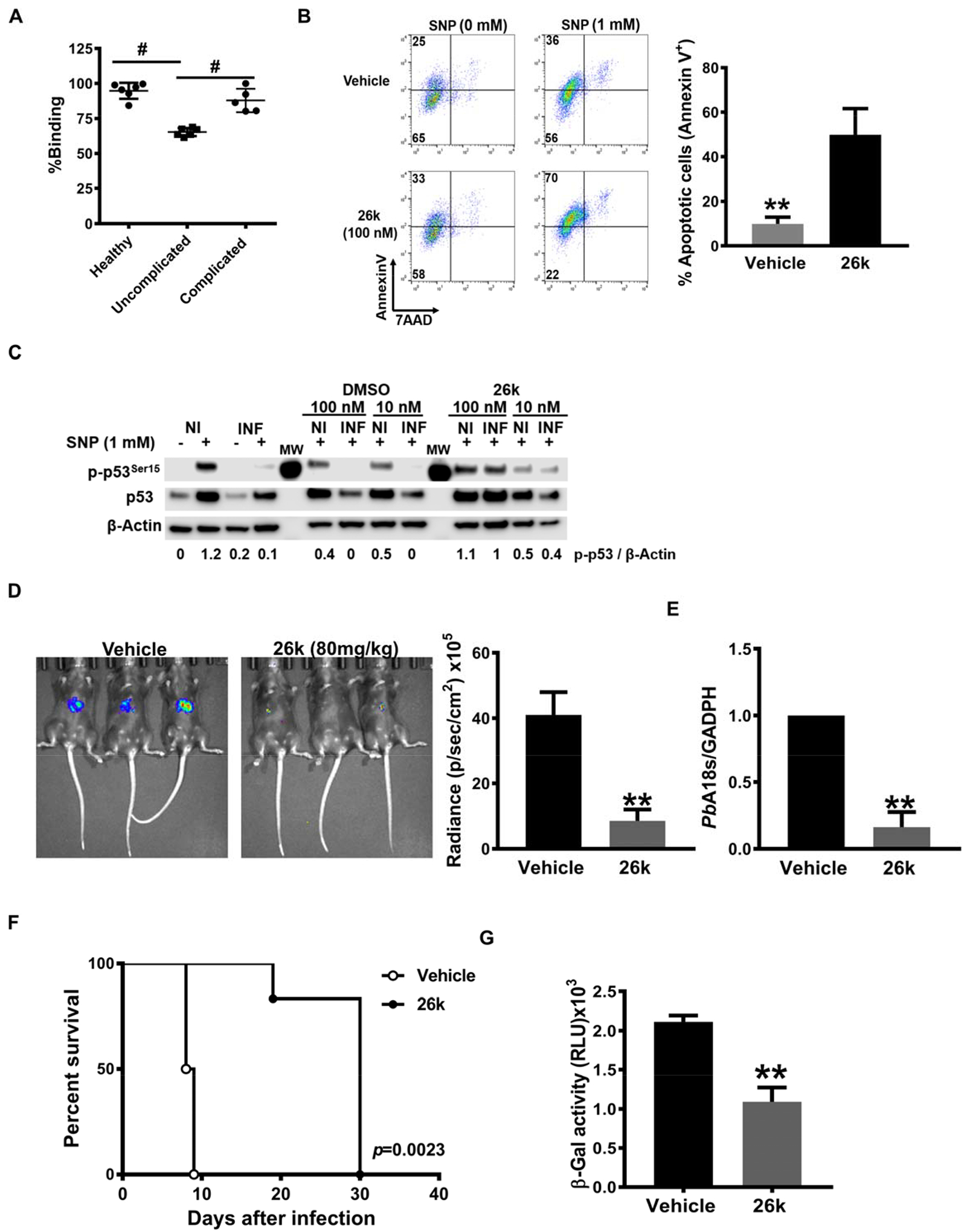
A. Effect of human serum on PMIF binding to the immobilized human CD74 ectodomain (aa 134–232). Measured values are relative to control without serum for each condition (n=6 healthy uninfected controls, n=6 uncomplicated malaria, n=6 complicated malaria. Mean±SD; #p<0.0001 by 1-way ANOVA. B, Percentage of apoptotic PbAWT infected HepG2 cells measured by AnnexinV and 7AAD staining after 26k or vehicle treatment. 1×105 PbAWT infected hepatocytes treated with 26K (100 nM) or vehicle were cultured for 48 h followed by the addition of the NO donor SNP (1 mM) for 4 h to induce apoptosis. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; *p=0.0011; by two-tailed Mann-Whitney test. C, PbAWT infected HepG2 cells were lysed and assessed for total p53 and p53Ser15 by Western blotting with β-actin as loading control. Numerals represent the mean densitometric scanning ratios. Data are representative of two independent replicate experiments for the western blot analysis. NI: non-infected, INF: PbA infected. C57BL/6J mice were treated with vehicle or 26k (80 mg/kg, ip) before (0 h), 24 h, and 48 h after i.v. infection with 2×103 PbA-luciferase sporozoites. D, Liver PbA-luc burden was quantified by luminescence and E, by qPCR of liver PbA 18S rRNA relative to host GAPDH 48 h after infection. Bars represent the mean ± SD; **p=0.0043 (D), **p=0.0079 (E); by Mann-Whitney test. F, Kaplan–Meier survival plots for vehicle and 26k treated mice following infection with PbA-luc sporozoites. Data are from two pooled independent experiments with 6 animals per group; **p = 0.0023 by log-rank (Mantel Cox) test. G, Brain microvessel cross-presentation of PbAGAP50 from vehicle and 26k treated mice following infection with PbA-luc sporozoites. Brains microvessels were isolated when vehicle-treated, infected mice exhibited neurological signs at 9 days after infection and co-incubated with LR-BSL8.4a reporter cells for 24 h and then assessed for β-galactosidase activity. Data are shown as mean ± SD of n=6 mice per group and pooled from two independent biological replicates; **p=0.0022 by two-tailed Mann-Whitney test.
