


Abstract
Haploinsufficiency drives Darwinian evolution. Siblings, while alike in many aspects, differ due to monoallelic differences inherited from each parent. In cancer, solid tumors exhibit aneuploid genetics resulting in hundreds to thousands of monoallelic gene-level copy-number alterations (CNAs) in each tumor. Aneuploidy patterns are heterogeneous, posing a challenge to identify drivers in this high-noise genetic environment. Here, we developed Shifted Weighted Annotation Network (SWAN) analysis to assess biology impacted by cumulative monoallelic changes. SWAN enables an integrated pathway-network analysis of CNAs, RNA expression, and mutations via a simple web platform. SWAN is optimized to best prioritize known and novel tumor suppressors and oncogenes, thereby identifying drivers and potential druggable vulnerabilities within cancer CNAs. Protein homeostasis, phospholipid dephosphorylation, and ion transport pathways are commonly suppressed. An atlas of CNA pathways altered in each cancer type is released. These CNA network shifts highlight new, attractive targets to exploit in solid tumors.
Graphical Abstract
Graphical Abstract.
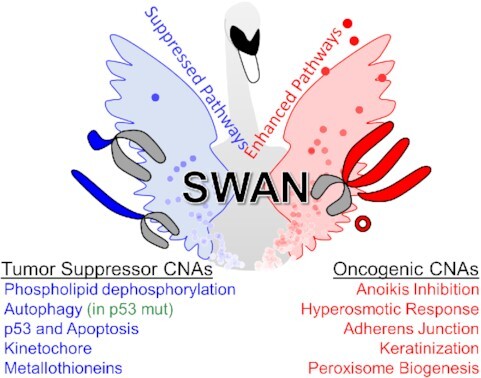
SWAN is a general-use pathway analysis web tool capable of integrating three independent data types. Here, it is released along with a pan-cancer copy-number alteration analysis.
INTRODUCTION
Efforts to establish personalized medicine in cancer therapies have led to curative success in specific cancer types (1). However, progress in most lethal cancer types has been limited by the paucity of eligible patients available for testing in clinical trials. The National Cancer Institute - Molecular Analysis for Therapy Choice (NCI-MATCH) group estimated ∼9% of all cancer patients may be administered therapy based on mutation or amplification targeted data, although this already low inclusion rate does not estimate patient benefit (2). We previously demonstrated autophagy-loss copy-number alterations (CNAs) are druggable in high-grade serous ovarian cancer (OV). While 85–99% of OV primary tumors have a mutation in p53, there are few other canonical tumor suppressor or oncogene mutations, and none reached >10% of patients (3). It remains possible that extremely rare mutations may drive tumors like OV (4). The treatment feasibility issue with such exceedingly rare driver mutations is well-known: with so few patients worldwide, how can drugs be reasonably developed and approved for patient care?
CNAs are another driving factor. One seminal study in the early –omics era for cancer research showed that tumor suppressor mutations were enriched on deletion CNAs while oncogene mutations were enriched on gain or amplification CNAs (5). We similarly found that networks built from molecular pathways and scored by CNA data were suppressed, with known tumor suppressors as network hubs, or elevated, with established oncogenes as network hubs (6). Within the most suppressed OV pathway network, the autophagy pathway, we identified BECN1 and LC3B as the most influential gene hubs. Suppression of either autophagy gene sensitized cells to autophagy inhibitors chloroquine phosphate or nelfinavir mesylate. In a platinum-resistant patient-derived xenograft (PDX) model, we found autophagy targeted drugs could completely abolish observable tumor burden, even when dual platinum-taxane combination therapy had no effect on the PDX model. These results demonstrate tumor CNAs, even monoallelic CNAs, are pharmacologically targetable. Pharmacologic treatment of CNAs may be amenable to removal of early pre-cancerous cells for some tumor types. For example, clear cell renal cell cancers often lose chromosome 3p years prior to development of disease (7). CNA losses persist during disease progression, suggesting that they remain as biological drivers or at least persistent vulnerabilities (8–10).
CNAs rarely encompass a single gene. Entire chromosomes are often altered within solid tumors, creating CNAs across hundreds of genes with a single genetic alteration. Few studies have adequately addressed this background noise problem of thousands of gene-level CNAs in each tumor, many of which are passengers, for both logistical and conceptual reasons. It is arduous or infeasible to model aneuploid events in cellular and mouse models, precluding causal genetic studies. However, a handful of well-controlled studies have been completed. TP53-adjacent genes, EIF5A and ALOX15B, contribute to tumor formation and progression. In a lymphoma Eμ-Myc pre-B cell mouse model, suppression of these genes by shRNA independently increased lethality (11). Chromosome 8p is often lost in cancers while chromosome 8q is often gained. Here, we adopt the definition of aneuploidy in our discussion as changes encompassing entire chromosome arms (12); 8p loss or 8q gain are both independent aneuploid events. These aneuploid changes often give rise to the observed gene-level CNAs assessed in the current study, due to RNA changes of affected genes. To model 8p loss in breast cancer, 8p loss was engineered in non-malignant MCF10A cells (13). While 8p deletion did not induce transformation, cells exhibited increased invasiveness and elevated mevalonate metabolism. In most cases, a single aneuploid chromosome causes a cell cycle delay. Accordingly, the 8p deletion exhibited this phenotype. Furthermore, a 3p deletion commonly found in lung cancers decreased proliferation in an immortalized lung epithelium AALE cell line, although cells eventually adapted (14). Aneuploidy itself leads to increased usage of the proteasome and autophagosome machinery (15), indicating metabolic inefficiency. Transcriptomic compensation for aneuploidy is rare (16) and protein levels correlate well with CNAs (17). Aneuploidy can lead to slowed tumor growth in RAS mutant xenografts (18) and immune surveillance (19). However, under specific nutrient or signaling conditions, select aneuploid events increase cellular fitness. In serum starved cells, trisomy 13 cells exhibit greater fitness than control cells (20). These examples directly demonstrate CNAs drive cancer in specific selective conditions.
Here, we developed a new pathway network algorithm, Shifted Weighted Annotation Network (SWAN), designed to handle high-noise biological data, such as monoallelic CNAs spread across the genome. We report SWAN analysis of 10,395 tumors studied by The Cancer Genome Atlas (TCGA) from 31 cancer types and 4,925 pathways. We demonstrate SWAN prioritizes known tumor suppressors and oncogenes within CNAs. SWAN further characterized 24 high-confidence novel multi-cancer oncogenes. Molecular pathway suppression caused by loss of tumor suppressor genes were prevalent across tumor types, representing potentially targetable vulnerabilities. We show biological validation of a tumor-specific elevated pathway, peroxisome biogenesis, and a multi-cancer suppressed pathway, cadmium response. We release an online CNAlysis Atlas and easily accessible web-based SWAN pathway analysis tools.
MATERIALS AND METHODS
Key resources table
See supplement.
Experimental model and subject details
Cell lines
Established cell lines were purchased from the American Type Culture Collection (ATCC) and validated by short tandem repeat (STR) profiling (Duke University and ATCC).
Human high-grade serous ovarian cancer samples
Flash frozen samples were requested from biorepositories. All samples were stage 2C or higher high-grade serous fallopian or ovarian cancer, with the exception of a single control Caucasian sample with paired uterus normal control used for bioinformatic quality control. Cancer stage, self-reported race and ethnicity (either non-Hispanic black or non-Hispanic white), and age at diagnosis is reported in Supplementary Table S5.
Method details
SWAN
SWAN is pathway analysis software used to explore the cumulative impact of gene changes within complex sets of data. SWAN is encoded in R, but available on the web as a browser-based fully-functional analysis application. As inputs, SWAN can use data from paired or unpaired controls along with experimental data, or strictly experimental sample data when control samples cannot be acquired. SWAN is capable of utilizing up to three types of data in a single analysis: CNA data, RNA data, and/or mutation data. However, SWAN runs with one type of data (e.g. CNA data), and does not require layering of different data types. All data must be numeric. SWAN first assigns genes into pathways and then builds a network based on established protein-protein interactions between the gene products within the pathway. SWAN then scores the pathway's network using the input data and haploinsufficiency data. If the input data within a particular pathway are negative, for example, this pathway's network will have a negative score. By default, SWAN uses curated haploinsufficiency data to improve prioritization of genes, but including this phenotype is optional. SWAN builds an estimate of noise for each sample, using all genes containing data for each sample, to compare network scores, creating ‘network shift’ scores for all samples. Then, SWAN generates a P-value for each pathway using these network shift scores. As outputs, SWAN yields magnitudes, statistical factors such as nominal P-values and false-discovery-rate (FDR) corrected values, and the genes which most impacted a negative or positive shift within each pathway. The web application of SWAN produces rich, customizable plots for data interpretation.
The precise form of SWAN calculations can be found in Supplemental Methods.
For the pan-cancer analysis presented in all figures unless otherwise noted, CNA data utilized was gene-level copy number alterations normalized to -2 (homozygous deletion), -1 (heterozygous deletion), 0 (no change), 1 (gain of one copy) and 2 (gain of two or more copies). TCGA data were downloaded from cBioPortal using ‘Provisional’ datasets (21). For layering of mutation data in specific figure panels, as denoted in the results section, mutation data included TCGA mutation data with a ‘1’ marking a non-synonymous mutation or a ‘0’ for no non-synonymous somatic mutation. For layering of RNA data in specific figure panels, RNA data were log2 per-gene normalized TCGA microarray data from the pan-cancer TCGA set, ‘EBPlusPlusAdjustPANCAN_IlluminaHiSeq_ RNASeqV2.geneExp.tsv’ (22). Pathway sets used were the Hallmark, KEGG, Reactome, and GO.
Quality control of SWAN
The cancer types used in the tumor suppressor gene (TSG) and oncogene (OG) quality control (QC) analysis were ACC, BLCA, BRCA, CESC, COAD, ESGA, GBM, HNSC, KICH, KIRC, LGG, LIHC, LUAD, LUSC, MESO, OV, PAAD, PRAD, READ, SARC, SKCM, STAD, TGCT, THCA, UCEC and UVM.
To test whether SWAN was able to enrich for known oncogenes and tumor suppressor genes in its list of prioritized genes, tabular results from SWAN were queried. COSMIC Cancer Gene Census Tier 1 was used as the list of tested OGs and TSGs. As a possible alternative, STOP and GO genes were also tested. Each STOP and GO gene used was from two of three sources from the Elledge lab (5,23,24). For ‘gene prioritization enrichment,’ genes from pathways called as ‘haploinsufficient’ or ‘triploproficient’ (FDR ≤ 0.0001) marked by SWAN as in the top five negatively or positively scoring genes, respectively, were tabulated. The enrichment ratio is calculated as the ratio of the sum of TSGs in the top five prioritized genes within a haploinsufficient pathway divided by the sum of those which are not known TSGs, over a similar ratio within neutral-called (‘no selection’) pathways from SWAN. An equivalent calculation was performed for OGs. A fisher's exact test was performed on these counts for each cancer type separately for TSGs and OGs (Figure 2B and Supplementary Figure S3). To determine the loss of enrichment, pan-cancer SWAN analysis was performed with 1,000 iterations across these 26 QC cancer types while adjusting single parameters. QC analysis was then performed again, and the percent difference in enrichment from the null 1 ratio value was calculated.
Figure 2.

Design of Shifted Weighted Annotation Network (SWAN) pathway analysis tool and pan-cancer results. (A) A conceptual diagram of SWAN calculations. Raw data in this pan-cancer analysis is CNAs. Pathway networks are then built utilizing protein-protein interaction data and haploinsufficiency data. Sample network scores are compared to paired-shuffled control data. Details are found in SWAN documentation and Methods. (B) Plot of the number of pathways which are identified among 31 cancer types as elevated or suppressed relative to the sum of SWAN shifts. (C) Plot of the statistical enrichment of known OGs and TSGs on elevated or suppressed pathways in each of the 26 QC-compatible tumor types studied.
