

Summary
The ability to precisely edit the genome of human induced pluripotent stem cell (iPSC) lines using CRISPR/Cas9 has enabled the development of cellular models that can address genotype to phenotype relationships. While genome editing is becoming an essential tool in iPSC-based disease modeling studies, there is no established quality control workflow for edited cells. Moreover, large on-target deletions and insertions that occur through DNA repair mechanisms have recently been uncovered in CRISPR/Cas9-edited loci. Yet the frequency of these events in human iPSCs remains unclear, as they can be difficult to detect. We examined 27 iPSC clones generated after targeting 9 loci and found that 33% had acquired large, on-target genomic defects, including insertions and loss of heterozygosity. Critically, all defects had escaped standard PCR and Sanger sequencing analysis. We describe a cost-efficient quality control strategy that successfully identified all edited clones with detrimental on-target events and could facilitate the integrity of iPSC-based studies.
KEYWORDS: CRISPR/Cas9, iPSCs, disease modeling, isogenic control lines, genome editing, on-target insertions/deletions, mtDNA, KCNQ2, WGS
Graphical abstract
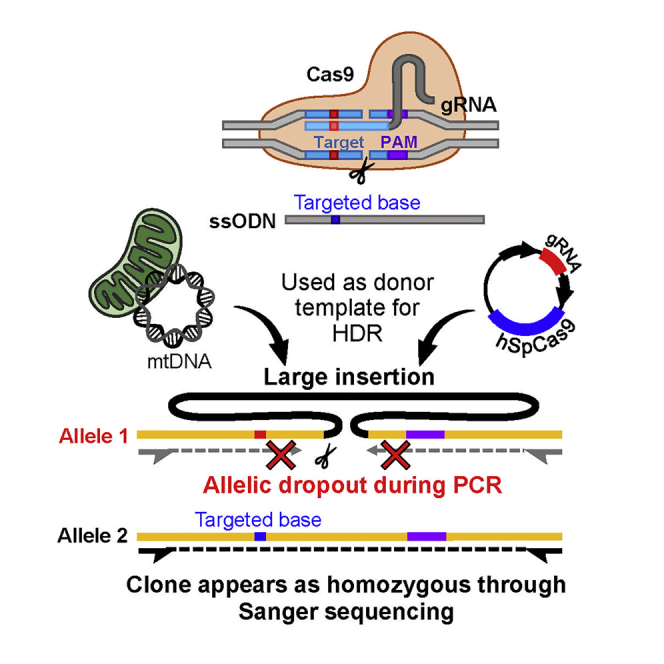
Highlights
-
•
Examination of 27 iPSC clones generated after editing 9 genomic loci across 4 genes
-
•
CRISPR/Cas9-edited iPSC lines exhibit high incidence of large on-target defects
-
•
Majority of damaging on-target insertions are undetectable by PCR/Sanger sequencing
-
•
Description of cost-efficient quality control assays to detect on-target defects
In this study, Simkin and colleagues investigate the occurrence of CRISPR/Cas9-mediated deleterious on-target effects in human iPSC clones deemed to be accurately edited. In 33% of cases, edited clones had acquired large on-target structural variants, including insertions and microdeletions. These defects caused allelic dropout and escaped detection using standard PCR/Sanger sequencing. They describe methods to accurately screen for on-target effects.
Introduction
The ability to correct disease-associated genetic variants in patient-specific induced pluripotent stem cells (iPSCs) or to insert pathogenic variants into healthy control iPSCs by CRISPR/Cas9 has enabled the development of cellular models that can be used to interrogate the relationship between genotype and phenotype in differentiated, disease-relevant cells. Correcting a disease-associated mutation in a patient iPSC line can demonstrate a causal relationship between the mutation and a cellular phenotype, while introducing a mutation in a healthy control iPSC line can assess its sufficiency for a particular phenotype. The generation of isogenic pairs of iPSC lines is quickly becoming an essential prerequisite in disease modeling studies, as the genetic background of reprogrammed iPSC lines may confound phenotypic analysis of differentiated disease-relevant cells (Ichida and Kiskinis, 2015; Merkle and Eggan, 2013). However, editing genes without undesired genomic effects in iPSCs remains difficult (Kwart et al., 2017) and requires significant resources and expertise that often challenge research laboratories that are focused on using these cells as a tool to study human development or disease mechanisms.
Precise genome editing is achieved by inducing CRISPR/Cas9-mediated double-stranded DNA breaks (DSBs) targeted to a specific location by a single-guide RNA (sgRNA; Sander and Joung, 2014). While the exact repair mechanisms in human cells remain poorly defined, DSBs are likely repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR). HDR can occur through the supply of an exogenous donor sequence as a template or through homologous recombination with an endogenous allele on the sister chromatid (which can lead to copy-neutral loss of heterozygosity [LOH]; Bibikova et al., 2003; Liang et al., 1998; Mao et al., 2008; Shrivastav et al., 2008). The efficiency of successful HDR in human iPSCs is highly variable (e.g., 2%–20%), and more than 80% of selected clones following genotype screening contain unwanted genomic defects, such as insertions, deletions, inversions, and rearrangements (Byrne et al., 2014; Maguire et al., 2019; Martin et al., 2019; Santos et al., 2016). Many approaches have been employed to improve the odds of achieving the intended edit and reduce the burden of screening hundreds of clones (Blair et al., 2016; Hockemeyer and Jaenisch, 2016; Ikeda et al., 2018; Kwart et al., 2017; Merkle et al., 2015; Popp et al., 2018; Steyer et al., 2018). These include strategic design of sgRNAs and single-stranded oligo donors (ssODNs), and the use of positive selection markers for flow cytometry sorting or antibiotic selection (for extended reviews, see Mianne et al., 2020; Wang et al., 2018). Nevertheless, most protocols still require large-scale screening of 25–100 isolated clones (Paquet et al., 2016). This labor-intensive step is typically done by PCR amplification of a few hundred bases spanning the target locus, followed by Sanger sequencing.
