


Abstract
METTL8 has recently been identified as the methyltransferase catalyzing 3-methylcytidine biogenesis at position 32 (m3C32) of mitochondrial tRNAs. METTL8 also potentially participates in mRNA methylation and R-loop biogenesis. How METTL8 plays multiple roles in distinct cell compartments and catalyzes mitochondrial tRNA m3C formation remain unclear. Here, we discovered that alternative mRNA splicing generated several isoforms of METTL8. One isoform (METTL8-Iso1) was targeted to mitochondria via an N-terminal pre-sequence, while another one (METTL8-Iso4) mainly localized to the nucleolus. METTL8-Iso1-mediated m3C32 modification of human mitochondrial tRNAThr (hmtRNAThr) was not reliant on t6A modification at A37 (t6A37), while that of hmtRNASer(UCN) critically depended on i6A modification at A37 (i6A37). We clarified the hmtRNAThr substrate recognition mechanism, which was obviously different from that of hmtRNASer(UCN), in terms of requiring a G35 determinant. Moreover, SARS2 (mitochondrial seryl-tRNA synthetase) interacted with METTL8-Iso1 in an RNA-independent manner and modestly accelerated m3C modification activity. We further elucidated how nonsubstrate tRNAs in human mitochondria were efficiently discriminated by METTL8-Iso1. In summary, our results established the expression pattern of METTL8, clarified the molecular basis for m3C32 modification by METTL8-Iso1 and provided the rationale for the involvement of METTL8 in tRNA modification, mRNA methylation or R-loop biogenesis.
INTRODUCTION
Transfer RNA (tRNA) is one of the most abundant RNA species in the cell (1,2). It functions as an adaptor during the translation of the genetic code at the ribosome. tRNAs exhibit a cloverleaf secondary structure with several conserved domains, including amino acid accepting stem, D-stem and loop, anticodon stem and loop, variable stem or loop and TψC stem and loop.
After transcription by RNA polymerase, tRNA precursors usually undergo posttranscriptional processing, including numerous base or sugar modifications, by various tRNA modifying enzymes (2,3). In fact, tRNAs harbor the most extensive posttranscriptional modification among all cellular RNA species (1). Specifically, bases in the anticodon loop are the most heavily modified. These modifications play various critical roles in maintaining tRNA structure and stability, limiting or expanding codon-anticodon base pair capacity, restricting the occurrence of frameshifting and promoting tRNA aminoacylation (2–4).
3-Methylcytidine modification at position 32 (m3C32) is present only in eukaryotic cytoplasmic tRNAThr, tRNASer, tRNAArg (only in mammals) and mitochondrial tRNAThr and tRNASer species (5–9). The biological function of tRNA m3C32 modification in mRNA translation has not been fully established. Considering its location in the anticodon loop, we suggest that it likely participates in regulating loop conformation, codon-anticodon base pairing and/or the biogenesis of tRNA-derived small regulatory RNAs. Loss of cytoplasmic tRNASer m3C formation was found to influence pluripotency and cancer cell growth (10).
With respect to the biogenesis of tRNA m3C32 in budding yeast, Trm140 is responsible for m3C32 modification of both tRNAThr and tRNASer (5,6); however, two enzymes, Trm140 and Trm141, catalyze tRNAThr and tRNASer m3C32 modification, respectively, in fission yeast (11). The rationale for the evolution of the additional gene (Trm141) in fission yeast is unclear. In vivo genetic data have shown that N6-threonylcarbamoyladenosine (t6A) or N6-isopentenyladenosine (i6A) modification at position 37 (t6A37 or i6A37) of yeast tRNAThr or tRNASer functions as a positive determinant in catalyzing m3C32 biogenesis by Trm140 and Trm141 (11,12). In mice, Mettl2 has been identified as the m3C32 methyltransferase for tRNAThr and tRNAArg; Mettl6 has been identified as the methyltransferase for tRNASer, based on the observation that m3C32 formation on these specific tRNAs was impaired after deleting the corresponding genes. Interestingly, it has been shown that mRNA also undergoes m3C modification, which is mediated by Mettl8. Moreover, Mettl8 knockout has little effect on the m3C32 abundance of tRNAs, leading to the hypothesis that Mettl8 is not a tRNA but rather a mRNA m3C methyltransferase (13). A recent study reported that METTL8 is a potential regulator of R-loop stability in the ribosomal RNA (rRNA) gene locus (14). Indeed, another recent study reported that m3C abundance in mRNAs is lower than that in tRNAs (15). In human cells, two Mettl2 homologs, METTL2A and METTL2B, have further evolved, with amino acid differences at only six positions, for unclear reasons (16). Recently, we purified human METTL2A, METTL2B and METTL6 and established the m3C32 modification activities of all these enzymes. We found that t6A37 in human cytoplasmic tRNAThr is a critical determinant of m3C32 biogenesis by METTL2A/2B; however, t6A37 in human cytoplasmic tRNASer(GCU) plays little role in modification by METTL6, in assistance with cytoplasmic seryl-tRNA synthetase (SerRS, encoded by SARS1) (16). Very recently, we further determined the crystal structure of METTL6, defining its active site and substrate binding patterns (17).
Among the 22 human mitochondrial tRNAs (hmtRNAs), only tRNAThr (hmtRNAThr) and tRNASer(UCN) (hmtRNASer(UCN)) contain m3C32 modification (9). Meanwhile, hmtRNAThr and hmtRNASer(UCN) harbor a t6A37 modification, catalyzed by YRDC and OSGEPL1 (18,19), and a i6A37 modification mediated by TRIT1 (20,21), respectively. Of the four known m3C modifying enzymes (METTL2A, METTL2B, METTL6 and METTL8), METTL2A/2B and METTL6 are distributed in the cytoplasm but not the mitochondria, suggesting that they do not function as mitochondrial m3C32-modifying enzymes (16). Therefore, the enzyme responsible for mammalian tRNAThr and tRNASer(UCN) m3C32 biogenesis has long been a mystery. However, a recent mitochondrial proteome screening found that METTL8 is located in the mitochondrial fraction, eliciting the question of whether METTL8 is the missing mitochondrial tRNA m3C32 methyltransferase (22). Indeed, METTL8 has very recently been proven to be the mitochondrial tRNA m3C32 methyltransferase (23).
Despite being revealed as a multifunctional protein, however, the localization of METTL8 in mitochondria as a tRNA m3C32 modifying enzyme (23) seemingly contradicts the findings that Mettl8 potentially mediates mRNA m3C formation in mouse cells (13) and regulates R-loop biogenesis and stability in human cells (14), which require the presence of METTL8 in the cytoplasm and/or nucleolar fraction. To more clearly understand these seemingly inconsistent findings about the multiple roles of METTL8, we initially analyzed all potential mRNA and protein isoforms in detail and identified several protein variants that were generated via alternative splicing of METTL8 mRNA. The longest variant, METTL8-Iso1, harbors an N-terminal mitochondrial targeting sequence (MTS), which facilitates its import into mitochondria. We further precisely established the mature form of METTL8-Iso1 and reconstituted both hmtRNAThr and hmtRNASer(UCN) m3C32 modification activity of METTL8-Iso1. Our results showed that i6A37 of hmtRNASer(UCN) but not t6A37 of hmtRNAThr constituted the key determinant in modification and further established a tRNA recognition mechanism by METTL8-Iso1 using hmtRNAThr transcript. METTL8-Iso1 interacted with SARS2 (mitochondrial SerRS) but exhibited SARS2-independent activity.
In summary, our results significantly deepen our understanding of METTL8 gene expression and the mitochondrial tRNA m3C32 modification mechanism of METTL8-Iso1. In combination with our recent elucidation of the cytoplasmic tRNA m3C32 biogenesis mechanism involving METTL2A and METTL6 (16), these results collectively provide valuable insights into both the similarity and divergence between cytoplasmic and mitochondrial tRNA m3C32 modifications.
MATERIALS AND METHODS
Materials
S-Adenosylmethionine (SAM) was obtained from New England Biolabs (Ipswich, MA, USA). Protease inhibitor cocktail, horseradish peroxidase (HRP)-conjugated secondary antibodies, protein standards for gel filtration chromatography, anti-FLAG (F7425), anti-Myc (M4439) and anti-GAPDH (G8795) antibodies were purchased from Sigma (St. Louis, MO, USA). Anti-GFP (4B10) antibody was obtained from Cell Signaling Technology (Boston, MA, USA). Dynabeads Protein G, MitoTracker, RNase I (RNase) and restriction enzymes were obtained from Thermo Scientific (Waltham, MA, USA). Lipofectamine 8000 transfection reagent and 4’,6-diamidino-2-phenylindole (DAPI) were obtained from Beyotime Biotech (Shanghai, China). Polyvinylidene fluoride (PVDF) membranes were purchased from Merck (Darmstadt, Germany). Primers were synthesized by Biosune (Shanghai, China), and DNA sequencing was performed by Tsingke (Shanghai, China). PrimeScript RT Master Mix was obtained from Takara Bio (Kusatsu, Japan). Dimethylallyl diphosphate (DMAPP) was obtained from APExBIO Technology (Houston, TX, USA). The BCA Protein Assay Kit was obtained from TIANGEN Biotech (Beijing, China). [3H] SAM and [3H] DMAPP were obtained from Perkin Elmer, Inc. (Waltham, MA, USA). t6A (sc-286478) and m3C (HY-111645) standard samples for mass spectrometry were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and MedChemExpress (Monmouth Junction, NJ, USA), respectively.
