

Abstract

Metal-free boron Lewis acids, tris(pentafluorophenyl)borane B(C6F5)3, have the advantages of low toxicity and low cost and are a promising catalyst. A density functional theory (DFT) calculation was used to clarify the mechanism and the origin of the diastereoselective cyclopropanation of aryldiazodiacetate and styrene derivatives catalyzed by B(C6F5)3. Four pathways were calculated: B(C6F5)3-catalyzed N-, C-, and O-bound boron-activated aryldiazodiacetate and without B(C6F5)3 catalysis. By calculating and comparing the energy barriers, the most possible reaction mechanism was proposed, that is, first, B(C6F5)3 catalyzed O-bound boron to activate aryldiazodiacetate, followed by the removal of a N2 molecule, and finally, styrene nucleophilic attack occurred to produce [2+1] cyclopropane products. N2 removal is the rate-limiting step, and this step determines the preference of a given mechanism. The calculated results are in agreement with experimental observations. The origin of diastereoselectivity is further explained on the basis of the favorable mechanism. The steric hindrance interference between the styrene aryl group and the large tri(pentafluorophenyl)borane B(C6F5)3 and the favorable π–π stacking interaction between the benzene rings combined to cause the high diastereoselectivity, which resulted in lower energy of the transition state (TS) corresponding to the reaction mechanism. The calculated results not only provide a more detailed explanation of the mechanism for the experimental study but also have certain reference and guiding significance for other catalytic cyclopropanation reactions.
1. Introduction
Cyclopropane, a widely used discordant carbocyclic ring, is usually synthesized by [2+1] cycloaddition.1−6 Methods for synthesizing cyclopropane are crucial in the fields of drug discovery, chemical biology, and total synthesis. Some of the most effective cyclopropane synthesis methods depend on the activation of carbene precursors, such as diazoacetic acid, and transient metal carbene species have been generated using transition-metal catalysts.7 Despite the success of transition-metal catalysts, more sustainable and environmentally friendly systems for cyclopropanation/functionalization have attracted increasing attention. Compared with transition-metal catalysts, metal-free boron Lewis acids, such as tris(pentafluorophenyl)borane B(C6F5)3 and B(C6F5)nHm, have the advantages of low toxicities and low costs.8,9 Moreover, reactions catalyzed by such metal-free boron Lewis acids exhibit different characteristics compared to mature transition-metal catalytic reactions.10
In recent years, Lewis acid and Brønsted acid (containing H bonds) catalytically activated diazocarbonyl compounds have attracted significant attention. However, these transformations are mainly confined to X–H insertion (X = N, O, and C).11 Boron Lewis acids have proven to be effective metal-free catalysts for highly selective reactions of donor and acceptor diazo compounds with a series of substrates. In 2014, Mattson and co-workers12 reported the N–H insertion reaction between aniline and α-nitrodiazide activated by a thiourea catalyst. In 2016, Yu et al.13 reported the B(C6F5)3-activated ortho-selective C–H insertion reaction of aryldiazoacetate and phenol, and Zhang et al.14 subsequently clarified the root of chemical selectivity and regioselectivity through theoretical calculations. In 2017, Tang et al.15 conducted research on the 1,1-hydroboration reaction of Ph2CN2 with HB(C6F5)2. However, using inactivated alkenes to produce cyclopropane with boron Lewis acids has rarely been reported.