A major concern has been the potential off-target edits that CRISPR/Cas9 might introduce because of sequence homology of the sgRNA to other regions in the genome (Cho et al., 2014; Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013). Recent studies have highlighted the incidence of large deletions, insertions, and intricate genomic rearrangements at or within a short distance from the targeted site (Adikusuma et al., 2018; Kosicki et al., 2018; Leibowitz et al., 2021; O'Keefe et al., 2010; Owens et al., 2019; Shin et al., 2017). These deleterious on-target events, which likely occur through NHEJ, microhomology-mediated end joining (MMEJ), and homologous recombination (Fu et al., 2021; Wang and Xu, 2017), have been extensively described in mice (Kosicki et al., 2018) and human embryos (Alanis-Lobato et al., 2021; Liang et al., 2020; Ma et al., 2017; Zuccaro et al., 2020) and have also been shown to occur in human iPSCs (Blondal et al., 2021; Weisheit et al., 2020). The frequency and nature of on-target defects in CRISPR/Cas9-edited iPSC lines have not been widely interrogated by multiple groups. In the most comprehensive study to date, Weisheit et al. (2020) examined 35 iPSC clones edited across 3 genomic locations and identified on-target defects in 18%–40% of cases. Additionally, CRISPR/Cas9 can cause chromosomal instability, which can result in copy loss or copy-neutral LOH (Alateeq et al., 2018; Gorter de Vries et al., 2019; Hajiahmadi et al., 2019; Korablev et al., 2020; Ledford, 2020; Prat et al., 2020; Przewrocka et al., 2020; Rayner et al., 2019; Weisheit et al., 2020; Yang et al., 2017; Zischewski et al., 2017). Importantly, genotyping using PCR and Sanger sequencing of a short DNA fragment serves well as an initial screen, but may lead to false identification of edited clones, as large on-target insertions/deletions may cause exclusive amplification of only one allele. Such allelic dropout events constitute a known limitation of PCR-based approaches (Shestak et al., 2021).
Here, we evaluated 27 iPSC clones generated from 9 different CRISPR/Cas9 targeting events across 4 disease-associated genes (KCNQ2, SCN1A, NEK1, and DNAJC7). Critically, all these clones were deemed to be “correctly” edited during initial screening, as judged by PCR amplification of short ∼200 bp fragments and Sanger sequencing of the targeted region. We found that 33% had acquired complex, on-target genomic alterations, including large insertions and copy-neutral LOH. A combination of simple-to-execute assays including allele copy number quantitative genotyping PCR (qgPCR) and assessment of heterozygosity through sequencing of nearby heterozygous single nucleotide polymorphisms (SNPs) successfully identified all lines that had appeared to be correctly edited, but were, in fact, defective. Our findings are in accordance with recent reports (Weisheit et al., 2020, 2021) and further highlight the need for standardized quality control (QC) of CRISPR/Cas9-edited iPSC lines (for an overview of QC practices for iPSCs, see Steeg et al., 2021).
Results
CRISPR/Cas9 editing can cause detrimental on-target insertions in iPSCs: a case study of apparently correctly edited clones
We reprogrammed peripheral blood mononuclear cells (PBMCs) isolated from a pediatric epilepsy patient with a heterozygous mutation (c.821C > T; p.T274M) in the KCNQ2 gene to generate a patient-specific iPSC line. We produced several isogenic mutation-corrected control lines by CRISPR/Cas9 editing. In parallel to correcting the disease-causing mutation, we introduced a silent Cas9-blocking mutation at the cut site to reduce CRISPR/Cas9 re-cutting (Figure 1A and Table S1; Paquet et al., 2016). CRISPR/Cas9-blocking mutations have been shown to increase the efficiency of on-target editing up to 10-fold (Okamoto et al., 2019; Paquet et al., 2016). To avoid any unintended effects of the silent mutation on protein expression, we consulted the human codon usage frequency table to select a codon of similar usage frequency (Dhindsa et al., 2020). To screen for clones with the desired edit, we used PCR amplification and Sanger sequencing of 264 bp around the targeted region (Table S2). The initial screening of more than 50 single isolated edited clones revealed several that appeared to be homozygous for the wild-type (WT) KCNQ2 allele sequence, one of which carried the designed Cas9-blocking mutation (Figures 1B and 1C). This type of initial screening is common practice and allows for discarding clones with short deleterious edits, including insertions/deletions or mutations less than 200 bp in length. We selected 2 “corrected” clones for further characterization (KCNQ2-01-G6 and KCNQ2-01-A6). To identify any short duplications or inversions and to further validate the correction of the patient mutation, we amplified a larger PCR fragment (∼1.2 Kb) encompassing the targeted region and performed Sanger sequencing (Figure S1). Analysis of the larger PCR fragment confirmed the apparent correction of the disease-causing variant (Figure S1). Both corrected clones exhibited a normal karyotype as determined by standard G banding and digital KaryoStat analysis (Figure S2) and had DNA short tandem repeat (STR) profiles that matched the parental untargeted patient lines as determined by DNA fingerprinting (Figure S3). Moreover, evaluation by PCR and either T7E1 or Sanger sequencing analysis of the top 11 genomic regions with homology to the sgRNA used for targeting the KCNQ2 locus did not reveal any evidence of off-target edits (Figure S4 and Table S3).
Figure 1.

Standard QC methods fail to detect HDR-mediated large on-target defects in human iPSCs
(A) Illustration of Cas9 with sgRNA and ssODN and editing strategy for correction of KCNQ2 heterozygous mutation in patient-specific iPSCs (KCNQ2-01-het parental).
(B) Genotype screening of single CRISPR/Cas9 targeted iPSC clones was done by PCR amplification of 264 bp spanning the targeted site. Bottom: Sanger sequencing of gDNA PCR amplicons show KCNQ2-01-het parental iPSC line with a heterozygous mutation and mutation “correction” in 2 edited clones (KCNQ2-01-G6 and KCNQ2-01-A6). The absence of larger indels around the target site was validated by PCR amplification and Sanger sequencing of a larger fragment.
(C) A 264 bp fragment flanking the KCNQ2-01 target locus was PCR amplified using standard PCR extension times. A single band of expected size is visible when PCRs were run on an agarose gel.
(D) KCNQ2-01-het-par and corrected clones -G6 and -A6 iPSCs were differentiated into cortical excitatory neurons and transduced with CheRiff and QuasAr3 for all-optical electrophysiology (Optopatch) experiments. The well-wide average firing frequency of neurons (∼85 neurons per well, ∼4,000 neurons total) upon blue light stimulation with increasing intensity steps was analyzed across one independent experiment and two others (see Figure S5). KCNQ2-01-G6 and -A6 neurons exhibited significantly lower and higher firing frequencies, respectively, relative to KCNQ2-01-het parental patient-derived neurons across the stimulation protocol and independent experiments. ∗Indicates significant difference between KCNQ2-01-A6 and KCNQ2-01-G6 neurons (∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.0005). All data are reported as mean ± SEM (n = 48 wells analyzed per cell line, N = 1 experiment; 2 additional independent experiments are presented in Figure S5).
(E) Left: Agarose gel electrophoresis of 4,495 bp PCR amplified fragment reveals a single band from KCNQ2-01-het parental iPSC line and isogenic corrected clones. Right: Sanger sequencing in ∼850 bp steps across the 4.5 Kb PCR fragment revealed heterozygous SNPs in the KCNQ2-01-het parental unedited iPSC line on both sides of the target locus that were not present in corrected clones.
(F) A 264 bp fragment flanking the KCNQ2-01 target locus was PCR amplified using long duration PCR extension times. Agarose gel electrophoresis revealed the presence of large (3–5 Kb) insertions in all edited clones that had appeared to be corrected using standard PCR/Sanger sequencing in (B) and (C) (see Figures S6–S8).
