

Abstract
Small ubiquitin-related modifiers (SUMOs) conjugated or bound to target proteins can affect protein trafficking, processing and solubility. SUMOylation has been suggested to play a role in the amyloid plaque and neurofibrillary tangle pathology of Alzheimer disease (AD) and related neurodegenerative diseases. The current study examines the impact of SUMO1 on processing of the amyloid precursor protein (APP) leading to the production and deposition of the amyloid-β (Aβ) peptide. An in vivo model of these pathways was developed by the generation of double transgenic mice over-expressing human SUMO1 and a mutant APP. The SUMO1-APP transgenics displayed normal APP processing but, at later ages, exhibited increased insoluble Aβ and plaque density accompanied by increased dendritic spine loss, more pronounced synaptic and cognitive deficits. These findings suggest a potential impairment in Aβ clearance as opposed to increased amyloid production. Examination of microglia indicated a reduction in the SUMO1-APP transgenics which is a possible mechanism for the SUMO1-mediated increase in amyloid load. These findings suggest an indirect activity of SUMO1 possibly in the removal of Aβ plaques rather than a direct impact on amyloid generation.
Keywords: SUMO, Amyloid, Amyloid precursor protein, Alzheimer disease transgenic mouse models, Learning and memory, Electrophysiology
1. Introduction
Small ubiquitin-related modifiers (SUMOs) are a group of polypeptides which can be linked onto lysine residues of target proteins. The outcome of SUMOylation varies according to the target protein and cell type which can result in changes to protein stability, activity, folding, aggregation, and subcellular location (for reviews see, (Hannoun et al., 2010; Schorova and Martin, 2016)). SUMOylation can also influence other post-translational modifications such as ubiquitination and phosphorylation to promote novel protein-protein interactions and degradation (Feligioni et al., 2015). With such a wide range of potential outcomes, it stands to reason that protein SUMOylation may represent an important factor in diseases where protein misfolding and aggregation are predominant features.
Alzheimer's disease (AD) is a progressive neurodegenerative disease characterized by extracellular amyloid plaques and intracellular tangles. The neurofibrillary tangles are composed primarily of a hyperphosphorylated form of the microtubule-associated tau protein. The proteolytic processing of the amyloid precursor protein (APP) by β-amyloid cleaving enzyme 1 (BACE1) and γ-secretase complexes generates various Aβ peptide fragments which assemble into soluble oligomers and fibrillar amyloid plaques (reviewed in, (Andrew et al., 2016)). There are several mechanisms for Aβ removal which include proteolysis by neprilysin and insulin-degrading enzyme as well as phagocytosis and clearance by activated microglia. It is thought that these clearance processes become overwhelmed in AD and methods to increase their activity are being considered for potential therapeutic development (Devi and Ohno, 2015; Michaud et al., 2013).
APP and tau have both been found to be SUMOylated which may affect protein processing, aggregation and/or degradation (reviewed in (Feligioni et al., 2015)). Studies show increased levels of free and conjugated SUMO1 in AD mouse models accompanied by elevated Ubc9 and SENP1 levels in both the cortex and hippocampus (Nistico et al., 2014; Yun et al., 2013; McMillan et al., 2011). This suggests increased SUMO1 levels may exacerbate AD pathology while other studies have demonstrated a potential protective effect for SUMO2/3 against amyloid synaptotoxicity (Lee et al., 2014). A direct effect of SUMO1 on APP processing has been supported by studies where the in vitro over-expression or knockdown of SUMO2 was inversely correlated with Aβ levels (Li et al., 2003a). A subsequent investigation identified two SUMOylation consensus sequences immediately adjacent to the BACE1 cleavage site suggesting that SUMO1 conjugation to APP may alter processing due to steric hindrance (Zhang and Sarge, 2008). Furthermore, over-expression of Ubc9 and SUMO1 in APP expressing cells resulted in decreased levels of aggregated Aβ. In contrast, other in vitro studies reported an increase in Aβ processing exclusively by SUMO3 which was independent of conjugation and no changes were observed for SUMO1 or SUMO2 under similar conditions (Dorval et al., 2007a). In the current study, double transgenic mice were generated to investigate further the links between amyloid pathology and SUMOylation.
Transgenic animals expressing a mutant form of the human APP695 isoform (TgCRND8) which have an aggressive amyloid pathology were crossed with mice over-expressing human SUMO1. No differences were observed in APP processing or the generation of Aβ peptides in this mouse model. However, the SUMO1-APP double transgenic mice exhibited an increase in insoluble Aβ and a correspondingly higher plaque load upon aging which resulted in a decrease in dendritic spine density, impaired synaptic function and memory. This increased amyloid pathology was associated with a reduction in the number of activated hippocampal microglial cells which normally contribute to amyloid removal. These findings suggest the SUMO1 may contribute to Aβ clearance and degradation as opposed to having a direct impact on APP processing and amyloid production.
2. Materials and methods
2.1. Mice
All mouse work was approved by the Animal Care Committee of the University of Toronto and the University Health Network in accordance with the regulations of the Canadian Council on Animal Care, as well as the IACUC committee of Columbia University. SUMO1 overexpressing and the APP (TgCRND8) transgenic mice were generated as described previously (Matsuzaki et al., 2015; Chishti et al., 2001). The SUMO1 transgenics were originally maintained on an FVB background and, when crossed with TgCRND8, mice were maintained on a mixed C57Bl6/C3H/FVB background. Correspondingly, TgCRND8 (C57Bl6/C3H) were crossed to non-transgenic FVB mice to generate TgCRND8 control mice on an identical background. Non-transgenic (Non-Tg) animals are the littermates of TgCRND8 mice (APP+/−, APP-Tg). SUMO1 +/+ APP +/− double Tg (SUMO1-APP) mice were produced from crosses of SUMO1+/+ (SUMO1-Tg) animals with APP+/− animals. Both male and female mice were used in equal numbers at the 14 or 22 week time points investigated. Genotyping was performed as described (Matsuzaki et al., 2015; Chishti et al., 2001). Mortality was calculated as the percentage of mice found deceased in cages over a period of approximately 24 months.
2.2. Electrophysiology and behavior
Basal synaptic transmission (BST), long term potentiation (LTP), contextual/cued fear conditioning, and sensory threshold assessment were conducted as previously described (Matsuzaki et al., 2015).
2.3. Protein extraction
Frozen half brains or dissected brain regions (cortex, hippocampus, cerebellum) were weighed for whole protein extraction. Tissues (100 mg) were homogenized in 500 μl of buffer containing 20 mM Tris, pH 7.4; 250 mM sucrose; 1 mM EDTA; 1 mM EGTA and EDTA-free protease inhibitors (Sigma-Aldrich) followed by sonication. Homogenates were centrifuged for 5 min at 27,000g and supernatants collected. Protein quantification was performed by Bradford assay (BioRad) using the microplate format, as per the manufacturer's instructions. Readings were performed on a Spectra Max i3 (Molecular Devices). Samples were diluted in homogenization buffer to 3 or 6 mg/ml concentration and stored in an equal volume of Laemmli buffer at −80 °C.
2.4. Immunoblotting
Samples (5–45 μg total protein) were run on 4–12% Novex trisglycine gels (Life Technologies) and immunoblotting was performed as previously described (Satoh et al., 2015). The antibodies used were: 1:1000 anti-Neprilysin (Millipore), 1:100 C1/6.1 (BioLegend), 1:1000 LC3 (NovusBiologicals NB600-1384), 1:2000 22C11 which detects full length and secreted APP fragments (Millipore), 1:10,000 anti-Actin (Sigma-Aldrich), 1:1000 SUMO1 (Cell Signaling Technology), 1:250 anti-SUMO2 (Life Technologies), 1:500 BACE1 (ProSci Inc.). After washing HRP-conjugated anti-mouse (1:3000), anti-rabbit (1:5000) or anti-goat (1:5000) was applied for 1 h at room temperature and bands were visualized by enhanced chemiluminescence (ECL).