To determine an appropriate default cutoff FDR value for SWAN, a tuning range of ‘0.2, 0.1, 0.05, 0.04, 0.03, 0.02, 0.01’ and then 10-fold less, down to ‘10−50’ was used. All 26 QC cancer types were again tested for a TSG and OG enrichment ratio. In this case, the fisher's exact test summed TSGs and OGs together and non-TSGs with non-OGs together, comparing pathways called as significant to pathways called as non-significant. This allowed for a single metric which balanced sensitivity and specificity; the statistical test would yield a larger P-value for lower N of significant pathways as well as when the neutral-called pathways began to have similar rates of OGs and TSGs as significantly-called pathways. Scrolling across this metric, an optimal FDR ≤ 0.0001 was determined for a default value (Supplementary Figure S1E).
Gene set enrichment analysis
Twenty-six cancer types were analyzed using Gene Set Enrichment Analysis (GSEA) version 4.0.3. Integer normalized TCGA data was used identically as in SWAN and diploid data was set as control. KEGG Pathway gene set was used with 1,000 permutations to the phenotype as the ‘on-the-fly’ setting. Since bench scientists are interested in follow-up molecular biology studies of altered pathways, for each pathway, the top five elevated genes and the bottom five downregulated genes were compared to the top five altered genes by SWAN analysis. The number and enrichment of tumor suppressor genes and oncogenes (defined by Tier 1 Cancer Gene Census COSMIC annotations) within these top five genes were calculated identically as defined in SWAN Quality Control. Note that not all genes with literature support of a tumor suppressor or oncogenic phenotype are in the Tier 1 COSMIC Cancer Gene Census.
BET inhibitor study analysis
Data sets used in each analysis were attained from Gene Expression Omnibus (GEO). Raw data files were formatted using SWAN Data Groomer and transformed to log base 2. Each resulting experimental and control data files were input into either pan-pathway or single pathway SWAN. Pan-pathway analysis calculated 200 control permutations and had a significance threshold of 0.05. Pathways including <10 or >200 genes were omitted. Single pathway analysis calculated 100 control permutations and had a significance threshold of 0.001
Identification of novel oncogenes and tumor suppressors
SWAN interactome summary data from GO pathway analysis was used as a starting point to classify genes as general CNA-influenced OGs or TSGs. Z-scores of each gene within the interactome dataset were averaged for OGs in cancers in which the Z-score was positive and a similar calculation was performed on negative values for TSGs. The number of times a gene was displayed on a cancer interactome priority plot was summed for each candidate OG and TSG. Alpha transparency values represent how much CNA influence originates from the gene itself (i.e. the gene is deleted or amplified) or from the interacting genes (i.e. its interactors are deleted or amplified) and were tabulated for each gene in each tumor type. Only OGs and TSGs which were detected through this method in ≥5 tumor types are reported in the figures and supplemental tables. COSMIC Tier 1 genes were marked as ‘known’ OGs and TSGs as long as they were not characterized as fusion-only TSGs or OGs. All genes outside these criteria were considered ‘novel’, although we caution this does not capture the entirety of the cancer literature. Essential gene data were downloaded from a CRISPR-Cas9 screen of 324 cell lines (25). OGs which were not hits within this screen were included in the Supplementary Figure S6A. TSGs as identified by SWAN interactome analysis not considered ‘known’ TSGs were plotted in Figure 4D and summarized within Supplementary Table S3.
Figure 4.

Pan-cancer suppressed CNA pathways. (A) Unusually pervasive suppressed CNA pathways. Violin histograms of SWAN scores with blue fill indicating significant (FDR < 0.0001) pathway suppression and red fill indicating significant pathway elevation. (B) SWAN Circos plot. Red and blue outer rings are frequency plots of gains or deletions and the inner ribbons represent genes within the selected pathway. Labeled gene symbols are enriched for CNA losses. (C) SWAN network generated, with edges represent protein-protein interactions. Blue nodes are enriched for loss CNAs and red nodes are enriched for gain CNAs. (D) Known and novel TSGs discovered by interactome analysis of all 31 cancer types analyzed, with those present in at least five tumor types displayed against z-score values. Green color indicates previously known COSMIC TSGs. Size is proportional to the mean z-score SWAN contribution across cancers with TSG-containing pathway suppression. Higher transparency indicates interacting protein genes influenced each gene's identification by SWAN, rather than CNAs of the gene itself. (E) RT-qPCR data of metallothioneins within 16q gene cluster. (F) Validation of shRNA-mediated knockdown of MT2A by RT-qPCR. (G) Genotoxic damage as measured by γH2AX immunofluorescence in the presence of 100 μM cadmium is shown for OVCAR3 cells and (H) in the presence of 50 μM cadmium for CAOV3 cells. (E–H) N = 3 experiments, with mean ± s.e.m. shown. (G, H) Scale bar is 10 μm.
Mutation association analysis
Genes mutated in >3% of all TCGA tumors were analyzed for differential SWAN shift distributions. Subsets were taken from each tumor type by splitting mutant versus non-mutant tumors by each putative driver gene. A Wilcoxon rank-sum test was performed on SWAN shifts in mutant tumors and compared to non-mutant tumors. If the mean SWAN shift of the mutant group was lower than the mean SWAN shift of the non-mutant group, the association was considered negative and conversely for positive. The final Supplemental Table 6 lists pathways reaching an FDR (by Benjamini–Hochberg correction of P-values) <0.05. It may be noted that most genes only yielded significant associations in limited cancer types due to the inadequate number of mutations in other tumor types.
Machine-learning prognostic analysis
Patients with both overall survival data and SWAN shift data were analyzed for prognostic SWAN pathways. Patient data were first separated by SWAN pathway shifts, with the ‘low’ and ‘high’ groups separated by 1 standard-deviation (SD) centered at the median shift. To consider the possibility of a false positive, 101 training and test sets were created for Cox-proportional hazard (CoxPH) models which utilize SWAN shift data. Each training set consists of a random selection of 67% of the tumors, and the test set is the remaining 33% of tumors. The training set is used to build a CoxPH model based on overall survival and SWAN shift scores and produce a hazard ratio (HR). This model is then applied to the test set to predict risk scores using SWAN shifts. The upper quartile risk is compared to the lower quartile risk group by log-rank test (survdiff of Surv function in the R package ‘survival’) to yield a P-value for the test group. Potential false positive prognostic pathways were removed by (i) determining significance in the applied CoxPH model risk for each test set and (ii) determining CoxPH HR direction in each training set. The most prognostic pathways are labeled as those with >80% of randomizations as significant and in same HR direction and provided as Supplementary Table S7. Kaplan–Meier curves and survival analysis on the entire cohort without machine-learned filtering is available using the Shiny SWAN Single-Pathway app online.
Cell culture and biologic quality control
Cells were cultured at 37°C with 5% CO2. Established cell lines (OVCAR3, SKOV3, CAOV3 and 293T) were purchased from the American Type Culture Collection (ATCC) and validated by short tandem repeat (STR) profiling. Routine microscopic morphology tests were performed prior to each experiment. All cells were grown in RPMI-1640 media supplemented with antibiotics (penicillin, streptomycin), sodium pyruvate, and 10% FBS (Gibco). Lentiviral constructs for PEX5 and PEX19 were purchased from Genecopoeia. PEX19 cDNA (NM_002857.3) was cloned into EX-G0621-LV242 with a C-Avi-FLAG tag and puromycin resistance. PEX5 (NM_001131023.1) was synthesized and cloned into the EX-Z6463-LV157 vector with C-3xHA Neomycin resistance. Lentivirus was produced in 293T cells and filtered through a 33 μm filter prior to transduction. Confirmation of cDNA insert was performed by Sanger sequencing using forward primer 5′ AGGCACTGGGCAGGTAAG 3′ and reverse primer 5′ CTGGAATAGCTCAGAGGC 3′ for LV242 and forward primer 5′ GCGGTAGGCGTGTACGGT 3′ and reverse primer 5′ ATTGTGGATGAATACTGCC 3′ for LV157. SKOV3 and OVCAR3 cells were selected for LV242 lentiviral integration by addition of 4μg/ml puromycin (Thermo Fisher) to the media or for LV157 integration by addition of 200μg/ml Geneticin (Fisher Scientific).
For determination of mRNA expression of metallothionein isoforms, 1 × 106 CAOV3 or 2 × 106 OVCAR3 cells per well were plated in a six-well dish. After 20 h, cells were rinsed once with PBS (phosphate-buffered saline), and RNA was isolated using the miRNeasy Mini Kit (Qiagen) according to the protocol of the manufacturer. One μg of total RNA was used to transcribe cDNA using the iScript cDNA Synthesis Kit (Biorad). For quantitative real-time reverse-transcriptase polymerase chain reaction (qPCR) 20 ng of cDNA per reaction and the iTaq Universal SYBR Green Supermix (Biorad) was employed. Primer sequences are available in Supplemental Table 8. Triplicate samples were normalized to TBP and relative gene expression was determined by the ΔΔCt method. To knock down metallothionein 2A (MT2A) mRNA expression, shRNA lentiviral vectors targeting MT2A and scrambled control were used to generate lentivirus in 293T cells and filtered through a 33 μm filter prior to transduction. CAOV3 and OVCAR3 cells were selected for lentiviral integration by addition of 4 μg/ml puromycin (Thermo Fisher). For γH2AX staining 2.5 × 103 CAOV3 or OVCAR3 cells were seeded onto black, optical bottom, 96-well plates (VWR). After allowing the cells to adhere for 16h, cells were treated with 50 μM (CAOV3) or 100 μM (OVCAR3) CdCl2 (Sigma-Aldrich) for 24 h. Then cells were fixed in 4% paraformaldehyde for 10min, permeabilized with 0.1% Triton X-100 for 2 min, and nonspecific binding was blocked with PBS containing 5% bovine serum albumin and 5% goat serum for 45min. Then cells were incubated with purified anti-γH2AX antibody (phospho-Ser139; BioLegend) overnight, primary antibody was removed with three PBS washes of 10 min, incubated in Hoechst 33342 (Fisher Scientific) and secondary goat anti-mouse Alexa Fluor 594 nm antibody (LifeTechnologies) for 1.5 h, and secondary was removed followed by three PBS washes of 10 min each. Finally, a Lionheart FX automated microscope (BioTek) was used to image the cells and ImageJ was employed to quantify γH2AX puncta number and intensity.
Metabolomics
Metabolomics were performed as previously described (26). Specific changes for the data presented here include: five replicates were used per genetic condition, cell line used was SKOV3. Otherwise, the methods are repeated and provided in Supplemental Methods.
Metabolomic data associated with the figures is provided in Supplementary Table S4.
Flow cytometry
SKOV3 and OVCAR3 cells transduced with LV157 or LV242 with or without PEX5 and PEX19 respectively were seeded at 25,000 cells per well in a 24-well TC plate in 1 ml media containing antibiotics. A day after seeding, cisplatin (10 μM in DMSO) or N-acetyl-cysteine (2 mM in ddH2O) were added to the media and the cells grown for 48 h prior to staining for flow cytometry. Staining was performed with 10 μM H2DCFDA (2′,7′-dichlorodihydrofluorescein diacetate, VWR #89138-260) for 1 h. Cells were then rinsed with PBS and 500 μl Trypsin 0.05% EDTA (Thermo Fisher Scientific #25300120) was added for 5 min. Trypsinized cells were added to 500 μl iced RPMI in 1.5 ml microcentrifuge tubes. Cells were centrifuged at 3,000 g for 1 min and media aspirated. 1 ml iced PBS was then added to cells and cells were briefly resuspended. Cells were centrifuged at 3,000 g for 1 min. PBS was aspirated, 300 μl fresh iced PBS was added to cells and cells were transferred to an iced 5 ml polypropylene flow cytometry tube (VWR #352063). Cells were analyzed for fluorescence in the 488 nm channel on a BD FACSCanto II cytometer and analyzed using BD FACSDiva software.