(G) Non-homologous or microhomology-mediated end joining can result in large insertions by using exogenously supplied Cas9 plasmid DNA as the donor template (see also Figures S1–S8).
We next proceeded to perform functional phenotyping assays on cortical excitatory neurons differentiated from the two corrected iPSC clones (KCNQ2-01-G6 and KCNQ2-01-A6) and the parental patient line (KCNQ2-01-het-par). To examine the firing frequency of large numbers of neurons, we used the Optopatch platform, a system for high-throughput, all-optical electrophysiology with single cell resolution (Hochbaum et al., 2014; Kiskinis et al., 2018; Werley et al., 2017). The combined expression of CheRiff (a blue-light-activated channelrhodopsin) and QuasAr3 (a fluorescent voltage indicator) allows for the simultaneous stimulation and recording from multiple neurons within an elaborate network (Figure 1D). Using Synapsin promoter-driven expression constructs for CheRiff and QuasAr3, we imaged and analyzed the firing frequency of more than 10,000 neurons per cell line under a blue-light illumination protocol (Figures 1D and S5). After 35 days in culture, neurons were monitored for 2 s without stimulation followed by five 500-ms pulses of blue light of increasing intensity (Figure 1D, middle). As shown by the average firing rates of neurons (Figures 1D and S5), KCNQ2-01-G6 and KCNQ2-01-A6 exhibited opposing behavior in reference to parental patient-derived neurons. KCNQ2-01-G6 exhibited a significantly lower firing frequency, whereas KCNQ2-01-A6 neurons exhibited higher firing frequency relative to parental-derived neurons across the protocol (Figures 1D and S5).
This divergent electrophysiological behavior prompted us to investigate its origin. Close inspection of Sanger sequencing data of long-range PCR fragments (∼4.5 Kb) around the edited locus revealed that the corrected clones were homozygous for SNPs and short intronic indels flanking the edited locus, which were heterozygous in the parental untargeted cell line (Figure 1E). This apparent LOH suggested the occurrence of either copy-neutral LOH through homologous recombination with an endogenous allele or allelic dropout resulting from introduction of a monoallelic structural variant such as a large insertion or deletion that would compromise primer binding or PCR amplification. Indeed, PCR amplification of the targeted region using longer extension times revealed the presence of large monoallelic insertions ranging from 3 to 5 Kb within the 2 corrected clones as well as within 2 additional clones from the same targeting experiment (Figure 1F). Gel extraction, purification, and Sanger sequencing of the inserted DNA revealed inserted segments corresponding to the plasmid used to express Cas9 (Figures 1G and S6). Each of the 4 edited clones (KCNQ2-01-G6, -A6, -A3, and -H3) had incorporated different segments of Cas9 plasmid DNA at the targeted locus. Thus, standard PCR and Sanger sequencing protocols were misleading because they failed to distinguish between a true homozygously edited clone and one where a monoallelic structural variant caused allelic dropout (Figure S7). Critically, our inability to initially detect the deleterious structural variants within the KCNQ2 gene confounded our subsequent electrophysiological phenotyping experiments.
Large on-target genomic defects are a common feature of CRISPR/Cas9-edited iPSC lines
Our discovery of falsely corrected clones for KCNQ2-01 prompted us to investigate a larger set of CRISPR/Cas9-edited iPSC lines that we had generated. We examined 27 iPSC clones produced after CRISPR/Cas9 editing of 9 separate genomic regions within 4 genes (KCNQ2, SCN1A, DNAJC7, and NEK1). For 7 out of 9 regions, CRISPR/Cas9 was used to correct a disease-associated mutation, while in the remainder, CRISPR/Cas9 was used to introduce a disease-associated mutation. As recently recommended by Weisheit et al. (2020), we used qgPCR assays to evaluate the targeted allele copy number as an initial screening approach (Figure 2A). This assay is more scalable compared with long-range PCR, which in some cases requires troubleshooting with different primer sets to amplify the desired fragment with high specificity. We designed 2 independent sets of primers with probes that amplified 300–400 bp around each respective targeted locus that in most cases were at least 100 bp away from the cut site (Table S4). In the qgPCR assay, large deletions or insertions in the target region prohibit amplification of the affected allele, and thus such clones exhibit higher cycle threshold (Ct) values corresponding to a reduced allele copy number (Figure 2A). After normalization to an internal reference gene (TERT), the Ct value detected in each edited clone was compared with the value of its own parental, untargeted cell line. Of the 27 clones that were deemed to be correctly edited by short PCR/Sanger sequencing, we identified 8 that exhibited significantly lower allele copy numbers (Figure 2B). As an additional QC assay, we evaluated all targeted clones that appeared to be biallelic with the qgPCR assay for the presence of heterozygous SNPs within long-range PCR fragments (4–11.5 Kb) spanning the targeted locus. We identified one clone (DNAJC7-01-hom-43) that lacked a nearby upstream heterozygous SNP (dbSNP:rs6503679; −68 bp away from the target site), which was present in its respective parental iPSC line (Figure 2C). Critically, we found that a downstream heterozygous SNP (dbSNP:rs12952314; +1979 bp from target) was present in both the parental untargeted iPSC line and the DNAJC7-01-hom-43 clone. The fact that this line had a normal allele copy number (Figure 2B) yet was homozygous for a parental heterozygous SNP suggests that CRISPR/Cas9 editing caused on-target and upstream copy-neutral LOH.
Figure 2.

Assessment of genomic allele copy number
(A) Illustration of qgPCR assay. Large deletions prevent primer or probe binding, and large insertions prevent PCR extension and thus reveal amplification of only one allele.
(B) qgPCR assay was used to assess allele copy numbers in 27 CRISPR/Cas9 edited clones. Eight out of 27 (30%) clones were found to be monoallelic. Data are represented as mean ± SEM (n = 3 technical replicates).
(C) A 4.4 Kb fragment spanning the targeted locus was PCR amplified and subjected to Sanger sequencing to identify the presence of heterozygous SNPs. Sequencing of DNAJC7-01-WT parental line revealed presence of upstream and downstream heterozygous SNPs. Sequencing of DNAJC7-01-hom-43 homozygously edited clone revealed absence of upstream heterozygous SNP indicating a copy neutral LOH (see Figure 6).
(D) Donut chart of edited clones representing 9 targeting events and numbers of clones found to be correctly edited (blue) and clones with unwanted monoallelic editing (red) (see also Figures 6 andS8).