2.5. ELISA
Using the hippocampal, cortical and cerebellar lysates described above both soluble and insoluble forms of Aβ40/42 and secreted APPβ/WT were measured using commercially available ELISA kits (IBL international) as previously described (Satoh et al., 2015). Briefly, the soluble Aβ40/42 is extracted from a 10% (w/v) tissue homogenate (20 mM Tris-HCl; 0.25 M sucrose; 1 mM EDTA/EGTA) using an equal volume of 4% diethylamine in 100 mM NaCl. The insoluble amyloid is obtained by centrifugation (100,000g for 1 h) and extracted by sonication using cold formic acid. Quantification of the endogenous murine Aβ40/42 was performed by ELISA (Invitrogen) as previously described (Satoh et al., 2015).
2.6. Immunohistochemistry and morphology
Brain hemispheres were fixed in 10% formalin (Sigma-Aldrich) overnight at 4 °C then immersed in 70% ethanol. Serial sections (5 μm) of paraffin embedded tissue were stained for amyloid plaques using an Aβ specific antibody (4G8, BioLegend). Plaque density and morphological assessment of spine length and density were determined as previously described (Satoh et al., 2015). Staining for microglia was performed using an anti-Iba1 antibody (Wako Chemicals) at a 1:200 dilution. Sections were incubated with primary antibody overnight at 4 °C after antigen retrieval by boiling in Tris EDTA buffer (10 mM Tris, 2 mM EDTA, pH 9.0) for 10 min, peroxidase block in 3% H2O2 for 15 min and blocking in 5% serum for 1 h at room temperature. Secondary HRP-conjugated anti-rabbit antibodies (Vector Laboratories) were applied for 1 h at room temperature followed by DAB staining (Vector Laboratories) and sections were counterstained with hematoxylin. Images were taken at 20 × magnification using an Axiovision Slide scanner. Three representative sections of the same size per sample were analyzed using Volocity find objects by intensity function and excluding objects < 20 μm.
2.7. Statistical analysis
Values are presented as averages ± standard error of the mean. Independent sample t-test was used to determine differences in plaque density, Aβ40 and Aβ42 ELISAs and secreted APPβ (sAPPβ) ELISAs between SUMO1-APP and APP-Tg mice. Two-way ANOVA and Sidak multiple comparisons post-hoc tests were used to determine the differences in spine numbers and densities and dendritic length. ANOVA followed by post hoc analysis (multiple comparisons was used for electrophysiology and behavioral studies. Analysis of the microglia densities in the non-transgenic, SUMO1 transgenic and SUMO1-APP double transgenes utilized an independent two-tailed t-test. Statistical analyses of the mortality displayed by the APP and SUMO1 transgenic lines were conducted using the Comprehensive R Archive Network (CRAN Software) chi-squared tests that have previously been employed for Kaplan-Meier plots (please see, https://cran.r-project.org/).
3. Results
3.1. Over-expression of SUMO1 does not alter APP processing
To determine if SUMO1 overexpression affects cleavage of the APP protein, the cortex, hippocampus and cerebellum of 12–14 (onset of plaque pathology) and 22 (abundant plaque pathology) week old APP-Tg and SUMO1-APP mice were examined. Immunoblotting for SUMO1 revealed that the SUMO1-APP mice not only overexpress SUMO1 monomers but have abundant high molecular weight SUMO1 conjugates (Fig. 1A and Supplemental Fig. S1). This was consistent with observations in over-expressing SUMO1 transgenic animals and suggests that the same high levels of SUMOylated target proteins are maintained in the SUMO1-APP double transgenics (Matsuzaki et al., 2015). No changes in the levels of SUMO2/3 monomers or conjugates were observed in the APP-Tg or SUMO1-APP transgenic animals (Fig. 1B).
Fig. 1.
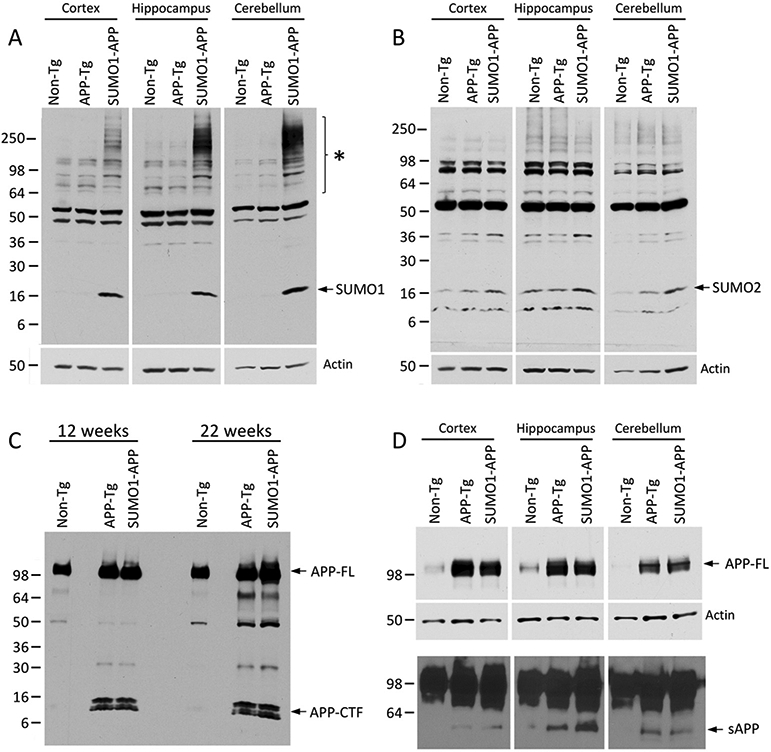
Representative Western blots of (A) SUMO1 showing increased high molecular weight conjugates in dissected brain regions of 22 week old SUMO1-APP and no significant changes in APP-Tg mice. (B) SUMO2 with long exposure to demonstrate no changes in conjugates or monomers in dissected brain regions from 22 week old mice. Similar levels were observed for (C) APP full-length (APP-FL) and C-terminal fragments (APP-CTF) from whole brains of 12 and 22 week old mice and (D) full-length APP and total secreted APP (sAPP, lower panel) from 22 week old mice.
Whole brain lysates from young (12 weeks) and old (22 weeks) mice were also assessed for any changes associated with SUMO1 over-expression. Probing with a monoclonal antibody directed towards the APP C-terminal tail indicated comparable levels of full-length APP (APP-FL) and the C-terminal fragment (APP-CTF; α/β cleavages) in both age groups (Fig. 1C). Examination of the APP species in different brain regions revealed similar levels of APP-FL protein in the cortex, hippocampus and cerebellum of the APP-Tg and SUMO1-Tg mice (Fig. 1D). As with whole brain extracts, all of these regions also displayed no significant changes in the APP-CTF levels as determined by quantitative immunoblot analysis (Supplemental Fig. S2). Immunoblotting for the total secreted forms of cleaved APP (sAPP) was also performed using an antibody directed towards an epitope in the APP ectodomain. This indicated some variation in overall levels in the cortex, hippocampus and cerebellum but no significant differences were observed between the APP-Tg and SUMO1-APP transgenics (Fig. 1D, lower panel). Previous in vitro investigations have indicated that APP is modified by SUMO1 in the presence of high levels of the Ubc9 ligase (Zhang and Sarge, 2008). Immunoprecipitation was used to examine if similar SUMOylation of APP occurs in the double transgenic mice. However, there was no indication of an APP-SUMO1 conjugated species in the mice over-expressing both proteins (Supplemental Fig. S3). APP was also not identified as a SUMO1 target as determined by proteomic analysis of whole brain lysates from the SUMO1 over-expression animals (Matsuzaki et al., 2015). Cumulatively, these findings suggest no major changes in the APP transgene expression or its processing as a result of high expression levels of the human SUMO1.
Intriguingly, SUMO1 over-expression had a significant positive impact on mortality rates in APP-Tg mice. The majority of APP transgenic lines exhibit a high mortality rate that is thought to be caused by spontaneous seizures (Palop et al., 2007; Del Vecchio et al., 2004). This affects males in particular which display a highly aggressive behavior and have nearly twice the mortality rate of females. The addition of a SUMO1 over-expressing allele reduces the mortality in both genders of APP-Tg mice (Fig. 2). The effect is particularly notable and reached statistical significance in males where there appears to be a dose-response related to SUMO1 over-expression. The mortality in the female APP-Tg mice was also reduced when on the SUMO1 background but the differences did not reach statistical significance. A complete breakdown of the statistical analyses and p-values are presented in Supplemental Table S1 and Fig. S4. The SUMO1 over-expressing heterozygotes have the same low mortality rates as our background strain (Non-Tg), suggesting that the modulating effect of the SUMO1 allele on mortality is only in the context of increased APP expression. The mechanism(s) for this SUMO1-dependent decrease in the mortality of the APP transgenics is potentially due to the modulation of the off-pathway non-amyloid pathology effects related to APP over-expression (e.g., spontaneous nonconvulsive seizures (Palop et al., 2007)).
