


Abstract
There is considerable interest in creating a precise and sensitive strategy for in situ visualizing and profiling intracellular miRNA. Present here is a novel photocaged amplified FRET nanoflare (PAFN), which spatiotemporal controls of mRNA-powered nanomachine for precise and sensitive miRNA imaging in live cells. The PAFN could be activated remotely by light, be triggered by specific low-abundance miRNA and fueled by high-abundance mRNA. It offers high spatiotemporal control over the initial activity of nanomachine at desirable time and site, and a ‘one-to-more’ ratiometric signal amplification model. The PAFN, an unprecedented design, is quiescent during the delivery process. However, upon reaching the interest tumor site, it can be selectively activated by light, and then be triggered by specific miRNA, avoiding undesirable early activation and reducing nonspecific signals, allowing precise and sensitive detection of specific miRNA in live cells. This strategy may open new avenues for creating spatiotemporally controllable and endogenous molecule-powered nanomachine, facilitating application at biological and medical imaging.
Graphical Abstract
Graphical Abstract.
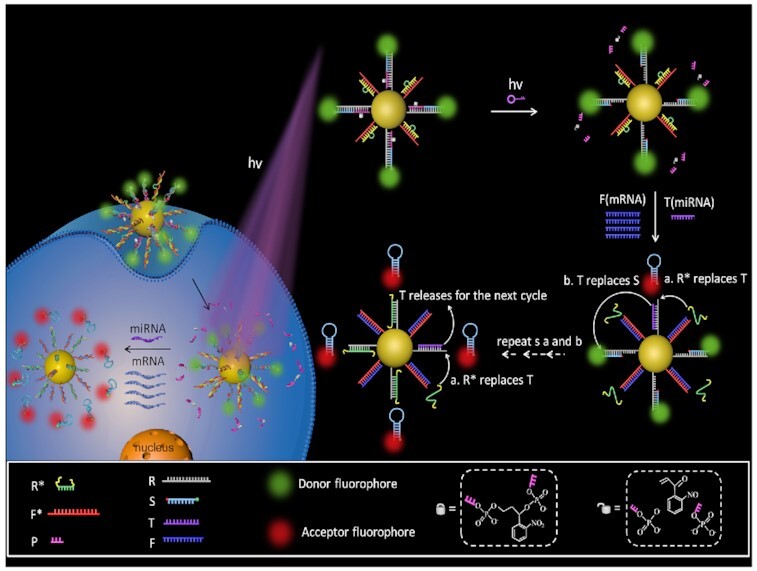
Spatiotemporal controllable of amplified FRET nanoflare is activated by illumination, triggered by specific low-abundance miRNA and fueled by high-abundance mRNA in live cells.
INTRODUCTION
MicroRNA (miRNA) is a small non-coding RNA that generally involves in RNA silencing and post-transcriptional regulation by targeting mRNA for degradation or transcriptional repression (1–3). Some miRNAs play key roles in cell proliferation, differentiation and apoptosis, and have the function similar to oncogenes or tumor suppressors (4). Emerging evidence indicates that aberrant expression of miRNA is also closely associated with some human diseases such as cancers (5–7). More importantly, miRNA has been reported as a powerful and potential biomarker for diagnostics and prognostics (8–10). Therefore, visualizing and quantifying intracellular miRNA is of great significance to better understand of their function and regulatory mechanisms related to diseases. However, the inherent characteristics of miRNA, involving its small size, high homology, ready degradation and trace amount in cytoplasm make it a challenge to profile miRNA in live cells (11–13).
Nucleic acid probes have the potential to provide powerful tools for RNA detection in living cells (14–16). Among these technologies, molecular beacons have been developed to detect target nucleic acids based on the switch of the stem–loop structure with high signal-to-background ratio (17). However, bare molecular beacons may suffer from limitations, including poor cell permeability, requirement microinjection or additional transfection reagents and instability in cellular environments, susceptible to enzymatic degradation, requirement chemical modification. Of course, to address the limitations of permeability, a variety of nanocarriers have been constructed for safe and effective delivery of molecular beacons (18–20). However, free molecular beacons are susceptible to degradation by nucleases (e.g. cytosolic DNase I) after cellular delivery (21). To further improve the stability of molecular beacons, Dong et al. proposed polyethylenimine-grafted graphene nanoribbon as an effective vector for delivery of locked nucleic acid modified molecular beacons for recognition of miRNA, which protect locked nucleic acid modified molecular beacon probes from nuclease digestion (22). In addition, Zhu et al. also developed a probe using nanoparticles of molecular beacon self-assembled with chitosan to image miRNA-155 in cancer cells, which employed locked nucleic acid to modify molecular beacons and chitosan was used as an effective transfection for safe delivery of molecular beacons for improving stability and targeting specificity to miRNA-155 (23). These reports have successfully solved the problems of permeability and stability of molecular beacons to a certain extent, but the cost of chemical modification is high. Thus, there is a class of probes that is the oligonucleotide-functionalized gold nanoparticle (AuNP), and is referred to as ‘nanoflare’, for visualizing and quantifying intracellular targets (24). Nanoflares offer substantial properties, including efficient fluorescence quenching, excellent cell permeability and enzymatic stability without additional chemical modification, thus, overcoming the challenges of efficient delivery and stability to create promising intracellular probes. However, single-dye-labeled nanoflares fail to effectively avoid false positive signals from chemical interferences and cause low detection accuracy. Here, a solution is to create a strategy for ratiometric fluorescent output based on fluorescence resonance energy transfer (FRET) to avoid false positive signal. This strategy based on dual-fluorophore-labeled flare has been developed by our group, termed ‘FRET nanoflares’, which allow avoiding false positive signal through ratiometric fluorescent measurement compared to single-dye nanoflares (25–27). However, when the expression level of target is extremely low in cytoplasm, abovementioned nanoflares are often unable to sensitively detect trace amount target, due to ‘one-to-one’ signal-triggered model. So, it is necessary to establish a ‘one-to-more’ signal amplification model. This approach has been positively pursued by many research groups, including ours. The amplified nanoflares with ‘one-to-more’ signal-triggered model have been constructed with the fueling of auxiliary additives (28–31), or endogenous stimulus (32–34). However, the initial activity of these probes is not controllable. In other words, these probes may directly interact with targets, once they meet in transit, before they reach interest tumor site, making it impossible to selectively activate the targets detecting at a chosen time and position, leading to unwanted early activation and nonspecific signals, allowing poor detection accuracy. Thus, it is desirable to fabricate a sensing platform that has the potential to achieve spatiotemporally control over the probe's initial activity. It needs to design the probes that will not exert function to targets until selectively be activated by external stimulation at interest location and time.
Light is often used as a powerful external stimulus for bioorthogonal regulation, because it can be manipulated easily and precisely in space and time, allowing precise regulation of biosensor activity (35–38). Here, we present a photocaged amplified FRET nanoflare (PAFN), which spatiotemporally controls of mRNA-powered nanomachine for precise and sensitive miRNA imaging in live cell. It employs illumination as external stimulus to spatiotemporally control over initial activity of nanomachine and endogenous GAPDH mRNA as fuel to power the nanomachine work, which represents, to our knowledge, the first example of a nanomachine with these properties to spatiotemporally controllable and sensitive sensing of specific miRNA in living cells. The designed nanomachine, powered by mRNA, is silent during the delivery process. However, upon reaching the interest tumor site, it can be selectively activated in a remotely controllable way and actuate function to miRNA with high precision and spatiotemporal resolution.
MATERIALS AND METHODS
Materials and instruments
Chloroauric acid (HAuCl4•4HO2) were purchased from Sinopharm Chemical Reagent Co., Ltd. (China). Deoxyribonuclease I (DNase I) was obtained from New England Biolabs LTD (Beijing, China). Glutathione (GSH), Dulbecco's Modified Eagle Medium (DMEM), Lipofectamine™ 3000 transfection reagent, Lysotracker Blue DND-22 and cell viability assay (MTS) were ordered by Thermo Fisher Scientific Co., Ltd. (USA). Thirty percent acrylamide and Hoechst 33342 were bought by Sangon Biotechnology Co., Ltd. (Shanghai, China). SYBR Gold was purchased from Invitrogen (USA). Six times loading buffer was bought from TaKaRa Bio Inc. (Dalian, China). Mito-Tracker Green was purchased from Beyotime Biotechnology Co., Ltd. (Shanghai, China). All other reagents were analytically grade. All ultrapure water (≥18 MΩ, Milli-Q, Millipore) used throughout experiment was sterilized. All the cell lines used came from Cell Bank of the Committee on Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All oligonucleotides were synthesized by Sangon Biotechnology Co., Ltd. (Shanghai, China) and Tsingke Biological Technology (Beijing, China). These sequences are listed as Supplementary Table S1.
