

Abstract
The linker histone H1 is a highly prevalent protein that compacts chromatin and regulates DNA accessibility and transcription. However, the mechanisms behind H1 regulation of transcription factor (TF) binding within nucleosomes are not well understood. Using in vitro fluorescence assays, we positioned fluorophores throughout human H1 and the nucleosome, then monitored the distance changes between H1 and the histone octamer, H1 and nucleosomal DNA, or nucleosomal DNA and the histone octamer to monitor the H1 movement during TF binding. We found that H1 remains bound to the nucleosome dyad, while the C terminal domain (CTD) releases the linker DNA during nucleosome partial unwrapping and TF binding. In addition, mutational studies revealed that a small 16 amino acid region at the beginning of the H1 CTD is largely responsible for altering nucleosome wrapping and regulating TF binding within nucleosomes. We then investigated physiologically relevant post-translational modifications (PTMs) in human H1 by preparing fully synthetic H1 using convergent hybrid phase native chemical ligation. Both individual PTMs and combinations of phosphorylation and citrullination of H1 had no detectable influence on nucleosome binding and nucleosome wrapping, and had only a minor impact on H1 regulation of TF occupancy within nucleosomes. This suggests that these H1 PTMs function by other mechanisms. Our results highlight the importance of the H1 CTD, in particular, the first 16 amino acids, in regulating nucleosome linker DNA dynamics and TF binding within the nucleosome.
Introduction
The eukaryotic genome is repeatedly wrapped in nucleosomes along chromosomal DNA to form chromatin.1 Nucleosomes contain 147 bp of DNA wrapped ∼1.65 times around a histone octamer comprising two copies each of the core histones H2A, H2B, H3, and H4.2 These repeating units are further condensed into higher order structures via the abundant linker histone H1 found at ∼1 H1 per nucleosome in human somatic cells.3 H1 binds the nucleosomal core and linker DNA to form the chromatosome and alters the linker DNA angle to condense mononucleosomes and arrays of nucleosomes.4−6 Compaction of the genome into chromatin plays an important role in regulating DNA processing including transcription and DNA repair by influencing the accessibility of DNA binding proteins to their sites.
Chromatin compaction and dynamics regulate transcription factor (TF) accessibility in promoters and enhancers.7,8 H1 is known for its role in repression of transcription due to its ability to compact chromatin. The linker histone tends to be depleted at transcription start sites (depending on the isoform) and has been found in vitro to reduce TF binding to single nucleosomes and long arrays of nucleosomes.9−13 H1 can regulate TF binding to nucleosomes by altering the spontaneous site exposure of binding sites within nucleosomes.12 Nucleosomal DNA spontaneously unwraps, which exposes binding sites that are sterically blocked by the histone octamer. This reduces TF occupancy at target sites within nucleosomes as compared to naked DNA.14 H1 shifts the equilibrium toward the wrapped state and increases the length of DNA wrapped around the histone octamer. This results in H1 reducing but not completely blocking TF occupancy.5,12 Our previous work showed H1.0 remains bound to nucleosomes as TFs bind partially unwrapped nucleosomes.12 However, it remains unclear how H1.0 remains in complex with nucleosomes as TFs bind to their sites and what are the implications for regulating TF binding to nucleosomes.
H1 has three domains: a short (∼30 amino acids) unstructured N terminal domain (NTD), a globular winged helix domain (WHD) (∼80 amino acids), and a long (∼100 amino acids) unstructured C terminal domain (CTD).15 The WHD binds the nucleosome dyad and the first 10 bp of the linker DNA utilizing lysines and arginines that interact with the DNA phosphate backbone.5,16 The WHD is sufficient for nucleosome specific recognition, while the NTD has little effect on nucleosome binding in vitro(15−17) In contrast, the CTD is critical for compaction of chromatin into higher structures and has varying effects on in vitro binding affinity depending on the nucleosomes and the H1 isoform.18−21 The CTD varies significantly between isoforms, but as a consensus, it contains a few S/TPXK phosphorylation target motifs and has 30–50 positively charged, mostly lysine, amino acids throughout the domain.22 The domain is intrinsically disordered but appears to become more structured and compact upon nucleosome binding.20 Interestingly, two separate regions of the CTD were shown to be important for nucleosome array compaction, while one of these regions was also shown to be important for H1 affinity.19,20 The H1 CTD is potentially critical to nucleosome wrapping, but the regulatory role of the domain in nucleosome wrapping and the effects on altering TF binding remain largely unexplored.
Like the core histones, H1 is extensively modified by post-translational modifications (PTMs) including phosphorylation, citrullination, acetylation, and methylation.23−26 H1 phosphorylation occurs frequently at CDK consensus motifs (S/T-P-X-K/R) in mostly the CTD of H1 in a cell cycle-dependent matter.24 Among the phosphorylation sites, different residues in different isoforms are modified at distinct stages of the cell cycle and associated with specific cellular events. H1.2 S172 is modified during the interphase and the S phase and is associated with sites of active transcription.27,28 In contrast, H1.2 T145 is modified during mitosis and involved in regulation of p53 transcription and DNA repair.29,30 In addition, citrullination has been shown to be related to transcription regulation. H1.2 R54 citrullination results in a loss of positive charge and displacement of the isoform from chromatin and subsequent chromatin decondensation.31 While some PTMs are linked to changes in chromatin condensation and regulation of transcription, the mechanisms behind these changes are largely unexplored, including the influence of H1 PTMs on nucleosome wrapping dynamics and TF binding.
We investigated the role of the H1.0 CTD and multiple physiologically relevant H1.2 PTMs on nucleosome wrapping in regulating TF binding to nucleosomes. We found that during a TF binding event within a partially unwrapped nucleosome, H1.0 remains bound to the nucleosome dyad, while the H1.0 CTD is released from the linker DNA. In addition, a small region of the CTD is important for altering nucleosome wrapping and reducing TF binding. Lastly, H1.2 PTMs individually and in combination had essentially no impact on H1.2 binding to nucleosomes but modestly influences TF binding to H1.2 bound nucleosomes. Overall, this work reveals that the H1.0 CTD is important for regulating TF binding within nucleosomes, while the PTMs of H1.2 functions by a mechanism that is distinct from H1’s ability to bind to nucleosomes, influence nucleosome partial unwrapping, and regulate TF binding within nucleosomes.
Materials and Methods
DNA Preparation and Purification
All DNA constructs were produced using PCR with Cy3- or Cy5-labeled oligonucleotides and a plasmid containing the Widom 601 nucleosome positioning sequence (NPS) with a Gal4 binding site at bases 8–26 of the 601 NPS as done previously.12,32,33 DNA constructs were a total of 272 bp long with 50 bp on the one side of the 147 bp 601 NPS and 75 bp on the other side containing a 3′ attached biotin for future single molecule studies. Either Cy3 or Cy5 was attached to the eighth bp outside the 601 NPS on the 50 bp linker DNA side (Figure 1A). Oligonucleotides were labeled with Cy3 or Cy5 NHS ester attached to an amine modified thymine (amino-modifier-C6-dT) and then purified with a reverse phase C18 column. For attachment, Cy3 or Cy5 NHS ester was resuspended with anhydrous dimethylformamide (DMF) to 15 mM final concentration of dye. The fluorophore was then slowly added to the oligonucleotide in a 5:1 (dye/DNA) ratio and mixed ∼100 times with a micropipette taking care to not introduce bubbles. The reaction was slowly rotated in a rotisserie at room temperature overnight. After the reaction was complete, ethanol precipitations were used to remove most of the free dye and then purified by reverse phase high-performance liquid chromatography (HPLC). PCR was performed with primers using PFU polymerase in a 96-well plate. DNA was purified via anion exchange chromatography and then buffer exchanged and concentrated into 0.5× TE (Tris-EDTA buffer) with a 30 kDa centrifugal filter.
Figure 1.

Nucleosome/H1 constructs and nucleosome purification gels. (A) Schematic of the DNA used in nucleosome experiments. DNA is 272 bp long and consists of the Widom 601 sequence (light grey) flanked by a 50 bp linker DNA (left, dark grey) and a 75 bp linker (right, dark grey) with biotin attached to the 3′ end of the 75 bp linker. Within the 601 sequence is a 19 bp Gal4 binding site (blue) at 8–26 bp in the 601 NPS. Cy3 (light green) is attached to a modified thymine 8 bp into the 50 bp linker DNA away from the 601 NPS. (B) Schematic of the H1.0 domains: NTD (1–22 amino acids, grey), WHD (23–100 amino acids, blue), and CTD (101–194 amino acids, grey). (C) Image of native PAGE analysis of nucleosomes. DNA lane contains only DNA used to make nucleosomes. Pre-purification lane contains nucleosomes before sucrose gradient purification. Final lane contains nucleosomes after sucrose gradient purification. The top band is the center-positioned nucleosome and the center band contains end-positioned nucleosomes. The white lines between the gel on the right indicate non-adjacent lanes from the same gel being placed together in the figure. (D) Schematic of the H1/nucleosome constructs used in experiments. DNA-OctFRET is designed for FRET-detected distance changes between Cy3-labeled linker DNA and Cy5-labeled histone octamer at H2A K119C. H1-OctFRET is designed for FRET-detected distance changes between Cy3-labeled H1.0 at either S22C, S98C, or S131C and Cy5-labeled H2A K119C. H1-DNAquench is designed for fluorescence quenching-detected distance changes between TQ3-labeled H1.0 at either S22C, S98C, or S131C and Cy3-labeled linker DNA. (E) Nucleosome structure with human H1.0 (PDB ID 7K5X) indicating the estimated distances between Cy3 on the DNA (green), Cy5 on H2A (red), H1.0 D24 (purple), and H1.0 K97 (orange). The histone octamer is in gray, DNA is in black, the Gal4 binding site is in dark blue, and H1.0 is in light blue.
Histone H1.0 Expression, Purification, and Labeling
H1.0 expression vectors were mutated using site-directed mutagenesis. All H1.0 mutants were expressed in Bl21 DE3 pLyseS cells. Transformed cells were grown in LB to an OD600 of 0.6 at 37 °C and induced with 0.4 mM IPTG. After 3 h of expression, cells were spun at 4000g for 15 min, supernatant was removed, and cell pellets were flash frozen with liquid nitrogen.