Fig. 2.
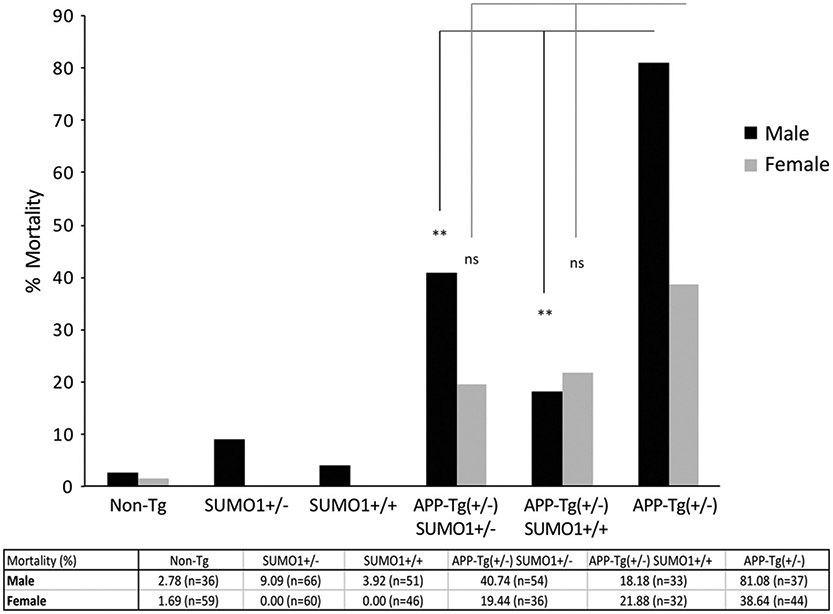
Mortality rates in SUMO1-APP mice. High mortality rates observed in the APP-Tg mice were significantly decreased on the SUMO1 +/+ and +/− backgrounds in male group. Mortality of Non-Tg, SUMO1 +/− and SUMO1 +/+ mice were also significantly decreased compared to APP-Tg in both male and female groups. All data were analyzed by R (CRAN Software) using chi-squared test. The p-values displayed on the figure are **, p < 0.01; ns, not significant by chi-squared test.
To examine APP processing in more detail, extracts from dissected brain regions (cortex and hippocampus) were assessed for their respective amounts of secreted APPβ (sAPPβ). Quantitative ELISA analysis indicated there were no statistically significant differences in secreted APPβ (sAPPβ) were found in the cortex or hippocampus from SUMO1-APP transgenics as compared to animals expressing only the APP transgene (Fig. 3A, B). Previous studies have indicated that SUMO1 can impact the expression levels of BACE1 (Yun et al., 2013). It was observed that BACE1 in primary neuronal cultures was elevated following Aβ treatments which occurred in a SUMO1-dependent manner. This effect was not dependent on SUMOylation of target proteins since conjugation-deficient mutants (SUMO1-AA) induced comparable increases in BACE1.
Fig. 3.
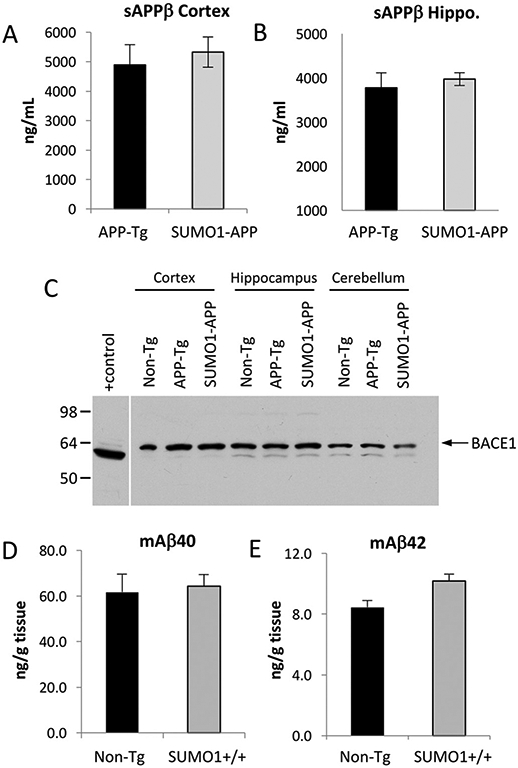
Quantification of secreted APPβ (sAPPβ) for 22 week old APP-Tg and SUMO1-APP mice in the (A) cortex shows a slight but not significant elevation of β-secretase cleavage and comparable levels were observed in (B) the hippocampus (N = 3–4/group). (C) Immunoblotting for BACE 1 from total brain lysates (45 μg) from 22 week old indicted that SUMO1-APP mice expressed comparable levels of the protease as that observed in APP-Tg and Non-Tg animal. HEK293 cells transiently transfected with BACE1 was used as a positive control. (D, E) Quantification of the endogenous murine Aβ40 and Aβ42 (mAβ40/42) indicated no significant differences between non-transgenic (Non-Tg) and the SUMO1 (SUMO1-Tg) over-expressing animals (n = 5 mice/group).
To determine if similar events occurred in the transgenic mouse models, BACE1 levels were assessed by immunoblotting of whole brain extracts. However, no significant differences in BACE1 were observed in the SUMO1-APP transgenics or the APP-Tg mice as compared to non-transgenic animals (Fig. 3C). This observation is also consistent with a lack of change in the secreted APPβ ectodomain in the SUMO1-APP transgenics. The reasons for these different observations are unclear but may be due to variations in cultured neurons as opposed to in vivo models.
It is possible that over-expression of APP may lead to issues with detecting small changes in the levels of Aβ that may be induced in the SUMO1 transgenics. To examine this further, the endogenous murine Aβ (mAβ) was quantified in Non-Tg and SUMO1 transgenics (SUMO1-Tg) animals. Quantification of the mouse Aβ peptides by ELISA indicated there were no significant differences between the Non-Tg and SUMO1-Tg mice for mAβ40 or mAβ42 (Fig. 3D, E). These findings are consistent with the lack of any discernable effect of SUMO1 over-expression on APP processing to generate Aβ even under physiological conditions.
In addition to changes in BACE1, it has also been reported that Aβ processing can be increased via SUMO1-induced autophagy (Cho et al., 2015). It was shown that overexpression of SUMO1 in H4 cells resulted in increased cleavage to LC3-II and the formation of autophagosomes. The SUMO-mediated increase in autophagy was accompanied by an elevation in Aβ40 levels by ~4-fold. The link between macroautophagy and APP processing to generate Aβ has been supported by previous investigations (Haung et al., 2005). However, examination of the LC3-II levels in the SUMO1-APP transgenics indicated there was no significant increase in these animals as compared to APP transgenics mice (Fig. 4A-B). Moreover, no difference was observed between SUMO1-Tg and non-Tg animals (Fig. 4C). Quantitative analyses of these and additional samples revealed no statistically significant differences in the levels of LC3-II in the APP-Tg mice and the SUMO1-APP transgenics (Fig. S5). This is consistent with the absence of any substantive changes of Aβ levels in the SUMO1-APP mice. The reasons for these different observations in terms of lack of BACE1 and autophagic changes in the transgenic mice is unclear but may be due to variations in cultured neurons as opposed to the in vivo models used in the current study.
Fig. 4.