Small molecule MIF inhibitors have been developed and are in clinical evaluation (37, 38). We recently identified a small molecule PMIF antagonist, termed 26k, that shows a 2500-fold greater selectivity for PMIF than for host MIF (Ki = 40 nM for PMIF versus Ki >100 μM for MIF) (19) (20) and blocks downstream ERK1/2 MAPK signaling (Fig. S4A). We treated PbA sporozoite infected HepG2 cells with 26k in vitro and measured parasite content by expression of PbA18s RNA together with sensitivity to apoptosis induction. Parasite burden decreased in the cells treated with 26k compared with vehicle (Fig. S4B). Hepatocytes infected with PbA and treated with 26k also were more susceptible to apoptosis, as evidenced by Annexin V staining (Fig. 5B). Moreover, 26k-treated cells showed increased p53 phosphorylation and intracellular accumulation, a known consequence of CD74 blockade (Fig. 5C) (9).
To determine the potential in vivo action of 26k, we treated C57BL/6J mice with 26k before infection with 2×103 PbA sporozoites and then continued treatment once daily for 2 days. We assessed liver-stage infection, blood-stage patency, and the development of ECM and lethality. Treatment with 26k markedly decreased parasite burden in the liver at 48 h after infection compared with vehicle controls (Fig. 5D, E). Sporozoite infection led to blood-stage patency after 3 days in vehicle-treated mice but not until day 5 in the 26k treated group (Fig. S4C). All vehicle-treated mice developed ECM symptoms (head deviation, ataxia, and paraplegia) 8 days after sporozoite infection, and all mice succumbed to cerebral malaria by days 9–10. By contrast, all mice treated with 26k were spared from cerebral malaria symptoms and did not succumb until after day 20 (Fig. 5F, S3D). We also examined the impact of 26k on Plasmodium antigen cross-presentation by brain microvascular endothelial cells. Brain microvessels from PbA infected WT mice were isolated at the time of ECM and treated in vitro with 26k (or vehicle) together with LR-BSL8.4a reporter T cells. Microvessels treated with 26k showed a reduction in PbA antigen cross-presentation as quantified by LacZ expression (Fig. 5G). Finally, we examined if 26k administration prevented ECM when mice were inoculated directly with iRBCs. We observed only partial protection from ECM in mice treated with 26k in this model (Fig. S4E).
Taken together, these results support the conclusion that pharmacologic inhibition of the PMIF/CD74 interaction may be a promising approach to protect from liver infection and, consequently, ECM.
Discussion
Plasmodium parasites have evolved highly specialized invasion strategies that function to evade immune destruction, sustain infection, and ensure completion of their life cycle. Our study highlights the importance of the pre-erythrocytic phase of infection in the development of the immune response and the subsequent progression of ECM. The present findings also emphasize the importance of investigating Plasmodium genes whose pathologic relevance may be underestimated based on the stage of the infection.
Our precedent studies reported that PMIF promotes inflammatory signaling through the host MIF receptor CD74 (16). Prior work has only implicated intracellular form of CD74 (e.g., the invariant chain) in MHC II expression, where its genetic deficiency, is associated with impaired B cell maturation and reduced CD4+ T cell numbers (27)
We show herein that PMIF increases the expression of CD74 in the brain of infected mice and serves a previously unforeseen role in the cross-presentation of Plasmodium antigens to promote a CD8+ T cell-mediated, pathologic inflammatory response. Increased IFNγ expression also is a feature of ECM pathology (26) and may further contribute to brain endothelial CD74 expression (39). While dendritic cells are considered the primary antigen-presenting cells responsible for activating CD4 and CD8 T cell responses against Plasmodium (40), endothelial cell activation contributes importantly to blood-brain barrier breakdown and neurological disease (6, 21). Our data further implicate PMIF, which appears to be universally expressed by the Plasmodium genus (30, 31), and its interaction with hepatocyte CD74 as an adaptive mechanism for sporozoites to usurp a host-protective apoptosis pathway to prevent their destruction and enable differentiation and patent infection.