Centrifuge was carried out by Beckman Coulter Allegra 25R centrifuge (Brea, CA, USA). Gel imaging was performed on Azure C600 (America). All buffer pH measurements were performed on Orion 3 Star pH meter (Thermo Scientific, USA). The UV–vis absorption was obtained by a Biospec-nano UV-vis spectrophotometer (Shimadzu, Japan). Morphology characterization of AuNPs was performed on JEM-2100 Plus transmission electron microscope (JEOL Ltd., Japan). All the fluorescence was measured on near infrared steady state fluorescence spectroscopy system (QM40-NIR, PTI Inc., America). An UV lamp (LUYOR-365, China) is used to cleave PC-linker (365 nm, 7000 μW/cm2). Nikon A1 confocal laser scanning microscopy (Japan) was used for confocal fluorescence imaging. All cells were cultured in a humidified HF90 CO2 incubator (Shanghai Lishen Scientific Equipment Co. Ltd.). MTS cell viability assays were obtained from an Epoch 2 microplate reader (Bio Tek Instruments Co., Ltd., USA).
Polyacrylamide gel electrophoresis
Firstly, all hybrid complex required were heated to 95°C for 5 min, respectively, then cooled down to room temperature for the following use. Above R/S/P triple mixtures were treated with UV light irradiation for 5 min, target (T) or R*, UV and T/R* for 3 h at 37°C in 10 mM PBS (137 mM NaCl, 10 mM phosphate, 2.72 mM KCl, pH 7.4), respectively. According to the above methods, R*/F* duplex and R/T duplex were incubated with F and R* for 3 h at 37°C in 10 mM PBS, respectively. The same method as above was used for the total reaction. Secondly, 12% polyacrylamide gel was prepared for running. 10 μl above samples were mixed with 2 μl 6× loading buffer and 2 μl 100× SYBR Gold. Finally, it run at 95 V for 1.5 h in 1× TBE on ice and the result was analyzed by Azure C600 gel imaging system.
Preparation and characterization of AuNPs
13 nm AuNPs were synthetized by classical sodium citrate reduction method (39). Firstly, all glassware used was soaked for at least 8 h in aqua regia (HCl/HNO3 3:1), washed for three times and then oven dry for the following use. Secondly, 100 ml 0.01% HAuCl4•4HO2 was heated to boiling in 250 ml conical flask in the case of uniform stirring. When fully boiling, the freshly prepared 3.5 ml trisodium citrate (1%) was rapidly added into its vortex center and observed the color change of the above solution until it became a stable wine red. Finally, it was kept slightly boiling for 15 min, removed the heat source and cooled down to room temperature under stirring evenly. The prepared AuNPs were stored at 4°C for the following use. Concentration quantification and characterization of AuNPs were measured on UV absorption at 519 nm (ϵ = 2.7 × 108 l mol−1 cm−1) and TEM imaging.
Preparation of PAFN
PAFN were prepared by reported freezing-based labeling method (40–43). 3 μl 100 μM thiolated R/S/P complex and 3 μl 100 μM thiolated R*/F* duplex were mixed well with 200 μl AuNPs, and then the mixtures were frozen at −20°C or −80°C for at least 2 h. After that, they were thawed at room temperature. Subsequently, the mixtures solution was centrifugated for removing the free DNA at 4°C for 10 min at 13 000 rpm/min. The red precipitates were washed three times with buffer (0.1 M NaCl in 0.01 M phosphate buffer, pH 7.4). The functionalized AuNPs were resuspended in 10 mM PBS buffer for the further use.
Quantification of DNA duplex loading density on each AuNP
The modification density of two types of DNA probes were quantitated by previous reported methods (44). 5 nM PAFN were incubated with mercaptoethanol (ME) with a final concentration of 20 mM at 25°C for overnight under shaking. Subsequently, centrifugation for 30 min at 25°C was conducted to collect supernatant and its fluorescence intensity was measured on steady state near infrared fluorescence spectroscopy system (QM40-NIR). S labeled with Cy5 and R* labeled with FAM were diluted with different concentrations, measured their fluorescence change versus concentrations and then draw standard curve of fluorescence intensity versus concentrations, respectively. FAM was excited at 488 nm, recording from 505 to 550 nm. Cy5 was excited at 630 nm and recording from 650 to 700 nm with 10 nm excitation/emission slit width. The fluorescence intensity was converted to molar concentration of DNA by standard curve. All experiments were repeated three times in parallel.
Study on sensing performance of PAFN in tube
Firstly, various P strands were designed to obtain a low background. Corresponding PAFN were equally divided into three groups and treated with T + F and UV + T + F, respectively for 3 h at 37°C in 10 mM PBS. In addition, one group required no treatment as control.
Secondly, in order to test sensing performance, different ratios of R to S and P were investigated by fluorescence experiment according to the above methods. For optimizing the ratio of R to S, fixing the ratio of R to P was 1:2 and gradually changed the ratio of R to S. In the same way, the ratio of R to S was kept 1:1.2 and gradually changed the ratio of R to P. In addition, the concentration of F might affect the amplification efficiency. The PAFN was equally divided into two groups and treated with UV illumination in advance, and then added T and without T. Followed by incubated with various concentrations F (100, 200, 300, 400, 500 and 600 nM) for 3 h at 37°C in 10 mM PBS. After measurement, the above samples, treated with 600 nM F, but not T, were recovered and further incubated with 100 nM T and monitored the fluorescence intensity.
Finally, the sensing feasibility of PAFN was tested. The samples were divided into four groups on average, one group required no treatment as control and other three groups were treated with UV, T, UV + T, respectively and incubated with 300 nM F for 3 h at 37°C in 10 mM PBS. To verify that PAFN was controllable for sensing miRNA in a concentration-dependent manner, various concentrations T (0, 10, 25, 50, 75 and 100 nM) and 300 nM F were added into PAFN with or without UV light irradiation and incubated for 3 h at 37°C in 10 mM PBS. For verification temporal control of PAFN, they were equally divided into two groups and treated with various UV illumination time (0, 1, 2, 3, …, 10 min) and 300 nM F, and then incubated with T or without T for 3 h at 37°C in 10 mM PBS. All fluorescence measurement was performed on QM40-NIR, exciting at 530 nm and recording emission from 550 to 750 nm with 10 nm excitation/emission slit width. All experiments were repeated three times in parallel.
Sensitivity and specificity assays
According to the optimal experimental condition, PAFN were equally divided into two groups. One group was treated with 5 min UV illumination and the other group required no UV illumination as control. Subsequently, they were incubated with various concentrations T (10, 50, 100, 500 pM, 10, 25, 50, 75, 100, 150 and 200 nM) and 300 nM F for 3 h at 37°C in 10 mM PBS.
For specificity assays, PAFN were treated with 5 min UV illumination in advance, and incubated with 100 nM target miRNA and 200 nM negative control miRNAs (homologous family, let-7e, let-7i and let-7d; non-homologous family, miRNA-200b and miRNA-429) and 300 nM F for 3 h at 37°C in 10 mM PBS.
For testing the ability to distinguish one-base mismatched, PAFN were pretreated with UV light irradiation and incubated with no target, one-base mismatched target and perfectly target, respectively and 300 nM F for 3 h at 37°C in 10 mM PBS. All fluorescence intensity was monitored on QM40-NIR, exciting at 530 nm and recording emission from 550 nm to 750 nm with 10 nm excitation/emission slit width. All experiments were repeated three times in parallel.
Verification of the ability to avoiding false positive signals
For DNase I hydrolysis, probes that single-Cy5 labeled S replaced dual-dye labeled S served as control. The same concentration of dual-dye labeled PAFN and single-Cy5 labeled PAFN were pretreated with illumination and added into 1 cm quartz cell. After reaching balance of samples, 3 μl 50 U/l DNase I was rapidly added into them and monitored their fluorescence on QM40-NIR, and then scanned every 10 for 60 min, respectively. For GSH reduction, same concentration of dual-dye labeled PAFN and single-Cy5 labeled PAFN were irradiated with light and incubated with final concentration of 2 mM GSH for 10 h at 37°C in 10 mM PBS. Followed by collected their supernatants at various time points (0, 1, 3, 5, 7 and 9 h) and measured their fluorescence intensity on QM40-NIR. The dual-dye labeled fluorescence intensity was excited at 530 nm, recording from 550 to 750 nm and the single-Cy5 labeled fluorescence intensity was excited at 630 nm, recording from 650 to 750 nm with 10 nm excitation/emission slit width. All experiments were repeated three times in parallel.
