


Abstract
Proteins encoded by newly-emerged genes (‘orphan genes’) share no sequence similarity with proteins in any other species. They provide organisms with a reservoir of genetic elements to quickly respond to changing selection pressures. Here, we systematically assess the ability of five gene prediction pipelines to accurately predict genes in genomes according to phylostratal origin. BRAKER and MAKER are existing, popular ab initio tools that infer gene structures by machine learning. Direct Inference is an evidence-based pipeline we developed to predict gene structures from alignments of RNA-Seq data. The BIND pipeline integrates ab initio predictions of BRAKER and Direct inference; MIND combines Direct Inference and MAKER predictions. We use highly-curated Arabidopsis and yeast annotations as gold-standard benchmarks, and cross-validate in rice. Each pipeline under-predicts orphan genes (as few as 11 percent, under one prediction scenario). Increasing RNA-Seq diversity greatly improves prediction efficacy. The combined methods (BIND and MIND) yield best predictions overall, BIND identifying 68% of annotated orphan genes, 99% of ancient genes, and give the highest sensitivity score regardless dataset in Arabidopsis. We provide a light weight, flexible, reproducible, and well-documented solution to improve gene prediction.
INTRODUCTION
Eukaryotic and prokaryotic genomes contain genes (‘orphan genes’) whose proteins are recognizable only in a single species. Some of these have emerged de novo from the genome, while others have diverged from their cousins so quickly that no sequence similarity is detectable (1–5).
As encoders of completely novel proteins, orphan genes provide a disruptive force in evolution. Orphans play a crucial role in adaptation to new biological niches. Studies from vertebrates, annelids, insects, fungi and plants show that many extant orphan proteins mitigate novel biotic challenges (prey, predators, hosts) or emergent environmental shifts (2,6–9).
Some proteins encoded by orphan genes (e.g. toxins (6)) act externally, while others integrate into internal metabolic and developmental pathways (7). Phylostratigraphic reconstructions, which determine the phylogenic origin of every gene based on homology, have implicated orphans in the evolution of new reproductive and neural structures (10,11). Thus, the advent of orphan genes may provide a critical enabler of speciation. The ability to accurately predict orphan genes and other young lineage-specific genes conveys unique percipience about evolution and ecology (2,11,12).
A subset of orphans are retained as genes and continue to evolve, such that each genome contains a mixture of genes of different ages (phylostrata) (1,3,13,14). Thus, the age of each gene can be considered the time since its deepest ancestor emerged, as opposed to its most recent duplication. Even the most ancient genes were orphan genes once (for protein-coding genes, these would be genes whose proteins trace back to early eukaryotic organisms or to prokaryotes). Of the estimated billions of extant protein-coding orphan genes across all eukaryotic species (conservatively calculated as 8.7 million extant eukaryotic species (15) × 1000 orphans per eukaryote) (16), the functions of only a few hundred have been elucidated (2,3,7,8,17–21). In contrast, about two thirds of the annotated genes of Arabidopsis are very ancient (2), tracing back to a very early eukaryotic or a prokaryotic origin. In part because they have common motifs across species, these ancient genes are more likely to be at least partially characterized. A model of the transcriptome, which illustrates some of the nuances of gene prediction research, is shown in Figure 1.
Figure 1.

The dynamic transcriptome: Dark and annotated. Genes form, evolve, remodel, and recede in a continuum across evolutionary time. These dynamics result in a significant challenge to gene prediction. The expressed genome, i.e., the transcriptome, is comprised in part by annotated genes. However, the transcriptome also includes a vast, uncharacterized but expressed body of sequences that can be termed the ‘dark transcriptome’. Within this dark transcriptome is low abundance non-genic ‘noise’, as well as (as-yet-unannotated) protein-coding genes, pseudogenes, and non-coding genes (5,22–26). Protein coding orphan genes can be formed from expressed non-functional sequence, extant non-coding genes, and to a lesser extent from existing genes whose proteins have rapidly evolved beyond recognition (4,12,27). Over evolutionary time, some orphans will be retained in the species and become members of deeper phylostrata. In general, an older, more conserved gene is more likely to have a known molecular function. The proportion of functional genes that are unannotated in any given species is unclear; we posit that, depending on the species, a sizable proportion of orphan genes remain unannotated. This is because many are sparsely expressed (16,26,28,29), by definition none have homologs, many may have not yet evolved the canonical features by which a gene can be recognized ab initio, and there is a grey area in evolution between ‘noise’ and ‘gene’. Black arrows, evolutionary transitions; red font, protein-coding genes; green font, non-[protein]-coding genes; grey font, non-genic transcripts; blue oval, annotated protein-coding genes; green oval, annotated non-[protein]-coding genes.
Orphan genes as well as genes of deeper origin can be inferred by phylostratigraphy. This technique identifies genes based on the earliest common ancestor to which a protein homolog can be traced (1,30). Thus, phylostratigraphy classifies genes independently of their origin, including those that emerged de novo from non-genic sequence or within an existing gene, and those that evolved so rapidly that they cannot be recognized even in genomes of their closest (genus-level) relatives (1–3). In phylostatigraphic inference, a phylogenetic tree is assembled, individual representative of the tree are selected as target species,and the proteins of each target species are compared to those of the species of interest generally using BLAST. Challenges are that phylostratal inferences are sensitive to false positives, incorrect or ambiguous phylogentic trees, and the quality of protein prediction in each target species differ across time and by species. Historically, the process was time consuming and not easily reproduced; however, the advent of the R tool phylostratr (13) standardizes and automates the entire process, renders it reproducible, and provides detailed diagnostics at each step of the analysis.
Genes of ‘hybrid’ phylostrata, i.e. genes encoding proteins that contain regions of more than one phylostratal origin, are not uncommon; the presence of such genes been examined in relation to evolutionary mechanisms such as recombination (31) and elongation of 3’ UTR into protein coding (32). (Indeed, the later mechanism is an almost universal aspect of gene evolution- older genes become longer as they ‘mature’.) ‘Hybrid’ genes are typically classified according to the phylostratum of their most ancient domain (1,30), and that is the approach we use herein. This approach necessarily misses evolutionary nuances. However, even if the exact evolutionary back-trajectory of each gene is determined, rules for classification of such genes become arbitrary (13). Also, requiring the entire protein to be species-specific provides a stringent classification of orphans.
An inescapable diversion to the entire field of gene prediction is that it is predicated on the definition of a ‘gene’. The same massive high-throughput sequencing that has enabled the identification and functional characterizations of genes has raised havoc with the traditional definition of a ‘gene’ (3,5,26,33,34). There are a surprising diversity of definitions of ‘gene’ in current literature; an entire philosophy surrounds the concept of the nature of a gene (see Supplementary Table S1 and references therein).
To be consistent with current understanding of coding and non-coding genes, we will consider a gene as ‘an expressed, selected DNA sequence that confers a chemical, developmental, morphological or biochemical phenotype under one or more conditions’. This definition requires function, an expressed product (RNA or protein), and that the sequence has been selected for based on its function. It also emphasizes the context-dependency of some genes–a sequence might confer a function that is essential only under particular conditions (e.g. exposure to a draught or a novel virus), or contribute a minor survival advantage (35). An expressed sequence would be considered a protein-coding gene if it is translated and its protein product effects a phenotype under some condition(s), and hence under that condition has been under selection (inherited). Under conditions in which function and heritability are weak, sequence evidence of selective constraints could be muddied. Because many sequences are in transition to (or from) ‘gene-ness’, this murkiness is inescapable.
Perhaps most problematic for predicting genes is that genes themselves are on a continuum of ‘gene-ness’ (Figure 1). A given gene may emerge, remodel, and recede in a continuum across evolutionary time. some novel gene predictions might be ‘transcriptional noise’ (25)—fodder for de novo evolution of protein-coding genes; others might be ncRNAs with a non-translated or non-functional ORF (3,5,27,36). Other novel gene predictions may be on the continuum between orphan gene and non-genic transcript or the path from gene to pseudogene (3,5). And yet other novel gene predictions might be bonafide functional genes. No clear criteria articulate these products of the dark transcriptome. However, a first step is to predict them, and provide evidence-based metadata.
Gene prediction is a fundamental step in genome sequencing projects. However, no standard best practice has been established, predictions are often based on very limited RNA-Seq evidence, protocols are diverse, and pipelines are rarely well-documented (Supplementary Table S2). Prevailing methods often combine homology-based analysis, which compares a new genome to previously-identified genes from other species, and ab initio prediction of genes from the genome sequence (37). Each approach may have inherent bias against orphan genes (38). Homology-based methods assume that genes have identifiable orthologs in other species. Because orphan genes are species-specific, homology-based methods are not useful in predicting them. Ab initio-based predictors assume a pre-defined gene structure for all protein-coding genes of an organism (39). Nucleic acid signatures by which genes are predicted ab initio can include sequence motifs of untranslated regions, translation start sites, termination sites, and intron-exon boundaries. However, canonical sequence signatures may be less well-defined in young genes (29), in which case ab initio approaches might be less likely to detect them. The ability of ab initio approaches to detect genes of recent origin had not been directly evaluated.
A more straightforward evidence-based approach to identify genes is to directly align RNA-Seq data to genomes(25,27). This approach is key for predicting non-coding RNAs (40) and young genes(3). Over 50% of novel transcriptional active regions in rice were identified by transcriptome profiling early in 2010 (41). However, it has been less widely adopted to predict protein coding genes, in part because of the challenge of distinguishing ‘noise’ from true genetic signal (25,27). One approach to reduce ‘noise’ and other false positive predictions is to combine direct inference of genes with sequence similarity (25,39); this approach excludes orphan genes (42).
Here, we compare predictions of five prediction pipelines: MAKER (44), BRAKER (43), Direct Inference- an evidence-based pipeline we developed to predict genes by genome-guided alignment of RNA-Seq data, and two novel pipelines that combine ab initio and the Direct Inference approaches: MAKER-Inferred Directly (MIND), and BRAKER-Inferred Directly (BIND) (Figure 2).
Figure 2.