Independent genetic studies indicate that relative increases in liver CD74 expression correlate with enhanced susceptibility to hepatocyte infection by sporozoites (17). Immunostaining studies suggest that PMIF is expressed on the surface of infective sporozoites and within the parasitophorous vacuole during liver stage development (13, 15). The precise localization of PMIF interaction with host CD74 is currently unclear; however, we suggest two possible scenarios: 1) by contact between PMIF on the invading sporozoite and CD74 expressed on the hepatocyte cell surface, or 2) after sporozoite internalization and contact between PMIF in the parasitophorous vacuole and endosomal-expressed CD74 during later-stage Plasmodium development (41). Either pathway could initiate activation of Akt and cellular pro-survival pathways (34). It is also notable that PMIF vaccination is associated with a more robust liver resident memory CD8+ T cell response, suggesting that the apoptotic destruction of infected hepatocytes promotes protective immunity (14).
We additionally show that the PMIF/CD74 interaction pathway is amenable to pharmacologic targeting. The PMIF selective antagonist 26k (28,29) recapitulates the experimental effects of genetic PMIF or CD74 deficiency, and short-term administration of 26k reduced PbA intrahepatic development and provided complete protection against cerebral malaria. Together with precedent co-crystallization studies supporting the selectivity of 26k for PMIF versus host MIF, these results provide proof-of-concept for pharmacologic PMIF antagonism as a tractable approach for malaria prophylaxis or liver-stage treatment, and potentially across a range of Plasmodium species and strains (29). Several studies have suggested a role for NLRP3-dependent IL-1β release in the pathogenesis of malaria (42, 43) and its inhibition protects against ECM (44). Interestingly, host MIF is required for NLRP3-dependent inflammasome activation in (45, 46). Whether PMIF hast a role in the inflammasome activation during malaria infection remains unexplored and has been suggested that exogenous MIF does not act on NLRP3 inflammasome (45). Nevertheless, our results show that PMIF induces inflammation and thus, its role in inflammasome activation and the impact of its inhibition by 26k needs further study. PMIF is highly conserved among Plasmodium species, with only five single nucleotide polymorphisms in PMIF among the 202 sequenced strains of P. falciparum present in the PlasmoDB resource (9, 10). This high degree of structural conservation may be auspicious for therapeutic targeting, particularly in a genetically complex pathogen prone to resistance development. Additional studies to optimize the absorption, distribution, metabolism, and excretion properties of 26k will be necessary to advance PMIF selective inhibitors such as 26K into clinical utility.
Supplementary Material
Fig. S1. CD74 is necessary for the development of ECM. A, Peripheral blood parasitemia for CD74 deficient (Cd74−/−) and WT C57BL6/J mice infected i.p with 106 PbA iRBC. B, Peripheral blood parasitemia during the 6 first days of infection. Data are shown as mean ± SD of n=10 WT and n=10 Cd74−/− mice and pooled from three independent experiments. Wild type (WT) and Cd74−/− C57BL6/J mice were infected i.p with 106 PbAmif− iRBC C, Kaplan–Meier survival plots for WT and Cd74−/− mice following infection with PbAmif−. Data are from two pooled independent experiments with 10 animals per group; p < 0.0001 by log-rank (Mantel Cox) test D, peripheral blood parasitemia. E, ECM malaria score was assessed as described before. Data are shown as mean ± SD of n=7 WT and n=10 Cd74−/− mice and pooled from 2 independent experiments. F, PbA18s transcript expression in the brain of infected WT and Cd74−/− measured by quantitative real-time PCR 7 days after infection was measured in brains tissue of PbA infected WT and Cd74−/− mice on day 7 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; n.s.: non-significant by Mann-Withney test.
Fig. S2. PMIF induces systemic inflammation in WT and Mif−/− mice. C57BL6/J mice were infected i.p with 106 PbAWT or PbAmif- iRBC and serum levels of IL-12 and IFNγ content were detected by specific ELISA 7 days after infection in A, WT and B, Mif−/− mice. Data are form two pooled independent experiments with a total of 6 animals per group. **p=0.0022 and *p=0.0152, by two-tailed Mann-Whitney test.