Cell culture and illumination toxicity assessment
RPMI 1640 medium and DMEM with 10% fetal bovine serum (FBS) and 4 ml 10 000/units streptomycin/penicillin antibiotics solution were used for A-549 cells (human lung cancer cell line) and 7721 cells (human liver cancer cell line), MCF-7 (human breast cancer cell line) culture, respectively. All cells were grown in humidified HF90 cell incubator containing 5% CO2 at 37°C. When grown a certain density, MCF-7 cells were seeded in a 96-well plates and total volume was 100 μl every well and further incubated at 37°C in CO2 incubator for 24 h. When MCF-7 cells grown well, old medium was removed and replaced it with fresh medium. Various UV illumination times (0, 1, 2, 3,…, 10 min) were exposed on MCF-7 cells and further incubated for 12 h. Subsequently, old medium was discarded, 10 μl MTS solution was added and then supplemented 90 μl fresh medium to each well for 4 h. The absorption was obtained from Epoch 2 microplate reader at 490 nm. All experiments were repeated three times in parallel.
Study on the internalization time of activated PAFN
Well-grown MCF-7 cells were seeded in 35-mm confocal dish and cultured for 24 h. MCF-7 cells were incubated with an activated PAFN (R/S duplex replaced R/S/P triplex where S was labeled with single-dye Cy5 at 5′ end) for various times (2, 4, 6, 8 h) and then washed three times with PBS to remove the non-internalization probes. Subsequently, the fluorescence imaging was performed on Nikon A1 confocal laser scanning microscopy (CLSM) with excitation at 633 nm and recording from 663 to 738 nm. The mean fluorescence intensity was measured by Image J.
Investigation of the PAFN’s stability in living cells
MCF-7 cells were incubated with PAFN for various times (2, 4, 6, 8 and 10 h) under no UV irradiation. They were washed three times with PBS to remove the non-internalization probes. Subsequently, the fluorescence imaging was performed on Nikon A1 confocal laser scanning microscopy (CLSM) with excitation at 633 nm and recording from 663 to 738 nm. The mean fluorescence intensity was measured by Image J.
Monitoring incubation time of PAFN with cells after light activation
Well-grown MCF-7 cells were planted on 35-mm confocal dish for 24 h. Firstly, PAFN were treated with MCF-7 cells for 4 h to internalize. Secondly, the old medium was discarded and cells were washed for three times with PBS to remove the non-internalization DNA. Thirdly, fresh medium was added, exposed on UV light irradiation for 5 min (1 min break after 1 min illumination) and further incubated for various times (1, 2, 3, 4, 5 h). Finally, all cells were confocal fluorescence imaged on Nikon A1 confocal laser scanning microscopy (CLSM) with excitation at 561 nm and recording from 570 to 620 nm and 663 to 738 nm. Data analysis was processed by Nikon A1 analysis software.
Light-activated intracellular sensing performance and co-localization analysis
MCF-7 cells were grown on 35-mm confocal dish for 24 h and equally divided four groups. Two groups treated with photocaged FRET nanoflares (PFN) without amplification, respectively and other two groups respectively incubated with PAFN for 4 h. After that, old DMEM was discarded and washed with PBS for three times to remove non-internalization probes, and then added fresh DMEM. One group PFN treated and other group PAFN treated was exposed on UV light irradiation for 5 min (1 min break after 1 min illumination) and all cells were further incubated for 3 h, respectively.
For co-localization analysis, MCF-7 cells were seeded on 35 nm confocal dish for 24 h and pretreated with PAFN for 4 h to internalize, and then washed for three times with PBS to remove the non-internalization DNA. Fresh medium was supplemented and exposed on UV illumination for 5 min (1 min break after 1 min illumination) to activate the probes and continue to incubate for 3 h. Before confocal imaging, 2 μl 1 mg/mL Hoechst 33342/Lyso-Tracker Blue DND-22/Mito-Tracker Green were treated for 15–30 min with a total volume of 200 μl. Confocal fluorescence imaging was performed on Nikon A1 confocal laser scanning microscopy (CLSM) with excitation at 561 nm and recording the fluorescence emission channel of Cy3 and Cy5.
Cellular TEM images
MCF-7 cells were seeded on six-well plate to 90% confluence and incubated with 2 nM PAFN for 8 h. After being trypsinized and centrifuged, the cells were fixed for 1 h with 2.5% glutaraldehyde and 1% osmic acid in PBS. Then three washing steps (10 min each) with PBS followed. Subsequently, the samples were dehydrated in increasing acetone (30, 50, 70, 90%) for 10 min each. In order to resin embedding, samples were then treated with resin and ethanol (50:50) for 40 min followed by infiltrate into the cell pellet overnight with 100% resin. The cell pellet was infiltered with 100% epon-araldite and stored at 55°C for 48 h to allow resin polymerization. Then, the sectioned samples were finally deposited on grids and stained with 3% uranyl acetate and lead nitrate.
Confocal fluorescence imaging of three types cell lines
MCF-7 cells, A-549 cells and 7721 cells were seeded on 35-mm confocal dish for 1–2 days, respectively and treated with PAFN for 4 h in advance. Afterwards, old medium was removed and washed for three times with PBS, and then exposed on UV light irradiation for 5 min (1 min break after 1 min illumination) and further incubated for 3 h. Cell fluorescence imaging was performed on Nikon A1 confocal laser scanning microscopy with excitation at 561 nm and recording the fluorescence emission channel of Cy3 and Cy5.
Sensing the change of miRNA expression level in MCF-7 cells
Well-grown MCF-7 cells were planted on 35-mm confocal dish for 24 h and divided into three groups on average. Let-7a mimics or various concentrations let-7a mimics (0, 100, 200 and 300 nM) and anti-let-7a miRNAs were first transfected into above cells for 3 h by lipofectamine™ 3000 transfection reagent. In addition, one group required no pretreatment as control. After pretreatment, all cells were washed for three times with PBS to remove the excessive DNA and incubated with PAFN for 4 h. Next, MCF-7 cells were exposed on UV light irradiation for 5 min (1 min break after 1 min illumination) and further incubated for 3 h. Fluorescence imaging of all cells was carried out on Nikon A1 confocal laser scanning microscopy with excitation at 561 nm and recording the fluorescence emission channel of Cy3 and Cy5.
Specificity analysis of PAFN for sensing miRNA let-7a in MCF-7 cells
Well-grown MCF-7 cells were incubated with PAFN and scramble probes for 4 h, respectively. Then, above cells were washed for three times with PBS and were exposed on UV light irradiation for 5 min (1 min break after 1 min illumination) and further incubated for 3 h. Confocal microscopy imaging was carried out on Nikon A1 confocal laser scanning microscopy with excitation at 561 nm and recording the fluorescence emission channel of Cy3 and Cy5.
PAFN for spatiotemporally controlled miRNA imaging in vivo
All animal experiments were performed in accordance with the relevant laws and institutional guidelines and approved by the Institutional Animal Care and Use Committee of Hunan University. Female BALB/c mice aged 5–6 weeks (18–21 g) were purchased from Hunan SJA Laboratory Animal Co., LTD and maintained in a sterile environment. Xenograft tumor models were established by inoculating MCF-7 cells (5 × 106 cells/100 μl in saline) into the left flank of mice. When the tumors volume reached to 200–400 mm3, the mice were intratumorally injected with PAFN or AFN (amplified FRET nanoflare without photoresponsive strand). In light-activation experiments, the tumor site was irradiated for 5 min (1 min break after 1 min irradiation). Finally, mice were sacrificed and collected for frozen section, and then the slides were stained with Hoechst 33342 for 15–30 min before imaging on Nikon A1 confocal laser scanning microscopy with excitation at 561 nm and recording the fluorescence emission channel of Cy3 and Cy5.
Quantification the expression levels of intracellular let-7a miRNA and GAPDH mRNA by qRT-PCR
Firstly, well-grown cell suspension was counted and centrifuged to collect cell precipitates. Next, cell precipitates was treated with 1 ml Trizol reagent (Sangon Co. Ltd., Shanghai, China) to extract intracellular total RNA. Then it was quantitated by micro-spectrophotometer and reverse-transcribed to cDNA, and then PCR analysis was conducted. Finally, standard curves of standard samples threshold cycle (Ct) versus copies were draw. All primers involved in experiments as the following.
