

Abstract
Alzheimer’s disease is a neurodegenerative disease which severely impacts the health of the elderly. Current treatments are only able to alleviate symptoms, but not prevent or cure the disease. The neurofibrillary tangles formed by tau protein aggregation are one of the defining characteristics of Alzheimer’s disease, so tau protein has become a key target for the drug design. In this study, we show that fisetin, a plant-derived polyphenol compound, can inhibit aggregation of the tau fragment, K18, and can disaggregate tau K18 filaments in vitro. Meanwhile it is able to prevent the formation of tau aggregates in cells. Both experimental and computational studies indicate that fisetin could directly interact with tau K18 protein. The binding is mainly created by hydrogen bond and van der Waal force, prevents the formation of β-strands at the two hexapeptide motifs, and does not perturb the secondary structure or the tubulin binding ability of tau protein. In summary, fisetin might be a candidate for further development as a potential preventive or therapeutic drug for Alzheimer’s disease.
Keywords: Alzheimer’s disease, tau protein, protein aggregation, fisetin, compound-protein interaction, molecular dynamic simulation
Introduction
Alzheimer’s disease (AD), the most common dementia, places a severe burden on patients, family and society. AD patients are characterized by memory loss, cognitive decline and personality changes [1]. The molecular mechanisms of AD are not clear yet, but there are several hypotheses including the amyloid hypothesis, tau hypothesis, cholinergic hypothesis, mitochondrial cascade hypothesis, calcium homeostasis and NMDA hypotheses, inflammatory hypothesis, and others [2]. Senile plaques consisting of the amyloid beta (Aβ) peptide and neurofibrillary tangles (NFTs) containing tau protein are the two major pathological characteristics of AD [3]. Tau protein fibrils are also deposited in other neurodegenerative diseases including frontotemporal lobar dementia, Pick’s disease, progressive supranuclear palsy, corticobasal degeneration and others, collectively called tauopathies [4]. Tau is a microtubule associated protein that is able to interact with tubulin, promote microtubule assembly, and regulate microtubule stability and dynamics [5]. When tau protein is post-translationally modified by hyperphosphorylation, it detaches from microtubules leading to its self-aggregation and microtubule disassembly [4]. There are six isoforms of tau protein in the human central nervous system due to the alternative splicing of exons 2, 3 and 10. The full-length tau protein has 441 amino acids, containing an N-terminal projection domain, a proline-rich domain, and a C-terminal microtubule binding domain (MTBD). The MTBD includes four imperfect 31- or 32-residue repeats, namely R1, R2, R3 and R4. The differential splicing of exon 10 leads to tau species that contain either three repeats (3R-tau) or four repeats (4R-tau). The MTBD is the main region to bind tubulin and to promote microtubule assembly [6]. Moreover, the first six residues in R2 and R3 are essential for heparin induced tau fibrilization in vitro [7]. Recent cryo-EM structures of tau filaments from AD brains show that the filament cores are made of two identical protofilaments comprising residues 306-378 of tau protein, located in the MTBD [8]. Here tau K18, a tau fragment containing all four repeats domain, is used as the experimental subject. Tau K18 is a widely used model for the full-length tau protein as they exhibit very similar physiological and pathological functions [9–14].
Although most drug trials for AD have targeted Aβ or pathways related to Aβ like β-and γ-secretase cleavage, tau protein has gained increasing interest as another important target. The current strategy for drugs targeting tau protein can be summarized as inhibition of tau aggregation, inhibition of tau phosphorylation, reduction of tau levels, and tau immunization[15]. In AD patient’s brains, tau was found to contain 6-8 mol phosphate/mol protein while in healthy human, tau proteins contained 1.9 mol phosphate/mol protein [16]. The phosphorylation sites related to pathological tau are not clear, and whether this process is necessary or sufficient for aggregation is unknown. Therefore, screening for tau aggregation inhibitors might be more practical and promising. Some tau aggregation inhibitors have been reported such as methylene blue [17, 18], myricetin (similar to fisetin in structure) [18], N744 [19], azure A [20], xanthohumol [21] and others. But most of these results are laboratory-based and additional experiments addressing drug toxicity, ultimately followed by clinical trials on human are needed. Notably one compound named TRx0237 inhibits tau aggregation and phase III clinical trials show that the brain atrophy rate of AD patients declined after 9 months of treatment [22]. Here, we identify the natural product compound fisetin (3,3′,4′,7-Tetrahydroxyflavone) as a novel tau aggregation inhibitor.
Fisetin, a polyphenol flavonoid, is a yellow acicular crystal. Its structure is shown in Figure 1A. It is abundant in vegetables, fruits, teas and Anacardiaceae plants [23]. Fisetin has been reported to possess anti-oxidant, anti-angiogenic, anti-neoplastic, anti-inflammatory and neuroprotective properties [24–27]. Fisetin inhibits the proliferation of various activated neuroinflammatory mediators and glial cells [24, 28], and has a strong neuroprotective effect on neurotoxicity induced by Aβ1-42 [28]. Fisetin is able to reduce the accumulation of Aβ peptide in vivo [27–29], improve cognitive dysfunctions [28, 30, 31] and promote the function of the hippocampus [31]. Moreover, it can reduce the level of tau protein phosphorylation in AD mouse models [28, 32]. In this study, we discover two new activities of fisetin, inhibition of tau aggregation and disaggregation of tau filaments. We further reveal the molecular mechanisms for the interaction between fisetin and tau protein. The results may assist in the development of fisetin or fisetin-derived compounds as novel therapies for AD.
Figure 1.
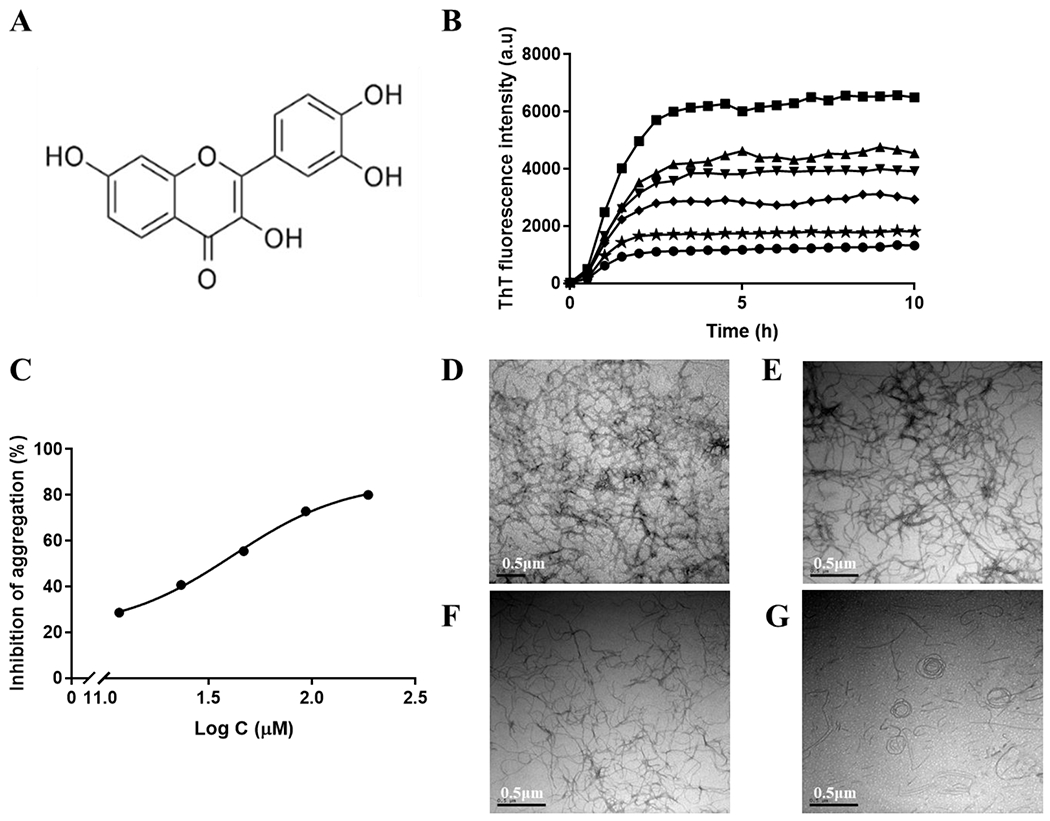
(A) The chemical structure of fisetin. (B) Curves of ThT fluorescence as a function of time with different concentrations of fisetin (0 μM (■), 11.7 μM (▴), 23.3 μM (▼), 46.7 μM (♦). 93.3 μM (★) and 186.7 μM (•)) Each sample contains 75 μM tau K18, 24 μM heparin, and 60 μM ThT in 5% DMSO, 50mM Tris and 100mM NaCl buffer, pH 7.4. (C). Curve of percentages of inhibition as a function of logarithm of fisetin concentration. (D) TEM image of tau K18 final aggregates without fisetin. (E) TEM image of tau K18 final aggregates with 11.7 μM fisetin. (F) TEM image of tau K18 final aggregates with 46.7 μM fisetin. (G) TEM image of tau K18 final aggregates with 186.7 μM fisetin. Scale bar = 0.5 μm.
