

Abstract
As the design of genetic circuitry for synthetic biology becomes more sophisticated, diverse regulatory bioparts are required. Despite their importance, well-characterized 3′-untranslated region (3′-UTR) bioparts are limited. Thus, transcript 3′-ends require further investigation to understand the underlying regulatory role and applications of the 3′-UTR. Here, we revisited the use of Term-Seq in the Escherichia coli strain K-12 MG1655 to enhance our understanding of 3′-UTR regulatory functions and to provide a diverse collection of tunable 3′-UTR bioparts with a wide termination strength range. Comprehensive analysis of 1,629 transcript 3′-end positions revealed multiple 3′-termini classes generated through transcription termination and RNA processing. The examination of individual Rho-independent terminators revealed a reduction in downstream gene expression over a wide range, which led to the design of novel synthetic metabolic valves that control metabolic fluxes in branched pathways. These synthetic metabolic valves determine the optimal balance of heterologous pathways for maximum target biochemical productivity. The regulatory strategy using 3′-UTR bioparts is advantageous over promoter- or 5′-UTR-based transcriptional control as it modulates gene expression at transcription levels without trans-acting element requirements (e.g. transcription factors). Our results provide a foundational platform for 3′-UTR engineering in synthetic biology applications.
INTRODUCTION
Synthetic biology aims to design and construct novel genetic systems capable of unprecedented biological functions. This is accomplished by combining numerous reliable and standardized bioparts. In particular, considerable progress has been made with genetic engineering and synthetic biology of transcript 5′-ends using tunable promoters and ribosome binding sites (1,2), which were extensively identified via rapid amplification of cDNA ends and differential RNA-sequencing (dRNA-Seq) (3,4). These techniques have identified diverse regulatory elements, including promoters, secondary RNA structures, and ribosome-binding sites (5–9). Although 5′-untranslated region (5′-UTR) bioparts are powerful gene expression controlling tools, they are limited in the requirement of precious molecules for induction and noise-level expression due to the trans-acting element. Multiple genetic systems have been developed to circumvent these limitations (10,11). However, this introduces additional noise, which is not compatible with industrial scaling. Additionally, transcriptional terminators are limited and may play an important role in reducing genetic complexity. The genetic circuits constructed thus far are dependent on a few powerful terminators or their derivatives. Moreover, whether strong termination is the best solution for various applications, including metabolic engineering and genetic circuit construction, remains uncertain. Thus, emphasizing the need to develop a reliable terminator biopart repertoire and its characterization, along with an examination of the influence of various terminators on genetic circuit functionality.
Unlike eukaryotic mRNAs, which have specific molecular features on both ends (5′-caps and polyadenylated 3′-ends), bacterial mRNAs possess a molecular feature only on their 5′-end (tri-phosphorylation). Thus, a detailed genome-wide investigation of bacterial transcription 3′-ends is limited. Consequently, a sequencing technique, called Term-Seq, was developed to capture the 3′-ends of RNA transcripts (12–15). Although previous studies revealed various regulations related to transcriptional termination, further genome-wide surveys of various 3′-ends are required to understand the underlying regulatory role of the bacterial 3′-untranslated region (3′-UTR) (16–18) and its downstream applications.
Here, we identified multiple 3′-end classes encoded in the E. coli K-12 MG1655 genome associated with transcription termination, transcript processing, and regulation, using Term-Seq and machine-learning analysis. This comprehensive characterization of transcriptional terminators provides a valuable collection of terminator bioparts for synthetic biological applications. Terminator bioparts produce a 4.1-fold lower noise at controlling gene expression when compared to a conventional inducible promoter-based regulation system because of the reduced intrinsic variation of the system. Finally, we demonstrated robust metabolic pathway control using the synthetic terminator parts, which led to the discovery of optimal solutions for flux distribution between heterologous and endogenous pathways in different E. coli strains. These findings enhanced our understanding of 3′-UTR regulation and expanded the range of regulatory elements in synthetic biology applications, providing a novel transcriptional control strategy for metabolic engineering and synthetic biology applications (10,19,20).
MATERIALS AND METHODS
E. coli strains, media, and culture
The E. coli strains, K-12 MG1655, genome-reduced E. coli eMS57, and their derivatives were used in this study. The E. coli MG1655 ΔpfkA Δzwf double-knockout strain was constructed using two sequential lambda recombination steps, as previously described (21). Briefly, the zwf coding sequence was replaced with a kanamycin resistance cassette amplified from pKD13, using the pKD46 helper plasmid. The kanamycin resistance cassette was removed by flippase recombination, using the pCP20 plasmid. Phosphofructokinase A (pfkA) was then sequentially deleted using the same method. The double knockout strain was constructed, followed by pKD46 and pCP20 curing at a non-permissive temperature (42°C). For the fluorescence reporter assay, overnight E. coli MG1655 culture, harboring the pDRA1, pDRA2, or pGFP plasmids, was inoculated into 400 μL M9 glucose medium (47.75 mM Na2HPO4, 22.04 mM KH2PO4, 8.56 mM NaCl, 18.70 mM NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, and 2 g L–1 glucose), with an initial optical density at 600 nm (OD600nm) of 0.05. Various concentrations of IPTG were added, as required. The orthogonality of transcript 3′-ends (T3PEs) was tested under M9 glycerol (2 g L–1), M9 high-glucose (10 g L–1), LB broth (BD Biosciences, San Jose, CA, USA), and TB (BD Biosciences). For the 2,3-butanediol (BDO) production experiment, E. coli MG1655 harboring pBDO was inoculated into 60 mL M9 high-glucose medium containing 10 g L–1 glucose, 100 μg mL–1 ampicillin, and 0.1 mM IPTG, with an initial OD600nm of 0.05. The culture was grown aerobically in a 300 mL Erlenmeyer flask at 37°C for 24 h in a rotary shaker. For the myo-inositol (MI) production experiment, E. coli MG1655 or eMS57 ΔpfkA Δzwf double knockout strains harboring the pMI plasmid was inoculated into 60 mL M9 high-glucose medium containing 10 g L–1 glucose, 100 μg mL–1 ampicillin, and 0.1 mM IPTG, with an initial OD600nm of 0.05. The culture was grown at 30°C, as previously described, and aerobically in a 300 mL Erlenmeyer flask for up to 48 h in a rotary shaker (10). Cell density (OD600nm) was monitored non-invasively using an OD-Monitor System (Taitec Corporation, Saitama, Japan) composed of an ODSensor-S and ODBox-A.
Term-Seq
Term-Seq libraries were constructed as previously described, with slight modifications (13,22). Briefly, RNA was extracted from E. coli MG1655 cultures sampled at the mid-log phase using the RNAsnapTM method (23). Next, 5 μg of total RNA was treated with 2 U of RNase-free DNase I (NEB, Ipswich, MA, USA) for 15 min at 37°C. DNase I-treated RNA was purified using a phenol-chloroform-isoamyl alcohol extraction, followed by ethanol precipitation. Ribosomal RNA (rRNA) was depleted from purified DNA-depleted RNA samples using the RiboZero rRNA Removal Kit for bacteria (Illumina, San Diego, CA, USA), according to the manufacturer's instructions. The RNA 3′ adaptor was ligated to 1 μg of rRNA-depleted RNA at 23°C for 2.5 h, in 25 μL of a 3′ adaptor ligation reaction mixture containing 1 μL of 150 μM RNA 3′ adaptor, 2.5 μL 10 × T4 RNA Ligase 1 Buffer (NEB), 25 U T4 RNA Ligase 1 (NEB), 2.5 μL of 10 mM ATP, 2 μL dimethyl sulfoxide (DMSO), and 9.5 μL of 50% polyethylene glycol 8000 (PEG8000). The 3′-adaptor-ligated RNA was purified using Agencourt AMPure XP Beads (Beckman Coulter, Brea, CA, USA) and fragmented using the RNA Fragmentation Reagent (Ambion, Austin, TX, USA) at 72°C for 90 s. The fragmented RNA was purified using Agencourt AMPure XP Beads and reverse transcribed using a SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA), as described by the manufacturer, with 10 pmol RT primer. Complementary DNA (cDNA) was purified using Agencourt AMPure XP beads, and the cDNA 3′ adaptor was ligated at 23°C for 8 h in 25 μL of cDNA 3′ adaptor ligation reaction mixture containing 1 μL of 150 μM cDNA 3′ adaptor, 2.5 μL of 10 × T4 RNA Ligase 1 Buffer (NEB), 25 U T4 RNA Ligase 1 (NEB), 2.5 μL of 10 mM ATP, 2 μL DMSO, and 9.5 μL of 50% PEG8000. Adaptor-ligated cDNA was purified using Agencourt AMPure XP beads. The final sequencing library was amplified and indexed using PCR amplification in 50 μL of a PCR reaction mixture containing 1 U Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA), 10 μL of 5 × Phusion HF Buffer (Thermo Fisher Scientific), 37.5 pmol of each primer (Amp_F and Amp_Index#_R), and 1 μL of 10 mM dNTP mix. The PCR reaction was completed at a semi-plateau and included the following steps: initial activation at 98°C for 30 s; 10 cycles of 98°C for 30 s, 52°C for 30 s, and 72°C for 15 s; and final elongation at 72°C for 30 s. The amplified sequencing library was subjected to two consecutive purifications using Agencourt AMPure XP beads. Sequencing was conducted for 50 cycles of a single-ended recipe using a HiSeq 2500 sequencer (Illumina). The primer sequences are summarized in Supplementary Table S1.