Let-7a-5p RT: 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACTATA-3′
Let-7a-5p F: 5′-ACACTCCAGCTGGGTGAGGTAGTAGGTTG -3′
All R: 5′-TGGTGTCGTGGAGTCG-3′
H-GAPDH-F: 5′-TGGGTGTGAACCATGAGAAGT-3′
H-GAPDH-R: 5′-TGAGTCCTTCCACGATACCAA-3′
RESULTS AND DISCUSSION
Principle of the PAFN
The PAFN is established by conjugating reporter probes and fuel probes onto AuNP as illustrated in Figure 1, detailed amplification mechanism and structure design see Supplementary Figure S1 and Supplementary Figure S2. The reporter probe, a triplex R/P/S, consists of a recognition strand (R), a photo-responsive strand (P) with a photocleavable linker group, and a signal strand (S) labeled with fluorophore Cy3 and Cy5 at 5′ and 3′ termini, respectively. The fuel probe is composed of F*/R* duplex, in which F* is designed complementary to fuel strand F (mRNA) and R* is designed complementary to R. The toehold of R, which is designed specifically complementary to T (miRNA), is masked by P strand, thus blocking toehold-mediated strand displacement reaction. Upon exposure to illumination, the P strand is quickly cleaved to be two free DNA fragments and released from the R strand, exposing the toehold domain available for hybridization with T. When a small quantity of T strands (miRNAs) and a great quantity F strands (mRNAs) appear concurrently, T binds to R and releases S, F hybridizes with F* to liberate R* via toehold-mediated strand displacement. Here, a FRET signal will manifest as S is separated from R and self-folds a stronger hairpin structure that brings the donor (Cy3) and acceptor (Cy5) into close proximity. In addition, the released R* can replace T as shown in step a and the T continues to replace the next S with higher FRET signal as depicted in step b. In such a way, step a and step b are repeatedly and continuously happened to generate multiplex S strands from the AuNP, fulfilling ‘one-to-more’ signal amplification model. In short, the initial activity of the PAFN could be selectively activated by illumination. The intracellular low-abundance specific miRNA is employed to start the nanomachine and the endogenous high-abundance mRNA is introduced to drive the nanomachine repeatedly and continuously work, resulting in significantly amplified FRET signals.
Figure 1.

Schematic illustration of the operation mechanism of the PAFN. The AuNP is functionalized with reporter probes (R/P/S) and fuel probes (F*/R*). Firstly, the PAFN is inert until light irradiation. Secondly, in the presence of T (miRNA) and F (mRNA), F competitive binds to F*and releases R*, T recognizes with R and displaces S, and the free S enable self-folding a stronger hairpin structure that brings Cy3 and Cy5 into close proximity, resulting in a FRET signal. Thirdly, the released R* replaces T (step a) and T replaces S (step b), generating higher FRET signals. In such a way, step a and step b are repeatedly and continuously work to generate multiplex S strands from the AuNP, fulfilling ‘one-to-more’ signal amplification model. Note: the length of all oligonucleotides is not the actual length.
Validation of the PAFN
The principle of PAFN is essentially a series of toehold-mediated strand displacement reactions. Necessarily, the working performance of PAFN is verified step by step using polyacrylamide gel electrophoresis (PAGE) in buffer, respectively. The detailed strand displacement mechanism of every step and the PAGE analysis are showed in Figure 2. As shown in Figure 2A, the reporter probe (R/S/P) is inactive until UV illumination. When UV illumination is applied to the system, reporter probe can specifically respond to T and release S via toehold-mediated strand displacement. There are four strands (P, S, R and T) in lane 1–4 and R, S, P are annealed to form reporter probe in lane 6. Upon reporter probe exposure on UV illumination, a new R/S duplex band (lane 5) is clearly visualized and old band disappears in lane 7, which is attributed to effectively cleavage of P by light irradiation. However, no any new band will generate upon adding T alone to reporter probe in lane 8, suggesting that reporter probe is silent in the absence of illumination, even in the presence of T. Interestingly, when light irradiation and T are simultaneously applied to reporter probe, it will result in a R/T duplex (lane 10) and a S band (lane 2) in lane 9, confirming that reporter probe can be effectively activated by UV illumination and subsequent interact with T to release S.
Figure 2.

Schematic illustration and verification of the strand displacement reaction of each step using 12% polyacrylamide gel electrophoresis (PAGE). (A) T can release S out of the R/P/S complex under UV illumination. (B) R* can’t release S out of the R/P/S complex in the absence of T, even with UV illumination. (C) F can release R* out of the F*/R* duplex. (D) R* can release T out of the R/T duplex. (E) The whole system of cascaded strand displacement reactions of the T-triggered, R*-powered DNA nanomachine. Note: the length of all oligonucleotides is not the actual length.
In order to verify whether R* could directly destroy the reporter probe to release S in the absence of T, as shown in Figure 2B, corresponding PAGE analysis is conducted to prove the scheme. Here, four strands (P, S, R and R*) are included in lane 1–4, respectively. It is clear that no new bands will generate in lane 8 when the addition of only R* to reporter probe. Moreover, the co-stimulation of UV illumination and R* will not induce new R/R*duplex (lane 10) in lane 9, demonstrating that R* will not destroy reporter probe to release S, even in the presence of light.
The scheme of Figure 2C is confirmed by the result of PAGE, three strands (F*, R* and F) are included in lane 1–3. R*and F* anneals to form R*/F*duplex in lane 4. It can be found that the addition of F to R*/F* duplex will result in F/F* duplex (lane 6) and free R* (lane 2) in lane 5. Similarly, the scheme of Figure 2 (D) is demonstrated by the result of PAGE. Lane 1–3 represent R, T and R*, respectively. R/R* duplex band (lane 6) with higher molecular weight and T band (lane 2) are visualized when the addition of R* to R/T duplex in lane 5. Based on the PAGE analysis of Figure 2C and D, we could conclude that F could replace R* in F*/R*, and then R* could replace T in R/T duplex.
As shown in Figure 2E, the whole system of cascaded reactions of the T-triggered, R*-fuelled DNA nanomachine is verified by PAGE analysis. Lane 1–3 are R*, S and T, respectively. It is found that the addition of T and R* to system will not result in any new band in lane 7, due to in the absence of UV irradiation. On the contrary, upon exposure on UV illumination in advance and subsequent the addition of T and R* to system, it will release S (lane 2) and result in R/R* duplex (lane 9) in lane 8. All the above results clearly evidenced that spatiotemporal control of toehold-mediated strand displacement is able to progressively work as expected.
Preparation and characterization of the PAFN
The 13 nm AuNPs and DNA functionalized AuNPs via Au-S bound form are first characterized by transmission electron microscopy (TEM) imaging and UV-via absorption spectra in Supplementary Figure S3. TEM imaging present good dispersibility and there are blurry borders of DNA functionalized AuNPs, however, AuNPs are only clear edges in TEM imaging. Besides, the maximum absorption peak of DNA functionalized AuNPs red-shift compared with AuNPs. These results indicate an average size of 13 nm AuNPs and DNA functionalized AuNPs are well-prepared. The amounts of two types DNA probes loaded on each AuNP are estimated based on classical methods (in Supplementary Figure S4). The result shows that each AuNP surface approximately modified on 38 ± 1 reporter probes and 40 ± 1 R*/F* fuel probes.
Prior to fluorescent test, we optimize the length of P strand (Supplementary Figure S5) and the ratio of R, S, to P (Supplementary Figure S6 and Supplementary Figure S7). By comparison, higher signal-to-noise ratio can be obtained with P-5 and 1:1.2:2. Using fluorescent experiments, sensing feasibility of PAFN responding to T in tube is assessed with the results shown in Figure 3A. When only T is present, no strong FRET signal enhancement can be detected, thus suggesting that PAFN is silent in the absence of UV illumination. However, once exposure on UV irradiation in advance and then with the addition of T, it shows a significant FRET signal enhancement, indicating that UV irradiation enables selectively activation of PAFN and actuating function to T. It should be emphasized that no enhancement in FRET signal is detected when in the presence of UV irradiation alone. This means that the sensing performance of PAFN is remotely controllable. Among other things, we further verify that the fluorescence response of PAFN is miRNA-dependent. The PAFN is first pretreated with UV irradiation and subsequently incubates with various concentrations T. As a control, PAFN is the same treated but lacks the UV irradiation. As a result, when PAFN is irradiated with UV, FRET signal enhanced gradually with increasing T concentrations (red line). In contrast, if in the absence of UV illumination, the FRET signal still remains at a very low level upon the addition of different concentrations T (black line) in Figure 3B. These results also confirm that PAFN is feasible and controllable for sensing miRNA targets in tube. In addition, because the toehold domain is exposed depending on photocleavage, a progressive FRET signal will increase with the light irradiation time increase and reach equilibrium after 5 min in the presence of T. Nevertheless, almost no appreciable change in FRET signal is seen as illumination times increased in the absence of T shown in Figure 3C. These results imply that the sensing performance of PAFN is highly temporal control.