The qgPCR assays revealed that 33% of targeted clones acquired complex on-target genomic defects that escaped detection by standard PCR/Sanger sequencing (Figure 2D). We next employed a longer PCR extension strategy of short (200–500 bp) amplicons coupled to Sanger sequencing to further examine the defects in the same cohort of iPSC clones (Figure S8). We found that clone KCNQ2-03-C47 had a large (>10 Kb) insertion of mtDNA, while clone SCN1A-02-B6 had acquired a complex rearrangement. While long-range PCR and qgPCR assays suggested monoallelic PCR amplification and the potential presence of structural variants in clones KCNQ2-04-55 and SCN1A-02-B3, we were not able to detect any abnormalities through long extension PCR (Figure S8). Our collective QC analyses revealed that most compromised clones appeared to have large on-target insertions, which included parts of the Cas9 plasmid used for targeting (Figures 1G and S6) or portions of mtDNA (Figure S8). Critically, we found that deleterious on-target defects occurred independently of the method used to deliver Cas9, as there were problematic clones edited with a plasmid (KCNQ2-01 and SCN1A-02) as well as ones edited with recombinant proteins (KCNQ2-03, KCNQ2-04, and DNAJC7-01) (Figure 2D).
Whole-genome sequencing identifies deleterious on-target defects
We next investigated whether whole-genome sequencing (WGS) would allow us to identify deleterious on-target effects including large insertions of exogenous (e.g., Cas9 plasmid) and endogenous DNA (e.g., mtDNA or other chromosomal DNA) and copy-neutral LOH. WGS does not rely on targeted PCR-based primer binding for amplification and therefore is not subject to allelic dropout. Instead, DNA libraries are prepared using complete genomic DNA (gDNA) that is fragmented into short segments and ligated to adaptor sequences at each end (Figure 3A). Primers corresponding to the adaptor sequences are used to unbiasedly amplify the gDNA fragments by PCR for library preparation and sequencing from each end of the fragments (typically 150 bp is sequenced from each end in paired-end WGS). However, large insertions such as the ones we found here are difficult to identify using typical short read paired-end WGS because reads that do not align within the reference genome are routinely discarded during the alignment to a reference genome assembly (Van der Auwera et al., 2013; Van der Auwera and O'Connor, 2020).
Figure 3.

WGS can help identify specific deleterious on-target effects of CRISPR/Cas9 editing and detect clones falsely identified as corrected
(A) Illustration of paired-end WGS. gDNA is fragmented into random size fragments, and then 150 bp are sequenced from both ends; the insert between the sequenced reads is unknown. Read mate sequences are aligned to a reference genome assembly.
(B) Illustration of paired-end WGS alignments in the presence of a monoallelic structural variant such as the large on-target insertion in KCNQ2-01-G6. A short 17 bp deletion was also introduced around the cut site.
(C) WGS Integrative Genomic Viewer (IGV) plot showing human genome (T2T) reference assembly mapped sequencing reads around the targeted locus of KCNQ2-01-het-parental and -G6 edited line. WGS analysis revealed the presence of the heterozygous patient mutation in both KCNQ2-01-G6 and KCNQ2-01-A6 clones (see Figure S9) that had appeared to be corrected by Sanger sequencing in Figure 1B. When alignments are grouped by concordance to reference assembly, partially mapped reads are displayed at the top. Reads with unmapped mates are outlined in red. Sequence alignment stops abruptly near the Cas9 cut site, with soft clipped bases and unmapped read mate sequences that are homologous to Cas9 carrying plasmid used to target this locus (see also Figure S9).
We sequenced or re-analyzed (Simkin et al., 2021) WGS data from 13 iPSC lines including parental untargeted and edited clones, with or without defects (Table S5). Sequence reads were aligned to the new CHM13 T2T human genome reference assembly (Nurk et al., 2021), in which long read sequencing was used to fill in the gaps of the previous reference assembly. Using Integrated Genome Viewer (IGV) software, we identified deleterious on-target effects of CRISPR/Cas9 editing in all the falsely edited clones examined. By grouping the sequencing alignment reads by “reference concordance,” reads that were partially mapped (split reads), or reads with mates that did not map to the reference sequence owing to exogenous DNA insertions (in paired-end sequencing), became easy to spot (Figures 3, 4, 5, and 6, Figure S9). The presence of discordant reads signifies the potential presence of a structural variant.
Figure 4.

WGS of KCNQ2-04 clones and detection of on-target mtDNA insertion in KCNQ2-04-55 that was falsely identified as corrected
(A) Illustration of paired-end WGS alignments in the presence of a monoallelic structural variant such as the large on-target mtDNA insertion in KCNQ2-04-55.
(B) WGS IGV plot showing human genome (T2T) reference assembly mapped sequencing reads around the targeted locus of KCNQ2-04-het-parental line, correctly edited KCNQ2-04-4 clone, and falsely corrected KCNQ2-04-55 clone. WGS analysis revealed the presence of the heterozygous patient mutation in KCNQ2-04-55 clone that had appeared to be corrected by Sanger sequencing. When alignments are grouped by concordance to reference assembly, partially mapped reads are displayed at the top. Sequence alignment stops abruptly at the Cas9 cut site, with soft clipped bases and read mates from both sides corresponding to mitochondrial DNA.
Figure 5.

WGS of KCNQ2-03 clones and detection of on-target mtDNA insertion in KCNQ2-03-C47 that was falsely identified as corrected
(A) Illustration of paired-end WGS alignments in the presence of a monoallelic structural variant such as the large on-target mitochondrial DNA insertion in KCNQ2-03-C47. A short 10 bp deletion was also introduced at the Cas9 cut site.
(B) WGS IGV plot showing human genome (T2T) reference assembly mapped sequencing reads around the targeted locus of KCNQ2-03-het-parental line, correctly edited KCNQ2-03-C12 clone, and falsely corrected KCNQ2-03-C47 clone. When alignments are grouped by concordance to reference assembly, partially mapped reads are displayed at the top. Sequence alignment stops abruptly at the Cas9 cut site, with soft clipped bases and read mates from both sides corresponding to mtDNA.
Figure 6.

WGS of DNAJC7 clones and detection of heterozygous SNPs to identify copy-neutral LOH
WGS IGV plot showing human genome (T2T) reference assembly mapped sequencing reads around the targeted locus and nearby heterozygous SNPs present in DNAJC7-01-WT-parental line. The presence of parental line heterozygous SNPs was assessed in DNAJC7-01-hom-43, DNAJC7-01-het-30, and DNAJC7-01-het-18 edited clones. DNAJC7-01-hom-43 was missing a single het SNP site 68 bp from the targeted locus, while other het SNPs from both sides were still present. This indicates that copy-neutral LOH affects only this one het SNP site and not the rest of the gene.