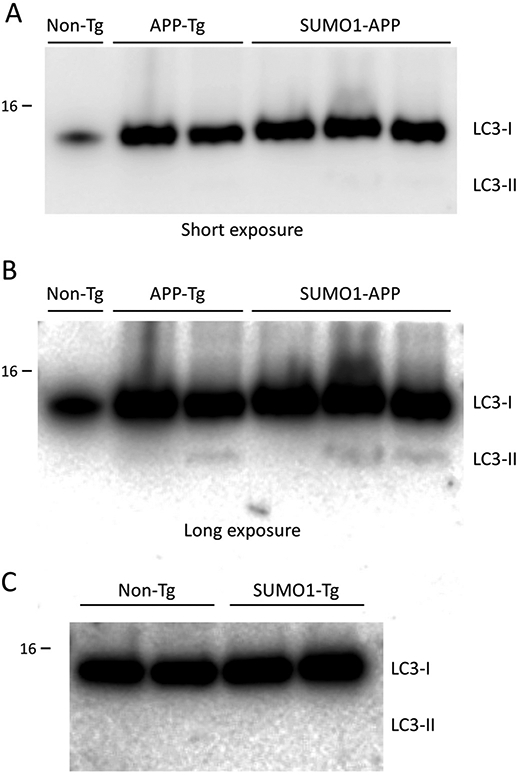
Autophagy in SUMO1 over-expressing transgenics. (A) Immunoblotting indicated signals for LC3-I and not detectable LC3-II in the APP transgenic (APP-Tg) or the SUMO1-APP double transgenic animals (at 22 weeks of age). (B) Longer exposures of the immunoblots did reveal the presence of small levels of LC3-II but no major differences were observed in the SUMO1-APP double transgenic animals. (C) Examination of the SUMO1 only transgenics (SUMO1-Tg) also indicated no increases in autophagy as determined by LC3-II immunoreactivity as compared to non-transgenic (Non-Tg) controls (22 weeks of age). Protein loading for APP-Tg and SUMO1-APP lysates was 45 μg/lane; Non-Tg and SUMO1-Tg lysates were 60 μg/lane.
3.2. Overexpression of SUMO1 increases amyloid deposition in APP transgenic mice
The level of amyloid pathology in the transgenics was determined by a combination of immunohistochemistry with image analysis and ELISA-based quantification of the soluble and insoluble Aβ40/42. The amyloid plaque pathology in the SUMO1-APP and APP-Tg transgenics was assessed at 14 and 22 weeks of age. The APP-Tg (TgCRND8) animals typically begin to display amyloid deposition at ~12 weeks and, at the younger ages (14 weeks), there were no differences in the levels of soluble Aβ40 and Aβ42 extracted from total brain lysates of the APP-Tg and SUMO1-APP animals (Fig. 5A). However, a decrease in the insoluble Aβ40 and 42 was observed in the SUMO1-APP transgenics during this early stage in the development of amyloid plaques (Fig. 5B). This variation may be due to the small amounts of deposited Aβ in this early stage of the pathology as shown by immunohistochemistry (Supplemental Fig. S6). Examination of whole brain extracts at the later time-point (22 weeks) revealed comparable levels of soluble and insoluble Aβ with a slight trend towards an increase in the insoluble Aβ42 in the SUMO1-APP transgenics (Fig. 5C, D). Analysis of specific brain regions affected by amyloid pathology indicated no significant differences in the Aβ levels from the cortex of the SUMO1-APP and APP-Tg mice (Fig. 5E). One possible explanation for the lack of a change in cortical insoluble Aβ as determined by ELISA is that the extent of the amyloid pathology has plateaued at the 22 week period. However, differences in overall amyloid loads in the cortex of the SUMO1-APP transgenes was detectable by immunocytochemical and image analysis approaches (see below). However, a substantial increase in the insoluble Aβ42 was observed in the hippocampus of the SUMO1-APP animals (Fig. 5F). These findings suggest that the SUMO1-APP double transgenics may have more extensive amyloid pathology as a result of elevated SUMOylation of targets in pathways related to APP processing and/or amyloid clearance.
Fig. 5.
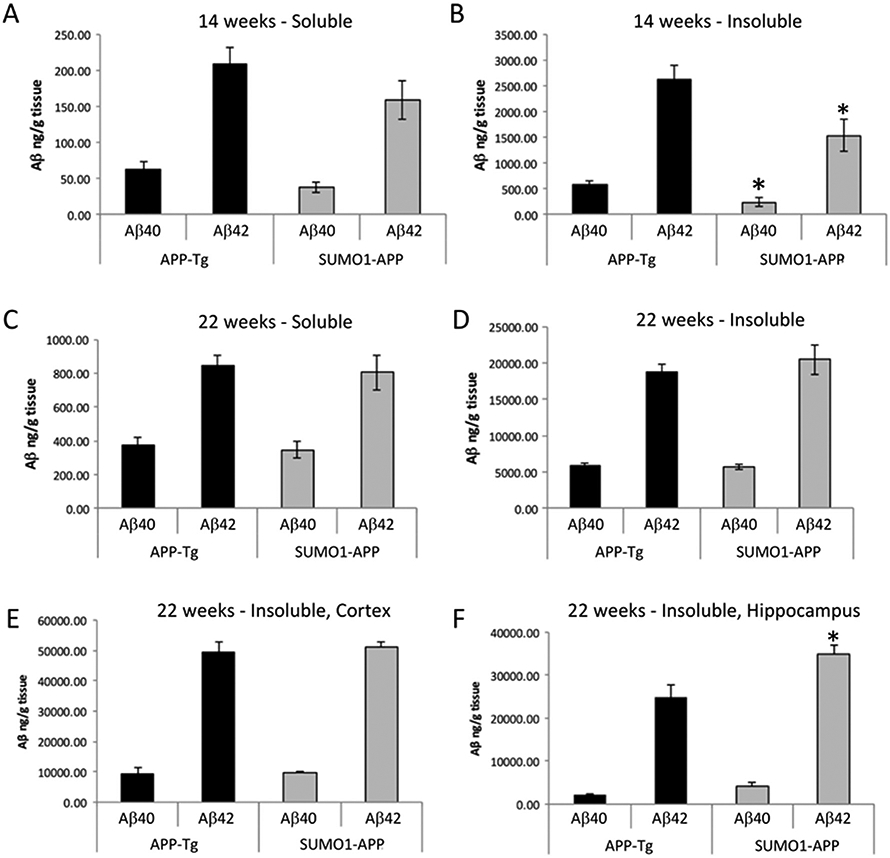
Assessing soluble and insoluble Aβ in SUMO1-APP mice. (A, B) Quantification of total brain Aβ levels in APP-Tg and SUMO1-APP mice at 14 weeks of age for soluble and insoluble Aβ40 and Aβ42. (C, D) Similar ELISA quantification at 22 weeks of age for soluble and insoluble Aβ40 and Aβ42 showing modest increases in the SUMO1-APP mice at later stages. (E, F) Quantification of Aβ in different brain regions indicates a trend towards increased levels in the cortex and significant increases in insoluble Aβ42 in the hippocampus of the SUMO1-APP mice. *p < 0.05, N = 3–4.
To examine the pathology in more detail, Aβ immunohistochemistry was used to assess amyloid plaque density in both young and old animals. Staining of brain tissue from the 14 week old animals indicated only modest numbers of amyloid plaques for both groups of animals (Supplemental Fig. S6). At the later ages (22 weeks), extensive numbers of plaques were observed in the cortex and hippocampus of APP-Tg and SUMO1-APP mice (Fig. 6A, B). Quantification of the dense and diffuse plaques by image analysis indicated similar levels in both the APP-Tg and SUMO1-APP transgenics at 14 weeks of age (Fig. 6C, D). The diffuse amyloid refers to the halo of Aβ-positive material surrounding the dense core of the plaque. Consistent with the immunohistochemistry, quantitative analysis of the stained tissue from 22 week old animals indicated significantly increased amyloid loads (dense and diffuse) in the cortex and hippocampus of the SUMO1-APP transgenics as compared to APP-Tg only mice (Fig. 6E, F). The elevated levels of the amyloid deposits, particularly in the hippocampus, of the SUMO1-APP transgenics at 22 weeks is also consistent with the higher amount of insoluble Aβ40/42 as determined by ELISA.
Fig. 6.

Amyloid pathology and plaque density in SUMO1-APP mice. (A, B) Representative anti-Aβ staining in 22 week old animals showing amyloid pathology in APP-Tg and SUMO1-APP mice with hematoxylin counterstaining. (C, D) Quantification by image analysis of amyloid plaque densities in younger mice (14 weeks of age) indicated comparable levels in the APP-Tg and SUMO1-APP mice. (E, F) At later stages of pathology development (22 week), significant increases in the dense and diffuse plaques in the cortex and hippocampus of the SUMO1-APP double were observed. Scale bar = 500 μm, *p < 0.05, **p < 0.01 N = 4–6.