Plasmid construction and gene expression
Genes encoding different METTL8 (UniProt No. Q9H825) isoforms were amplified from cDNA obtained by reverse transcription of total RNA from human embryonic kidney 293T (HEK293T) cells and inserted between the XhoI and EcoRI sites of pEGFP-N2 with a C-terminal EGFP tag. The initiating Met codon (ATG) of EGFP was mutated to the Ile codon (ATC) to prevent the potential generation of free EGFP. The gene encoding METTL8-Iso1 was also inserted between the NotI and XhoI sites of pCMV-3Tag-3A with C-terminal 3 × FLAG tags. Mature METTL8-Iso1 and METTL8-Iso4 were inserted between the NcoI and XhoI sites of pET28a with a C-terminal His6-tag for expression and purification in an E. coli expression system. A plasmid expressing mature SARS2 (Thr35-Ser518) was constructed and expressed as described in a previous report (19). Full-length SARS2 was inserted between the EcoRI and NotI sites of pcDNA3.1 with a C-terminal Myc-tag. The gene encoding Escherichia coli MiaA (EcMiaA) (UniProt No. P16384) was amplified by PCR from the E. coli genome and inserted between the NcoI and XhoI sites of pET28a with a C-terminal His6-tag. The plasmid expressing Saccharomyces cerevisiae Sua5 (ScSua5) (UniProt No. P32579) was constructed and expressed as reported previously (19,24). All primers used in gene cloning are listed in Supplementary Table 1.
For gene expression in E. coli, constructs were transformed into E. coli BL21 (DE3) cells. METTL8-Iso1, METTL8-Iso4 and EcMiaA gene expression was induced with a final concentration of 200 μM isopropyl β-d-1-thiogalactopyranoside (IPTG), and S. cerevisiae KEOPS (ScKEOPS) expression from pJ241-ScKEOPS (25) was induced with a final concentration of 500 μM IPTG when the initial cell culture reached an absorbance at 600 nm (A600) of 0.6–0.8. Transformants were cultured overnight at 18°C. Proteins were purified by Ni2+-NTA Superflow resin according to the manufacturer's protocol. The purified METTL8 proteins were concentrated and further purified on a HiTrap Q FF column and dialyzed against storage buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 2 mM DDT). The purified EcMiaA proteins were concentrated and dialyzed against storage buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 2 mM DDT). The purified ScKEOPS was concentrated and further purified via ion exchange chromatography (Mono Q column) and then dialyzed against storage buffer (25 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM β-ME). All purified proteins were mixed with an equal volume of glycerol and then stored at –20°C. The protein concentrations were determined by using a BCA Protein Assay Kit according to the manufacturer's instructions.
tRNA gene cloning and transcription
Plasmids for in vitro transcription of hmtRNAThr, hmtRNALys-Ki and hmtRNATrp were constructed as previously reported (19,24,26). hmtRNASer(UCN), hmtRNATyr, hmtRNALeu(UUR), hmtRNAPhe and hmtRNAMet were engineered into the pTrc99b plasmid. A hammerhead ribozyme was added between the T7 promoter and hmtRNAPhe gene to improve transcription efficiency (27). Note that a mutant of hmtRNALys, hmtRNALys-Ki (U50:A64) (with A50:U64 in wild-type hmtRNALys replaced with U50:A64), which has been shown to substitute well for wild-type hmtRNALys (28), was transcribed and used in this study to avoid misfolding. tRNA gene mutagenesis was performed according to the protocol provided with the KOD-plus mutagenesis kit. All tRNAs were obtained by in vitro tRNA transcription, which was performed as previously described (19,29). The primers used for template preparation are listed in Supplementary Table 1.
Determination of in vitro m3C and i6A modification
The m3C modification reaction was performed at 37 °C in a 40 μl reaction mixture containing 50 mM Tris–HCl (pH 7.5), 20 mM KCl, 10 mM MgCl2, 1 mM DTT, 1 mM spermidine, 100 μM [3H] SAM, 5 μM hmtRNAs (or variants) and 1 μM mature METTL8-Iso1 (or METTL8-Iso1 and SARS2 in different molar ratios). The i6A modification reaction was performed at 37 °C in a 10 μl reaction mixture containing 50 mM Tris-HCl (pH 7.5), 3.5 mM MgCl2, 1 mg/ml BSA, 5 mM β-ME, 30 μM [3H] DMAPP, 5–15 μM hmtRNAs (or variants) and 1 μM EcMiaA. Aliquots of the reaction solution were added to filter pads at various time points (for m3C modification) or at 15 min (for i6A modification) and quenched with cold 5% TCA. The pads were washed three times for 15 min each with cold 5% TCA and then three times for 10 min each with 100% ethanol. Finally, the pads were dried under a heat lamp, and the radioactivity of the precipitates was quantified using a scintillation counter (Beckman Coulter, Atlanta, GA, USA).
Reverse transcription and identification of METTL8 mRNAs
Total RNA was extracted from HEK293T cells using TRIzol reagent. cDNAs were reverse-transcribed using PrimeScript RT Master Mix according to the manufacturer's protocol. After cDNA synthesis, PCR was performed with different primers using 2 × Taq Master Mix according to the manufacturer's protocol. PCR products were separated via agarose gel electrophoresis, the relevant bands were excised, and the DNA was extracted with a HiPure Gel Pure Micro Kit and sequenced by Tsingke. The DNA sequencing results were then compared with various exon sequences to define alternative splicing patterns. The locations of the primers and exons are described in the Results section, and the primer sequences are listed in Supplementary Table 1.
Cell culture, transfection and coimmunoprecipitation (Co-IP)
HEK293T cells were cultured in DMEM supplemented with 10% FBS in a 37°C incubator with 5% CO2 at a confluence of 70% before transfection using Lipofectamine 8000 transfection reagent according to the manufacturer's protocol. For co-IP, at 24 h after transfection, cells were harvested, washed with ice-cold PBS three times, and lysed for 30 min at 4 °C with rotation in 1 ml of ice-cold lysis buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% NP-40 and 0.25% sodium deoxycholate) supplemented with a protease inhibitor cocktail. The supernatant was collected by centrifugation at 12,000 g for 20 min. Whole cell lysates were supplemented with anti-FLAG antibodies, incubated overnight with rotation, and then treated with Dynabeads protein G for an additional 4 h. The recovered immune complexes were washed three times with ice-cold PBS containing 0.05% Tween-20 (PBST). Protein was eluted from the beads in 2 × protein loading buffer comprising 100 mM Tris–HCl (pH 7.0), 4% SDS, 0.2% bromophenol blue, 20% glycerol and 200 mM DTT.
Fluorescence assays
After cell culture and transfection, the cells were incubated with MitoTracker at a 1:20,000 dilution for 30 min in a 37 °C incubator. After washing 3 times, the cells were fixed in 4% paraformaldehyde for 30 min, washed with PBS 3 times, and then permeabilized in 0.2% Triton X-100 and 1% BSA for 5 min on ice. After washing with PBS 3 times again, the fixed and permeabilized cells were blocked in PBS containing 4% BSA, washed with PBS 3 times, and then immunolabeled with the nuclear counterstain DAPI in PBS at a 1:1000 dilution for 5 min at RT. Fluorescent images were taken using a Leica TCS SP8 STED confocal microscope and analyzed.
Preparation of t6A- and i6A-modified tRNAs
For preparation of t6A-modified hmtRNAThr, the t6A modification reaction was performed at 37 °C for 1 h in a 200 μl reaction mixture containing 50 mM Tris–HCl (pH 8.0), 200 mM NaCl, 15 mM MgCl2, 4 mM ATP, 5 mM DTT, 5 mM MnCl2, 50 mM NaHCO3, 1 mM L-Thr, 10 μM hmtRNAThr and 5 μM yeast Sua5 (30) and KEOPS complex (31). For the preparation of i6A-modified hmtRNAs, an i6A modification reaction was performed at 37°C for 30 min in a 200 μl reaction mixture containing 50 mM Tris–HCl (pH 7.5), 3.5 mM MgCl2, 1 mg/ml BSA, 5 mM β-ME, 0.5 mM DMAPP, 20 μM hmtRNAs and 5 μM EcMiaA (32). The t6A- or i6A-modified tRNA was purified by phenol/chloroform and precipitated by EtOH with NaAc overnight at –20°C. The modified tRNA concentration was determined by denaturing urea–PAGE based on linear curves from tRNA transcript samples with known concentrations.
Ultra-performance liquid chromatography–mass spectrometry/mass spectrometry (UPLC–MS/MS) analysis
For UPLC–MS/MS, 500 ng of unmodified or m3C-, t6A- or i6A-modified tRNA sample was digested with 1 U of nuclease P1 and 0.5 U of benzonase nuclease in a 20 μl solution including the nuclease P1 buffer supplied with the kit and 20 mM NH4OAc (pH 5.2) at 37°C for 6 h. Then, 0.5 U of calf alkaline phosphatase (CAP) and 0.5 U of bacterial alkaline phosphatase (BAP) with CAP buffer were added to the reaction solution. After complete hydrolysis at 37°C overnight and filtration, 1 μl of product was subjected to UPLC–MS/MS. m3C and t6A were detected with Poroshell 120 PFP column (Agilent) and i6A was detected with Hypersil GOLD aQ column (Thermo Scientific).
Western blotting
After co-IP or protein purification, protein samples were separated on a 10% separating gel via SDS–PAGE. Western blotting was performed as described in a previous report (33).
Gel filtration analyses of mature METTL8-Iso1 and METTL8-Iso4
Purified mature METTL8-Iso1 and METTL8-Iso4 were analyzed by gel filtration chromatography on a Superdex 200 column using running solution (50 mM Tris–HCl (pH 8.0), 150 mM NaCl) at a rate of 0.5 ml/min. Protein standards were also loaded and eluted under the same conditions. A linear calibration curve was obtained by plotting the logarithms of the known molecular masses of the standards versus their elution times.