For purification, cells were thawed and resuspended with 25 mL of H1 buffer 500 (50 mM Tris-HCl pH 8, 500 mM NaCl, 10% glycerol, 1 mM EDTA, 5 mM BME, and 0.5 mM PMSF) with one complete protease inhibitor cocktail tablet (Roche) added. Cells were sonicated, spun at 23,000g for 15 min, and lysate containing the protein was poured off. Lysate was purified using Bio-Rex 70; 50–100 mesh (Bio-Rad) cation exchange resin packed into a column for use on an FPLC. A salt gradient from 0.5 to 0.8 M was used to elute the full length H1.0 while a gradient from 0.1 to 0.5 M was used for C terminal deletions using the same H1 buffer 500 mentioned above, except with increased/decreased NaCl concentrations. After collected fractions were analyzed on a 16% acrylamide sodium dodecyl sulfate (SDS) gel, fractions were pooled and diluted with H1 buffer 0 (0 NaCl) so the salt concentration was reduced to either 500 mM for full length H1 or 100 mM for H1 C terminal deletions for an additional round of ion exchange purification. The protocol for the second round of purification was the same as the first, except Bio-Rex 70; 100–200 mesh resin was used for improved resolution near the full-length products. Fractions containing the correct length protein were pooled and dialyzed into water with 2 mM BME and 0.5 mM PMSF and then lyophilized to concentrate and buffer exchange samples. Lyophilized H1.0 was resuspended in H1 storage buffer (20 mM sodium phosphate pH 7, 300 mM NaCl, and 1 mM EDTA), flash frozen, and stored at −80 °C. The final yield was ∼1 mg/L of culture grown.
H1.0 was labeled with either Cy3 maleimide or Tide Quencher 3 (TQ3). H1.0 was incubated with 10 mM TCEP pH 7.1 for 30 min to reduce disulfides, and then dialyzed into 5 mM PIPES and 2 M NaCl pH 6.1 overnight. H1.0 was removed from the dialysis and purged with argon gas as well as another buffer of 2 M (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) pH 7.1 for 20 min. HEPES was quickly added to the H1.0 for a final concentration of 100 mM HEPES and then 10-fold molar excess of Cy3 or TQ3 at 22 mM dissolved in DMF was added to the H1.0 solution and reacted at room temperature for 1 h before overnight incubation at 4 °C. Labeled H1 was purified by diluting the protein with H1 buffer 250 (250 mM NaCl) and then mixing the protein with Bio-Rex 70; 50–100 mesh resin as before. Resin with H1 was washed with H1 buffer 250 to remove the unreacted fluorophore/quencher, and then H1 was eluted with H1 buffer 1000 (1,000 mM NaCl). Fractions containing the H1.0 protein were pooled and dialyzed in water with 2 mM BME and 0.5 mM PMSF and then lyophilized. Lyophilized H1.0 was resuspended in H1 storage buffer (20 mM sodium phosphate pH 7, 300 mM NaCl, and 1 mM EDTA), flash frozen, and stored at −80 °C.
H1.0 concentration was determined using a modified Lowry assay with H1.0 of a known concentration (New England Biolabs) to produce a standard curve and verified by loading equal amounts of the known and unknown H1.0 on a 16% acrylamide SDS gel and confirming similar band intensity.
Core Histone Expression, Purification, and Histone Octamer Preparation
Core histones were expressed and purified as previously described.12,34 Human H2A, H2A K119C, and H2B were expressed in Rosetta Bl21 DE3 pLyseS cells, while human H3 C110A and H4 were expressed in Bl21 DE3 pLyseS cells. Histones were purified separately under denaturing conditions using size exclusion and cation exchange chromatography. The histone octamer was also prepared as previously described.12,34 Lyophilized histones were resuspended in unfolding buffer (20 mM Tris-HCl pH 7.5, 7 M guanidinium, and 10 mM DTT) at 5 mg/mL and mixed together in a ratio of 1.2:1 of (H2A and H2B):(H3 and H4). The histone octamer was formed by performing double dialysis with the mixture into refolding buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl, and 5 mM BME). The octamer was removed from dialysis and labeled with Cy3 or Cy5 maleimide as previously described.35 The octamer was incubated with 10 mM TCEP pH 7.1 for 30 min to reduce disulfides and then dialyzed into 5 mM PIPES and 2 M NaCl pH 6.1 overnight. The octamer was removed from the dialysis and purged with argon gas as well as a separate buffer of 2 M HEPES pH 7.1 for 20 min. HEPES was quickly added to the octamer for a final concentration of 100 mM HEPES, and then, a 10-fold molar excess of fluorophore at 22 mM dissolved in DMF was added to the octamer solution and reacted at room temperature for 1 h before overnight incubation at 4 °C. Excess maleimide was quenched with 10 mM DTT before the octamer was purified via size exclusion chromatography to remove excess heterodimer, free histones, and excess fluorophore. The unlabeled octamer was purified with size exclusion chromatography directly after refolding dialysis.
Nucleosome Preparation
Nucleosomes were made and purified as previously described.12,34 DNA and histone octamers were mixed in a ratio of 1.25:1 in high salt refolding buffer (0.5× TE pH 8.0, 2 M NaCl, and 1 mM benzamidine–HCl) and reconstituted using double dialysis into 4 L of refolding buffer at 4 °C for 5–6 h and then changed to a new 4 L bucket of buffer at 4 °C overnight (0.5× TE pH 8.0, 1 mM benzamidine–HCl). Nucleosomes were added to a 5–30% w/v sucrose gradient and purified with an SW41 Ti (Beckman Coulter) rotor in an Optima L-90K ultracentrifuge (Beckman Coulter) spinning at 41,000 rpm for 22 h at 4 °C.36 Sucrose gradients were fractionated into 0.4 mL fractions and analyzed by native polyacrylamide gel electrophoresis (PAGE), and fractions with center positioned nucleosomes were pooled, concentrated, and buffer exchanged into 0.5× TE pH 8 with a 30 kDa centrifugal filter. Finally, purified nucleosomes were analyzed by a native 5% acrylamide and 0.3× TBE gel to confirm purity. Of note, multiple bands can be seen on these, and subsequent gels due to the imperfect nucleosome positioning of the Widom 601 NPS on DNA longer than 147 bp.
Three nucleosome constructs were used to monitor the movement between H1.0, the histone octamer, and the linker DNA (Figure 1D). DNA-OctFRET was labeled with Cy3 on the DNA with Cy5 attached to H2A K119C. H1-OctFRET has Cy3-labeled H1.0 with Cy5 attached to H2A K119C (Figure S1A). H1-DNAquench has Cy3-labeled DNA with H1.0 labeled with the TQ3 quencher (Figure S2A). We initially prepared a FRET system to monitor distance changes between H1.0 and the linker DNA (Figure S3A). However, while the FRET system accurately measured H1.0 binding to nucleosomes (Figure S3B), the FRET measurement of the Gal4 S1/2 to H1.0 bound nucleosomes (Figure S3C) was an order of magnitude higher than determined by EMSA (Figure S3D,E). Because of this difference, we relied on the H1-DNAquench construct to measure changes between H1 and the DNA, which did not have this difference.
Gal4 Expression and Purification
The Gal4 DNA binding domain (amino acids 1–147) was expressed in Rosetta Bl21 DE3 pLyseS cells. Cells were grown in 2xYT and induced at OD600 of 0.5 with 1 mM IPTG and 100 mM zinc acetate. After 3 h of expression, cells were spun at 4000g for 15 min, the supernatant was removed, and cell pellets were frozen with liquid nitrogen.
Cells were thawed and resuspended in 30 mL of buffer A (50 mM Tris-HCl pH 8, 200 mM NaCl, 10 mM imidazole, 10 mM BME, 20 μM zinc acetate, 1 mM DTT, and 1 mM PMSF) with leupeptin and pepstatin added at 20 μg/mL final concentration. Resuspended cells were sonicated and spun at 23,000g for 15 min, and the lysate containing the protein was poured off. The lysate was added to a Ni-NTA column, washed with buffer A, and the protein was eluted with buffer B (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 200 mM imidazole, 20 μM zinc acetate, 1 mM DTT, 1 mM PMSF, and 0.2% TWEEN 20). Fractions were run on a 12% acrylamide SDS gel, pooled, and dialyzed into buffer C with 200 mM NaCl (25 mM Tris-HCl pH 7.5, 200 mM NaCl, 20 μM zinc acetate, 1 mM DTT, and 1 mM PMSF). After dialysis, the protein was further purified using cation exchange chromatography with a gradient from 200 mM NaCl to 600 mM NaCl using buffer C with appropriate salt concentrations. Protein-containing fractions were pooled, concentrated, exchanged into buffer D (10 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol, 20 μM zinc acetate, 1 mM DTT, and 1 mM PMSF), and then flash frozen for storage at −80 °C. The final yield was ∼5 mg/L of culture grown.
Electrophoretic Mobility Shift Assays
1 nM nucleosomes were incubated with a range of H1 concentrations in T130 buffer (10 mM Tris-HCl pH 8.0, 130 mM NaCl, 10% glycerol, and 0.005% TWEEN20) in a total volume of 20 μL for 20 min at room temperature. Reactions were analyzed by gel electrophorsis with a 4% polyacrylamide, 0.3× TTE, 10% glycerol gel at 4 °C with 300V and imaged with a fluorescence imager.
Fluorescence Measurements
For nucleosome-H1 binding measurements, nucleosomes and H1 were incubated in T130 buffer in a 60 μL volume at room temperature for 20 min and then analyzed with a FluoroMax 4 fluorometer (Horiba). In Gal4 binding experiments, H1 and nucleosomes were incubated for 20 min and then Gal4 added and incubated for 10 min. We showed previously that these incubation times were sufficient to reach the equilibrium.12 Nucleosomes were at 1 nM in all experiments. Fluorescence spectra were measured with a FluoroMax 4 fluorimeter (Horiba) by exciting Cy3 at 510 nm and measuring emission from 530 to 750 nm and separately exciting Cy5 at 610 nm and measuring emission from 630 to 670 nm. FRET efficiency was calculated using the ratioA method.37 Quenching experiments were performed the same way except only Cy3 was excited at 510 nm, and emission was measured from 530 to 750 nm.
Synthesis of PTM-Containing H1.2
Modified histone H1.2 was synthesized using a convergent hybrid phase native chemical ligation (CHP-NCL) approach.38 Overall, H1.2 was split into eight peptide segments at 7 Ala sites (Ala24, Ala49, Ala67, Ala100, Ala134, Ala163, and Ala189), which were transiently mutated to cysteines for native chemical ligation. These eight peptide segments were grouped into two blocks by their sequence. Each block (N-block residues 1–99 and C-block residues 100–212) was assembled on a solid support, and the N-block was desulfurized on the resin to remove extraneous thiols that would interfere with subsequent reaction prior to cleavage. The two blocks were ligated in solution, after which desulfurization was carried out to convert the remaining cysteine back to alanine and to generate the full-length protein with the native sequence.
A 3-amino-4-methylaminobenzoic acid (MeDbz) linker39 was installed on the C-terminus of peptides H1-(1–23), H1-(24–48), H1-(49–66), H1-(100–133), H1-(134–162), and H1-(163–188) to generate a thioester through on-resin activation into the acylurea form MeNbz. Peptides H1-(67–99) and H1-(189–212) had additional attachments. Peptide H1-(67–99) was equipped with an internal 3,4-diaminobenzoic acid (Dbz) linker followed by a cysteine side chain linked to a lysine via an isopeptide bond on the C-terminus. An internal hydroxymethyl benzoic acid linker40 was installed in peptide H1-(189–212) as a C-terminal protecting group in order to release the carboxylic acid after base cleavage. l-4-Thiazolidinecarboxylic acid (Thz) was attached to the N-terminal of each peptide as a cysteine protecting group to avoid self-polymerization or cyclization during ligation.41 The group was later deprotected with methoxyamine. All peptides were prepared by Fmoc solid-phase peptide synthesis (Fmoc-SPPS) with or without microwave heating in high purity and yield.