Schematic diagram of MIND and BIND gene prediction pipelines. BRAKER (43) (top left) and MAKER (44) (top right) predict genes based solely on ab initio machine learning, while Direct Inference (top middle) is an evidence-based method we developed to predict genes based on alignment of RNA-seq data to an assembled transcriptome. BRAKER (darker turquoise area) automates most of the pipeline software; Direct Inference (lime green area) automates the full pipeline; MAKER requires user-intervention for each step. BRAKER parameters for training are hard-coded within script and difficult to manipulate; Direct Inference and MAKER enable parameter management. Direct Inference also enables software to be exchanged (e.g. STAR for HiSat2) (45), MAKER uses settings files and can be user-edited to change parameters. BRAKER, MAKER, and Direct Inference prerequisites and features are shown in Supplementary Figure S7. The BIND and MIND pipelines (bottom) use Mikado (46) to combine predictions from the Direct Inference pipeline with predictions of either MAKER (MIND) or BRAKER (BIND). The full MIND and BIND pipelines with clearly documented, open source code are at https://github.com/eswlab/orphan-prediction.
We compare our gene predictions to those of the highly-curated ‘gold-standard’ gene predictions in Arabidopsis thaliana (Arabidopsis) and Saccharomyces cerevisia (yeast) and apply these methods to the most recent and hence less-curated NCBI predictions for a genome of the staple crop, Oryza sativa (rice)(47). Our results reveal that ab initio prediction pipelines can vastly under-detect younger genes. We show that diverse RNA-Seq evidence significantly improves gene prediction, in particular for younger genes. We demonstrate that the novel BIND and MIND pipelines improve the number and performance of predictions.
To enable Findable, Accessible, Interoperable and Reusable (FAIR)(48) pipelines for MIND and BIND, we implemented the Direct Inference pipeline in an automated, reproducible manner using the python-based RNA-Seq processing workflow, pyrpipe (45) such that it can be easily customized; we included singularity containers for MAKER and BRAKER.
MATERIALS AND METHODS
Software and data
Complete methods, all scripts used in this study, and all result files are documented, and a fully automated version of the direct inference pipeline implemented with pyrpipe and snakemake (49) are available at https://github.com/eswlab/orphan-prediction. pyrpipe source code is available at https://github.com/urmi-21/pyrpipe. The pyrpipe package can be installed from bioconda (https://anaconda.org/bioconda/pyrpipe) or PyPi (https://pypi.org/project/pyrpipe).
RNA-Seq, genome and protein input data
Arabidopsis thaliana Col0 genome (version Araport11) and reference genomes, GFF3 files, annotated transcript and protein sequences for (Araport11 version) were downloaded from The Arabidopsis Informatics Resource (TAIR) (50). Saccharomyces cerevisiae analysis used genome (version R64-1-1) and annotated genes (version R64-1-1). Oryza sativa analysis used genome (GCA_009797565.1) and annotated genes (GCA_009797565.1) downloaded from NCBI.
We identified RNA-Seq datasets of various sizes and complexities for Arabidopsis, yeast and rice (see Supplementary Table S3, and Table 1). The smallest datasets we term ‘Typical’; they are of sizes at the upper end of those often used in many gene prediction projects (The ‘Pool’ datasets are about 10-fold larger than the ‘Typical’ datasets, and are more diverse. The ‘Orphan-rich’ datasets for Arabidosis and yeast are designed to maximize orphan representation. In developing this dataset, we reasoned that selecting samples which contain a breadth of orphan gene transcripts would be important because many, though by no means all, orphans are highly expressed under only a very limited set of conditions, such as a particular stress or a unique developmental stage (2,3,16). The ‘Orphan-rich’ datasets for A.thaliana and yeast were comprised of 38 RNA-Seq samples that each contained >60% of all annotated orphan transcripts. We compiled and predicted genes using three additional intermediate-sized datasets for Arabidopsis, and tested a ‘ground truth’ dataset, composed of models of all Arabidopsis genes/proteins as annotated in Araport11 (‘Ara11’ dataset) (The Arabidopsis Informatics Resource (TAIR) (50)) (Supplementary Table S3-A). RNA-Seq data and respective sample metadata were downloaded from NCBI-SRA as raw reads using the SRA-toolkit (v2.8.0) (51) or automatically via pyrpipe (45).
Table 1.
Gene prediction scenario used for A. thaliana. The ‘Typical’ RNA-Seq dataset is of a size similar to or greater than that used in many gene prediction projects. The ‘Pooled’ dataset is more diverse and includes the Typical dataset. The ‘Orphan-rich’ dataset is designed to maximize orphan representation
| Extrinsic evidence | ||||
|---|---|---|---|---|
| Scenario (abbreviation) | Analysis pipeline | Description | # SRR samples | Datasize (GB) |
| Ara11 | NA (gold-standard annotations from TAIR (50)) | Araport11 version of A. thaliana annotationsa | NA | NA |
| Maker-Typical | MAKER | Maker predictions using Typical dataset (assembled transcripts and translated proteins) | 12 | 12.8 |
| Maker-Pool | Maker predictions using Pooled dataset (assembled transcripts and translated proteins) | 77 | 241.4 | |
| Maker-Orphan | Maker predictions using Orphan-rich dataset (assembled transcripts and translated proteins) | 38 | 595.1 | |
| Braker-Typical | BRAKER | Braker predictions using Typical RNA-Seq dataset (raw RNA-Seq) | 12 | 12.8 |
| Braker-Pool | Braker predictions using Pooled RNA-Seq dataset (raw RNA-Seq) | 77 | 241.4 | |
| Braker-Orphan | Braker predictions using Orphan-rich RNA-Seq dataset (raw RNA-Seq) | 38 | 595.1 | |
| DirInf-Typical | Direct Inference | Direct Inference using Typical RNA-Seq dataset (transcripts assembled using multiple assemblers) | 12 | 12.8 |
| DirInf-Pool | Direct Inference using Pooled RNA-Seq dataset (transcripts assembled using multiple assemblers) | 77 | 241.4 | |
| DirInf-Orphan | Direct Inference using Orphan-rich RNA-Seq dataset (transcripts assembled using multiple assemblers) | 38 | 595.1 | |
| BIND-Typical | Combined (BIND or MIND) | Direct-Inference using Typical RNA-Seq dataset plus BRAKER Typical predictions | 12 | 12.8 |
| MIND-Typical | Direct-inference using Typical RNA-Seq dataset plus Maker Typical predictions | 12 | 12.8 | |
| BIND-Pool | Direct-Inference using Pooled RNA-Seq dataset plus BRAKER Pooled predictions | 77 | 241.4 | |
| MIND-Pool | Direct-inference using Pooled RNA-Seq dataset plus Maker Pooled predictions | 77 | 241.4 | |
| BIND-Orphan | Direct-Inference using Orphan-rich dataset plus BRAKER Orphan-riched predictions | 38 | 595.1 | |
| MIND-Orphan | Direct-inference using Orphan-rich RNA-Seq dataset plus Maker Orphan-riched predictions | 38 | 595.1 | |
aWe use the word ‘annotation’ to describe genes that have been predicted experimentally and/or computationally, and to which, as possible, a putative function has been assigned.
Protein sequence used as evidence in MAKER (44) were generated in one of two ways: (i) for Arabidopsis, yeast, and rice, RNA-Seq reads were assembled using Trinity (v2.6.6) (52), followed by open reading frame (ORF) prediction and translation using orfipy (42) or TransDecoder (v3.0.1) (52). (ii) For Arabidopsis only, data was downloaded from Phytozome (53) as predicted protein sequences for nine species: Arabidopsis thaliana, (Glycine max, Populus trichocarpa, Arabidopsis lyrata, Conradina grandiflora, Setaria italica, Oryza sativa, Physcomitrella patens, Chlamydomonas reinhardtii and Brassica rapa).
Ab initio prediction of genes by BRAKER
RNA-Seq raw reads were mapped to the indexed reference genome using HiSat2 aligner (v2.1.0) (54) (default settings). The resultant SAM files were sorted and converted to BAM format. BAM files from each set of RNA-Seq samples were combined using SAMTools (v1.9) (55) and provided as training for the BRAKER (v2.1.2) pipeline (43), along with the unmasked genome. BRAKER is an automated pipeline to predict genes using GeneMark-ET (v4.33) (56) and AUGUSTUS (v3.3.1) (57). Briefly, GeneMark-ET is used to iteratively train AUGUSTUS, by generating initial gene predictions. GeneMark-ET-predicted genes are filtered and provided for AUGUSTUS training, followed by AUGUSTUS prediction, integrating the RNA-Seq information, to generate the final gene predictions (Figure 2, upper left panel).
BRAKER (v2.1.2) permits use of protein sequence training data to supplement the RNA-Seq training data. However, results with RNA-Seq plus protein evidence and with RNA-Seq evidence alone were virtually identical (the BRAKER User Guide also notes that it is not always best to use all evidence, https://github.com/Gaius-Augustus/BRAKER#running-braker). Thereafter, we used RNA-Seq evidence but not protein sequence for input training data to BRAKER.
Ab initio prediction of genes by MAKER
To implement the MAKER (v2.31.10) (44) pipeline, RNA-Seq data was assembled into a transcriptome using Trinity (v2.6.6) (52); this CDS evidence, was supplied along with the unmasked genomes (Araport11). Depending on the case study and species, either CDS-only; CDS and translated proteins; or CDS and Phytozome proteins were supplied (Supplementary Tables S3 and S4).
MAKER was run in two successive rounds with default settings (Figure 2). In round one, transcriptome and protein data were aligned to the reference genome to generate crude gene predictions. These crude predictions were then used for training SNAP (release 2006-07-28)(58) and AUGUSTUS (v3.2.1) ab initio gene predictors with default options. In round two, the Hidden Markov Models (HMM) for ab initio gene predictors, along with self-trained HMM of GeneMark-ES (v4.32) were used within MAKER to predict genes. MAKER finalizes the comprehensive sets of genes from all three predictors by ranking using Annotation Edit Distance (AED) (59); the highest-ranking genes were retained for the final set of predictions. MAKER’s default output includes key metadata about gene predictions (evidence scores supporting each prediction, name of the component(s) within MAKER that generated the prediction).
Evidence-based prediction of genes by Direct Inference
Raw RNA-Seq reads were assembled using three genome-guided transcriptome assemblers: viz. Class2, StringTie and CuffLinks (60–62).