Insect cell culture, transfection, expression and determination of the N-terminal sequence of mature METTL8-Iso1
Sf9 and High Five cells were cultured in Sf-900 II SFM medium at 27°C. The pFastBac2-METTL8-Iso1 precursor was constructed with a C-terminal His6 tag for gene expression in insect cells by inserting the gene encoding the METTL8-Iso1 precursor between the BamHI and XhoI sites of pFastbac2. Recombinant baculoviruses were generated by transfecting the bacmid into Sf9 cells using Cellfectin II reagent according to the manufacturer's protocol. P1 virus stock was harvested after 6–7 days at 27 °C and used to infect Sf9 cells, which were then incubated for 5–6 days at 27°C to produce a higher-titer P2 virus stock. The same procedure was used to produce P3 virus stock, which was then stored at 4°C with protection from light. Gene expression was performed by infecting High Five cells in Sf-900 II SFM medium with P3 virus stock at a 1:200 ratio. Cells were harvested by centrifugation (500 g, 5 min) 66 h after infection, washed in PBS and stored at –80°C. The High Five cells were disrupted by sonication on ice, and the whole cell lysate was applied to Ni-NTA Superflow resin for mature METTL8-Iso1 purification according to the manufacturer's protocol. The eluted proteins were separated on a 10% separating gel via SDS–PAGE and transferred to PVDF membranes in CAPS transfer buffer (10 mM CAPS, 10% methanol), stained with Ponceau red for 25 min and washed with ultrapure water. The washing steps were repeated several times until the band of interest was clearly visible. The band was excised and sent to BiotechPack Scientific (Beijing, China) for identification of the N-terminal sequence through the Edman degradation method.
RESULTS
METTL8 encodes several isoforms via mRNA alternative splicing
Seven protein isoforms of the human METTL8 gene have been deposited in the NCBI database (Supplementary Figure 1). Primary sequence alignment of these isoforms, together with other m3C32 methyltransferases, including yeast Trm140 and Trm141 and mammalian METTL2A, METTL2B and METTL6, showed that isoforms 5–7 are truncated (Figure 1A). Comparison with our recently resolved crystal structure of METTL6 (PDB No. 7EZG) (17) reveals that isoforms 5–7 of METTL8 lack a complete SAM- and tRNA-binding domain. An absolutely conserved residue in METTL6, Thr217, has been shown to be critical for m3C32 methylation, likely due to impaired tRNA binding (17). Notably, its counterpart was absent in isoforms 5–7 but present in isoforms 1–4 of METTL8 (Thr337 in isoform 1) (Supplementary Figure 1). Therefore, isoforms 5–7 are unlikely to be m3C32-modifying enzymes. Based on these observations, we focus mainly on the presence or absence of isoforms 1–4 in this work. Notably, isoforms 3 and 4 (designated METTL8-Iso3 and -Iso4 hereafter) differ only in the presence of five additional amino acids (1MPRDH5) in the N-terminus of METTL8-Iso3. Compared with METTL8-Iso3 or -Iso4, isoforms 1 and 2 (METTL8-Iso1 and -Iso2) contain longer extensions of the N-terminus (Figure 1A, Supplementary Figure 1).
Figure 1.

Protein and mRNA analyses of METTL8. (A) Schematic showing NCBI-annotated METTL8 protein isoforms. Isoforms 5–7 lack a complete SAM and tRNA binding domain. Isoforms 3 and 4 differ only in the presence of five residues (MPRDH) in the former. The same difference exists between isoforms 5 and 6. A TLFI motif in isoforms 5–7, which was absent in isoforms 1–4 is also indicated. (B) Schematic showing NCBI-annotated METTL8 mRNA variants with each exon indicated. The black box represents intron 2. mRNAs 1v1-4 encoded the identical METTL8-Iso1 protein isoform, while mRNAs 2–7 encoded isoforms METTL8 Iso2-7. The pairing positions of primers used for mRNA identification are labeled at the top of the relevant exons. Bands that were successfully amplified using F1 and F2 forward primers and DNA sequenced are highlighted in red, while predicted bands not obtained by PCR are marked in black. (C, D) Agarose gel images of PCR products with different primer combinations as indicated. Each lane was confirmed by DNA sequencing. A nonspecific band not derived from the METTL8 gene is marked by an asterisk in (C). Three bold and underlined sizes represent DNA products that could be definitely assigned to specific mRNA isoforms in (B) and (C).
METTL8 contains 12 exons (sequence of each exon is shown in Supplementary Table 2). Retrieved mRNA sequences encoding these protein isoforms in the NCBI database showed that METTL8-Iso1 was potentially encoded by four mRNA sequences (mRNAs 1v1-4) produced by alternative splicing (Figure 1B). mRNA sequences (mRNAs 2–7), sequentially encoding METTL8-Iso2 to METTL8-Iso7, were also generated by mRNA alternative splicing (Figure 1B). To distinguish and confirm the sequences of these mRNAs, we designed three forward primers (F1 complementary to exon 1; F2 to exon 2 and F3 to exon 4) (Figure 1B). Using cDNA from HEK293T cells, in combination with a universal reverse primer (R1 complementary to exon 6), we amplified several fragments. In detail, using F1 and R1, a weak fragment was obtained, which was then sequenced and found to have 341 bp, derived from mRNA 3 and/or mRNA 6, encoding METTL8-Iso3 and/or METTL8-Iso6, respectively (Figure 1C). Two obvious fragments amplified with F2 and R1 were sequenced and found to be 505 and 350 bp, clearly showing the existence of mRNA 1v2 (encoding METTL8-Iso1) and mRNA 4 (encoding METTL8-Iso4). In addition, two weak but detectable fragments of 474 and 629 bp were also obtained and sequenced. The former was definitely derived from mRNA 7, while the latter fragment indicated the existence of mRNA 5 (encoding METTL8-Iso5) and/or mRNA 1v3 (still encoding METTL8-Iso1) (Figure 1C). A 411 bp fragment was obtained with F3 and R1 and sequenced, indicating that the cDNA template was qualified, but it could not be assigned to an mRNA isoform (Figure 1C). The presence of mRNAs 5–7 was further confirmed by amplification with forward primer F4 (complementary to exon 9) and reverse primer R2 (complementary to exon 11), which produced a 449 bp fragment showing that exon 10 was indeed skipped, in addition to a 556 bp fragment that included exon 10 (Figure 1D). Notably, amplification of DNA products of 505, 350 and 474 bp with F2 and R1 primers definitely showed the presence of mRNA 1v2, mRNA 4 and mRNA 7 in human cells.
In summary, the above results showed a complex mRNA alternative splicing landscape of the METTL8 gene in human HEK293T cells. In detail, at least two distinct mRNAs (including mRNAs 1v2 and 4) encode two full-length protein isoforms (METTL8-Iso1 and METTL8-Iso4). In addition, at least mRNA 7 encoded a truncated protein lacking a complete SAM- and tRNA-binding domain and was not further investigated in this study.
Cellular localization of METTL8 protein isoforms
As described above, METTL8-Iso3 and METTL8-Iso4 are nearly the same, with a difference of only five additional amino acids (1MPRDH5) in the N-terminus of METTL8-Iso3. Compared with METTL8-Iso4, METTL8-Iso1 has a longer N-terminal extension (Met1-His50, NTD50). MitoProt software (34) predicted no mitochondrial distribution of either METTL8-Iso3 or METTL8-Iso4; however, METTL8-Iso1 exhibited a higher probability (with a score of 0.8052) of import into mitochondria, suggesting that it is a mitochondrial protein, in accordance with a recent proteome analysis reporting mitochondrial localization of METTL8 in human mitochondria (22).
The open reading frames (ORFs) encoding METTL8-Iso1, -Iso3 and -Iso4 could all be readily amplified from HEK293T cDNA. Three ORFs were individually cloned into pEGFP-N2 to express C-terminal EGFP-fusion proteins. Fluorescence analysis showed that the main fractions of both METTL8-Iso3 and METTL8-Iso4 were located in the nucleolus, which was confirmed by their colocalization with fibrillarin (FBL), a key small nucleolar protein involved in pre-rRNA processing (Supplementary Figure 2A). However, METTL8-Iso1 was obviously distributed in the mitochondrial fraction, as stained by MitoTracker (Figure 2). The mitochondrial localization of METTL8-Iso1 was also confirmed by immunostaining of a C-terminal FLAG-tagged METTL8-Iso1 (METTL8-Iso1-FLAG) using anti-FLAG antibodies (Supplementary Figure 2B). Furthermore, the N-terminal extension of METTL8-Iso1, NTD50, was cloned upstream of EGFP. Consistently, the overexpressed NTD50-EGFP was exclusively targeted to mitochondria, suggesting a high efficiency of NTD50 in promoting the import of a foreign protein into the mitochondria (Figure 2).
Figure 2.
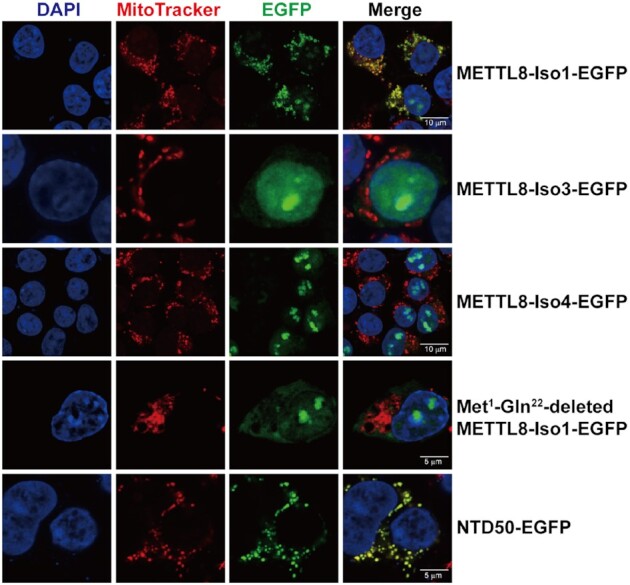
Cellular localization of various METTL8 protein isoforms. Different METTL8 protein isoforms and NTD50 were fused with a C-terminal EGFP and expressed, and the cellular distribution of individual proteins was observed. The nuclei and mitochondria were stained with DAPI and MitoTracker, respectively.
In summary, the above data clearly showed that two METTL8 isoforms (METTL8-Iso3 and METTL8-Iso4) are mainly located in the nucleolar fraction, while METTL8-Iso1 is present in the mitochondria, indicating that METTL8-Iso1 is an m3C methyltransferase in this organelle.