PEGA resin was used as a water compatible solid support for solid phase ligation. The resin was either equipped with a thioester moiety for capture of the cysteine-containing peptide H1-(67–99) to initiate the N-block assembly or functionalized with an N-terminal cysteine moiety for capture of MeNbz containing peptide H1-(189–212) to start the C-block assembly. Thz was deprotected with ring opening buffer (6 M guanidine, 0.4 M methoxyamine, and 0.1 M phosphate, pH 3–4) to release cysteine for the next round of ligation until all peptides were ligated. Ligation was carried out in ligation buffer (6 M guanidine, 50–100 mM 4-mercaptophenylacetic acid, 0.1 M phosphate, and 20 mM TCEP, pH 6–7) with peptidyl MeNbz. The ligated peptides were then cleaved with either TFA (N-block) or 0.05 M NaOH (C-block) for later solution phase ligation.
Ligated N-block peptide H1-(1–99)-Dbz-x (limiting reactant) and C-block peptide H1-(100–212)-OH were dissolved in a minimal amount of guanidine buffer (6 M guanidine, 0.1 M phosphate, pH 3) taking care to keep the peptide concentration greater than 1 mM and prechilled in an ice/salt bath (−15 to −20 °C). 10–20 equiv of sodium nitrite was added, and the mixture was incubated in an ice/salt bath for 20 min with stirring.42 50–100 equiv of 4-mercaptophenylacetic acid (MPAA) was added to the activated solution, and the pH was adjusted to 6–7 to initiate ligation. The ligation was allowed to proceed until completion (4–6 h). After ligation, the mixture was dialyzed against guanidine buffer to remove MPAA to prevent interference with desulfurization.43 Free-radical-mediated desulfurization was carried out to recover the native Ala residue.44 The final product was purified by reverse-phase high-performance liquid chromatography. The identity and purity of the purified protein was confirmed by bench-top matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) and SDS-PAGE (Figures S4 and S5). The concentration was determined as before with H1.0 using a modified Lowry assay with commercial H1.0 for the standard curve.
Results
Fluorescence Assays for Measuring H1 and Nucleosome Interactions
In a previous study, we showed that TFs can bind to their target sites within partially unwrapped nucleosomes, while H1 remains associated with the nucleosome.12 However, the structural changes that occur when a TF binds within the nucleosome including the relative movements of H1, the histone octamer, and the linker DNA remain undetermined. To investigate these structural changes, we developed three fluorophore systems that detect movements of the linker DNA, histone octamer, and domains of H1 relative to each other to monitor their movement during TF binding. We used the human H1.0 isoform for our first set of experiments because it is the most extensively studied isoform and is highly conserved.5,12,16,19
To monitor distance changes between the DNA and histone octamer, we utilized our previously developed system, referred to as DNA-OctFRET (Figure 1D).12 Cy3 is attached to an amine modified thymine at the eighth bp in the linker DNA outside the 601 NPS (Figure 1A) and Cy5 is attached to H2A K119C (Figure 1D). This construct indirectly measures H1 binding and TF binding through changes in nucleosomal DNA wrapping, leading to changes in FRET as measured by the ratioA method between the DNA and the histone octamer.37 When H1 binds, the FRET efficiency increases due to an increase in DNA wrapped around the histone octamer.5,12 We report H1 binding to nucleosomes as S1/2, the ligand concentration where half of the nucleosomes are bound. As previously reported, H1.0 titrations show that the DNA-OctFRET system detects H1.0 binding to nucleosomes at H1.0 concentrations of a few nM as measured by FRET (3 ± 1 nM) and EMSA (Figure 2A,B). Furthermore, EMSAs reveal that H1.0 induces nucleosome self-association above ∼10 nM H1.0 concentrations (Figure 2A), which is likely due to liquid–liquid phase separation.45,46
Figure 2.

Examples of H1.0 binding to the nucleosome constructs. (A) EMSA of full length H1.0 binding to DNA-OctFRET nucleosomes. Nucleosome band begins shifting at 1 nM and is fully shifted at 5 nM with nucleosome self-association beginning to occur at 10 nM H1.0. Nucleosomes are self-associated at 30,100, and 300 nM H1.0. (B) FRET efficiency of DNA-OctFRET increases with increasing concentrations of WT H1.0 and binds with an S1/2 of 3 ± 1 nM. Self-association occurs in the gray region. (C) RatioA calculation of H1-OctFRET nucleosomes with increasing concentration of Cy3 H1.0 S22C. RatioA begins to increase at low nM concentrations of H1.0 until self-association occurs in the gray region of the graph. RatioA is Cy5 emission when Cy3 is excited, divided by Cy5 emission when Cy5 is directly excited. (D) Normalized Cy3 emission using H1-DNAquench nucleosomes and increasing amounts of TQ3 H1.0 S22C. Cy3 emission was normalized by dividing measured Cy3 emission at each point by the Cy3 emission of nucleosomes with no H1 so that the binding curve begins at 1 and decreases as H1 is added. An S1/2 of 3.7 ± 0.4 nM was measured for this quencher position.
To measure the relative structural movements between the histone octamer and H1.0, we developed the H1-OctFRET system (Figure 1D). We prepared H1.0 with Cy3 attached to a cysteine at three separate locations (S22C, S98C, and S131C) and nucleosomes with Cy5 attached to H2A K119C. Locations H1.0(S22C) and H1.0(S98C) position the fluorophores adjacent to the WHD and within an estimated 35–40 Å distance of Cy5, resulting in FRET upon H1.0-nucleosome binding (Figure 1E). The H1.0(S131C) location is within the disordered CTD and will most likely have a more variable distance. However, we anticipated the Cy3–Cy5 distances to be within the R0 of ∼60 Å based on the H1-nucleosome Cryo-EM structure.5,47
H1.0 titrations reveal that Cy3-Cy5 undergoes significant energy transfer as measured by the ratioA method upon H1.0 binding for all three Cy3 locations within H1.0 (Figures 2C and S1B). This indicates that H1-OctFRET can be used to detect H1.0 binding to the nucleosome for H1.0 concentrations below 10 nM. However, at H1.0 concentrations that induce significant self-association, which occurs above 10 nM, the ratioA method inferred a FRET efficiency above 1, which is not physical. This is likely a result of changes to the fluorophore environment that are caused by H1.0-induced nucleosome self-association. Therefore, we focused on H1.0 concentrations below 10 nM to avoid this issue. Furthermore, we chose to report the stimulated emission of Cy5 by plotting the ratioA, which is the relative acceptor (Cy5) emission when Cy3 is excited by 510 nm to acceptor emission when directly excited by 610 nm. This avoided reporting an estimated FRET efficiency value above 1.
Finally, to monitor distance changes between DNA and H1.0, we generated a H1-DNAquench system (Figure 1C). Here, DNA was again labeled with Cy3 at the eighth base pair outside of the 601 NPS, which is the location used in the DNA-OctFRET system. H1.0 was labeled with TQ3, which has an R0 of ∼38 Å with Cy3.48 Titrations of TQ3-labeled H1.0 with Cy3-labeled nucleosomes show efficient Cy3 quenching as the H1.0 concentration is increased from 1 to 10 nM. We fit this to a binding isotherm with an S1/2 of 3.7 ± 0.4 nM for the H1.0 S22C location (Figure 2D). The other two label positions (S98C and S131C) had similar S1/2 measurements in the low nanomolar regime (Figure S2B,C). These results indicate that the H1-DNAquench system provides an accurate measurement of H1.0 binding to nucleosomes. We did initially attempt to use a Cy3–Cy5 FRET system for measuring H1-DNA interactions. However, this fluorophore system disrupted H1.0-regulated TF binding within the nucleosome, while the Cy3–TQ3 system did not (see Materials and Methods for details). We, therefore, focused on the H1-DNAquench system. Of note, there are multiple possible states where H1 is bound to the nucleosome. The CTD can interact with either one linker DNA arm or both.5,49 In addition, H1 can be bound on-dyad, where the L1 loop of H1 is interacting with either the Cy3-labeled or -unlabeled linker DNA. However, our experiments are ensemble experiments where measured FRET\ratioA and quenching are an average of all states; thus, we cannot determine the fraction of H1 bound in each state. We have chosen to fit these data and subsequent data as if there are two states, an unbound nucleosome and H1 bound nucleosome state.
Overall, the combined studies of these three constructs show that they can be used to track the location of different H1 domains relative to the histone octamer and nucleosomal DNA and to monitor changes in their relative locations during TF binding within the nucleosome.
H1.0 Remains Bound to the Nucleosome Dyad While the H1.0 CTD Releases the Linker DNA during TF Binding within Partially Unwrapped Nucleosomes
The structural fluctuations of H1 during nucleosome unwrapping and TF binding are largely unexplored yet are important for understanding H1 regulation of TF occupancy. The fluorescence constructs we developed here allow us to determine relative movements between the linker DNA, histone octamer, and H1.0 during TF binding. Our studies focused on the truncated model eukaryotic TF activator Gal4, using the DNA binding domain of Gal4 (amino acids 1–147) which binds DNA as a homodimer and comprised of a dimerization domain and Zn2Cys6 binuclear cluster DNA-binding motif.33,50 Gal4 binds a 19 bp target sequence and wraps about 270° around the DNA. We included a 19 bp Gal4 TF target sequence that starts at the eighth base pair of the nucleosome and extends 26 base pairs into the nucleosome (Figure 1A). As previously reported, we used the DNA-OctFRET system to detect Gal4 binding. As the concentration of Gal4 is increased, it binds to transiently unwrapped nucleosomes, trapping them in a partially unwrapped state that exhibits low FRET efficiency12,14 (Figure 3A). By comparing Gal4 S1/2 for binding to nucleosomes with and without H1.0, we can determine the impact of H1.0 on the site accessibility for Gal4 occupancy at its site within a partially unwrapped nucleosome. We found that 1 nM nucleosomes bound with 2.5 nM H1.0 increases Gal4 S1/2 from 15 to 56 nM and nucleosomes bound with 4 nM H1.0 increases Gal4 S1/2 to 90 nM, which implies a ∼4–6 fold reduction in binding affinity (Figure 3D). These results match our previous findings and show that H1.0 shifts the equilibrium of spontaneous nucleosome unwrapping to a more wrapped state but does not completely prevent unwrapping of nucleosomes and subsequent TF binding.12 Of note, in Gal4-binding experiments, we were careful to use H1 concentrations near the S1/2 for each purified H1 sample as even small amounts of nucleosome self-association have a large effect on Gal4 binding that make accurate Gal4-binding measurements difficult.