Figure 3.

Characterization and sensing performance of the PAFN in tube. (A) Normalized fluorescence spectra of PAFN under different conditions. (B) FRET signal changes of PAFN function as various T concentrations with (red line) or without UV irradiation (black line). (C) Corresponding curve of PAFN responds to various illumination times in the presence or in the absence of T. (D) Normalized fluorescence spectra of PAFN change function as different concentrations of T, with the fueling of 300 nM F at 365 nm light irradiation. (E) Selectivity analysis of PAFN toward let-7a targets over other control miRNAs. (F) A fluorescence kinetic study of PAFN responded to let-7a or not after UV activation. FA/FD (FRET efficiency) is the fluorescence intensity ratio of acceptor to donor. Error bars represent three independent repeated experiments.
The introduction of abundant Fs can cause T reuse and release multiplex Ss with high FRET signals. Subsequently, we test the sensing performance of PAFN responding to T upon the addition of Fs at the increasing concentrations. It is found that the signal-to-noise ratio gradually increased with the addition of Fs in the range of 100–300 nM with the results shown in Supplementary Figure S8. However, whether high concentration F can destroy PAFN to induce FRET signal leakage, PAFN is selectively activated by light irradiation in advance and directly treated with various concentrations F. As shown in Supplementary Figure S9, it exhibits no enhancement in FRET signal upon the addition of F at the increasing concentrations. Amazingly, after being treated with high concentration F, above PAFN is further incubated with T and a significant recovery in FRET signal is observed. These results fully demonstrate high concentration F will not destroy PAFN to produce signal leakage. It can be also explained by the feasibility and priority of strand displacement in principle design shown in Supplementary Figure S2 as expected, in which the toehold 2 for competitively binding to R* is exposed only if the T displaces S after UV irradiation.
We further inspect the sensing performance of PAFN response to T in tube. As expected, gradual enhancements in FRET signal as T concentration increased is obtained at 365 nm light irradiation (Figure 3D). However, as a control, there is no obvious change in FRET signal upon the addition of T at increasing concentrations in the absence of UV irradiation (in Supplementary Figure S10), suggesting that PAFN response to T is light-initiated and temporally controllable. In addition, the limit of detection (LOD) is defined as the concentration of target that yields a net signal equivalent to three times the standard deviation of a series of replicates of background. A good linear correlation of the FRET signal versus the logarithm of low miRNA concentrations is detected with a calculated detection limit of 3.5 pM based on the 3δ/slope rule (Supplementary Figure S11), which are approximately three orders of magnitude lower than that of traditional nanoflares without amplification (45–47). As shown in Figure 3E, PAFN allows for the available profiling of let-7a miRNA with high specificity, but other negative control miRNAs at UV illumination. Meanwhile, it also has a good ability to recognize one base-mismatched target as evidenced in Supplementary Figure S12. In the light of these results, the PAFN possesses excellent sensitivity and specificity, which is high enough to discriminate the miRNA let-7a from other negative miRNAs. In order to further explore the sensing performance of PAFN, a fluorescence kinetic study of PAFN responding to let-7a miRNA is investigated. It shows that the FRET efficiency of PAFN gradually increases with the increasing of incubation times and achieves the maximal signal approximately at 3 h after UV irradiation in Figure 3(F). On the whole, these results demonstrate that PAFN has the potential to detect miRNA in the test tube environment.
In addition, in order to confirm PAFN has excellent ability to avoid false positive signal, DNase I degradation and glutathione (GSH) reduction experiments are conducted, respectively. As expected, no strong enhancement in background signal of dual-dyes labeled PAFN is visualized compared to single-Cy5 labeled PAFN in Supplementary Figure S13. It clearly means that PAFN enable effectively avoiding false positive signal, which further prove its potential for the available sensing miRNA in live cells.
Feasibility of the PAFN in live cells
It is of great significance to detect intracellular miRNA with temporal control. However, prior to intracellular usage, the issue that whether UV illumination can cause potential photodamage to cells under the conditions studied must be considered. MCF-7 cells are irradiated with various times and further culture for 12 h, and then the toxicity is evaluated by MTS assay. As a result, the cell viabilities are measured to be higher than 90% within 5 min illumination (Supplementary Figure S14), suggesting almost no damage to cells under the conditions studied.
Cellular uptake time of PAFN is explored in the following experiment. An activated PAFN without photo-responsive strand is redesigned. R/S duplex replaces R/S/P triplex where S is labeled with single-dye Cy5 at 5′ end functionalized to AuNPs, and its fluorescence signal is always in ‘ON’ state. After being incubated with MCF-7 cells for different times, intracellular Cy5 signals increase gradually and reach plateau at 4 h with increasing incubation times shown in Supplementary Figure S15, indicating that PAFN enable effectively internalization within 4 h.
Free oligonucleotides are susceptible to rapid degradation by nucleases such as cytosolic DNase I. Therefore it was essential to investigate the stability of our PAFN towards enzymatic degradation in living cells. PAFN were incubated with MCF-7 cells for various times. Due to the close proximity of the fluorophore (Cy5) to the AuNP surface leads to quenching of the fluorescence. Prior to UV activation, PAFN will remain in an inactivated stage and no enhancement in fluorescence signal (Cy5) is observed with excitation at 633 nm. In turn, if the oligonucleotides are digested due to enzymatic degradation, the Cy5 dyes would be released in solution and quenching would no longer occur. However, there is no significant enhancement in red fluorescence signal (Cy5) with the increasing of incubation times within 8 h and only an extremely weak red fluorescence signal is observed at 10 h in Supplementary Figure S16. It reveals that the oligonucleotides remained on the nanoparticle surface without any signs of degradation under the conditions studied.
Co-localization fluorescence imaging is studied to investigate the intercellular distribution of PAFN. Nuclear co-localization analysis shows that the Cy3 (green) and Hoechst 33342 does not co-localize in MCF-7 cells, and then we quantified the Pearson's coefficients between red and green is 0.98 and green and blue (Hoechst 33342) is 0.18 in Figure 4A. In order to further identify the location of the PAFN within cells, co-localization analysis of PAFN with Lyso-Tracker Blue (lysosome dye) is investigated, it shows that only a small amount of PAFN exist in lysosome and is not co-localized well with it (in Supplementary Figure S17). These results reveal that PAFN are localized within the cytoplasm, as also evidenced by TEM imaging of cells incubated with nanoparticles (in Supplementary Figure S18). In fact, these ones are likely responsible for the highly specific signals we obtained. The mechanism underlying the endosome/lysosome escape capability of PAFN may correlate with the possible interaction between the 3D oligonucleotide architecture of PAFN and the resident proteins of lipid rafts on endosomal membrane (48,49). Nevertheless, we found that a fraction of fluorescence signal obtained from the probes displays mitochondrial co-localisation (in Supplementary Figure S19), which is consistent with previously findings by Mirkin and Kanaras (50,51). This observation is in agreement with previously reports that the oligonucleotides labelled with Cyanine dyes can selectively target cancer-cell mitochondria and be localized in it, due to the mitochondrial membrane potential (52–54). We can therefore assume that the released Cyanine-dye-tagged flare strand diffused away from the probes towards the mitochondria, resulting in the observed fluorescence signal localisation.
Figure 4.

Feasibility analysis of the PAFN in live cells. (A) Co-localization fluorescence imaging analysis of the PAFN and Hoechst 33342 and corresponding Z-axis scanning (shown in the image below) through entire MCF-7 cells. The green and red represents the position of the PAFN and the blue (Hoechst 33342) exhibits the nucleus. Pearson's coefficient between red and green is 0.98 and green and blue is 0.18. (B) Confocal fluorescence images of MCF-7 cells treated with PAFN and PFN in the presence of UV irradiation (+UV) or in the absence of UV irradiation (−UV). Local schematic illustration of PAFN and PFN treated with UV irradiation or without it at left. Scar bar is 20 μm.
We next monitor the intracellular reaction time of PAFN after UV light activation. MCF-7 cells are treated with PAFN for 4 h in advance and irradiated with 365 nm light for 5 min, and then further incubated for various times. As shown in Supplementary Figure S20, FRET signals reach saturation approximately at 3 h with increasing reaction times.