Purification of the mitochondrial form of METTL8-Iso1
To reconstitute potential mitochondrial tRNA m3C32 modification, it was necessary to purify METTL8-Iso1. The MitoProt database predicted that the cleavage site of METTL8-Iso1 after import into the mitochondrial matrix was between Phe43 and Glu44. However, we observed frequent inaccuracy in locating precise cleavage sites during studies of human mitochondrial aminoacyl-tRNA synthetases (such as LARS2, TARS2 and AARS2) (29,35–37) and tRNA modifying enzymes (such as GTPBP3) (38). Instead, we found that N-terminal protein sequencing by automatic Edman degradation after gene expression and purification of the mitochondrial proteins in a Bac-to-Bac baculovirus expression system is an excellent method for establishing the exact MTS (35,38). To this end, the gene encoding the METTL8-Iso1 precursor was expressed with a C-terminal His6 tag in the baculovirus expression system and subsequently purified by Ni-NTA affinity chromatography (Supplementary Figure 3). The N-terminus of purified METTL8-Iso1 was determined to be SGYHPV, clearly establishing that Met1-Gln22 is the MTS of METTL8-Iso1, in contrast to the predicted peptide (Met1-Phe43) (Supplementary Figure 1). To validate the mitochondrial targeting capacity of Met1-Gln22, a gene encoding mature METTL8-Iso1 (with Met1-Gln22 deleted) and a C-terminal EGFP fusion was overexpressed in HEK293T cells. Consistently, the EGFP-fused Met1-Gln22-truncated METTL8-Iso1 was distributed in both the nucleus and cytoplasm but no longer in the mitochondria (Figure 2), showing loss of mitochondrial localization of Met1-Gln22-truncated METTL8-Iso1 in the absence of the MTS.
Then, the gene encoding mature METTL8-Iso1 (with N-terminal Met1-Gln22 deleted) was expressed in E. coli and purified (Figure 3A). Both METTL2A and METTL6 are shown to be monomers in solution. To understand the quaternary structure of mature METTL8-Iso1, the protein was subjected to gel filtration analysis with Superdex S200 (Figure 3B). The results unexpectedly showed that METTL8-Iso1 eluted in a single peak with an elution volume between those of apoferritin (443 kDa) and yeast alcohol dehydrogenase (150 kDa). Based on the elution volume of the four protein standards, the calculated molecular mass was 346.6 kDa (Figure 3C). The theoretical molecular mass of purified METTL8-Iso1 with a C-terminal His6 tag is 45.3 kDa; these results suggested that mature METTL8-Iso1 exists as a polymer (likely octamer) in solution. We further purified METTL8-Iso4 from E. coli (Figure 3A), and the same analysis revealed that the molecular mass of purified METTL8-Iso4 was 60.47 kDa (Figure 3C), suggesting that METTL8-Iso4 (theoretical molecular mass 42.1 kDa) is possibly a monomer in solution. The clear difference between the quaternary structures of mature METTL8-Iso1 and METTL8-Iso4 is unanticipated considering that the former is only 28 aa longer than the latter at the N-terminus (Supplementary Figure 1). The reason for this difference is unclear and needs to be studied further.
Figure 3.

METTL8-Iso1 forms polymers. (A) SDS–PAGE analyses of purified recombinant mature METTL8-Iso1 and METTL8-Iso4 from the E. coli expression system. (B) Gel filtration analysis of purified mature METTL8-Iso1. (C) Determination of the molecular mass of mature METTL8-Iso1 and METTL8-Iso4 based on the elution volumes of four protein standards, namely, apoferritin (443 kDa), yeast alcohol dehydrogenase (150 kDa), bovine serum albumin (66 kDa) and carbonic anhydrase (29 kDa). (D) The METTL8-Iso1-FLAG and METTL8-Iso1-EGFP genes were coexpressed in HEK293T cells. METTL8-Iso1-EGFP was precipitated by METTL8-Iso1-FLAG in a co-IP assay.
To further confirm that METTL8-Iso1 forms a polymer in vivo, genes encoding METTL8-Iso1-FLAG and METTL8-Iso1-EGFP were coexpressed in HEK293T cells. Coimmunoprecipitation (co-IP) analysis using anti-FLAG antibodies showed that METTL8-Iso1-EGFP indeed coprecipitated with METTL8-Iso1-FLAG (Figure 3D), suggesting that METTL8-Iso1 forms polymers in vivo.
m3C32 biogenesis by METTL8-Iso1 is not reliant on t6A37 modification
After obtaining purified METTL8-Iso1, we subsequently explored whether METTL8-Iso1 is able to modify the hmtRNAThr transcript or requires a preformed t6A moiety as a determinant, as revealed by the modification of cytoplasmic tRNAThr by METTL2A (16). Indeed, METTL8-Iso1 obviously generated m3C modification in the hmtRNAThr transcript (Figure 4A). To understand whether position 32 was modified, we mutated C32 to three other nucleotides, generating the hmtRNAThr-C32A, -C32U and -C32G mutants (Figure 4B). The three mutants were unable to be modified by METTL8-Iso1, suggesting that position 32 was modified. The modified hmtRNAThr transcript was further hydrolyzed to mononucleosides and analyzed by UPLC–MS/MS, confirming the formation of m3C modification (Figure 4C). These data showed that METTL8-Iso1 indeed modified the hmtRNAThr transcript with m3C at position 32.
Figure 4.
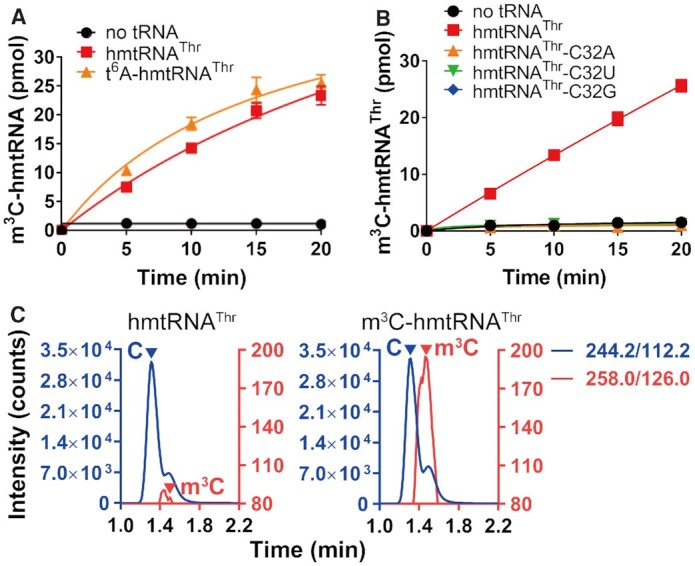
Reconstitution of m3C32 modification activity of METTL8-Iso1 for hmtRNAThr. Time-course curves of the m3C32 modification of hmtRNAThr and t6A-hmtRNAThr (A) or hmtRNAThr, hmtRNAThr-C32A, -C32U and -C32G (B) by mature METTL8-Iso1. (C) UPLC–MS/MS analysis of the digested product of METTL8-Iso1-modified hmtRNAThr. m/z value 244.2 to 112.2 (C) and m/z value 258.0 to 126.0 (m3C) are shown on the right. Controls in (A) and (B) (no tRNA) represent assays without tRNA added. The data represent the averages of three independent experiments and the corresponding standard error of the mean (SEM) in (A) and (B).
We further prepared t6A37-modified hmtRNAThr (t6A-hmtRNAThr) using yeast Sua5 and KEOPS complex (39), which was confirmed by UPLC–MS/MS analysis after digestion of t6A-hmtRNAThr into mononucleosides (Supplementary Figure 4). We found that METTL8-Iso1 introduced comparable amounts of the m3C moiety into t6A-hmtRNAThr and the hmtRNAThr transcript (Figure 4A). These data clearly showed that t6A formation at position 37 has little stimulatory effect on m3C32 biogenesis by METTL8-Iso1 in vitro, which is in sharp contrast to its role in modification by METTL2A (16).
Mechanism of hmtRNAThr recognition by METTL8-Iso1
After establishing that the hmtRNAThr transcript is a qualified METTL8-Iso1 substrate, we further studied how it is selected among the mitochondrial tRNAs. Our previous studies on the METTL8 homologs METTL2A and METTL6 revealed that the anticodon loop is a key element for m3C32 formation by both enzymes (16). Based on these results, we targeted the anticodon loop of hmtRNAThr. We mutated each nucleotide in the anticodon loop (U33, U34, G35, U36, A37 and A38) into three other nucleotides (Figure 5A). hmtRNAThr-U33G was not modified by METTL8-Iso1, while hmtRNAThr-U33A and hmtRNAThr-U33C were modified with comparable efficiency to wild-type hmtRNAThr (Figure 5B). For position 34, hmtRNAThr-U34A and hmtRNAThr-U34G were not modified, while the modification efficiency of hmtRNAThr-U34C was even higher than that of wild-type hmtRNAThr (Figure 5C). Strikingly, none of the G35 mutants were modified. Position 35 has no interaction with other nucleotides in tRNAs. Mutation at this residue has little possibility to influence tRNA folding and structure. Therefore, no modification of all G35 mutants clearly revealed that G35 functions as a positive determinant of m3C32 biogenesis by METTL8-Iso1 (Figure 5D). For position 36, only hmtRNAThr-U36A was obviously modified, but with a reduced (approximately half) efficiency (Figure 5E). The results from mutants of A37 showed that hmtRNAThr-A37G was only slightly modified, while hmtRNAThr-A37U and hmtRNAThr-A37C displayed higher or comparable modification efficiency than wild-type hmtRNAThr (Figure 5F). Finally, hmtRNAThr-A38G was not modified, while hmtRNAThr-A38U and hmtRNAThr-A38C exhibited reduced modification efficiency (Figure 5G).
Figure 5.