Cell death and proliferation assays
SKOV3 and OVCAR3 cells transduced with LV157 or LV242 with or without PEX5 and PEX19 respectively were seeded at 10,000 cells per well in a 96-well TC plate in 50 μl media containing antibiotics. Cells were allowed to adhere for 3 h prior to addition of 50 μl media containing 2× treatment solution (20 μM cisplatin, 4 mM NAC, and/or 0.2% DMSO control). Cells were then grown for 48 h prior to fixation. For fixation, cells were first rinsed in 125 μl PBS and then stained with crystal violet staining and fixation solution (0.11% crystal violet, 0.17 M NaCl and 22% methanol in ddH2O) for 15 min. Crystal violet stain was aspirated, 125 μl PBS wash performed twice, and then cells were dried for 30 min at 37°C in a dry incubator. 85 μl methanol was added to each well and absorbance was read in an absorbance spectrophotometer at 600 nm. Background consisting of cells killed to 100% penetrance using 1 mM H2O2 was subtracted from all reads. Growth inhibition was calculated as the fractional difference in absorbance of a treated well compared to the average control-treated well for an isogenic cell line on the same 96-well plate.
Whole-exome sequencing and data processing
Samples were processed using a Promega Maxwell RSC Instrument (AS4500) and Maxwell RSC Tissue DNA kit (AS1610) to obtain purified DNA. DNA was sent to GENEWIZ for whole-exome processing using an Agilent SureSelect Human All Exon V6 kit and next-generation sequencing on an Illumina HiSeq-4000.
SNVs and indels in TP53 were called using one of two methods. The first method was a default DRAGEN protocol used by GENEWIZ. The second, used to call mutations in the remaining half of samples, utilized a triple-tool calling method. FASTQ reads were aligned to hg38 to create BAM files. BAM files were removed of PCR duplicated using RmDup. The three variant callers used on the BAM files were: LoFreq, FreeBayes, and samtools followed by VarScan (27,28). Variant callers were run using the Galaxy platform (29). TP53 mutations called by all three tools were then filtered by those present in gnomAD v3 (30) at an allele frequency >0.0001 in any ethnicity group. Annovar was used to annotate variants (31). To query ethnicity at genome-scale, EthSEQ (32) was used on genome-wide called variants fitting the three-tool intersect threshold without a gnomAD cutoff (N = 7,695 variants assessed in the new cohort).
Control-FREEC (33) was used to call CNAs without using paired normal controls. To aid in estimation of stromal cell contamination, the TP53 mutation allele frequency was used as the estimated tumor cell fraction. Settings included: breakPointThreshold (1.2), readCountThreshold (50), window (500,000 bp), telocentromeric (100,000 bp), contaminationAdjustment (TRUE), sex (XX), contamination (using normal TP53 allele fraction), and a single control uterus was used as a control target capture region. To best match TCGA CNA data, all BAMs used were aligned to hg19. To create a –2 to 2 normalized file, negative CNAs were given a value of –1, positive CNAs a value of +1, and any positive CNAs exceeding 2 standard deviations above the median a value of +2. Gene-level CNAs were determined using the SWAN Data Groomer functions built for *.seg files. Genome conversion of CNAs or mutation variants between hg19 and hg38 used liftOver with UCSC chain files (34).
Quantification and statistical analysis
In all cell biology figures, P-values are calculated using a two-tailed Student's t-test, unless otherwise indicated. The description of SWAN describes SWAN statistical considerations in detail. Survival outcomes were assessed using Kaplan–Meier curves with log-rank tests.
RESULTS
Oncogenic CNAs drive cancer
Copy-number alterations function as drivers for oncogenesis. Previous studies of normal tissue found normal epithelium contains oncogenic driver single-nucleotide variants (SNVs) and insertion-deletion (indel) mutations, but few CNAs (35–39). Further drivers are necessary to escape local arrest and expand. Oncogenic mutations or CNAs allow for the slow development of clonal CNAs or other mutations over many cell division generations (Figure 1A). Our analysis of TCGA tumors reveals 5–50% of solid tumors (on average, 18%) contain only oncogenic mutations found in normal tissue clones, out of 251 previously identified driver SNVs (Figure 1B). Solid tumors have 15–70% of each tumor genome altered by CNAs, with a median alteration of 39% of the genome (Figure 1C). The scale of established oncogenes (OGs) and tumor suppressor genes (TSGs) on gain or loss CNAs, respectively, range from 20 to 30 of each in solid tumors (Figure 1D). CNAs of known driver genes are a hallmark of solid tumor genetics.
Figure 1.

Copy-number alteration drivers are present in tumors with insufficient SNV drivers. (A) Model for oncogene accumulation by CNAs to drive cancer initiation and progression. (B) TCGA tumors were analyzed for the number of tumors with insufficient SNV drivers. Tumors were queried for 251 Tier 1 COSMIC oncogenes and tumor suppressor gene mutations. Tumors with only p53, or with p53 and mutations commonly found in normal human epithelium (includes NOTCH1-3, FGFR3 and FAT1) and no other COSMIC OG or TSG mutation are plotted as a percent of all tumors queried. (C) Frequency of CNAs in the same cancer types. Stacked bars represent cumulative CNAs of any type. (D) COSMIC Tier1 cancer genes overlapping deletion CNAs (for TSGs) or amplification CNAs (for OGs).
Design of Shifted Weighted Annotation Network (SWAN) analysis
CNA analyses have previously focused on segments of DNA that are significantly altered in tumors. Exceptional amplifications of genes like MYC or EGFR and homozygous deletions of CDKN2A are highlighted in previous studies due to a consistent amplification pattern (40–42). These analyses often ignored the biological changes caused by the 90% of tumor CNAs: removal or duplication of a single allele. Cumulatively within the same molecular pathway, this can have dramatic effects on cell biology. Pathways contain multiple genes that are typically located on multiple chromosome arms. Thus, tumors with different chromosome content may nonetheless upregulate the same pathway if genes within the same pathway are altered in the same direction (either losses or gains). These individual genes may differ between patients, yet the pathway is nonetheless similarly altered in flux. Previous location-centric analyses miss these consistent biological changes occurring on more than one chromosome. To quantify and prioritize multi-chromosomal pathway changes in CNA data, we developed SWAN analysis.
SWAN analysis is broadly applicable to any gene-level data set and is similar conceptually to Gene Set Enrichment Analysis (GSEA), but SWAN includes the addition of phenotypic data to improve the testing of suppression or activation pathway hypotheses by forming weighted pathway networks. In SWAN, pathway-specific elevation and suppression hypotheses are independently tested and compared to a randomized null hypothesis. Permutations of gene-level data were done for each tumor (1,000 random pairs in this study) to generate null distributions specifically relevant to each sample and pathway (Figure 2A). The precise details of SWAN calculations can be found in Supplemental Methods, Supplementary Figure S1, the supplied code, Supplemental Videos 1–3, and the Supplemental Manual provided here.
After much fine tuning of the SWAN algorithm (see quality control section and supplemental methods), we performed a pan-cancer CNA analysis of 10,395 tumors studied by The Cancer Genome Atlas (TCGA) from 31 cancer types and 4,925 pathways. Pathways included Hallmark, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome. There was a wide distribution of highly to moderately suppressed or highly to moderately elevated pathways across all cancer types, which is the focus of the analytics presented in this publication (Figure 2B, Supplementary Table S1). Before exploring our pan-cancer analysis, we first describe features and quality control of this newly released SWAN software.
Similarities and differences with other pathway analysis software
SWAN was built to appropriately handle widely different input data and network structures to output pathway information at both the cohort-level and individual-sample level. It can combine CNA data with RNA and/or mutation data in a single analysis, a feature absent in any graphical user interface tool to our knowledge. Previous tools built and used for RNA data were incompatible with somatic CNA data. Normal tissue, on average, does not contain CNAs, resulting in the control dataset as strictly zero for all genes and samples. Pathifier is a widely used individual-sample pathway scoring software designed for RNA that relies on an estimate of noise from control samples (43). With CNAs, this noise is zero in normal bulk tissue, resulting in an unavoidable error to run an analysis. PARADIGM is another well-cited patient-specific pathway analysis tool, but at the time of this writing could not be tested or compared to SWAN as it is no longer available for public use (44). The zero-comparison problem is circumvented by SWAN using its randomized control methods, enabling an analyses of somatic CNAs.
Cohort-level pathway analysis tools can utilize CNA data, even with substantial zeroes in normal controls. One exceptionally well-cited tool, Gene Set Enrichment Analysis (GSEA), enabled a direct comparison of pathway alteration results with SWAN (45). Using GSEA, OV had only a single significantly elevated Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, ‘complement and coagulation cascades’. Further applied to 26 cancer types, very few, if any, pathway alterations were found by GSEA across all KEGG pathways (Supplementary Table S2, Supplementary Figure S2A). This is unlikely to be the real biological situation for tumors containing clear patterns across chromosome arms. SWAN was designed to circumvent this false-negative issue using phenotype-layered networks, and produces dozens of significant KEGG pathway changes per cancer type. GO term analysis is another popular method of cohort-level pathway analysis. GO term tools use a hypergeometric test to determine differences in an altered set of genes (for example, those with positive CNAs) compared to a background set of all genes analyzed. Hypergeometric tests do not allow for prioritization of which genes may be most relevant within the cohort, in contrast to the shifts SWAN calculates per gene.
There are limitations to SWAN. Its utilization of phenotype data, haploinsufficiency and protein-protein interactions, may be more informative in a context-specific manner. However, such data for each tissue, organism, or cancer type, are not currently available. In some cases, genetic interaction data may be more informative than protein-protein interaction data. To mitigate this limitation, there is an option in SWAN for the user to upload their own phenotype information, thereby replacing haploinsufficiency, protein–protein interactions, or both. Another limitation includes the scoring of haploinsufficiency in a non-linear scale. For CNAs, this makes sense, as haploinsufficiency is defined as a loss of an allele (an integer copy difference). However, it may not be as appropriate as other tools for RNA data, which most other pathway analysis tools specialize in. To reduce this limitation, the user has the option to omit haploinsufficiency scoring.
SWAN may provide more false positive pathway changes than GSEA but less false positives than iPath or Pathifier. While SWAN identified more pathways as significant in most cancer types than GSEA, it identified less as significant than iPath or Pathifier. Previously published analysis of BRCA RNA data using iPath yielded 168 of 186 KEGG pathways as nominally significantly dysregulated (P < 0.05) and 153 of 186 passing a q < 0.05 threshold. Pathifier yielded 180 of 186 KEGG pathways as nominally significantly dysregulated (P < 0.05) and also 180 of 186 passing a q < 0.05 threshold (46). In contrast, SWAN analysis of the same RNA data yielded 33 of 186 pathways as P < 0.05 and 6 of 186 pathways with FDR < 0.05.
Ease-of-use was prioritized with SWAN. GSEA, Pathifier, and others require users to download the software and dependencies to run locally, but have been widely used due to their intuitive graphical interface. SWAN is hosted online and dependencies are updated on the server, minimizing user hassle for first-time utilization. Other excellent pathway analysis tools, such as PathTracer (47), require R-programming or python-programming knowledge. SWAN is encoded in R, but is available online as a Shiny App, sheltering the complex code behind a convenient graphical interface.
Quality control of SWAN for cancer data
SWAN uses two major phenotypes to improve performance: known haploinsufficiency and protein-protein interactions. To quantitatively assess phenotype importance in appropriately defining tumor CNA genetics, we performed multiple pan-cancer SWAN calculations across 26 quality control (QC) compatible cancer types studied by TCGA with 4,925 pathways (KEGG, Gene Ontology [GO], Hallmark, and Reactome). As a positive control, we used TSGs and OGs from COSMIC’s Tier 1 Cancer Gene Census (48). SWAN identifies the most influential suppressed genes within suppressed pathways and the most influential enhanced genes within elevated pathways. If working appropriately in a cancer context, suppressed pathways should have TSGs prioritized and elevated pathways should have OGs prioritized. There was a significant (P ≤ 0.05) enrichment of TSG prioritization within suppressed pathways in 23 of 26 tumor types and a significant enrichment of OG prioritization amongst elevated pathways in 22 of 26 tumor types (Figure 2B, Supplementary Figure S2B). Cancer types without significant enrichment had unusually low CNAs, consistent with the hypothesis that CNAs are not strong drivers amongst all tumors in these cancer types. Applying identical quality control to GSEA and SWAN KEGG pathway analysis, SWAN outperformed GSEA in prioritizing driver genes in 25 of 26 tumor types (Supplementary Figure S2B). Amongst all 31 cancers studied, SWAN found no pathway was altered in one direction in every tumor type and there was a wide distribution of pan-cancer pathway shifts to single-cancer shifts (Figure 2C).