3.3. Defects in Aβ clearance may result in increased plaque density in SUMO1-APP transgenic mice
In the absence of an observed increase in APP levels or APP processing, the increased insoluble Aβ and increased plaque densities point to a potential failure of Aβ clearance. One possibility is a change in levels of the membrane-bound metallo-endopeptidase neprilysin which is one of the main proteases involved in the degradation of Aβ aggregates (Iwata et al., 2000; Miners et al., 2008). However, no differences in neprilysin expression was observed in whole brain lysates from the APP-Tg and SUMO1-Tg mice at 22 weeks of age (Fig. 7A). Since higher amounts of aggregate Aβ were seen in the hippocampus of the SUMO1-APP animals, different brain regions were examined. The cortex and hippocampus neprilysin levels were identical in the two groups as well as in the cerebellum where amyloid pathology is not observed (Fig. 7B). The insulin degrading enzyme (IDE) has also been identified a one of the primary protease capable of clearing Aβ aggregates and oligomers (Qiu et al., 1998; Farris et al., 2003). Examination of IDE levels by immunoblotting indicated there were similar levels in the APP-Tg and SUMO1-APP animals (Supplemental Fig. S7). While neprilysin and IDE were unaffected, it is possible that other Aβ-degrading proteases or clearance pathways could be impacted by the over-expression of SUMO1 in the APP transgenics.
Fig. 7.

Amyloid degradation and clearance in SUMO1-APP mice. (A) Representative immunoblot from whole brain lysates for neprilysin indicated comparable levels for APP-Tg, SUMO1-Tg and Non-Tg mice. (B) Immunoblotting for neprilysin in different brain regions (cortex, hippocampus, cerebellum) at 22 weeks of age yielded similar results. (C) Microglial response was assessed by number of Iba1-positive (Iba1 +) cells which were similar in the cortex and significantly decreased in the hippocampus of the SUMO1-APP transgenics (N = 5–7, *p < 0.05). (D,E) Representative immunostaining Iba1 + cells in the hippocampus of 22 week old APP-Tg and SUMO1-Tg mice. Scale bar is 200 μm.
Microglia-related pathways were also examined to determine if SUMO1 over-expression impacted their role in amyloid clearance. Reactive gliosis was assessed and quantified using the microglial marker Iba1. This indicated a significant decrease in the number of Iba1 + cells in the hippocampus of SUMO1-APP mice at 22 weeks of age as compared to APP-Tg mice (Fig. 7C). A decrease in microglia cells was also seen in the cortex of the SUMO1-APP animals but this did not reach statistical significance. Representative immunohistochemistry of the Iba1 + microglia indicates the reduced level of staining in the hippocampus of the SUMO1-APP transgenics as opposed to the APP-Tg animals (Fig. 7D, E). This reduced inflammatory response may also be a contributing factor to the higher degree of amyloid pathology in mice over-expressing SUMO1.
Quantification of microglia was also investigated in the SUMO1 only transgenic which indicated similar levels in both the hippocampus and cortex of these animals (Supplemental Fig. S8). This suggests that the differences seen in the SUMO1-APP mice may be the result of activation caused by the associated amyloid pathology. The prion cos-tet vector used to over-express SUMO1 is also active in microglia raising the possibility of higher levels of expression in these cells in addition to neurons. The upregulation of SUMO1-related pathways in the transgenic microglia may alter function under conditions of stress caused by the amyloid pathology.
3.4. Overexpression of SUMO1 decreases spine density in APP transgenic mice
Fibrillar Aβ in plaques can serve as a source of Aβ oligomers through shedding (Knowles et al., 2014). Oligomeric Aβ is toxic to neurons, disrupts synaptic function, and leads to widespread synaptic loss. Aβ-mediated synaptotoxicity and synaptic loss leading to cognitive impairments are significant features in APP transgenic mouse models of Alzheimer amyloid pathology (Steele et al., 2014; Sclip et al., 2011). Dendritic spine density and lengths were quantified in the APP-Tg and SUMO1-APP double transgenics using Golgi staining similar to the approaches used in the SUMO1-Tg animals (Matsuzaki et al., 2015). As we have reported previously, pyramidal neurons in the frontal cortex of non-transgenic animals (apical and basal) on the same background strain have a total of ~2000 dendritic spines (Supplemental Fig. S9A, B (Matsuzaki et al., 2015)). As expected, the APP-Tg and SUMO1-APP mice exhibited significantly reduced spine density (~70% reduction) as compared to Non-Tg mice (Fig. 8A). The over-expression of SUMO1 in the brain, even in the absence of APP, results in changes in synaptic development that leads to a failure in spine development and a decrease in density (Matsuzaki et al., 2015). However, the synapses which are formed still display a sensitivity to amyloid toxicity and the SUMO1-APP transgenics have a greater loss (~15–20) of spines than mice expressing only the mutant human APP (Fig. 8B). The significant decreases in spine densities between the APP-Tg and SUMO1-APP mice were observed for both apical and basal spines (Fig. 8C). This decrease was also consistently seen across most branch orders (Fig. 8D, E). The surviving spines exhibited no differences in length or morphology (Supplemental Fig. S9C, D). Cumulatively, these findings are consistent with the increase in Aβ aggregates in the SUMO1-APP mice which leads to more extensive synaptic loss.
Fig. 8.
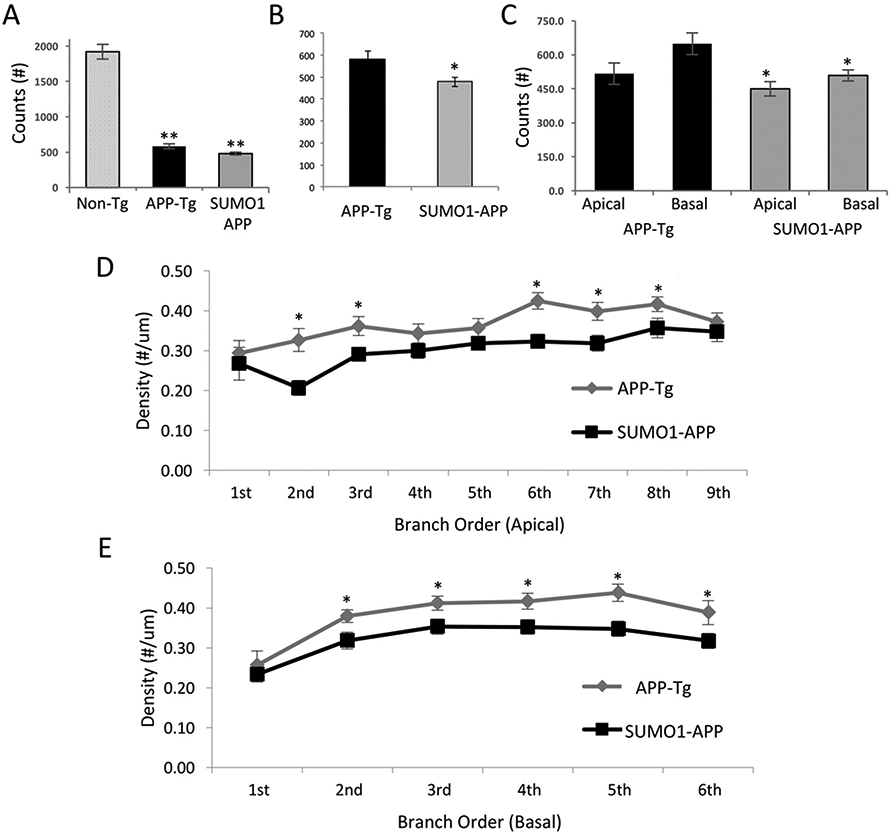
Morphological analysis of spines and dendrites in SUMO1-APP mice. (A) Total dendritic spine counts of pyramidal neurons revealed significant decreases in APP-Tg and SUMO1-APP transgenic animals as compared to Non-Tg mice. (B) Total dendritic spine counts in the SUMO1-APP indicated a significant decrease to levels observed in APP-Tg mice. (C) Apical and basal spine counts also displayed significant decreases in the 22 week SUM01-APP as compared to APP-Tg mice. (D, E) Apical and basal spine densities determined for branch orders indicates a consistent decrease in the SUMO-APP mice. N = 3, *p < 0.05.