Methods
Expression and purification of tau protein
The tau K18 proteins were expressed in E.coli BL-21 cells that were cultured in LB medium containing 100 μg/mL ampicillin for 12-16 hours at 37°C in an incubator shaker and transfected with a plasmid coding for the K18 polypeptide. When the OD600 value reached 0.8-1.0, 0.5mM IPTG (Aladdin, China) was added to the medium followed by another 4-hour incubation. The cells were harvested by centrifugation at 5000 rpm and then resuspended in lysis buffer by sonication at 4 °C. After centrifugation at 12,000 rpm for 30 min at 4°C, the supernatant was filtered by a 0.45 μm filter and injected into a C8 column in a high performance liquid chromatography instrument (Agilent, USA). Buffer A was 90% H2O, 10% acetonitrile, 0.1% TFA and buffer B was 90% acetonitrile, 10% H2O, 0.1% TFA. The purity of tau protein was analyzed by SDS-PAGE.
ThT fluorescence experiments
Heparin was selected to induce tau K18 aggregation in vitro [33, 34]. 75 μM tau K18 in buffer containing 5% dimethyl sulfoxide (DMSO), 50mM Tris and 100mM NaCl was incubated with 24 μM heparin and 60 μM thioflavin T (ThT) at pH 7.4, 37°C. Fluorescence signals were detected using a microplate reader (Biotek, U.S.) with excitation wavelength of 440 nm and emission wavelength of 485 nm. To measure whether fisetin inhibits tau K18 aggregation, different concentrations of fisetin (0 μM, 11.7 μM, 23.3 μM, 46.7 μM, 93.3 μM, and 186.7 μM respectively) were added at the beginning of incubation. To determine whether fisetin disaggregates tau filaments, 75 μM fisetin was added at 2 hours, 4 hours, and 6 hours respectively. The curves were analyzed using GraphPad Prism7 software.
Transmission electron microscope (TEM)
10 μL of the final solution from ThT fluorescence experiments was diluted 10 times and placed on a 300-mesh carbon-coated copper grid for 25min. The excess protein solution was inspired with a small piece of filter paper. The copper grid was washed by ddH2O and then stained with 1% uranyl acetate for 1 minute. Imaging was performed on a transmission electron microscope (JEM1230, Japan) at an acceleration voltage of 200 kV.
Bio-layer interferometry (BLI)
The binding kinetics were carried out by BLI technique on an Octet RED96e instrument (ForteBio, U.S.). Tau K18 protein was biotinylated by mixing with NHS-biotin at a ratio of 1:1 for 30 minutes. The free biotin was removed using a PD-10 gravity desalting column. The biotin-bound protein was immobilized on the super streptavidin biosensor with baseline, loading and baseline periods of 60 s, 300 s and 60 s respectively. Then, the biosensors were soaked in fisetin solution with different concentrations (200μM, 100μM, 50μM, and 25μM). The baseline, association and dissociation process were set to 60 s, 180 s and 180 s respectively. The association and dissociation curves were fitted with Fortebio biosystems (global fitting algorithm) to obtain the KD value.
Fisetin fluorescence measurements
20 μM tau K18 was incubated with different concentrations (0, 1, 2, 5, 10, 20, 40 and 80 μM) of fisetin in 50 mM Tris and 100 mM NaCl buffer, pH 7.4 for an hour. The fluorescence wavelength scan spectra (400–680nm) were monitored after exciting at 370 nm. The observed intensities were corrected for the inner filter effect using the following equation:
where Fcorr is the corrected fluorescence intensity, Fobs is the observed fluorescence intensity, Aexcitation is the absorbance of fisetin at its excitation wavelength, 370nm, and Aemission is the absorbance of fisetin at its emission wavelength, 530 nm. The corrected fluorescence intensity was used to calculate the binding affinity. A dissociation constant (KD) was estimated using the following equation:
where ΔF is the corrected fluorescence intensity change at 530nm by subtracting fluorescence of fisetin alone from the fluorescence of tau K18-fisetin complex, ΔFmax is the maximum difference in the fluorescence intensity, and C is the concentration of fisetin.
Circular dichroism (CD)
10 μM tau K18 was incubated in the absence or presence of 10 μM fisetin in 1% methanol, 20 mM potassium phosphate buffer, pH 7.4. The far-UV (190-260 nm) CD spectrum was measured on the J-815 CD instrument (Jasco, Japan) in a 1.0-mm-length quartz cuvette. For each sample, 3 scans were performed with a speed of 1 nm/s and averaged.
Tubulin assembly kinetics
30 μM tau K18 and 30 μM tubulin (Cytoskeleton, USA) were mixed in 5% DMSO, 80 mM PIPES, 0.5 mM EGTA, 2 mM MgCl2 buffer, pH 7.0 to monitor tubulin assembly. 30 or 60 μM fisetin was added to see its affect. The absorbance at 340 nm was recorded every minute for a total 40 minutes.
Modeling of tau K18 and fisetin
The initial coordinates of tau K18 protein were obtained from the QUARK webserver[35, 36] (https://zhanglab.ccmb.med.umich.edu/QUARK/), that uses an ab initio algorithm for protein structure prediction. The coordinate file of fisetin molecule was downloaded from the PubChem web server (https://pubchem.ncbi.nlm.nih.gov/) with a PubChem CID of 5281614.
Protein molecular dynamics (MD) simulation
Four simulation systems were established that include tau K18 alone, tau K18 with 2, 5 or 10 fisetin molecules respectively. The protein was placed in the center of the box filling with TIP3P water,[37] and different numbers of compound was randomly added around the protein. The minimum distance between the edges of the cubic periodic box and the fisetin or tau K18 molecules was 2.5 nm. Counterions (Na+ and Cl−) were added to neutralize the system and provided an additional 0.15 M salt concentration. All simulations were performed by the GROMACS 2018.7 software package[38, 39]. The intrinsically disordered protein-specific force field CHARMM36IDPSFF[40, 41] was used for tau K18 and CHARMM General Force Field (CGenFF) [42, 43] was parameterized for fisetin. The systems were subjected to 50000 steps of steepest descent minimization, or were stopped when all maximum force was less than 10.0 kJ/mol. Before the production simulations, 1 ns equilibration simulations were performed in isothermal isometric ensemble (NVT) and isothermal isobaric ensemble (NPT), respectively. All production simulations were conducted in NPT at a temperature of 310 K using the velocity-rescaling method[44], and a pressure of 1 bar using the Parrinello-Rahman method[45]. Constraints were applied on bond lengths by the LINCS algorithms[46]. The long-range electrostatics and the van der Waals interactions were calculated using a same cutoff of 1.2 nm. All production simulations were performed for 800 ns and the trajectories were saved every 10 ps.
Simulation analyses
The MD trajectory analyses were carried out by GROMACS facilities and third-party packages, including definition of secondary structure of proteins (DSSP) program[47, 48]. Open-Source PyMOL[49, 50], VMD program[51], and so on. The gmx rms, gmx gyrate and gmx hbond were used to calculate Cα root-mean-square deviation (RMSD), radius of gyration (Rg) and number of hydrogen bonds (Hbonds), respectively. The gmx cluster was used for cluster analysis by GROMOS method[52, 53] with a RMSD cutoff of 0.5 nm. In order to calculate the binding affinity between tau K18 and fisetin, and to estimate the energy contribution of every protein residue, the Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method[54, 55] was employed with the g_mmpbsa tool[56]. All data were calculated by averaging 100 snapshots taken from the last 100 ns of each simulation trajectory.
Cell culture
The human embryonic kidney 293 (HEK293) cells were stably transfected with flag-Tau441 by the lentiviral packaging method. The cells were cultured in dulbecco’s modified eagle medium (Hyclone, U.S.), 10% fetal bovine serum (Gibco, Brazil origin) and 100 U/mL of penicillin, 100 μg/mL of streptomycin ( Gibco, USA ) in the incubator with 5% CO2 at 37 °C.
Cell viability assay
When the HEK293 cells grew to around 60%, the culture medium was changed to a medium containing different concentrations of fisetin in the presence of 0.5% DMSO. The cells were cultured for another 24 hours. Then the medium was removed and 100 μl fresh medium containing 10 μl CCK-8 reagent (Beyotime, China) was added. After 0.5-hour, 1-hour, 2-hour and 4-hour incubation, absorbance at 450 nm was measured to detect the viability of the cells using the following formula:
Ablank is the absorbance of CCK-8 reagent at 450 nm, A0 dosing is the absorbance of cell culture plus 0.5% DMSO, and Adosing refers to absorbance of cell culture with fisetin.
Protein extraction and Western blot
The cells were seeded with 3x105 per well in a 6-well plate the day before treatment, then treated with 1 μM or 10 μM fisetin dissolved in vehicle DMSO, respectively. After 24-hour incubation, total cell lysates were scraped and collected with cell scraper by directly adding boiling sample loading buffer without dithiothreitol, 20 mM N-ethylmaleimide (NEM) was added in the sample loading buffer to prevent active sulfhydryls from forming disulfide bonds. To reduce protein disulfide bonds, 80 μM Tris(2-carboxyethyl)phosphine (TCEP) solution was added to the sample loading buffers. The cell lysates were sonicated (SCIENTZ-II D, China) to shear sticky DNA. The expression of tau was detected with tau5 antibody (ab80579, abcam) by western blot with 10% SDS-PAGE (EpiZyme, China). The immunogen of tau5 is full length native protein (purified) corresponding to Cow Tau according to the manufacturer’s instruction.