Term-Seq data processing and transcript 3′-end detection using machine-learning
Term-Seq data were processed using the CLC Genomics Workbench (CLC Bio, Aarhus, Denmark). Raw reads were trimmed using the Trim Sequence Tool, with a quality limit of 0.05. Reads with more than two ambiguous nucleotides were discarded. Two random nucleotides located at the 5′- and 3′-ends that were attached during adaptor ligation were removed from the trimmed reads. The reads were mapped onto the MG1655 reference genome (NC_000913.3) with a mismatch cost of two, indel cost of three, length fraction of 0.9, and similarity fraction of 0.9. Mapping data were converted to the GFF file format, as previously described for downstream processing (24). Briefly, the positions of 5′-ends of aligned reads that marked the RNA 3′-ends were extracted from the BAM file using Samtools and BEDTools. The 5′-ends in the genome were counted and written as a GFF file format using an in-house Python script. T3PEs were searched using an in-house Python script based on scikit-learn packages. To determine a specific genomic position is a stable T3PE, the Term-Seq signals from −10 to +11 nt, relative to the position, were submitted to a machine classifier as a dataset, which provided a determination call for the position as its output (Supplementary Figure S1). This was iterated throughout the genome for both strands to detect T3PE. The machine classifier was trained using a training set composed of 694 manually curated or RegulonDB termination sites, each comprising 191 positive and 503 negative calls. Two different machine classifiers, K-nearest neighbor (KNN) and a support vector machine, trained by the training set had a mean accuracy of 94.0% and 80.7%, respectively, upon cross-validation (trained with half of the randomly selected training set, with performance measured on the remaining half, iterated 1000 times). The KNN classifier was used for further termination site discovery. The machine was revised twice with a manual false call inspection. The Python script and KNN machine classifiers used in this study (pickled Python objects) are available in the GitHub repository (https://github.com/robinald/ML_Term-Seq).
Transcript 5′-end mapping using differential RNA-Seq
The 5′-end of the RNA was probed using dRNA-Seq, as previously described (5). Briefly, rRNA-subtracted RNA was split into two samples. One sample was treated with 20 U RNA 5′ polyphosphatase (Epicentre, Madison, WI, USA) at 37°C for 60 min. The other sample was treated with nuclease-free water. The dephosphorylated RNA adaptor was ligated to both samples using 5 U of T4 RNA ligase (Epicentre) at 37°C for 90 min. The adaptor-ligated RNA samples were purified, and cDNA was synthesized using the SuperScript III First-Strand Synthesis System with 3.125 pmol random nonamers. DNA libraries were amplified by PCR with Phusion High-Fidelity DNA Polymerase, using P5 and P7 index primers for 20 cycles. The amplified libraries were sequenced via the 50 cycles single-ended recipe on a HiSeq 2500 sequencer. The oligonucleotide sequences are summarized in Supplementary Table S1.
RNA structure prediction and the free energy of folding
RNA structure and the free energy of RNA folding were calculated from the 45 nt-long DNA sequence upstream of the T3PE using RNAfold software (14,25).
Plasmid construction
The dual-reporter plasmid, pDRA1_empty, was constructed by inserting egfp into the mrfp1-expressing iGEM plasmid BBa_J04450-pSB1C3. Briefly, egfp DNA was amplified from the pTrc-egfp plasmid (26) using the primers, egfp_F and egfp_R. The plasmid backbone was amplified using the pSB1C3_inv_F and pSB1C3_inv_R primers. The two DNA fragments were ligated using an In-Fusion Cloning Kit (TaKaRa Bio, Shiga, Japan), as described by the manufacturer. The pDRA1_terminator plasmid was constructed by inserting terminator fragments into pDRA1_empty. Briefly, a 60-nt primer pair was annealed together in a 10 μL PCR reaction mixture composed of 0.2 U Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific), 2 μL of 5 × Phusion HF Buffer, 2 pmol of each primer (terminator_F and terminator_R), and 0.1 μL of 10 mM dNTP mix. The annealing reaction was performed at an initial activation temperature of 98°C for 30 s, and then 12 cycles of 98°C for 30 s, 52°C for 30 s, 72°C for 15 s, with a final elongation at 72°C for 30 s. The annealed 100-bp dsDNA fragment was composed of a 15 nt sequence homologous to pDRA1_empty at each end and a 70 nt terminator fragment. All annealed products were inspected using gel electrophoresis and purified using a MinElute PCR Purification Kit (Qiagen, Hilden, Germany), as described by the manufacturer. The pDRA1_empty plasmid backbone was also PCR-amplified and linearized using the primers pSB1C3_inv_F and reporter_inv_R. The annealed terminator fragment (1.5 ng) and 10 ng of the pDRA1_empty plasmid backbone were incubated at 50°C for 15 min in a 1 μL cloning reaction volume containing 0.2 μL of 5 × In-Fusion HD Enzyme Premix.
The pDRA2_terminator plasmid was constructed by inserting the egfp-terminator-mrfp1 DNA fragment, amplified with the transfer_assay_F and transfer_assay_R primers from the pDRA1_terminator plasmid, into the pTrcHis2A plasmid (Invitrogen) backbone, which was also PCR-amplified using the backbone_F and backbone_R primers. Cloning was performed using 10 and 8 ng of egfp-terminator-mrfp1 and pTrcHis2A plasmid backbone, respectively, in a 1 μL cloning reaction volume containing 0.2 μL of 5 × In-Fusion HD Enzyme Premix. The pGFP_terminator plasmid was constructed by self-ligating the inverse PCR product that excluded the mrfp1 gene from the pDRA2_terminator plasmid, using the pGFP_F and pGFP_R primers. Self-ligation was performed in a 2.5 μL cloning reaction volume containing 0.5 μL of 5 × In-Fusion HD Enzyme Premix and 20 ng inverse PCR product. The pDRA2_DualP plasmid was constructed by inserting the araC-araBAD promoter cassette into the pDRA2 plasmid by In-Fusion cloning. The araC-araBAD promoter cassette was PCR amplified from the pBADMyc-His C plasmid (Invitrogen) using the ara_F and ara_R primers. The pBDO_empty plasmid was constructed by the sequential insertion of chemically synthesized (IDT gBlock Gene Fragment) B. subtilis alsS-RBS-Aeromonas hydrophila alsD and codon-optimized Thermoanaerobacter brockii bdh (27) into pTrcHis2A. First, the alsSD fragment was amplified using the alsS_pTrc_F and alsS_pTrc_R primers. The alsSD fragment was In-Fusion cloned into pTrcHis2A and amplified with the backbone_F and backbone_R primers. Then, bdh was PCR-amplified with the tbr_bdh_F and tbr_bdh_R primers, and In-Fusion cloned into PCR-amplified pTrcHis2A-alsSD using the alsSD_F and alsSD_R primers. The codon-optimized bdh sequence is available in the Supplementary Data. The pBDO_terminator plasmid was constructed by restriction ligation of the terminator fragment amplified from the pDRA1_terminator plasmid, using the terminator_BDO_F and terminator_BDO_R primers, into pBDO_empty using XhoI and SpeI sites. The pMI_empty plasmid was constructed by cloning yeast INO1, E. coli pfkA, and the terminator fragment into pTrcHis2A. First, the INO1 structural gene was amplified from Saccharomyces cerevisiae CEN.PK genomic DNA, using the INO1_F and INO1_R primers. The INO1 gene fragment was In-Fusion cloned into the pTrcHis2A plasmid, which was amplified using the backbone_F and backbone_R primers. Next, pfkA was amplified from E. coli MG1655 genomic DNA using the pMI_pfkA_F and pMI_pfkA_R primers. The gene was In-Fusion cloned into the pTrcHis2A-INO1 plasmid amplified using the alsSD_F and pMI_INO1_R primers. The terminator fragment was cloned using restriction ligation with the XhoI and SpeI sites in pMI_empty to generate the pMI_terminator. The primer sequences are summarized in Supplementary Table S1.
Fluorescence measurement and normalization
The fluorescence levels of the E. coli cultures were measured using a Synergy H1 Microplate Reader (BioTek Instruments, Winooski, VT, USA) 24 h after inoculation. For GFP, excitation with a 485 nm xenon flash and emission at 528 nm was measured with a gain of 90. For RFP, excitation with a 584 nm xenon flash and emission of 619 nm was measured with a gain of 120. Read-through values were calculated by dividing the RFP intensity by GFP and normalized by setting the read-through of pDRA1_empty as 1.