The BAM file generated by mapping reads to the Araport11-annotated indexed genome using HiSat2 (v2.1.0) (54) was provided as training for the assemblers. The resultant assembled transcripts were used to predict ORFs using Transdecoder(52) or orfipy (42). We selected those complete ORFs over 150 nt. (Other user requirements might include: transcript length, number of exons, exon length, intron length, expression value or presence of UTRs.) These data files, along with splicing junctions identified from the alignments using Portcullis (63), were provided as input to Mikado (46). Mikado pick was run with 28 threads, ‘nosplit’ mode, report all orfs, and other default parameters.
We have engineered our Direct Inference pipeline in an automated, reproducible, scalable, and flexible manner by implementing the steps from downloading data through transcriptome assembly in the python library, pyrpipe (45). Direct Inference requirements are relatively simple and are detailed in Singh et al. (45).
Package managers, like Conda, or containers like Singularity or Docker, control for the execution environment and tool versions. We have implemented Direct Inference such that it can be used as is, or easily customized by the user. All requirements for the Direct Inference pipeline can be easily installed via Bioconda (64) and the provided Conda environment file. Conda was built to install and update python packages and their non-python dependencies; it can also package software in other languages. The Conda environment enables a Direct Inference user to add or substitute software facilely to evaluate efficacy for different use cases. We selected Conda because it not only controls for the execution environment and tool versions, but it also works across platforms, and makes available a wide variety of bioinformatic software packages via the Bioconda channel. The Bioconda channel is a community driven repository to provide up-to-date bioinformatics software.
We used the Snakemake workflow manager (49) to integrate the Direct Inference pyrpipe pipeline with the meta-assembly steps. The Direct Inference pipeline is available from https://github.com/eswlab/orphan-prediction/tree/master/evidence_based_pipeline.
Combined gene predictions by MIND and BIND
The total predictions from Direct Inference were integrated with BRAKER predictions (BIND) or with MAKER predictions (MIND), using Mikado (46) to merge predictions. The process and parameters of Mikado were identical to those for Direct Inference, except that the input files were changed to BRAKER (or MAKER) predictions and Direct Inference predictions. The merged predictions were finalized in GFF3 format.
Implementation, computer allocations and ease of use: BRAKER, MAKER and Direct Inference
Supplementary Figure S7 gives an overview of BRAKER, MAKER and Direct Inference prerequisites and features. For this comparison, we ran the three pipelines using the Arabidopsis genome with the Typical and Pooled RNA-Seq datasets as input (Table 1). Without a container, installation is far easier for BRAKER and Direct Inference than for MAKER.
To run Direct Inference with pyrpipe and Snakemake requires a single step - the input of SRA accession IDs; BRAKER requires a user to execute three single command-line operations. In contrast, MAKER is a hands-on program, in which a user must manually perform most steps, including transcript assembly, translation, evidence collection, training ab initio gene prediction programs (snap, AUGUSTUS, GeneMark). Users must also manually track outputs of all these steps and use them to run multiple iterations of MAKER.
Being able to change software and software parameters in prediction pipelines is important because genomes of different organisms have different characteristics, and different quantities of RNA-Seq data will be available depending on the species. Because of its modular construction in python, the Direct Inference pipeline can be modified with respect to the software program parameters. Furthermore, the software programs themselves can be exchanged or added. The RNA-Seq processing components implemented via pyrpipe are simple to modify. Snakemake provides multiple options for executing and scaling the pipeline on different HPC systems. Running MAKER entails a higher manual overhead, but this design aspect allows for the parameters to be changed by the user (65) However, MAKER is not designed to enable changes of software programs. Because BRAKER is hard coded, changing parameters is difficult. However, some software programs that meet BRAKER’s core script requirements can be swapped or added.
Implementing computational pipelines that are easily reproducible can be a challenging task (45,66). Because bioinformatics pipelines run a number of software programs that interact with the operating system libraries and with each other, controlling for the version, execution environment, and parameters of each program is essential for reproducible pipelines. We have implemented the Direct Inference pipeline keeping this principle in mind. All the required dependencies for the Direct Inference pipeline are automatically installed inside an isolated Conda environment. Centralized parameter management makes it easy to share and modify pipeline parameters. To maximize reproducibility for the MAKER and BRAKER pipelines, we have provided Singularity containers; these containers execute the tool in a virtual environment.
Gene prediction using large RNA-Seq datasets may best be done using multiple nodes. Because we implemented Direct Inference using the Snakemake workflow manager, it can be conveniently managed and scaled for multiple nodes. The BRAKER container is also optimized for use on multiple nodes. The MAKER container was not optimized for running on multiple nodes. For MAKER, the user would need to correctly configure the MPI program on both host and on the container- which would be quite challenging, and eliminate the benefits of having a container. Further, the configuration would likely require admin privileges on the HPC; general users rarely have such privileges.
If running the pipelines on a single node, relative efficiency is dependent on data size. When run with the large Pooled or Orphan datasets, BRAKER is more efficient than Direct Inference or MAKER in terms of disk usage, disk I/O (input/output). In contrast, when run with the ‘Typical’ dataset (12.8 GB), Direct Inference is more efficient than BRAKER or MAKER in terms of disk usage.
Comparing prediction scenarios and estimating performance of gene prediction methods
The results of each gene prediction pipeline scenario were compared to the existing predictions using Mikado Compare (46). Gene structure prediction predictions were provided to Mikado as GFF3 files. Similarity statistics are reported for each gene locus individually. Performance calculations were based on sensitivity (Sn), precision (Pr), and the combined performance metric, F1 score (67). Sn is a measure of the percent of predicted genes matched to all reference annotated genes (true positives) (Equation 1). Precision (Pr) is a measure of of the specificity of the predictions, that is percent of reference annotated genes (true positives) matching all predicted genes (Equation 2). The F1 score combines the sensitivity and precision as a measure of performance (Equation 3).
 |
(1) |
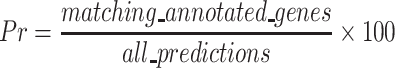 |
(2) |
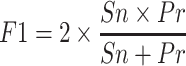 |
(3) |
Gene and transcriptome features for each prediction
The gene features were identified by Genome Annotation Generator (GAG) (68) based on the final GFF3 files (available at https://github.com/eswlab/orphan-prediction/tree/master/prediction_gff3). These include: number and length summary for genes, mRNAs, exons and introns; genome coverage by CDSs and genes; overlapping and contained genes in Supplementary Table S5. The coverage of transcribed sequence was calculated by bedtools2 (69) based on the predicted transcriptome.
RNA-Seq expression analysis
To investigate the expression level for A.thaliana predictions, we collected 5210 RNA-Seq samples from NCBI-SRA database. Criteria were: paired end reads (layout) and transcriptomic (source) and rna-seq (strategy) and illumina (platform) and organism (Arabidopsis thaliana). Salmon (70) (mapping-based mode) was used to quantify the expression of all genes predicted by Orphan-rich dataset and annotated in Araport11. We calculated the median of mean expression of all Araport11 genes across all samples; this median was 2.18 tpm. Then, those unannotated transcripts with expression of at least 2.18 tpm in at least 100 samples were used to plot Figure 5. The less-expressed transcripts were considered as low-expressed transcripts. The expression for all predicted transcripts was visualized by R and shown in Supplementary Figure S1.
Figure 5.

Upset plot of Arabidopsis protein-coding Araport11-annotated genes and novel genes for each prediction pipeline. The Orphan-rich dataset was used as input data. The resultant 41 078 non-redundant predictions were filtered for low expression across over 5000 diverse RNA-Seq samples before plotting (see Materials and Methods and Supplementary Figure S5). Top panel, percentage of genes, binned by five phylostrata. Middle panel, numbers of predictions; Bottom panel, non-redundant genes grouped by prediction method; Bottom right panel, total number of genes in Araport11 and predicted by each pipeline, colored by phylostrata. When the Orphan-rich dataset is used as evidence, about 89% of all annotated genes are predicted by any single method alone.
Ribo-Seq analysis
To investigate the translational activity of BIND predictions, we analysed 185 samples of Ribo-Seq data from 21 studies (Supplementary Table S8). Raw reads were downloaded from NCBI-SRA, and the SRA-toolkit (v2.8.0) was used to convert the raw reads to a FASTQ format. BBDuk was used to remove adapter sequences from the 3énd of reads, and rRNA reads were identified and removed using BBMap (71). The cleaned Ribo-Seq reads were aligned to the reference genome by STAR aligner (v2.5.3) (72). Ribosome-bound ORFs were detected and quantified by Ribotricer (73), which considers the periodicity of ORF profiles, using the recommended parameters for Arabidopsis. A gene with at least five codons with non-zero reads was considered to have translation signal in a Ribo-Seq sample (73).
Phylostratigraphy
Phylostratigraphic analysis based on the homology of predicted proteins to proteins in clades of increasing depth (age) was inferred using the R-platform phylostratr software (v0.2.0) (13). The focal species were set as ‘3702’ for A. thaliana, ‘4932’ for S. cerevisiae and ‘39947’ for O. sativa. With the exception that we incorporated additional user-specified target datasets for the three test species (See Supplementary Tables S7, S11 and S12), we used the default options in phylostratr. phylostratr automatically (i) creates a clade tree from the species represented in UniProt, based on the current NCBI tree of life; (ii) trims the clade tree, using an algorithm that maximizes the evolutionary diversity of species; (iii) creates a database of protein sequences from hundreds of species retrieved from Uniprot Proteome (74), which we supplemented by adding selected species of very high quality genomes (Supplementary Table S10); (iv) pairwise BLASTs the focal species proteome against the proteome of each species in the clade tree; (v) identifies the ‘best hits’ for each focal gene against each target species; (vi) phylostratr assigns each gene to the phylostratum associated with the deepest clade to which the gene has an inferred homolog. The genes present only in the focal species are inferred as orphan genes, and assigned to phylostratum ‘Arabidopsis thaliana’ (for A. thaliana), ‘Saccharomyces cerevisiae’ (for S. cerevisiae), and ‘Oryza sativa’ (for O. sativa). Phylostratal designations for TAIR-annotatated genes of Arabidopsis, as based on our previous analysis (13), are available in TAIR’s jbrowse ‘phylostratigraphy identification’ track (arabidopsis.org). We used the resultant phylostratal assignments to benchmark each gene prediction pipeline for its efficacy in predicting genes according to their inferred phylostrata.
RESULTS
MAKER is a popular software that combines a variety of ab initio approaches to predict genes (44). Our original impetus for this research was to assess MAKER’s ability to predict orphan genes. However, our initial analysis predicted only 11% of the orphan genes annotated in TAIR (Araport11 version) (50).