Fig. S3. PMIF influences PbA liver-stage development and promotes survival of Plasmodium-infected hepatocytes by inhibiting p53 activity. 1×105 HepG2 cells were infected with 2×103 PbAWT or PbAmif- sporozoites. Hepatocellular content of A, CSP and B, MSP-1 at 24 h and 48 h after infection with 2×103 PbAWT or PbAmif- sporozoites assessed by western blot relative to β-actin as loading control. Hepatocellular content of MSP-1 at 48 h after infection with 2×103 PbAWT or PbAmif- sporozoites was also assessed by western blot relative to PbAHSP70 as loading control. Cultured hepatocytes (1×105 HepG2 cells/well) infected with 2×103 PbAWT or PbAmif- sporozoites followed by the addition of 1 mM SNP at 4 h and 48 h after infection. C, Lysates were assessed for total Akt and pAktSer473 by Western blotting with β-actin as loading control. Numerals represent the densitometric scanning ratios. D, Transcriptional expression of IFNγ, Perforin and Granzyme B was measured in brain tissue of PbAWT or PbAmif- infected WT mice on day 8 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; **p=0.0022 for Perforin and IFNγ and **p=0.0317 for Granzyme B, by two-tailed Mann-Whitney test. 1×105 HepG2 cells were treated with 10 nM of shRNA directed at CD74 (shCD74) or a control shRNA (shCon), and infected 24 h later with 2×103 PbAWT sporozoites. E, Cd74 mRNA expression measured by qPCR in HepG2 after 48h treatment. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; **p= 0.002 by Mann-Whitney test. Wild type (WT) or Cd74−/− C57BL/6J mice were infected i.v. with 2×103 PbA-luciferase sporozoites and liver PbA-luc load quantified by luminescence at 48 h after infection. Bars represent the mean ± SD; **p=0.0022; by Mann-Whitney test. F, Kaplan-Meier plots showing the percentage of WT (●) and Cd74−/− (○) C57BL/6J mice with blood-stage patency following i.v. infection with 2×103 PbA-luc sporozoites. Patency was determined by microscopic enumeration of thin blood smears. Data are from two independent experiments, with 3–4 animals per group; p<0.0001 by Log-rank (Mantel Cox) test.
Fig. S4. The small molecule PMIF antagonist 26k reduces PMIF/CD74 signal transduction and protects from ECM. A. BMDM were treated with or without recombinant PMIF pre-incubated with vehicle control (DMSO) or small molecule 26k (10,50 or 100 nM) for 2h. Cells were lysed and the lysates assessed for total ERK and pERKThr202/Tyr204 by western blotting. Numerals represent the densitometric scanning ratios. HepG2 cells (1×105 cells/well) were infected with 2×103 PbAWT sporozoites and treated with 26k or vehicle. B, Hepatocellular parasite load was measured by quantitative PCR of PbA 18S rRNA relative to host GAPDH 48 h after treatment with 26k (0.5 nM to 50 μM) or vehicle. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; *p=0.0336, **p=0.0021, Ѱp=0.0008, #p<0.0001 by Kruskal-Wallis test. C57BL/6J mice were treated with vehicle or 26k (80 mg/kg, ip) before (0 h), 24 h, and 48 h after i.v. infection with 2×103 PbA-luciferase sporozoites. C. Kaplan-Meier plots showing the percentage of vehicle (o) and 26k (●) treated mice with blood-stage patency (determined by microscopic enumeration of thin blood smears) following i.v. infection with 2×103 PbA-luc sporozoites and D, ECM malaria score. Data are from two pooled independent experiments, with 6 animals per group p<0.0001 by Log-rank (Mantel Cox) test. C57BL/6J mice were treated with vehicle or 26k (80 mg/kg, ip) before (0 h) and then every two days after i.p infection with 106 PbAWT iRBCs. E, Kaplan–Meier survival plots for vehicle (o) and 26k (●) treated mice following infection with PbA iRBC. Data are from two pooled independent experiments with 8 total animals per group; p = 0.0137 by log-rank (Mantel Cox) test.
Significance statement.
Cerebral Malaria (CM) is the deadliest complication of Plasmodium infection. Therapies targeting CM symptoms have been unsuccessful. Thus, understanding the mechanisms underlying the pathology is necessary to find new therapies. We show that the Plasmodium ortholog of the cytokine MIF (PMIF) has a central role in Plasmodium’s ability to produce infection and cerebral malaria by activating the host MIF receptor, CD74. We demonstrate that inhibition of this interaction, with a selective PMIF inhibitor, leads to survival from cerebral malaria in mice.
Nonstandard abbreviations
- ECM
experimental cerebral malaria
- MIF
Macrophage Migration Inhibitory Factor
- BBB
Blood Brain Barrier
- PbA
Plasmodium berghei ANKA
- iRBCs
Infected Red Blood Cells
- CSP
CircumSporozoite Protein
- MSP-1
Merozoite Surface Protein-1
Footnotes
Conflict of Interest: The authors declare no competing interests.