Measurement of the transcript decay rate
Cells were inoculated in a 250 mL Erlenmeyer flask containing 50 mL M9 glucose medium supplemented with 100 μg mL–1 ampicillin and 1 mM IPTG. When the culture reached the exponential phase (OD600nm = 0.3), 50 μg mL–1 of rifampicin was added. Then, 10 mL of the rifampicin-treated culture was flash-frozen in liquid nitrogen at 0.5, 5, and 10 min after treatment. Each frozen culture was centrifuged at 4,000 × g at 4°C for 30 min. Total RNA was extracted from the cell pellet using the RNAsnap method (23). Next, 5 μg of total RNA was treated with 2 U RNase-free DNase I at 37°C for 1 h. DNA-subtracted RNA was purified using phenol-chloroform extraction. Complementary DNA was synthesized from 500 ng of purified RNA using the SuperScript III First-Strand Synthesis System, as described by the manufacturer. Briefly, 500 ng DNA-subtracted RNA, 25 ng random hexamer, 0.5 μL of 10 mM dNTP mix, and nuclease-free water were mixed in a 5 μL reaction volume. The mixture was incubated at 65°C for 5 min and immediately placed on ice after incubation. Then, 5 μL cDNA Synthesis Mix (containing 1 μL of 10 × RT buffer, 2 μL of 25 mM MgCl2, 1 μL of 0.1 M dithiothreitol, 20 U RNaseOUT recombinant ribonuclease inhibitor, and 100 U SuperScript III reverse transcriptase) was added and incubated at 25°C for 10 min. The mixture was incubated at 50°C for 50 min, and then at 85°C for 5 min. One unit of E. coli RNase H was incubated at 37°C for 20 min to remove the RNA. Quantitative PCR was performed in a 10 μL reaction volume containing 5 μL KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems, Wilmington, MA, USA), 5 pmol of the forward and reverse primers, 1 μL of 20 × diluted cDNA, and 0.2 μL of 50 × ROX High dye in 40 PCR cycles with the following steps: 95°C for 10 s, 58°C for 20 s, and 72°C for 10 s. PCR was performed and monitored using a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). When measuring RNA stability, 16S ribosomal RNA was assumed to be stable as its half-life was reported to exceed 10 min. The amount of egfp mRNA was measured using the -ΔΔCT method, with rrsA as an internal reference. All primers were designed using Primer-BLAST (28) without non-specific binding to the E. coli MG1655 genome sequence (NC_000913.3). The primer sequences are summarized in Supplementary Table S1.
Mathematical modeling of the mRNA levels
The change in GFP mRNA concentration per unit time in an E. coli population carrying the pGFP plasmid was expressed in a first-order decay model, as shown in Equation (1):
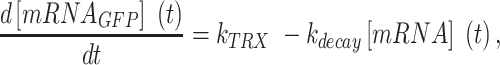 |
(1) |
where 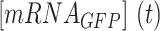 is the GFP mRNA concentration,
is the GFP mRNA concentration, 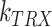 is the constant transcription rate, and
is the constant transcription rate, and 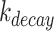 is the mRNA decay rate. Thus, mRNA concentration is expressed by Equation (2), by solving the first-order differential equation (Equation 1), with an initial value of
is the mRNA decay rate. Thus, mRNA concentration is expressed by Equation (2), by solving the first-order differential equation (Equation 1), with an initial value of 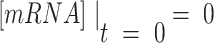 .
.
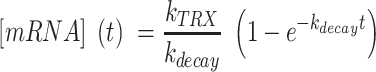 |
(2) |
GFP protein synthesis and fluorescence were assumed to be linearly proportional to the mRNA level. In the simulation of pDRA2, a model was built in a similar manner, considering transcription termination (Equation 3, and Supplementary Figure S2). The model was based on two mRNA species. One is mRNA terminated by the terminator, which encodes only GFP (Equation 3):
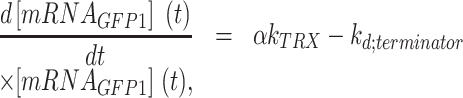 |
(3) |
where 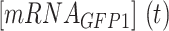 is the concentration of mRNA terminated by the terminator,
is the concentration of mRNA terminated by the terminator, 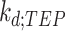 is the decay rate of the mRNA with a terminator on its 3′-UTR;
is the decay rate of the mRNA with a terminator on its 3′-UTR; 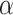 is a transcription termination strength of 0 for 100% read-through, and 1 for 100% transcription termination.
is a transcription termination strength of 0 for 100% read-through, and 1 for 100% transcription termination.
The other mRNA is transcribed by read-through and encodes both GFP and RFP (Equations 4 and 5):
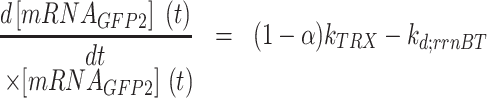 |
(4) |
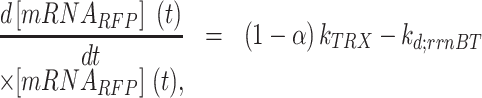 |
(5) |
where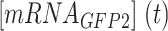 and
and 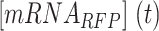 are the read-through mRNA concentrations, and
are the read-through mRNA concentrations, and 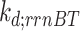 is the decay rate of the mRNA with the rrnB terminator at the end. Thus, the collective concentration of GFP-expressing mRNA was calculated using Equation (6), and the RFP-expressing mRNA was expressed by Equation (7).
is the decay rate of the mRNA with the rrnB terminator at the end. Thus, the collective concentration of GFP-expressing mRNA was calculated using Equation (6), and the RFP-expressing mRNA was expressed by Equation (7).
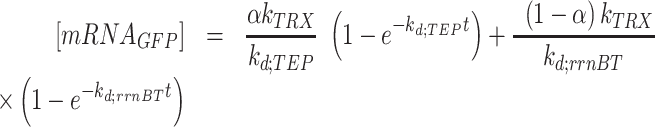 |
(6) |
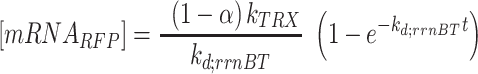 |
(7) |
The read-through (RFP/GFP) of the saturated culture can be expressed by dividing the saturated (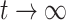 ) RFP concentration by GFP (Equation 8).
) RFP concentration by GFP (Equation 8).
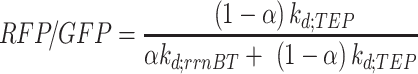 |
(8) |
Finally, the termination strength 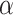 can be calculated using Equation (8), with the measured parameters (Equation 9).
can be calculated using Equation (8), with the measured parameters (Equation 9).
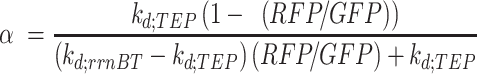 |
(9) |
Model parameters and calculated termination strengths are provided in Supplementary Tables S2 and S3.
Flow cytometry
Cells harboring pDRA2 or pDRA2_DualP were aerobically cultured in M9 glucose medium for 24 h at 37°C with IPTG (1 or 10 μM) and L-arabinose (1, 10, or 30 mM). The cell culture was diluted 10-fold in PBS and analyzed on an S3e Cell Sorter (Bio-Rad, Hercules, CA, USA). Approximately 100,000 events were collected and analyzed.
Noise definition and data parameterization
The GFP and RFP intensities of the entire population were plotted, and a linear regression line was obtained. Noise was defined as the coefficient of variation (CV; the standard deviation divided by the mean), as previously described (29). Noise can be further decomposed into extrinsic and intrinsic noise that are independent and orthogonal to each other (29). To calculate the two orthogonal noise components, the scatter plot showing the flow cytometry data was rotated by the slope of the linear regression line (Supplementary Figure S3). The CV of the extrinsic (along the horizontal axis) and intrinsic (along the vertical axis) components were then calculated.
Quantification of acetoin, BDO, and MI
One milliliter of a 24 h E. coli pBDO culture, or a 48-h E. coli pMI culture, was collected in 1.5 mL microcentrifuge tubes and centrifuged at 16,000 × g for 1 min. Supernatants were filtered using a syringe filter (0.2-μm pore size). The filtrate (200 μL) was transferred to a 2 mL high-performance liquid chromatography (HPLC) vial containing a 250 μL glass vial insert. Then, 20 μL of each sample was analyzed using HPLC with a system comprising a model 2414 refractive index detector (Waters Corporation, Milford, MA, USA), a 1525 binary HPLC pump (Waters), a Metacarb 87H (7.8 × 300 mm) HPLC column (Agilent Technologies, Santa Clara, CA, USA), and a 2707 Autosampler (Waters). The mobile phase was 0.007 N sulfuric acid, the flow rate was 0.6 mL min–1, and the oven and detector temperatures were 50°C.
Statistical analyses
Term-Seq was conducted with three biological replicates, and all 1,629 T3PE were supported by two or more replicates. Read-through value distributions according to the T3PE categories were compared using a two-sided t-test for unequal variance (Welch's t-test). All statistical analyses were performed using the SciPy package (30). Comparisons with a p-value < 0.01 were considered significant in this study.
RESULTS
Identification of multiple 3′ transcript end classes
Transcriptional terminators require characterization to be utilized in the rational design of genetic circuits in synthetic biology. Thus, we sought to identify transcript 3′-ends (T3PE) classes in the E. coli MG1655 transcriptome using Term-Seq. To avoid human bias and positional constraints with respect to known annotations, we employed machine-learning analysis to detect 1,583, 1,795, and 1,951 T3PEs from each biological replicate. Among these, 1,629 T3PEs observed from two or more biological replicates were stable T3PEs (Supplementary Table S4).