In 2020, Melen et al.16 first reported the regioselective and diastereoselective C–H insertion, cyclopropanation, and ring-opening reactions of aryldiazodiacetate with a series of substrates (including indole, benzofuran, indene, pyrrole, styrene, and furan) under mild conditions. They also reported the mechanism of these reactions using comprehensive density functional theory (DFT) research to fully understand the regioselectivity and diastereoselectivity they observed. Almost at the same time, Mancinelli and Wilkerson-Hill17 reported the diastereoselective cyclopropanation of aryldiazodiacetate and styrene derivatives catalyzed by B(C6F5)3 (Scheme 1a). Based on the available experimental and computational background,14,18 Mancinelli and Wilkerson-Hill17 proposed a possible reaction mechanism of aryldiazodiacetate O-bound boron activated by B(C6F5)3 (pathway C, Scheme 1b). In 2021, Yang et al.19 calculated and studied the boron–O binding mechanism of cyclopropanation of styrene and aryldiazodiacetate catalyzed by B(C6F5)3 using density functional theory (DFT) and explained the origin of diastereoselectivity, in which the steric hindrance interference between the styrene aryl group and the bulky tri(pentafluorophenyl)borane catalyst played an important role in determining diasteroselectivity. However, there are many active sites on aryldiazodiacetate, and the competition between aryldiazodiacetate N-, C-, and O-bound boron activated by B(C6F5)3 complicates the reaction mechanism. In this study, to probe the reaction mechanism in detail and clarify the origin of diastereoselectivity, various pathways were investigated using DFT calculations, including B(C6F5)3-catalyzed N-bound boron activation (pathway A), C-bound boron activation (pathway B), and O-bound boron activation (pathway C, Scheme 1b) as well as without B(C6F5)3 catalysis (pathway D). The most plausible reaction mechanism was proposed by comparing these pathways. The calculated results are in agreement with experimental observations.17 Based on the most favorable mechanism, combined with frontier orbital analysis and distortion/interaction analysis, the origin of diastereoselectivity was explained, in which the favorable π–π stacking between aryldiazodiacetate and styrene aryl plays an important role in determining diastereoselectivity. The calculated results not only verified the experimental findings but also provided a more reliable mechanistic explanation for this reaction, which can provide guidance for other catalytic cyclopropanation reactions.
Scheme 1. (a) Cyclopropanation of Styrene and Aryldiazodiacetate Catalyzed by B(C6F5)3 and (b) Proposed Mechanisms.

2. Computational Details
According to the experimental work, p-phenyldiazonium bromide and styrene were selected as the model substrates in this study. The reaction temperature is 323.15 K, and 1,2-dichloroethane (1,2-DCE) is used as the solvent for the reaction.17 All of the calculations in this paper are carried out in the Gaussian 0920 software package. All reactants and transition states are optimized in the solvent phase, and the B3LYP-D321 calculation method is adopted, in which nonmetallic atoms such as C, H, N, O, B, F, and Br are carried out at the 6-31G* basis group level. The single-point energy is calculated using the SMD solvation model, and the calculation method is B3LYP-D3, where the basis set used for nonmetallic atoms such as C, H, N, O, B, F, and Br is 6-311++G**. The internal coordinate of the reaction (IRC) is calculated,22 the selected key transition state structure relative to the corresponding reactants and products is determined, and the analytical resonance frequency indicates the transition state (one virtual frequency) and stable structure (no virtual frequency) at the same theoretical level. An optimized 3D structure image is made using CYLview23 software, and LUMO frontier orbits are made using GaussView 5.020 software. Noncovalent interaction analysis is performed with Multiwfn24 software.
3. Results and Discussion
3.1. Mechanism of the Cyclopropanation of Styrene and Aryldiazodiacetate Catalyzed by Tris(pentafluorophenyl)borane
3.1.1. Mechanism A of Activating N-Bound Boron
The mechanism for N-bound boron activation in the aryldiazodiacetate substrate was considered to involve styrene nucleophilic attack + N2 removal (pathway A1, red lines, Figure 1). The nucleophilic attack of styrene on IN1A proceeds through the transition state TS1A, which has a Gibbs free energy of 28.0 kcal/mol, resulting in a N-containing five-membered cyclic compound (IN2A) as an intermediate. Subsequently, the ternary carbocyclic ring is constructed through the transition state TS2A (Gibbs free energy of 41.0 kcal/mol). Finally, N2 and B(C6F5)3 are removed to produce 3a. We ruled out this process because of its high energy barrier (41.0 kcal/mol). Meanwhile, we also considered the N-bound boron activation mechanism involving N2 removal + styrene nucleophilic attack (pathway A2, green lines, Figure 1). As the free energy of the transition state TS1A-N2 is 36.9 kcal/mol, we also excluded this process. In conclusion, for aryldiazodiacetate 1a, the N-bound boron activation mechanisms are not feasible.