References
- 1.WHO. (2019) World malaria report 2019. [Google Scholar]
- 2.Langhorne J, Ndungu FM, Sponaas A-M, and Marsh K (2008) Immunity to malaria: more questions than answers. Nature Immunology 9, 725. [DOI] [PubMed] [Google Scholar]
- 3.Milner DA, Whitten RO, Kamiza S, Carr R, Liomba G, Dzamalala C, Seydel KB, Molyneux ME, and Taylor TE (2014) The systemic pathology of cerebral malaria in African children. Frontiers in Cellular and Infection Microbiology 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riggle BA, Manglani M, Maric D, Johnson KR, Lee M-H, Neto OLA, Taylor TE, Seydel KB, Nath A, Miller LH, McGavern DB, and Pierce SK (2020) CD8+ T cells target cerebrovasculature in children with cerebral malaria. The Journal of Clinical Investigation 130, 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw TN, Stewart-Hutchinson PJ, Strangward P, Dandamudi DB, Coles JA, Villegas-Mendez A, Gallego-Delgado J, van Rooijen N, Zindy E, Rodriguez A, Brewer JM, Couper KN, and Dustin ML (2015) Perivascular Arrest of CD8+ T Cells Is a Signature of Experimental Cerebral Malaria. PLOS Pathogens 11, e1005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howland SW, Poh CM, and Rénia L (2015) Activated Brain Endothelial Cells Cross-Present Malaria Antigen. PLOS Pathogens 11, e1004963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ Jr., Treatman C, and Wang H (2008) PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Research 37, D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aurrecoechea C, Barreto A, Basenko EY, Brestelli J, Brunk BP, Cade S, Crouch K, Doherty R, Falke D, Fischer S, Gajria B, Harb OS, Heiges M, Hertz-Fowler C, Hu S, Iodice J, Kissinger JC, Lawrence C, Li W, Pinney DF, Pulman JA, Roos DS, Shanmugasundram A, Silva-Franco F, Steinbiss S, Stoeckert CJ Jr, Spruill D, Wang H, Warrenfeltz S, and Zheng J (2016) EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Research 45, D581–D591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, and Bucala R (2002) Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: Regulatory role in the innate immune response. Proceedings of the National Academy of Sciences 99, 345–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, and Bucala R (2003) MIF Signal Transduction Initiated by Binding to CD74. The Journal of Experimental Medicine 197, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamir D, Zierow S, Leng L, Cho Y, Diaz Y, Griffith J, McDonald C, Merk M, Mitchell RA, Trent J, Chen Y, Kwong Y-KA, Xiong H, Vermeire J, Cappello M, McMahon-Pratt D, Walker J, Bernhagen J, Lolis E, and Bucala R (2008) A Leishmania Ortholog of Macrophage Migration Inhibitory Factor Modulates Host Macrophage Responses. The Journal of Immunology 180, 8250–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, and Weber C (2007) MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature medicine 13, 587–596 [DOI] [PubMed] [Google Scholar]
- 13.Miller JL, Harupa A, Kappe SHI, and Mikolajczak SA (2012) Plasmodium yoelii Macrophage Migration Inhibitory Factor Is Necessary for Efficient Liver-Stage Development. Infection and Immunity 80, 1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baeza Garcia A, Siu E, Sun T, Exler V, Brito L, Hekele A, Otten G, Augustijn K, Janse CJ, Ulmer JB, Bernhagen J, Fikrig E, Geall A, and Bucala R (2018) Neutralization of the Plasmodium-encoded MIF ortholog confers protective immunity against malaria infection. Nature Communications 9, 2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Augustijn KD, Kleemann R, Thompson J, Kooistra T, Crawford CE, Reece SE, Pain A, Siebum AHG, Janse CJ, and Waters AP (2007) Functional Characterization of the Plasmodium falciparum and P. berghei Homologues of Macrophage Migration Inhibitory Factor. Infection and Immunity 75, 1116–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun T, Holowka T, Song Y, Zierow S, Leng L, Chen Y, Xiong H, Griffith J, Nouraie M, Thuma PE, Lolis E, Janse CJ, Gordeuk VR, Augustijn K, and Bucala R (2012) A Plasmodium-encoded cytokine suppresses T-cell immunity during malaria. Proceedings of the National Academy of Sciences 109, E2117–E2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaushansky A, Austin LS, Mikolajczak SA, Lo FY, Miller JL, Douglass AN, Arang N, Vaughan AM, Gardner MJ, and Kappe SHI (2015) Susceptibility to Plasmodium yoelii Preerythrocytic Infection in BALB/c Substrains Is Determined at the Point of Hepatocyte Invasion. Infection and Immunity 83, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato Y, Ries S, Stenzel W, Fillatreau S, and Matuschewski K (2019) The Liver-Stage Plasmodium Infection Is a