G35 is a positive determinant of m3C32 biogenesis by METTL8-Iso1. (A) Point mutations targeting the anticodon loop of hmtRNAThr. Time-course curves of the m3C32 modification of hmtRNAThr, hmtRNAThr-U33A, -U33C and -U33G (B); hmtRNAThr, hmtRNAThr-U34A, -U34C and -U34G (C); hmtRNAThr, hmtRNAThr-G35A, -G35C and -G35U (D); hmtRNAThr, hmtRNAThr-U36A, -U36C and -U36G (E); hmtRNAThr, hmtRNAThr-A37U, -A37C and -A37G (F); or hmtRNAThr, hmtRNAThr-A38U, -A38C and -A38G (G). Negative controls (no tRNA) represent assays without tRNA added. Positive controls (hmtRNAThr) represent the modification of wild-type hmtRNAThr. The data in each graph represent averages of three independent experiments and the corresponding SEM.
Altogether, the observation of a lack of reliance on t6A37 enables us to establish a full understanding of the roles of individual nucleotides in the anticodon loop in m3C32 biogenesis of mitochondrial tRNA. Our results showed that G35 is a critical determinant, and the sequence requirement of the anticodon loop is determined as follows: C32-(U/A/C)33-(C > U)34-G35-(U > A)36-(U > A = C > G)37-(A > U > C)38.
The newly evolved A29-C41 mismatch fine-tunes the m3C32 level of hmtRNAThr
An evolutionary analysis in our previous study identified that an A29-C41 mismatch was newly evolved in the mitochondrial tRNAThr of human and gorilla but not those of other mammals, for unknown reasons (24). Based on the key role of the anticodon stem in m3C32 biogenesis by METTL2A, we wondered whether an A29-C41 mismatch in hmtRNAThr could influence m3C32 modification by METTL8-Iso1. We mutated either A29 to G29 or C41 to U41 (Supplementary Figure 5A). The resultant hmtRNAThr-A29G and hmtRNAThr-C41U have an anticodon stem with full Watson–Crick base pairing (G29–C41 or A29–U41). Tests of modification by METTL8-Iso1 indeed showed that both mutants were modified with a significantly higher efficiency (Supplementary Figure 5B).
In agreement with these observations, the disease-causing G30A mutation, leading to disruption of the anticodon stem (24), abolished m3C32 modification (Supplementary Figure 5C). Our previous result also showed that in the context of the G30A mutation, an additional C40U mutation is able to restore the tRNA structure, thermal stability and aminoacylation capacity (24); however, hmtRNAThr-G30A/C40U was still a poor substrate of METTL8-Iso1 (Supplementary Figure 5C). In addition, when we mutated A39 to G39, changing the U31–A39 Watson–Crick base pair to the U31–G39 wobble base pair, hmtRNAThr-A39G was modified, but with reduced efficiency (Supplementary Figure 5C).
Taken together, these results suggest that the anticodon stem plays an important role in determining m3C32 biogenesis and that the newly evolved A29-C41 mismatch is able to fine-tune the m3C32 levels in hmtRNAThr.
i6A37 is a positive determinant of m3C32 biogenesis in hmtRNASer(UCN)
After establishing that METTL8-Iso1 is active in catalyzing hmtRNAThr m3C32 formation, we investigated its modification activity toward hmtRNASer(UCN). However, the hmtRNASer(UCN) transcript was not modified (Figure 6A), suggesting that it is not a qualified substrate or that METTL8-Iso1 alone is insufficient in mediating hmtRNASer(UCN) m3C modification in vitro. Considering that t6A37 and i6A37 are frequently found to be prerequisites for m3C32 formation in yeast and human cytoplasmic tRNAs (11,12,16), we reasoned that METTL8-Iso1 probably requires i6A37 of hmtRNASer(UCN). We purified EcMiaA and found that it efficiently introduced an i6A moiety to hmtRNASer(UCN) (Figure 6B), consistent with a previous report that only A36-A37-A38 is required by EcMiaA (32). The i6A moiety of the EcMiaA-prepared hmtRNASer(UCN) was further confirmed by UPLC–MS/MS analysis (Figure 6C). We found that prepared i6A-hmtRNASer(UCN) was indeed a substrate of METTL8-Iso1 (Figure 6A). Therefore, these data clearly revealed that i6A37 functions as a positive determinant for m3C32 modification by METTL8-Iso1.
Figure 6.
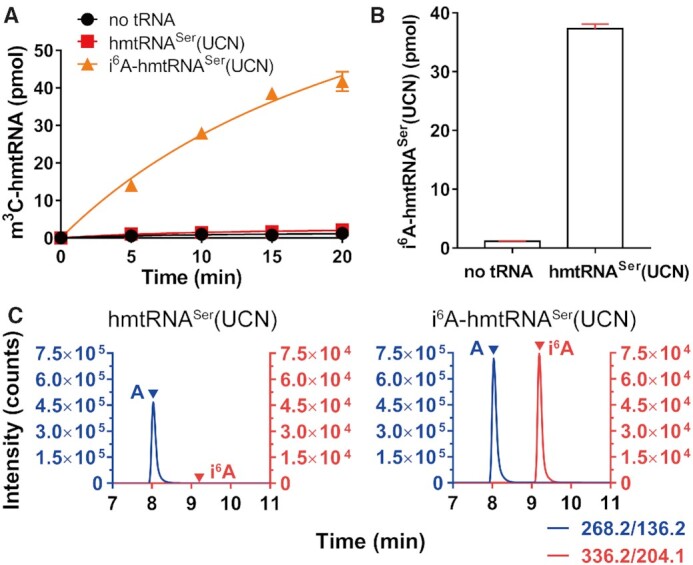
i6A modification is a prerequisite for m3C32 biogenesis in hmtRNASer(UCN) by METTL8-Iso1. (A) Time-course curves of the m3C32 modification of hmtRNASer(UCN) and i6A-hmtRNASer(UCN) by METTL8-Iso1. Negative controls (no tRNA) represent assays without tRNA added. (B) EcMiaA was able to efficiently modify hmtRNASer(UCN). (C) UPLC–MS/MS analysis of the digested product of EcMiaA-modified hmtRNASer(UCN). m/z value 268.2 to 136.2 (A) and m/z value 336.2 to 204.1 (i6A) are shown on the right. Data represent averages of three (A) or two (B) independent experiments and the corresponding SEM.
SARS2 interacts with METTL8-Iso1 directly but only slightly stimulates its modification activity
Modification of cytoplasmic tRNASer(GCU) by METTL6 critically relies on the presence of SARS1 due to augmented tRNA binding of the long variable arm of tRNASer(GCU) by SARS1. However, METTL6 does not bind SARS1 directly (16). Mitochondrial tRNASer(UCN) is unique in that it lacks the class II tRNA-defining long variable arm. This structural deviation prompted us to ask whether METTL8-Iso1 is able to interact with SARS2. To this end, both METTL8-Iso1-FLAG and a C-terminal Myc-tagged SARS2 (SARS2-Myc) were coexpressed in HEK293T cells. Co-IP purification showed that SARS2-Myc indeed coprecipitated with METTL8-Iso1-FLAG. Furthermore, the addition of RNase to the whole cell lysate prior to co-IP had no effect on the copurification of both enzymes (Figure 7A). These results revealed that METTL8-Iso1 interacted with SARS2 in an RNA-independent mode, in contrast to the METTL2A-SARS1 interaction, which requires tRNA as a scaffold (13,16).
Figure 7.

SARS2 interacts with METTL8-Iso1 directly but does not determine the m3C32 modification activity of METTL8-Iso1. (A) Genes encoding METTL8-Iso1-FLAG and SARS2-Myc were coexpressed in HEK293T cells. SARS2-Myc was precipitated by METTL8-Iso1-FLAG using anti-FLAG antibodies. The addition of RNase had no effect on the METTL8-Iso1 and SARS2 interaction. (B) Time-course curves of the m3C32 modification of i6A-hmtRNASer(UCN) by METTL8-Iso1 and different concentrations of SARS2 as indicated (METTL8-Iso1 to SARS2 mole ratio 1:1, 1:2, or 1:5). Two negative controls (modification by METTL8-Iso1 alone without (METTL8-Iso1 + no tRNA) or with (METTL8-Iso1 + hmtRNASer(UCN)) tRNA transcript added were included. (C) Time-course curves of the m3C32 modification of hmtRNASer(UCN) transcript by METTL8-Iso1 and SARS2 (1:2). Negative controls (METTL8-Iso1 + no tRNA) represent assays without tRNA added. Modification of i6A-hmtRNASer(UCN) by METTL8-Iso1:SARS2 (1:2) was included as a positive control. Data represent averages of two (B) or three (C) independent experiments and the corresponding SEM.
To understand whether the presence of SARS2 was able to stimulate the activity of METTL8-Iso1, mature SARS2 was purified (Supplementary Figure 6). We incubated increasing concentrations of SARS2 relative to METTL8-Iso1, and m3C32 modification activity assays showed that SARS2 only slightly elevated the modification of i6A-hmtRNASer(UCN) (Figure 7B). The highest activity of METTL8-Iso1 was observed at a relative molar ratio of 1:2 (METTL8-Iso1 to SARS2), and further elevation of the SARS2 concentration (molar ratio of METTL8-Iso1 to SARS2 of 1:5) provided little stimulatory activity. Again, the hmtRNASer(UCN) transcript was not a substrate of SARS2 and METTL8-Iso1 (Figure 7C), suggesting that the interaction of METTL8-Iso1 with SARS2 did not abrogate the prerequisite for i6A37 modification.
Altogether, these results showed that SARS2 interacted with METTL8-Iso1 directly but only slightly stimulated its modification activity.