Figure 1.
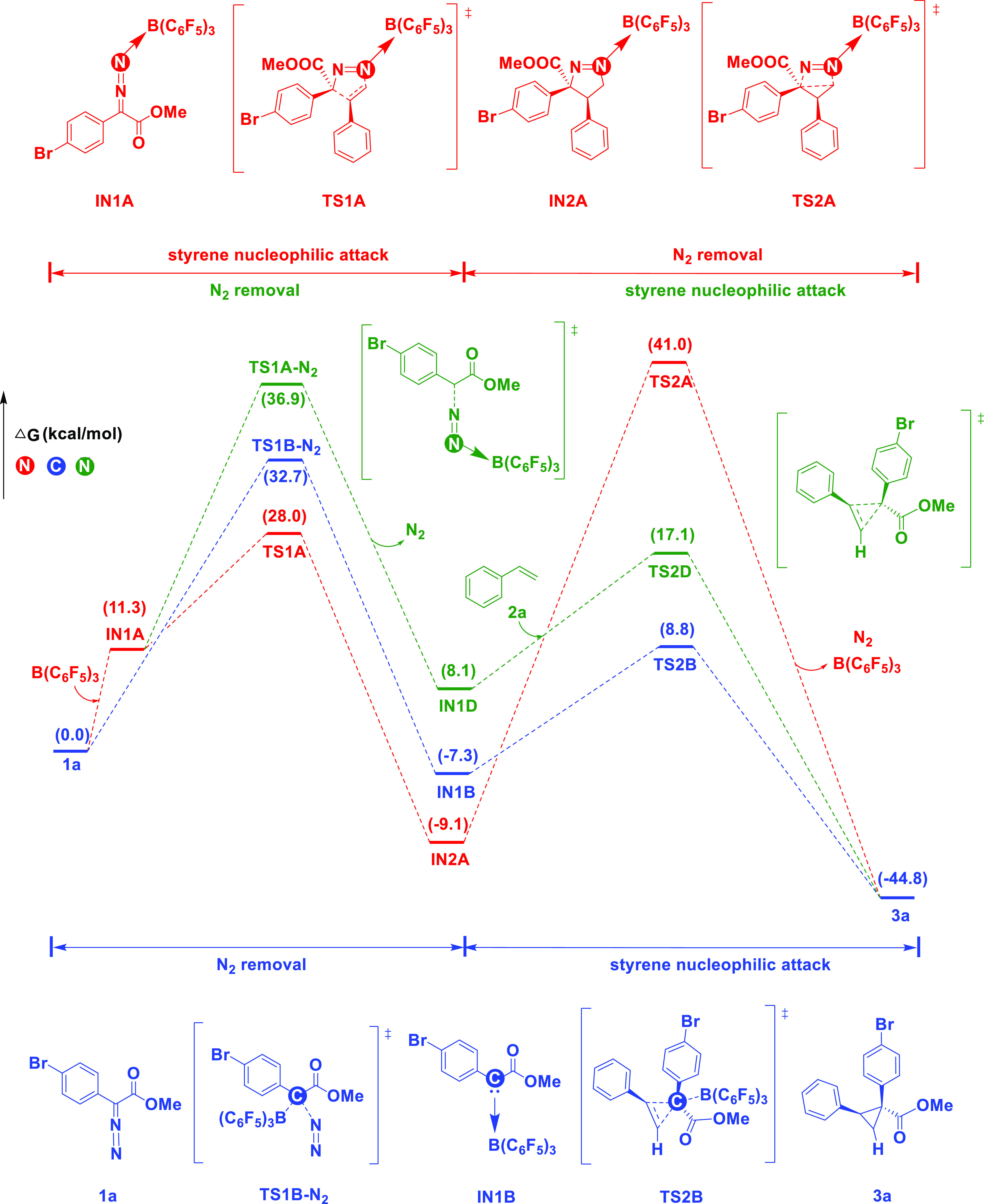
N/C-bound boron activation mechanisms for the aryldiazodiacetate substrate (pathways A/B). Free energies (kcal/mol) are relative to 1a.