To test if creation of phenotype networks aided in the prioritization of TSGs and OGs, three additional pan-cancer SWAN analyses were performed. First, removal of haploinsufficiency and triploproficiency scoring yielded a moderate and consistent decrease in SWAN’s ability to prioritize TSGs and OGs across tumors (Supplementary Figures S2C, S3). Second, the protein-protein interactions (PPIs) used to build pathway networks were removed. Removal of PPIs substantially reduced the ability of SWAN to correctly prioritize TSGs and OGs. Third, removal of both phenotypes from SWAN completely abrogated prioritization of TSGs and reduced prioritization of OGs by 87%. These data support the use of phenotype information, particularly haploinsufficiency and PPIs, to aid in the analysis of CNAs in cancer. Noting that COSMIC-annotated OGs were better prioritized in SWAN than TSGs, we postulate that true TSGs on CNA regions may be ‘moderate or low impact’ TSGs in that multiple TSG deletions are necessary for a stronger pro-proliferative effect (5,11). To test this, QC was also performed using STOP and GO genes as annotated from at least two sources (see materials). Unexpectedly, STOP genes were not more enriched than known TSGs, potentially due to a lack of tissue-specific information ((24), reference Supplementary Figure S2C). Taken together, these QC tests show SWAN appropriately prioritizes genes most likely to act as true TSGs or OGs within CNA data across cancers.
Integrative analysis
While the current pan-cancer study is primarily intended to provide new light into tumor genetics using solely CNA data, there is community interest in allowing for integrative analyses. SWAN enables the routine integration of RNA and mutation data with CNA data (Supplementary Figure S2D). By comparing SWAN shifts using CNA data alone to those shifts produced by RNA layered onto CNA data, outlier pathways with exceptional RNA modulation can be identified. In bladder cancer, we found an upregulation of xenobiotics and drug metabolism RNAs and fatty acid degradation relative to DNA copy number (Supplementary Figure S2E, red), whereas the spliceosome pathway had a reduction in RNA relative to DNA copy number (Supplementary Figure S2E, blue). Mutation shifts were less striking due to the infrequently consistent mutation events for driver genes in most cancer types. TP53 is a rare exception in that it is commonly mutated in entire cohorts, shifting the p53 signaling pathway away from the null in an otherwise well-correlated pan-pathway analysis (Supplementary Figure S2F). Overall transcription shift correlations across 4,912 pathways were high when RNA was scored only if in the same direction as CNAs, while specific cancer types had widely different RNA shifts when scored additively with CNA data (Supplementary Figure S4).
Development of point-and-click integrated network analysis web platform
SWAN was designed to be useful to statisticians, bioinformaticians, and molecular biologists alike. Specifically, SWAN is available in two forms: as an R package with standalone code (for statisticians and bioinformaticians, at https://github.com/jrdelaney/SWAN) and as a hosted website (for everyone, https://www.delaneyapps.com/#SWAN). R standalone code is streamlined for minimal memory use with fast computation time. The point-and-click applications are optimized for minimal user input with logical defaults and downloadable example input files. To enable use from non-programmers, all input data are designed to be simple tab or comma delimited spreadsheets readily manipulated in Excel or Google Sheets. CNA segment to gene mapping, mm9 to mm10 or hg19 to hg38 conversion, and basic –2 to 2 scaling and normalization capabilities were built into the SWAN Data Groomer. Online Shiny App versions of the statistical SWAN software were developed to enable molecular biologists with no programming experience to readily perform these advanced SWAN network analyses, including integrated RNA and mutation analyses. As such, a priority on graphical and intuitive outputs was made.
To demonstrate broad utility of the web SWAN App for bench scientists, we provide an example of SWAN use in a generic molecular biology context. We first used SWAN to analyze RNA-seq data from control or JQ1-treated human primary myofibroblasts (49). JQ1 inhibits the DNA localization of the BET family of proteins, traditionally identified as major epigenetic regulators. BET inhibition has become a strategy for tumor treatment; however, the fundamental biology of BET proteins (BRD2/3/4/T) is still being unraveled. By querying Reactome pathways in SWAN, we identified similar conclusions as the original authors: both collagen formation and extracellular matrix organization pathways were significantly down regulated in JQ1-treated cells (Supplementary Figure S5A). We also noted several other suppressed pathways that are related to each other, including DNA replication, activation of the pre-replicative complex, and DNA unwinding. To further investigate these results, we next analyzed RNA-seq data from three other publications using multiple forms of BET inhibition across several cell lines (50–52). In all of these independent datasets, we found that BET inhibition suppressed expression networks of genes involved in the activation of the pre-replicative complex (Supplementary Figure S5B). These results were recapitulated in a genetic knockdown of BRD4 (Supplementary Figure S5C). BET inhibition has been suggested to affect genes involved in DNA replication (53), however, there is little literature detailing these findings. Interestingly, several proteins involved in the pre-replication complex, including CDC6, MCM5 and MCM7, have been implicated as BRD4-interating partners (54). Indeed, BET inhibition has resulted in DNA replication stress (55) and replication re-initiation (54). BET proteins are also implicated in the regulation of proliferating cell nuclear antigen unloading (56). The SWAN analysis presented here underscores the importance of BET proteins in the biology of DNA replication and serves as an example of SWAN usage outside of cancer genetics. This molecular biology example complements the pan-cancer analysis by showing SWAN can be used in a variety of contexts and provides useful, accurate, and interpretable information.
Identification of known and novel oncogenic pathways
In our SWAN pan-cancer CNA analysis, the hyperosmotic response was the most commonly elevated pathway (25 of 31 cancers elevated) (Figure 3A, Supplementary Table S1). The most common SWAN impactful genes within the hyperosmotic response included ARHGEF2, AQP1, and RAC1. RAC1 is a tier 1 COSMIC oncogene and ARHGEF2 is required for RAS-mediated oncogenesis. AQP1 is best known for its role in enabling water transport along an osmotic gradient in kidney proximal tubules, but is also implicated in endothelial cell migration (57). The second most commonly elevated pathway was epidermal growth factor receptor signaling (24 of 31 tumor types), led by canonical oncogenes EGFR and SRC. Negative regulation of anoikis (23 cancers elevated), led by amplifications in caveolin (CAV1), SRC, PIK3CA, and FAK/PTK2 (Figure 3B), was the third highest. Among the most commonly altered was keratinization, which drives cell cycle progression in breast epithelial cells (24). Other frequently elevated pathways include amoebiasis, cAMP signaling, DNA methylation and female meiotic division.
Figure 3.

Pan-cancer elevated CNA pathways. (A) Unusually pervasive elevated CNA pathways. Violin histograms of SWAN scores with red fill indicating significant (FDR < 0.0001) pathway elevation and blue fill indicating significant pathway suppression. (B) SWAN Circos plot. Red and blue outer rings are frequency plots of gains or deletions and the inner ribbons represent genes within the selected pathway. Labeled gene symbols are enriched for CNA gains. (C) Impact summary of novel pan-cancer SWAN elevation-prioritized genes. Green color indicates COSMIC OGs. Size is proportional to the mean Z-score SWAN contribution across cancers with OG-containing pathway elevation. Higher transparency indicates interacting protein genes influenced each gene's identification by SWAN, rather than CNAs of the gene itself. (D) SWAN network generated, with edges represent protein-protein interactions. Blue nodes are enriched for loss CNAs and red nodes (such as PEX5 and PEX19) are enriched for gain CNAs. (E) Mean ± standard error of crystal violet viability assays comparing 48h cisplatin to control 0.1% DMSO treatment. N = 4 experiments with data combined from all experiments. (F) Flow cytometry of ROS indicator H2DCFDA following 48h cisplatin or control 0.1% DMSO treatment in PEX19 overexpressing or PEX5 overexpressing cells. Significance determined from N = 3 independent experiments. Dotted line marks peak control stain. (G) Mean ± standard error of crystal violet viability assays comparing 48h cisplatin to control 0.1% DMSO treatment with or without 2mM NAC. N = 2 experiments. (H) Summary of metabolite concentrations within a metabolomic study comparing N = 5 PEX19 overexpressing SKOV3 cells relative to control vector cells. Acetyl-CoA is highlighted as an outlier. *P < 0.05, **P < 0.01, ns P > 0.05.
Modern cancer therapies are often developed to target genes that are overexpressed or constitutively active in cancer. To evaluate novel CNA targets, we compared SWAN interactome prioritization data with sgRNA screens of 324 cancer cell lines (25). Figure 3C depicts putative (defined here as not Tier 1 COSMIC) OGs which were identified as a dependency gene in at least 25% of cancer cell lines (Supplementary Table S3). Sixty-five additional prioritized OG nodes were found which were not analyzed in the 324-cell line screen (Supplementary Figure S6A, Supplementary Table S3). Included within these putative novel OG CNAs are emergent targets for cancer therapy. Of note, ADORA2A encodes adenosine receptor A2a, which negatively regulates inflammatory immune response (58), and its blockade enhances pre-clinical syngeneic models of PD-1, TIM-3 or CTLA-4 therapies (59). Another, PTK2, encodes focal adhesion kinase (FAK), which enables cells to survive a loss of adhesion (60). Two Phase II oncology trials target FAK with a small molecule inhibitor defactinib (NCT02465060 and NCT03727880). Future studies may consider the SWAN prioritized CNA-altered OGs as therapeutic targets.
To investigate the oncogenic potential of a novel hit, we next analyzed pathway disruptions which were unique to a single or a handful of cancers. Such pathways may represent unusually selective pathways for targeted treatment or early diagnosis. One largely unexplored but selectively elevated pathway involved peroxisome biogenesis. Second to testicular cancer, OV was most affected by pathway CNA elevation (Supplementary Figure S6B), but not by mutation (Supplementary Figure S6C). SWAN networks highlighted PEX5 and PEX19 as the most relevant amplified genes in serous ovarian cancer (Figure 3D, Supplementary Figure S6D). PEX5 is within an elevated CNA in 53.6% of OV tumors and PEX19 in 57.8% of OV tumors (Supplementary Figure S6E–G). PEX5 and PEX19 function to properly import peroxisome membrane proteins to the organelle (61). Overexpression (-OE) of either PEX5 or PEX19 modestly reduced cisplatin lethality within SKOV3 and OVCAR3 ovarian cancer cells (Figure 3E), neither of which contain amplifications of either gene (Supplementary Figure S6H). To investigate if this phenotype was due to a reduction of intracellular reactive oxygen species (ROS), which peroxisome metabolism contributes to (62), we stained cells for cisplatin-stimulated ROS. PEX5-OE or PEX19-OE reduced the amount of cisplatin-stimulated ROS (Figure 3F). Scavenging ROS with N-acetyl cysteine (NAC) similarly abrogated cisplatin toxicity in the control cell lines (Figure 3G). These results were not initially expected since peroxisomes can be a source of ROS, primarily through catabolic Acyl-CoA oxidase function for lipid β-oxidation (63). To test directly if lipid metabolism was disrupted, we performed unbiased ultra-performance liquid chromatography mass-spectrometry metabolomics. An outlier metabolite in PEX19-OE SKOV3 cells was a 92% reduction in Acetyl-CoA, a product of β-oxidation (Figure 3H, P < 0.051). Overall lipid content of the cells was unchanged (Supplementary Figure S6I and Supplementary Table S4), suggesting that PEX19-OE cells may need to replace Acetyl-CoA via exogenous sources of lipids, a known phenotype of ovarian cancer (64). PEX5 expression correlated with poor prognosis, whereas PEX19 did not (Supplementary Figures S6J and S6K). In summary, SWAN identified two potential OGs within the peroxisome KEGG pathway, and overexpression of each was sufficient to reduce ROS generation in chemotherapy-stressed ovarian cancer cells.