G35 is important but no longer a determinant in hmtRNASer(UCN)
Considering the divergence in the requirement of t6A37 or i6A37 between hmtRNAThr and hmtRNASer(UCN) by METTL8-Iso1, we wanted to know whether there is difference in the role of nucleotides in the anticodon loop of hmtRNASer(UCN) in m3C32 biogenesis. Due to the preparation of the i6A37 modification, A36-A37-A38 could not be mutated (32). Therefore, we constructed six mutants of hmtRNASer(UCN), including hmtRNASer(UCN)-U33A, -U33G, -U34C, -U34G, -G35U and -G35C (Figure 8A). EcMiaA was unable to efficiently introduce i6A to hmtRNASer(UCN)-U33A and -U33G for unknown reasons; therefore, these two mutants were further excluded from the following assays. hmtRNASer(UCN)-U34C, -U34G, -G35U and -G35C exhibited slightly decreased i6A37 modification levels (within 2-fold) (Supplementary Figure 7A). After obtaining i6A-modified tRNA mutants, assays of m3C32 modification by mature METTL8-Iso1 showed that hmtRNASer(UCN)-U34C and -G35C were obviously modified, albeit with reduced efficiencies, which might be attributable to a small decrease in i6A modification levels (Supplementary Figure 7A). In sharp contrast, both hmtRNASer(UCN)-U34G and -G35U were not modified (Figure 8B), in spite of their comparable i6A modification efficiencies with those of hmtRNASer(UCN)-U34C and -G35C (Supplementary Figure 7A). Notably, hmtRNAThr-U34C displayed a higher modification efficiency than wild-type hmtRNAThr, and none of the hmtRNAThr G35 mutants were modified (Figure 5C and D). These data collectively revealed divergent recognition of hmtRNAThr and hmtRNASer(UCN) by METTL8-Iso1 and showed that G35 in hmtRNASer(UCN), while important, is not a determinant of m3C32 modification.
Figure 8.
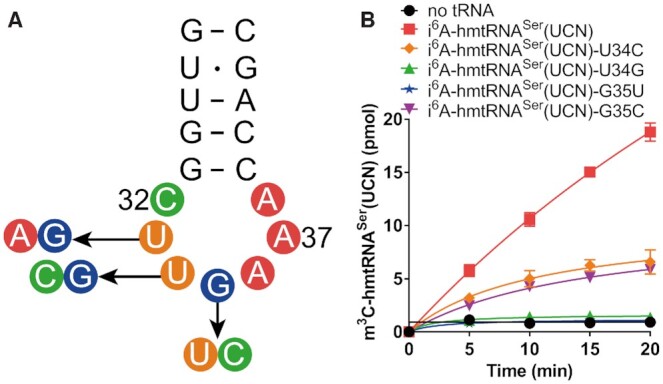
G35 is important but is not a determinant in i6A-hmtRNASer(UCN). (A) Point mutations targeting the anticodon loop of hmtRNASer(UCN). (B) Time-course curves of the m3C32 modification of i6A-hmtRNASer(UCN)-U34C, -U34G, -G35U and -G35C. Negative controls (no tRNA) represent assays without tRNA added. Positive controls (i6A-hmtRNASer(UCN) represent modification of i6A-hmtRNASer(UCN). Data represent averages of two independent experiments and the corresponding SEM.
Converting other mitochondrial tRNAs to be substrates of METTL8-Iso1
Based on the requirement for C32-(U/A/C)33-(C > U)34-G35-(U > A)36-(U > A = C > G)37-(A > U > C)38 in the anticodon of hmtRNAThr for m3C32 formation by METTL8-Iso1, we checked the mitochondrial tRNA sequences and found that several tRNAs harbor a similar anticodon loop sequence lacking only the G35 nucleotide, such as hmtRNALys, hmtRNALeu(UUR) and hmtRNAMet (Figure 9A). Thus, we mutated base 35 of these tRNAs to G35 to produce hmtRNALys-Ki-G35, hmtRNALeu(UUR)-G35 and hmtRNAMet-G35 (Figure 9A). The wild-type tRNAs and their mutants were subsequently modified by METTL8-Iso1, and the results showed that hmtRNAMet-G35 was modified with an obviously higher efficiency than hmtRNAThr (Figure 9B). In parallel, hmtRNALys-Ki-G35 was also modified, but the efficiency was quite low (Figure 9B and C). However, METTL8-Iso1 was unable to introduce m3C modification into tRNALeu(UUR)-G35 (Figure 9B). These results suggested that METTL8 discriminates among some mitochondrial tRNAs via the presence or absence of G35, at least for hmtRNALys and hmtRNAMet. Differences in modification efficiency among various tRNAs were probably due to differences in the sequences and/or conformations of the anticodon stem, which has been proven to be an important element for m3C biogenesis in hmtRNAThr. On other hand, modifications in the anticodon loop of hmtRNALys (5-taurinomethyl-2-thiouridine at position 34, τm5s2U34) and hmtRNAMet (5-formylcytidine at position 34, f5C34) (9) may also facilitate discrimination in vivo.
Figure 9.

Discrimination of hmtRNAThr and hmtRNASer(UCN) from nonsubstrate tRNAs by METTL8-Iso1. Point mutations targeting the anticodon loop of hmtRNALys, hmtRNALeu(UUR) and hmtRNAMet (A) or hmtRNAPhe, hmtRNATrp and hmtRNATyr (D). Time-course curves of the m3C32 modification of hmtRNAThr (positive control), hmtRNALys-Ki, hmtRNALys-Ki-U35G, hmtRNALeu(UUR), hmtRNALeu(UUR)-A35G, hmtRNAMet, and hmtRNAMet-A35G (B); Modification of hmtRNALys-Ki and hmtRNALys-Ki-U35G are shown independently in (C) for clarity. Time-course curves of the m3C32 modification of i6A-hmtRNASer(UCN) (positive control), i6A-hmtRNAPhe, i6A-hmtRNAPhe-G34U/A35G, i6A-hmtRNATrp, i6A-hmtRNATrp-C35G, i6A-hmtRNATyr, and i6A-hmtRNATyr-G34U/U35G (E). Negative controls (no tRNA) represent assays without tRNA added in (B, C, E). Data represent averages of three (B and C) or two (E) independent experiments and the corresponding SEM.
Several human mitochondrial tRNAs harbor the i6A37 modification, including hmtRNATyr, hmtRNAPhe and hmtRNATrp, in addition to hmtRNASer(UCN) (9,21) (Figure 9D). This poses the question of how METTL8-Iso1 discriminates hmtRNASer(UCN) from these i6A37-harboring tRNAs. EcMiaA was able to modify hmtRNATyr, hmtRNAPhe and hmtRNATrp albeit with variable efficiencies (Supplementary Figure 7B). We obtained i6A-hmtRNATyr, i6A-hmtRNAPhe and i6A-hmtRNATrp using EcMiaA and found that METTL8-Iso1 was indeed unable to modify i6A-hmtRNATyr and i6A-hmtRNAPhe (Figure 9E). However, i6A-hmtRNATrp was modified, albeit with a much lower efficiency than i6A-hmtRNASer(UCN). Among these three tRNAs, only hmtRNATrp harbors C35 (A35 in hmtRNAPhe and U35 in hmtRNATyr). These results were consistent with our earlier data showing that i6A-modified tRNASer(UCN)-G35C (with C35) could be modified by METTL8-Iso1 (Figure 8B). Furthermore, we constructed hmtRNAPhe-G34U/A35G, hmtRNATyr-G34U/U35G and hmtRNATrp-C35G mutants to mimic the anticodon loop of hmtRNASer(UCN) (Figure 9D). All these mutants were efficiently i6A-modified by EcMiaA compared with wild-type tRNAs (Supplementary Figure 7B). Indeed, METTL8-Iso1 generated even more m3C32 in i6A-hmtRNAPhe-G34U/A35G than in i6A-hmtRNASer(UCN). Likewise, the modification efficiency of i6A-hmtRNATrp-C35G was significantly higher than that of i6A-hmtRNATrp (Figure 9E). Notably, hmtRNAPhe-G34U/A35G exhibited only a small increase in i6A modification level in comparison with wild-type hmtRNAPhe; similarly, hmtRNATrp-C35G displayed only a small decrease in i6A modification level when compared with wild-type hmtRNATrp (Supplementary Figure 7B). Additionally, despite the hypomodification of i6A-hmtRNATyr, the modification of i6A-hmtRNATyr-G34U/U35G was obvious. Possibly, the significant increase in i6A modification of hmtRNATyr-G34U/U35G (Supplementary Figure 7B) also partially contributed to stimulated m3C modification (Figure 9E). All these results clearly revealed how METTL8-Iso1 efficiently discriminates hmtRNASer(UCN) from other tRNAs in the presence of i6A37, mainly based on the tRNA anticodon loop. In vivo, m3C32 is absent in hmtRNATrp (9). Our observation that i6A-hmtRNATrp could be m3C-modified by METTL8-Iso1 in vitro suggested that other modifications in hmtRNATrp, especially those at the anticodon loop, including τm5s2U at position 34 and/or downstream modification of i6A at position 37 (2-methylthio-N6-isopentenyladenosine, ms2i6A) (9), may prevent hmtRNATrp from being modified by METTL8-Iso1 in vivo.
DISCUSSION
Although the types of tRNA modifications in both mammalian cytoplasmic and mitochondrial tRNAs have been remarkably expanded due to the rapid development of RNA mass spectrometry methodologies in recent years, the identification and confirmation of the corresponding tRNA-modifying enzymes has lagged behind. Indeed, genes have not yet been identified for ∼23% of the >100 human tRNA modifying enzymes that are predicted to exist (40). Among these missing genes, the mitochondrial tRNA m3C32 methyltransferase has long been uncertain. The identification of Mettl2 and Mettl6 as mouse tRNA m3C32-modifying enzymes and the exclusion of Mettl8 from tRNA modification led to the hypothesis that METTL2A, METTL2B and METTL6 in human cells are missing enzymes (13,40). However, the exclusive cytoplasmic localization of METTL2A/2B and METTL6 makes them unlikely to be responsible for mitochondrial tRNA modification (16). A recent study revealed that METTL8 is the missing mitochondrial m3C modifying enzyme (23). Here, we provide further evidence, based on mRNA and protein analyses and reconstituted activity, that an METTL8 isoform mediates hmtRNAThr and hmtRNASer(UCN) m3C32 biogenesis.