3.5. SUMO1 overexpression worsens the impairment of synaptic function and memory in APP transgenic mice
SUMO1 over-expressing transgenic mice display a defect in synaptic function which results in cognitive impairments based on contextual and cued fear conditioning (Matsuzaki et al., 2015). Using the same approaches, comparisons between APP-Tg, SUMO1-APP and Non-Tg mice were made to examine the effects of increased SUMO1 levels when combined with amyloid pathology. As reported previously, the APP-Tg mice have impairments in basal synaptic transmission (BST) and LTP as compared to Non-Tg animals (Fig. 9A-C) (Kimura et al., 2012; Jolas et al., 2002). The SUMO1-APP transgenics showed a more profound reduction in BST than APP-Tg mice. However, this defect was not associated with a similar reduction in LTP as the APP-Tg mice has already a profound deficit in potentiation compared to mice expressing only SUMO1 (Fig. 9B-C).
Fig. 9.
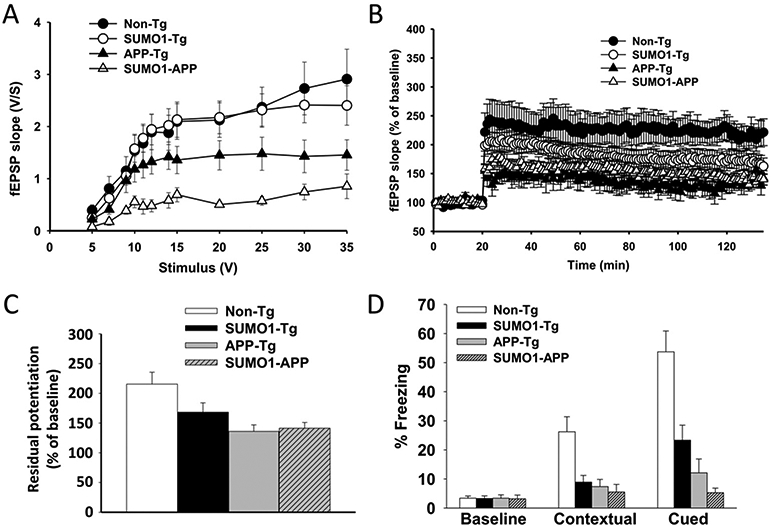
Synaptic and memory effects in SUMO1-APP mice. (A) Basal synaptic transmission (BST) was reduced in APP-Tg mice compared with WT mice and SUMO1 Tg mice. The impairment was more profound in the SUMO1-APP double Tgs. N = 7–14, p < 0.001 comparing SUMO1-APP vs. non-Tgs. (B) Long-term potentiation (LTP) was reduced both in SUMO1 and APP-Tg mice. SUMO1-APP double Tgs exhibited impairments similar to those seen in single APP-Tg mice. N = 7–14, p < 0.001. (C) Quantification of the last 5 min of LTP recordings shown in B. (D) Contextual and cued fear conditioning demonstrated impairments in SUMO1, APP and SUMO1-APP Tg mice. The impairments in SUMO1-APP double Tg mice were more severe than in SUMO1-Tg and APP-Tg mice in the cued conditioning task. (N = 7–14, p < 0.0005 for contextual conditioning and p < 0.0001 for cued conditioning).
These changes in BST and LTP were also reflected in alterations in learning and memory to different degrees in transgenic models. Contextual fear conditioning reflects hippocampal and amygdala function. The three Tg groups including SUMO1, APP and SUMO1-APP mice had clear defects in contextual and cued memory compared to the non-Tg group (Fig. 9D). Most importantly, cued fear conditioning, an associative behavioral test of amygdala function, indicated substantial impairments in the SUMO1-APP double transgenic mice (Fig. 9D), compared to APP-Tg mice and SUMO1-Tg animals. Sensory threshold analysis (Supplemental Fig. S10) revealed no difference in response to stimuli between the groups demonstrating no differences in animal capability to perceive the electric shock among different groups of mice.
Taken together these results suggest that overexpression of the SUMO1 transgene in APP-Tg mice results in worsening of the basal neurotransmission without further deterioration of LTP. This probably reflects a plateauing of the detrimental effects on synaptic plasticity by concurrent overexpression of SUMO1 and APP transgenes. Most importantly, although the SUMO1 transgenics already exhibit learning and memory impairments, the introduction of amyloid pathology in these animals resulted in a worsening of the phenotype with respect to cued memory.
4. Discussion
SUMOylation and SUMO-related pathways have been linked to a number of neurodegenerative disorders including Alzheimer and Parkinson disease, amyotrophic lateral sclerosis (ALS), and Huntington disease (reviews see, (Dorval and Fraser, 2007; Lee et al., 2013; Krumova and Weishaupt, 2013; Feligioni et al., 2013)). Several studies have specifically demonstrated the involvement of SUMOylation in the neurofibrillary tangle and amyloid and neurofibrillary tangle pathology which characterizes AD. Conjugation of SUMO1 to the tangle-related tau protein induces hyperphosphorylation and inhibits tau degradation by the ubiquitin proteasome pathway (Luo et al., 2014). This connection is supported by the observed co-localization of SUMO-immunoreactivity with phosphorylated tau aggregates in transgenic mouse models (Takahashi et al., 2008). Several investigations have also demonstrated a link between SUMO1 and SUMO2 in the amyloid pathology and APP processing pathways in AD. An initial in vitro study revealed that reduced SUMOylation or mono-SUMO2 conjugation resulted in increased Aβ production (Li et al., 2003a; Li et al., 2003b). In contrast, the SUMO2 K11R mutant which prevents poly-SUMOylation caused a reduction in Aβ levels. SUMO1 has also been reported to be conjugated to APP in cells over-expressing the E2 ligase, Ubc9 (Zhang and Sarge, 2008). This direct SUMOylation of APP negatively impacts processing leading to lower Aβ levels. Similar investigations have reported conflicting findings with no effect of SUMO1 or SUMO2 on APP processing while SUMO3 appeared to specifically increase Aβ, possibly by altering APP substrate levels (Dorval et al., 2007b). However, SUMO3 did not affect APP via direct SUMOylation as similar increases in Aβ could be achieved by the GG-AA conjugation-deficit mutants. The debate on the connections between SUMO and amyloid pathology have expanded to investigate potential mechanisms to determine if direct or indirect pathways are involved.
The current study examines the specific effects of SUMO1 on APP processing, Aβ production and amyloid plaque pathology using in vivo transgenic models. This was achieved by cross-breeding a SUMO1 over-expressing transgenic with an aggressive model of AD-related amyloid pathology (TgCRND8) (Matsuzaki et al., 2015; Chishti et al., 2001). A modest increase in the insoluble Aβ and plaque densities were observed in the SUMO1-APP double transgenic as compared to mice expressing only the human APP. This was particularly evident in the hippocampus and cortex of older SUMO1-APP animals (22 weeks of age and older). However, processing of the APP transgene was not dramatically altered as evidenced by comparable levels of soluble Aβ40 and 42 and the secreted APP ectodomain generated by β-cleavage, sAPPβ. Previous in vitro studies have indicated that SUMO1 is capable of increasing BACE1 expression leading to elevated Aβ levels (Yun et al., 2013). However, changes in BACE1 protein levels were not observed in either the SUMO1 or SUMO1-APP transgenic mice. This would be consistent with unaltered β-secretase cleavage products, Aβ and sAPPβ, in these animals. These findings suggest that the observed increase in amyloid plaque density was not primarily due to alterations in APP processing or Aβ production.
One possible explanation is a decrease in the clearance of Aβ aggregates which can occur through transcytosis across the blood-brain barrier, proteolytic degradation or phagocytosis by microglia [reviewed in (Bohm et al., 2015)]. Neprilysin is one of the main proteases involved in Aβ degradation which is negatively regulated by the amyloid intracellular domain (AICD/AID) generated by the γ-secretase complex (Pardossi-Piquard et al., 2005; Pardossi-Piquard et al., 2006). A decrease in neprilysin could account for the increase plaque density in the SUMO1-APP mice. However, this was not the case in our studies in which we found that the levels were comparable with the APP-Tg animals. Similarly, the levels of the other primary Aβ proteases, insulin degrading enzyme (IDE), remained unchanged in the SUMO1-APP double transgenics. Microglia also have the ability to clear fibrillar Aβ by phagocytosis and large numbers activated cells are seen surrounding plaques (El Khoury et al., 2007). This may be a factor in the transgenic animals as a decrease in the number of microglia was observed, specifically in the hippocampus, of the SUMO1-APP mice. Changes in microglia numbers were not observed in the SUMO1 only transgenic (SUMO1-Tg) mouse model suggesting that the amyloid-related stress may be involved in this process.