This poor performance of MAKER in identifying orphans and other young genes led us to consider other gene prediction scenarios. First, we reasoned that using more RNA-Seq evidence for training MAKER’s algorithms might improve predictions. However, there was little guidance in the literature on how to formulate the training datasets for ab initio gene predictions (43,44,75); many genome predictions use very limited datasets (See Supplementary Table S2). Second, we considered that other gene prediction programs might improve prediction outcomes.
Thus, we evaluated the efficacy of different gene prediction software and the extrinsic evidence of varied sizes on predictions. Specifically, raw reads from diverse RNA-Seq datasets were used by ab initio (MAKER and BRAKER), Direct Inference or the combined (MIND and BIND) approaches to predict genes.
We focused specifically on predicting protein-coding genes; the pipelines we applied—MAKER, BRAKER, and Direct Inference- each require the presence of an ORF to indicate the potential for translation. Protein evidence for each ORF would confirm its translation.
Arabidopsis gene predictions by MAKER
MAKER (44) is a widely-used gene prediction pipeline. Reads are aligned to the genome and assembled into transcripts before being provided to MAKER, which uses assembled transcripts of RNA-Seq datasets along with predicted protein sequence exclusively as training data.
We assembled seven combinations of transcript and protein evidence (Table 1 and Supplementary Tables S3-A and S4-A and provided these to MAKER as training data. Gene predictions corresponding to annotated genes were greater when RNA-Seq and protein data were both provided Supplementary Table S11-A. Regardless of input evidence, MAKER’s ability to predict genes (Figure 3 and Supplementary Table S11-A) was greatest for the genes of the oldest phylostratum (Cellular Organisms, PS1) and progressively decreased for younger phylostrata.
Figure 3.

Arabidopsis Araport11-annotated gene predictions, shown by phylostratal designation (see Supplementary Table S4 for predictions of all tested scenario). For each prediction scenario, the ability to predict genes was greater for the genes of oldest phylostratum (Cellular Organisms) and gradually decreased for the younger phylostrata. More annotated genes were predicted when pipelines were supplied with a more diverse dataset, an exception being BRAKER pipeline predictions. Overall, BIND with the Orphan-rich dataset as input predicted the most genes matching Araport11 annotations.
Predictions by MAKER were highly dependent on the training dataset supplied. Total predictions varied between 80% and 96% of the annotated genes. For example, 22 065 of the annotated genes were predicted when the Typical RNA-Seq dataset plus its predicted proteins were used as input, whereas 25 649 of the annotated genes of Arabidopsis were predicted with training data from the Pooled dataset plus its predicted proteins (Figure 3 and Supplementary Table S11-A).
This sensitivity to input evidence was much more pronounced for genes of the younger phylostrata. Twenty-one percent of the annotated orphan genes were predicted if MAKER was provided the Typical RNA-Seq dataset, versus 53% predicted from the Pooled dataset, and 68% for the Orphan-rich dataset (Figure 3 and Supplementary Table S11-A). Even when provided a ‘gold-standard gene set’ comprised of all annotated genes and their proteins (including all orphans) as training data, only 77% of the annotated orphans were predicted by MAKER (Supplementary Table S11-A).
The greater the diversity of the RNA-Seq evidence provided to MAKER, the more novel genes MAKER predicted that did not match any current gene annotation, for example, the Typical dataset predicted 4035 novel genes (1,183 of which were inferred by phylostratigraphy to be orphans), while the Orphan-rich dataset predicted 13 657 novel genes (7194 of which were inferred by phylostratigraphy to be orphans) (Supplementary Table S12-A).
Arabidopsis gene predictions by BRAKER
BRAKER differs from MAKER in that it uses RNA-Seq alignments to the genome for unsupervised training of GeneMark-ES/ET (Figure 2). Then, BRAKER selects a subset of the predicted protein coding genes to train AUGUSTUS and predict genes. BRAKER (43) predicted 96–97% of all annotated genes (Figure 3 and Supplementary Table S11-A). Regardless of the training dataset provided (Supplementary Table S4-A), gene predictions differed in quantity and performance by less than 2% (Supplementary Tables S7-A and S13-A).
BRAKER’s ability to predict genes was greatest for the genes of the oldest phylostratum (‘Cellular organisms’) and progressively decreased for younger phylostrata (Figure 3 and Supplementary Table S11-A). For example, when provided the Pooled dataset as training data, BRAKER predicted 100% of ancient genes that traced back to Cellular Organisms, but only 41% of the orphan genes (Figure 3); 8,204 of the genes predicted by BRAKER were not annotated in TAIR (1,498 of these encode orphans) (Supplementary Table S12-A).
Arabidopsis gene predictions by direct inference
Prediction by direct alignment of transcriptomic evidence to the genome is rarely used to annotate genes in newly sequenced genomes (e.g. Supplementary Table S2) (76). That said, the use of cDNA and EST evidence-based prediction was a mainstay for early predictions (50,77).
We considered that genes with non-canonical sequence features might be difficult to identify with machine learning algorithms. To mitigate this possibility, in addition to using transcript evidence indirectly as training data only, we developed an evidence-based approach that generates gene predictions, ‘Direct Inference’ (Figure 2; detailed in Methods). Briefly, the pipeline uses a genome-guided assembly, concatenates the transcripts, removes redundant transcripts, and determines ORF(s) in each inferred transcript. Because this approach directly relies exclusively on RNA-Seq alignments, only those RNAs that are expressed under the conditions sampled will be detected. Thus, we anticipated that providing RNA-Seq evidence collected from a wide variety of developmental stages, tissues and environmental conditions would be critical to maximize predictions when using Direct Inference. The overall performance of Direct Inference improved with larger dataset size Supplementary Table S13-A. Specifically, the diverse Orphan-rich dataset predicted nearly 96% of all annotated genes, and 63% of annotated orphans, while the Typical dataset predicted about 71% of all annotated genes, and 13% of the orphans (Figure 3, Supplementary Table S11-A).
BIND and MIND: gene predictions combining ab initio (BRAKER or MAKER) with direct inference
Because ab initio methods can identify very high proportions of the conserved genes, while direct inference can identify genes that are represented in the RNA-Seq data evidence without regard to canonical structure or homology to genes in other organisms, we evaluated whether combining both approaches would maximize gene predictions across phylostrata (Figure 2).
BIND increases the number of genes matching annotated genes compared to BRAKER or Direct Inference alone; MIND performed better than MAKER or Direct Inference alone (Figure 3 and Supplementary Table S11-A). Using the Orphan-rich dataset as input, BIND, and to a lesser extent, MIND, predicted more TAIR-annotated orphan genes than did either ab initio predictor alone. Base level F1 scores for overall prediction performance were comparable for BIND and MIND (∼75%). Regardless of the RNA-Seq data input, BIND predicted the most accurate representation of all genes, and young genes in particular (Figure 4, Supplementary Table S13-A). MIND predicted 18 114 genes that did not match any TAIR-annotated genes; BIND predicted 14 739 such genes.
Figure 4.
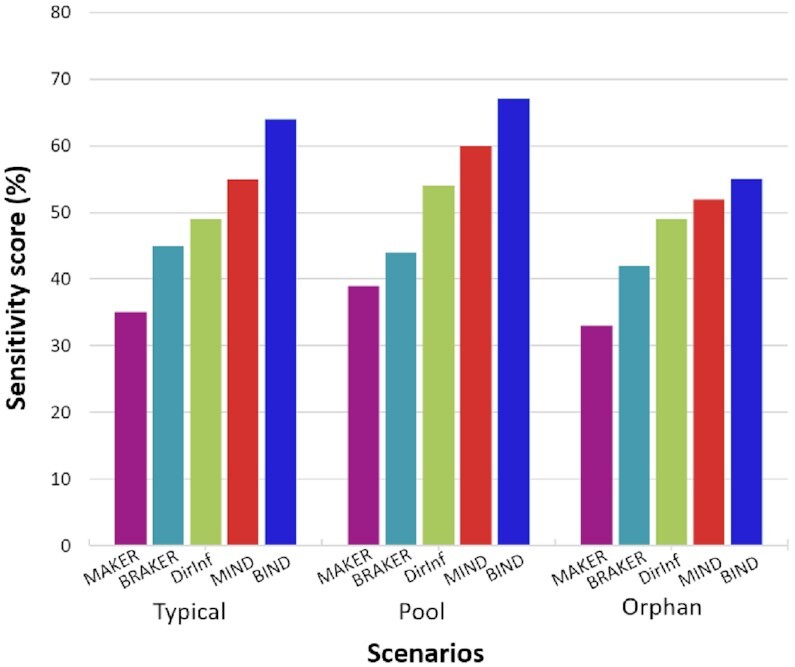
Sensitivity scores of gene predictions scenarios for A. thaliana at gene level. Results are compared to the annotations in TAIR (Araport11 version) (50). Regardless of the RNA-Seq dataset supplied, BIND gives the highest sensitivity score.
Multiple low-expression transcripts are predicted by the ab initio methods, MAKER or BRAKER (Supplementary Figure S1). To remove low expressed novel predictions from predictions using the Orphan-rich dataset, we combined all the annotated and novel transcripts predicted; then, we filtered those novel transcripts with low-expression (see methods). When using the Orphan-rich dataset as input, ∼89% of the transcripts annotated in Araport11 are predicted by each pipeline; 6% more of the Araport11-annotated transcripts are predicted by one or more pipelines (Figure 5). BIND and MIND also predicted more short genes than other methods (Supplementary Table S14).
Over 85% of annotated orphan genes were predicted by combining BIND and MIND, using the Orphan-rich dataset or the Pooled dataset. In contrast, using the Typical dataset, only 51% of annotated orphan genes were predicted by combining BIND and MIND, (Supplementary Figure S2).
We used ribosome footprinting data to assess the translation evidence for those genes predicted by BIND with the Orphan-rich dataset. (See Supplementary Table S6-A for all predictions) Predictions were filtered to remove those of low expression (see Materials and Methods). A limitation of this analysis was that the 185 Ribo-Seq samples that were publicly available did not represent diverse developmental and environmental conditions (Supplementary Table S6-B). Overall, 97% of the genes predicted by BIND had translation evidence. Translation evidence was greatest for proteins of the most ancient phylostratum (Cellular Organisms), decreasing for younger phylostrata (Figure 6). Ninety-eight percent of the predictions that matched annotated genes had translation evidence, while about 56% of the novel genes had translation evidence (Supplementary Table S6-A).