To investigate the tau protein solubility, we used radio immunoprecipitation assay (RIPA) buffer that is 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS added with PMSF, protease/phosphatase inhibitor cocktail (Cell Signaling Technology, US) and 20mM NEM to lyse cells. The cell lysates were sonicated and then centrifugated with 50,000 g for 20 min at 4 °C. After centrifugation, the supernatant and pellet were collected separately as RIPA-soluble and RIPA-insoluble fraction for further western blot analysis.
Immunofluorescence
Cells were seeded on the coverslip in a 6-well plate with the number of 6x105 cells per well the day before experiment. On the next day, cells were fixed in 4% PFA for 15 min, then washed with PBS, followed by permeabilization in 0.5% of PBS-Triton for 10 min. After 1-hour blocking in immunofluorescence blocking buffer (Beyotime, China), cells were stained with the primary antibody tau T22 (ABN454, Sigma-Aldrich, US) diluting 1:500 at 4°C overnight on the shaker. Its immunogens are recombinant human Tau-441 (Tau-F, Tau-4, 2N4R isoform) oligomers[57]. The secondary antibody Alexa Fluor 488-goat ani-rabbit was used at 1:500 dilution. Cells were mounted with vectashield mounting medium. Images were acquired with a Carl Zeiss LSM710 laser scanning confocal microscope with a 63X oil immersion (1.4 NA) objective, and then analyzed with Image J.
Statistical analysis
Data were analyzed using GraphPad Prism5. The one-way ANOVA analysis followed by Tukey’s multiple comparison test was used to determine the statistical significance of differences of the means. P<0.05 was accepted as statistically significant.
Results
Fisetin inhibits tau K18 aggregation
ThT fluorescence is a commonly-used method to detect the aggregation kinetics of amyloid proteins since the fluorescence signal is proportional to the amount of β-sheet structure. The fibrillation of tau protein is the organization of β-strands in the core of the fibrils[58]. As shown in Figure 1B, in the presence of heparin, tau K18 starts to aggregate at 30 minutes and forms mature fibrils at around 4 hours. After 4 hours, the fluorescence intensity remains stable indicating the fibrilization process of tau K18 is complete. The addition of fisetin results in a decrease in the final ThT fluorescence intensity. The more fisetin added, the larger the intensity decrease. This suggests that fisetin can limit the extent of tau K18 fibril formation by inhibiting tau K18 aggregation. However, the compound does not affect the time when tau K18 starts to aggregate. As fisetin inhibits the aggregation in a concentration-dependent manner, we analyzed a plot of inhibition rate versus log fisetin concentration (Figure 1C). The half-maximal aggregation inhibitory concentration (IC50), 41.45 μM, could be obtained from fitting the curve.
We then utilized TEM to directly observe how fisetin affects the morphology of tau fibrils. Without fisetin, tau assembles into long and straight fibrils and the fibrils are prone to form clusters (Figure 1D). The addition of fisetin causes the tau fibrils to become shorter, thinner and less prevalent (Figure 1E, 1F and 1G). Even at high concentration of fisetin, a number of curved or round fibrils are observed (Figure 1G). Thus, fisetin is able to inhibit, but not eliminate, in vitro tau K18 aggregation.
Fisetin disaggregates tau K18 fibrils
Besides its inhibitory effects, we examined whether fisetin could depolymerize tau aggregates after their formation. We monitored ThT fluorescence when fisetin (equivalent to tau K18 concentration) was added to aggregation reactions at 0 h, 2 h, 4 h, and 6 h, respectively (Figure 2A). No matter when fisetin was added, the fluorescence intensity drops to about three-fold immediately and stays at that value. TEM was again used to observe the morphology of protein aggregates (Figure 2B and 2C). Because the fluorescence intensities of all fisetin-containing solution were very similar, we show the TEM image of sample with fisetin added at 6 hours as representative (Figure 2C). Compared to the long and bundled fibrils formed without fisetin, treatment with fisetin results in shorter dispersed aggregates which may not be actual fibrils. Therefore, we conclude that addition of fisetin can inhibit the formation of tau K18 fibrils at any time during the aggregation process and can disaggregate mature tau K18 filaments as well.
Figure 2.
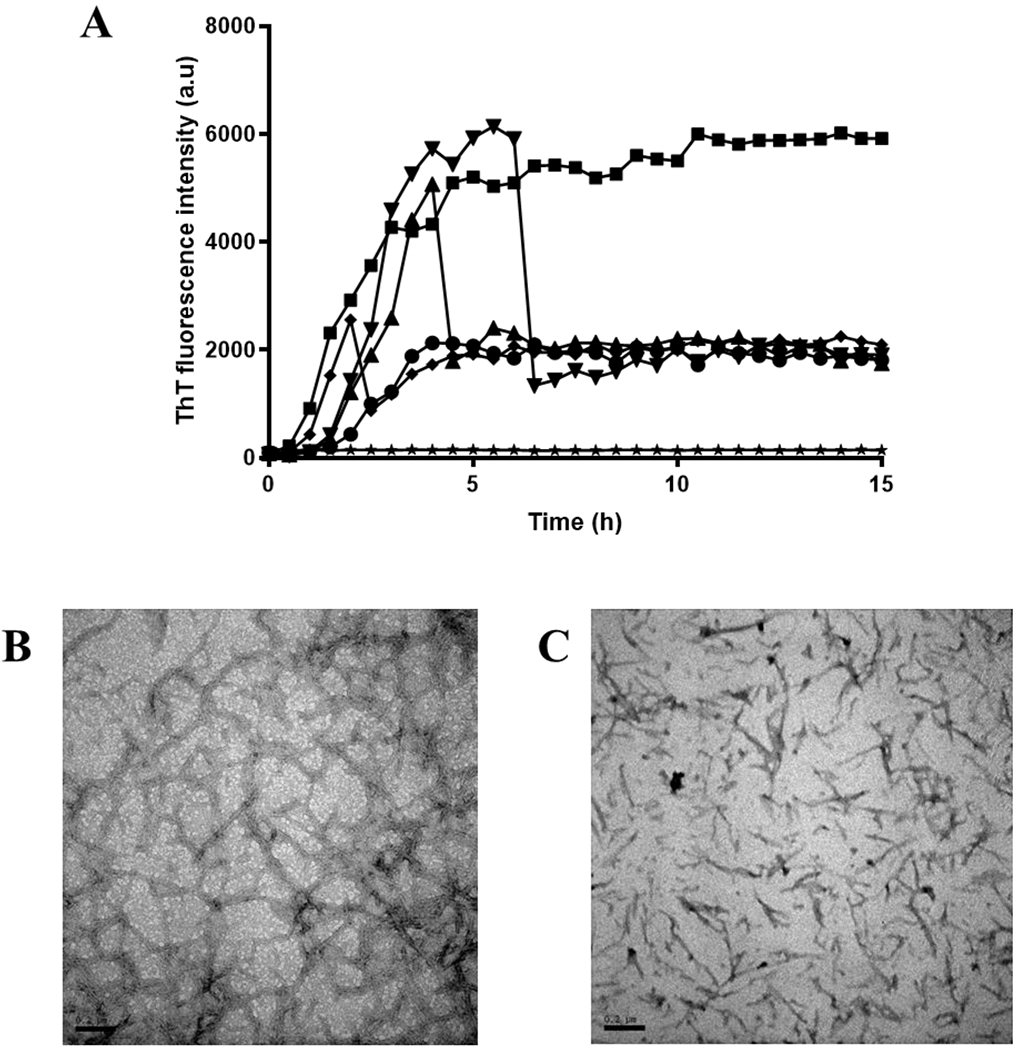
(A) Curves of ThT fluorescence as a function of time with fisetin added at different times (0 hour (•), 2 hours (♦). 4 hours (▴), 6 hours (▼)) while ■ and ★ represent the tau K18 alone and fisetin alone respectively. Each sample contains 75 μM tau K18, 24 μM heparin, and 60 μM ThT in 5% DMSO, 50mM Tris and 100mM NaCl buffer, pH 7.4. (B) TEM image of tau K18 final aggregates without fisetin. (C) TEM image of tau K18 final aggregates with fisetin added at 6 hours. Scale bar = 0.2 μm.
Fisetin interacts with tau K18 protein
The interaction between fisetin and tau K18 was monitored using two different approaches, fluorescence and BLI technique. The wavelength-scan emission spectra of fisetin excited at 370 nm are shown in Figure 3A. The largest emission signal appears at 530 nm. The relationship between fluorescence intensity change as a function of fisetin concentration in the presence of a fixed amount of tau K18 is plotted in Figure 3B. A binding affinity value, KD, of 22.60 ± 1.81 μM is obtained by fitting the line according to the equation shown in the Method section. BLI is useful to measure binding kinetics and affinity between two molecules [59, 60]. The association (first 180 seconds) and dissociation (from 180 to 360 seconds) kinetics between fisetin and tau K18 are shown in Figure 3C. Fisetin gradually binds to tau K18 as time goes on in the association step while in the dissociation step after oscillation fisetin detaches from the protein. A KD value of 61.5 μM is estimated by fitting the curves. The two KD values approximations differ but are nevertheless of the order of magnitude, suggesting that the true value is likely in the tens of micromolar. CD spectra were recorded to measure whether binding of fisetin changes tau K18’s secondary structure (Figure 3D). The results show that tau K18 primarily consists of random coil structures, in agreement with previous reports [61, 62]. The CD spectrum of fisetin-bound tau K18 overlaps very well with that of tau K18 alone, indicating that fisetin does not change the secondary structure of tau K18.