First, we identified 407 T3PEs with a conserved sequence motif of rho-independent terminators (RIT) (Figure 1A). The sequences upstream of these T3PEs contained conserved GC-rich sequences that form the stem of a hairpin structure and a consecutive U-stretch (Figure 1B), characteristic of intrinsic terminators (31,32). The RITs had a strong folding free energy (ΔGfolding) compared to random genomic positions (Figure 1C), consistent with a previous study (Figure 1C; Dar) (14). Second, the remaining 1,222 T3PEs without a conserved sequence motif (motif-less T3PEs) were unlikely to form a stem-loop structure because of their relatively high ΔGfolding. Among them, 413 motif-less T3PEs were located on the ribosomal RNA (rRNA)-transfer RNA (tRNA) operons, which matched the rRNA-tRNA processing sites (Figure 1D) (33–38). The processive nuclease activity of RNase D was also observed (Figure 1D, inset), indicating nucleotide-level accuracy of the Term-Seq results. The remaining 809 motif-less T3PEs may represent non-canonical types of transcriptional terminators, or stable transcript ends that are independent of termination, such as intermediates of RNA processing. However, these T3PEs are unlikely to be footprints of RNA degradation during the experimental procedure because they were observed in more than two biological replicates.
Figure 1.

Discovery of diverse classes of transcript 3′-ends in Escherichia coli using Term-Seq. (A) Conserved sequence motif of rho-independent terminators upstream of 407 transcript 3′-ends in the presence of a conserved sequence motif (RITs). (B) Predicted secondary structure of one of the RITs, T3PE-713, which includes a stable stem-loop structure. (C) Comparison of the predicted folding energies of the different RNA termini classes. Random indicates the folding energy of 1,000 random genomic positions. Dar indicates the folding energy of 1,095 previously reported T3PEs (14); three duplicated T3PEs were removed. Box limits, whiskers, and center lines indicate 1st and 3rd quartiles, 10th and 90th percentiles, and the median of the distribution, respectively. (D) Term-Seq revealed the nucleotide-level footprint of ribosomal RNA and transfer RNA processing in the r/tRNA operon. The different RNase cleavage sites are marked by arrowheads. (E) Composition of the different T3PE classes in this study and a comparison to previous reports (12,14). (F) Meta-analysis of the RNA-Seq profile showing the RNA density near the T3PEs. Profiles were normalized at their maximum peak intensity within the analysis window (−200 to + 200 nt). (G) Nucleotide occupancy of the conserved Rho-dependent terminator motif found upstream of motif-less T3PEs.
To cross-validate the classification, we compared 1,629 T3PEs with previously reported T3PEs in E. coli BW25113 (14) or MG1655 (12). The 1,095 T3PEs in E. coli BW25113 were detected with positional constraints (within 150 bp downstream of genes), while the other 2,073 T3PEs in E. coli MG1655 were reported without any constraints (12). With 3 nt accuracy, 459 T3PEs were found in both E. coli MG1655 and BW25113 (Figure 1E) (14). These shared T3PEs were mostly located in the intergenic region, and more than half were RITs (n = 268, 58.4%). Conversely, motif-less T3PEs (n = 1,031, 88.1%) formed a large proportion of the remaining 1,170 T3PEs detected in our study. Considering the tendency of detecting transcriptional terminators due to the positional constraint in the previous report, our unbiased detection method enabled the determination of T3PEs that were not transcriptional terminators. Another report on E. coli MG1655 showed greater agreement with T3PE detected in this study (Figure 1E) (12). Of the 1,170 T3PEs that did not overlap with E. coli BW25113, 318 T3PEs agreed with those previously detected in the same strain. Interestingly, these were mainly motif-less T3PEs (n = 273, 85.5%), indicating that these were consistently observed in multiple experiments. Finally, we identified 852 novel T3PEs comprising 87 RITs and 765 motif-less T3PEs. To assess the functions of the various T3PEs, including motif-less T3PEs, we investigated the RNA-Seq profiles in close vicinity. A meta-analysis of 100,000 random genome positions presented no fluctuation in transcript signals within the 400 nt sequence window (Figure 1F). The RNA-Seq signal was clearly depleted after RITs, indicating strong transcriptional termination. In contrast, the RNA-Seq profile of neighboring motif-less T3PEs displayed no significant transcript depletion and formed a stable profile. A few motif-less T3PE (n = 41) matched with previously reported Rho-dependent terminators (14). Although these had sequence features of Rho-dependent terminators (Figure 1G), a weakly-conserved Rho-dependent terminator motif was not discovered in this case. Thus, the majority of motif-less T3PEs are related to biological processes other than transcriptional termination.
Determination of transcription termination strength
We devised a dual reporter assay system (pDRA1) to further characterize the identified T3PEs, which contained a 70 nt DNA fragment obtained upstream of a T3PE between two different fluorescence reporters (Figure 2A). We constructed pDRA1 plasmids containing one of the 50 randomly selected RITs and 100 motif-less T3PEs (excluding those located in the rRNA operons). Different T3PEs led to different expression levels of fluorescent proteins located downstream of T3PE (Figure 2B). A fraction of the two different fluorescence signals was used to quantitatively measure the terminator strengths, hereafter referred to as the read-through fraction. The strong rrnB T1 terminator (rrnBT) displayed a read-through fraction of 0.009, indicating termination of 99.1% of the transcription (Figure 2C). The RITs located downstream of tRNA, asnV (T3PE-605) and leuW (T3PE-211), mediated termination as effectively as rrnBT. The read-through fractions from E. coli cultures carrying one of the 150 T3PE-carrying pDRA1 plasmids ranged from 0.004 to 91.4 (Supplementary Table S5).
Figure 2.

Dual fluorescence reporter assay measurements of the transcription termination by upstream sequences of transcript 3′-ends. (A) Plasmid map of the dual reporter assay plasmid pDRA1. (B)Escherichia coli harboring different pDRA1 plasmids displayed different red fluorescent protein (RFP) expressions when grown on agar media. Low RFP expression indicates strong termination strength. (C) Monitoring the two different fluorescence signals revealed diverse termination activities of 150 T3PEs. Two signals were normalized by setting the green fluorescent protein (GFP) and RFP signals of pDRA1_empty to the same value. Error bars indicate the standard deviation of two biological replicate cultures. (D) Read-through distributions of T3PEs in the absence of a motif (motif-less T3PEs) revealed that T3PEs differed significantly from RITs. Read-through indicates RFP intensity divided by GFP intensity. *p-value: 0.0478 (Welch's t-test). Box limits, whiskers, and center lines indicate 1st and 3rd quartiles, 10th and 90th percentiles, and the median of the distribution, respectively. Circles indicate individual T3PEs. Colored horizontal lines denote read-throughs of pDRA1_empty and pDRA1_rrnBT. (E-G) Transcript profiles of T3PEs that are independent of canonical transcriptional termination. Transcripts with 3′-termini are products of (E) attenuation, (F) RNA stabilizing element, and (G) RNase III cleavage. Transcript 5′-end mapping supports the formation of an RNase III cleavage footprint. (H) Secondary structure of the 5′-untranslated region of proP mRNA. Arrows indicate the RNase III cleavage sites discovered elsewhere (42) and locations of T3PE-1523, which coincide with each other.
The RITs displayed an average read-through fraction of 0.294 (Figure 2D). Conversely, motif-less T3PEs showed significantly weaker transcription termination than RITs (Figure 2D). Further analyses indicated that the 545 motif-less T3PEs could be the products of biological processes other than transcriptional termination (Supplementary Table S6). For example, T3PE-26 was located downstream of a leuABCD operon leader peptide, LeuL (Figure 2E). LeuL is responsible for the post-transcriptional attenuation of leucine biosynthesis in response to excess leucine (39). Furthermore, the location of T3PE-226 coincides with that of the bacterial interspersed mosaic element (BIME) (Figure 2F), a palindromic repeat sequence throughout the E. coli genome (40). BIMEs are occasionally located in the middle of polycistronic mRNAs and play a role in differential gene expression that are co-transcribed as a single mRNA by stabilizing upstream genes (41). T3PE-1523 perfectly matched a well-known RNase III digestion site on the 5′-UTR of the proline transporter (ProP) mRNA, which modulates ProP expression in response to hyperosmotic stress (Figure 2G and H) (42). RNA digestion resulted in the generation of 5′- and 3′-ends at the cleavage site. We conducted transcript 5′-end mapping using dRNA-Seq to assess the other cleavage end. A stable transcript 5′-end generated by RNase III processing was also observed in transcript 5′-end mapping, supporting the cleavage event (Figure 2G). The 5′-end was not generated by transcription initiation on the promoter, as the transcription start site can be distinguished by differential RNA-Seq (5,8). T3PE-1523 cleavage on GFP-RFP mRNA in pDRA1 led to a marked increase (54.0 times) in RFP intensity compared to GFP, possibly due to exposure of the ribosome binding site of RFP and the nucleolytic degradation of GFP mRNA at its 3′-end after RNase III cleavage. Thus, we concluded that motif-less T3PEs are independent of transcriptional termination. Using the pDRA1 assay, we confirmed the capability of Term-Seq to detect multiple E. coli T3PE classes produced by transcriptome reshaping, RNA processing, cis-regulation, and transcriptional terminators.