Identification of known and novel tumor suppressor pathways
Focal deletion regions of PPP2R2A, CDKN2A, ATM, NOTCH1, TP53, PTEN and the BRCA1/2 genes have been previously highlighted in CNA studies (41). In our pan-cancer SWAN analysis, these tumor suppressors were often highly influential in determining suppression of pathways; PTEN, TP53 and BRCA1/2 are highlighted in the top 1% of suppressed pathways (Supplementary Table S1). Phospholipid dephosphorylation was the most commonly suppressed pathway, found as significantly suppressed in 22 of the 31 tumor types studied (Figure 4A). Along with PTEN, losses in phosphatidylinositol 4,5-bisphosphate 5-phosphatases (INPP5E/J/K), lipid phosphate phosphohydrolase (PPAP2A/C), lipid phosphate phosphatase-related protein (LPPR1/3/4), and synaptic inositol 1,4,5-trisphosphate 5-phosphatase (SYNJ2) were commonly dysregulated by deletions. Replicative senescence (Figure 4B) and apoptotic regulators (Figure 4C) are suppressed as expected. Phospho-STAT signaling was suppressed in 17 tumor types, led by deletions in Type I interferon genes. Attachment of spindle microtubules to the kinetochore was suppressed in 20 tumor types, led by deletions connected to Aurora kinases (AURKB/C), kinetochore microtubule motor CENPE, and anaphase promoting complex regulator BUB3. OV, a highly aneuploid tumor type, was amongst the most suppressed for protein localization to the kinetochore, due to distributed gene losses on Chr1p, Chr15q, and Chr17. Chromatin organization was suppressed in 21 cancer types, with deletions in p53, ATM, β-catenin, sterol regulatory element-binding transcription factor 1 (SREBF1), lysine (K)-specific demethylase 1A (KDM1A), and histone acetyltransferase p300 (Supplementary Table S1). The most commonly identified TSGs were amongst the best-established tumor suppressors. SWAN interactome analysis predicts p53 as the most significant and common TSG (Figure 4D). Altogether, 170 novel TSGs were identified (Supplementary Table S3).
Protein quality control and cellular homeostasis was commonly disrupted across cancers. Protein deglycosylation was suppressed (19 of 31 tumor types) most commonly through deletions related to EDEM1 and DERL2, proteins involved in extracting misfolded glycoproteins as part of endoplasmic reticulum associated degradation. Protein processing in the endoplasmic reticulum was a commonly suppressed KEGG pathway (14 of 31 tumor types). We previously found autophagy, by MAP1LC3B and BECN1 gene loss, to be suppressed and therapeutically targetable in OV (6). BECN1 is a bona-fide tumor suppressor in ovarian cancer and contributes to genome instability (26,65). In this pan-cancer analysis, autophagy was suppressed in many other tumor types as well (14 of 31 tumor types), as was protein ubiquitination and degradation (9–14 tumor types). Ion homeostasis was commonly disrupted; negative regulation of potassium transport (9 tumor types), manganese ion transport (8 tumor types), copper ion homeostasis (7 tumor types), and zinc or cadmium ion response (10 or 15 tumor types, respectively) were pervasively suppressed by CNAs.
To confirm if SWAN identified novel tumor suppressor pathways which are biologically relevant, we selected the zinc and cadmium response pathways. These are dominated by concomitant loss of metallothionein genes in a cluster on Chr16q. Ovarian tumors lose this gene cluster in 60% of high-grade serous tumors. Metallothioneins are cysteine-rich proteins which chelate divalent cations within the cell: particularly Zn2+ and toxic heavy metals such as Cd2+ (66). Cadmium is an environmental toxin thought to increase lung, pancreatic, and endometrial cancer risk (67). Using ovarian cancer cell lines, metallothionein-2A (MT2A) mRNA was most highly expressed amongst all isoforms (Figure 4E). Therefore, MT2A was knocked down in ovarian cancer cells (Figure 4F). Loss of the metallothionein gene cluster may contribute to cadmium-mediated oncogenesis by allowing for genomic instability. To test this hypothesis, knockdown cells were evaluated for γH2AX foci following cadmium exposure. MT2A knockdown cells contained more γH2AX foci than control cells (Figure 4G and H), consistent with the hypothesis that these metallothionein genes protect cells against cadmium-mediated genotoxic damage.
Cancer-specific pathway alterations
Dysregulated pathways in distinct cancer types may be particularly informative. Glioblastoma multiforme (GBM) is the only tumor type elevated in ‘positive regulation of neuron death,’ while 19 tumor types are haploinsufficient (Figure 5A). This was due to SRPK2 elevation, which has recently been implicated in RNA dysregulation in GBM (68) via phosphorylation of SRSF3 (69), and elevation of PTPRZ1, which macrophages stimulate for GBM stem cell growth (70), CDK5, which is involved in neuronal migration, and canonical oncogenes MAP2K7 and ABL1 (Figure 5B). GBM cells may alter this pathway in a single chromosome gain event, as SRPK2, CDK5 and PTPRZ1 are all encoded on Chr7q (Figure 5C). By CAIRN analysis of CNAs (26), these genes are co-incidentally gained in 45% of GBM tumors.
Figure 5.

Cancer-specific CNA patterns. (A) Rare pathways had opposite SWAN shifts relative to the majority of cancer types. Shown is the example of ‘GO: Positive regulation of neuron death’ which was uniquely upregulated in the brain cancer GBM, as illustrated in a (B) SWAN feather plot and (C) Circos plot. (D) Overall survival (OS), P is from Kaplan–Meier analysis. (E) Progression-free survival (PFS) plot. (F) SWAN Circos plot. (G) Progression-free survival (PFS) plot. (H) SWAN analysis scored by HPNE CRISPR-proliferation screen hits in place of haploinsufficiency. The most suppressed pathway by magnitude is highlighted. (I) The most frequently deleted genes from (H) pathway. (J) Integrative Genomics Viewer cohort summary plots for the new African-American enriched OV cohort (SCTR) compared to The Cancer Genome Atlas (TCGA) OV cohort. Noted genes indicate SWAN prioritized genes within indicated pathway. (K) EthSEQ analysis and principal component clustering of variants in the SCTR cohort. Self-identified race is labeled for black and white patients. (L) Kaplan–Meier analysis of TCGA tumors separated by self-identified race and plotted for overall survival. (M) Kaplan–Meier plot of TCGA data separated by SWAN shifts.
In Low-Grade Glioma (LGG), Hallmark Myc Targets V1 was marked as ‘suppressed’ whereas 19 cancer types were marked as ‘elevated’. Interestingly, the subset of tumors with CNA losses within MYC targets both had greater overall survival (Figure 5D) and progression free survival (Figure 5E) in LGG. These genes are enriched on Chr1p and Chr19q, which was identified in the TCGA publication as prognostic of IDH1 mutant tumors (71). LGG is often driven by MYC or IDH1/2 mutations, suggesting that the tumors which have spontaneously lost an array of MYC targets have attenuated their oncogenic potential.
Uveal melanoma (UVM) exhibits an unusual mode of initial extravasation which first involves intercalation with endothelial cells (72). While 11 cancers are elevated in the KEGG pathway melanogenesis, which usually involves gains in Wnt-β-catenin regulating factors FZD1, GNAI1 and WNT3A, UVM was the sole cancer suppressed in this pathway due to Chr1 and Chr3 losses overlapping MITF, GSK3B and DVL1 (Figure 5F). Patients with these losses are in the poor prognosis group, particularly for progression free survival (Figure 5G). Poor prognosis is associated with increased immunosuppressive profiles (73).
To address tissue specificity in an unbiased fashion, the top 1% variable pathways were K-means clustered. There were clearly different cancer subsets with regard to phospho-STAT signaling, keratinization, epigenetic regulation of rDNA, protein carboxylation, and serine peptidase (Supplementary Figure S7). Cancer types did not strongly cluster together. The remaining cancer-specific altered pathways can be found in Supplementary Table S1.
Tissue-specific gene weighting reveals a suppression of cytosolic DNA response
Whole-genome CRISPR-Cas9 screens of non-transformed normal tissue have identified tissue-specific drivers of proliferation (24). In lieu of haploinsufficiency data, we instead applied scoring weights on pancreatic cancer (PAAD) CNAs in SWAN using genes enriched for proliferation changes from a CRISPR-Cas9 screen in primary immortalized pancreatic HPNE cells. In a KEGG pathway analysis, the cytosolic DNA-sensing pathway was the most suppressed pathway in PAAD (-9.4 SWAN shift, FDR ≤ 8.1 × 10−8, Figure 5H). This was led by the IFNA genes, which produce type I interferons and act as positive feedback inducers of a central dsDNA sensor cGAS (74) (Figure 5I). Another suppressed gene was DDX58, a primarily dsRNA sensor which can also detect some types of dsDNA (75). Chromosome instability often results in micronuclei, which normally activate cGAS-STING signaling. However, in some cancer cells this pathway was found to be attenuated by an unknown mechanism (76), allowing for cell survival and increased metastasis. CNAs may be one mechanism cancer cells use to reduce cGAS-STING pathway signaling, particularly in pancreatic cancer. Weighting by tissue-specific sgRNA screens thus yielded further insights into CNA patterns and tumor biology.
SWAN case study on race-specific CNA patterns
African American data represents only 6% of the tumors present in TCGA OV data. Expansion of data and analysis in this group is warranted. We obtained 12 tumors from African American high-grade serous ovarian cancer patients and 8 non-Hispanic white patient controls and performed whole-exome sequencing (Supplementary Table S5). Since normal tissue was not available for these unique samples, confident somatic SNV analysis was complicated by rare but normal variants. CNAs, conversely, are uncommon in bulk normal tissue and ascertainment of CNAs was possible using Control-FREEC software (33). This method was remarkably similar in overall cohort CNA calls to TCGA analyzed tumors (Figure 5J), demonstrating technical consistency. To investigate the possible race-specific CNAs in African American patients, SWAN was used to compare African American patients to white patients from this study as well as combined with the TCGA study. The cytokine production pathway was found to be significantly elevated in African American patient tumors relative to white patient tumors (Figure 5J gene labels). Self-reported race matched race-defining variants found by EthSEQ analysis on these tumors, with expected higher admixture present in African American patients (Figure 5K). Black OV patients respond poorly to therapy relative to white patients, even when taking into account socioeconomic factors and comorbidities (77). This trend, albeit not significant, was seen in TCGA survival data (Figure 5L). SWAN shifts mapping to elevation of cytokine production were associated with poor prognosis in OV overall (Figure 5M). Existing socioeconomic factors which lead to persistent inflammation in black patients may allow for de-repressed cytokine production in black patient tumor cells, which would otherwise allow T-cell responses to clear tumors. Low-dose rapamycin treatment may re-enable T-cell clearance within these patients (78,79).
Pathways associated with SNV mutant drivers
Each SNV/indel driver mutation may be predicted to require its own set of CNAs to assist in cancer development. To test this hypothesis, we analyzed which tumor types had CNA-altered pathways within the subset of specific mutant tumors, relative to non-mutant tumors of the same histotype. We tested commonly mutated TSGs/OGs: TP53, CDKN2A, KRAS or NRAS or HRAS, BRAF, BRCA1 or BRCA2, EGFR, PTEN, HIF1A or VHL, RB1, ATM, APC and MSHs (MSH2,3,4,5, or 6). Of all of these possible driver mutations, TP53 had the most pathways commonly affected in multiple cancers (Supplementary Table S6). This is consistent with the observation that TP53 mutation is the most significantly associated with aneuploidy by multiple orders of magnitude (14). A suppression of KEGG: autophagy in p53 mutant subsets of tumors was observed in 8 cancer types (Figure 6A). Uterine/endometrial cancer is known to have worse prognosis with p53 mutation, and these tumors were severely reduced in autophagy gene content (Figure 6B). OV, the cancer type we have thoroughly investigated for its loss of autophagy (6,26,65), was not found in this set due to the ubiquity of p53 mutations, precluding a non-mutant control comparison. Mutation in p53 is also commonly associated with elevation of GO: Regulation of Cell Adhesion Mediated by Integrin (14 tumor types elevated, led by FAK/PTK2 and LYN).
Figure 6.