Figure 3.
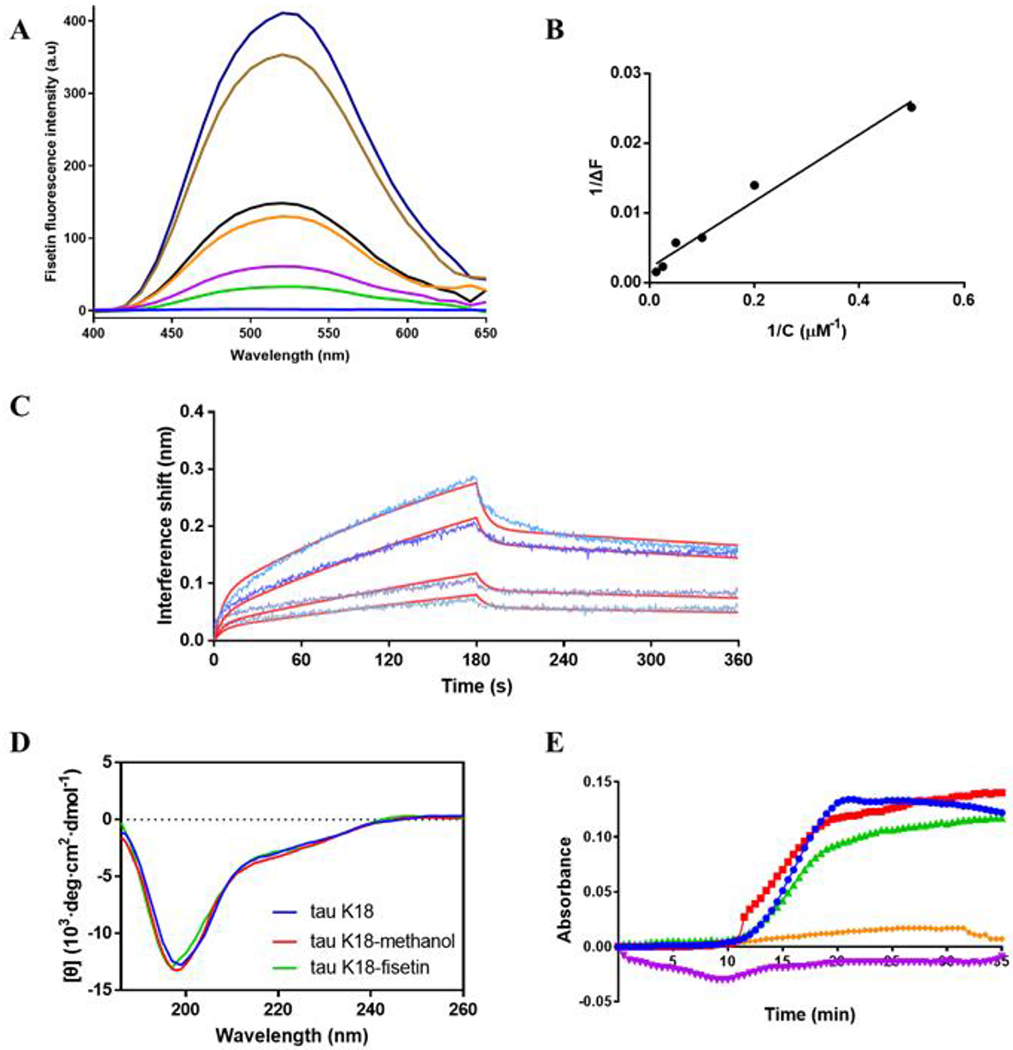
(A) Fluorescence emission spectra of 5 μM tau K18 and different concentrations of fisetin (from top to bottom, 80, 40, 20,10, 5, 2, and 0 μM) excited at 370 nm. (B) Relationship of change of fluorescence intensity at 530 nm and fisetin concentration. The fitting equation is given as Y= 0.04779*X + 0.002114. (C) BLI curves of the binding kinetics of tau K18 and different concentrations of fisetin (from top to bottom, 200, 100, 50, and 25 μM). First 180 seconds represent the association process while the second 180 seconds the dissociation. The red lines are the fittings to the original curves. (D) CD spectra of tau K18 in buffer (blue), tau K18 in buffer with 1% methanol (red), and tau K18 plus fisetin in buffer with 1% methanol (green). Buffer contains 20 inM potassium phosphate, pH 7.4. Both tau K18 and fisetin concentrations are 10 μM. (E) Curves of absorbance at 340 nm as a function of time showing the tubulin assembly kinetics. Samples are tau K18+tubulin (•), tau K18+tubulin+30 μM fisetin (■), tau K18+tubulin+60 μM fisetin (▴), tau K18 alone (♦). and fisetin alone (▼) respectively in buffer including 5% DMSO, 80 mM PIPES, 0.5 mM EGTA, 2 mM MgCl2 buffer, pH 7.0. Both tau K18 and tubulin concentrations are 30 μM.
Furthermore, we explored the mechanisms of the interaction between tau K18 and fisetin by computational analysis. Since there is no defined structure for monomeric tau protein, it is not possible to simply use molecular docking using a static protein structure. MD simulation is currently an appropriate way to study intrinsic disordered protein like tau. Four simulation systems including K18 alone, K18 & two fisetin molecules, K18 & five fisetin, and K18 & ten fisetin, were performed. As shown in Figure 4A, the RMSD values achieved stable values at around 600 ns, indicating that the simulations have reached convergence. The radial probability density function (PDF) was calculated in each simulation (Figure 1B). The width of the PDF curve in the K18 alone group is the broadest, extending from 1.5 to 1.9 nm. While in all the systems containing fisetin, the peak of the distribution curve became narrower. Especially in the K18 & 5 fisetin group, the width of the distribution is the smallest, ranging from 1.47 to 1.65 nm. This indicates that the treatment of fisetin induces more compact conformations of the K18 protein.
Figure 4.
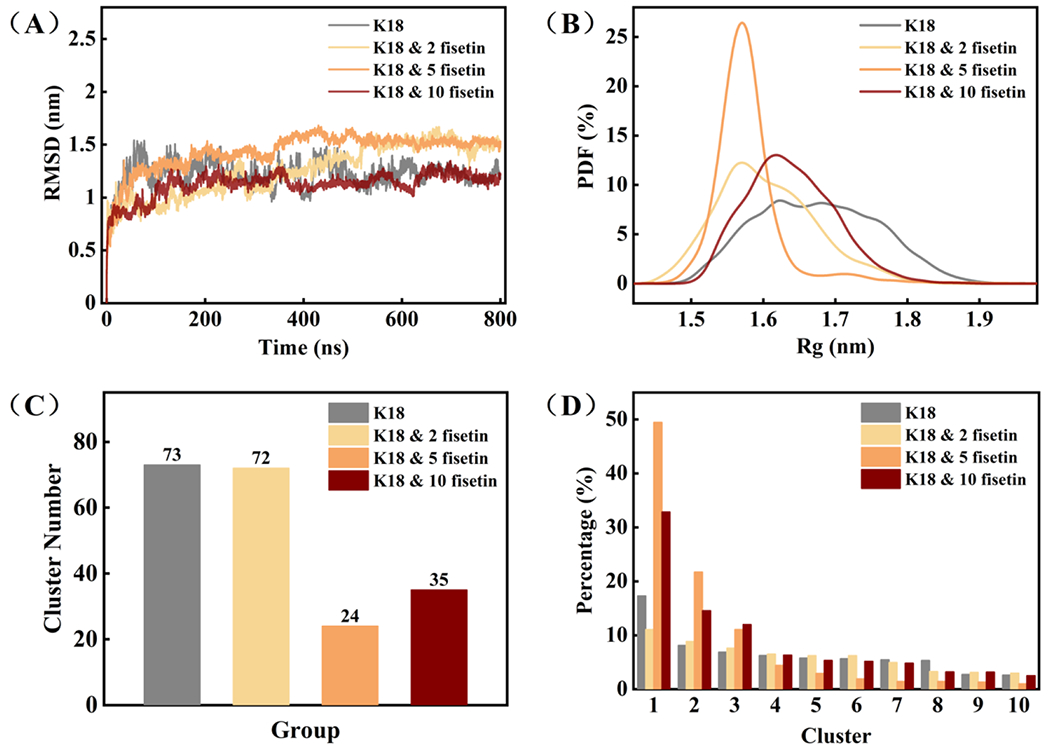
(A) Time evolution of Cα RMSD relative to the initial structure of tau K18; (B) The probability density function (PDF) curves of the protein radius; (C) The histogram of cluster number in each group; (D) The percentage of the top ten clusters in each group.
Similar results were obtained in the cluster analysis (Figure 4C and 4D). The protein conformations in the K18 alone and K18 & 2 fisetin trajectories could be divided into 73 and 72 clusters respectively. The percentage of the top ten clusters is equal. But with more addition of fisetin, the number of clusters becomes much smaller, while the top two clusters account for over 50% of the total conformations. Therefore, the binding of fisetin increases the stability of certain conformations of K18 in solution, inducing a more compact and less flexible protein structure, which appears preventing protein self-aggregation.