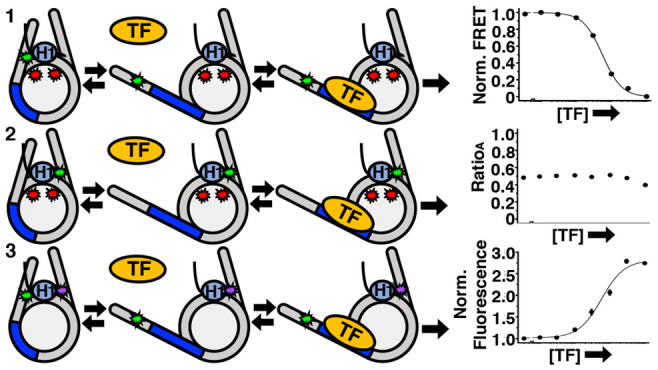
Figure 3.

Gal4 binding to H1.0 bound nucleosomes with the nucleosome constructs. (A–C) Models of TF binding within the DNA-OctFRET, H1-OctFRET, and H1-DNAquench nucleosome constructs, respectively, to illustrate the changes in fluorescence. (D) Normalized ΔFRET (the asymptote of the beginning of the hill plot fit set to 1 and the asymptote of the ending of the fit is set to 0) of DNA-OctFRET nucleosomes with increasing amounts of Gal4. Black is Gal4 binding to nucleosomes without H1.0, green is Gal4 binding to nucleosomes bound with 2.5 nM H1.0, and gray is Gal4 binding to nucleosomes bound with 4 nM H1.0. The model in panel A illustrates Gal4 binding this construct. (E) RatioA (Cy5 emission when Cy3 is excited divided by Cy5 emission when Cy5 is directly excited) of H1-OctFRET nucleosomes bound with Cy3 H1.0 S22C with increasing amounts of Gal4. The model in panel B illustrates Gal4 binding this construct. (F) H1-DNAquench nucleosomes bound with TQ3 H1.0 S22C with increasing amounts of Gal4. Normalized Cy3 emission is calculated by dividing the Cy3 emission at each point by the emission of nucleosomes alone. The model in panel C illustrates Gal4 binding this construct. (H) RatioA of H1-OctFRET nucleosomes bound with Cy3 H1.0 S131C with increasing amounts of Gal4. (I) Normalized Cy3 emission of H1-DNAquench nucleosomes bound with TQ3 H1.0 S131C with increasing amounts of Gal4. (G) Measured S1/2 values for data in panel D, F, I, and Figure S2D. Values for panel D are Gal4 binding to nucleosomes only (black), 15 ± 2 nM, Gal4 binding to nucleosomes with 2.5 nM H1.0 (green), 56 ± 3 nM, and Gal4 binding to nucleosomes with 4 nM H1.0 (gray), 90 ± 30 nM. Values for panel F are Gal4 binding to 2.5 nM TQ3 H1.0 S22C bound nucleosomes (green), 50 ± 10 nM, and Gal4 binding to 5 nM TQ3 H1.0 S22C bound nucleosomes (red), 60 ± 10 nM. Values for panel I are Gal4 binding to 5 nM TQ3 H1.0 S131C bound nucleosomes (red), 50 ± 8 nM, and Gal4 binding to 9 nM TQ3 H1.0 S131C bound nucleosomes (orange), 64 ± 9 nM.
To determine how H1.0 accommodates nucleosome partial unwrapping and Gal4 binding, we performed Gal4 titrations with H1.0-bound nucleosomes using the H1-OctFRET and H1-DNAquench systems. To determine how H1.0 moves relative to the histone octamer, we used the H1-OctFRET system. We titrated Gal4 to nucleosomes in the presence of a constant concentration of H1.0 to observe the change in Cy5-stimulated emission as measured by ratioA. Multiple H1.0 concentrations below 10 nM were used to assure nucleosome self-association induced by H1.0 was not contributing to the measurement. At all H1.0 concentrations investigated, the ratioA between either the WHD (S22C and S98C) or the CTD (S131C) of H1.0 and the histone octamer showed little change over the concentrations that Gal4 binds nucleosomes (Figures 3E,H and S1C). Interestingly, there is a slight increase in the ratioA with H1.0(S131C) indicating potential movement of the H1.0 CTD toward to the histone octamer as Gal4 binds within nucleosomes trapping it in a partially unwrapped state. The little to no change in the ratioA indicates that the H1.0 WHD and CTD remain in similar positions during nucleosome partial unwrapping and TF binding. This implies that (i) H1.0 WHD does not dissociate from the nucleosome dyad during unwrapping and (ii) H1.0 CTD releases the linker DNA so the nucleosome can partially unwrap to accommodate TF binding.
To directly investigate how the linker DNA moves relative to H1.0 as the nucleosome unwraps and Gal4 binds its target site, we utilized the H1-DNAquench system. Titrations of Gal4 with H1.0 bound nucleosomes resulted in an increase in Cy3 fluorescence indicating that as Gal4 binds within partially unwrapped nucleosomes, the Cy3-labeled linker DNA moves away from the H1.0 labeling positions in the WHD (S22C and S98C) and the CTD (S131C) (Figures 3F,I, and S2D). Data were normalized to the starting fluorescence, so all binding curves start at 1. Because of this, higher concentrations of H1 lead to a larger increase in normalized Cy3 emission as these nucleosomes start out more quenched than those with less H1. The non-normalized data can be found in the supplement (Figure S2D). Furthermore, measured Gal4 S1/2 values with H1-DNAquench are all within the values of the Gal4 S1/2 measurements with the DNA-OctFRET system, showing that the reduction in Cy3 quenching quantitatively agrees with Gal4 concentrations that trap H1.0 bound nucleosomes in a partially unwrapped state (Figure 3G). The observation that the quencher on H1.0 has moved away from the linker DNA implies that both the H1.0 WHD and CTD do not remain near the linker DNA as Gal4 binds partially unwrapped nucleosomes. Overall, the combined results from the three fluorescence systems indicate that as H1.0-bound nucleosomes partially unwrap and are trapped in this state by Gal4 binding, H1.0 remains bound to the nucleosome dyad and the H1.0 CTD releases the linker DNA.
Small Region of the H1.0 CTD Is Largely Responsible for Altering Nucleosome Wrapping and Subsequent TF Binding
Our results reveal that the H1.0 CTD dissociates from the linker DNA during nucleosome unwrapping, which indicates that the CTD is important for H1.0 regulation of nucleosome unwrapping/rewrapping equilibrium. Previous work reported that the H1.0 CTD becomes ordered upon nucleosome binding, and the first eight amino acids of the Xenopus laevis H1.0 CTD (amino acids 101–108) are important for H1 affinity.20 In addition, the first 24 amino acids of the mouse H1.0 CTD was reported to be important for nucleosome array compaction.19 Combined, these results suggest that the beginning region of the CTD could be a key region for H1.0 regulation of nucleosome unwrapping and TF binding within nucleosomes.
To investigate the importance of the H1.0 CTD in regulating nucleosome unwrapping and TF binding to the nucleosome, we prepared two H1.0 mutants. The first mutant, H1.0ΔC, had the entire CTD deleted (amino acids 101–194), while the second mutant, H1.0 ΔCpartial, retained the first 16 amino acids of the CTD (amino acids 117–194 were deleted) (Figure 4A). We decided to retain the first 16 amino acids of the CTD in H1.0 ΔCpartial because it is the intermediate length between the 8 and 24 amino acids of the Xenopus and mouse H1.0 CTD, respectively, that were critical for H1.0 binding and array compaction.19,20 We hypothesized that a length between 8 and 24 amino acids would be important for regulating nucleosome wrapping. EMSAs were used to investigate the impact of these deletions on H1.0 binding (Figure 4). Both H1.0ΔC and H1.0 ΔCpartial bind nucleosomes at a concentration of about 30 nM (Figure 4B,C), while full length H1.0 binds nucleosomes at about 3 nM (Figure 2A). This implies that both deletion mutants have about a 10-fold lower affinity to nucleosomes than full length H1.0. Interestingly, H1.0 ΔCpartial initially induces an increase in electrophoretic mobility in contrast to H1.0 and H1.0ΔC, which reduces electrophoretic mobility. These results are consistent with the dominant impact of H1.0 and H1.0ΔC on electrophoretic mobility being due to the overall reduction in negative charge, while the dominant impact of H1.0 ΔCpartial is due to an increase in DNA wrapped into the nucleosome.
Figure 4.
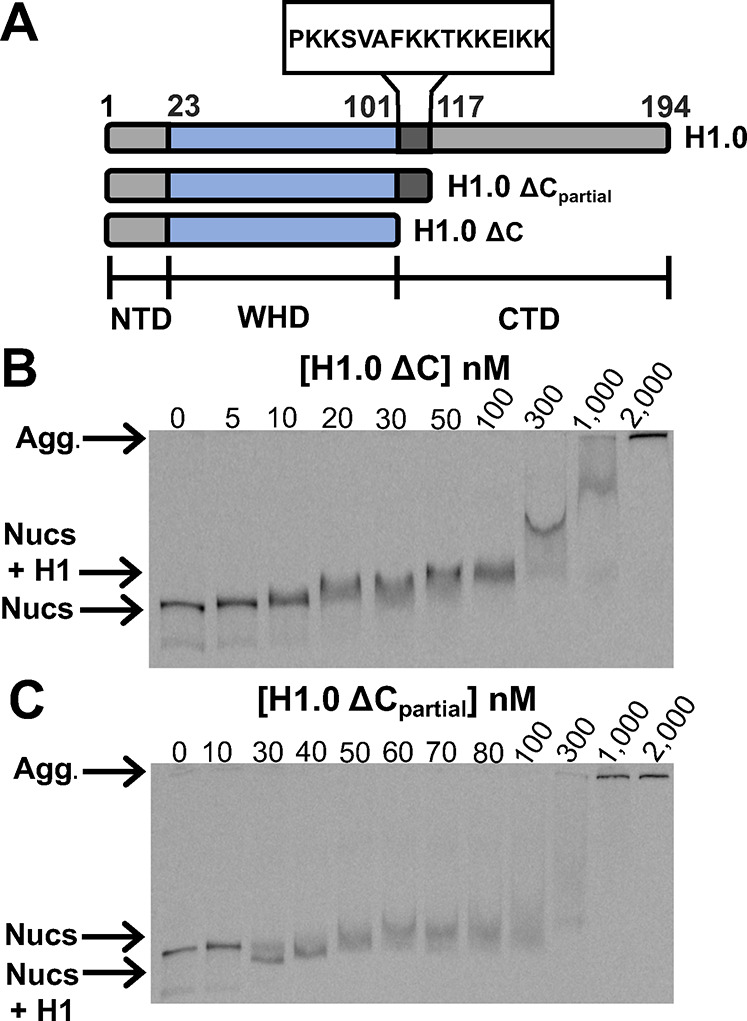
EMSAs of H1.0 CTD mutants binding to DNA-OctFRET nucleosomes. (A) Diagrams of full length and partially deleted H1.0. The NTD ranges from 1 to 22 (gray), the WHD domain is from 23 to 100 (blue), and the CTD is from 101 to 194 (dark gray and gray). H1.0 ΔCpartial contains amino acids 101–116 of the CTD. (B) EMSA of H1.0 ΔC binding to nucleosomes. The first band shift occurs at 10 nM H1.0 with most nucleosomes at 20 nM H1.0 being shifted. Increasing shifts can be seen as H1.0 reaches 50–1000 nM, probably corresponding to multiple H1.0 molecules binding to a nucleosome. Self-association begins at 1000 nM. (C) EMSA of H1.0 ΔCpartial binding to nucleosomes. The first band shift occurs at 30 nM H1.0, and most nucleosomes are shifted at this concentration. The H1.0-bound band runs faster through the gel likely because roughly half the mass of H1.0 has been removed but is still able to compact nucleosomes similarly to WT H1.0. Slower moving bands occur at 50 nM and above, likely corresponding to multiple H1.0 molecules binding to nucleosomes while significant self-association begins at 1000 nM.