The relationship between termination and expression level
As multiple RITs exhibit a wide range of transcription termination strengths, we further examined terminators at different gene expression levels to assess whether the termination strength remained the same. Two control sequences (empty and rrnBT) and 49 RITs were tested under the control of the inducible Lac promoter (pDRA2; Figure 3A). The fluorescence level increased logarithmically at different isopropyl β-D-1-thiogalactopyranoside (IPTG) concentrations (Supplementary Figure S4). Strikingly, the termination levels of many RITs varied according to the transcription level (Figure 3B and Supplementary Table S7). Transcription termination was generally stronger at higher expression levels, likely because of local resource depletion, such as NTPs, which lowers the elongation rate so that RNA polymerase can easily disassociate from its template (43,44). This variation may explain why precise termination strength prediction is difficult, thereby limiting the standardization of terminator bioparts (Supplementary Figure S5) (44,45).
Figure 3.

Differential termination strengths of rho-independent terminators (RITs) at different transcription levels. (A) Design of the dual reporter assay plasmid, pDRA2, with transcription initiation levels regulated by an isopropyl β-d-1-thiogalactopyranoside-inducible lac promoter. (B) Diverse read-throughs of RITs at different transcription initiation levels. (C) Plasmid design to measure the decay rate of messenger RNAs (mRNAs) with different 3′-untranslated regions (UTRs). (D) The fluorescence intensity of green fluorescent protein (GFP) differed depending on the 3′-UTR. Error bars indicate the standard deviation of two biological replicates. (E) Exponential decay of mRNA from pGFP. Error bars indicate the standard deviation of two biological replicates, each composed of technically triplicate qRT-PCR reactions. T1/2 indicates the mRNA half-life. (F) Fluorescence intensity was directly correlated with the half-life of mRNA encoding GFP. (G) Computational modeling of fluorescence protein expression showed a reliable prediction of the experimental measurements. Circles indicate experimental values from pGFP_empty and pGFP_rrnBT. Shades are ranges of the possible fluorescence intensity set by the standard error of the measured half-lives.
We also found that the termination strength reported by the two fluorescent proteins (45–48) may be different from the actual transcriptional termination event. A thorough inspection of the data revealed considerable variations in GFP intensity. For example, the GFP intensity of pDRA1_rrnBT was 127,583 arbitrary units (AU), whereas that of pDRA1_empty was only 70,548 AU (Figure 2C and Supplementary Table S5). We hypothesized that the T3PE sequence at the 3′-end of the transcript is involved in transcript stability. In such cases, the differential degradation of the terminated gfp-only and read-through gfp-rfp transcripts causes the ratio of the two fluorescence signals to differ from the initial transcript ratio set by termination. To test this hypothesis, rfp was removed from the pDRA2 plasmid to construct a plasmid designated pGFP (Figure 3C). Because transcripts generated by different pGFP plasmids have the same 5′-UTR, coding sequence, and plasmid backbone, the difference in GFP intensity should originate solely from the T3PE sequence located in the 3′-UTR. The pGFP assay indicated that E. coli harboring different pGFP plasmids displayed a maximum 5.0-fold difference in the fluorescence intensity (Figure 3D). Time-course measurement of mRNA abundance at 0.5, 5, and 10 min after transcription inhibition through treatment with the transcription inhibitor, rifampicin, revealed an exponential cellular mRNA decay (Figure 3E) (49). The mRNA half-life varied from 3.0 to 14.8 min, consistent with the magnitude of GFP intensity variance. Moreover, the GFP intensity was directly proportional to the mRNA half-life (Figure 3F).
We then constructed a first-order decay model of the GFP transcript to determine whether differential mRNA decay resulted in a difference in fluorescence. Computational modeling confirmed that the measured decay rate of the mRNAs was sufficient to induce a large difference in the GFP intensity of pGFP (Figure 3G). Next, we constructed a mathematical pDRA2 model using the same decay model to examine whether the differential decay of gfp-only and read-through transcripts resulted in a significant discrepancy between the measured read-through and termination strengths (Supplementary Figure S2). Regarding pDRA2_rrnBT, where the terminators on gfp and rfp mRNA were the same, the strengths of the measured termination and modeled termination were identical (Supplementary Table S3). However, the termination strength calculated from the model, which considered the mRNA decay rates, differed significantly from the strength measured using the two fluorescence signals (Supplementary Table S3). For example, T3PE-692 showed a moderate read-through with a strength of 0.31 in the pDRA2 assay but was considered a strong terminator based on the model-calculated termination strength of 0.91. This example highlights the difficulties in characterizing terminators. This discrepancy is based on one of the complex processes governing transcriptional termination. These processes include the binding strength of RNA polymerase to template DNA, RNA decay, the energy required to denature dsDNA to maintain the transcription bubble, poly-U-stretch, partial rho-factor overlap, and secondary stem-loop structures (31,50,51).
Consistency and noise levels of transcription terminator bioparts
Next, we examined whether the termination strengths of RIT were reliable under different experimental conditions. In addition to the M9 glucose medium tested in the previous section, an additional incubation temperature (30°C) and four different culture media, M9 glycerol, M9 high glucose (10 g L–1), Luria-Bertani (LB), and Terrific Broth (TB), were tested. As RITs are independent of trans-acting elements, changes in transcription termination under various experimental conditions should be negligible. As expected, the read-through fractions of RITs under the five additional conditions strongly correlated with the M9 glucose (2 g L–1) medium (Supplementary Table S8). Specifically, read-through fractions of the tested RITs in high glucose (10 g L–1) M9 medium were almost identical to those in normal M9 glucose, with a Pearson's R2 correlation of 0.93 and an average fold-change of 0.96 (Figure 4A). Similarly, RITs had an average fold-change of 1.09 in LB medium. Unlike the high-glucose M9 and LB media, read-through fractions of RITs grown in M9 glycerol, TB media, and at 30°C deviated from those grown in M9 glucose medium with mean fold-changes of 0.66, 1.84, and 1.34, respectively (Supplementary Figure S6). These deviations reflect different growth rates and energy levels, which may change the transcription kinetics, such as elongation rates, and could result in a transcriptome-wide termination shift (43,44). The transcription elongation rate of E. coli depends on the growth rate, as evidenced by the slower elongation rate of E. coli grown in glycerol minimal medium compared to that grown in minimal glucose medium (52,53). The slower elongation rate in M9 glycerol medium increases susceptibility of RNA polymerase to a transcriptional pause at the terminator, resulting in stronger termination. In contrast, growth in TB resulted in a high elongation rate and reduced transcription termination. Cells grown in M9 glucose medium at 30°C had a 1.34-fold increase in read-through, compared to cells grown at 37°C. The pDRA2 assay results obtained under several experimental conditions indicate that rho-independent transcription terminators in E. coli present consistent relative strength. However, the overall absolute strength of the entire termination shifts according to the global cellular changes in transcription kinetics and trans-acting elements, such as the elongation rate and possibly the alarmone level. This complex termination behavior must be considered in synthetic genetic circuitry.
Figure 4.

Consistency and noise level of transcription terminator bioparts. (A) Read-through fraction of rho-independent terminators (RITs) in different experimental conditions. Scatter plots of the correlation between RIT read-throughs in diverse experimental conditions with M9 glucose medium. Error bars indicate the standard deviation of biological replicates. R2 indicates the Pearson's correlation coefficient. (B) Design of a genetic system expressing two fluorescence proteins using two independent promoters. (C) Flow cytometry measurements of fluorescence proteins expressed by two independent promoters with combinations of inducer concentrations. Each point represents a single cell. a indicates the slope of the linear regression line. ηext and ηint indicate the extrinsic and intrinsic noise components in RNA expression, respectively. (D) Flow cytometry measurement of the fluorescence proteins expressed by terminator bioparts. RT indicates the read-through of pDRA2 observed from overnight induced (with 10 μM IPTG) cell cultures (Supplementary Table S7).
Furthermore, we compared the experimental fluctuations of a genetic system composed of terminator bioparts with those of conventional systems. Previous assessments of stochastic gene expression revealed two independent noise components (defined by the coefficient of variation, which is the standard deviation divided by the mean) that contribute to the overall fluctuation of cellular behavior (29). Extrinsic noise (ηext) is induced by different states, such as transcriptional/translational machinery concentrations and cellular energy states that cause variation in overall protein expression. The other component, intrinsic noise (ηint), is caused by molecular events, such as random binding of RNA polymerases, ribosomes, and transcription factors, even when cells are assumed to have the same cellular state. Thus, we constructed an expression system that expressed two different fluorescent proteins from two different promoters to set a reference (Figure 4B). Cells expressing GFP and RFP by the lac promoter and araBAD promoter had ηext and ηint values of 0.451 and 18.4, respectively, yielding a total noise (ηtot) of 18.4 (Figure 4C). When terminator bioparts were used, ηint was 4.08-fold lower than that of the dual promoter system (mean of 4.53) (Figure 4D), whereas ηext remained the same (1.04-fold increase). The noise assessment indicated a 4.07-fold lower total system noise by reducing the sources of biological variation in transcription initiation (e.g. RNAP binding) and transcription factor regulation. This highlights a significant advantage of terminator bioparts over conventional regulatory systems that use multiple promoters and ribosome-binding sites.