Figure 6.
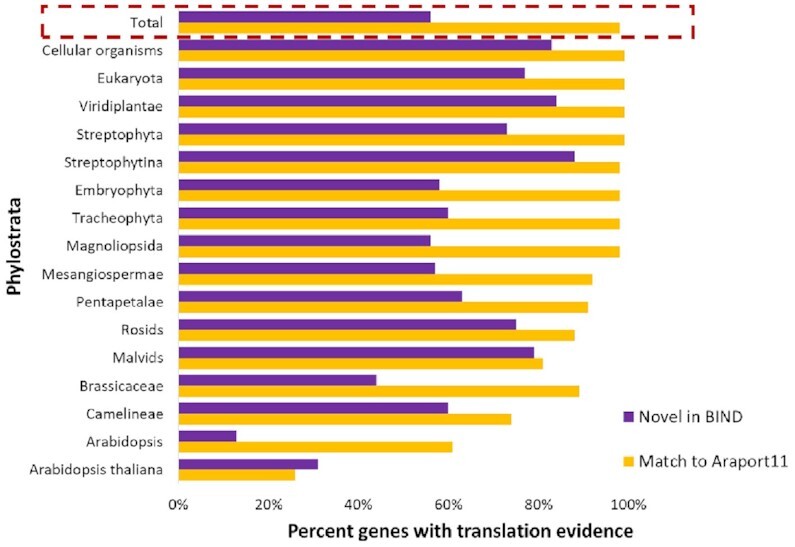
Translation evidence for highly expressed BIND-predicted genes of A. thaliana. Novel genes predicted by BIND were filtered for expression (see Supplementary Figure S6 for all predicted genes). Predicted proteins are binned by phylostratal designation. Translation signal was evaluated from the available ribosome profiling data. Translation evidence is consistent, but not sufficient, to indicate a protein-coding gene. Regardless of whether predictions matched to Araport11 annotations or were novel, younger genes had less translation evidence than ancient genes, as might be expected based on the sparser transcription patterns of younger genes.
Gene predictions for Saccharomyces cerevisiae
We evaluated the efficacy of the gene prediction pipelines on a disparate genome, that of the model system fungus, S. cerevisiae, using the highly curated Saccharomyces Genome Database (SGD) (77) gene annotations for benchmarking. As for Arabidopsis, yeast genes have been manually annotated through experimental evidence over tens of years. We assembled three datasets of varied sizes and compositions: a ‘Typical’ dataset; a ‘Pooled’ dataset, consisting of the ‘Typical’ dataset plus other RNA-Seq data from samples from varied conditions; and an ‘Orphan-rich’ dataset, comprising 38 RNA-Seq samples (selected from 3,457 high-quality samples (26)) that are highly represented in SGD-annotated orphan genes (Supplementary Tables S3-B and S4-B). We partitioned the SGD-annotated genes according to the phylostratigraphic inferences from a previous study (26).
Yeast genes were predicted using the MAKER, BRAKER, Direct Inference, MIND and BIND gene annotation pipelines, each in combination with ‘Typical’, Pooled’ and ‘Orphan-rich’ datasets as extrinsic training data, thus providing a total of 15 gene prediction scenarios.
MAKER’s ability to predict genes was greater for more ancient genes (e.g. Cellular Organisms and Fungi, PS1-4) than genes of younger phylostrata, (Saccharomyces, PS10; and orphans, PS11), regardless of the extrinsic evidence provided (Supplementary Table S11-B). MAKER predicted more SGD-annotated genes when the Pooled or Orphan-rich data was provided as input, matching 74% and 71% of SGD-annotated genes, respectively.
BRAKER’s ability to predict genes was greater for genes of more ancient phylostrata than those of younger phylostrata (Supplementary Table S11-B). BRAKER yielded predictions that differed in quantity and F1 score by less than one percent for each of the three datasets. BRAKER predicted 82% of all SGD-annotated genes (Supplementary Table S11-B, Figure S8) with an F1 score of 97% (Supplementary Table S13-B, base level). However, BRAKER did not predict a single orphan gene in yeast, even when supplied the Orphan-rich dataset.
Using the Orphan-rich dataset as input, the Direct-Inference pipeline predicted nearly 83% of all annotated genes and 13% of SGD-annotated orphans. In contrast, using the Typical dataset, Direct-Inference predicted only 33% of all SGD-annotated genes and 6% of the annotated orphan genes (Supplementary Table S11-B, Figure S8).
BIND and MIND, using the Orphan-rich dataset as input, predicted more SGD-annotated orphan genes than did either of the ab initio predictors or the Direct Inference pipeline alone (Supplementary Table S11-B, Figure S8). Similar as Arabidopsis, BIND gives the highest sensitivity score than other method (Supplementary Figure S10). Three quarters of the SGD-annotated orphan genes were missed even using the Orphan-rich RNA-Seq datasets as input. However, novel orphan genes were predicted using the Orphan-rich dataset; over 90% of these were predicted by Direct Inference (Supplementary Figure S3). BIND and MIND predicted 206 and 223 novel orphan genes, respectively.
Gene predictions for Oryza sativa
The monocot, rice, is a staple crop for over half the world’s population, with a complex genome where 35% of the sequence is made up of transposable elements (47). We predicted rice genes in Oryza sativa subsp. japonica cv. Kitaake used the MAKER, BRAKER, Direct Inference, BIND, and MIND pipelines to and compared these predictions to those of NCBI (GCA_009797565.1). The recent high-quality KitaakeX genome (78) was used by NCBI and by us. The NCBI in silico gene predictions were obtained via the standard NCBI ab initio and homology pipeline; it is note on that they were enriched with EST evidence, however, there is no indication of which EST evidence was used (Supplementary Table S2). japonica cv. Kitaake rice annotations have not undergone the manual community curation as have Arabidopsis and yeast, thus do not provide the same gold standard on which to base our interpretation of the efficacy of the methodologies. Oryza phylogenetic research (79,80) has predicted multiple orphan genes; however, to our knowledge, these gene predictions have not been integrated into the NCBI annotations. Thus, because the gene annotations we are using for rice have not yet been highly curated, they do not represent the same gold standard as do the Arabidopsis and yeast gene annotations. It is more likely that the annotations include many false-positive predictions and are missing multiple true genes.
We assembled two RNA-Seq datasets to input to the gene prediction pipelines. The first was a ‘Typical’ dataset; the second was a larger ‘Pooled’ dataset consisting of diverse tissues and stress conditions (Supplementary Tables S3-C and S4-C). Because the rice gene predictions have not yet been extensively curated manually, we did not assemble an ‘orphan-rich’ RNA-Seq dataset. We partitioned the NCBI-annotated genes, as well as the resultant predictions made by each of the pipelines, according to their inferred phylostrata (Supplementary Tables S7-C and S9).
Similar to the results with Arabidopsis and yeast, regardless of prediction scenario, the ability to predict genes was greatest for the genes of the most ancient phylostratum and gradually decreased for the younger phylostrata. As observed for Arabidopsis and yeast, more annotated genes were predicted when pipelines were supplied with a more diverse dataset, with the exception being the BRAKER pipeline predictions (Supplementary Table S11-C, Figure S9). Similar to Arabidopsis and yeast, MIND predicted more genes matching annotations than did the MAKER or Direct Inference methods alone.
Unlike Arabidopsis and yeast, BRAKER predicted more genes that match to the NCBI annotations compared to any other pipeline (Supplementary Table S11, Figure S9). However, 13 178 of the transcripts that BRAKER predicted contained incorrect fusions of splice junctions (Supplementary Table S5-C), that’s why BRAKER predict over 9000 novel orphan genes which were not in the annotation or predicted in any other method (Supplementary Figure S4). These 13,178 incorrect fusions were removed by BIND in the step combining BRAKER and Direct Inference predictions using Mikado. MAKER predicted 2302 incorrect fusions for the typical rice dataset, while NCBI annotations include 1374 incorrect fusions. MIND and BIND predicted the correct splicing. (In contrast, for Arabidopsis, no incorrect fusions were predicted by BRAKER, BIND or MIND, though MAKER predict several hundred; unsurprisingly, no pipeline tested predicted incorrect junctions for yeast (Supplementary Tables S5-A and S5-B)). About 28% of BRAKER-predicted rice genes (a number of which contain incorrect splice junctions) and 13% of BIND-predicted rice genes, are inferred by phylostratal analysis to be orphans; most were not annotated by NCBI. Sensitivity scores were under 13% for MAKER and BRAKER; DI, MIND and BIND were between 17 and 36% depending on the dataset analyzed (Supplementary Figure S11). BIND gives a somewhat lower sensitivity score than Direct Inference, however, BIND correctly predicts more genes.
To enable BIND and MIND in a best practices format, we have implemented all Direct Inference core RNA-Seq processing steps in python using pyrpipe (45) and the pipeline in Snakemake (49) (see methods for details), we provide MAKER and BRAKER in singularity containers, and we have developed full, documented MIND and BIND pipelines in a versatile reproducible open source framework.
DISCUSSION
Structural prediction of genes in a genome is critical to making genomics data useful to the research community. However, the multiple protocols used to annotate genes, some of which exclusively rely on ab initio predictions; the wide variation in amount of training data input; and the dearth of reproducible methods (Supplementary Table S2) do not make for best-practice. Also importantly, they are rarely set up to easily compare across gene prediction methods. For example, EuGene (81) gene prediction program can be configured to run either completely ab initio, completely homology-based or as a hybrid. It is typically run using a single configuration file, where all the settings and inputs are passed to the predictor. The manual suggests that the program is quite resource-heavy, requiring almost a week for 500Mb plant genome, or almost 2 weeks for 3Gbp plant genome on a cluster with 500 cores. Running it with custom datasets will most likely increase the run-time.
As another example, the NCBI Eukaryotic Genome Annotation Pipeline (82) offered by NCBI provides comprehensive annotation starting from fetching raw and curated data from public repositories, alignment of sequences, and the prediction of genes, to the submission of the accessioned annotation products to public databases. However, the components of this pipeline are tightly integrated into the NCBI infrastructure, and external testing is not possible. In order to predict genes using this pipeline, the user will need to submit the genome, track the progress on the website; then, the results are made available on their databases upon completion. Due to this limitation, extensive tests with varying datasets are not feasible.