Critical Checkpoint for Development of Experimental Cerebral Malaria. Frontiers in Immunology 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahlgren MK, Garcia AB, Hare AA, Tirado-Rives J, Leng L, Bucala R, and Jorgensen WL (2012) Virtual Screening and Optimization Yield Low-Nanomolar Inhibitors of the Tautomerase Activity of Plasmodium falciparum Macrophage Migration Inhibitory Factor. Journal of Medicinal Chemistry 55, 10148–10159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pantouris G, Rajasekaran D, Garcia AB, Ruiz VG, Leng L, Jorgensen WL, Bucala R, and Lolis EJ (2014) Crystallographic and Receptor Binding Characterization of Plasmodium falciparum Macrophage Migration Inhibitory Factor Complexed to Two Potent Inhibitors. Journal of Medicinal Chemistry 57, 8652–8656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, and Rénia L (2013) Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Molecular Medicine 5, 984–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howland SW, Claser C, Poh CM, Gun SY, and Rénia L (2015) Pathogenic CD8+ T cells in experimental cerebral malaria. Seminars in Immunopathology 37, 221–231 [DOI] [PubMed] [Google Scholar]
- 23.Thuma PE, van Dijk J, Bucala R, Debebe Z, Nekhai S, Kuddo T, Nouraie M, Weiss G, and Gordeuk VR (2011) Distinct clinical and immunologic profiles in severe malarial anemia and cerebral malaria in Zambia. The Journal of infectious diseases 203, 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, and Jorgensen WL (2009) Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J Med Chem 52, 416–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belnoue E, Kayibanda M, Vigario AM, Deschemin J-C, Rooijen N. v., Viguier M, Snounou G, and Rénia L (2002) On the Pathogenic Role of Brain-Sequestered αβ CD8+ T Cells in Experimental Cerebral Malaria. 169, 6369–6375 [DOI] [PubMed] [Google Scholar]
- 26.Villegas-Mendez A, Strangward P, Shaw TN, Rajkovic I, Tosevski V, Forman R, Muller W, and Couper KN (2017) Gamma Interferon Mediates Experimental Cerebral Malaria by Signaling within Both the Hematopoietic and Nonhematopoietic Compartments. Infection and immunity 85, e01035–01016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shachar I, Elliott EA, Chasnoff B, Grewal IS, and Flavell RA (1995) Reconstitution of invariant chain function in transgenic mice in vivo by individual p31 and p41 isoforms. Immunity 3, 373–383 [DOI] [PubMed] [Google Scholar]
- 28.Topilski I, Harmelin A, Flavell RA, Levo Y, and Shachar I (2002) Preferential Th1 immune response in invariant chain-deficient mice. Journal of immunology (Baltimore, Md. : 1950) 168, 1610–1617 [DOI] [PubMed] [Google Scholar]
- 29.Cresswell P (1994) Assembly, Transport, and Function of MHC Class II Molecules. Annual Review of Immunology 12, 259–291 [DOI] [PubMed] [Google Scholar]
- 30.Basha G, Omilusik K, Chavez-Steenbock A, Reinicke AT, Lack N, Choi KB, and Jefferies WA (2012) A CD74-dependent MHC class I endolysosomal cross-presentation pathway. Nature Immunology 13, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDevitt MA, Xie J, Ganapathy-Kanniappan S, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, Keefer J, David J, Leng L, and Bucala R (2015) A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med 212, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van de Sand C, Horstmann S, Schmidt A, Sturm A, Bolte S, Krueger A, Lütgehetmann M, Pollok J-M, Libert C, and Heussler VT (2005) The liver stage of Plasmodium berghei inhibits host cell apoptosis. Molecular Microbiology 58, 731–742 [DOI] [PubMed] [Google Scholar]
- 33.Leirião P, Albuquerque SS, Corso S, Van Gemert G-J, Sauerwein RW, Rodriguez A, Giordano S, and Mota MM (2005) HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cellular Microbiology 7, 603–609 [DOI] [PubMed] [Google Scholar]
- 34.Kaushansky A, Ye Albert S., Austin Laura S., Mikolajczak Sebastian A., Vaughan Ashley M., Camargo N, Metzger Peter G., Douglass Alyse N., MacBeath G, and Kappe Stefan H. I. (2013) Suppression of Host p53 Is Critical for Plasmodium Liver-Stage Infection. Cell Reports 3, 630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prudêncio M, Mota MM, and Mendes AM (2011) A toolbox to study liver stage malaria. Trends in Parasitology 27, 565–574 [DOI] [PubMed] [Google Scholar]