Development of metabolic flux valves based on terminator bioparts
Various elements are used to regulate the expression of multiple genes in synthetic biology. As genetic circuits become more complicated to accommodate multiple connections and regulations, more bioparts are required. Thus, we examined whether RITs with different termination strengths can function as reliable regulatory bioparts to provide additional means of regulating synthetic pathways with fewer bioparts. Acetoin and 2,3-butanediol (2,3-BDO) were selected as target products. Acetoin is produced by several bacteria to store the carbon flux overflow for later use and prevent the accumulation of acidic cellular metabolites. Acetolactate synthase (AlsS) and acetolactate decarboxylase (AlsD) catalyze the condensation of two pyruvate molecules into a single acetoin molecule (Figure 5A). Additionally, 2,3-BDO is an important organic molecule used to produce synthetic rubber. Butanediol dehydrogenase (Bdh) catalyzes the one-step conversion of acetoin to 2,3-BDO (Figure 5A).
Figure 5.

Stable terminator bioparts can control metabolic pathways in a reliable and scalable manner. (A) Heterologous pathways for acetoin and 2,3-butanediol (2,3-BDO) biosynthesis from pyruvate. Abbreviations are as follows: alsS, acetolactate synthase; alsD, acetolactate decarboxylase; and bdh, butanediol dehydrogenase. (B) The expression of bdh is dependent on the read-through of RITs located upstream. (C) Altered production of acetoin and 2,3-BDO in response to the RIT strengths. Titers of extracellular metabolites were measured after 24 h batch fermentation. Error bars indicate the standard deviations of the two biological replicate cultures. Pie charts indicate the relative expression of alsSD and bdh. (D) Metabolic flux valve system controlling the glycolytic flux for cellular resources and myo-inositol (MI) production. The expression of pfkA involves the metabolic valve of stable terminator bioparts. Abbreviations are as follows: zwf, glucose-6-phosphate dehydrogenase; pgi, glucose-6-phosphatate isomerase; pfkA, 6-phosphofructokinase 1; and INO1, inositol-3-phosphate synthase. (E) Design of the flux-valved MI production pathway. The E. coli ΔpfkA Δzwf strain was used for maximum production and control efficiency. (F) Glucose consumption, MI production, and MI yield from the glucose (g/g) of E. coli harboring one of the pMI plasmids. The conv. sys. indicates the E. coli MG1655 wild-type (both pfkA and zwf were not perturbed) strain heterologously expressing INO1 without a metabolic valve. Error bars indicate the standard deviations of the two biological replicate cultures.
Three RIT groups were selected to control the system expression. The first group, Group A, contained five RITs (T3PE-211, rrnBT, 383, 529, and 1498) that had read-throughs of < 1 and relatively stable read-through fractions at different transcription rates (Figure 3B). The RITs in Group B (T3PE-1345, 936, 390, and 948) had read-through values > 1. Therefore, downstream gene expression was higher than the upstream genes. The last group, Group C, was composed of three RITs that shortened the mRNA half-life when located at the 3′-UTR of an mRNA, as shown in the pGFP assay (Figure 3D). They were located downstream of alsSD in the polycistronic 2,3-BDO synthetic plasmid pBDO (Figure 5B). Bdh expression was controlled by the RIT read-through fraction, which determined the relative flux between acetoin and 2,3-BDO from pyruvate. Then, 24 h after the addition of IPTG (0.1 mM), E. coli strains harboring one of the pBDO plasmids with RITs from Group A showed various 2,3-BDO titers due to various expression levels of bdh by different terminator read-through fractions (Figure 5C). The amount of 2,3-BDO produced by pBDO_rrnBT, which harbors the rrnB T1 terminator upstream of bdh, was 0.126 g L–1. The BDO titer increased as the termination strength decreased, such that the fully induced negative control cells (pBDO_empty) produced 0.382 g L–1 of 2,3-BDO, the maximum titer of Group A RITs. More importantly, Group B RITs could support greater 2,3-BDO production, even greater than pBDO_empty, which has no transcription termination activity. In theory, bdh expression should always be lower than that of the upstream genes because the transcriptional terminator is located upstream of bdh. However, we observed higher 2,3-BDO production with the Group B RITs (maximum 0.513 g L–1). This was consistent with the pDRA2 assay of the Group B RITs with read-through values > 1. This phenomenon was not due to a promoter sequence that might overlap with T3PE. However, RFP expression was not observed in the pDRA2 assays of Group B RITs when they were not induced (Supplementary Table S9). The behavior of Group B terminators, which increases the expression of downstream genes compared to that of upstream genes, was counter intuitive. Thus, we conducted transcript 5′-end mapping, which captures the stable transcript 5′-end in a cell. Among the 13 Group B terminators examined by the pDRA2 assay, five had stable transcript 5′-ends that were not produced by transcription initiation at their close vicinity, indicating active post-transcriptional RNA processing near the terminator (Supplementary Figure S7). Thus, increased ribosome entry and RBS availability induced by post-transcriptional cleavage, generating a new 5′-UTR, may be responsible for the high expression of downstream genes. Regardless of the reason, 2,3-BDO production continuously increased as the read-through fraction increased. Production reached a plateau at a read-through of 2.343 (T3PE-390).
Saturation of the 2,3-BDO titer at the high read-through fraction was possibly due to the saturation of the cellular resource to convert acetoin to 2,3-BDO, that is, NADH. This illustrates the ability of T3PE bioparts to support a wide range of transcription ratios among genes. Because read-through values can exceed 1, the system requires no additional modification to change the gene order to accomplish higher downstream gene expression. Overall, as the read-through of T3PE increased, a greater metabolic flux was directed toward 2,3-BDO from acetoin. The 2,3-BDO to acetoin ratio was logarithmically proportional to the read-through strength of the T3PEs with high correlation, indicating the reliability of T3PEs as synthetic bioparts for modifying metabolic flux (Supplementary Figure S8). Finally, we tested Group C RITs that produced short-lived mRNAs. As expected, E. coli harboring pBDO with Group C RITs produced less acetoin and 2,3-BDO than cells with pBDO consisting of RITs in other groups (Figure 5C). This highlights the importance of selecting an appropriate terminator biopart to achieve the desired functionality of a genetic circuit.
We used this flux regulation mechanism to control the flux between central carbon metabolism and myo-inositol (MI) production. When a biological system is used for production, the metabolic fluxes directed toward production and biomass must be balanced, as they are interconnected and constrained by a finite amount of cellular resources. We combined MI-1-phosphate synthase (INO1) and phosphofructokinase I (PfkA, an effective rate-limiting enzyme of glycolysis (54)) as an interconnection point of production and cellular growth (Figure 5D). An RIT located between the two genes acts as a metabolic valve to direct glycolytic resources (Figure 5E). Five MI production plasmids encoding different valves were tested in glucose-6-phosphate 1-dehydrogeanse (zwf) and pfkA double-knockout strains. Plasmids overexpressing INO1 without flux control displayed severe growth retardation and marginal MI production (Figure 5F and Supplementary Figure S9). As pfkA expression increased in accordance with the increase in transcription read-through, glucose consumption and MI production also increased. At a read-through rate of 0.519 (T3PE-383), pfkA expression supported an MI production titer of 0.682 g L–1, which was 8.92-times higher than that of the conventional system (pTrc-INO1) with a 38% MI yield (g g–1) increase from glucose (Figure 5F). When pfkA expression exceeded T3PE-383, the MI titer was reduced. At this point, excessive flux was used to promote cellular growth and biomass formation instead of MI production. Thus, we successfully identified an optimally rapid growth rate to support resources and energy for producing MI while not overconsuming a given nutrient (Supplementary Figure S10). We then examined the valves in genome-reduced E. coli that might have possessed a different optimum metabolic balance (55). The genome-reduced strain reached its optimum metabolic balance at strong termination. At optimal balance, where expression is terminated by a strong rrnB terminator, it produced 0.864 g L–1 of MI (1.27-fold higher than the maximum titer of MG1655). The MI titer was reduced after the plateau, where expression was higher with weaker terminators compared to that with rrnB terminator. This varying optimum metabolic balance indicates a lower carbon cost for the cellular growth of the genome-reduced strain compared to that for the growth of its wild-type counterpart (Supplementary Figure S9). These collective results demonstrate the novel use of transcription terminators in metabolic engineering. Terminators and metabolic valves can be operated in a scalable and reliable manner, regardless of the host strain.
DISCUSSION
Here, we reported various classes of T3PEs in E. coli and discovered 1,629 T3PEs using machine learning to avoid analytic or human bias. Of these T3PEs, 407 were immediately identified as RITs based on their conserved sequence motifs. The remaining 1,222 appeared to be products of various types of RNA processing. More than 100 T3PEs coincided with various biological processes that can generate stable RNA 3′-ends, including RNA processing, post-transcriptional attenuation, small RNA targeting, and RNA stabilization on BIME. The remaining motif-less T3PEs were predicted to be produced by processes that remain to be characterized as key starting points for investigating dynamic transcript regulation.