Careful analysis of the edited locus of line KCNQ2-01-G6 confirmed that it had a monoallelic novel DNA insertion of Cas9 plasmid DNA at the target site that was flanked by short deletions and that the heterozygous disease-associated mutation was not corrected (Figures 3B and 3C). In stark contrast to the parental line, KCNQ2-01-G6 showed many discordant alignments. These included a drop in coverage around the deletion and several partially mapped reads with unmapped mate reads (Figures 3B and 3C). WGS of clone KCNQ2-01-A6 revealed that the patient mutation was also not corrected and that it had similarly acquired a novel monoallelic DNA insertion (Figure S9). This was apparent by the frequency of discordant alignments that included partially mapped reads with unmapped mate reads as well as ones that partially mapped to a region on human chromosome 4. Analysis of clone KCNQ2-04-55 revealed a monoallelic insertion of mtDNA (Figure 4), while clone KCNQ2-03-C47 had a short 10 bp deletion adjacent to another monoallelic mtDNA insertion (Figure 5). Lastly, WGS enabled us to verify that CRISPR/Cas9 editing had caused copy-neutral LOH in clone DNAJC7-01-hom-43 as well as define the boundaries of this loss through inspecting for the presence of heterozygous SNPs (Figure 6).
A cost-efficient strategy for evaluating edited iPSC lines
Given the high incidence of complex on-target genomic defects we detected in edited human iPSC lines, there is a need to design a time- and cost-efficient QC strategy to successfully identify detrimental events and facilitate the integrity of subsequent experiments. Because CRISPR/Cas9 gene editing efficiency is low and there is a need to screen many clones, initial selection is typically based on PCR coupled to Sanger sequencing (Figure 7). During this first selection process, the presence of any heterozygous bases (whether a heterozygous SNP or an introduced silent heterozygous mutation) within the PCR fragment sequence demonstrates biallelic amplification. Thus, when nearby heterozygous SNPs are present near the targeted locus in parental gDNA, the most efficient way to confirm on-target editing and biallelic amplification is to amplify a gDNA fragment that spans heterozygous SNPs on both sides of the targeted locus (Figure 7; step 1). Correctly edited clones with evidence of biallelic amplification can be nominated for further use. Clones that appear to be correctly edited for the variant of interest but that exhibit no apparent evidence of biallelic amplification require further evaluation by an allele copy number qgPCR assay (Figure 7; step 2). Clones with significantly altered allele copy numbers should be discarded. Any clones that appear to have a normal allele copy number but that lack clear evidence of heterozygosity based on initial sequencing can be further assessed by screening for SNPs that are heterozygous in the parental line, around the targeted region using Sanger sequencing or SNP microarrays (Figure 7; step 3). Lastly, although it is more costly and requires expertise for analysis, WGS can be employed to validate correctly edited clones or even replace several QC measures, such as validation of cell line identity, genetic stability, and detection of any on-target and off-target defects.
Figure 7.

A cost-efficient strategy for evaluating edited iPSC lines
Steps to confirm biallelic PCR amplification and eliminate clones with unwanted on-target defects after CRISPR/Cas9 editing. PCR and Sanger sequencing of the target locus is not sufficient unless there is evidence of biallelic amplification.
Discussion
The goal of using human iPSC lines to model monogenic or polygenic diseases is to understand how genetic variation can lead to cellular dysfunction. Owing to genetic variability between different human subjects, it has become critically important to make phenotypic comparisons within pairs of isogenic iPSC lines that differ genetically only at the intended sequence. CRISPR/Cas9 technology has enabled the generation of such controls with relative ease. However, while CRISPR/Cas9 enables precise genome editing, it can also cause significant unintended effects that may be hard to detect. In this study we focused on assessing the frequency of aberrant on-target genomic effects and alarmingly found that 33% of them had acquired on-target genetic aberrations.
As we demonstrate, allelic dropout leading to false identification of edited iPSC clones can severely compromise the validity of phenotyping experiments. The majority of falsely corrected clones found in our cohort had large on-target DNA insertions. The “foreign” DNA identified in 4 cases was part of the Cas9 plasmid and in 2 other cases was a large segment of mtDNA. The integration of a Cas9 plasmid gene cassette as well as parts of mtDNA has previously been reported in CRISPR/Cas9 editing systems (Bachu et al., 2015; Blondal et al., 2021; Ono et al., 2019; Strecker et al., 2019, 2020). These events are likely detrimental and can lead to haploinsufficiency or to the production of a dysfunctional protein. For example, in a recent study, a CRISPR/Cas9-edited clone was falsely identified as homozygous, while, in fact, a small deletion prevented proper sequencing of targeted region (Herai et al., 2021; Maricic et al., 2021; Trujillo et al., 2021). Moreover, the genetic consequences of copy-neutral LOH, which we identified in another falsely corrected clone, are not limited to the target locus, as LOH can uncover recessive alleles and elicit long-range transcriptional consequences. In fact, large stretches of genomic homozygosity have been identified in the normal human population and have also been associated with a large number of clinical phenotypes (Clark et al., 2019; Gibson et al., 2006; Keller et al., 2012).
The frequency of on-target defects we describe is in strong accordance with the only other comprehensive study focused on the incidence of these events in iPSCs (Weisheit et al., 2020). Weisheit et al. (2020) examined 35 iPSC clones edited across 3 genomic locations and identified on-target defects in 18%–40% of cases, whereas we examined 27 iPSC clones edited across 9 genomic locations and found defects in 33%–43% of cases. Interestingly, most defects in our cohort included large insertions (Figure S6), while the study by Weisheit et al. (2020) and several others have reported mostly large deletions (Adikusuma et al., 2018; Korablev et al., 2020; Kosicki et al., 2018; Owens et al., 2019; Yang et al., 2017). The reason for this difference is hard to discern but could be related to editing methodology or the genomic context of the targeted regions. Critically, we and Weisheit et al. highlight the difficulty in identifying on-target genomic defects by standard PCR/Sanger sequencing. Lastly, we were able to showcase the utility of WGS analysis in detecting on-target genomic defects simply by carefully examining sequence alignments. While de novo assembly strategies could be used to reconstruct the genomic sequence, we found that careful examination of sequence alignments can be effective. Clear red flags include a sharp reduction in sequencing coverage (suggesting deletions), or a significant proportion of mapped on-target reads having mates that are only partially, or not at all mapped to the reference genome (suggesting insertions). In conclusion, we propose that it is necessary to incorporate QC strategies to detect deleterious on-target editing errors that are typically lacking in studies utilizing gene-edited iPSCs for in vitro disease models.