- 36.Han C, Lin Y, Shan G, Zhang Z, Sun X, Wang Z, Wei C, Deng Y, Zhang L, Bu L, Shao D, and Wang H (2010) Plasma concentration of malaria parasite-derived macrophage migration inhibitory factor in uncomplicated malaria patients correlates with parasitemia and disease severity. Clin Vaccine Immunol 17, 1524–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, Bucala R, Cappello M, Gross M, Gaeta F, Johnson K, and Lolis EJ (2010) Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci U.S.A 107, 11313–11318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fox RJ, Coffey CS, Cudkowicz ME, Gleason T, Goodman A, Klawiter EC, Matsuda K, McGovern M, Conwit R, Naismith R, Ashokkumar A, Bermel R, Ecklund D, Koepp M, Long J, Natarajan S, Ramachandran S, Skaramagas T, Thornell B, Yankey J, Agius M, Bashir K, Cohen B, Coyle P, Delgado S, Dewitt D, Flores A, Giesser B, Goldman M, Jubelt B, Lava N, Lynch S, Miravalle A, Moses H, Ontaneda D, Perumal J, Racke M, Repovic P, Riley C, Severson C, Shinnar S, Suski V, Weinstock-Gutman B, Yadav V, and Zabeti A (2016) Design, rationale, and baseline characteristics of the randomized double-blind phase II clinical trial of ibudilast in progressive multiple sclerosis. Contemp Clin Trials 50, 166–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanese K, Hashimoto Y, Berkova Z, Wang Y, Samaniego F, Lee JE, Ekmekcioglu S, and Grimm EA (2015) Cell Surface CD74-MIF Interactions Drive Melanoma Survival in Response to Interferon-γ. The Journal of investigative dermatology 135, 2775–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez-Ruiz D, Lau LS, Ghazanfari N, Jones CM, Ng WY, Davey GM, Berthold D, Holz L, Kato Y, Enders MH, Bayarsaikhan G, Hendriks SH, Lansink LIM, Engel JA, Soon MSF, James KR, Cozijnsen A, Mollard V, Uboldi AD, Tonkin CJ, de Koning-Ward TF, Gilson PR, Kaisho T, Haque A, Crabb BS, Carbone FR, McFadden GI, and Heath WR (2017) Development of a Novel CD4(+) TCR Transgenic Line That Reveals a Dominant Role for CD8(+) Dendritic Cells and CD40 Signaling in the Generation of Helper and CTL Responses to Blood-Stage Malaria. Journal of immunology (Baltimore, Md. : 1950) 199, 4165–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopes da Silva M, Thieleke-Matos C, Cabrita-Santos L, Ramalho JS, Wavre-Shapton ST, Futter CE, Barral DC, and Seabra MC (2012) The host endocytic pathway is essential for Plasmodium berghei late liver stage development. Traffic (Copenhagen, Denmark) 13, 1351–1363 [DOI] [PubMed] [Google Scholar]
- 42.Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza Mdo C, Franklin BS, Elian S, Martins FS, Pereira D, Reed G, Fitzgerald KA, Golenbock DT, and Gazzinelli RT (2014) Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog 10, e1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, and Fitzgerald KA (2014) Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep 6, 196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strangward P, Haley MJ, Albornoz MG, Barrington J, Shaw T, Dookie R, Zeef L, Baker SM, Winter E, Tzeng T-C, Golenbock DT, Cruickshank SM, Allan SM, Craig A, Liew FY, Brough D, and Couper KN (2018) Targeting the IL33–NLRP3 axis improves therapy for experimental cerebral malaria. 115, 7404–7409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, Fan H, Creed SJ, Deen NS, Traore DAK, Mueller I, Stanisic D, Baiwog FS, Skene C, Wilce MCJ, Mansell A, Morand EF, and Harris J (2018) Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat Commun 9, 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shin MS, Kang Y, Wahl ER, Park HJ, Lazova R, Leng L, Mamula M, Krishnaswamy S, Bucala R, and Kang I (2019) Macrophage Migration Inhibitory Factor Regulates U1 Small Nuclear RNP Immune Complex-Mediated Activation of the NLRP3 Inflammasome. Arthritis & rheumatology (Hoboken, N.J.) 71, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CD74 is necessary for the development of ECM. A, Peripheral blood parasitemia for CD74 deficient (Cd74−/−) and WT C57BL6/J mice infected i.p with 106 PbA iRBC. B, Peripheral blood parasitemia during the 6 first days of infection. Data are shown as mean ± SD of n=10 WT and n=10 Cd74−/− mice and pooled from three independent experiments. Wild type (WT) and Cd74−/− C57BL6/J mice were infected i.p with 106 PbAmif− iRBC C, Kaplan–Meier survival plots for WT and Cd74−/− mice following infection with PbAmif−. Data are from two pooled independent experiments with 10 animals per group; p < 0.0001 by log-rank (Mantel Cox) test D, peripheral blood parasitemia. E, ECM malaria score was assessed as described before. Data are shown as mean ± SD of n=7 WT and n=10 Cd74−/− mice and pooled from 2 independent experiments. F, PbA18s transcript expression in the brain of infected WT and Cd74−/− measured by quantitative real-time PCR 7 days after infection was measured in brains tissue of PbA infected WT and Cd74−/− mice on day 7 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; n.s.: non-significant by Mann-Withney test.