Observations of mRNA modification (13), the involvement of METTL8 in rRNA-mediated R-loop biogenesis (14), and the presence of METTL8 in the mitochondrial proteome (22) led to confusion about the exact localization of METTL8 and its function in RNA posttranscriptional modification. We demonstrated that the METTL8 gene generates multiple protein isoforms through alternative mRNA splicing and subsequent translation. The longest isoform, METTL8-Iso1, contains a 22-amino acid MTS that targets it to the mitochondria. Other isoforms, including METTL8-Iso4, are localized to the nucleolus, consistent with results obtained by others (14). The distribution of METTL8 in the nucleolus suggests its possible role in R-loop biogenesis by modifying some yet unidentified nucleic acid constituents (Figure 10). It is puzzling that METTL8-Iso3, if present, differs from METTL8-Iso4 in only the very N-terminal five residues (1MPRDH5). Both localized in the nucleolus, but whether they exhibit similar or differential methyltransferase activity is unclear at present. Moreover, METTL8-Iso1 contains an in-frame Met residue (Met51) when compared with the initiating Met of METTL8-Iso4. We cannot absolutely exclude the possibility that mRNA 1v2 (encoding METTL8-Iso1) is able to generate METTL8-Iso4 via translational re-initiation. However, fluorescence reporter assays show that ectopic expression of mRNA 1v2 in HEK293T cells drives mitochondrial localization, suggesting that this possibility, if it occurs, is low. In addition, it is worth noting that truncated METTL8 isoforms terminating in the middle of the SAM-binding site due to skipping of exon 10 were also potentially translated in human cells. The biological significance of these noncanonical METTL8 isoforms, which likely cannot catalyze m3C32 biogenesis due to their incomplete SAM- and tRNA-binding domain, needs to be further explored. Possibly, METTL8-Iso4 and/or the noncanonical METTL8 isoforms are involved in mRNA m3C modification.
Figure 10.

Expression of human METTL8 and its selective m3C modification of hmtRNAThr and hmtRNASer(UCN).METTL8 generates multiple mRNA isoforms via alternative splicing. Some protein isoforms, including METTL8-Iso4, are distributed in the nucleolus with unclear function. A longer isoform, METTL8-Iso1, containing a 22-amino acid MTS, is imported into mitochondria. Mature METT8-Iso1 selects hmtRNAThr mainly via recognition of the tRNA anticodon loop and stem, particularly G35, and selects hmtRNASer(UCN) via a preformed i6A37 moiety, in addition to the anticodon loop. Non-substrate tRNAs are discriminated by mature METTL8-Iso1 due to unfavorable sequences and/or modifications in the anticodon loop.
Regarding the modification mechanism, METTL8-Iso1 exhibits both similarity to and divergence from both METTL2A and METTL6. For the modification of cytoplasmic tRNAThr, METTL2A is completely reliant on the preformed t6A37 modification (16) catalyzed by YRDC and KEOPS in the cytoplasm (31,39,41); however, METTL8-mediated mitochondrial m3C32 biogenesis is independent of the formation of the t6A modification. On the other hand, both METTL8 and METTL2A require an anticodon loop, especially the positive determinant G35, for efficient m3C32 formation. Anticodon stems also contribute to m3C32 modification by both enzymes (16). For the modification of cytoplasmic tRNASer(GCU), METTL6 requires the presence of SARS1 to generate m3C32. SARS1 binds the long variable stem and loop region of cytoplasmic tRNASer(GCU). Indeed, Ue3 and the U-Ae12 base pair of tRNASer(GCU) are two elements in the variable stem that are critical determinants for efficient m3C32 modification by METTL6-SARS1 (16). However, we showed here that METTL8-Iso1 alone is able to mediate mitochondrial tRNA modification, suggesting that the activity of METTL8-Iso1 is not dependent on other helper proteins. Furthermore, METTL6 requires cytoplasmic tRNASer(GCU) as a scaffold to interact with SARS1; indeed, SARS2 interacts with METTL8-Iso1 but in a tRNA-independent manner, and the presence of SARS2 did not significantly stimulate METTL8-Iso1 activity. Notably, both mitochondrial tRNASer isoacceptors are unique due to the absence of the long variable arm, suggesting that SARS2 probably does not play a determinative role by binding to a long variable arm. This might explain why METTL8-Iso1 activity is independent of the presence of SARS2. Finally, we have proposed that the lack of the N-terminal domain of METTL6 is responsible for its reliance on the presence of SARS1 (16). METTL8-Iso1, like METTL2A, contains a long N-terminal domain compared with METTL6. The presence of this domain also likely explains why METTL8-Iso1 alone is an active m3C32 methyltransferase.
We also revealed an obvious divergence between the recognition of hmtRNAThr and hmtRNASer(UCN) by METTL8-Iso1. METTL8-Iso1-mediated m3C32 biogenesis depends on i6A37 in hmtRNASer(UCN) but not its equivalent t6A37 in hmtRNAThr. On the other hand, G35 plays a determinative role in hmtRNAThr but not in hmtRNASer(CUN) (Figure 10). Based on these observations, we further determined how METTL8-Iso1 recognized only hmtRNAThr and hmtRNASer(UCN) in human mitochondria. The most important discriminator seems to be G35. After the introduction of G35, both hmtRNAMet and hmtRNALys were found to be good substrates of METTL8-Iso1, albeit with variable modification efficiencies. On the other hand, with a determinative i6A37 modification, METTL8-Iso1 selected hmtRNASer(UCN), among other i6A-modified tRNAs, mainly using the anticodon loop. Indeed, when the anticodon loops of hmtRNAPhe and hmtRNATyr are changed, they become excellent substrates of METTL8-Iso1.
Identification of METTL8 being a mitochondrial m3C methyltransferase was first reported by Schöller and colleagues. They further reported that lack of m3C impaired mitochondrial translation and respiratory chain complex assembly and activity. In addition, increased METTL8 level was observed in pancreatic cancer (23). After submission of this study, a more recent work by Kleiber et al. also reported that METTL8 was responsible for catalyzing m3C32 biogenesis of hmtRNAThr and hmtRNASer(UCN). They revealed the tRNA recognition and selective mechanisms of METTL8 and also found METTL8 knockout led to impaired mitochondrial translation. Both U34 and G35 were found to be recognition elements of METTL8 for hmtRNAThr; however, only the U34A mutant was constructed (42). Our study indeed found hmtRNAThr-U34A was not modified by mature METTL8-Iso1; however, multiple mutations at U34 suggested that it was not a determinant in m3C32 modification, because hmtRNAThr-U34C was modified with even a higher efficiency. Notably, both studies used the 407-aa METTL8 (23,42), exactly the METTL8-Iso1 identified in this work. However, our study investigated the splicing pattern of METTL8 mRNA in detail and observed alternative splicing modes generating several protein isoforms. Besides the METTL8-Iso1, METTL8-Iso4 was localized in nucleolus. Our study experimentally identified the MTS (Met1-Gln22) of METTL8-Iso1, different from the predicted MTS (Met1-Arg20) in both studies (23,42). In addition, our in vitro study showed that t6A37 modification of hmtRNAThr only slightly stimulated m3C32 modification activity of mature METTL8-Iso1; however, Kleiber et al. reported that t6A37 increased m3C32 modification about 2-fold (42). It is possible that the stimulatory role of t6A37 is more pronounced in vivo. It is notable that all three studies demonstrated m3C32 modification of hmtRNASer(UCN) was critically reliant on i6A37 modification (23,42); however, our data revealed different recognition mechanisms of mature METTL8-Iso1 for hmtRNAThr and hmtRNASer(UCN). Moreover, we found that SARS2 interacted with METTL8-Iso1 directly but did not determine m3C32 modification activity of the latter.
In summary, our results clearly revealed that METTL8 generated a mitochondria-localized isoform to mediate human mitochondrial tRNA m3C32 modification. Reconstitution of mitochondrial tRNA m3C32 modification activity provides valuable insights into the substrate recognition and mechanism of mitochondrial m3C32 biogenesis and a full understanding of both the similarities and differences between cytoplasmic and mitochondrial m3C32 modification by multiple m3C32 transferases (16). The roles of the other METTL8 isoforms would benefit from further in-depth investigation.
DATA AVAILABILITY
All data presented in this study are available within the Figures and in the Supplementary Data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Howard Gamper (Thomas Jefferson University) for carefully reading the manuscript. We are grateful to Dr Liang Cheng (Institute of Chemistry, CAS) for providing i6A standard sample. We thank Dr Mo-Fang Liu in our institute for providing FBL antibody. We also thank the molecular biology core facility of our institute for technical support in UPLC–MS/MS analysis.
Contributor Information
Meng-Han Huang, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Gui-Xin Peng, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; School of Life Science and Technology, ShanghaiTech University, 393 Middle Hua Xia Road, Shanghai 201210, China.
Xue-Ling Mao, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Jin-Tao Wang, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Jing-Bo Zhou, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Jian-Hui Zhang, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
Meirong Chen, School of Pharmacy, China Pharmaceutical University, 639 Longmian Avenue, Nanjing 211198, Jiangsu, China.
En-Duo Wang, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China; School of Life Science and Technology, ShanghaiTech University, 393 Middle Hua Xia Road, Shanghai 201210, China.
Xiao-Long Zhou, State Key Laboratory of Molecular Biology, CAS Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, University of Chinese Academy of Sciences, 320 Yue Yang Road, Shanghai 200031, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2017YFA0504000, 2021YFA1300800, 2021YFC2700903]; Natural Science Foundation of China [91940302, 31870811, 31822015, 81870896, 31670801, 32000889]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDB19010203]; Committee of Science and Technology in Shanghai [22ZR1481300]; Shanghai Key Laboratory of Embryo Original Diseases [Shelab201904]. Funding for open access charge: Natural Science Foundation of China [31822015, 81870896, 31670801].