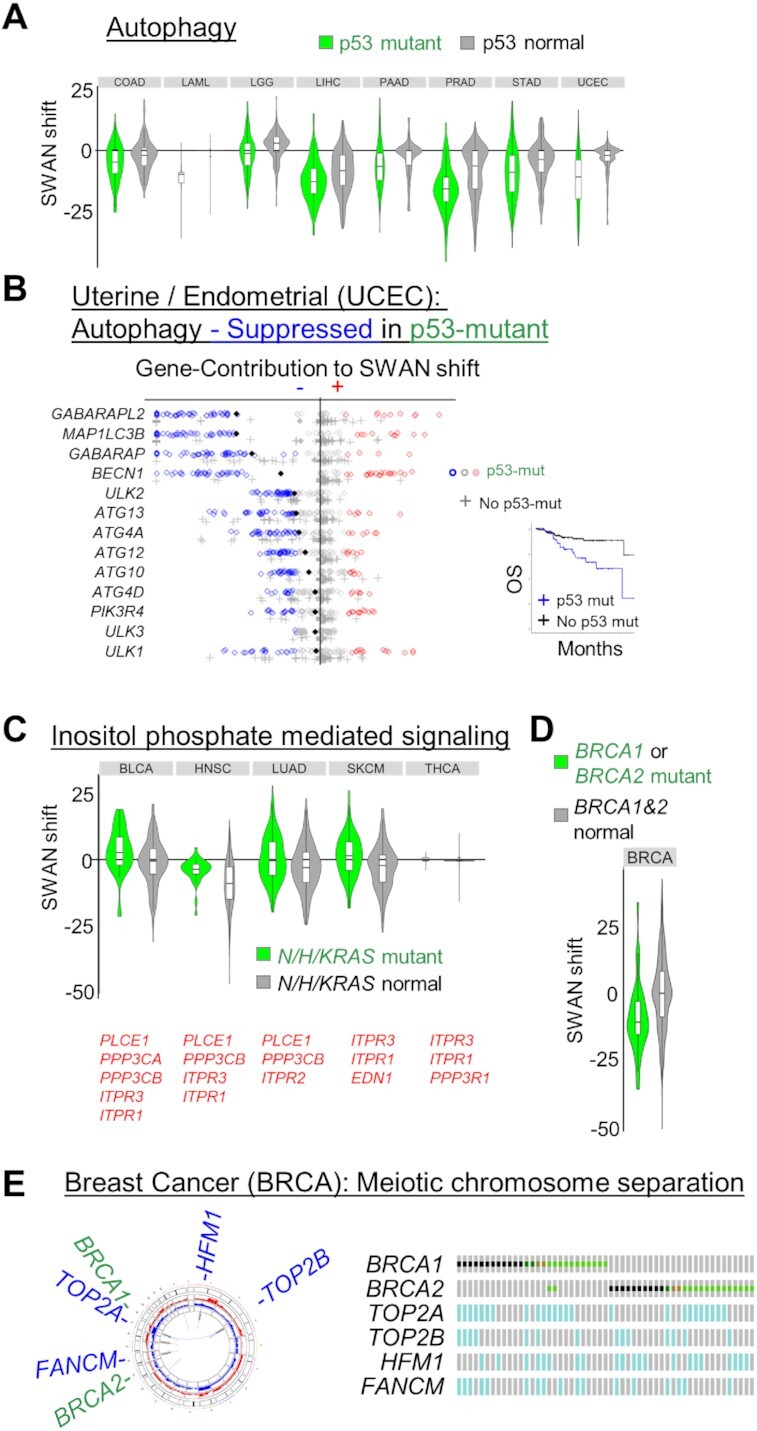
Mutation associated SWAN shifts. (A) All cancers with a significantly different SWAN shift spectrum for p53 mutant tumors compared to p53 wild-type tumors are shown for the KEGG: Autophagy pathway. (B) SWAN feather plot of suppressed autophagy genes. UCEC samples with p53 mutation are plotted as circles, samples with no p53 mutation as crosses. Filled diamonds represent mean SWAN shifts per gene. Inset panel shows overall survival of p53 mutant UCEC compared to non-mutant UCEC. (C) All cancers with a significantly different SWAN shift spectrum for RAS (NRAS, HRAS or KRAS) mutant tumors compared to RAS wild-type tumors are shown for the GO: Inositol phosphate mediated signaling pathway. (D) Breast was the only cancer with a significantly different SWAN shift spectrum for BRCA1 or BRCA2 mutant tumors compared to BRCA wild-type tumors for the GO: meiotic chromosome separation pathway, with (E) pathway genes highlighted for regions of gene deletion by Circos plot (left) or Oncoprint (right).
The next most consistently altered pathway sets occurred in RAS mutant cancers. In these cases, the commonly dysregulated pathways may further enhance the activation of the RAS pathway. RAS mutation was associated with an increase in inositol phosphate mediated signaling (Figure 6C), likely increasing activation of PI3K (80). The remainder of known, common driver mutations were distinct to individual cancer types. For example, the BRCA1/2 driven cancers were associated with a decrease in meiotic chromosome condensation specifically in breast cancer (Figure 6D). This pathway was suppressed via allelic loss in FANCM, TOP2A/B and HFM1 (Figure 6E). Since CNAs are far more common than individual driver SNV mutations, more samples are needed to provide the statistical threshold for CNA pathway differences associated with other individually rare SNV drivers. Overall, these data support the model that SNV/indel mutations alter biology in a manner which is further exacerbated by CNAs.
Prognostic alterations
If SWAN-identified CNA pathways drive the biology of tumors, then it would be expected that some pathways influence patient prognosis. Both cBioPortal and ProteinAtlas offer survival curves comparing ‘low’ and ‘high’ expression of individual genes for this purpose. To highlight the utility of SWAN-based pathway analysis for prognosis comparisons, we provide one use-case study of the OV dataset as an example.
An active area of investigation in OV is how homology directed repair (HDR) processes affect chemotherapy response. Clinical data associated with genomics from the past two decades have assessed platinum-based survival, and current trials continue to assess the interaction with PARP inhibitors. Haploinsufficiency of the HDR genes BRCA1 and BRCA2 is well established (81–83). Yet, in the platinum-based therapy TCGA cohort, single allele loss of either gene is not prognostic (P < 0.52 for either, P < 0.98 for BRCA1 or P < 0.081 for BRCA2, Supplementary Figure S8A). Analyzing CNAs within the HDR pathway with SWAN and comparing with prognosis yields nominal significance with chemotherapy response (P < 0.051, Supplementary Figure S8B). Finally, layering RNA data consistent with CNA data captures the best separation (P < 0.010), with losses of HDR genes conferring better response. This effect was not found with RNA alone (P < 0.25). Suppression of HDR conferring chemotherapy sensitivity is consistent with clinical practices with OV (84,85).
There is an important limitation to pathway-based prognostic approaches. Sometimes a single gene does indeed drive biology in a dominant fashion. We note two publications both implicate SMAD2 loss as negatively prognostic in colorectal cancers (86,87). SMAD2 is a regulator of the TGF-beta pathway, which is comprised of both positive and negative regulators (88). When two significantly suppressed pathways including SMAD2 are analyzed for prognosis by SWAN analysis of CNAs, ‘GO Organ Growth’ and ‘GO Regulation of Cellular Response to TGF-beta Signaling’, neither are positively or negatively prognostic in COAD. These findings, taken together, indicate a pathway approach may be useful in those situations wherein multiple genes contribute in the same direction to pathway efficiency, whereas single-gene approaches remain useful when pathways contain both positive and negative regulators or a gene produces sufficient effects on its own.
We next explored whether pathway scores can separate prognostic groups. In a log-rank test analysis using whole-cohort data, 12,781 pathways from 18 cancers were found to be significantly prognostic by comparing upper and lower tertiles of SWAN shifts. Survival estimates can be misleading (89), and we accordingly found an ability to erroneously call a single pathway as positively or negatively prognostic, depending on which patients were randomly selected (Figure 7A, Supplementary Figure S8C). Desiring to prioritize the 12,781 pathway hits into those most likely to be medically informative, we developed a machine learning approach. A random subset of two-thirds of tumors was used to build Cox-proportional hazard (Cox-PH) models from SWAN network shift data. The remaining one-third of tumors per random selection was used to generate hazard ratios using the Cox-PH model. 101 random patient selections were used to determine which pathways scored as similarly prognostic in the test set. While Cox-PH is traditionally used for multiple covariates, its predictive capacity for single variables was herein used. The machine-learning approach here required an identical hazard ratio direction and subsequent test group log-rank significance in >80% of patient selections to call a pathway as prognostic (Figure 7B). This strategy was amenable to 18 cancer types with sufficient survival data and reduced the prognostic pathways from 12,781 pathways within the pan-cancer cohort to 1,696 pathways (Supplementary Figure S8D-F). 1,079 were from the highly predictable LGG data, leaving 617 machine-learned prognostic pathways from 17 other cancer types. While an ideal machine-learning approach would include an independent dataset, data of sufficient size and comparable form across cancer types were unavailable. Our approach nonetheless sharply narrowed the scope of likely prognostic CNA pathways. Kaplan–Meier curves of the entire cohort confirm that machine learned pathways were negatively prognostic (Figure 7B) and positively prognostic (Figure 7C), suggesting that SWAN data may be used to categorize patients by prognosis. Machine-learned prognostic pathways for each cancer type are supplied as Supplementary Table S7.
Figure 7.

Machine-learning approach for improved prognostic estimates of CNA influenced pathways. (A) An example of a nominally-significant prognostic pathway, corrected by a machine learning approach. Patient data are first separated by SWAN pathway shifts, with the ‘low’ and ‘high’ groups separated by 1 SD centered at the median shift. With the entire patient cohort considered, a Kaplan–Meier analysis yields P < 0.047. 101 training sets of SWAN shift data build risk scores, then the upper quartile risk is compared to the lower quartile risk group by Kaplan–Meier analysis to yield a P-value for the test group. Colored circles indicate HR at least 1 SD from the null 1 value. Potential false positive prognostic pathways were removed by (1) determining significance in the applied CoxPH model risk for each test set, and (2) determining CoxPH HR direction in each training set. The most prognostic pathways are labeled as those with >80% of randomizations as both significant and in same HR direction, such as in example (B). A Kaplan–Meier overall survival curve with 95% confidence interval is shown for the whole patient cohort (right panel). (C) A negatively prognostic Kaplan–Meier overall survival curve with 95% confidence interval is shown. In this case, 98% of patient picks were significant.
Online atlas
We uploaded our SWAN analysis of 31 cancer types studied by TCGA into an online portal. The accessible data include SWAN CNA network-level scores and per-patient scores for all cancer types for four pathway sets: GO, KEGG, Hallmark and Reactome. This database is provided online at https://delaney.shinyapps.io/cnalysis_tcga_atlas/. Summary data for all analyses discussed here are also provided as Supplementary Tables S1–S7.
DISCUSSION
Copy-number alterations represent a wealth of unexplored oncogene and tumor suppressor gene data within the cancer genome landscape. Expanse and heterogeneity of these data have previously hindered identification of biologically relevant CNAs. SWAN demonstrably improves the identification of TSGs and OGs on CNAs within a pan-cancer dataset. In addition to this computational validation, wet-lab validation was performed. SWAN identified PEX5 and PEX19 as elevated genes within the peroxisome biogenesis pathway. These genes each conferred platinum resistance by oxidative control. SWAN marked cadmium response as suppressed in 15 tumor types, and knockdown of metallothionein 2A conferred increased γH2AX foci formation in response to cadmium. Newly sequenced tumors analyzed by SWAN determined that elevation of cytokine production genes occurs within tumors from an underrepresented racial group. SWAN was developed with an emphasis on ease-of-use and widespread applicability so that SWAN may be utilized in future experiments with complex genetics. While optimized for oncology, SWAN may be considered for use as generally applicable pathway analysis software.
A limitation with modern genetically-targeted cancer treatments is the small percentage of patients who may benefit from a specific genetic alteration. Some estimates are that only 8.3% of patients are eligible for genetically-targeted therapy (2018), compared to 5.1% over a decade prior (2006) (90). In OV, which lacks canonically targetable mutant drivers, alternative tests have been developed to test for functional deficiency in homology directed repair, thereby enabling PARP inhibitor targeted therapy even in the absence of BRCA1/2 mutations (91). This HDR deficiency is influenced by the ubiquitous CNAs described here. Our analysis of CNAs represents a wealth of potentially targetable pathways that are altered in double-digit percentages of patients. Moreover, therapeutic windows as a result of tumor CNAs are of high likelihood since functional normal cells do not contain clonal somatic CNAs. Targeting pathways, rather than individual genes, may reduce the likelihood of resistance mutation development and resurgent cancer, especially if multiple drugs are used (92).