Experimental procedures
Detailed methods are provided in the supplemental experimental procedures
Generation, maintenance, and CRISPR/Cas9 genome editing of human iPSC lines
Patient iPSC lines were generated from PBMCs isolated from whole blood following informed consent under protocols approved both by Ann & Robert H. Lurie Children's Hospital of Chicago and Northwestern University Institutional Review Board (#2015-738) as previously described (Simkin et al., 2021). Healthy control iPSC lines CS20002 (DNAJC7-01-WT) and HPSI0114i-KOLF2 (NEK1-02-WT) were acquired from Cedar Sinai and HipSci, respectively. All iPSCs were grown on Matrigel with mTeSR1 media, maintained at 37 °C in 5% CO2, and passaged weekly using Accutase (MilliporeSigma). The NEK1-02-WT iPSC line was edited by Jackson Laboratory. All other iPSC lines were edited by Applied StemCell Inc. (Milpitas, CA). For details on editing strategy, genomic DNA PCRs and Sanger sequencing, genomic integrity and pluripotency assays, analysis of off-target Cas9 sites, and quantitative genotyping PCR-based copy number assays, please see Supplemental experimental procedures.
Whole-genome sequencing
WGS was outsourced to Novogene Corporation Inc., as previously described (Simkin et al., 2021). We performed sequencing analysis after aligning the reads to the complete telomere-to-telomere human reference genome T2T-CHM13 v.1.1 (Nurk et al., 2021). The T2T-CHM13 reference genome improves coverage of complex regions and variant calling (Aganezov et al., 2021), delivering consensus sequences without the use of alternative contigs. IGV software was used to visualize alignment tracks and assess read coverage (Robinson et al., 2011). For details on WGS analysis, please see Supplemental experimental procedures.
Cortical neuron differentiation and Optopatch measurements
iPSCs were differentiated into glutamatergic neurons using a previously described protocol based on Ngn2 overexpression (Simkin et al., 2021). Cryo-stocks of Ngn2 neurons differentiated from KCNQ2-01-het parental and KCNQ2-01-G6 and -A6 iPSC lines were co-plated with primary mouse glial cells onto custom-made ibidi cell culture plates. Neurons were transduced with lentiviral particles encoding all-optical electrophysiology (Optopatch) components CheRiff-BFP and QuasAr3-Citrine and recorded 2 weeks later on DIV 35 with Optopatch imaging (Hochbaum et al., 2014; Kiskinis et al., 2018; Werley et al., 2017). Additional details regarding cell culture and imaging can be found in Supplemental experimental procedures. Statistical significance was determined using t tests or Mann-Whitney tests with a custom MATLAB routine.
Statistical analysis
Statistical differences between groups were evaluated using t tests or Mann-Whitney test for nonparametric data where appropriate. All data are reported as means ± SEM.
Data availability
All data generated or analyzed during this study are included in the article and supporting files. WGS data for all patient samples are available upon request. WGS data cannot be deposited on public repositories owing to lack of patient consent.
Author contributions
Conceptualization, D.S. and E.K.; methodology, D.S. and V.P.; software, B.I.B. and S.J.L.; formal analysis, D.S., B.I.B., C.M.A, S.J.R., and V.B.; investigation: D.S., V.P., B.I.B., C.M.A., and L.A.W.; resources: J.E.L., A.L.G., and E.K.; Writing – original draft: D.S., V.P., and E.K.; Writing – review & editing: D.S., V.P., B.I.B., C.M.A., S.J.R., V.B., L.A.W., G.T.D., O.B.M., J.E.L., S.J.L., A.L.G., and E.K.; visualization: D.S., V.P., and E.K; supervision: G.T.D., O.B.M., S.J.L., A.L.G., and E.K; project administration: E.K.; funding acquisition: A.L.G. and E.K.
Conflict of interest
J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics, is a consultant for Biogen, Inc., and provides expert testimony for Perkins Coie LLP. E.K. holds Q-State Biosciences stock. C.M.A., S.J.R., V.B., L.A.W., G.T.D., and O.B.M. are employees of Q-State Biosciences.
Acknowledgments
We are grateful to the following funding sources: US National Institutes of Health (NIH) National Institute on Neurological Disorders and Stroke (NINDS) U54NS108874 (A.L.G. and E.K.), NIH/NINDS and National Institute on Aging (NIA) R01NS104219 (E.K.), NIH/NINDS R01NS073873 (J.E.L.) and R56NS073873 (J.E.L.), the Les Turner ALS Foundation (E.K.), and the New York Stem Cell Foundation (E.K.). We are grateful to Bill Skarnes and Justin McDonough for generation of the NEK1-02 iPSC isogenic line. E.K. is a Les Turner ALS Center Investigator and a New York Stem Cell Foundation – Robertson Investigator.
Published: March 10, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2022.02.008.
Supplemental information
References
- Adikusuma F., Piltz S., Corbett M.A., Turvey M., McColl S.R., Helbig K.J., Beard M.R., Hughes J., Pomerantz R.T., Thomas P.Q. Large deletions induced by Cas9 cleavage. Nature. 2018;560:E8–E9. doi: 10.1038/s41586-018-0380-z. [DOI] [PubMed] [Google Scholar]
- Aganezov S., Yan S.M., Soto D.C., Kirsche M., Zarate S., Avdeyev P., Taylor D.J., Shafin K., Shumate A., Xiao C., et al. A complete reference genome improves analysis of human genetic variation. bioRxiv. 2021;2007 doi: 10.1101/2021.07.12.452063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanis-Lobato G., Zohren J., McCarthy A., Fogarty N.M.E., Kubikova N., Hardman E., Greco M., Wells D., Turner J.M.A., Niakan K.K. Frequent loss of heterozygosity in CRISPR-Cas9-edited early human embryos. Proc. Natl. Acad. Sci. United States America. 2021;118:202004832. doi: 10.1073/pnas.2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alateeq S., Ovchinnikov D., Tracey T., Whitworth D., Al-Rubaish A., Al-Ali A., Wolvetang E. Identification of on-target mutagenesis during correction of a beta-thalassemia splice mutation in iPS cells with optimised CRISPR/Cas9-double nickase reveals potential safety concerns. APL Bioeng. 2018;2:046103. doi: 10.1063/1.5048625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachu R., Bergareche I., Chasin L.A. CRISPR-Cas targeted plasmid integration into mammalian cells via non-homologous end joining. Biotechnol. Bioeng. 2015;112:2154–2162. doi: 10.1002/bit.25629. [DOI] [PubMed] [Google Scholar]
- Bibikova M., Beumer K., Trautman J.K., Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Sci. (New York, N.Y. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Blair J.D., Bateup H.S., Hockemeyer D.F. Establishment of genome-edited human pluripotent stem cell lines: from targeting to isolation. J. visualized experiments : JoVE. 2016:e53583. doi: 10.3791/53583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondal T., Gamba C., Moller Jagd L., Su L., Demirov D., Guo S., Johnston C.M., Riising E.M., Wu X., Mikkelsen M.J., et al. Verification of CRISPR editing and finding transgenic inserts by Xdrop indirect sequence capture followed by short- and long-read sequencing. Methods. 2021;191:68–77. doi: 10.1016/j.ymeth.2021.02.003. [DOI] [PubMed] [Google Scholar]