Funannotate (83), a fungal genome annotation pipeline, is probably one the most user-friendly gene prediction programs. It uses similar components of the MAKER gene prediction pipeline and provides scripts that automate the training and predicting steps. It also integrates the function annotation and comparison utilities. Many of the default settings are preset to fungal gene prediction and users are expected to manually change them before running it on the target genome (eg: default intron size, default busco profile etc). We tested Funannotate on the ‘Typical’ dataset for Arabidopsis genome, it performed better in predictions than MAKER, however, it was not good as BRAKER, MIND or BIND.
Quality gene prediction is particularly important for expression studies; this is because standard practice is to align RNA-Seq reads directly to reference transcriptomes consisting exclusively of annotated genes, or to align transcripts to the reference genome but count only the annotated genes. Both practices completely miss the detection of any genes that have not been annotated.
Prediction of young genes is improved by a pipeline that combines ab initio/evidence-based methods and by supplying diverse RNA-Seq training data
By deploying the manually-curated, community-based gene annotations from the model species, Arabidopsis and yeast, as gold standards(50,77) as well as the more recently annotated genes of Kitaake rice, we have explicitly illustrated some of the challenges of annotating young genes. The efficacy of every gene prediction scenario we tested, regardless of the pipeline used for prediction, the amount or quality of evidence supplied, or the species annotated, was strongly dependent on the phylostratal origin of the gene. As reflected by their lower visibility to ab initio prediction, younger genes appear to generally have a less recognizable sequence signature than do their more ancient counterparts, which have undergone hundreds of millions of years of selection. The clear trend is that the younger a gene is, the less likely it is to be predicted. This finding supports previous speculation (79).
Ab initio pipelines infer genes by their structural motifs. In contrast, the Direct Inference pipeline uses the RNA-Seq evidence provided to directly infer genes. We show that combining ab initio predictions with evidence-based, direct inference of genes improves predictions compared to either method alone.
Our analyses reveal the extent to which selection of training data can significantly affect gene predictions by the MAKER, Direct Inference, MIND, and BIND pipelines. For these pipelines, extrinsic data from samples drawn from diverse environmental and developmental conditions improve the prediction of annotated genes. Although BRAKER is less affected by the quantity of training data, it has a ceiling on predictions. A possible explanation for this phenomenon is that, unlike MAKER, the extrinsic RNA-Seq evidence is filtered by BRAKER prior to making its final predictions. This filtering of the RNA-Seq evidence enables BRAKER to make reasonable predictions from even small RNA-Seq datasets, but also means it cannot take full advantage of the extra information offered by large, diverse RNA-Seq training datasets. The integration of Direct Inference predictions via BIND also removed most mis-spliced (artifactual) predictions made by BRAKER when annotating the transposon-rich rice genome (Supplementary Table S5). In the absence of extended diverse RNA-Seq data, BIND provides the best option for gene prediction.
If highly diverse RNA-Seq data is available, the BIND and MIND pipelines leverage this information. Including samples that often express high levels of young genes,such as reproductive tissues and stressed tissues (9,16,28,84–87), is particularly critical for prediction of young genes.
Challenges and limitations of gene prediction
Compelling evidence from bacteria, to yeast, to humans, indicates that many sequences that are not annotated as genes are transcribed and translated (3,22–27,88–90). This ‘pervasive transcription’ does not appear to occur randomly across genomes. Alignment to the genomes of all transcripts (with or without ORFs) from the RNA-Seq data summed from the diverse developmental, environmental conditions and genetic perturbations used in this study, indicates about 17% of the yeast genome and 24% of the Arabidopsis genome is not detectably transcribed. (Thirty-five percent of the rice genome was not detectably transcribed, but this result is based on more limited RNA-Seq evidence).
A major quandary in any gene prediction pipeline, and especially for evidence-based pipelines like Direct Inference (26,33,35,91), is to determine which of the many gene predictions to retain and which to filter out. Each scenario that we tested predicted thousands of genes that were not annotated. One approach to provide more evidence for each prediction of a protein coding gene is to obtain translation evidence (Ribo-Seq and proteomics). This approach is limited by the available translation data, which may be non-existant for newly-sequenced species. In this study, over half of the unannotated ancient genes predicted by BIND for Arabidopsis also have translational evidence– even though the samples represented in the available Ribo-Seq data were limited as to the diversity of conditions represented.
Indeed, the same massive high-throughput sequencing that has enabled the identification and functional characterizations of genes has raised havoc with the traditional definition of a ‘gene’ (3,5,26,33). Perhaps most problematic for predicting genes is that genes themselves are on a continuum of ‘gene-ness’ (Figure 1). A given gene may emerge, remodel, and recede in a continuum across evolutionary time.
As can be deduced from Figure 1, almost any of the methods that predicts genes from genomic and other omic data will include false positives. Some predictions might be ‘transcriptional noise’ (25)—fodder for de novo evolution of protein-coding genes; others might be ncRNAs with a non-translated or non-functional ORF (3,5,27,36). Other gene predictions may actually be transcripts that are on the continuum between orphan gene and non-genic transcript or the path from gene to pseudogene (3,5). And yet other novel gene predictions might be bonafide functional genes. No clear criteria articulate these products of the dark transcriptome. However, a first step is to predict them, and provide evidence-based metadata.
Most protein-coding orphans appear to have arisen de novo from non-protein coding sequence, although various scenarios of defunctionalization and refunctionalization of existing genes provide another origin (1,3,4,27,92). These youngest, most recently-formed protein-coding genes, encoding proteins with no amino acid sequence similarity to proteins of any other species, are among the least likely to have been functionally characterized (2). A few orphan genes will persist over vast evolutionary time- as newer, younger genes arise in the organism the former orphans will become more and more ancient. Such genes will trace to deeper evolutionary origins (e.g., in the extant species Arabidopsis a gene that arose as an orphan at the inception of it’s Magnoliophyta ancestor would be classified in the Magnoliophyta phylastratum). About two thirds of the annotated genes of Arabidopsis trace back to a very early eukaryotic or a prokaryotic origin (2). The most complex molecular process, catalysis appears predominantly restricted to genes of these ancient phylostrata(2).
Application of gene prediction
In practice (see Supplementary Table S2), gene prediction is often biased against the prediction of young protein-coding genes. prediction protocols that contribute to bias against young genes include: (i) using RNA-Seq evidence from only a few conditions, which may not detect sparsely-expressed genes; (ii) relying exclusively on ab initio predictions, which miss many annotated orphans and other young genes; (iii) filtering out gene predictions that have only one exon, thereby excluding the many orphan genes with a single exon; (iv) removing gene predictions that do not have homologs in other species, which eliminates all orphan genes; (v) removing predicted genes based on low overall expression or have many missing values, which eliminates the many orphan genes are expressed only under limited conditions. Thus, although these filtering strategies may minimize false positives, they also can exclude thousands of bonafide genes.
The challenge of confirming that young genes are annotated efficiently is not addressed by evaluating the extent that the prediction identifies Benchmarking Universal Single-Copy Orthologs (BUSCO, http://busco.ezlab.org), highly-conserved genes among the gene predictions. This method, though highly useful in determining how well a pipeline annotates more ancient genes, does not capture the efficacy of a pipeline in identifying orphans or other young genes. Recently, Scalizati et al (93) have developed a benchmarking approach that takes phylostrata into account; herein we provide a different approach to benchmarking that considers phylostrata, and enables customization of phylostrata to include line-specific genes.
There is no substitute for manual curation in providing gene annotations that are useful to the community. An inclusive approach to predicting and annotating genes that would benefit both curators and researchers would be to make accessible in the annotations the confirmed, curated genes along with predicted genes, as inferred based on expression and/or ab initio analysis, together with the available evidence for each. Thus, the signal of each predicted gene would be retained in the reference annotations. To minimize gene predictions that are actually ‘transcriptional noise’, filtering predictions based on transcript accumulation may provide a useful approach; however, applying a cutoff should take into consideration the RNA-Seq data that is available as well as the sparsity of expression of many orphan genes. Ultimately, providing broad, straightforward access to metadata on predicted genes will facilitate understanding of genome evolution and gene function.
The importance of retaining a broad view of gene expression is highlighted by the crucial functions that have been experimentally demonstrated for proteins encoded by both annotated and unannotated orphan genes. This is true particularly in the realm of orphan genes that confer resistance to abiotic and biotic stresses. Such genes may provide disruptive genetic elements that fundamentally reposition metabolic and regulatory networks (8,36,94,95). The potential for transcripts that encode orphan proteins to be beneficial or essential has been reinforced by findings from synthetic biology research. This growing body of research reveals that even randomly-generated or evolutionarily-selected peptides with no clear similarity to any known proteins are often able to bind small molecules, such as ATP and amino acids, in vivo (96). Furthermore, such ‘synthetic’ orphan genes can have beneficial consequences, including developmental, stress-resistance, and longevity phenotypes, when expressed in vivo (96,97). Thus, although a gene has only ‘recently’ been subjected to selection pressure, it may be important to the organism. If, as we advocate here, information on predicted non-canonical genes was easily accessible, experiments could be designed to prioritize these inferred genes for experimental study and to elucidate the potential roles of these transcripts (45,49,66) and to validate new pipelines by benchmarking against well-sequenced, well-annotated genomes. Furthermore, gene expression studies would include these predicted genes, and experimental biologists would gain a sense of how the genes might be acting.
FAIR-ness of gene prediction protocols
Best practice for gene prediction is to use pipelines that can be easily reproduced and have been validated in model species. Unfortunately, neither have been standard practice (eg., Supplementary Table S2).
Two key factors to advance the field of gene prediction are reproducibility and the ability to modify the pipeline and its parameters. Our aim is implementation of Best Practice, reproducible pipelines for the methods we have deployed and developed in this research. To enhance reproducibility of the MIND and BIND pipelines, the workflow utilizes a package manager (for Direct Inference) and singularity containers (for MAKER and BRAKER) to install and run the bioinformatics tools. By automating the Direct Inference pipeline using pyrpipe (45) and Snakemake (49), we provide an end-to-end prediction solution that requires minimal user intervention.This automation, combined with extensive step-by-step documentation, enable a researcher aiming to annotate a novel genome to apply the methods to her/his own dataset.