In the following experiments, we focus on intracellular amplification performance and controllability of PAFN response to miRNA powered by endogenous mRNA with precise control. Photocaged FRET nanoflare (PFN) without amplification, which we developed previously (47), was established as a control. As shown in Figure 4B, only a faint FRET signal of both PAFN and PFN is visualized, which is attributed to the lack of UV light irradiation. In contrast, upon exposure on UV light irradiation, it exhibits FRET signals in both PAFN and PFN, but a much stronger FRET signal of PAFN than PFN in cells and corresponding normalized FRET efficiency is quantified by imageJ in Supplementary Figure S21. The most likely reason is that endogenous mRNA can result in strand displacement events and target-recycling to achieve signal amplification. Subsequently, we quantify the expression level of GAPDH mRNA in three cell lines by absolute qRT-PCR to confirm our speculation (Supplementary Figure S22). We assume that cell is an ideal sphere model and the diameter is 10 μm. According the copies of GAPDH mRNA in three cell lines, mRNA in three cell lines are estimated to be approximately 12 nM/cell in MCF-7, 9 nM/cell in A-549 and 16 nM in 7721. It shows that the concentration is lower than in the tube used. However, the true local concentration of GAPDH mRNA will be much higher than the calculated results, because the intracellular environment is not homogeneous and mRNAs are not evenly distributed throughout the cell. In addition, the distribution space of the mRNA in cell should exclude the nucleus and other organelles. Taken together, these results fully confirm that light irradiation is the prerequisite for intracellular miRNA imaging and endogenous mRNA as fuel strand indeed can cause strand displacement events to accomplish signal amplification as expected.
Specificity of the PAFN in live cells
In order to test the specificity of the PAFN in live cells, we employ PAFN to detect miRNA let-7a in different cell lines (MCF-7, A-549, 7721). According to previous report (55,56), MCF-7 and A-549 cells express relatively high levels let-7a and 7721 cells have a relatively low expression level of let-7a. We chose above-mentioned three cells as test models. Various FRET signals are observed in above three cells incubated with PAFN and followed by UV illumination. MCF-7 cells result in the strongest FRET signal while 7721 cells generate the lowest FRET signal in Figure 5A, B. A further analysis has been performed for quantification of the relative expression levels of miRNA let-7a in three cell lines using standard measurement method qRT-PCR (Supplementary Figure S23), which is basically consistent with the result of confocal imaging and there is still a small difference between q-PCR data and imaging FRET ratio. It might be explained by the cell-to-cell individual variations. On the one hand, q-PCR detection techniques are developed for miRNAs profiling following extraction from cell lysate, it detect the average miRNAs in cell populations. However, in situ detection of miRNAs in live cell can reveal the cell-to-cell variations. On the other hand, probes are unevenly distributed among cells with various internalization, it may result in various imaging results. In order to further validate the specificity of PAFN for sensing miRNA let-7a in living cells, a non-targeting scramble probe is designed by non-targeting recognition sequence, for which no miRNA target existed in MCF-7 cells. Confocal microscopy revealed that no significant FRET signal could be observed in control group treated with scramble probe (in Supplementary Figure S24), demonstrating that PAFN is high specificity for sensing miRNA let-7a and capable of distinguishing the expression levels of miRNA let-7a in different cell lines.
Figure 5.

Specificity of the PAFN in live cells. (A) Confocal fluorescence images of MCF-7 cells, A-549 cells and 7721 cells incubated with PAFN under UV light irradiation. (B) Normalized FRET efficiency value is quantified by ImageJ in three cell lines. (C) Confocal fluorescence images of MCF-7 cells treated with let-7a inhibitor or let-7a mimic by lipofectamine™ 3000 transfection reagent for 3 h in advance, followed by incubation with PAFN under UV light irradiation. (D) Normalized FRET efficiency value is quantified by Image J in MCF-7 cells treated with different condition. Values are mean ± SD (n = 3), statistical significance was determined by two-tailed Student's t-test, the symbol ** indicates the statistical significance at levels of P < 0.01. Scar bar is 20 μm.
As intracellular miRNA expression levels are highly dynamic in different development stage of tumor, we apply PAFN to discriminate expression level changes of miRNA let-7a in live cells with spatiotemporal control. We first regulate the let-7a expression profiles by anti-let-7a and let-7a mimics where anti-let-7a can selectively bind and decrease intracellular let-7a level and let-7a mimics enable enhancement let-7a level (57–59). The control groups do not need any pretreatment. As shown in Figure 5C, D, in up-regulation group, there is a significantly higher FRET signal in MCF-7 cells while a very weak FRET signal is detected in down-regulation group compared with those in the control group. These results are basically consistent with the relative quantitative by qRT-PCR in Supplementary Figure S25. In addition, when various concentration let-7a mimics were transfected into MCF-7 cells in advance, then detected with PAFN after illumination, we found that the FRET signal gradually increased with the addition of let-7a mimics at increasing concentrations (in Supplementary Figure S26). These results actually demonstrate that our proposed PAFN can be employed for the profiling of the fluctuations of miRNA let-7a level in live cells.
Light-gated PAFN for spatiotemporally controlled miRNA imaging in vivo
In order to demonstrate spatiotemporally resolved of PAFN in tumor site, MCF-7-breast-tumor-bearing mice were intratumorally injected with PAFN. In addition, AFN, an amplified FRET nanoflare without photo-responsive strand and proposed by us previously, is selected as a control (33). In light-activation experiments, the tumor site was irradiated for 5 min (1 min break after 1 min irradiation). Mice were sacrificed and the tumor was immediately collected for frozen section. The schematic illustration of the tumor tissue imaging workflow are shown in Figure 6A. Fluorescence imaging of tissue section displayed a strong FRET signal throughout the tumor tissue, whereas no obvious intratumoral FRET signal was observed for the group without irradiation in Figure 6B. Quantitative results showed that there was a 2.6-fold higher FRET signal in the tumor tissues treated with PAFN and light irradiation than in the control group treated with PAFN but no light irradiation (see Figure 6C). As a control, AFN showed obvious intratumoral FRET signal upon UV light irradiation or not, due to the uncontrollability and directly interaction with targets (Figure 6D and Figure 6E). In addition, 7721-liver-tumor tissue showed a weaker FRET signal than MCF-7-breast-tumor tissue upon light illumination, attributed to the low expression level of let-7a miRNA in 7721 liver tumor tissue (Figure 6F and G). Therefore, it is conceivable that remotely light-triggered PAFN allows for precise miRNA imaging in tumor tissues.
Figure 6.

Study on the spatiotemporally resolved of PAFN in tumor tissues. (A) Schematic illustration of PAFN for spatiotemporally controlled miRNA imaging in vivo. (B) Confocal imaging of MCF-7 tumor tissues treated with PAFN and irradiated with or without light at the tumor site. (C) Normalized FRET efficiency of MCF-7 tumor tissues pretreated with PAFN and followed by light irradiation or not at tumor site. (D) Representative fluorescence imaging of MCF-7 tumor tissues after treatment with AFN with or without light irradiation at the tumor site. (E) Normalized FRET efficiency of MCF-7 tumor tissues treated with AFN and subsequent light irradiation or not at tumor site. (F) Fluorescence imaging of 7721 liver cancer tissues and MCF-7 breast cancer tissue treated with PAFN, respectively and subsequently light irradiated at the tumor site. (G) Corresponding normalized FRET efficiency of 7721 liver cancer tissue and MCF-7 breast cancer tissue treated with PAFN and irradiated by light at tumor site, repectively. Data are represented as means ± SD (n = 3), statistical significance was determined by two-tailed Student's t-test, the symbol ** indicates the statistical significance at levels of P < 0.01. NS indicates no significance (P > 0.05). Scar bar is 20 μm. Note: the size of all objects is not the actual size.
Generality of PAFN
Since the recognition sequence can be changed according to detection target, we supposed that our system could be as a general strategy for sensing a variety of intracellular RNA molecules. To demonstrate this, we designed another PAFN for detecting miRNA-21 both in tube and in living cells. MiRNA-21, one kind of oncogene, is significantly overexpressed in a wide variety of cancers including breast, lung and liver (60). We selected it as a target model. When in the presence of miRNA-21 and illumination, a significant enhancement FRET signal was observed. However, when there is only miRNA-21 or illumination, almost no detectable FRET signal was obtained (in Supplementary Figure 27). Then, the probes were used for intracellular miRNA-21 imaging. MCF-7 cells treated with PAFN under UV irradiation showed remarkable intracellular FRET signal where red fluorescence intensity (Cy5) was really conspicuous. However, when in the absence of UV irradiation, there was almost no Cy5 fluorescence shown on the red channel, revealing that it was inactivated (in Supplementary Figure 28). These results demonstrated that our system is suitable for miRNA-21 imaging in live cells. Overall, this system proposed by us could be as a versatile platform for detection of miRNAs in living cells.