There are several lines of evidence connecting SUMOylation with microglia and macrophage pathways. For example, SUMO conjugation to the IFN regulatory factor 8 (IRF8) is one of the steps in macrophage activation to trigger innate immune responses (Chang et al., 2012). This is part of much wider effect of SUMO post-translational modifications of nuclear receptors and the peroxisome proliferator-activated receptor-γ (PPAR-γ) which can repress the transcriptional regulation of several inflammatory response genes (Pascual et al., 2005). SUMOylation can also alter miRNA levels in lung macrophages which can be rescued by blocking Ubc9 activity (Gross et al., 2014). In addition, SUMO1 conjugation of the myeloid transcription factor, MafB, leads to an increase in macrophage differentiation (Tillmanns et al., 2007). However, since this is a transgenic model it raises questions as to whether or not the SUMO1 transgene will impact microglia by increasing cellular expression.
SUMO1 expression is driven by the prion (PrP) cos-tet promoter which is typically considered to be a neuronal housekeeping gene and expressed at lower levels in astrocytes (DeArmond et al., 1997). However, PrP expression in microglia has been shown to be important for their activation as demonstrated by changes associated with superoxide exposure (Brown et al., 1998). This was confirmed using transgenic models that express PrP in multiple tissue types using the same cos-tet promoter as that in the SUMO1 transgenic mice (Race et al., 2000). Transgenics with wide expression driven by the cos-tet promoter (Tg7-HaPrP) displayed high levels of microglia activation and retinal degeneration following treatment with infections prions (Kercher et al., 2007). No effect was observed in transgenics expressing PrP (Tg-NSE) under a neuron-specific promoter. These findings indicate that the SUMO1 and SUMO1-APP transgenics generated with the prion cos-tet promoter are expected to increase expression in microglia. This represents a potentially novel aspect of SUMO1 with respect to its involvement in amyloid pathogenesis.
Aβ aggregates and oligomers are associated with synaptic dysfunction and loss of dendritic spines which results in the AD-related cognitive impairments. Our previous study demonstrated that SUMO1 by itself has detrimental effects on spine development that results in transgenics mice with inherent learning and memory problems, even without the addition of Aβ pathology (Matsuzaki et al., 2015). The current study has indicated that the SUMO1-APP double transgenics display a worsening of the overall synaptic and behavioral phenotypes with increased amyloid loads. Interestingly, the decrease in fEPSP slope was slightly less than the decrease in total spine number. One possibility is that the presence of a spine does not necessary mean that a synapse is at that level which would be in keeping with the fact that functioning synapses are more than just the isolated spine. Alternatively, it is possible that there is some compensation in the strength of the synapse when there are less spines. Nevertheless, the findings that synaptic and behavioral phenotypes are worsened in SUMO1-APP mice is relevant and consistent with the overall negative role of SUMO1 onto amyloid load.
While SUMO1 appears to have a negative impact, SUMO2 can be beneficial with respect to Aβ toxicity and synaptic function. This was originally proposed following the observed increase in SUMO2/3 conjugates after induction of LTP (Lee et al., 2014). Subsequent studies revealed that by enhancing SUMOylation through an increase in Ubc9 levels or a decrease in SENP were able to prevent Aβ-mediated impairments in LTP and behavior. Other studies have also indicated that SUMO2 is neuroprotective and is elevated in response to stress such as ischemic stroke and possibly mitigate some of the detrimental aspects of these cellular insults (Lee and Hallenbeck, 2013; Wang et al., 2013; Datwyler et al., 2011).
Cumulatively, our findings indicate that SUMO1 does not have a major on APP processing in the in vivo transgenic model but may impact Aβ clearance, possibly by altering microglial activity. These findings also support the notion that maintenance of a constitutive degree of SUMO1 conjugations is important for neuronal development and function. As a consequence, increased expression or failure to de-conjugate SUMO1 negatively impacts a number of neuronal pathways, resulting in no effect or further worsening of pathology in amyloid depositing animals. Intriguingly, SUMO2 may be more critical in a beneficial response to Aβ insults and it would be of interest to determine if SUMO2 over-expressing transgenes when crossed to APP mice result in the rescue of the AD-like phenotype.
Supplementary Material
Acknowledgements
This investigation was supported by funding from the Canadian Institute of Health Research (TAD-117950) (http://www.cihr-irsc.gc.ca), the Alzheimer's Society of Ontario (http://www.alzheimer.ca) to PEF and a National Institutes of Health grant NIH-NS049442 to OA (http://www.nih.gov). Support was also provided by a Grant-in-Aid for Young Scientists B (23790224), the Ichiro Kanehara Foundation, the Mitsui Sumitomo Insurance Welfare Foundation and the Nakatomi Foundation (SM). Additional funding was provided by the Japanese Society for the Promotion of Science Program (JSPS) for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (S2603) (SM, HT, TK), the Nakayama Foundation for Human Science and a Grant-in-Aid for Young Scientists B (No. 26860133) to HT. We also wish to thank Rosemary Ahrens, Kyung Han, Kathy Ha and Kelvin So for their invaluable technical assistance.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2017.11.015.
References
- Andrew RJ, Kellett KAB, Thinakaran G, Hooper NM, 2016. A Greek tragedy: the growing complexity of Alzheimer amyloid precursor protein proteolysis. J. Biol. Chem 291 (37), 19235–19244. 10.1074/jbc.R116.746032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm C, Chen F, Sevalle J, Qamar S, Dodd R, Li Y, Schmitt-Ulms G, Fraser PE, St George-Hyslop PH, 2015. Current and future implications of basic and translational research on amyloid-beta peptide production and removal pathways. Mol. Cell. Neurosci 66 (Pt A), 3–11. 10.1016/j.mcn.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Besinger A, Herms JW, Kretzschmar HA, 1998. Microglial expression of the prion protein. NeuroReport 9 (7), 1425–1429. [DOI] [PubMed] [Google Scholar]
- Chang TH, Xu S, Tailor P, Kanno T, Ozato K, 2012. The small ubiquitin-like modifier-deconjugating enzyme sentrin-specific peptidase 1 switches IFN regulatory factor 8 from a repressor to an activator during macrophage activation. J. Immunol 189 (7), 3548–3556. 10.4049/jimmunol.1201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D, 2001. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem 276 (24), 21562–21570. 10.1074/jbc.M100710200. . [DOI] [PubMed] [Google Scholar]
- Cho SJ, Yun SM, Jo C, Lee DH, Choi KJ, Song JC, Park SI, Kim YJ, Koh YH, 2015. SUMO1 promotes Aβ production via the modulation of autophagy. Autophagy 11 (1), 100–112. 10.4161/15548627.2014.984283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datwyler AL, Lättig-Tünnemann G, Yang W, Paschen W, Lee SLL, Dirnagl U, Endres M, Harms C, 2011. SUMO2/3 conjugation is an endogenous neuroprotective mechanism. J. Cereb. Blood Flow Metab 31 (11), 2152–2159. 10.1038/jcbfm.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeArmond SJ, Sánchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB, 1997. Selective neuronal targeting in prion disease. Neuron 19 (6), 1337–1348. 10.1016/S0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA, 2004. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci. Lett 367 (2), 164–167. 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- Devi L, Ohno M, 2015. A combination Alzheimer's therapy targeting BACE1 and neprilysin in 5XFAD transgenic mice. Mol. Brain 8, 19. 10.1186/s13041-015-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval V, Fraser PE, 2007. SUMO on the road to neurodegeneration. Biochim. Biophys. Acta 1773 (6), 694–706. 10.1016/j.bbamcr.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Dorval V, Mazzella MJ, Mathews PM, Hay RT, Fraser PE, 2007a. Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem. J 404 (2), 309–316. 10.1042/BJ20061451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorval V, Mazzella MJ, Mathews PM, Hay RT, Fraser PE, 2007b. Modulation of Abeta generation by small ubiquitin-like modifiers does not require conjugation to target proteins. Biochem. J 404 (2), 309–316. 10.1042/BJ20061451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD, 2007. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med 13 (4), 432–438. 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guénette S, 2003. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. U. S. A 100 (7), 4162–4167. 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feligioni M, Mattson MP, Nisticò R, 2013. SUMOylation in neuroplasticity and neurological disorders. NeuroMolecular Med. 15 (4), 637–638. 10.1007/s12017-013-8267-5. [DOI] [PubMed] [Google Scholar]