- Byrne S.M., Mali P., Church G.M. Genome editing in human stem cells. Methods Enzymol. 2014;546:119–138. doi: 10.1016/B978-0-12-801185-0.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D.W., Okada Y., Moore K.H.S., Mason D., Pirastu N., Gandin I., Mattsson H., Barnes C.L.K., Lin K., Zhao J.H., et al. Associations of autozygosity with a broad range of human phenotypes. Nat. Commun. 2019;10:4957. doi: 10.1038/s41467-019-12283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhindsa R.S., Copeland B.R., Mustoe A.M., Goldstein D.B. Natural selection shapes codon usage in the human genome. Am. J. Hum. Genet. 2020;107:83–95. doi: 10.1016/j.ajhg.2020.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.W., Dai X.Y., Wang W.T., Yang Z.X., Zhao J.J., Zhang J.P., Wen W., Zhang F., Oberg K.C., Zhang L., et al. Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 2021;49:969–985. doi: 10.1093/nar/gkaa1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J., Morton N.E., Collins A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- Gorter de Vries A.R., Couwenberg L.G.F., van den Broek M., de la Torre Cortes P., Ter Horst J., Pronk J.T., Daran J.G. Allele-specific genome editing using CRISPR-Cas9 is associated with loss of heterozygosity in diploid yeast. Nucleic Acids Res. 2019;47:1362–1372. doi: 10.1093/nar/gky1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajiahmadi Z., Movahedi A., Wei H., Li D., Orooji Y., Ruan H., Zhuge Q. Strategies to increase on-target and reduce off-target effects of the CRISPR/Cas9 system in plants. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20153719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herai R.H., Szeto R.A., Trujillo C.A., Muotri A.R. Response to Comment on “Reintroduction of the archaic variant of NOVA1 in cortical organoids alters neurodevelopment”. Science. 2021;374:eabi9881. doi: 10.1126/science.abi9881. [DOI] [PubMed] [Google Scholar]
- Hochbaum D.R., Zhao Y., Farhi S.L., Klapoetke N., Werley C.A., Kapoor V., Zou P., Kralj J.M., Maclaurin D., Smedemark-Margulies N., et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods. 2014;11:825–833. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell. 2016;18:573–586. doi: 10.1016/j.stem.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J.K., Kiskinis E. Probing disorders of the nervous system using reprogramming approaches. EMBO J. 2015;34:1456–1477. doi: 10.15252/embj.201591267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K., Uchida N., Nishimura T., White J., Martin R.M., Nakauchi H., Sebastiano V., Weinberg K.I., Porteus M.H. Efficient scarless genome editing in human pluripotent stem cells. Nat. Methods. 2018;15:1045–1047. doi: 10.1038/s41592-018-0212-y. [DOI] [PubMed] [Google Scholar]
- Keller M.C., Simonson M.A., Ripke S., Neale B.M., Gejman P.V., Howrigan D.P., Lee S.H., Lencz T., Levinson D.F., Sullivan P.F., et al. Runs of homozygosity implicate autozygosity as a schizophrenia risk factor. Plos Genet. 2012;8:e1002656. doi: 10.1371/journal.pgen.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E., Kralj J.M., Zou P., Weinstein E.N., Zhang H., Tsioras K., Wiskow O., Ortega J.A., Eggan K., Cohen A.E. All-optical electrophysiology for high-throughput functional characterization of a human iPSC-derived motor neuron model of ALS. Stem Cell Rep. 2018;10:1991–2004. doi: 10.1016/j.stemcr.2018.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korablev A., Lukyanchikova V., Serova I., Battulin N. On-target CRISPR/Cas9 activity can cause undesigned large deletion in mouse zygotes. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21103604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwart D., Paquet D., Teo S., Tessier-Lavigne M. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 2017;12:329–354. doi: 10.1038/nprot.2016.171. [DOI] [PubMed] [Google Scholar]
- Ledford H. CRISPR gene editing in human embryos wreaks chromosomal mayhem. Nature. 2020;583:17–18. doi: 10.1038/d41586-020-01906-4. [DOI] [PubMed] [Google Scholar]
- Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D., Gutierrez N.M., Chen T., Lee Y., Park S.-W., Ma H., Koski A., Ahmed R., Darby H., Li Y., et al. Frequent Gene Conversion in Human Embryos Induced by Double Strand Breaks. bioRxiv. 2020;2006 doi: 10.1101/2020.06.19.162214. [DOI] [Google Scholar]
- Liang F., Han M., Romanienko P.J., Jasin M. Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc. Natl. Acad. Sci. U S A. 1998;95:5172–5177. doi: 10.1073/pnas.95.9.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H., Marti-Gutierrez N., Park S.W., Wu J., Lee Y., Suzuki K., Koski A., Ji D., Hayama T., Ahmed R., et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- Maguire J.A., Cardenas-Diaz F.L., Gadue P., French D.L. Highly efficient CRISPR-cas9-mediated genome editing in human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2019;48:e64. doi: 10.1002/cpsc.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z., Bozzella M., Seluanov A., Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008;7:1765–1771. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricic T., Helmbrecht N., Riesenberg S., Macak D., Kanis P., Lackner M., Pugach-Matveeva A.D., Paabo S. Comment on “Reintroduction of the archaic variant of NOVA1 in cortical organoids alters neurodevelopment”. Science. 2021;374:eabi6060. doi: 10.1126/science.abi6060. [DOI] [PubMed] [Google Scholar]
- Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M., et al. Highly efficient and marker-free genome editing of human pluripotent stem cells by CRISPR-cas9 RNP and AAV6 donor-mediated homologous recombination. Cell Stem Cell. 2019;24:821–828.e825. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Eggan K. Modeling human disease with pluripotent stem cells: from genome association to function. Cell Stem Cell. 2013;12:656–668. doi: 10.1016/j.stem.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Neuhausser W.M., Santos D., Valen E., Gagnon J.A., Maas K., Sandoe J., Schier A.F., Eggan K. Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep. 2015;11:875–883. doi: 10.1016/j.celrep.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mianne J., Bourguignon C., Nguyen Van C., Fieldes M., Nasri A., Assou S., De Vos J. Pipeline for the generation and characterization of transgenic human pluripotent stem cells using the CRISPR/Cas9 technology. Cells. 2020;9 doi: 10.3390/cells9051312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., Altemose N., Uralsky L., Gershman A., et al. The complete sequence of a human genome. bioRxiv. 2021;2005 doi: 10.1101/2021.05.26.445798. [DOI] [Google Scholar]