CONCLUSIONS
In summary, we here propose a novel photocaged amplified FRET nanoflare (PAFN), which is a spatiotemporally controllable nanomachine for sensing intracellular specific miRNA powered by endogenous mRNA. It is, to our knowledge, the first report that integrating external stimulus for temporal and precise control over the initial activity of nanomachine with endogenous mRNA molecules for driving the nanomachine operation, it possesses the following merits. Firstly, the introduction of endogenous high-abundant mRNA molecules, first proposed by us, enables driving nanomachine work and fulfillment target cycle for signal amplification without any extra auxiliary, avoiding the dilution of the system or the change of intracellular environment due to the necessity of externally adding DNA strands for displacement. Secondly, light irradiation is employed as external stimulus to temporally control over the nanomachine's initial activity, it would not work until selectively activated with illumination at a desirable time and site, avoiding undesirable early activation of the nanomachine before reaching the interest tumor sites (eg. during internalization or delivery process), reducing unwanted nonspecific signals and improving detection reliability and accuracy. Thirdly, through combining FRET ratiometric measurement with nanomachine, it offers low background signal and effectively avoid false positive signal from glutathione (GSH) competition and nuclease (DNase I) degradation. Fourthly, due to the participation of endogenous mRNA, the detection limit of PAFN is calculated to 3.5 pM, which are approximately three orders of magnitude lower than that of traditional nanoflares without amplification in tube (45–47). Finally, the PAFN is highly specific for miRNA detection, enabling differentiation of the target from other negative miRNAs. We believe, the nanomachine, with different external stimulus and endogenous high-abundant molecules as fuels, will potentially be applied to create a new strategy for detection of trace amount targets in live systems.
DATA AVAILABILITY
Derived data supporting the findings of this study are available from the corresponding author (J.H.) on request.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: J.L. and J.H. designed the study, J.L. wrote the manuscript, J.H. revised the manuscript, J.L., S.L., J.W. and R.L. performed the experiments. J.H., X.Y. and K.W. supervised and analyzed the data. All the authors discussed the results.
Contributor Information
Jing Li, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China; School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, P.R. China.
Shiyuan Liu, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
Jiaoli Wang, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
Ruiting Liu, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
Xiaohai Yang, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
Kemin Wang, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
Jin Huang, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Hunan University, Changsha, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [21735002, 21874036]; Natural Science Foundation for Distinguished Young Scholars of Hunan Province [2021JJ10011]. Funding for open access charge: National Natural Science Foundation of China [21874036].
Conflict of interest statement. None declared.
REFERENCES
- 1. Wienholds E., Kloosterman W.P., Miska E., Alvarez-Saavedra E., Berezikov E., Bruijin E., Horvitz H.R., Kauppinen S., Plasterk R.A.. MicroRNA expression in zebrafish embryonic development. Science. 2005; 309:310–311. [DOI] [PubMed] [Google Scholar]
- 2. Filipowicz W., Bhattacharyya S.N., Sonenberg N.. Mechanisms of post-transcriptional regulation by micrornas: are the answers in sight?. Nat. Rev. 2008; 9:102–114. [DOI] [PubMed] [Google Scholar]
- 3. Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 4. Glavac D., Hrasovec S.. MicroRNAs as novel biomarkers in colorectal cancer. Front. Genet. 2012; 3:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ruan K., Fang X., Ouyang G.. MicroRNAs: novel regulators in the hallmarks of human cancer. Cancer Lett. 2009; 285:116–126. [DOI] [PubMed] [Google Scholar]
- 6. Esquela-Kerscher A., Slack F.J.. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer. 2006; 6:259–269. [DOI] [PubMed] [Google Scholar]
- 7. Ma L., Teruya-Feldstein J., Weinberg R.A.. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007; 449:682–688. [DOI] [PubMed] [Google Scholar]
- 8. Lin S., Gregory R.I.. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015; 15:321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Calin G.A., Croce C.M.. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006; 6:857–866. [DOI] [PubMed] [Google Scholar]
- 10. Volinia S., Calin G.A., Liu C., Ambs S., Cimmino A.. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A. 2006; 103:2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li S., Xu L., Ma W., Wu X., Sun M., Kuang H., Wang L., Kotov N.A., Xu C.. Dual-mode ultrasensitive quantification of microRNA in living cells by chiroplasmonic nanopyramids self-assembled from gold and upconversion nanoparticles. J. Am. Chem. Soc. 2016; 138:306–312. [DOI] [PubMed] [Google Scholar]
- 12. Li L., Feng J., Liu H., Li Q., Tong L., Tang B.. Two-color imaging of microRNA with enzyme-free signal amplification via hybridization chain reaction in living cells. Chem. Sci. 2016; 7:1940–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peng W., Zhao Q., Chen M., Piao J., Gao W., Gong X., Chang J.. An innovative “unlocked mechanism” by a double key avenue for one-pot detection of microRNA-21 and microRNA-141. Theranostics. 2019; 9:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nitin N., Santangelo P.J., Kim G., Nie S., Bao G.. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kloosterman W.P., Wienholds E., de Bruijn E., Kauppinen S., Plasterk R.H.A.. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat. Methods. 2006; 3:27–29. [DOI] [PubMed] [Google Scholar]
- 16. Santangelo P.J., Nix B., Tsourkas A., Bao G.. Dual FRET molecular beacons for mRNA detection in living cells. Nucleic Acids Res. 2004; 32:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng J., Yang R., Shi M., Wu C., Fang X., Li Y., Li J., Tan W.. Rationally designed molecular beacons for bioanalytical and biomedical applications. Chem. Soc. Rev. 2015; 44:3036–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu J., Zhang L., Lei J., Ju H.. MicroRNA-responsive cancer cell imaging and therapy with functionalized gold nanoprobe. ACS Appl. Mater. Interfaces. 2015; 7:19016–19023. [DOI] [PubMed] [Google Scholar]
- 19. Yang F., Liu P., Meng X., Lu H., Cao Y., Dai W., Wang C., Dong H.. Functional MoS2 nanosheets for precursor and mature microRNA detection in living cells. Anal. Bioanal. Chem. 2019; 411:4559–4567. [DOI] [PubMed] [Google Scholar]
- 20. Dai W., Lu H., Yang F., Dong H., Zhang X.. Accurate detection of intracellular microRNAs using functional Mo2C quantum dots nanoprobe. Chem. Commun. 2019; 55:10615–10618. [DOI] [PubMed] [Google Scholar]
- 21. Seferos D.S., Prigodich A.E., Giljohann D.A., Patel P.C., Mirkin C.A.. Polyvalent DNA nanoparticle conjugates stabilize nucleic acids. Nano Lett. 2009; 9:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong H., Ding L., Yan F., Ji H., Ju H.. The use of polyethylenimine-grafted graphene nanoribbon for cellular delivery of locked nucleic acid modified molecular beacon for recognition of microRNA. Biomaterials. 2011; 32:3875–3882. [DOI] [PubMed] [Google Scholar]
- 23. Zhu H., An J., Yao Q., Han J., Li X., Jiang F., Chen G., Peng L., Li Y., Sun J., Chen Z.. Chitosan combined with molecular beacon for mir-155 detection and imaging in lung cancer. Molecules. 2014; 19:14710–14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seferos D.S., Giljohann D.A., Hill D.H., Prigodich A.E., Mirkin C.A.. Nano-flares: probes for transfection and mRNA detection in living cells. J. Am. Chem. Soc. 2007; 129:15477–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang Y., Huang J., Yang X., Quan K., Wang H., Ying L., Xie N., Ou M., Wang K.. FRET nanoflares for intracellular mRNA detection: avoiding false positive signal and minimizing effects of system fluctuations. J. Am. Chem. Soc. 2015; 137:8340–8343. [DOI] [PubMed] [Google Scholar]