We then investigated the impact of the full and partial CTD deletions on nucleosomal DNA wrapping. Titration of H1.0 ΔC over the range of binding observed in EMSAs to DNA-OctFRET nucleosomes shows that deletion of the entire H1.0 CTD nearly abolishes the FRET efficiency increase induced by H1.0 implying that the CTD is required for H1.0 to increase nucleosomal DNA wrapping when bound to nucleosomes (Figure 5A). In contrast, titration of H1.0 ΔCpartial reveals that the inclusion of the first 16 amino acids of the H1.0 CTD results in a FRET efficiency increase that is similar to full length H1.0 (Figure 5A). These results indicate that while the full-length CTD is important for the overall H1.0 binding affinity to nucleosomes, only the first 16 amino acids of the CTD are required for bound H1.0 to increase nucleosomal DNA wrapping.
Figure 5.

FRET measurements of H1.0 CTD truncations binding to nucleosomes and regulating Gal4 binding within DNA-OctFRET nucleosomes. (A) FRET efficiency of nucleosomes with increasing amounts of H1.0. WT H1.0 increases FRET efficiency as seen before and binds with an S1/2 of 3 ± 1 nM (black circles). H1.0 ΔC increases nucleosome FRET efficiency only slightly over the range of concentrations that bind in an EMSA, indicating that the mutant has little effect on linker DNA wrapping (gray triangles). H1.0 ΔCpartial increases the FRET efficiency of nucleosomes to a similar degree as WT H1.0 with a reduced S1/2 of 30 ± 10 nM showing that it alters the nucleosomal DNA wrapping to a similar degree as WT H1.0 but binds nucleosomes with a reduced affinity (gray squares). (B) Normalized ΔFRET efficiency of nucleosomes bound to H1.0 with increasing amounts of Gal4. Gal4 binding to nucleosomes alone (black) and nucleosomes bound with two different H1.0 ΔC concentrations (red and green circle) is similar, showing H1.0 ΔC does not alter Gal4 binding. In the presence of WT H1.0 (pink and gray circles) or H1.0 ΔCpartial (red and green triangles), Gal4 binding to nucleosomes is reduced with S1/2 values reported in panel C, indicating H1.0 ΔCpartial is able to alter Gal4 binding to nucleosomes. (C) S1/2 values for Gal4 binding data in panel B. Gal4 binding to nucleosomes alone (black): 15 ± 2 nM. Gal4 binding to nucleosomes with 2.5 nM WT H1.0 (pink): 56 ± 3 nM. Gal4 binding to nucleosomes with 4 nM WT H1.0 (gray): 90 ± 30 nM. Gal4 binding to nucleosomes with 30 nM H1.0 ΔC (red): 15 ± 2 nM. Gal4 binding to nucleosomes with 100 nM H1.0 ΔC (green): 19 ± 3 nM. Gal4 binding to nucleosomes with 30 nM H1.0 ΔCpartial (red): 43 ± 1 nM. Gal4 binding to nucleosomes with 100 nM H1.0 ΔCpartial (green): 89 ± 5 nM.
Since the impact of H1.0 on nucleosomal DNA wrapping correlates with the influence of H1.0 on Gal4 binding to a partially unwrapped nucleosome, we investigated the impact of the H1.0 deletion mutants on Gal4 binding to DNA-OctFRET nucleosomes. Gal4 titrations with H1.0 ΔC held at constant concentrations where H1.0 ΔC binding to nucleosomes (30 nM and 100 nM) reveals that H1.0 ΔC does not impact the S1/2 of Gal4 binding (Figure 5B,C). In contrast, Gal4 titrations with H1.0 ΔCpartial held at the same constant concentrations cause an increase in the Gal4 S1/2 that is similar to full length H1.0. These results show that amino acids 101–116 are not only necessary and sufficient to increase nucleosomal DNA wrapping; this region is required for reducing TF binding to partially unwrapped nucleosomes.
Phosphorylation and Citrullination of H1.2 Modestly Influence H1.2 Regulation of TF Binding within Nucleosomes
In addition to the WHD and CTD of H1 being key regulators of chromatin function, the PTMs of these H1 domains are strongly correlated with H1 function, while disruption of H1 PTMs are connected to disease.24,28,30,31 Given the importance of H1 PTMs, we investigated the influence of physiologically relevant H1 PTMs on H1-nucleosome binding, nucleosomal DNA wrapping, and TF binding within the nucleosome. We decided to focus on the H1.2 linker histone isoform because it contains a number of PTMs that are established to be biologically relevant.25,28,31 Furthermore, H1.0 and H1.2 bind to nucleosomes, increase nucleosome wrapping, and restrict TF binding within nucleosomes to a similar degree.12 In order to study H1.2 PTMs, we used a CHP-NCL strategy to create two peptides on a solid phase that are then ligated in solution to create full length H1.238 (Figure 6A). We focused on citrullination (cit) within the WHD at R54 and phosphorylation (ph) of the CTD at T145, T153, and S172. All four of the H1.2 PTMs have been identified in both human and mouse, while both the citrullination of R54 and phosphorylation of S172 are correlated with active transcription and located in regions of the genome that appear to be decondensed chromatin.28,31 We decided to prepare H1.2 with not only separate PTMs but combinations to determine if they functioned synergistically. In total, we prepared H1.2(R54cit), H1.2(S172ph), H1.2(R45cit,S172ph), H1.2(T147ph,T153ph), and H1.2(T147ph,T153ph,S172ph). Each construct was verified by both SDS-PAGE and MALDI-TOF mass spectrometry (Figures S4 and S5). We hypothesized these PTMs may alter H1.2 binding affinity and/or the ability of H1.2 to regulate nucleosome wrapping and thus allow for enhanced TF binding because they impact the charge of the amino acid and the interaction of H1 with nucleosomes is largely electrostatic.5
Figure 6.
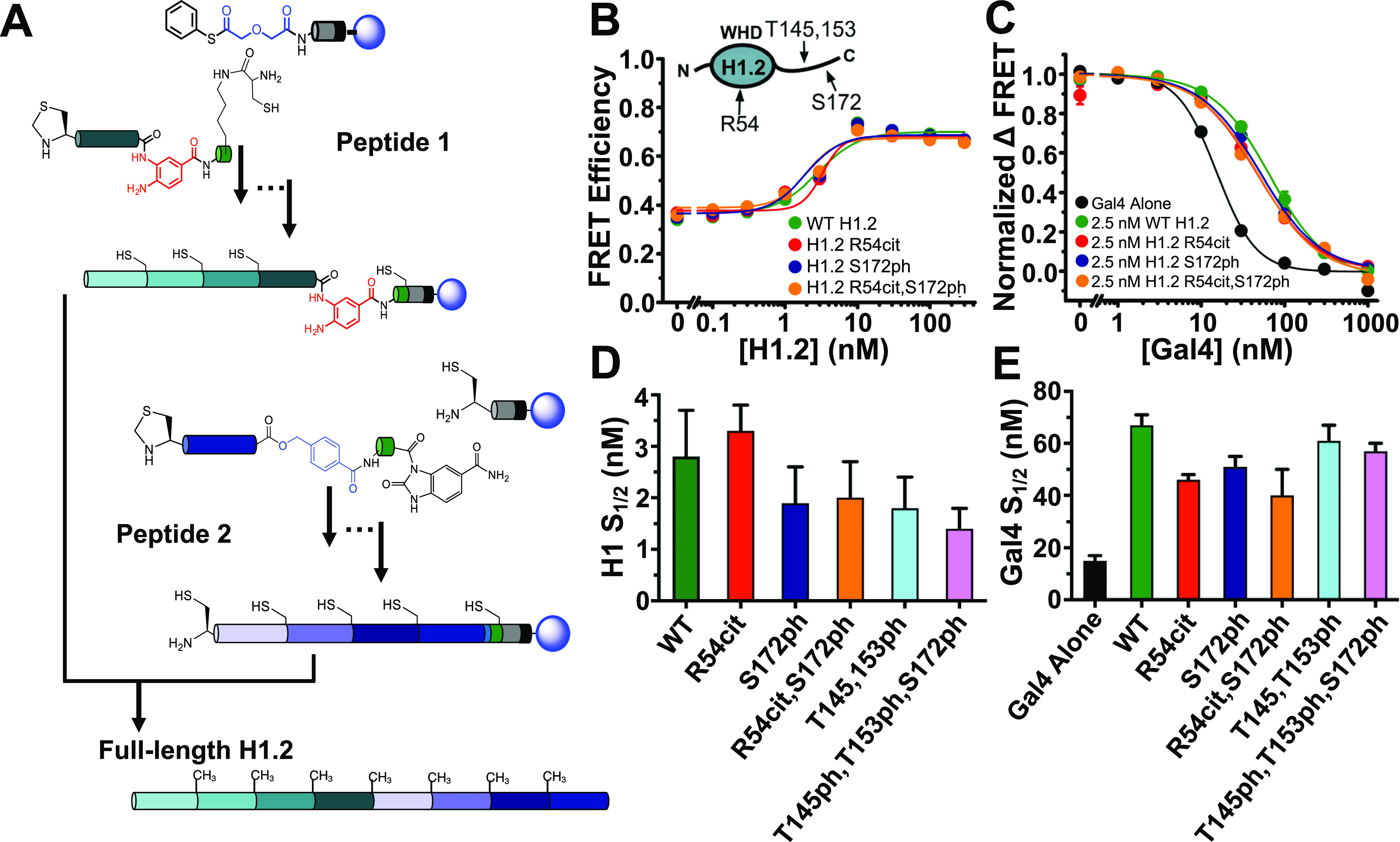
H1.2 with PTMs and Gal4 binding to DNA-OctFRET nucleosomes. (A) H1.2 synthesis strategy. Peptide 1 and peptide 2 were synthesized on a solid support and then cleaved and ligated together in solution to create full length H1.2. (B) Selected examples of FRET efficiency of nucleosomes with increasing amounts of H1.2 containing various PTMs. All H1.2 PTMs tested induced the same increase in FRET efficiency indicating that they alter nucleosome wrapping to a similar degree. Measured S1/2 values for each H1.2 as seen in panel D were similar. (C) Selected examples of normalized ΔFRET efficiency of various H1.2 bound nucleosomes with increasing amounts of Gal4. Gal4 binds nucleosomes alone with a higher affinity than when H1.2 is bound to the nucleosomes (black compared to other colors). Addition of 2.5 nM H1.2 with and without various PTMs decreases Gal4 binding affinity to nucleosomes to various degrees depending on the PTM. (D) S1/2 values for H1.2 binding data in panel B, WT H1.2 (green): 2.8 ± 0.9 nM; H1.2(R54cit) (red): 3.3 ± 0.5 nM; H1.2(S172ph) (dark blue): 1.9 ± 0.7 nM; H1.2(R54cit,S172ph) (orange): 2.0 ± 0.7 nM; H1.2(T145ph,T153ph) (light blue): 1.8 ± 0.6 nM; H1.2(T145ph,T153ph,S172ph) (pink): 1.4 ± 0.4 nM. (E) S1/2 values for Gal4 binding data in panel C, nucleosomes alone (black): 15 ± 2 nM; WT H1.2 (green): 67 ± 4 nM; H1.2(R54cit) (red): 46 ± 2 nM; H1.2(S172ph) (dark blue): 51 ± 4 nM; H1.2(R54cit,S172ph) (orange): 40 ± 10 nM; H1.2(T145ph,T153ph) (light blue): 61 ± 6 nM; H1.2(T145ph,T153ph,S172ph) (pink): 57 ± 3 nM.