While previous CNA studies have thoroughly studied focal homozygous deletions and arm-level aneuploidy in cancer, these studies do not provide a comprehensive statistical assessment of what patterns of changes can alter biological pathways. This is critical because a pathway may be suppressed via multiple chromosome arms or monoallelic changes which may differ tumor-to-tumor. An example of this is the keratinization pathway. While it was previously shown that single gene overexpression of keratinization factors can promote cell cycle progression, these effects were cell type specific (24). However, those cell-type specificity results must be a consequence of other factors within the cell which ameliorate or exacerbate the effects of single gene overexpression. Networks, conversely, take into account these disparate interacting factors that differ between individual tumor samples. An analysis of focal-amplification regions consistently altered across tumors identified regulators of Nf-κB, Wnt/β-catenin, MYC, AKT, ERBB2, Cyclin-D1 and -E1, and TERC (41). Pathway alterations within these commonly amplified segments centered on chromatin modifiers such as BRD4, KDM2A, and KDM5A (41). These genes are well-known oncogenes and were often identified as impactful by SWAN. However, SWAN was able to additionally identify cancer addictions depleted in sgRNA screens and further prioritized 65 novel oncogenes. Pan-cancer and cancer-specific CNA pathways were identified and their relevance to primary chemotherapy can be referenced to the machine-learned prognostic pathways released here.
A caveat of the pan-cancer analysis presented here is the deliberate absence of RNA data. This choice was made because many previous RNA pathway analyses have been performed and CNA patterns are more exclusive to tumor tissue than normal tissue. For example, a tSNE plot of a pan-cancer RNA analysis by Fang et al. shows a clear delineation of tumor site clustering (93), while our clustering of CNA pathway data results in clusters containing a handful of tumor sites (Supplementary Figure S7). Prognostic interpretations also differ but are likely complementary; the Fang et al. study highlights an elevated DAP12/TYROBP-containing pathway as negatively prognostic in BRCA due to IGFBP and Akt signaling. Here, we found elevated carbohydrate transport was most negatively prognostic in BRCA, which may be related. The Frost lab, like our current study, has found gene-centric estimates of prognosis are conceptually limited by strong co-regulation of other genes within the same pathway (for example, E2F targets) (94). Like the Frost CNA study (95), the LGG cancer type was most predictable in our study due to high concordance of CNA data. Pathway-level views of noisy, low gene-gene expression correlations, in contrast, enable a more reliable predictive performance. Nonetheless, gene-level expression remains valuable to analyze prognosis, as evidenced by the development and use of iPath (46), SurvExpress (96), and KmPlot (97). Future studies are needed to ascertain if CNA-inferred prognosis and other biological estimates of pathway changes improve through integration with RNA data.
DATA AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joe Delaney, PhD (delaneyj@musc.edu).
Materials Availability
Cell lines and plasmids generate in this study are maintained at the Medical University of South Carolina and will be made available upon request.
Data and Code Availability
Protected human datasets generated during this study are available in dbGaP, accession phs002313. Flow cytometry data are available in FlowRepository under accession FR-FCM-Z3V2.
The code generated during this study are available at Github (https://github.com/jrdelaney/SWAN) and web software at https://www.delaneyapps.com/#SWAN.
Supplementary Material
ACKNOWLEDGEMENTS
We thank our molecular biology collaborators for feedback on SWAN analysis for their projects, which led to several substantial improvements prior to manuscript submission. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, NCI, or NCATS.
Author Contributions: J.R.D., E.A.P., D.T.L. and R.R.B. designed the research. K.E.A. advised statistical considerations. J.R.D., R.R.B. and C.M.J. wrote the paper. J.R.D. scripted SWAN. All authors performed the research and analyzed the data.
Contributor Information
Robert R Bowers, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA.
Christian M Jones, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA.
Edwin A Paz, Departments of Neurology, Neurobiology, and Cell Biology, and the Duke Center for Neurodegeneration & Neurotherapeutics, Duke University School of Medicine, Durham, NC, USA.
John K Barrows, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA.
Kent E Armeson, Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA.
David T Long, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA.
Joe R Delaney, Department of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
South Carolina Clinical & Translational Research Institute with an academic home at the Medical University of South Carolina CTSA NIH NCATS [UL1TR001450 to J.R.D., TL1TR001451 to J.K.B.]; NIH [CA207729 to J.R.D., CA256104 to J.R.D., GM119512 to D.T.L., GM132055 to C.M.J.]; Rivkin Center for Ovarian Cancer (to J.R.D., in part); the MUSC Proteogenomics Facility was used and is supported by GM103499 and MUSC’s Office of the Vice President for Research; supported in part by the Biostatistics Shared Resource, Hollings Cancer Center, Medical University of South Carolina [P30 CA138313]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gambacorti-Passerini C. Part I: milestones in personalised medicine–imatinib. Lancet Oncol. 2008; 9:600. [DOI] [PubMed] [Google Scholar]
- 2. Flaherty K.T., Gray R., Chen A., Li S., Patton D., Hamilton S.R., Williams P.M., Mitchell E.P., Iafrate A.J., Sklar J.et al.. The molecular analysis for therapy choice (NCI-MATCH) trial: lessons for genomic trial design. J. Natl. Cancer Inst. 2020; 112:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Research, N Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar S., Warrell J., Li S., McGillivray P.D., Meyerson W., Salichos L., Harmanci A., Martinez-Fundichely A., Chan C.W.Y., Nielsen M.M.et al.. Passenger mutations in more than 2,500 cancer genomes: overall molecular functional impact and consequences. Cell. 2020; 180:915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davoli T., Xu A.W., Mengwasser K.E., Sack L.M., Yoon J.C., Park P.J., Elledge S.J.. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013; 155:948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delaney J.R., Patel C.B., Willis K.M., Haghighiabyaneh M., Axelrod J., Tancioni I., Lu D., Bapat J., Young S., Cadassou O.et al.. Haploinsufficiency networks identify targetable patterns of allelic deficiency in low mutation ovarian cancer. Nat. Commun. 2017; 8:14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchell T.J., Turajlic S., Rowan A., Nicol D., Farmery J.H.R., O’Brien T., Martincorena I., Tarpey P., Angelopoulos N., Yates L.R.et al.. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell. 2018; 173:611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patch A.M., Christie E.L., Etemadmoghadam D., Garsed D.W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P.J.et al.. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489–494. [DOI] [PubMed] [Google Scholar]
- 9. Wedge D.C., Gundem G., Mitchell T., Woodcock D.J., Martincorena I., Ghori M., Zamora J., Butler A., Whitaker H., Kote-Jarai Z.et al.. Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat. Genet. 2018; 50:682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mamlouk S., Childs L.H., Aust D., Heim D., Melching F., Oliveira C., Wolf T., Durek P., Schumacher D., Blaker H.et al.. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat. Commun. 2017; 8:14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y., Chen C., Xu Z., Scuoppo C., Rillahan C.D., Gao J., Spitzer B., Bosbach B., Kastenhuber E.R., Baslan T.et al.. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature. 2016; 531:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ben-David U., Amon A.. Context is everything: aneuploidycancer. Nat. Rev. Genet. 2019; 21:44–62. [DOI] [PubMed] [Google Scholar]
- 13. Cai Y., Crowther J., Pastor T., Abbasi Asbagh L., Baietti M.F., De Troyer M., Vazquez I., Talebi A., Renzi F., Dehairs J.et al.. Loss of chromosome 8p governs tumor progression and drug response by altering lipid metabolism. Cancer Cell. 2016; 29:751–766. [DOI] [PubMed] [Google Scholar]
- 14. Taylor A.M., Shih J., Ha G., Gao G.F., Zhang X., Berger A.C., Schumacher S.E., Wang C., Hu H., Liu J.et al.. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018; 33:676–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santaguida S., Vasile E., White E., Amon A.. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015; 29:2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Torres E.M., Springer M., Amon A.. No current evidence for widespread dosage compensation in s. cerevisiae. Elife. 2016; 5:e10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E., Zhou J.Y., Petyuk V.A., Chen L., Ray D.et al.. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016; 166:755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheltzer J.M., Ko J.H., Replogle J.M., Habibe Burgos N.C., Chung E.S., Meehl C.M., Sayles N.M., Passerini V., Storchova Z., Amon A. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell. 2017; 31:240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang R.W., Vigano S., Ben-David U., Amon A., Santaguida S.. Aneuploid senescent cells activate NF-kappaB to promote their immune clearance by NK cells. EMBO Rep. 2021; 22:e52032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rutledge S.D., Douglas T.A., Nicholson J.M., Vila-Casadesus M., Kantzler C.L., Wangsa D., Barroso-Vilares M., Kale S.D., Logarinho E., Cimini D. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci. Rep. 2016; 6:22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E.et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Group P.T.C., Calabrese C., Davidson N.R., Demircioglu D., Fonseca N.A., He Y., Kahles A., Lehmann K.V., Liu F., Shiraishi Y.et al.. Genomic basis for RNA alterations in cancer. Nature. 2020; 578:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Solimini N.L., Xu Q., Mermel C.H., Liang A.C., Schlabach M.R., Luo J., Burrows A.E., Anselmo A.N., Bredemeyer A.L., Li M.Z.et al.. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012; 337:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sack L.M., Davoli T., Li M.Z., Li Y., Xu Q., Naxerova K., Wooten E.C., Bernardi R.J., Martin T.D., Chen T.et al.. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell. 2018; 173:499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Behan F.M., Iorio F., Picco G., Goncalves E., Beaver C.M., Migliardi G., Santos R., Rao Y., Sassi F., Pinnelli M.et al.. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019; 568:511–516. [DOI] [PubMed] [Google Scholar]
- 26. Delaney J.R., Patel C.B., Bapat J., Jones C.M., Ramos-Zapatero M., Ortell K.K., Tanios R., Haghighiabyaneh M., Axelrod J., DeStefano J.W.et al.. Autophagy gene haploinsufficiency drives chromosome instability, increases migration, and promotes early ovarian tumors. PLoS Genet. 2020; 16:e1008558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilm A., Aw P.P., Bertrand D., Yeo G.H., Ong S.H., Wong C.H., Khor C.C., Petric R., Hibberd M.L., Nagarajan N.. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012; 40:11189–11201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koboldt D.C., Chen K., Wylie T., Larson D.E., McLellan M.D., Mardis E.R., Weinstock G.M., Wilson R.K., Ding L.. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009; 25:2283–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Eberhard C.et al.. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alfoldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P.et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Romanel A., Zhang T., Elemento O., Demichelis F.. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics. 2017; 33:2402–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boeva V., Popova T., Bleakley K., Chiche P., Cappo J., Schleiermacher G., Janoueix-Lerosey I., Delattre O., Barillot E.. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012; 28:423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuhn R.M., Haussler D., Kent W.J.. The UCSC genome browser and associated tools. Brief. Bioinform. 2013; 14:144–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M.et al.. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015; 348:880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martincorena I., Fowler J.C., Wabik A., Lawson A.R.J., Abascal F., Hall M.W.J., Cagan A., Murai K., Mahbubani K., Stratton M.R.et al.. Somatic mutant clones colonize the human esophagus with age. Science. 2018; 362:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee-Six H., Olafsson S., Ellis P., Osborne R.J., Sanders M.A., Moore L., Georgakopoulos N., Torrente F., Noorani A., Goddard M.et al.. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019; 574:532–537. [DOI] [PubMed] [Google Scholar]