- 26. Huang J., Ying L., Yang X., Yang Y., Quan K., Wang H., Xie N., Ou M., Zhou Q., Wang K.. Ratiometric fluorescent sensing of pH value in living cells by dual-fluorophore-labeled i-motif nanoprobes. Anal. Chem. 2015; 87:8724–8731. [DOI] [PubMed] [Google Scholar]
- 27. Yang Y., Huang J., Yang X., Quan K., Xie N., Ou M., Tang J., Wang K.. Aptamer-based FRET nanoflares for imaging potassium ions in living cells. Chem. Commun. 2016; 52:11386–11389. [DOI] [PubMed] [Google Scholar]
- 28. Liang C., Ma P., Liu H., Guo X., Yin B., Ye B.. Rational engineering of dynamic, entropy-driven DNA nanomachine for intracellular microRNA imaging. Angew. Chem. Int. Ed. 2017; 129:9205–9209. [DOI] [PubMed] [Google Scholar]
- 29. He X., Zeng T., Li Z., Wang G., Ma N.. Catalytic molecular imaging of MicroRNA in living cells by DNA programmed nanoparticles disassembly. Angew. Chem. Int. Ed. 2016; 55:3073–3076. [DOI] [PubMed] [Google Scholar]
- 30. Li D., Zhou W., Yuan R., Xiang Y.. A DNA-fuel and catalytic molecule machine lights up trace under-expressed microRNA in living cells. Anal. Chem. 2017; 89:9934–9940. [DOI] [PubMed] [Google Scholar]
- 31. Li B., Liu Y., Liu Y., Tian T., Yang B., Huang X., Liu J., Liu B.. Construction of dual-color probe with target-triggered signal amplification for in situ single-molecule imaging of microRNA. ACS Nano. 2020; 14:8116–8125. [DOI] [PubMed] [Google Scholar]
- 32. Xian L., Ge H., Xu F., Xu N., Fan J., Shao K., Peng X.. Intracellular microRNA imaging using telomerase-catalyzed fret ratioflare with signal amplification. Chem. Sci. 2019; 10:7111–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li J., Wang J., Liu S., Xie N., Quan K., Yang Y., Yang X., Huang J., Wang K.. Amplified FRET nanoflares: an endogenous mRNA-powered nanomachine for intracellular microRNA imaging. Angew. Chem. Int. Ed. 2020; 59:20104–20111. [DOI] [PubMed] [Google Scholar]
- 34. Ma P., Liang C., Zhang H., Yin B., Ye B.. A highly integrated DNA nanomachine operating in living cells powered by an endogenous stimulus. Chem. Sci. 2018; 9:3299–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riggsbee C.W., Deiters A.. Recent advance in the photochemical control of protein function. Trends Biotechnol. 2010; 28:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deiters A. Principles and applications of the photochemical control of cellular processes. ChemBioChem. 2010; 11:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan Z., Feagin T.A., Heemstra J.M.. Temporal control of aptamer biosensors using covalent self-caging to shift equilibrium. J. Am. Chem. Soc. 2016; 138:6328–6331. [DOI] [PubMed] [Google Scholar]
- 38. Di Z., Liu B., Zhao J., Gu Z., Zhao Y., Li L.. An orthogonally regulatable DNA nanodevice for spatiotemporally controlled biorecognition and tumor treatment. Sci. Adv. 2020; 6:9381–9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grabar K.C., Freeman R.G., Hommer M.B., Natan M.J.. Preparation and characterization of Au colloid monolayers. Anal. Chem. 1995; 7:735–743. [Google Scholar]
- 40. Liu B., Wu T., Huang Z., Liu Y., Liu J.. Freezing-directed stretching and alignment of DNA oligonucleotides. Angew. Chem. Int. Ed. 2019; 58:2109–2113. [DOI] [PubMed] [Google Scholar]
- 41. Hu M., Yuan C., Wang X., Sun J., Xiong E., Zhou X.. Single-step, salt-aging-free, and thiol-free freezing construction of aunp-based bioprobes for advancing crispr-based diagnostics. J. Am. Chem. Soc. 2020; 142:7506–7513. [DOI] [PubMed] [Google Scholar]
- 42. Qing Z., Luo G., Xing S., Zou Z., Lei Y., Liu J., Yang R.. Pt-S bond-mediated nanoflares for high-fidelity intracellular applications by avoiding thiol cleavage. Angew. Chem. Int. Ed. 2020; 59:14148–14152. [DOI] [PubMed] [Google Scholar]
- 43. Liu B., Liu J.. Freezing directed construction of bio/nano interfaces: reagentless conjugation, denser spherical nucleic acids, and better nanoflares. J. Am. Chem. Soc. 2017; 139:9471–9474. [DOI] [PubMed] [Google Scholar]
- 44. Demers L.M., Mirkin C.A., Mucic R.C., Reynolds R.A., Letsinger R.L., Elghanian R., Viswanadham G.A.. Fluorescence-based method for determining the surface coverage and hybridization efficiency of thiol-capped oligonucleotides bound to gold thin films and nanoparticles. Anal. Chem. 2000; 72:5535–5541. [DOI] [PubMed] [Google Scholar]
- 45. Li J., Huang J., Yang X., Yang Y., Quan K., Xie N., Wu Y., Ma C., Wang K.. Two-color-based nanoflares for multiplexed microRNAs imaging in live cells. Nanotheranostics. 2018; 2:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li N., Chang C., Pan W., Tang B.. A multicolor nanoprobe for detection and imaging of tumor-related mRNA in living cells. Angew. Chem. Int. Ed. 2012; 51:7426–7430. [DOI] [PubMed] [Google Scholar]
- 47. Li J., Cai S., Zhou B., Meng X., Guo Q., Yang X., Huang J., Wang K.. Photocaged FRET nanoflares for intracellular microRNA imaging. Chem. Commun. 2020; 56:6126–6129. [DOI] [PubMed] [Google Scholar]
- 48. Yan R., Chen J., Wang J., Rao J., Du X., Liu Y., Zhang L., Qiu L., Liu B., Zhao Y., Jiang P., Chen C., Li Y.. A nanoflare-based strategy for in situ tumor margin demarcation and neoadjuvant gene/photothermal therapy. Small. 2018; 14:1802745. [DOI] [PubMed] [Google Scholar]
- 49. Choi C.H.J., Hao L., Narayan S.P., Auyeung E., Mirkin C.A.. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Briley W.E., Bondy M.H., Randeria P.S., Dupper T.J., Mirkin C.A.. Quantification and real-time tracking of RNA in live cells using sticky-flares. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9591–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heuer-Jungemann A., El-Sagheer A.H., Lackie P.M., Brown T., Kanaras A.G.. Selective killing of cells triggered by their mRNA signature in the presence of smart nanoparticles. Nanoscale. 2016; 8:16857–16861. [DOI] [PubMed] [Google Scholar]
- 52. Rhee W.J., Bao G.. Slow non-specific accumulation of 2′-deoxy and 2′-O-methyl oligonucleotide probes at mitochondria in live cells. Nucleic Acids Res. 2009; 38:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jiang T., Zhou L., Liu H., Zhang P., Liu G., Gong P., Li C., Tan W., Chen J., Cai L.. Monitorable mitochondria-targeting DNAtrain for image-guided synergistic cancer therapy. Anal. Chem. 2019; 91:6996–7000. [DOI] [PubMed] [Google Scholar]
- 54. Zhou L., Wu Y., Luo Y., Li H., Meng X., Liu C., Xiang J., Zhang P., Gong P., Cai L.. Mitochondria-localized self-reporting small-molecule-decorated theranostic agents for cancer-organelle transporting and imaging. ACS Appl. Bio Mater. 2019; 2:5164–5173. [DOI] [PubMed] [Google Scholar]
- 55. Nie W., Wang Q., Zou L., Zheng Y., Liu X., Yang X., Wang K.. Low-fouling surface plasmon resonance sensor for highly sensitive detection of microRNA in a complex matrix based on the DNA tetrahedron. Anal. Chem. 2018; 90:12584–12591. [DOI] [PubMed] [Google Scholar]
- 56. Tang Y., He X., Zhou Z., Tang J., Guo R., Feng X.. Highly sensitive and selective miRNA detection based on a closed ring probe and multiple signal amplification. Chem. Commun. 2016; 52:13905–13908. [DOI] [PubMed] [Google Scholar]
- 57. Wang L., Wang Y., Zhang D., Fang X., Sun P., Xue H.. Let-7a mimic attenuates CCL18 induced breast cancer cell metastasis through Lin 28 pathway. Biomed. Pharmacother. 2016; 78:301–307. [DOI] [PubMed] [Google Scholar]
- 58. Liu K., Zhang C., Li T., Ding Y., Tu T., Zhou F., Qi W., Chen H., Sun X.. Let-7a inhibits growth and migration of breast cancer cells by targeting HMGA1. Int. J. Oncol. 2015; 46:2526–2534. [DOI] [PubMed] [Google Scholar]
- 59. Liu C., Chen Z., Fang M., Qiao Y.. MicroRNA let-7a inhibits proliferation of breast cancer cell by downregulating USP32 expression. Transl. Cancer Res. 2019; 8:1736–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Krichevsky A.M., Gabriely G.. MiR-21: a small multi- faceted RNA. J. Cell. Mol. Med. 2009; 13:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Derived data supporting the findings of this study are available from the corresponding author (J.H.) on request.