3.1.2. Mechanism B of Activating C-Bound Boron
Unlike pathway A, in pathway B with C-bound boron activation, the removal of N2 is assisted by B(C6F5)3 (blue lines, Figure 1). In this process, the carbene intermediate IN1B is formed via the transition state TS1B-N2 (Gibbs free energy of 32.7 kcal/mol), and then nucleophilic attack by styrene proceeds through the transition state TS2B (Gibbs free energy of 8.8 kcal/mol) to produce the ternary cyclic compound 3a. As this process has a high energy barrier of 32.7 kcal/mol, it was also excluded.
3.1.3. Mechanism C of Activating O-Bound Boron
For O-bound boron activation in the aryldiazodiacetate substrate, we considered a mechanism involving styrene nucleophilic attack + N2 removal (pathway C1, yellow lines, Figure 2). The first step in pathway C1 is similar to that in pathway A1. Substrate aryldiazodiacetate 1a reacts with the catalyst, B(C6F5)3, to form complex IN1C. Subsequently, styrene nucleophilically attacks to form a five-membered cyclic intermediate IN2C via the transition state TS1C (Gibbs free energy of 33.6 kcal/mol). As this intermediate is unstable, a N2 molecule is easily removed to form the ternary cyclic cyclopropane product 3a. Because this process has a high energy barrier (33.6 kcal/mol), it was also excluded. On the other hand, we also considered an O-bound boron activation mechanism involving N2 removal + styrene nucleophilic attack (pathway C2, black and purple lines, Figure 2). Initially, aryldiazodiacetate 1a and the catalyst, B(C6F5)3, form intermediate E-IN1C. Thereafter, a N2 molecule leaves via the transition state E-TS3C (Gibbs free energy of 24.5 kcal/mol) to form a carbene intermediate, E-IN3C, which cyclizes with styrene to form the ternary cyclic product 3a (purple lines, Figure 2). Aryldiazodiacetate 1a and the catalyst can also form intermediate Z-IN1C, which is relatively stable with a free energy of 0.3 kcal/mol. Subsequently, a molecule of N2 leaves via the transition state Z-TS3C (Gibbs free energy of 22.1 kcal/mol) to form Z-IN3C, a more stable carbene intermediate with a free energy of 0.2 kcal (black lines, Figure 2). The “Z” and “E” refer to the stereochemistry of the oxygen coordination to boron. The energy of the black pathway is 2.4 kcal/mol lower than that of the purple pathway (22.1 kcal/mol vs 24.5 kcal/mol). Moreover, this black reaction pathway has the smallest energy barrier at present.
Figure 2.