We have facilitated future research in gene prediction by making all pipelines, containers, scripts, benchmark data, output data, and extensive step-by-step documentation open source and available (https://github.com/eswlab/orphan-prediction). Researchers can easily add/swap in new software for Direct Inference, and to some extent for BRAKER, and can optimize parameters of the software modules for Direct Inference and MAKER. A new pipeline can be compared to existing pipelines using benchmark data for model organisms, such as the TAIR and SGD gene annotations and the RNA-Seq data we provide. Researchers can compare and contrast the prediction pipelines by using the post-prediction analysis tools provided, or other tools selected by the researcher.
CONCLUSION
We demonstrate that orphans and other young genes often elude prediction, and illustrate challenges and best practices in gene prediction. Our analyses showcase the importance of including diverse transcriptomic evidence and incorporating an evidence-based approach. BIND and MIND provide improved, user-friendly gene prediction, identifying sequences for further study and curation. In addition, the BIND and MIND platforms will facilitate future research on gene prediction approaches.
DATA AVAILABILITY
RNA-Seq used as training data input to ab initio predictors and for Direct Inference alignments are specified in Supplementary Tables S3 and S4 and were obtained from National Institutes of Health Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra. Gene predictions for each scenario and species are provided as GFF files at https://github.com/eswlab/orphan-prediction/tree/master/prediction_gff3. The full phylostratal designations and other metadata for annotated and predicted genes in Arabidopsis and rice are provided in Supplementary Tables S8 and S9. All Supplementary Tables are provided at https://github.com/eswlab/orphan-prediction/tree/master/SuppTables.
Overview with links to source code is available at https://orphan-prediction.readthedocs.io/en/latest/; recommended for analyses. All source code including for pipelines, downstream analyses, and creation of figures is available and documented at https://github.com/eswlab/orphan-prediction.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Nathan Weeks, USDA Agricultural Research Service, for development of singularity containers for MAKER and BRAKER. We thank Ethalinda Cannon, USDA Agricultural Research Service, for her prescient suggestions on gene metadata, Andrew J. Severin, Basil J. Nikolau, and Gene Prediction Summit members for valuable discussion. Linor Vaknin kindly provided expert design consultation on Figure 1. In addition to National Science Foundation Grant No. IOS 1546858 to E.S.W. This research was supported in part by the Center for Metabolic Biology at Iowa State University and by the United States. Department of Agriculture, Agricultural Research Service. USDA is an equal opportunity provider and Employer.
Contributor Information
Jing Li, Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014, USA; Center for Metabolic Biology, Iowa State University, Ames, IA 50014, USA; Genetics and Genomics Graduate Program, Iowa State University, Ames, IA 50014, USA.
Urminder Singh, Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014, USA; Center for Metabolic Biology, Iowa State University, Ames, IA 50014, USA; Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50014, USA.
Priyanka Bhandary, Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014, USA; Center for Metabolic Biology, Iowa State University, Ames, IA 50014, USA; Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50014, USA.
Jacqueline Campbell, Corn Insects and Crop Genetics Research Unit, US Department of Agriculture Agriculture Research Service, Ames, IA 50014, USA.
Zebulun Arendsee, Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014, USA; Center for Metabolic Biology, Iowa State University, Ames, IA 50014, USA; Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50014, USA.
Arun S Seetharam, Genome Informatics Facility, Iowa State University, Ames, IA 50014, USA.
Eve Syrkin Wurtele, Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50014, USA; Center for Metabolic Biology, Iowa State University, Ames, IA 50014, USA; Genetics and Genomics Graduate Program, Iowa State University, Ames, IA 50014, USA; Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA 50014, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Extreme Science and Engineering Discovery Environment (XSEDE) (National Science Foundation [ACI-1548562]) via Bridges HPC environment allocation TG-MCB190098; Iowa State University cyberinfrastructure; National Science Foundation [IOS 1546858 to E.S.W.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Tautz D., Domazet-Loso T.. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011; 12:692–702. [DOI] [PubMed] [Google Scholar]
- 2. Arendsee Z.W., Li L., Wurtele E.S.. Coming of age: orphan genes in plants. Trends Plant Sci. 2014; 19:698–708. [DOI] [PubMed] [Google Scholar]
- 3. Van Oss S.B., Carvunis A.-R.. De novo gene birth. PLoS Genet. 2019; 15:e1008160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vakirlis N., Carvunis A.-R., McLysaght A.. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. Elife. 2020; 9:e53500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh U., Wurtele E.S.. Genetic novelty: how new genes are born. Elife. 2020; 9:e55136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calvete J.J. Venomics: integrative venom proteomics and beyond. Biochem. J. 2017; 474:611–634. [DOI] [PubMed] [Google Scholar]
- 7. Qi M., Zheng W., Zhao X., Hohenstein J.D., Kandel Y., O’Conner S., Wang Y., Du C., Nettleton D., MacIntosh G.C.et al.. QQS orphan gene and its interactor NF-YC 4 reduce susceptibility to pathogens and pests. Plant. Biotechnol. J. 2019; 17:252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiao W., Liu H., Li Y., Li X., Xu C., Long M., Wang S.. A rice gene of de novo origin negatively regulates pathogen-induced defense response. PloS One. 2009; 4:e4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li G., Wu X., Hu Y., Muñoz-Amatriaín M., Luo J., Zhou W., Wang B., Wang Y., Wu X., Huang L.et al.. Orphan genes are involved in drought adaptations and ecoclimatic-oriented selections in domesticated cowpea. J. Exp. Bot. 2019; 70:3101–3110. [DOI] [PubMed] [Google Scholar]
- 10. Šestak M.S., Božičević V., Bakarić R., Dunjko V., Domazet-Lošo T.. Phylostratigraphic profiles reveal a deep evolutionary history of the vertebrate head sensory systems. Front. Zool. 2013; 10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lei L., Steffen J.G., Osborne E.J., Toomajian C.. Plant organ evolution revealed by phylotranscriptomics in Arabidopsis thaliana. Sci. Rep.-UK. 2017; 7:7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neme R., Tautz D.. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genomics. 2013; 14:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arendsee Z., Li J., Singh U., Seetharam A., Dorman K., Wurtele E.S.. phylostratr: a framework for phylostratigraphy. Bioinformatics. 2019; 35:3617–3627. [DOI] [PubMed] [Google Scholar]
- 14. Schmitz J.F., Ullrich K.K., Bornberg-Bauer E.. Incipient de novo genes can evolve from frozen accidents that escaped rapid transcript turnover. Nat. Ecol. Evol. 2018; 2:1626–1632. [DOI] [PubMed] [Google Scholar]
- 15. Mora C., Tittensor D.P., Adl S., Simpson A.G., Worm B.. How many species are there on Earth and in the ocean?. PLoS Biol. 2011; 9:e1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhandary P., Seetharam A.S., Arendsee Z.W., Hur M., Wurtele E.S.. Raising orphans from a metadata morass: a researcher’s guide to re-use of public’omics data. Plant Sci. 2018; 267:32–47. [DOI] [PubMed] [Google Scholar]
- 17. Li L., Foster C.M., Gan Q., Nettleton D., James M.G., Myers A.M., Wurtele E.S.. Identification of the novel protein QQS as a component of the starch metabolic network in Arabidopsis leaves. Plant J. 2009; 58:485–498. [DOI] [PubMed] [Google Scholar]
- 18. Li D., Dong Y., Jiang Y., Jiang H., Cai J., Wang W.. A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res. 2010; 20:408–420. [DOI] [PubMed] [Google Scholar]
- 19. Mayer M.G., Sommer R.J.. Nematode orphan genes are adopted by conserved regulatory networks and find a home in ecology. Worm. 2015; 4:e1082029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hahnel S., Parker-Manuel R., Dissous C., Cailliau K., Grevelding C.G.. First characterization of SmOPG1, a novel protein involved in gonad-associated processes in Schistosoma mansoni. Mol. Biochem. Parasitol. 2017; 213:22–25. [DOI] [PubMed] [Google Scholar]
- 21. Zhuang X., Cheng C.H.. ND6 gene ‘lost’ and found: evolution of mitochondrial gene rearrangement in Antarctic notothenioids. Mol. Biol. Evol. 2010; 27:1391–1403. [DOI] [PubMed] [Google Scholar]
- 22. Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L., et al.. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007; 316:1484–1488. [DOI] [PubMed] [Google Scholar]
- 23. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M.. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009; 457:1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009; 10:833–844. [DOI] [PubMed] [Google Scholar]
- 25. Pertea M., Shumate A., Pertea G., Varabyou A., Breitwieser F.P., Chang Y.-C., Madugundu A.K., Pandey A., Salzberg S.L.. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018; 19:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J., Singh U., Arendsee Z., Wurtele E.S.. Landscape of the dark transcriptome revealed through re-mining massive RNA-Seq data. Fronti. Genet. 2021; 12:1495–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ruiz-Orera J., Albà M.M.. Conserved regions in long non-coding RNAs contain abundant translation and protein–RNA interaction signatures. NAR Genom. Bioinformat. 2019; 1:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doughty T.W., Domenzain I., Millan-Oropeza A., Montini N., De Groot P.A., Pereira R., Nielsen J., Henry C., Daran J.-M.G., Siewers V.et al.. Stress-induced expression is enriched for evolutionarily young genes in diverse budding yeasts. Nat. Commun. 2020; 11:2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blevins W.R., Ruiz-Orera J., Messeguer X., Blasco-Moreno B., Villanueva-Cañas J.L., Espinar L., Díez J., Carey L.B., Albà M.M.. Uncovering de novo gene birth in yeast using deep transcriptomics. Nat. Commun. 2021; 12:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Domazet-Lošo T., Carvunis A.-R., Albà M., Šestak M.S., Bakarić R., Neme R., Tautz D.. No evidence for phylostratigraphic bias impacting inferences on patterns of gene emergence and evolution. Mole. Biol. Evol. 2017; 34:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Apic G., Gough J., Teichmann S.A.. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J. Mole. Biol. 2001; 310:311–325. [DOI] [PubMed] [Google Scholar]
- 32. Giacomelli M.G., Hancock A.S., Masel J.. The conversion of 3′ UTRs into coding regions. Mol. Biol. Evol. 2007; 24:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gerstein M.B., Bruce C., Rozowsky J.S., Zheng D., Du J., Korbel J.O., Emanuelsson O., Zhang Z.D., Weissman S., Snyder M.. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007; 17:669–681. [DOI] [PubMed] [Google Scholar]
- 34. Doolittle W.F. Is junk DNA bunk? A critique of ENCODE. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rancati G., Moffat J., Typas A., Pavelka N.. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018; 19:34. [DOI] [PubMed] [Google Scholar]
- 36. Andrews S.J., Rothnagel J.A.. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014; 15:193. [DOI] [PubMed] [Google Scholar]