We then examined the protein conformations in each cluster. The conformation of the most populated cluster in each simulation trajectory is shown in Figure 5. The initial structure of K18 obtained from the QUARK Webserver is unstructured with a few short and likely transient helixes. After MD simulation, K18 still retains mostly random coiled but forms two antiparallel β-strands at R2 and R3 repeats domain. Coincidentally, these two β-strands covers exactly the PHF6* (residue 275-280) and PHF6 (residue 306-311) regions, that are believed to play a key role in the initial process of tau self-aggregation [7],[63]. Upon addition of fisetin, the compound interacts with the protein and prevents the formation the β-strands. This observation is derived from a single conformational cluster, so next we examined the alterations of every type of secondary structure during the whole MD simulation (Figure 6). The β-strand structures at the two hexapeptide motifs are clearly evident once they are formed during the 800 ns simulation in the absence of fisetin (Figure 6A). However, in the fisetin groups, these two β-strands sometimes or do not appear at all. Interestingly, some α-helixes are produced that are not observed in the K18 alone trajectory. The probabilities of each type of secondary structure are measured and shown in Figure 7A and those of helix, β-sheet, and coil per residue in Figure 7B–D. The treatment of fisetin induces detectable formation of helixes at residue 268-275, 300-306, and 345-355 and prevents the formation of β-sheet at residue 272-280 and 303-312. However, these local conformational alterations do not cause significant changes in the total secondary structure content values (Figure 7A). This is consistent with the CD result that fisetin does not lead to significant change of the protein secondary structures.
Figure 5.
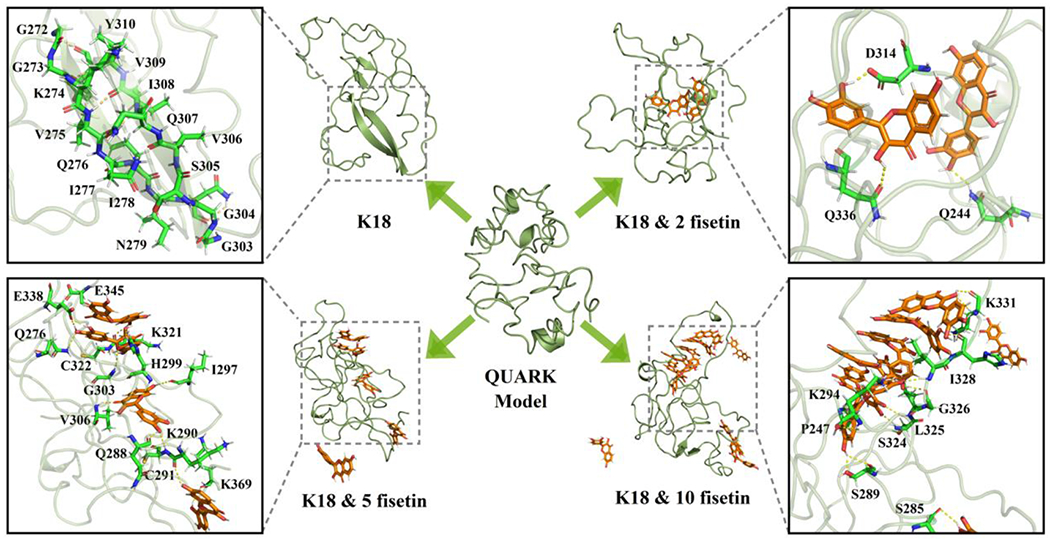
The representative conformation of the most populated clusters in each simulation trajectory. QUARK model represents the initial protein structure given by the Webserver.
Figure 6.
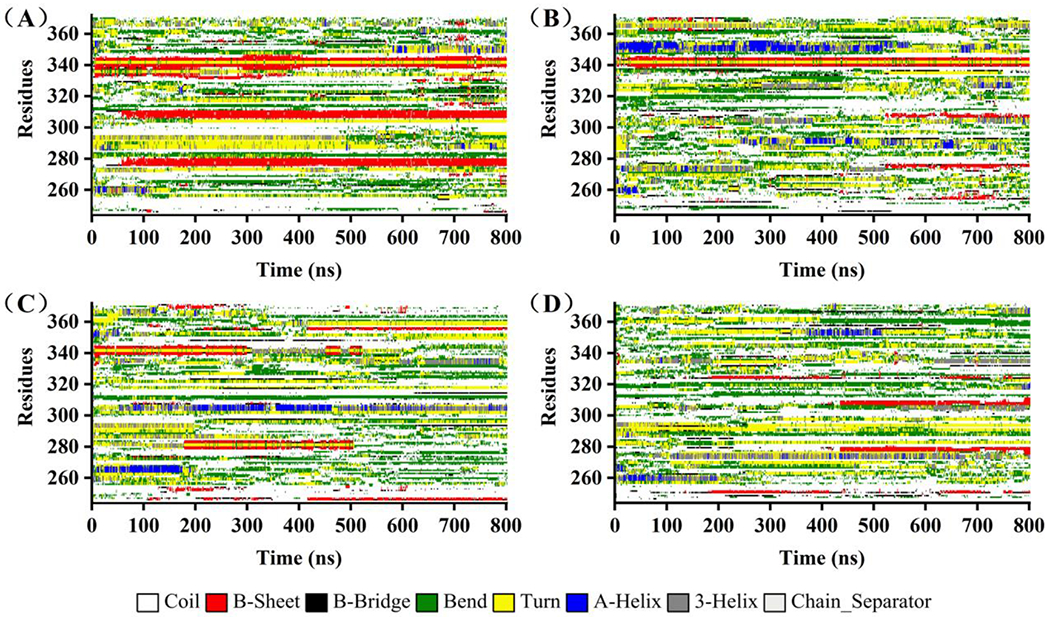
The secondary structure of every residue as a function of time in the simulation trajectories: (A) K18, (B) K18 & 2 fisetin, (C) K18 & 5 fisetin, (D) K18 & 10 fisetin. Different color labels different kind of secondary structure.
Figure 7.
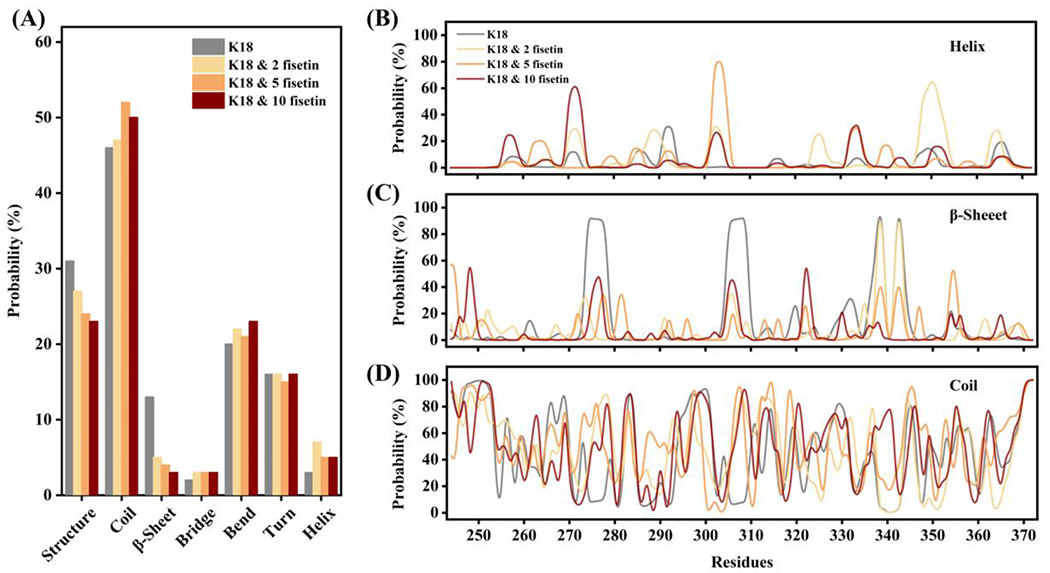
(A) The histogram of probability of each kind of secondary structure in the four simulation systems; (B) the probability of helix at each residue in the four simulations; (C) the probability of β-sheet at each residue in the four simulations; (D) the probability of coil at each residue in the four simulations.
Hydrogen bonds play an important role in the formation and maintenance of the secondary structures of proteins. Therefore, we analyzed the number of hydrogen bond within K18 and between K18 and fisetin to see the effects of fisetin on hydrogen bond formation. As shown in Figure 8B, the peak of the curve of percentage of the intrinsic hydrogen bond number is at around 60 in the K18 simulation trajectory. When treating with fisetin, the peak of the curve shifts to the left in the other three trajectories denoting less hydrogen bonds within the protein. As shown in Figure 8D, the compound can form hydrogen bonds with the K18 protein, and the bond number is significantly dose-dependent. Thus, fisetin is able to generate hydrogen bonds with K18 resulting in the loss of the intrinsic hydrogen bonds within the protein.
Figure 8.