Examination of 50 RITs revealed different terminator and termination strengths. Interestingly, the gene expression was weakly correlated with read-through (positive correlation with termination strength) (Supplementary Figure S11). It appears that genes with high expression levels require strong terminators. For example, terminators of tRNAs and stress-response sigma factor RpoS showed strong termination strength, presumably required to reduce the transcriptional interference of highly expressed genes on downstream genes and tight regulation. In contrast, T3PEs related to post-transcriptional processing, such as T3PE-1523 (RNase III processing site on proP) and T3PE-1238 (ivbL_t; translational attenuation of ilvBN), presented weak termination strengths (read-through of 4.653 and 1.560 in the pDRA2 assay, respectively) and provided a margin for the regulation of cellular stresses. This indicates a possible evolutionary pressure of terminator selection to reduce the nutrient waste generated by transcriptional read-through to ensure the optimal 3′-UTR regulatory efficiency. Individual terminators also have variable termination strengths, depending on different transcription initiation rates. Although the reason for the variation remains poorly understood, prior results on the termination strength of a terminator with different elongation rates suggest that this may be caused by a slower elongation rate (43,44). Resource depletion at high expression levels of some pDRA2 systems, followed by a lowered elongation rate, resulted in elevated dissociation of the elongation complex.
Bioparts must be reliable and stable for synthetic biological applications. Thus, this simulation and characterization of terminator activities at various expression levels provide a valuable reference for synthetic genetic system designs. In addition, bioparts may not be limited to E. coli. Previous reports indicated considerable conservation of sequence motif, such as U-tract, hairpin structure, and free energy of folding in bacterial rho-independent terminators (13,56,57). For example, the prediction of RIT in 57 Firmicutes, based on 463 experimentally determined terminators of Bacillus subtilis, was extremely successful (with an average sensitivity of 94%) (57). Prediction of E. coli terminators based on B. subtilis terminators also presented a considerably high sensitivity (67%). Thus, the terminators in this study may be applicable to other bacterial species without significant difficulty. Also, recent advances in high-throughput quantitative Term-Seq will enable rapid quantification and characterization of terminator bioparts in other bacterial species (47).
Furthermore, terminator bioparts showed relatively consistent read-through levels when examined in different media and experimental conditions. The read-through deviation in cells grown under different conditions could be explained by the different growth and transcriptional elongation rates. According to our observations, read-through decreases as the susceptibility of RNA polymerase to termination increases at lower transcriptional elongation speeds. Considering the relationship between the termination strength and elongation rate, RITs in cells grown at 30°C likely have slower elongation rates and stronger terminations. However, this opposed initial predictions. This unexpected result may reflect the low alarmone diphosphate guanosine (ppGpp) level, which can alter RNA chain elongation and promote intrinsic termination of cells grown at low temperatures (50,52,58). The possible role of ppGpp in transcription termination remains poorly understood and requires further characterization.
The dual reporter assay enables a robust transcriptional termination measure but presents an inherent limitation. Different 3′-end structures and sequences manifest different transcript stabilities. Thus, the ratio of the two fluorescence reporters is an inaccurate representation of the ratio that is initially set by transcription termination. The half-life of the tested RITs differed by up to 5-fold, and computational modeling of the assay system revealed that the measured read-through fraction deviated from its true termination rate by 27%. Previous studies using similar assay systems did not consider this error, possibly because the effect of transcript decay was marginal in highly conserved synthetic terminator sequences (45,46). Precise investigation of transcription termination requires a more advanced assay. The use of single T3PEs at the end of two reporters and the insertion of RNA stabilizers that normalize the decay rate may be a possible solution.
Furthermore, terminator bioparts presented significantly lower noise when expressing multiple proteins compared to the multiple promoter-based system. The low noise was due to the reduced number of components that induce fluctuations in protein expression, which is intrinsic to the molecular events in a cell. The complexity of the biological systems should be reduced to increase the fidelity of a genetic circuit. Examination of noise components demonstrated an underrated importance of terminator bioparts over regulatory elements in the 5′-UTR when designing synthetic genetic systems with low intrinsic noise.
Taken together, we demonstrated that terminators are reliable and scalable regulatory elements in the synthetic 2,3-BDO production pathway. The production of acetoin and 2,3-BDO was tunable with the termination strength tested in the pDRA2 assay. Acetoin and 2,3-BDO production using T3PEs, which generate mRNAs with short half-lives, resulted in a poor titer owing to low transcript levels. This demonstrates the underestimated importance of 3′-UTR engineering and selecting an appropriate terminator. However, these short-lived transcripts may be useful when temporal and reversible expression control is required. RITs that have short mRNA half-life can be effective post-transcriptional regulatory elements that can replace protein level control. Unlike acetoin production, heterologous MI production induced systematic failure. Similar to MI, many heterologous production pathways fail because they disrupt endogenous metabolism. To prevent such failures, a heterologous pathway needs to be tightly controlled to balance the flux entering the target production and the endogenous metabolism that sustains cellular energy and precursor generation. Determination of optimal conditions is challenging but is aided by the present construction of metabolic flux valves from stable terminators. Synthetic 3′-UTR valves can be used to rapidly determine the optimal metabolic flux distribution between heterologous and endogenous pathways in multiple strains with different optima. This design strategy using metabolic valves is an attractive alternative to conventional promoter-based systems as it only requires a short DNA fragment and is independent of trans-acting elements. This provides a novel and simple approach for metabolic engineering.
DATA AVAILABILITY
Term-Seq data generated in this study is available in the Bioproject/SRA and EMBL Nucleotide Sequence Database (ENA) with the primary accession number PRJEB36932. Trained K-nearest neighbor machine classifiers are available as pickled python objects through GitHub repository (https://github.com/robinald/ML_Term-Seq). RNA-Seq dataset was reported previously (45) and is available through Bioproject/SRA and EMBL Nucleotide Sequence Database (ENA) with the primary accession number PRJEB21199.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: B.-K.C. designed and supervised the project. D.C., K.K., M.K., and S.C. performed the experiments. D.C. S.-G.L., S.C., B.P., and B.-K.C. analyzed the data. D.C. S.C., B.P., and B.-K.C. wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Donghui Choe, Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA.
Kangsan Kim, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Minjeong Kang, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Seung-Goo Lee, Synthetic Biology & Bioengineering Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon 34141, Republic of Korea.
Suhyung Cho, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea; KI for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Bernhard Palsson, Department of Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; Department of Pediatrics, University of California San Diego, La Jolla, CA 92093, USA.
Byung-Kwan Cho, Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea; KI for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the C1 Gas Refinery Program [2018M3D3A1A01055733 to B.-K.C.], the Korea Bio Grand Challenge [2018M3A9H3024759 to B.-K.C.], and the Basic Science Research Program [2018R1A1A3A04079196 to S.C.] through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT. This work was also supported by the Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program [KGM5402221 to S.-G.L.] and a grant from the Novo Nordisk Foundation [NNF10CC1016517 to B.P.]
Conflict of interest statement. None declared.