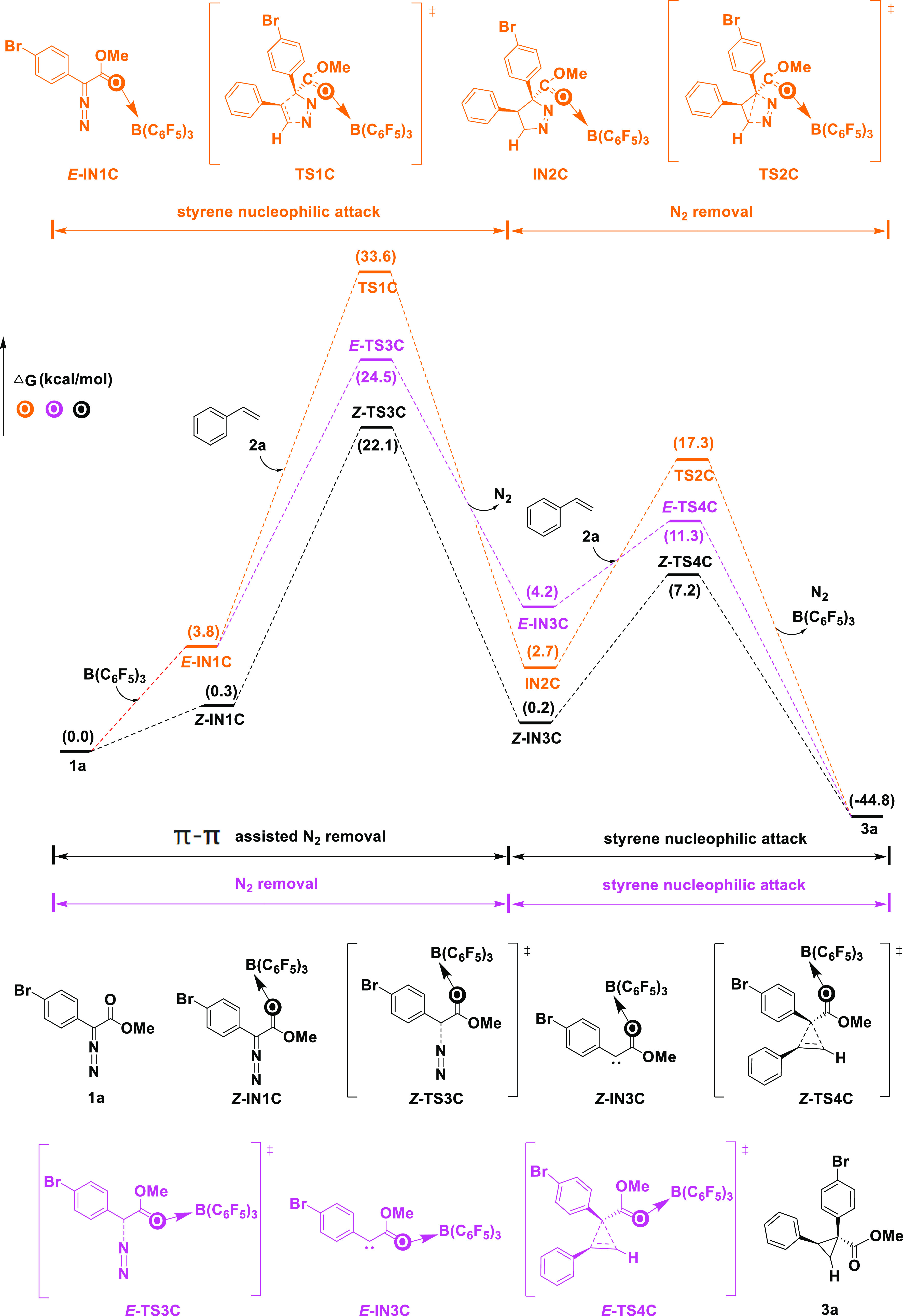
O-bound boron activation mechanisms for the aryldiazodiacetate substrate (pathway C). Free energies (kcal/mol) are relative to 1a.
3.1.4. Mechanism D of Catalyst Activation without B(C6F5)3
We also calculated the diastereoselective cyclopropanation reaction mechanism of aryldiazodiacetate 1a and styrene 2a without B(C6F5)3 catalysis, as shown in Figure 3. The reaction mechanisms without B(C6F5)3 catalysis, whether N2 removal or the styrene nucleophilic attack transition state, have a higher Gibbs free energy, which are 28.3, 28.8, and 41.7 kcal/mol, respectively. Therefore, the reaction mechanism without B(C6F5)3 as a catalyst is not feasible.
Figure 3.