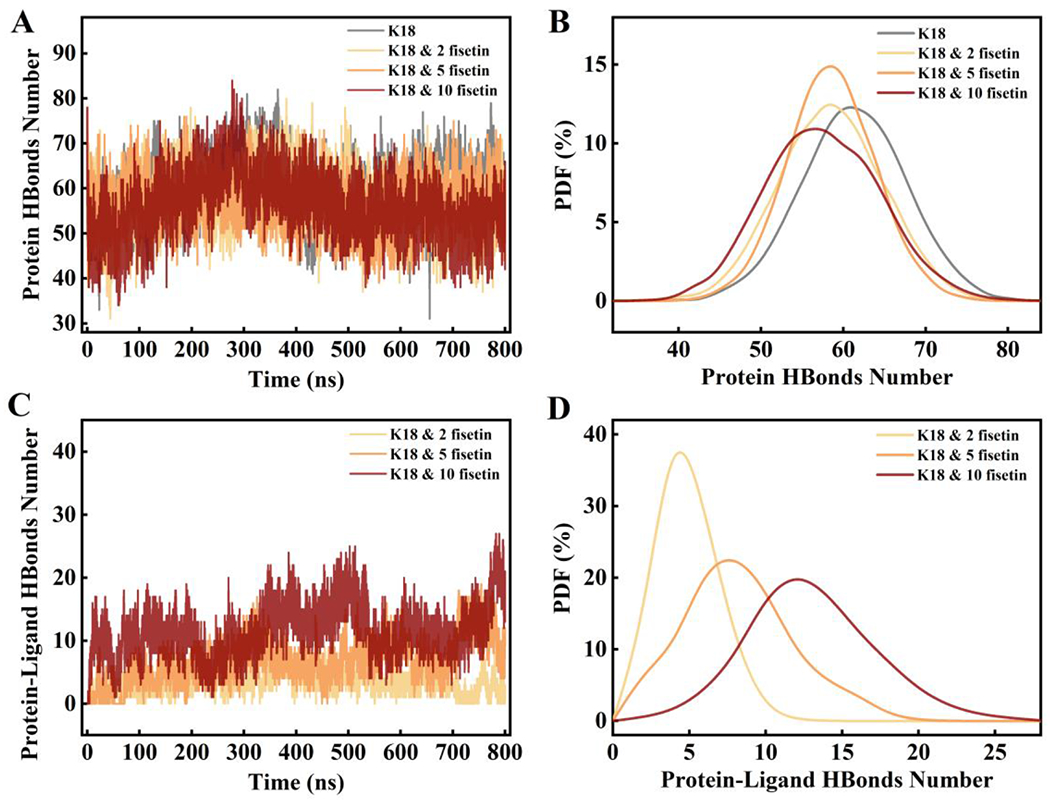
(A) The number of hydrogen bonds formed within the protein during the simulations in the four systems; (B) the probability density distribution curves of hydrogen bond number from trajectories in A; (C) The number of hydrogen bonds formed between K18 and fisetin during the simulations; (D) the probability density distribution curves of hydrogen bond number from trajectories in C. A cutoff of 3.5 Å in distance (donor-acceptor) and 30° in angle (hydrogen-donor-acceptor) was used to define the hydrogen bond.
Finally, the binding energies including van der Waal, electrostatics, polar solvation, and solvent accessible surface area (SASA) energy were calculated and summarized in Table 1. The van der Waal, electrostatic and SASA energy were favorable for the interaction of K18 and fisetin, while the polar solvation energy provided unfavorable contributions to the binding energies. In all groups, the values of the final binding energy are negative, indicating that the binding of K18 and fisetin is thermodynamically favorable. The largest contributor was Van der Waal energy which might be attributed to π-π stacking. It is worth noting that all fisetin molecules were considered as one object in the energy calculation. Therefore, the result does not represent the relationship between K18 and one single fisetin molecule, and the values of the binding energies are not proportional to the number of fisetin molecules.
Table 1.
The Binding Free Energy between K18 and fisetin
| Energy (kJ/mol) | K18 & 2 fisetin | K18 & 5 fisetin | K18 & 10 fisetin |
|---|---|---|---|
| van der Waal energy | −176.882 ± 47.778 | −452.113 ± 63.044 | −557.411 ± 89.625 |
| Electrostatic energy | −39.642 ± 24.174 | −246.164 ± 52.970 | −270.664 ± 79.890 |
| Polar solvation energy | 160.090 ± 55.951 | 554.831 ± 69.535 | 625.158 ± 121.528 |
| SASA energy | −19.390 ± 5.132 | −51.314 ± 5.930 | −63.376 ± 9.059 |
| Binding energy | −75.826 ± 51.769 | −194.760 ± 47.568 | −266.293 ± 74.808 |
Fisetin does not impact tau-induced tubulin assembly
One important physiological function of tau protein is to promote tubulin assembly into microtubules. We therefore tested whether fisetin-bound tau protein still induces tubulin assembly. Figure 3E shows tubulin assembly kinetics in different conditions. Under the promotion of tau K18, tubulin begins to assemble at about 10 minutes and forms stable microtubules by 20 minutes as monitored using absorbance at 340 nm. Fisetin itself has negligible absorbance at 340 nm. When fisetin was added at one- or two-fold the concentration of tau K18, tubulin is able to assemble into microtubule equally well. Therefore, fisetin does not affect tau-induced tubulin assembly including the kinetics or the final optical absobance as an indicatior of the amounts of the formed microtubules.
Fisetin decreases tau aggregates in cells
Besides the in vitro and computational studies, we performed cell-based experiments to further check the effects of fisetin. First, the CCK-8 method was used to test the toxicity of fisetin on the cells. Cell viability as a function of fisetin concentration is shown in Figure 9A. More than 60% of cells proliferate normally when fisetin concentrations as high as 271 μM were added. Less than 5% of cells were affected by 9 μM fisetin. In the following experiments, 1 μM and 10 μM fisetin were used. Then a HEK293 cell line stably transfected with a gene conding for flag-tau441 was constructed by lentiviral packaging. The western blot analysis of tau protein recognized by Tau5 antibody in the non-reduced condition shows a clear band in the high molecular weight region representing oligomeric tau (Figure 9B). Treatment with fisetin results in a decrease in the quantity of the tau oligomers especially in the 10 μM fisetin group, illustrating that fisetin can inhibite tau aggregation in cells (Figure 9C). When a reducing loading buffer with TCEP was used, the band for tau oligomer completely disappeared indicating the oligomer was produced by disulfide bonds as reported before [64](Figure 9B).
Figure 9.
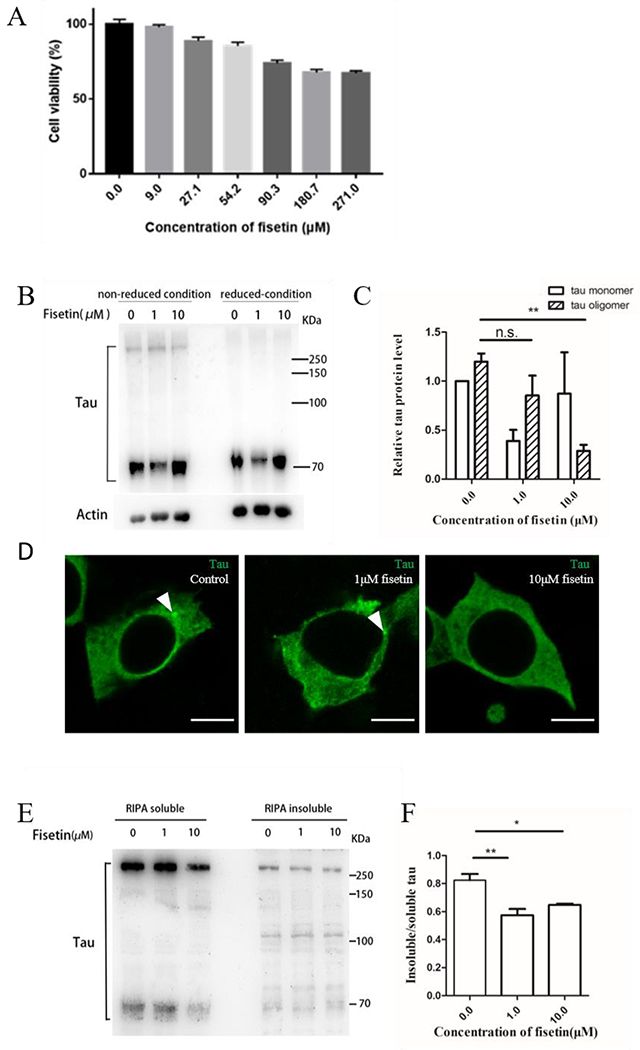
(A) Histogram of cell viability as a function of fisetin concentration; (B) western blot analysis of tau protein in the cell lysates treated without fisetin or with 1 μM and 10 μM respectively. Tau5 antibody was used. Non-reduced condition means no DTT while reduced condition 100mM TCEP in sample loading buffer. (C) quantitative comparison of tau monomers and tau oligomers in the non-reduced condition. Actin was used as a loading control. Error bar represents standard deviation from three repeated experiments, n.s. represents no significant differences, **P= 0.005. (D) immunofluorescence staining of tau in HEK293/tau441 cells after treatment of 1 μM or 10 μM fisetin for 24 hours. T22 antibody was used. White arrows indicate tau aggregates in cell cytosol. Scale bar =10 μm. (E) western blot analysis of tau protein in the RIPA-soluble and RIPA-insoluble proportion of the cells treated without fisetin or with 1 μM and 10 μM respectively. Tau5 antibody was used. (F) quantitative comparison of insoluble/soluble tau protein in the three groups. Error bar represents standard deviation from three repeated experiments, *P =0.0269, **P =0.0056.
We also used the T22 antibody, which recognizes oligomeric tau species, to investigate the distribution and morphology of tau in HEK293/tau441 (Figure 9D). The results show that tau aggregation could be observed in the control group, while after fisetin treatment, the aggregated tau puncta become smaller in the 1 μM fisetin group, and do not show up in the 10 μM fisetin group, demonstrating again that fisetin has the ability to repress tau aggregation.