- 37. Yandell M., Ence D.. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 2012; 13:329–342. [DOI] [PubMed] [Google Scholar]
- 38. Klasberg S., Bitard-Feildel T., Mallet L.. Computational identification of novel genes: current and future perspectives. Bioinformat. Biol. Insigh. 2016; 10:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keilwagen J., Hartung F., Paulini M., Twardziok S.O., Grau J.. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics. 2018; 19:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vivek A., Kumar S.. Computational methods for annotation of plant regulatory non-coding RNAs using RNA-seq. Brief. Bioinform. 2020; 22:bbaa322. [DOI] [PubMed] [Google Scholar]
- 41. Lu T., Lu G., Fan D., Zhu C., Li W., Zhao Q., Feng Q., Zhao Y., Guo Y., Li W.et al.. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010; 20:1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Singh U., Wurtele E.S.. orfipy: a fast and flexible tool for extracting ORFs. Bioinformatics. 2021; 37:3019–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoff K.J., Lange S., Lomsadze A., Borodovsky M., Stanke M.. BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2016; 32:767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holt C., Yandell M.. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011; 12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh U., Li J., Seetharam A., Wurtele E.S.. pyrpipe: a Python package for RNA-Seq workflows. NAR Genom. Bioinformat. 2021; 3:lqab049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Venturini L., Caim S., Kaithakottil G., Mapleson D.L., Swarbreck D.. Leveraging multiple transcriptome assembly methods for improved gene structure annotation. GigaScience. 2018; 7:giy093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sasaki T. The map-based sequence of the rice genome. Nature. 2005; 436:793–800. [DOI] [PubMed] [Google Scholar]
- 48. Wilkinson M.D., Dumontier M., Aalbersberg I.J., Appleton G., Axton M., Baak A., Blomberg N., Boiten J.-W., da Silva Santos L.B., Bourne P.E.et al.. The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data. 2016; 3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Köster J., Rahmann S.. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 50. Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., Huala E.. The Arabidopsis information resource: making and mining the ‘gold standard’ annotated reference plant genome. genesis. 2015; 53:474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leinonen R., Sugawara H., Shumway M.. The sequence read archive. Nucleic Acids Res. 2010; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haas B.J., Papanicolaou A., Yassour M., Grabherr M., Blood P.D., Bowden J., Couger M.B., Eccles D., Li B., Lieber M.et al.. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013; 8:1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N.et al.. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012; 40:D1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.et al.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lomsadze A., Ter-Hovhannisyan V., Chernoff Y.O., Borodovsky M.. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005; 33:6494–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stanke M., Diekhans M., Baertsch R., Haussler D.. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008; 24:637–644. [DOI] [PubMed] [Google Scholar]
- 58. Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004; 5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eilbeck K., Moore B., Holt C., Yandell M.. Quantitative measures for the management and comparison of annotated genomes. BMC Bioinformatics. 2009; 10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song L., Sabunciyan S., Florea L.. CLASS2: accurate and efficient splice variant annotation from RNA-seq reads. Nucleic Acids Res. 2016; 44:e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mapleson D., Venturini L., Kaithakottil G., Swarbreck D.. Efficient and accurate detection of splice junctions from RNA-seq with Portcullis. GigaScience. 2018; 7:giy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J.. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018; 15:475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang L., Lu Z., Van Buren P., Ware D.. SciApps: a cloud-based platform for reproducible bioinformatics workflows. Bioinformatics. 2018; 34:3917–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grüning B., Chilton J., Köster J., Dale R., Soranzo N., van den Beek M., Goecks J., Backofen R., Nekrutenko A., Taylor J.. Practical computational reproducibility in the life sciences. Cell Systems. 2018; 6:631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Powers D.M. Evaluation: from precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Tech. 2011; 2:37–63. [Google Scholar]
- 68. Geib S.M., Hall B., Derego T., Bremer F.T., Cannoles K., Sim S.B.. Genome Annotation Generator: a simple tool for generating and correcting WGS annotation tables for NCBI submission. GigaScience. 2018; 7:giy018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C.. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bushnell B. BBMap: A Fast, Accurate, Splice-Aware Aligner. 2014; Berkeley, CA (United States)Lawrence Berkeley National Lab. (LBNL)Technical Report LBNL-7065E. [Google Scholar]
- 72. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England). 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Choudhary S., Li W., Smith A.D.. Accurate detection of short and long active ORFs using Ribo-seq data. Bioinformatics. 2020; 36:2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mudge J.M., Harrow J.. The state of play in higher eukaryote gene annotation. Nat. Rev. Genet. 2016; 17:758–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pilkington S.M., Crowhurst R., Hilario E., Nardozza S., Fraser L., Peng Y., Gunaseelan K., Simpson R., Tahir J., Deroles S.C.et al.. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genomics. 2018; 19:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cherry J.M., Hong E.L., Amundsen C., Balakrishnan R., Binkley G., Chan E.T., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R.et al.. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012; 40:D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jain R., Jenkins J., Shu S., Chern M., Martin J.A., Copetti D., Duong P.Q., Pham N.T., Kudrna D.A., Talag J.et al.. Genome sequence of the model rice variety KitaakeX. BMC Genomics. 2019; 20:905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stein J.C., Yu Y., Copetti D., Zwickl D.J., Zhang L., Zhang C., Chougule K., Gao D., Iwata A., Goicoechea J.L.et al.. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018; 50:285. [DOI] [PubMed] [Google Scholar]
- 80. Guo W.-J., Li P., Ling J., Ye S.-P.. Significant comparative characteristics between orphan and nonorphan genes in the rice (Oryza sativa L.) genome. Comp. Funct. Genom. 2007; 2007:021676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sallet E., Gouzy J., Schiex T.. EuGene: an automated integrative gene finder for eukaryotes and prokaryotes. Gene Prediction. 2019; Springer; 97–120. [DOI] [PubMed] [Google Scholar]
- 82. Thibaud-Nissen F., DiCuccio M., Hlavina W., Kimchi A., Kitts P., Murphy T., Pruitt K., Souvorov A.. P8008 the NCBI eukaryotic genome annotation pipeline. J. Anim. Sci. 2016; 94:184–184. [Google Scholar]
- 83. Palmer J., Stajich J.. Funannotate: Funannotate v1.8.1: eukaryotic genome annotation. Zenodo. 2020; 10.5281/zenodo.4054262. [DOI] [Google Scholar]
- 84. Khraiwesh B., Qudeimat E., Thimma M., Chaiboonchoe A., Jijakli K., Alzahmi A., Arnoux M., Salehi-Ashtiani K.. Genome-wide expression analysis offers new insights into the origin and evolution of Physcomitrella patens stress response. Sci. Rep. 2015; 5:17434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Colbourne J.K., Pfrender M.E., Gilbert D., Thomas W.K., Tucker A., Oakley T.H., Tokishita S., Aerts A., Arnold G.J., Basu M.K.et al.. The ecoresponsive genome of Daphnia pulex. Science. 2011; 331:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhao L., Saelao P., Jones C.D., Begun D.J.. Origin and spread of de novo genes in Drosophila melanogaster populations. Science. 2014; 343:769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dion-Cote A.-M. A hotspot for new genes. eLife. 2019; 8:e50136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ji Z., Song R., Regev A., Struhl K.. Many lncRNAs, 5′ UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife. 2015; 4:e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Delcourt V., Staskevicius A., Salzet M., Fournier I., Roucou X.. Small proteins encoded by unannotated ORFs are rising stars of the proteome, confirming shortcomings in genome annotations and current vision of an mRNA. Proteomics. 2018; 18:1700058. [DOI] [PubMed] [Google Scholar]
- 90. Wang X., You X., Langer J.D., Hou J., Rupprecht F., Vlatkovic I., Quedenau C., Tushev G., Epstein I., Schaefke B.et al.. Full-length transcriptome reconstruction reveals a large diversity of RNA and protein isoforms in rat hippocampus. Nat. Commun. 2019; 10:5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pertea M., Shumate A., Pertea G., Varabyou A., Breitwieser F.P., Chang Y.-C., Madugundu A.K., Pandey A., Salzberg S.L.. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018; 19:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Arendsee Z., Li J., Singh U., Bhandary P., Seetharam A., Wurtele E.S.. Fagin: synteny-based phylostratigraphy and finer classification of young genes. BMC Bioinformatics. 2019; 20:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Scalzitti N., Jeannin-Girardon A., Collet P., Poch O., Thompson J.D.. A benchmark study of ab initio gene prediction methods in diverse eukaryotic organisms. BMC Genomics. 2020; 21:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ji Z., Song R., Regev A., Struhl K.. Many lncRNAs, 5’UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife. 2015; 4:e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Li L., Wurtele E.S.. The QQS orphan gene of Arabidopsis modulates carbon and nitrogen allocation in soybean. Plant. Biotechnol. J. 2015; 13:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bao Z., Clancy M.A., Carvalho R.F., Elliott K., Folta K.M.. Identification of novel growth regulators in plant populations expressing random peptides. Plant Physi. 2017; 175:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Neme R., Amador C., Yildirim B., McConnell E., Tautz D.. Random sequences are an abundant source of bioactive RNAs or peptides. Nat. Ecol. Evol. 2017; 1:0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq used as training data input to ab initio predictors and for Direct Inference alignments are specified in Supplementary Tables S3 and S4 and were obtained from National Institutes of Health Sequence Read Archive https://www.ncbi.nlm.nih.gov/sra. Gene predictions for each scenario and species are provided as GFF files at https://github.com/eswlab/orphan-prediction/tree/master/prediction_gff3. The full phylostratal designations and other metadata for annotated and predicted genes in Arabidopsis and rice are provided in Supplementary Tables S8 and S9. All Supplementary Tables are provided at https://github.com/eswlab/orphan-prediction/tree/master/SuppTables.
Overview with links to source code is available at https://orphan-prediction.readthedocs.io/en/latest/; recommended for analyses. All source code including for pipelines, downstream analyses, and creation of figures is available and documented at https://github.com/eswlab/orphan-prediction.