Conflict of interest statement. None declared
REFERENCES
- 1. McCown P.J., Ruszkowska A., Kunkler C.N., Breger K., Hulewicz J.P., Wang M.C., Springer N.A., Brown J.A.. Naturally occurring modified ribonucleosides. Wiley Interdiscip. Rev. RNA. 2020; 11:e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suzuki T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell. Biol. 2021; 22:375–392. [DOI] [PubMed] [Google Scholar]
- 3. El Yacoubi B., Bailly M., de Crecy-Lagard V.. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012; 46:69–95. [DOI] [PubMed] [Google Scholar]
- 4. Zhou J.B., Wang E.D., Zhou X.L.. Modifications of the human tRNA anticodon loop and their associations with genetic diseases. Cell Mol. Life Sci. 2021; 78:7087–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noma A., Yi S., Katoh T., Takai Y., Suzuki T., Suzuki T.. Actin-binding protein ABP140 is a methyltransferase for 3-methylcytidine at position 32 of tRNAs in saccharomycescerevisiae. RNA. 2011; 17:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Silva S., Haider S.J., Phizicky E.M.. A domain of the actin binding protein abp140 is the yeast methyltransferase responsible for 3-methylcytidine modification in the tRNA anti-codon loop. RNA. 2011; 17:1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lentini J.M., Alsaif H.S., Faqeih E., Alkuraya F.S., Fu D. DALRD3 encodes a protein mutated in epileptic encephalopathy that targets arginine tRNAs for 3-methylcytosine modification. Nat. Commun. 2020; 11:2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suzuki T., Suzuki T.. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2014; 42:7346–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki T., Yashiro Y., Kikuchi I., Ishigami Y., Saito H., Matsuzawa I., Okada S., Mito M., Iwasaki S., Ma D.et al.. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020; 11:4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ignatova V.V., Kaiser S., Ho J.S.Y., Bing X., Stolz P., Tan Y.X., Lee C.L., Gay F.P.H., Lastres P.R., Gerlini R.et al.. METTL6 is a tRNA m3C methyltransferase that regulates pluripotency and tumor cell growth. Sci. Adv. 2020; 6:eaaz4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arimbasseri A.G., Iben J., Wei F.Y., Rijal K., Tomizawa K., Hafner M., Maraia R.J.. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA. 2016; 22:1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Han L., Marcus E., D'Silva S., Phizicky E.M. S. cerevisiae trm140 has two recognition modes for 3-methylcytidine modification of the anticodon loop of tRNA substrates. RNA. 2017; 23:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu L., Liu X., Sheng N., Oo K.S., Liang J., Chionh Y.H., Xu J., Ye F., Gao Y.G., Dedon P.C.et al.. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J. Biol. Chem. 2017; 292:14695–14703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang L.H., Zhang X.Y., Hu T., Chen X.Y., Li J.J., Raida M., Sun N., Luo Y., Gao X.. The SUMOylated METTL8 induces R-loop and tumorigenesis via m3c. iScience. 2020; 23:100968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cui J., Liu Q., Sendinc E., Shi Y., Gregory R.I.. Nucleotide resolution profiling of m3C RNA modification by HAC-seq. Nucleic Acids Res. 2021; 49:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mao X.L., Li Z.H., Huang M.H., Wang J.T., Zhou J.B., Li Q.R., Xu H., Wang X.J., Zhou X.L.. Mutually exclusive substrate selection strategy by human m3C RNA transferases METTL2A and METTL6. Nucleic Acids Res. 2021; 49:8309–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen R., Zhou J., Liu L., Mao X.L., Zhou X., Xie W.. Crystal structure of human METTL6, the m3C methyltransferase. Commun. Biol. 2021; 4:1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin H., Miyauchi K., Harada T., Okita R., Takeshita E., Komaki H., Fujioka K., Yagasaki H., Goto Y.I., Yanaka K.et al.. CO2-sensitive tRNA modification associated with human mitochondrial disease. Nat. Commun. 2018; 9:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou J.B., Wang Y., Zeng Q.Y., Meng S.X., Wang E.D., Zhou X.L.. Molecular basis for t6A modification in human mitochondria. Nucleic Acids Res. 2020; 48:3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golovko A., Hjalm G., Sitbon F., Nicander B.. Cloning of a human tRNA isopentenyl transferase. Gene. 2000; 258:85–93. [DOI] [PubMed] [Google Scholar]
- 21. Lamichhane T.N., Mattijssen S., Maraia R.J.. Human cells have a limited set of tRNA anticodon loop substrates of the tRNA isopentenyltransferase TRIT1 tumor suppressor. Mol. Cell Biol. 2013; 33:4900–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rath S., Sharma R., Gupta R., Ast T., Chan C., Durham T.J., Goodman R.P., Grabarek Z., Haas M.E., Hung W.H.W.et al.. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021; 49:D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scholler E., Marks J., Marchand V., Bruckmann A., Powell C.A., Reichold M., Mutti C.D., Dettmer K., Feederle R., Huttelmaier S.et al.. Balancing of mitochondrial translation through METTL8-mediated m3C modification of mitochondrial tRNAs. Mol. Cell. 2021; 81:4810–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Zeng Q.Y., Zheng W.Q., Ji Q.Q., Zhou X.L., Wang E.D.. A natural non-Watson-Crick base pair in human mitochondrial tRNAThr causes structural and functional susceptibility to local mutations. Nucleic Acids Res. 2018; 46:4662–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collinet B., Friberg A., Brooks M.A., van den Elzen T., Henriot V., Dziembowski A., Graille M., Durand D., Leulliot N., Saint Andre C.et al.. Strategies for the structural analysis of multi-protein complexes: lessons from the 3D-Repertoire project. J. Struct. Biol. 2011; 175:147–158. [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Zhou J.B., Zeng Q.Y., Wu S.Q., Xue M.Q., Fang P.F., Wang E.D., Zhou X.L.. Hearing impairment-associated KARS mutations lead to defects in aminoacylation of both cytoplasmic and mitochondrial tRNALys. Sci. China Life Sci. 2020; 63:1227–1239. [DOI] [PubMed] [Google Scholar]
- 27. Fechter P., Rudinger J., Giege R., Theobald-Dietrich A.. Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett. 1998; 436:99–103. [DOI] [PubMed] [Google Scholar]
- 28. Sissler M., Helm M., Frugier M., Giege R., Florentz C.. Aminoacylation properties of pathology-related human mitochondrial tRNA(Lys) variants. RNA. 2004; 10:841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeng Q.Y., Peng G.X., Li G., Zhou J.B., Zheng W.Q., Xue M.Q., Wang E.D., Zhou X.L.. The G3-U70-independent tRNA recognition by human mitochondrial alanyl-tRNA synthetase. Nucleic Acids Res. 2019; 47:3072–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El Yacoubi B., Lyons B., Cruz Y., Reddy R., Nordin B., Agnelli F., Williamson J.R., Schimmel P., Swairjo M.A., de Crecy-Lagard V.. The universal yrdc/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res. 2009; 37:2894–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Yacoubi B., Hatin I., Deutsch C., Kahveci T., Rousset J.P., Iwata-Reuyl D., Murzin A.G., de Crecy-Lagard V.. A role for the universal Kae1/Qri7/YgjD (COG0533) family in tRNA modification. EMBO J. 2011; 30:882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soderberg T., Poulter C.D.. Escherichia coli dimethylallyl diphosphate:tRNA dimethylallyltransferase: essential elements for recognition of tRNA substrates within the anticodon stem-loop. Biochemistry. 2000; 39:6546–6553. [DOI] [PubMed] [Google Scholar]
- 33. Zhou X.L., Chen Y., Zeng Q.Y., Ruan Z.R., Fang P., Wang E.D.. Newly acquired N-terminal extension targets threonyl-tRNA synthetase-like protein into the multiple tRNA synthetase complex. Nucleic Acids Res. 2019; 47:8662–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Claros M.G., Vincens P.. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996; 241:779–786. [DOI] [PubMed] [Google Scholar]
- 35. Yao Y.N., Wang L., Wu X.F., Wang E.D.. The processing of human mitochondrial leucyl-tRNA synthetase in the insect cells. FEBS Lett. 2003; 534:139–142. [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Zhou X.L., Ruan Z.R., Liu R.J., Eriani G., Wang E.D.. A human disease-causing point mutation in mitochondrial threonyl-tRNA synthetase induces both structural and functional defects. J. Biol. Chem. 2016; 291:6507–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carapito C., Kuhn L., Karim L., Rompais M., Rabilloud T., Schwenzer H., Sissler M.. Two proteomic methodologies for defining N-termini of mature human mitochondrial aminoacyl-tRNA synthetases. Methods. 2017; 113:111–119. [DOI] [PubMed] [Google Scholar]
- 38. Peng G.X., Zhang Y., Wang Q.Q., Li Q.R., Xu H., Wang E.D., Zhou X.L.. The human tRNA taurine modification enzyme GTPBP3 is an active GTPase linked to mitochondrial diseases. Nucleic Acids Res. 2021; 49:2816–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang J.T., Zhou J.B., Mao X.L., Zhou L., Chen M., Zhang W., Wang E.D., Zhou X.L.. Commonality and diversity in tRNA substrate recognition in t6A biogenesis by eukaryotic KEOPSs. Nucleic Acids Res. 2022; 50:2223–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. de Crecy-Lagard V., Boccaletto P., Mangleburg C.G., Sharma P., Lowe T.M., Leidel S.A., Bujnicki J.M.. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res. 2019; 47:2143–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srinivasan M., Mehta P., Yu Y., Prugar E., Koonin E.V., Karzai A.W., Sternglanz R.. The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 2011; 30:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kleiber N., Lemus-Diaz N., Stiller C., Heinrichs M., Mai M.M., Hackert P., Richter-Dennerlein R., Hobartner C., Bohnsack K.E., Bohnsack M.T.. The RNA methyltransferase METTL8 installs m3C32 in mitochondrial tRNAs(Thr/Ser(UCN)) to optimise tRNA structure and mitochondrial translation. Nat. Commun. 2022; 13:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this study are available within the Figures and in the Supplementary Data.