To further clarify the role of fisetin in tau protein aggregation, we isolated the RIPA-soluble and RIPA-insoluble protein fractions from the HEK293/tau441 cells. The western blot analysis of tau protein demonstrats that the ratio of insoluble/soluble tau proteins is significantly reduced upon treatment with 1 μM or 10 μM fisetin compared to the control group (Figure 9E and 9F). In summary, fisetin is able to prevent tau aggregation and/or lower the amount of tau aggregates in cell models.
Discussion
Recently, more attention has been brought to focus on tau protein for AD drug development. In this study, we report that fisetin is able to inhibit tau K18 aggregation and disaggregate tau K18 filaments in vitro, and it could prevent or reduce the formation of full-length tau aggregates in the HEK293/tau441 cells as well. A direct interaction between fisetin and tau K18 is observed by fluorescence experiments and BLI technique. Studies of MD simulation illustrate that the addition of fisetin prevents the formation of β-sheets at PHF6* and PHF6 motifs that have been identified as the primary aggregation nucleation site [7],[63], thus inhibiting the protein aggregation. Meanwhile the fisetin-bound tau protein exhibits more compact and less flexible structures as indicated by lower radial distribution and conformational cluster values. In general, the radius reflects the conformational flexibility of the protein in solution. A larger radial value means that the conformation of the protein is more extended and looser, which increases the probability of contact between proteins and the propensity of aggregation as well. Further analysis of the simulation trajectories shows that hydrogen bond and Van der Waal forces greatly contribute to the interaction between K18 and fisetin.
One of the physiological functions of tau protein is to interact with tubulin and promote its assembly. Isothermal titration calorimetry experiments display two types of tau-tubulin binding modes: one corresponding to the participation of all four repeats in a high affinity binding site with a tau: tubulin stoichiometry of 0.2 and the other one to the participation of one repeat in a low affinity binding site with a stoichiometry of 0.8 [65], Residues 224-398 of tau protein are involved in tau-microtubule interaction and residues Lys225, Lys240, Lys257, Lys311, and Lys383 in tau can be cross-linked to the side chains of Lys336 and Lys338 of α-tubulin by amine-directed cross-linking [66]. Here we observed that fisetin-bound tau K18 is still able to interact with tubulin and promote its assembly (Figure 3E). The interpretations could be that the fisetin binding sites in tau K18 may not be essential for the tau-tubulin interaction or that tubulin outcompetes fisetin in binding to tau protein. We estimated the binding affinity between tau K18 and fisetin as tens of micromolar by two different methods. The Rhoades group calculated the binding affinity between tau K16 and tubulin to be 2.5 μM by fluorescence correlation spectroscopy [67]. Tau K16 contains the residues 198-372 while tau K18 includes the residues 244-372. It is possible that tau preferentially binds to tubulin rather than fisetin. As a microtubule stabilizer, tau detachment results in neuronal cytoskeleton instability. In the presence of the compound, the fisetin-bound tau would not self-aggregate into oligomers, but it could also induce tubulin assembly. Thus, fisetin could inhibit the gain of toxicity of tau protein but not alter the physiological function. Usually, the structure determines the function. As the CD experiments and MD simulation analysis both show that the binding of fisetin causes few changes on the secondary structure of tau protein.
AD is a complicated brain disease that might be caused by multiple factors. This might be the reason why many clinical trials of AD drugs failed as they focus on a single target. The use of plant-derived polyphenols as therapeutic or preventive agents is gaining increasing interest due to their multifactorial effects which have shown beneficial outcomes for a wide range of diseases [68]. Oxidative stress plays an essential role in the early stage of AD due to the disequilibrium between pro-oxidant and antioxidant [69]. One study showed that fisetin played a critical role in neuroprotection through the activation of the nuclear factor erythroid 2-related factor 2 (Nrf2)-antioxidant response element (ARE) pathway in a mouse model of traumatic brain injury [70]. Another report provided evidence that fisetin markedly abrogated the LPS-induced elevated ROS/oxidative stress and activated phosphorylated c-JUN N-terminal kinase (p-JNK) in the adult mouse hippocampus [71]. Inflammation is another important factor in the progression of AD. Some references indicate that fisetin inhibited LPS-induced activation of the inflammatory Toll-like receptors (TLR4)/cluster of differentiation 14 (CD14)/phospho-nuclear factor κ (NF-κB) signaling and attenuated other inflammatory mediators like tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), and cyclooxygenase (COX-2) [71, 72]. Fisetin has also been reported to inhibit Aβ fibril formation in vitro [73], and protect against Aβ-induced cognitive/synaptic dysfunction, neuroinflammation, and neurodegeneration in adult mice [28]. Reduction of tau phosphorylation by fisetin has been observed as well, through the autophagy pathway activated by transcription factor EB (TFEB) and Nrf2 [32]. In this study, we establish a direct relationship between fisetin and tau aggregation. In consideration of its inhibitory effects on tau aggregation discovered here and other neuroprotective effects reported elsewhere, fisetin could be evaluated further as a potential preventive and therapeutic drug candidate or food product for AD.
Highlights.
Fisetin inhibits tau aggregation in vitro and in cells
Fisetin interacts with tau protein through favorable hydrogen bonds and van der Waals
Fisetin prevents the formation of β-strands at the two critical hexapeptide motifs
Acknowledgments
The authors thank the Instrumental Analysis Center of Shenzhen University for technical assistance.
Funding
This work was financially supported by the Shenzhen Science and Technology Innovation Commission [JCYJ20180507182417779, JCYJ20200109110001818], Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions [2019SHIBS0003], and NIH grant R37AG019391 to D.E.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no conflicts of interest in regards to this manuscript.
References
- 1.Blennow K, de Leon MJ, Zetterberg H: Alzheimer’s disease. Lancet 2006, 368(9533):387–403. [DOI] [PubMed] [Google Scholar]
- 2.Liu PP, Xie Y, Meng XY, Kang JS: History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther 2019, 4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloom GS: Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 2014, 71(4):505–508. [DOI] [PubMed] [Google Scholar]
- 4.Arendt T, Stieler JT, Holzer M: Tau and tauopathies. Brain Res Bull 2016, 126(Pt 3):238–292. [DOI] [PubMed] [Google Scholar]
- 5.Cleveland DW, Hwo SY, Kirschner MW: Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 1977, 116(2):227–247. [DOI] [PubMed] [Google Scholar]
- 6.Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E: Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell 1995, 6(12):1887–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E: Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. P Natl Acad Sci USA 2000, 97(10):5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW: Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547(7662):185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albert M, Mairet-Coello G, Danis C, Lieger S, Caillierez R, Carrier S, Skrobala E, Landrieu I, Michel A, Schmitt M et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019, 142(6):1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elbaum-Garfinkle S, Cobb G, Compton JT, Li XH, Rhoades E: Tau mutants bind tubulin heterodimers with enhanced affinity. Proc Natl Acad Sci U S A 2014, 111(17):6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karikari TK, Nagel DA, Grainger A, Clarke-Bland C, Crowe J, Hill EJ, Moffat KG: Distinct conformations, aggregation and cellular internalization of different tau strains. Front Cell Neurosci 2019, 13:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar H, Udgaonkar JB: Mechanistic and structural origins of the asymmetric barrier to prion-like cross-seeding between tau-3R and tau-4R. J Mol Biol 2018, 430(24):5304–5312. [DOI] [PubMed] [Google Scholar]
- 13.Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, Mandelkow E, Knowles TP, Mandelkow E, Klenerman D: A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 2015, 6:7025–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ait-Bouziad N, Lv G, Mahul-Mellier A-L, Xiao S, Zorludemir G, Eliezer D, Walz T, Lashuel HA: Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat commun 2017, 8(1):1678–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Congdon EE, Sigurdsson EM: Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 2018, 14(7):399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ksiezak-Reding H, Liu WK, Yen SH: Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res 1992, 597(2):209–219. [DOI] [PubMed] [Google Scholar]
- 17.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR: Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 1996, 93(20):11213–11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M: Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem 2005, 280(9)7614–7623. [DOI] [PubMed] [Google Scholar]
- 19.Necula M, Chirita CN, Kuret J: Cyanine Dye N744 Inhibits Tau Fibrillization by Blocking Filament Extension: Implications for the Treatment of Tauopathic Neurodegenerative Diseases. Biochemistry 2005, 44(30): 10227–10237. [DOI] [PubMed] [Google Scholar]
- 20.Rane JS, Bhaumik P, Panda D: Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J Aizheim Dis 2017, 60(3):999–1014. [DOI] [PubMed] [Google Scholar]
- 21.Zhang M, Wu Q, Yao X, Zhao J, Zhong W, Liu Q, Xiao S: Xanthohumol inhibits tau protein aggregation and protects cells against tau aggregates. Food Func 2019, 10(12)7865–7874. [DOI] [PubMed] [Google Scholar]