Fig. S2. PMIF induces systemic inflammation in WT and Mif−/− mice. C57BL6/J mice were infected i.p with 106 PbAWT or PbAmif- iRBC and serum levels of IL-12 and IFNγ content were detected by specific ELISA 7 days after infection in A, WT and B, Mif−/− mice. Data are form two pooled independent experiments with a total of 6 animals per group. **p=0.0022 and *p=0.0152, by two-tailed Mann-Whitney test.
Fig. S3. PMIF influences PbA liver-stage development and promotes survival of Plasmodium-infected hepatocytes by inhibiting p53 activity. 1×105 HepG2 cells were infected with 2×103 PbAWT or PbAmif- sporozoites. Hepatocellular content of A, CSP and B, MSP-1 at 24 h and 48 h after infection with 2×103 PbAWT or PbAmif- sporozoites assessed by western blot relative to β-actin as loading control. Hepatocellular content of MSP-1 at 48 h after infection with 2×103 PbAWT or PbAmif- sporozoites was also assessed by western blot relative to PbAHSP70 as loading control. Cultured hepatocytes (1×105 HepG2 cells/well) infected with 2×103 PbAWT or PbAmif- sporozoites followed by the addition of 1 mM SNP at 4 h and 48 h after infection. C, Lysates were assessed for total Akt and pAktSer473 by Western blotting with β-actin as loading control. Numerals represent the densitometric scanning ratios. D, Transcriptional expression of IFNγ, Perforin and Granzyme B was measured in brain tissue of PbAWT or PbAmif- infected WT mice on day 8 after infection by quantitative real-time PCR. Results are expressed as mean ± SD of n=6 mice per group pooled from two experiments; **p=0.0022 for Perforin and IFNγ and **p=0.0317 for Granzyme B, by two-tailed Mann-Whitney test. 1×105 HepG2 cells were treated with 10 nM of shRNA directed at CD74 (shCD74) or a control shRNA (shCon), and infected 24 h later with 2×103 PbAWT sporozoites. E, Cd74 mRNA expression measured by qPCR in HepG2 after 48h treatment. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; **p= 0.002 by Mann-Whitney test. Wild type (WT) or Cd74−/− C57BL/6J mice were infected i.v. with 2×103 PbA-luciferase sporozoites and liver PbA-luc load quantified by luminescence at 48 h after infection. Bars represent the mean ± SD; **p=0.0022; by Mann-Whitney test. F, Kaplan-Meier plots showing the percentage of WT (●) and Cd74−/− (○) C57BL/6J mice with blood-stage patency following i.v. infection with 2×103 PbA-luc sporozoites. Patency was determined by microscopic enumeration of thin blood smears. Data are from two independent experiments, with 3–4 animals per group; p<0.0001 by Log-rank (Mantel Cox) test.
Fig. S4. The small molecule PMIF antagonist 26k reduces PMIF/CD74 signal transduction and protects from ECM. A. BMDM were treated with or without recombinant PMIF pre-incubated with vehicle control (DMSO) or small molecule 26k (10,50 or 100 nM) for 2h. Cells were lysed and the lysates assessed for total ERK and pERKThr202/Tyr204 by western blotting. Numerals represent the densitometric scanning ratios. HepG2 cells (1×105 cells/well) were infected with 2×103 PbAWT sporozoites and treated with 26k or vehicle. B, Hepatocellular parasite load was measured by quantitative PCR of PbA 18S rRNA relative to host GAPDH 48 h after treatment with 26k (0.5 nM to 50 μM) or vehicle. Data are from three independent experiments performed in duplicate. Bars represent the mean ± SD; *p=0.0336, **p=0.0021, Ѱp=0.0008, #p<0.0001 by Kruskal-Wallis test. C57BL/6J mice were treated with vehicle or 26k (80 mg/kg, ip) before (0 h), 24 h, and 48 h after i.v. infection with 2×103 PbA-luciferase sporozoites. C. Kaplan-Meier plots showing the percentage of vehicle (o) and 26k (●) treated mice with blood-stage patency (determined by microscopic enumeration of thin blood smears) following i.v. infection with 2×103 PbA-luc sporozoites and D, ECM malaria score. Data are from two pooled independent experiments, with 6 animals per group p<0.0001 by Log-rank (Mantel Cox) test. C57BL/6J mice were treated with vehicle or 26k (80 mg/kg, ip) before (0 h) and then every two days after i.p infection with 106 PbAWT iRBCs. E, Kaplan–Meier survival plots for vehicle (o) and 26k (●) treated mice following infection with PbA iRBC. Data are from two pooled independent experiments with 8 total animals per group; p = 0.0137 by log-rank (Mantel Cox) test.
Data Availability Statement
All study data are included in the article and SI Appendix.