- 38. Yoshida K., Gowers K.H.C., Lee-Six H., Chandrasekharan D.P., Coorens T., Maughan E.F., Beal K., Menzies A., Millar F.R., Anderson E.et al.. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020; 578:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moore L., Leongamornlert D., Coorens T.H.H., Sanders M.A., Ellis P., Dentro S.C., Dawson K.J., Butler T., Rahbari R., Mitchell T.J.et al.. The mutational landscape of normal human endometrial epithelium. Nature. 2020; 580:640–646. [DOI] [PubMed] [Google Scholar]
- 40. Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M.et al.. The landscape of somatic copy-number alteration across human cancers. Nature. 2010; 463:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhang C.Z., Wala J., Mermel C.H.et al.. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013; 45:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith J.C., Sheltzer J.M.. Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. Elife. 2018; 7:e39217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Drier Y., Sheffer M., Domany E.. Pathway-based personalized analysis of cancer. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vaske C.J., Benz S.C., Sanborn J.Z., Earl D., Szeto C., Zhu J., Haussler D., Stuart J.M.. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics. 2010; 26:i237–, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Su K., Yu Q., Shen R., Sun S.Y., Moreno C.S., Li X., Qin Z.S.. Pan-cancer analysis of pathway-based gene expression pattern at the individual level reveals biomarkers of clinical prognosis. Cell Rep Methods. 2021; 1:100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nygard S., Lingjaerde O.C., Caldas C., Hovig E., Borresen-Dale A.L., Helland A., Haakensen V.D.. PathTracer: High-sensitivity detection of differential pathway activity in tumours. Sci. Rep. 2019; 9:16332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A.. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer. 2018; 18:696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Suzuki K., Kim J.D., Ugai K., Matsuda S., Mikami H., Yoshioka K., Ikari J., Hatano M., Fukamizu A., Tatsumi K.et al.. Transcriptomic changes involved in the dedifferentiation of myofibroblasts derived from the lung of a patient with idiopathic pulmonary fibrosis. Mol Med Rep. 2020; 22:1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nagarajan S., Bedi U., Budida A., Hamdan F.H., Mishra V.K., Najafova Z., Xie W., Alawi M., Indenbirken D., Knapp S.et al.. BRD4 promotes p63 and GRHL3 expression downstream of FOXO in mammary epithelial cells. Nucleic Acids Res. 2017; 45:3130–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Garcia-Carpizo V., Ruiz-Llorente S., Sarmentero J., Grana-Castro O., Pisano D.G., Barrero M.J.. CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC regulatory axis in proliferation. Epigenet. Chromatin. 2018; 11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ren C., Zhang G., Han F., Fu S., Cao Y., Zhang F., Zhang Q., Meslamani J., Xu Y., Ji D.et al.. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:7949–7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Du Z., Song X., Yan F., Wang J., Zhao Y., Liu S.. Genome-wide transcriptional analysis of BRD4-regulated genes and pathways in human glioma U251 cells. Int. J. Oncol. 2018; 52:1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang J., Dulak A.M., Hattersley M.M., Willis B.S., Nikkila J., Wang A., Lau A., Reimer C., Zinda M., Fawell S.E.et al.. BRD4 facilitates replication stress-induced DNA damage response. Oncogene. 2018; 37:3763–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bowry A., Piberger A.L., Rojas P., Saponaro M., Petermann E.. BET inhibition induces HEXIM1- and RAD51-Dependent conflicts between transcription and replication. Cell Rep. 2018; 25:2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang M.S., Kim J., Ryu E., Ha N.Y., Hwang S., Kim B.G., Ra J.S., Kim Y.J., Hwang J.M., Myung K.et al.. PCNA unloading is negatively regulated by BET proteins. Cell Rep. 2019; 29:4632–4645. [DOI] [PubMed] [Google Scholar]
- 57. Verkman A.S. Aquaporins at a glance. J. Cell Sci. 2011; 124:2107–2112. [DOI] [PubMed] [Google Scholar]
- 58. Ohta A., Gorelik E., Prasad S.J., Ronchese F., Lukashev D., Wong M.K., Huang X., Caldwell S., Liu K., Smith P.et al.. A2A adenosine receptor protects tumors from antitumor t cells. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leone R.D., Emens L.A.. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer. 2018; 6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sulzmaier F.J., Jean C., Schlaepfer D.D.. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer. 2014; 14:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim P.K., Hettema E.H.. Multiple pathways for protein transport to peroxisomes. J. Mol. Biol. 2015; 427:1176–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schrader M., Fahimi H.D.. Peroxisomes and oxidative stress. Biochim. Biophys. Acta. 2006; 1763:1755–1766. [DOI] [PubMed] [Google Scholar]
- 63. Poirier Y., Antonenkov V.D., Glumoff T., Hiltunen J.K.. Peroxisomal beta-oxidation–a metabolic pathway with multiple functions. Biochim. Biophys. Acta. 2006; 1763:1413–1426. [DOI] [PubMed] [Google Scholar]
- 64. Motohara T., Masuda K., Morotti M., Zheng Y., El-Sahhar S., Chong K.Y., Wietek N., Alsaadi A., Karaminejadranjbar M., Hu Z.et al.. An evolving story of the metastatic voyage of ovarian cancer cells: cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene. 2019; 38:2885–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kumar M., Bowers R.R., Delaney J.R.. Single-cell analysis of copy-number alterations in serous ovarian cancer reveals substantial heterogeneity in both low- and high-grade tumors. Cell Cycle. 2020; 19:3154–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Klaassen C.D., Liu J., Choudhuri S.. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu. Rev. Pharmacol. Toxicol. 1999; 39:267–294. [DOI] [PubMed] [Google Scholar]
- 67. McElroy J.A., Kruse R.L., Guthrie J., Gangnon R.E., Robertson J.D.. Cadmium exposure and endometrial cancer risk: a large midwestern U.S. population-based case-control study. PLoS One. 2017; 12:e0179360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Song X., Wan X., Huang T., Zeng C., Sastry N., Wu B., James C.D., Horbinski C., Nakano I., Zhang W.et al.. SRSF3-regulated RNA alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res. 2019; 79:5288–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Long Y., Sou W.H., Yung K.W.Y., Liu H., Wan S.W.C., Li Q., Zeng C., Law C.O.K., Chan G.H.C., Lau T.C.K.et al.. Distinct mechanisms govern the phosphorylation of different SR protein splicing factors. J. Biol. Chem. 2019; 294:1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi Y., Ping Y.F., Zhou W., He Z.C., Chen C., Bian B.S., Zhang L., Chen L., Lan X., Zhang X.C.et al.. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017; 8:15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cancer Genome Atlas Research, N. Brat D.J., Verhaak R.G., Aldape K.D., Yung W.K., Salama S.R., Cooper L.A., Rheinbay E., Miller C.R., Vitucci M.et al.. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 2015; 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Onken M.D., Li J., Cooper J.A.. Uveal melanoma cells utilize a novel route for transendothelial migration. PLoS One. 2014; 9:e115472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Figueiredo C.R., Kalirai H., Sacco J.J., Azevedo R.A., Duckworth A., Slupsky J.R., Coulson J.M., Coupland S.E.. Loss of BAP1 expression is associated with an immunosuppressive microenvironment in uveal melanoma, with implications for immunotherapy development. J. Pathol. 2020; 250:420–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ma F., Li B., Liu S.Y., Iyer S.S., Yu Y., Wu A., Cheng G.. Positive feedback regulation of type i IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 2015; 194:1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zevini A., Olagnier D., Hiscott J.. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017; 38:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bakhoum S.F., Ngo B., Laughney A.M., Cavallo J.A., Murphy C.J., Ly P., Shah P., Sriram R.K., Watkins T.B.K., Taunk N.K.et al.. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018; 553:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hildebrand J.S., Wallace K., Graybill W.S., Kelemen L.E.. Racial disparities in treatment and survival from ovarian cancer. Cancer Epidemiol. 2019; 58:77–82. [DOI] [PubMed] [Google Scholar]
- 78. Mannick J.B., Del Giudice G., Lattanzi M., Valiante N.M., Praestgaard J., Huang B., Lonetto M.A., Maecker H.T., Kovarik J., Carson S.et al.. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014; 6:268ra179. [DOI] [PubMed] [Google Scholar]
- 79. Mannick J.B., Morris M., Hockey H.P., Roma G., Beibel M., Kulmatycki K., Watkins M., Shavlakadze T., Zhou W., Quinn D.et al.. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018; 10:eaaq1564. [DOI] [PubMed] [Google Scholar]
- 80. Castellano E., Downward J.. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011; 2:261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. King T.A., Li W., Brogi E., Yee C.J., Gemignani M.L., Olvera N., Levine D.A., Norton L., Robson M.E., Offit K.et al.. Heterogenic loss of the wild-type BRCA allele in human breast tumorigenesis. Ann. Surg. Oncol. 2007; 14:2510–2518. [DOI] [PubMed] [Google Scholar]
- 82. Pathania S., Bade S., Le Guillou M., Burke K., Reed R., Bowman-Colin C., Su Y., Ting D.T., Polyak K., Richardson A.L.et al.. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nat. Commun. 2014; 5:5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tan S.L.W., Chadha S., Liu Y., Gabasova E., Perera D., Ahmed K., Constantinou S., Renaudin X., Lee M., Aebersold R.et al.. A class of environmental and endogenous toxins induces BRCA2 haploinsufficiency and genome instability. Cell. 2017; 169:1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Birkbak N.J., Wang Z.C., Kim J.Y., Eklund A.C., Li Q., Tian R., Bowman-Colin C., Li Y., Greene-Colozzi A., Iglehart J.D.et al.. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012; 2:366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Takaya H., Nakai H., Takamatsu S., Mandai M., Matsumura N.. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020; 10:2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Poulogiannis G., Ichimura K., Hamoudi R.A., Luo F., Leung S.Y., Yuen S.T., Harrison D.J., Wyllie A.H., Arends M.J.. Prognostic relevance of DNA copy number changes in colorectal cancer. J. Pathol. 2010; 220:338–347. [DOI] [PubMed] [Google Scholar]
- 87. Kim M.Y., Yim S.H., Kwon M.S., Kim T.M., Shin S.H., Kang H.M., Lee C., Chung Y.J.. Recurrent genomic alterations with impact on survival in colorectal cancer identified by genome-wide array comparative genomic hybridization. Gastroenterology. 2006; 131:1913–1924. [DOI] [PubMed] [Google Scholar]
- 88. Chaudhury A., Howe P.H.. The tale of transforming growth factor-beta (TGFbeta) signaling: a soigne enigma. IUBMB Life. 2009; 61:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Crespo I., Gotz L., Liechti R., Coukos G., Doucey M.A., Xenarios I.. Identifying biological mechanisms for favorable cancer prognosis using non-hypothesis-driven iterative survival analysis. NPJ Syst. Biol. Appl. 2016; 2:16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Prasad V. Our best weapons against cancer are not magic bullets. Nature. 2020; 577:451. [DOI] [PubMed] [Google Scholar]
- 91. Longo D.L. Personalized medicine for primary treatment of serous ovarian cancer. N. Engl. J. Med. 2019; 381:2471–2474. [DOI] [PubMed] [Google Scholar]
- 92. Bozic I., Reiter J.G., Allen B., Antal T., Chatterjee K., Shah P., Moon Y.S., Yaqubie A., Kelly N., Le D.T.et al.. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013; 2:e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fang J., Pian C., Xu M., Kong L., Li Z., Ji J., Chen Y., Zhang L.. Revealing prognosis-related pathways at the individual level by a comprehensive analysis of different cancer transcription data. Genes (Basel). 2020; 11:1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zheng X., Amos C.I., Frost H.R.. Comparison of pathway and gene-level models for cancer prognosis prediction. BMC Bioinf. 2020; 21:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zheng X., Amos C.I., Frost H.R.. Cancer prognosis prediction using somatic point mutation and copy number variation data: a comparison of gene-level and pathway-based models. BMC Bioinf. 2020; 21:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Aguirre-Gamboa R., Gomez-Rueda H., Martinez-Ledesma E., Martinez-Torteya A., Chacolla-Huaringa R., Rodriguez-Barrientos A., Tamez-Pena J.G., Trevino V.. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013; 8:e74250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gyorffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z.. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010; 123:725–731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joe Delaney, PhD (delaneyj@musc.edu).
Materials Availability
Cell lines and plasmids generate in this study are maintained at the Medical University of South Carolina and will be made available upon request.
Data and Code Availability
Protected human datasets generated during this study are available in dbGaP, accession phs002313. Flow cytometry data are available in FlowRepository under accession FR-FCM-Z3V2.
The code generated during this study are available at Github (https://github.com/jrdelaney/SWAN) and web software at https://www.delaneyapps.com/#SWAN.