- Feligioni MM, Serena M, Knock Erin, Nadeem Urooba, Arancio Ottavio, Fraser Paul, 2015. SUMO modulation of protein aggregation and degradation. AIMS Mol. Sci 2 (4), 382–410. [Google Scholar]
- Gross TJ, Powers LS, Boudreau RL, Brink B, Reisetter A, Goel K, Gerke AK, Hassan IH, Monick MM, 2014. A Microrna processing defect in smokers' macrophages is linked to sumoylation of the endonuclease DICER. J. Biol. Chem 289 (18), 12823–12834. 10.1074/jbc.M114.565473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannoun Z, Greenhough S, Jaffray E, Hay RT, Hay DC, 2010. Post-translational modification by SUMO. Toxicology 278 (3), 288–293. 10.1016/j.tox.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Haung Yu W., Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA, 2005. Macroautophagy - a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J. Cell Biol 171 (1), 87–98. 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC, 2000. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med 6 (2), 143–150. 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Jolas T, Zhang XS, Zhang Q, Wong G, Del Vecchio R, Gold L, Priestley T, 2002. Long-term potentiation is increased in the CA1 area of the hippocampus of APP < inf > swe/ind < /inf > CRND8 mice. Neurobiol. Dis 11 (3), 394–409. 10.1006/nbdi.2002.0557. [DOI] [PubMed] [Google Scholar]
- Kercher L, Favara C, Striebel JF, LaCasse R, Chesebro B, 2007. Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. J. Virol 81 (19), 10340–10351. 10.1128/JVI.00865-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, MacTavish D, Yang J, Westaway D, Jhamandas JH, 2012. Beta amyloid-induced depression of hippocampal long-term potentiation is mediated through the amylin receptor. J. Neurosci 32 (48), 17401–17406. 10.1523/JNEUROSCI.3028-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM, 2014. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol 15 (6), 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Krumova P, Weishaupt JH, 2013. Sumoylation in neurodegenerative diseases. Cell. Mol. Life Sci 70 (12), 2123–2138. 10.1007/s00018-012-1158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Hallenbeck JM, 2013. SUMO and ischemic tolerance. NeuroMolecular Med. 15 (4), 771–781. 10.1007/s12017-013-8239-9. [DOI] [PubMed] [Google Scholar]
- Lee L, Sakurai M, Matsuzaki S, Arancio O, Fraser P, 2013. SUMO and alzheimer's disease. NeuroMolecular Med. 15 (4), 720–736. 10.1007/s12017-013-8257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L, Dale E, Staniszewski A, Zhang H, Saeed F, Sakurai M, Fa M, Orozco I, Michelassi F, Akpan N, Lehrer H, Arancio O, 2014. Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer's disease. Sci. Rep 4, 7190. 10.1038/srep07190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Wang S, Quon D, Liu Y-W, Cordell B, 2003a. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc. Natl. Acad. Sci. U. S. A 100 (1), 259–264. 10.1073/pnas.0235361100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Wang S, Quon D, Liu YW, Cordell B, 2003b. Erratum: positive and negative regulation of APP amyloidogenesis by sumoylation (proceedings of the National Academy of Sciences of the United States of America (January 7, 2003) 100:1 (259–264)). Proc. Natl. Acad. Sci. U. S. A 100 (15), 9102. 10.1073/pnas.1533444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H-B, Xia Y-Y, Shu X-J, Liu Z-C, Feng Y, Liu X-H, Yu G, Yin G, Xiong Y-S, Zeng K, Jiang J, Ye K, Wang X-C, Wang J-Z, 2014. SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc. Natl. Acad. Sci. U. S. A 111 (46), 16586–16591. 10.1073/pnas.1417548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki S, Lee L, Knock E, Srikumar T, Sakurai M, Hazrati LN, Katayama T, Staniszewski A, Raught B, Arancio O, Fraser PE, 2015. SUMO1 affects synaptic function, spine density and memory. Sci. Rep 5, 10730. 10.1038/srep10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan LE, Brown JT, Henley JM, Cimarosti H, 2011. Profiles of SUMO and ubiquitin conjugation in an Alzheimer's disease model. Neurosci. Lett 502 (3), 201–208. 10.1016/j.neulet.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, Rivest S, 2013. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc. Natl. Acad. Sci. U. S. A 110 (5), 1941–1946. 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S, 2008. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 18 (2), 240–252. 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico R, Ferraina C, Marconi V, Blandini F, Negri L, Egebjerg J, Feligioni M, 2014. Age-related changes of protein SUMOylation balance in the AbetaPP Tg2576 mouse model of Alzheimer's disease. Front. Pharmacol 5, 63. 10.3389/fphar.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, GQ Yu, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L, 2007. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55 (5), 697–711. 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Da Costa CA, Vincent B, Ring S, D'Adamio L, Shen J, Müller U, Hyslop PSG, Checler F, 2005. Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron 46 (4), 541–554. 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Dunys J, Yu G, St. George-Hyslop P, Alves Da Costa C, Checler F, 2006. Neprilysin activity and expression are controlled by nicastrin. J. Neurochem 97 (4), 1052–1056. 10.1111/jM471-4159.2006.03822.x. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK, 2005. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437 (7059), 759–763. 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ, 1998. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem 273 (49), 32730–32738. 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Race R, Oldstone M, Chesebro B, 2000. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. J. Virol 74 (2), 828–833. 10.1128/JVI.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Abe-Dohmae S, Yokoyama S, St George-Hyslop P, Fraser PE, 2015. ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J. Biol. Chem 290 (40), 24152–24165. 10.1074/jbc.M115.655076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorova L, Martin S, 2016. Sumoylation in synaptic function and dysfunction. Front. Synaptic Neurosci 8 10.3389/fnsyn.2016.00009. (APR). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclip A, Antoniou X, Colombo A, Camici GG, Pozzi L, Cardinetti D, Feligioni M, Veglianese P, Bahlmann FH, Cervo L, Balducci C, Costa C, Tozzi A, Calabresi P, Forloni G, Borsello T, 2011. c-Jun N-terminal kinase regulates soluble Aβ oligomers and cognitive impairment in AD mouse model. J. Biol. Chem 286 (51), 43871–43880. 10.1074/jbc.M111.297515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JW, Brautigam H, Short JA, Sowa A, Shi M, Yadav A, Weaver CM, Westaway D, Fraser PE, St George-Hyslop PH, Gandy S, Hof PR, Dickstein DL, 2014. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer's disease mouse model. J. Comp. Neurol 522 (10), 2319–2335. 10.1002/cne.23536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ishida M, Komano H, Takahashi H, 2008. SUMO-1 immunoreactivity co-localizes with phospho-Tau in APP transgenic mice but not in mutant tau transgenic mice. Neurosci. Lett 441 (1), 90–93. 10.1016/j.neulet.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Tillmanns S, Otto C, Jaffray E, Du Roure C, Bakri Y, Vanhille L, Sarrazin S, Hay RT, Sieweke MH, 2007. SUMO modification regulates MafB-driven macrophage differentiation by enabling Myb-dependent transcriptional repression. Mol. Cell. Biol 27 (15), 5554–5564. 10.1128/MCB.01811-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang R, Sheng H, Sheng SP, Paschen W, Yang W, 2013. Transient ischemia induces massive nuclear accumulation of SUMO2/3-conjugated proteins in spinal cord neurons. Spinal Cord 51 (2), 139–143. 10.1038/sc.2012.100. [DOI] [PubMed] [Google Scholar]
- Yun SM, Cho SJ, Song JC, Song SY, Jo SA, Jo C, Yoon K, Tanzi RE, Choi EJ, Koh YH, 2013. SUMO1 modulates Abeta generation via BACE1 accumulation. Neurobiol. Aging 34 (3), 650–662. 10.1016/j.neurobiolaging.2012.08.005.. [DOI] [PubMed] [Google Scholar]
- Zhang Y-Q, Sarge KD, 2008. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun 374 (4), 673–678. 10.1016/j.bbrc.2008.07.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.