- O'Keefe C., McDevitt M.A., Maciejewski J.P. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010;115:2731–2739. doi: 10.1182/blood-2009-10-201848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S., Amaishi Y., Maki I., Enoki T., Mineno J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019;9:4811. doi: 10.1038/s41598-019-41121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono R., Yasuhiko Y., Aisaki K.I., Kitajima S., Kanno J., Hirabayashi Y. Exosome-mediated horizontal gene transfer occurs in double-strand break repair during genome editing. Commun. Biol. 2019;2:57. doi: 10.1038/s42003-019-0300-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens D.D.G., Caulder A., Frontera V., Harman J.R., Allan A.J., Bucakci A., Greder L., Codner G.F., Hublitz P., McHugh P.J., et al. Microhomologies are prevalent at Cas9-induced larger deletions. Nucleic Acids Res. 2019;47:7402–7417. doi: 10.1093/nar/gkz459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D., Kwart D., Chen A., Sproul A., Jacob S., Teo S., Olsen K.M., Gregg A., Noggle S., Tessier-Lavigne M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533:125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp B., Krumbiegel M., Grosch J., Sommer A., Uebe S., Kohl Z., Plotz S., Farrell M., Trautmann U., Kraus C., et al. Need for high-resolution genetic analysis in iPSC: results and lessons from the ForIPS consortium. Sci. Rep. 2018;8:17201. doi: 10.1038/s41598-018-35506-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat F., Toutain J., Boutin J., Amintas S., Cullot G., Lalanne M., Lamrissi-Garcia I., Moranvillier I., Richard E., Blouin J.M., et al. Mutation-specific guide RNA for compound heterozygous porphyria on-target scarless correction by CRISPR/Cas9 in stem cells. Stem Cell Rep. 2020;15:677–693. doi: 10.1016/j.stemcr.2020.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przewrocka J., Rowan A., Rosenthal R., Kanu N., Swanton C. Unintended on-target chromosomal instability following CRISPR/Cas9 single gene targeting. Ann. Oncol. 2020;31:1270–1273. doi: 10.1016/j.annonc.2020.04.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner E., Durin M.A., Thomas R., Moralli D., O'Cathail S.M., Tomlinson I., Green C.M., Lewis A. CRISPR-Cas9 causes chromosomal instability and rearrangements in cancer cell lines, detectable by cytogenetic methods. CRISPR J. 2019;2:406–416. doi: 10.1089/crispr.2019.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander J.D., Joung J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos D.P., Kiskinis E., Eggan K., Merkle F.T. Comprehensive protocols for CRISPR/Cas9-based gene editing in human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2016;38:5B 6 1–5B 6 60. doi: 10.1002/cpsc.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestak A.G., Bukaeva A.A., Saber S., Zaklyazminskaya E.V. Allelic dropout is a common phenomenon that reduces the diagnostic yield of PCR-based sequencing of targeted gene panels. Front Genet. 2021;12:620337. doi: 10.3389/fgene.2021.620337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:15464. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M., De Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Simkin D., Marshall K.A., Vanoye C.G., Desai R.R., Bustos B.I., Piyevsky B.N., Ortega J.A., Forrest M., Robertson G.L., Penzes P., et al. Dyshomeostatic modulation of Ca(2+)-activated K(+) channels in a human neuronal model of KCNQ2 encephalopathy. Elife. 2021;10:e64434. doi: 10.7554/eLife.64434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeg R., Mueller S.C., Mah N., Holst B., Cabrera-Socorro A., Stacey G.N., De Sousa P.A., Courtney A., Zimmermann H. EBiSC best practice: how to ensure optimal generation, qualification, and distribution of iPSC lines. Stem Cell Rep. 2021;16:1853–1867. doi: 10.1016/j.stemcr.2021.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer B., Bu Q., Cory E., Jiang K., Duong S., Sinha D., Steltzer S., Gamm D., Chang Q., Saha K. Scarless genome editing of human pluripotent stem cells via transient puromycin selection. Stem Cell Rep. 2018;10:642–654. doi: 10.1016/j.stemcr.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J., Ladha A., Gardner Z., Schmid-Burgk J.L., Makarova K.S., Koonin E.V., Zhang F. RNA-guided DNA insertion with CRISPR-associated transposases. Science. 2019;365:48–53. doi: 10.1126/science.aax9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J., Ladha A., Makarova K.S., Koonin E.V., Zhang F. Response to Comment on “RNA-Guided DNA Insertion with CRISPR-Associated Transposases”. Science. 2020;368 doi: 10.1126/science.abb2920. [DOI] [PubMed] [Google Scholar]
- Trujillo C.A., Rice E.S., Schaefer N.K., Chaim I.A., Wheeler E.C., Madrigal A.A., Buchanan J., Preissl S., Wang A., Negraes P.D., et al. Reintroduction of the archaic variant of NOVA1 in cortical organoids alters neurodevelopment. Science. 2021;371 doi: 10.1126/science.aax2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11. doi: 10.1002/0471250953.bi1110s43. 11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., O'Connor B.D. O'Reilly Media; 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. [Google Scholar]
- Wang H., Xu X. Microhomology-mediated end joining: new players join the team. Cell Biosci. 2017;7:6. doi: 10.1186/s13578-017-0136-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Liu K.I., Sutrisnoh N.B., Srinivasan H., Zhang J., Li J., Zhang F., Lalith C.R.J., Xing H., Shanmugam R., et al. Systematic evaluation of CRISPR-Cas systems reveals design principles for genome editing in human cells. Genome Biol. 2018;19:62. doi: 10.1186/s13059-018-1445-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisheit I., Kroeger J.A., Malik R., Klimmt J., Crusius D., Dannert A., Dichgans M., Paquet D. Detection of deleterious on-target effects after HDR-mediated CRISPR editing. Cell Rep. 2020;31:107689. doi: 10.1016/j.celrep.2020.107689. [DOI] [PubMed] [Google Scholar]
- Weisheit I., Kroeger J.A., Malik R., Wefers B., Lichtner P., Wurst W., Dichgans M., Paquet D. Simple and reliable detection of CRISPR-induced on-target effects by qgPCR and SNP genotyping. Nat. Protoc. 2021;16:1714–1739. doi: 10.1038/s41596-020-00481-2. [DOI] [PubMed] [Google Scholar]
- Werley C.A., Brookings T., Upadhyay H., Williams L.A., McManus O.B., Dempsey G.T. All-optical electrophysiology for disease modeling and pharmacological characterization of neurons. Curr. Protoc. Pharmacol. 2017;78:11. doi: 10.1002/cpph.25. 11 20 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.J., Wang Y., Li Z.F., Gong Y., Zhang P., Hu W.C., Sheng D.H., Li Y.Z. Increasing on-target cleavage efficiency for CRISPR/Cas9-induced large fragment deletion in Myxococcus xanthus. Microb. Cell Fact. 2017;16:142. doi: 10.1186/s12934-017-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zischewski J., Fischer R., Bortesi L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017;35:95–104. doi: 10.1016/j.biotechadv.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020;183:1650–1664.e1615. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in the article and supporting files. WGS data for all patient samples are available upon request. WGS data cannot be deposited on public repositories owing to lack of patient consent.