To investigate the influence of these PTMs on the H1.2 binding affinity to the nucleosome, we titrated each PTM containing H1.2 with nucleosomes that are prepared with the DNA-OctFRET system and detected binding via EMSAs (Figure S6). We found that each PTM containing H1.2 bound to nucleosomes at nearly the same concentration as unmodified H1.2 indicating that these PTM combinations did not impact nucleosome binding. We then investigate the influence of PTMs on H1.2-induced nucleosome wrapping with the DNA-OctFRET system (Figure 6B,D). In H1.2 titrations, we find that the change in FRET efficiency as H1.2 binds the nucleosome is essentially identical between unmodified and PTM containing H1.2. Furthermore, the change in FRET efficiency occurred at the H1.2 concentration that induced electromobility shifts in the EMSAs. These results confirm that these H1.2 PTMs do not influence nucleosome binding nor the impact of H1.2 on nucleosomal DNA wrapping.
While PTMs did not influence H1.2-nucleosome binding and nucleosomal DNA wrapping, we considered the possibility that PTMs still impacted the probability of TF binding to its target sequence within a partially unwrapped nucleosome. To investigate this, we again used nucleosomes constructed with the DNA-OctFRET system and a Gal4 binding site that is located between 8 and 26 base pairs within the nucleosome. We titrated Gal4 to nucleosomes with a fixed concentration of 2.5 nM of either unmodified or PTM-containing H1.2. As previously reported, unmodified H1.2 shifts the Gal4 S1/2 from 15 ± 2 to 67 ± 4, which implies a 4.5-fold reduction in Gal4 binding affinity (Figure 6C,E).12 We found that the S1/2 of Gal4 binding to nucleosomes bound with PTM-containing H1.2 was reduced relative to unmodified H1.2. H1.2(R54cit) had the largest impact reducing the H1.2 influence on Gal4 binding from 4.5- to 3-fold (Figure 6E). This is about a 1.5-fold reduction in the influence of H1.2 on Gal4 binding. Therefore, we conclude that all five of the H1.2 PTM combinations have at most a very modest impact on the influence of H1.2 regulation of TF occupancy within nucleosomes.
Discussion
Using FRET and quenching measurements, we investigated the role of the H1.0 CTD on nucleosome wrapping, TF binding to nucleosomes, and the relative movements between H1.0, the histone octamer, and nucleosomal DNA in a partially unwrapped nucleosome. We determined that the H1.0 WHD remains bound to the nucleosome dyad while the H1.0 CTD releases the linker DNA to facilitate nucleosome unwrapping during TF binding (Figure 7). We also found a small, 16 amino acid region at the beginning of the H1.0 CTD is critical for altering nucleosome wrapping and the suppression of TF binding within nucleosomes by H1.0. These results highlight the particular importance of the H1.0 CTD on altering nucleosome wrapping to regulate TF binding within the entry/exit region of nucleosomes. This implies that H1.0 could impact the occupancy of many TFs since TF target sites are often located within the entry-exit region of nucleosomes in vivo(51−53) This function is synergistic with additional mechanisms by which H1 regulates TF occupancy in chromatin. H1 condensation of chromatin reduces binding of TFs to linker DNA between nucleosomes. Restriction enzyme digestion of DNA between nucleosomes in Mg2+-condensed nucleosome arrays was inhibited ∼50–300 fold when H1 was bound to nucleosomes.13 H1 may also compete directly with TF binding in the linker DNA and nucleosome dyad where H1 binds. The H1 CTD, in particular, is likely an important modulator of these mechanisms. In addition, the H1 CTD is integral for chromatin condensation and binds the linker DNA making it directly involved with TF binding competition to the linker DNA.5,16 Our results add to the mechanisms H1 employs to regulate TF binding and ultimately transcription.
Figure 7.

Model of H1 binding during nucleosome partial unwrapping during TF binding. State A: the nucleosome is not bound by H1 or Gal4. State B: H1 binds to nucleosomes increasing the wrapping of the nucleosome with the CTD of H1 interacting with the linker DNA. State C: nucleosomes bound with H1 spontaneously partially unwrap. H1 remains bound to the nucleosome dyad while the H1 CTD releases from the linker DNA to accommodate unwrapping. State D: a TF binds to the partially unwrapped nucleosome while H1 remains on the nucleosome dyad.
Our result that the first 16 amino acids of the H1.0 CTD are integral for altering nucleosome wrapping aligns well with the previous literature. The H1 CTD is required for condensation of nucleosome arrays and the formation of the stem loop DNA structure by H1.16,19 Electron microscope images of trinucleosome arrays show that H1 does not condense or form the stem loop structure when the histone lacks the CTD.16 This same study also showed 7 amino acids at the beginning of the human H1.5 CTD are critical for forming the stem loop structure. Structural evidence is also consistent with our findings. A recent study found that NMR chemical shifts of the alpha carbon of amino acids in the beginning of the human H1.0 CTD deviate from random coil values, indicating that this region of the CTD may interact with nucleosomal linker DNA.49 Analytical ultracentrifugation also showed that removal of the entire CTD removed the ability to condense 12mer nucleosome arrays.19 Interestingly, work on specific regions of the CTD highlighted the beginning of the CTD as important for condensing nucleosome arrays. Again, analytical ultracentrifugation with H1 CTD deletions showed the first 24 amino acids of the CTD were necessary for nucleosome array folding/condensation and self-association of nucleosomes in folded arrays, while another 24 amino acid region in the middle played a role only in array folding.19 Subsequent experiments showed that the amino acid composition of the beginning of the CTD is important for its function. Strikingly, either randomizing the primary sequence of the CTD beginning or swapping the beginning with another region of the CTD while maintaining amino acid sequence composition had no effect on the ability of the mutant H1 to condense nucleosome arrays.6 However, mutating just 3 valines to asparagine and 1 threonine to proline in the beginning of the CTD decreased the ability of the H1 mutant to condense nucleosome arrays. Our results are consistent with the amino acid composition being important. Comparing the first 7 amino acids of the human H1.0 CTD with the 7 amino acids of human H1.5 important in the electron microscopy data discussed above and the mouse H1.0 in the ultracentrifugation studies above shows they have similar amino acid compositions.6,16 These studies have led to the hypothesis that the H1 CTD is an intrinsically disordered domain that folds somewhat upon nucleosome binding with specific regions of the CTD-forming structures necessary for different roles.19,20 Our results are synergistic with this model. They show specifically that the beginning of the H1.0 CTD is also necessary for altering nucleosome wrapping which reduces TF accessibility to DNA target sites within nucleosomes. Furthermore, our results are consistent with the beginning of the CTD being responsible for the ability of H1 to condense chromatin as this region alters wrapping in the entry/exit region of the nucleosome.
We also found when a nucleosome is partially unwrapped during TF binding, H1.0 remains bound to the dyad, while the H1.0 CTD dissociates from the linker DNA. Structural studies have found that the H1 CTD is disordered but appears to be bound to the linker DNA of the nucleosome.5,16,49 H1 also appears to adopt two potential binding “modes”, either the on dyad mode, where the WHD binds the nucleosome dyad in the middle symmetrically, or the off dyad binding mode, where the WHD is closer to one of the two linker DNA arms.54−56 The binding mode adopted depends on key residues in the WHD, making it isoform dependent.54 This also has implications on the CTD as the domain may favor binding to one linker DNA over the other.5,16 In fact, recent cryo-EM structural studies reveal that H1.0 binds on dyad, where the H1.0 WHD interacts with one DNA linker through its α3 helix, while its L1 loop interacts with the other DNA linker.5,49 Bednar et al. reported that the CTD interacts with the L1 DNA linker when it interacts with only one DNA linker.5 In addition, Zhou et al. reported that the CTD can interact with not only the L1 DNA linker, but also with the α3 DNA linker.49 In our nucleosome constructs, the Gal4 binding site is on one side of the nucleosome, which results in a proximal and distal DNA linker relative to the Gal4 binding site. H1.0 can bind to nucleosomes with two different on dyad orientations where the L1 region interacts with linker DNA that is proximal or distal to the Gal4 binding site. Therefore, when the nucleosome is fully wrapped and Gal4 is not bound to its site, the nucleosome–H1.0 complex has the potential to be in four states: (i) the Gal4 proximal linker DNA is bound to L1 and the CTD is only bound to the Gal4 proximal linker DNA, (ii) Gal4 proximal linker DNA is bound to L1 and the CTD is bound to both the Gal4 proximal and distal linker DNAs, (iii) Gal4 distal linker DNA is bound to L1 and the CTD is only bound to the Gal4 distal linker DNA, and (iv) Gal4 distal linker DNA is bound to L1 and the CTD is bound to both the Gal4 distal and proximal linker DNAs. Our study probes only the side of the nucleosome where our Gal4 binding site is located. However, we cannot differentiate between these different H1.0-bound nucleosome states. Therefore, our Gal4 accessibility measurements are an average of these four different states transitioning to partially unwrapped states that can be bound by Gal4. Our results do reveal that the H1.0 CTD is required for H1.0 suppression of nucleosome unwrapping, which indicates that the WHD L1 and the α3 interactions with linker DNA alone do not suppress unwrapping. In addition, since there are up to four different fully wrapped H1.0-bound nucleosome states and we only quantify the average impact of these states, it is likely that the H1.0 orientation and the CTD binding of one or linker DNA results in different reductions in nucleosome accessibility. Further investigation into the possible asymmetric effects of the CTD binding to the linker DNA will be valuable in adding to the interpretation of our results.