REFERENCES
- 1. Seo S.W., Yang J.S., Kim I., Yang J., Min B.E., Kim S., Jung G.Y.. Predictive design of mRNA translation initiation region to control prokaryotic translation efficiency. Metab. Eng. 2013; 15:67–74. [DOI] [PubMed] [Google Scholar]
- 2. Mutalik V.K., Guimaraes J.C., Cambray G., Lam C., Christoffersen M.J., Mai Q.A., Tran A.B., Paull M., Keasling J.D., Arkin A.P.et al.. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013; 10:354–360. [DOI] [PubMed] [Google Scholar]
- 3. Sharma C.M., Hoffmann S., Darfeuille F., Reignier J., Findeiss S., Sittka A., Chabas S., Reiche K., Hackermuller J., Reinhardt R.et al.. The primary transcriptome of the major human pathogen Helicobacterpylori. Nature. 2010; 464:250–255. [DOI] [PubMed] [Google Scholar]
- 4. Frohman M.A., Dush M.K., Martin G.R.. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA. 1988; 85:8998–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choe D., Szubin R., Dahesh S., Cho S., Nizet V., Palsson B., Cho B.K.. Genome-scale analysis of Methicillin-resistant Staphylococcusaureus USA300 reveals a tradeoff between pathogenesis and drug resistance. Sci. Rep. 2018; 8:2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Babski J., Haas K.A., Nather-Schindler D., Pfeiffer F., Forstner K.U., Hammelmann M., Hilker R., Becker A., Sharma C.M., Marchfelder A.et al.. Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferaxvolcanii based on differential RNA-Seq (dRNA-Seq). BMC Genomics. 2016; 17:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Illingworth R.S., Gruenewald-Schneider U., Webb S., Kerr A.R., James K.D., Turner D.J., Smith C., Harrison D.J., Andrews R., Bird A.P.. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLos Genet. 2010; 6:e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh N., Wade J.T.. Identification of regulatory RNA in bacterial genomes by genome-scale mapping of transcription start sites. Methods Mol. Biol. 2014; 1103:1–10. [DOI] [PubMed] [Google Scholar]
- 9. Valen E., Pascarella G., Chalk A., Maeda N., Kojima M., Kawazu C., Murata M., Nishiyori H., Lazarevic D., Motti D.et al.. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE. Genome Res. 2009; 19:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta A., Reizman I.M., Reisch C.R., Prather K.L.. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol. 2017; 35:273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murphy K.F., Balazsi G., Collins J.J.. Combinatorial promoter design for engineering noisy gene expression. Proc. Natl. Acad. Sci. USA. 2007; 104:12726–12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adams P.P., Baniulyte G., Esnault C., Chegireddy K., Singh N., Monge M., Dale R.K., Storz G., Wade J.T.. Regulatory roles of Escherichiacoli 5′ UTR and ORF-internal RNAs detected by 3′ end mapping. Elife. 2021; 10:e62438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dar D., Shamir M., Mellin J.R., Koutero M., Stern-Ginossar N., Cossart P., Sorek R.. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science. 2016; 352:aad9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dar D., Sorek R.. High-resolution RNA 3′-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res. 2018; 46:6797–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dar D., Sorek R.. Extensive reshaping of bacterial operons by programmed mRNA decay. PLos Genet. 2018; 14:e1007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ren G.X., Guo X.P., Sun Y.C.. Regulatory 3′ untranslated regions of bacterial mRNAs. Front. Microbiol. 2017; 8:1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Menendez-Gil P., Caballero C.J., Catalan-Moreno A., Irurzun N., Barrio-Hernandez I., Caldelari I., Toledo-Arana A.. Differential evolution in 3′UTRs leads to specific gene expression in Staphylococcus. Nucleic Acids Res. 2020; 48:2544–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuo Y., Deutscher M.P.. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001; 29:1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nielsen A.A., Der B.S., Shin J., Vaidyanathan P., Paralanov V., Strychalski E.A., Ross D., Densmore D., Voigt C.A.. Genetic circuit design automation. Science. 2016; 352:aac7341. [DOI] [PubMed] [Google Scholar]
- 20. Brophy J.A., Voigt C.A.. Principles of genetic circuit design. Nat. Methods. 2014; 11:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Datsenko K.A., Wanner B.L.. One-step inactivation of chromosomal genes in Escherichiacoli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000; 97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee Y., Lee N., Jeong Y., Hwang S., Kim W., Cho S., Palsson B.O., Cho B.K.. The transcription unit architecture of Streptomyceslividans TK24. Front Microbiol. 2019; 10:2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stead M.B., Agrawal A., Bowden K.E., Nasir R., Mohanty B.K., Meagher R.B., Kushner S.R.. RNAsnap: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res. 2012; 40:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choe D., Palsson B., Cho B.K.. STATR: a simple analysis pipeline of Ribo-Seq in bacteria. J. Microbiol. 2020; 58:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho S., Choe D., Lee E., Kim S.C., Palsson B., Cho B.K.. High-level dCas9 expression induces abnormal cell morphology in Escherichiacoli. ACS Synth Biol. 2018; 7:1085–1094. [DOI] [PubMed] [Google Scholar]
- 27. Oliver J.W., Machado I.M., Yoneda H., Atsumi S.. Cyanobacterial conversion of carbon dioxide to 2,3-butanediol. Proc. Natl. Acad. Sci. USA. 2013; 110:1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L.. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012; 13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elowitz M.B., Levine A.J., Siggia E.D., Swain P.S.. Stochastic gene expression in a single cell. Science. 2002; 297:1183–1186. [DOI] [PubMed] [Google Scholar]
- 30. Oliphant T.E. Python for scientific computing. Comput. Sci. Eng. 2007; 9:10–20. [Google Scholar]
- 31. Gusarov I., Nudler E.. The mechanism of intrinsic transcription termination. Mol. Cell. 1999; 3:495–504. [DOI] [PubMed] [Google Scholar]
- 32. Ray-Soni A., Bellecourt M.J., Landick R.. Mechanisms of bacterial transcription termination: all good things must end. Annu. Rev. Biochem. 2016; 85:319–347. [DOI] [PubMed] [Google Scholar]
- 33. Gordon G.C., Cameron J.C., Pfleger B.F.. RNA sequencing identifies new RNase III cleavage sites in Escherichiacoli and reveals increased regulation of mRNA. Mbio. 2017; 8:e00128-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tock M.R., Walsh A.P., Carroll G., McDowall K.J.. The CafA protein required for the 5′-maturation of 16 S rRNA is a 5′-end-dependent ribonuclease that has context-dependent broad sequence specificity. J. Biol. Chem. 2000; 275:8726–8732. [DOI] [PubMed] [Google Scholar]
- 35. Li Z., Pandit S., Deutscher M.P.. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J. 1999; 18:2878–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Z., Pandit S., Deutscher M.P.. Maturation of 23S ribosomal RNA requires the exoribonuclease RNase T. RNA. 1999; 5:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reuven N.B., Deutscher M.P.. Multiple exoribonucleases are required for the 3′ processing of Escherichiacoli tRNA precursors in vivo. FASEB J. 1993; 7:143–148. [DOI] [PubMed] [Google Scholar]
- 38. Ow M.C., Kushner S.R.. Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev. 2002; 16:1102–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wessler S.R., Calvo J.M.. Control of leu operon expression in Escherichiacoli by a transcription attenuation mechanism. J. Mol. Biol. 1981; 149:579–597. [DOI] [PubMed] [Google Scholar]
- 40. Bachellier S., Clement J.M., Hofnung M., Gilson E.. Bacterial interspersed mosaic elements (BIMEs) are a major source of sequence polymorphism in Escherichiacoli intergenic regions including specific associations with a new insertion sequence. Genetics. 1997; 145:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khemici V., Carpousis A.J.. The RNA degradosome and poly(A) polymerase of Escherichiacoli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol. Microbiol. 2004; 51:777–790. [DOI] [PubMed] [Google Scholar]
- 42. Lim B., Lee K.. Stability of the osmoregulated promoter-derived proP mRNA is posttranscriptionally regulated by RNase III in Escherichiacoli. J. Bacteriol. 2015; 197:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McDowell J.C., Roberts J.W., Jin D.J., Gross C.. Determination of intrinsic transcription termination efficiency by RNA polymerase elongation rate. Science. 1994; 266:822–825. [DOI] [PubMed] [Google Scholar]
- 44. von Hippel P.H., Yager T.D.. Transcript elongation and termination are competitive kinetic processes. Proc. Natl. Acad. Sci. USA. 1991; 88:2307–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Y.J., Liu P., Nielsen A.A., Brophy J.A., Clancy K., Peterson T., Voigt C.A.. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods. 2013; 10:659–664. [DOI] [PubMed] [Google Scholar]
- 46. Curran K.A., Morse N.J., Markham K.A., Wagman A.M., Gupta A., Alper H.S.. Short synthetic terminators for improved heterologous gene expression in yeast. ACS Synth Biol. 2015; 4:824–832. [DOI] [PubMed] [Google Scholar]
- 47. Hudson A.J., Wieden H.J.. Rapid generation of sequence-diverse terminator libraries and their parameterization using quantitative Term-Seq. Synth. Biol. 2019; 4:ysz026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nojima T., Lin A.C., Fujii T., Endo I.. Determination of the termination efficiency of the transcription terminator using different fluorescent profiles in green fluorescent protein mutants. Anal. Sci. 2005; 21:1479–1481. [DOI] [PubMed] [Google Scholar]
- 49. Munchel S.E., Shultzaberger R.K., Takizawa N., Weis K.. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol. Biol. Cell. 2011; 22:2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Epshtein V., Cardinale C.J., Ruckenstein A.E., Borukhov S., Nudler E.. An allosteric path to transcription termination. Mol. Cell. 2007; 28:991–1001. [DOI] [PubMed] [Google Scholar]
- 51. Larson M.H., Greenleaf W.J., Landick R., Block S.M.. Applied force reveals mechanistic and energetic details of transcription termination. Cell. 2008; 132:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ryals J., Little R., Bremer H.. Temperature dependence of RNA synthesis parameters in Escherichiacoli. J. Bacteriol. 1982; 151:879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vogel U., Jensen K.F.. The RNA chain elongation rate in Escherichiacoli depends on the growth rate. J. Bacteriol. 1994; 176:2807–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brockman I.M., Prather K.L.J.. Dynamic knockdown of E. coli central metabolism for redirecting fluxes of primary metabolites. Metab. Eng. 2015; 28:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Choe D., Lee J.H., Yoo M., Hwang S., Sung B.H., Cho S., Palsson B., Kim S.C., Cho B.K.. Adaptive laboratory evolution of a genome-reduced Escherichiacoli. Nat. Commun. 2019; 10:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ermolaeva M.D., Khalak H.G., White O., Smith H.O., Salzberg S.L.. Prediction of transcription terminators in bacterial genomes. J. Mol. Biol. 2000; 301:27–33. [DOI] [PubMed] [Google Scholar]
- 57. de Hoon M.J., Makita Y., Nakai K., Miyano S.. Prediction of transcriptional terminators in Bacillussubtilis and related species. PLoS Comput. Biol. 2005; 1:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Furman R., Sevostyanova A., Artsimovitch I.. Transcription initiation factor DksA has diverse effects on RNA chain elongation. Nucleic Acids Res. 2012; 40:3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Term-Seq data generated in this study is available in the Bioproject/SRA and EMBL Nucleotide Sequence Database (ENA) with the primary accession number PRJEB36932. Trained K-nearest neighbor machine classifiers are available as pickled python objects through GitHub repository (https://github.com/robinald/ML_Term-Seq). RNA-Seq dataset was reported previously (45) and is available through Bioproject/SRA and EMBL Nucleotide Sequence Database (ENA) with the primary accession number PRJEB21199.