- 22.Wilcock GK, Gauthier S, Frisoni GB, Jia J, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Schelter BO, Wischik DJ et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J Aizheim Dis 2018, 61(1):435–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundarraj K, Raghunath A, Perumal E: A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed Pharmacother 2017, 97:928–940. [DOI] [PubMed] [Google Scholar]
- 24.Chuang JY, Chang PC, Shen YC, Lin C, Tsai CF, Chen JH, Yeh WL, Wu LH, Lin HY, Liu YS et al. : Regulatory effects of fisetin on microglial activation. Molecules 2014, 19(7):8820–8839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Touil YS, Seguin J, Scherman D, Chabot GG: Improved antiangiogenic and antitumour activity of the combination of the natural flavonoid fisetin and cyclophosphamide in Lewis lung carcinoma-bearing mice. Cancer Chemoth Pharm 2011, 68(2):445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishige K, Schubert D, Sagara Y: Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radical Biol Med 2001, 30(4):433–446. [DOI] [PubMed] [Google Scholar]
- 27.Akaishi T, Morimoto T, Shibao M, Watanabe S, Sakai-Kato K, Utsunomiya-Tate N, Abe K: Structural requirements for the flavonoid fisetin in inhibiting fibril formation of amyloid beta protein. Neurosci Lett 2008, 444(3):280–285. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad A, Ali T, Park HY, Badshah H, Rehman SU, Kim MO: Neuroprotective Effect of Fisetin Against Amyloid-Beta-Induced Cognitive/Synaptic Dysfunction, Neuroinflammation, and Neurodegeneration in Adult Mice. Mol Neurobiol 2017, 54(3):2269–2285. [DOI] [PubMed] [Google Scholar]
- 29.Prakash D, Sudhandiran G: Dietary flavonoid fisetin regulates aluminium chloride-induced neuronal apoptosis in cortex and hippocampus of mice brain. J Nutr Biochem 2015, 26(12):1527–1539. [DOI] [PubMed] [Google Scholar]
- 30.Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P: Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 2014, 13(2):379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He W-b, Abe K, Akaishi T: Oral administration of fisetin promotes the induction of hippocampal long-term potentiation in vivo. J Pharmacol Sci 2018, 136(1):42–45. [DOI] [PubMed] [Google Scholar]
- 32.Kim S, Choi KJ, Cho SJ, Yun SM, Jeon JP, Koh YH, Song J, Johnson GV, Jo C: Fisetin stimulates autophagic degradation of phosphorylated tau via the activation of TFEB and Nrf2 transcription factors. Sci Rep 2016, 6:24933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khosravi Z, Nasiri Khalili MA, Moradi S, Hassan Sajedi R, Zeinoddini M: The Molecular Chaperone Artemin Efficiently Blocks Fibrillization of TAU Protein In Vitro. Cell J 2018, 19(4):569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedhoff P, Schneider A, Mandelkow E-M, Mandelkow E: Rapid Assembly of Alzheimer-like Paired Helical Filaments from Microtubule-Associated Protein Tau Monitored by Fluorescence in Solution. Biochemistry 1998, 37(28):10223–10230. [DOI] [PubMed] [Google Scholar]
- 35.Xu D, Zhang Y: Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80(7): 1715–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D, Zhang Y: Toward optimal fragment generations for ab initio protein structure assembly. Proteins 2013, 81(2):229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML: Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79(2):926–935. [Google Scholar]
- 38.Berendsen HJC, Vanderspoel D, Vandrunen R: Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation. Comput Phys Commun 1995, 91(1-3):43–56. [Google Scholar]
- 39.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B: GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2(C):19–25. [Google Scholar]
- 40.Liu H, Song D, Lu H, Luo R, Chen HF: Intrinsically disordered protein-specific force field CHARMM36IDPSFF. Chem Bioi Drug Des, 92(4):1722–1735. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Song D, Zhang YP, Yang S, Luo RA, Chen HF: Extensive tests and evaluation of the CHARMM36IDPSFF force field for intrinsically disordered proteins and folded proteins. Phys Chem Chem Phys 2019, 21(39):21918–21931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanommeslaeghe K, MacKerell AD: Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J Chem Inf Model 2012, 52(12):3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanommeslaeghe K, Raman EP, MacKerell AD: Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J Chem Inf Model 2012, 52(12):3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bussi G, Donadio D, Parrinello M: Canonical sampling through velocity rescaling. J Chem Phys 2007, 126(1):014101. [DOI] [PubMed] [Google Scholar]
- 45.Nose S, Klein ML: Constant Pressure Molecular-Dynamics for Molecular-Systems. Mol Phys 1983, 50(5):1055–1076. [Google Scholar]
- 46.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM: LINCS: A linear constraint solver for molecular simulations. J Comput Chem 1997, 18(12):1463–1472. [Google Scholar]
- 47.Joosten RP, Beek TAHT, Krieger E, Hekkelman ML, Hooft RWW, Schneider R, Sander C, Vriend G: A series of PDB related databases for everyday needs. Nucleic Acids Res 2011, 39:D411–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabsch W, Sander C: Dictionary of Protein Secondary Structure - Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22(12):2577–2637. [DOI] [PubMed] [Google Scholar]
- 49.DeLano WL: Use of PYMOL as a communications tool for molecular science. Abstr Pap Am Chem S 2004, 228:U313–U314. [Google Scholar]
- 50.DeLano WL, Lam JW: PyMOL: A communications tool for computational models. Abstr Pap Am Chem S 2005, 230:U1371–U1372. [Google Scholar]
- 51.Humphrey W, Dalke A, Schulten K: VMD: Visual molecular dynamics. J Mol Graph Model 1996, 14(1):33–38. [DOI] [PubMed] [Google Scholar]
- 52.Friedemann R, Naumann S: GROMOS-MD simulations on 1,2-diol water clusters. J Mol Struc-Theochem 1997, 398:405–410. [Google Scholar]
- 53.Friedemann R, Fengler A, Naumann S: GROMOS-MD simulations on biamphiphilic tetraol clusters. Z Phys Chem 1999, 209:133–139. [Google Scholar]
- 54.Genheden S, Ryde U: The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Dis 2015, 10(5):449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang EC, Sun HY, Wang JM, Wang Z, Liu H, Zhang JZH, Hou TJ: End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem Rev 2019, 119(16):9478–9508. [DOI] [PubMed] [Google Scholar]
- 56.Kumari R, Kumar R, Lynn A, Consort OSDD: g_mmpbsa-A GROMACS Tool for High-Throughput MM-PBSA Calculations. J Chem Inf Model 2014, 54(7):1951–1962. [DOI] [PubMed] [Google Scholar]
- 57.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R: Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J 2012, 26(5):1946–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chiti F, Dobson CM: Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem 2017, 86:27–68. [DOI] [PubMed] [Google Scholar]
- 59.Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, Popovych N, Helgason E, Schoeffler A, Jeet S et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528(7582):370–375. [DOI] [PubMed] [Google Scholar]
- 60.Chen P, Tao L, Wang T, Zhang J, He A, Lam KH, Liu Z, He X, Perry K, Dong M et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 2018, 360(6389):664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barre P, Eliezer D. Structural transitions in tau k18 on micelle binding suggest a hierarchy in the efficacy of individual microtubule-binding repeats in filament nucleation. Protein Sci 2013, 22(8):1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.von Bergen M, Barghorn S, Jeganathan S, Mandelkow EM, Mandelkow E. Spectroscopic approaches to the conformation of tau protein in solution and in paired helical filaments. Neurodegener Dis 2006, 3(4-5):197–206. [DOI] [PubMed] [Google Scholar]
- 63.Goux WJ, Kopplin L, Nguyen AD, Leak K, Rutkofsky M, Shanmuganandam VD, Sharma D, Inouye H, Kirschner DA. The formation of straight and twisted filaments from short tau peptides. J Biot Chem 2004, 279(26):26868–26875. [DOI] [PubMed] [Google Scholar]
- 64.Chen H, Liu SM, Li SM, Chen JR, Ni JZ, Liu Q: Blocking the Thiol at Cysteine-322 Destabilizes Tau Protein and Prevents Its Oligomer Formation. ACS Chem Neurosci 2018, 9(7):1560–1565. [DOI] [PubMed] [Google Scholar]
- 65.Tsvetkov PO, Makarov AA, Malesinski S, Peyrot V, Devred F. New insights into tau-microtubules interaction revealed by isothermal titration calorimetry. Biochimie 2012, 94(3):916–919. [DOI] [PubMed] [Google Scholar]
- 66.Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M: Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A 2015, 112(24):7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elbaum-Garfinkle S, Cobb G, Compton JT, Li XH, Rhoades E: Tau mutants bind tubulin heterodimers with enhanced affinity. Proc Natl Acad Sci U S A 2014, 111(17):6311–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andrade S, Ramalho MJ, Loureiro JA, Pereira MDC: Natural Compounds for Alzheimer’s Disease Therapy: A Systematic Review of Preclinical and Clinical Studies. Int J Mol Sci 2019, 20(9):2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uttara B, Singh AV, Zamboni P, Mahajan RT: Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009, 7(1):65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L, Wang H, Zhou Y, Zhu Y, Fei M: Fisetin alleviates oxidative stress after traumatic brain injury via the Nrf2-ARE pathway. Neurochem Int 2018, 118:304–313. [DOI] [PubMed] [Google Scholar]
- 71.Ahmad A, Ali T, Rehman SU, Kim MO: Phytomedicine-Based Potent Antioxidant, Fisetin Protects CNS-Insult LPS-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment. J Clin Med 2019, 8(6): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh S, Singh AK, Garg G, Rizvi SI: Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci 2018, 193:171–179. [DOI] [PubMed] [Google Scholar]
- 73.Kim H, Park BS, Lee KG, Choi CY, Jang SS, Kim YH, Lee SE: Effects of naturally occurring compounds on fibril formation and oxidative stress of beta-amyloid. J Agr Food Chem 2005, 53(22):8537–8541. [DOI] [PubMed] [Google Scholar]