Our findings also raise the question of what happens to H1 binding orientation and dynamics when the CTD dissociates from the linker DNA during partial nucleosome unwrapping. The binding mode of H1 may switch and/or the CTD may either remain unbound from nucleosomal DNA, or given the unstructured and flexible nature of the CTD, it may bind to the other DNA linker. One study using an optical trap to measure DNA unzipping found that mouse H1.0 binds on dyad, but when one side of the nucleosome is repeatedly unzipped without causing nucleosome dissociation, H1 may switch to an off dyad binding mode.57 Unfortunately, the design of our FRET experiments and the nature of FRET make quantitative determination of the population of H1 in each binding mode difficult due to multiple affects. Our S22C and S98C labeling positions on the N terminal and C terminal side of the WHD are within 30–50 Å of both H2A K119C residues, meaning any FRET we measure is a complex mixture of FRET between both positions. In addition, Cy5 attached to K119C is a relatively large molecule that is connected via a single bond meaning Cy5 likely has a large amount of rotational freedom further complicating our determination of distance. However, our results that FRET between S22C and S98C and the histone octamer does not change when Gal4 binds (Figure S1C) are consistent with H1 remaining bound in the on dyad binding mode as changes in binding mode are unlikely to result in the same FRET efficiency as when H1 binds a fully wrapped nucleosome. In addition, this study found that the H1 CTD forms contacts with the linker DNA that are disturbed when partial unzipping occurs, agreeing with our finding that the CTD dissociates during nucleosome partial unwrapping. Further investigation into these asymmetric effects on linker binding and changes in H1 binding mode during nucleosome unwrapping are warranted to answer these questions.
For the physiologically relevant H1.2 PTMs investigated here, we found that they had no effect on H1 binding and nucleosome wrapping, while R54cit and S172ph had a modest impact on TF binding. The observation that these H1.2 PTMs induce a modest increase in TF binding to nucleosomes is qualitatively consistent with previous studies. H1.2 R54 citrullination was found to cause dissociation of H1.2 from chromatin in vivo, and H1.2 S172ph is localized at sites of active transcription.28,31 However, our modest results suggest that mechanisms other than altering nucleosome wrapping are likely responsible for the function of these PTMs. In addition, one study found that mouse H1.0 T153E does reduce CTD contacts with the linker DNA during DNA unzipping experiments, while we found that human H1.2 T153ph has little effect on nucleosome wrapping.57 Differences in the isoform used and our native PTM compared to mutation to glutamate may be a reason for our different findings. It is also important to note our H1 binding assay may not be fully sensitive to changes in H1 binding as our nucleosomes were limited to 1 nM for all experiments. Given that H1 affinity has been previously measured to be pM, our assays may not have been sensitive enough to detect modest changes in H1.2-nucleosome affinity.21 Future experiments with more sensitive methods will better determine whether these PTMs cause differences in H1 affinity to nucleosomes. There are other factors these PTMs may alter that contribute to their chromatin function. Interactions between histone chaperones, such as Nap1 and ProTα, and H1 could be disrupted by these PTMs.58,59 These H1.2 PTMs could provide binding sites for histone readers, which is an established mechanism of the core histone tail PTMs. In addition, liquid–liquid phase separation (LLPS) of chromatin by H1 may be altered by these PTMs. The H1 CTD contains many charges and has multivalent interactions as seen in proteins that undergo phase separation, and the CTD has been shown to be responsible for causing LLPS of nucleosome arrays.60 Furthermore, phosphorylation of S157, S175, and S193 of chicken H1.11L reduces the ability of H1 to cause LLPS with DNA.45 S157 and S175 correspond to the human H1.2 T153 and S172 phosphorylations we tested here, so these PTMs in particular may alter LLPS. Overall, our work rules out a role for these PTMs in significantly altering wrapping in a single nucleosome and instead points to future investigations into alternative mechanisms by which H1 PTMs regulate chromatin function.
Acknowledgments
We are grateful to Ralf Bundschuh, his lab members, and the Poirier Lab members for insightful discussions and feedback on this work. We would also like to thank Mark Parthun for providing the H1 expression vectors. This work was supported by the National Institutes of Health [R01 GM121966, R01 GM131626, R35 GM139564 to M.G.P., and T32 GM141955 to N.L.B.] and the NSF [MCB1715321].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00001.
Complete fluorescence data for all H1.0 labeling positions, mass spectrometry of final H1.2 PTM proteins, polyacrylamide SDS gels of final H1.2 PTM proteins, EMSAs of H1.2 PTM proteins binding to nucleosomes, and complete fluorescence data for all H1.2 PTMs tested (PDF)
Accession Codes
Human H1.0: NP_005309. Human H1.2: NP_005310. Human H2A: NP_003501. Human H2B: CAB02542. Human H3.2: NP_066403. Human H4: NP_001029249. Yeast Gal4: NP_015076.
The authors declare no competing financial interest.
Supplementary Material
References
- Kornberg R. D. Chromatin Structure : A Repeating Unit of Histones and DNA Chromatin Structure Is Based on a Repeating Unit of Eight. Science 1974, 184, 868–871. 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mäder A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Bates D. L.; Thomas J. O. Histones H1 and H5: One or Two Molecules per Nucleosome?. Nucleic Acids Res. 1981, 9, 5883–5894. 10.1093/nar/9.22.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson R. T. Structure of the Chromatosome, a Chromatin Particle Containing 160 Base Pairs of DNA and All the Histones. Biochemistry 1978, 17, 5524–5531. 10.1021/bi00618a030. [DOI] [PubMed] [Google Scholar]
- Bednar J.; Garcia-Saez I.; Boopathi R.; Cutter A. R.; Papai G.; Reymer A.; Syed S. H.; Lone I. N.; Tonchev O.; Crucifix C.; Menoni H.; Papin C.; Skoufias D. A.; Kurumizaka H.; Lavery R.; Hamiche A.; Hayes J. J.; Schultz P.; Angelov D.; Petosa C.; Dimitrov S. Structure and Dynamics of a 197 Bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 2017, 66, 384–397. 10.1016/j.molcel.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Hamkalo B.; Parseghian M. H.; Hansen J. C. Chromatin Condensing Functions of the Linker Histone C-Terminal Domain Are Mediated by Specific Amino Acid Composition and Intrinsic Protein Disorder. Biochemistry 2009, 48, 164–172. 10.1021/bi801636y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns B. R. The Logic of Chromatin Architecture and Remodelling at Promoters. Nature 2009, 461, 193–198. 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Maharana S.; Wang M. D.; Shivashankar G. V. Super-Resolution Microscopy Reveals Decondensed Chromatin Structure at Transcription Sites. Sci. Rep. 2015, 4, 4477. 10.1038/srep04477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R.; Thiel C.; et al. Reciprocal Binding of PARP-1 and Histone H1 at Promoters Specifies Transcriptional Outcomes. Science 2008, 319, 819–821. 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- Millán-Ariño L.; Izquierdo-Bouldstridge A.; Jordan A. Specificities and Genomic Distribution of Somatic Mammalian Histone H1 Subtypes. Biochim. Biophys. Acta, Gene Regul. Mech. 2016, 1859, 510–519. 10.1016/j.bbagrm.2015.10.013. [DOI] [PubMed] [Google Scholar]
- Juan L.-J.; Utley R. T.; Vignali M.; Bohm L.; Workman J. L. H1-Mediated Repression of Transcription Factor Binding to a Stably Positioned Nucleosome. J. Biol. Chem. 1997, 272, 3635–3640. 10.1074/jbc.272.6.3635. [DOI] [PubMed] [Google Scholar]
- Bernier M.; Luo Y.; Nwokelo K. C.; Goodwin M.; Dreher S. J.; Zhang P.; Parthun M. R.; Fondufe-Mittendorf Y.; Ottesen J. J.; Poirier M. G. Linker Histone H1 and H3K56 Acetylation Are Antagonistic Regulators of Nucleosome Dynamics. Nat. Commun. 2015, 6, 10152. 10.1038/ncomms10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra L. N.; Hayes J. J. A Nucleosome-Free Region Locally Abrogates Histone H1–Dependent Restriction of Linker DNA Accessibility in Chromatin. J. Biol. Chem. 2018, 293, 19191–19200. 10.1074/jbc.ra118.005721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach K. J.; Widom J. Mechanism of Protein Access to Specific DNA Sequences in Chromatin: A Dynamic Equilibrium Model for Gene Regulation. J. Mol. Biol. 1995, 254, 130–149. 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- Allan J.; Hartman P. G.; Crane-Robinson C.; Aviles F. X. The Structure of Histone H1 and Its Location in Chromatin. Nature 1980, 288, 675–679. 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- Syed S. H.; Goutte-Gattat D.; Becker N.; Meyer S.; Shukla M. S.; Hayes J. J.; Everaers R.; Angelov D.; Bednar J.; Dimitrov S. Single-Base Resolution Mapping of H1-Nucleosome Interactions and 3D Organization of the Nucleosome. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9620–9625. 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendzel M. J.; Lever M. A.; Crawford E.; Th’ng J. P. H. The C-Terminal Domain Is the Primary Determinant of Histone H1 Binding to Chromatin in Vivo. J. Biol. Chem. 2004, 279, 20028–20034. 10.1074/jbc.m400070200. [DOI] [PubMed] [Google Scholar]
- Allan J.; Mitchell T.; Harborne N.; Bohm L.; Crane-Robinson C. Roles of H1 Domains in Determining Higher Order Chromatin Structure and H1 Location. J. Mol. Biol. 1986, 187, 591–601. 10.1016/0022-2836(86)90337-2. [DOI] [PubMed] [Google Scholar]
- Lu X.; Hansen J. C. Identification of Specific Functional Subdomains within the Linker Histone H10 C-Terminal Domain. J. Biol. Chem. 2004, 279, 8701–8707. 10.1074/jbc.m311348200. [DOI] [PubMed] [Google Scholar]
- Caterino T. L.; Fang H.; Hayes J. J. Nucleosome Linker DNA Contacts and Induces Specific Folding of the Intrinsically Disordered H1 Carboxyl-Terminal Domain. Mol. Cell. Biol. 2011, 31, 2341–2348. 10.1128/mcb.05145-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. E.; Hieb A. R.; Luger K. A Quantitative Investigation of Linker Histone Interactions with Nucleosomes and Chromatin. Sci. Rep. 2016, 6, 19122. 10.1038/srep19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterino T. L.; Hayes J. J. Structure of the H1 C-Terminal Domain and Function in Chromatin Condensation. Biochem. Cell Biol. 2011, 89, 35–44. 10.1139/o10-024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A.; Schneider R. The Role of Linker Histone H1 Modifications in the Regulation of Gene Expression and Chromatin Dynamics. Biochim. Biophys. Acta, Gene Regul. Mech. 2016, 1859, 486–495. 10.1016/j.bbagrm.2015.09.003. [DOI] [PubMed] [Google Scholar]
- Andrés M.; García-Gomis D.; Ponte I.; Suau P.; Roque A. Histone H1 Post-Translational Modifications: Update and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 5941. 10.3390/ijms21165941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B. A.; Busby S. A.; Barber C. M.; Shabanowitz J.; Allis C. D.; Hunt D. F. Characterization of Phosphorylation Sites on Histone H1 Isoforms by Tandem Mass Spectrometry. J. Proteome Res. 2004, 3, 1219–1227. 10.1021/pr0498887. [DOI] [PubMed] [Google Scholar]
- Wiśniewski J. R.; Zougman A.; Krüger S.; Mann M. Mass Spectrometric Mapping of Linker Histone H1 Variants Reveals Multiple Acetylations, Methylations, and Phosphorylation as Well as Differences between Cell Culture and Tissue. Mol. Cell. Proteomics 2007, 6, 72–87. 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- Sarg B.; Helliger W.; Talasz H.; Förg B.; Lindner H. H. Histone H1 Phosphorylation Occurs Site-Specifically during Interphase and Mitosis: Identification of a Novel Phosphorylation Site on Histone H1. J. Biol. Chem. 2006, 281, 6573–6580. 10.1074/jbc.m508957200. [DOI] [PubMed] [Google Scholar]
- Talasz H.; Sarg B.; Lindner H. H. Site-Specifically Phosphorylated Forms of H1.5 and H1.2 Localized at Distinct Regions of the Nucleus Are Related to Different Processes during the Cell Cycle. Chromosoma 2009, 118, 693–709. 10.1007/s00412-009-0228-2. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; John S.; Pesavento J. J.; Schultz-Norton J. R.; Schiltz R. L.; Baek S.; Nardulli A. M.; Hager G. L.; Kelleher N. L.; Mizzen C. A. Histone H1 Phosphorylation Is Associated with Transcription by RNA Polymerases I and II. J. Cell Biol. 2010, 189, 407–415. 10.1083/jcb.201001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Jeong K. W.; Kim H.; Choi J.; Lu W.; Stallcup M. R.; An W. Functional Interplay between P53 Acetylation and H1.2 Phosphorylation in P53-Regulated Transcription. Oncogene 2012, 31, 4290–4301. 10.1038/onc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou M. A.; Castelo-Branco G.; Halley-Stott R. P.; Oliveira C. S.; Loos R.; Radzisheuskaya A.; Mowen K. A.; Bertone P.; Silva J. C. R.; Zernicka-Goetz M.; Nielsen M. L.; Gurdon J. B.; Kouzarides T. Citrullination Regulates Pluripotency and Histone H1 Binding to Chromatin. Nature 2014, 507, 104–108. 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. D.; Widom J. Sequence and Position-Dependence of the Equilibrium Accessibility of Nucleosomal DNA Target Sites. J. Mol. Biol. 2000, 296, 979–987. 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- Liang S. D.; Marmorstein R.; Harrison S. C.; Ptashne M. DNA Sequence Preferences of GAL4 and PPR1: How a Subset of Zn2 Cys6 Binuclear Cluster Proteins Recognizes DNA. Mol. Cell Biol. 1996, 16, 3773–3780. 10.1128/mcb.16.7.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K.; Rechsteiner T. J.; Richmond T. J. Preparation of Nucleosome Core Particle from Recombinant Histones. Methods Enzymol. 1999, 304, 3–19. 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- Shimko J. C.; North J. A.; Bruns A. N.; Poirier M. G.; Ottesen J. J. Preparation of Fully Synthetic Histone H3 Reveals That Acetyl-Lysine 56 Facilitates Protein Binding within Nucleosomes. J. Mol. Biol. 2011, 408, 187–204. 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar M.; Mooney A. M.; North J. A.; Nakkula R. J.; Picking J. W.; Edon A.; Fishel R.; Poirier M. G.; Ottesen J. J. Acetylation of Histone H3 at the Nucleosome Dyad Alters DNA-Histone Binding. J. Biol. Chem. 2009, 284, 23312–23321. 10.1074/jbc.m109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg R. M. Fluorescence Resonance Energy Transfer and Nucleic Acids. Methods Enzymol. 1992, 211, 353–388. 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- Hong Z. Z.; Yu R. R.; Zhang X.; Webb A. M.; Burge N. L.; Poirier M. G.; Ottesen J. J. Convergent Hybrid Phase Ligation Strategy for Efficient Total Synthesis of Large Proteins Demonstrated for 212-Residue Linker Histone H1.2. bioRxiv 2019, 661744. 10.1101/661744. [DOI] [Google Scholar]
- Blanco-Canosa J. B.; Nardone B.; Albericio F.; Dawson P. E. Chemical Protein Synthesis Using a Second-Generation N-Acylurea Linker for the Preparation of Peptide-Thioester Precursors. J. Am. Chem. Soc. 2015, 137, 7197–7209. 10.1021/jacs.5b03504. [DOI] [PubMed] [Google Scholar]
- Yu R. R.; Mahto S. K.; Justus K.; Alexander M. M.; Howard C. J.; Ottesen J. J. Hybrid Phase Ligation for Efficient Synthesis of Histone Proteins. Org. Biomol. Chem. 2016, 14, 2603–2607. 10.1039/c5ob02195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang D.; Kent S. B. H. A One-Pot Total Synthesis of Crambin. Angew. Chem., Int. Ed. 2004, 43, 2534–2538. 10.1002/anie.200353540. [DOI] [PubMed] [Google Scholar]
- Wang J.-X.; Fang G.-M.; He Y.; Qu D.-L.; Yu M.; Hong Z.-Y.; Liu L. Peptide o -Aminoanilides as Crypto-Thioesters for Protein Chemical Synthesis. Angew. Chem. 2015, 127, 2222–2226. 10.1002/ange.201408078. [DOI] [PubMed] [Google Scholar]
- Rohde H.; Schmalisch J.; Harpaz Z.; Diezmann F.; Seitz O. Ascorbate as an Alternative to Thiol Additives in Native Chemical Ligation. ChemBioChem 2011, 12, 1396–1400. 10.1002/cbic.201100179. [DOI] [PubMed] [Google Scholar]
- Wan Q.; Danishefsky S. J. Free-Radical-Based, Specific Desulfurization of Cysteine: A Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chem., Int. Ed. 2007, 46, 9248–9252. 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- Turner A. L.; Watson M.; Wilkins O. G.; Cato L.; Travers A.; Thomas J. O.; Stott K. Highly Disordered Histone H1–DNA Model Complexes and Their Condensates. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 11964–11969. 10.1073/pnas.1805943115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya A.; Park S.; Rana N.; King J. T. Liquid-Liquid Phase Separation of Histone Proteins in Cells: Role in Chromatin Organization. Biophys. J. 2020, 118, 753–764. 10.1016/j.bpj.2019.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal A.; Arslan S.; Okumus B.; Wilson T. J.; Giraud G.; Norman D. G.; Ha T.; Lilley D. M. J. Orientation Dependence in Fluorescent Energy Transfer between Cy3 and Cy5 Terminally Attached to Double-Stranded Nucleic Acids. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 11176–11181. 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. H.; Zheng J.; Feldman J. L.; Klein M. A.; Kuznetsov V. I.; Peterson C. L.; Griffin P. R.; Denu J. M. Multivalent Interactions Drive Nucleosome Binding and Efficient Chromatin Deacetylation by SIRT6. Nat. Commun. 2020, 11, 5244. 10.1038/s41467-020-19018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.-R.; Feng H.; Kale S.; Fox T.; Khant H.; de Val N.; Ghirlando R.; Panchenko A. R.; Bai Y. Distinct Structures and Dynamics of Chromatosomes with Different Human Linker Histone Isoforms. Mol. Cell 2021, 81, 166–182. 10.1016/j.molcel.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M.; Fitzgerald M. X.; Harper S.; Luo C.; Speicher D. W.; Marmorstein R. Structural Basis for Dimerization in DNA Recognition by Gal4. Structure 2008, 16, 1019–1026. 10.1016/j.str.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J. A.; Shimko J. C.; Javaid S.; Mooney A. M.; Shoffner M. A.; Rose S. D.; Bundschuh R.; Fishel R.; Ottesen J. J.; Poirier M. G. Regulation of the Nucleosome Unwrapping Rate Controls DNA Accessibility. Nucleic Acids Res. 2012, 40, 10215. 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac K. D.; Wang T.; Gordon D. B.; Gifford D. K.; Stormo G. D.; Fraenkel E. An Improved Map of Conserved Regulatory Sites for Saccharomyces Cerevisiae. BMC Bioinf. 2006, 7, 113. 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C.; Pugh B. F. A Compiled and Systematic Reference Map of Nucleosome Positions across the Saccharomyces Cerevisiae Genomes. Genome Biol. 2009, 10, R109. 10.1186/gb-2009-10-10-r109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.-R.; Feng H.; Ghirlando R.; Li S.; Schwieters C. D.; Bai Y. A Small Number of Residues Can Determine If Linker Histones Are Bound On or Off Dyad in the Chromatosome. J. Mol. Biol. 2016, 428, 3948–3959. 10.1016/j.jmb.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F.; Chen P.; Sun D.; Wang M.; Dong L.; Liang D.; Xu R.-M.; Zhu P.; Li G. Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science 2014, 344, 376–380. 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- Fyodorov D. V.; Zhou B.-R.; Skoultchi A. I.; Bai Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. 10.1038/nrm.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnizky S.; Khamis H.; Ginosar Y.; Goren E.; Melamed P.; Kaplan A. Extended and Dynamic Linker Histone-DNA Interactions Control Chromatosome Compaction. Mol. Cell 2021, 81, 3410–3421. 10.1016/j.molcel.2021.06.006. [DOI] [PubMed] [Google Scholar]
- Kepert J. F.; Mazurkiewicz J.; Heuvelman G. L.; Tóth K. F.; Rippe K. NAP1 Modulates Binding of Linker Histone H1 to Chromatin and Induces an Extended Chromatin Fiber Conformation. J. Biol. Chem. 2005, 280, 34063–34072. 10.1074/jbc.m507322200. [DOI] [PubMed] [Google Scholar]
- Karetsou Z.; Sandaltzopoulos R.; Frangou-Lazaridis M.; Lai C.-Y.; Tsolas O.; Becker P. B.; Papamarcaki T. Prothymosin α Modulates the Interaction of Histone H1 with Chromatin. Nucleic Acids Res. 1998, 26, 3111–3118. 10.1093/nar/26.13.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B. A.; Doolittle L. K.; Schneider M. W. G.; Jensen L. E.; Gamarra N.; Henry L.; Gerlich D. W.; Redding S.; Rosen M. K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484. 10.1016/j.cell.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.