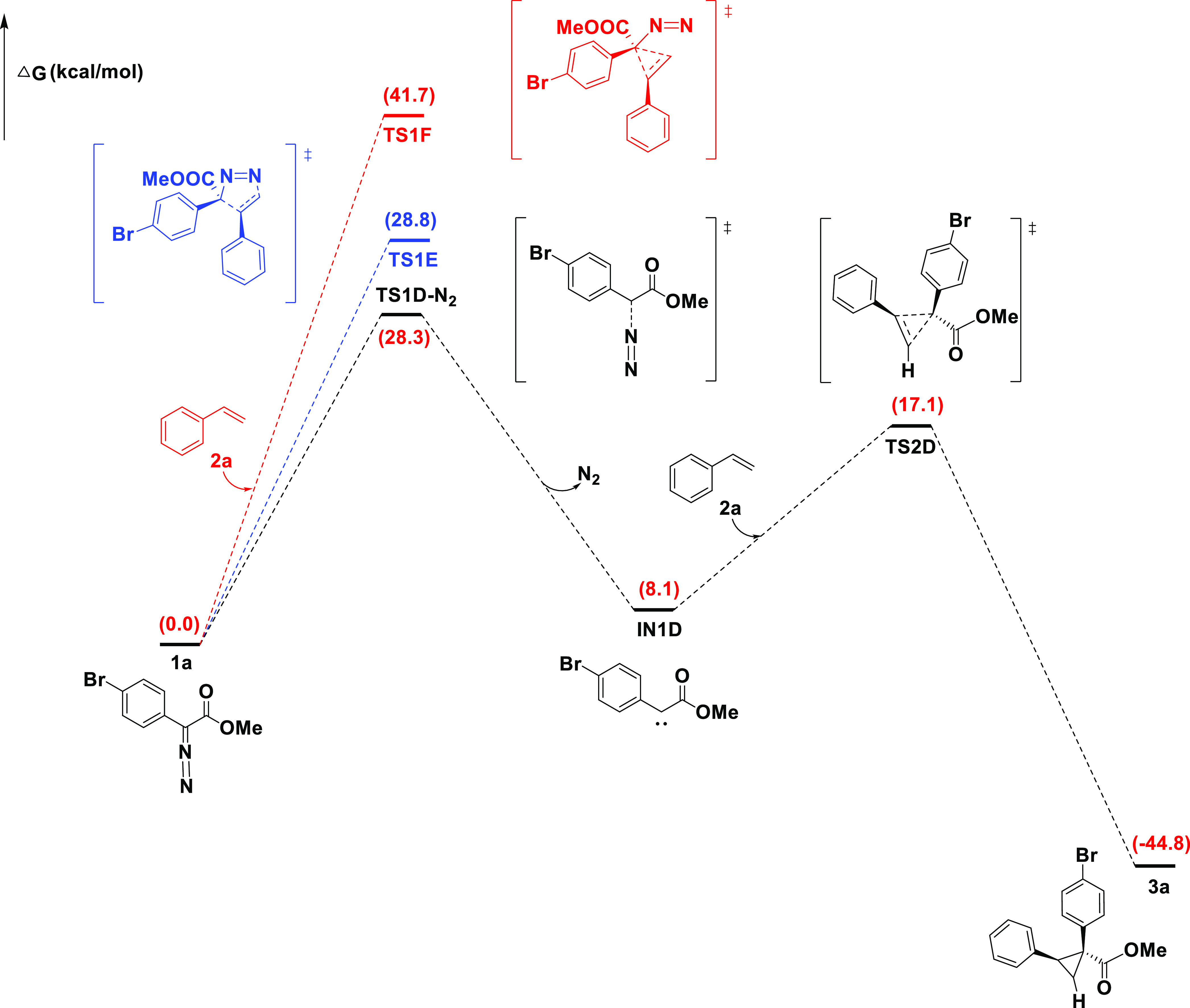
Mechanism D of catalyst activation without B(C6F5)3. Free energies (kcal/mol) are relative to 1a.
3.2. Analysis and Comparison of Key Transition States for the Four Mechanisms
The key transition state for each mechanism was analyzed to further clarify the reaction mechanism. Compared with pathways A and B with B(C6F5)3-catalyzed N- and C-bound boron activation and pathway D without B(C6F5)3 catalysis, pathway C with B(C6F5)3-catalyzed O-bound boron activation has a lower reaction energy barrier of 22.1 kcal/mol (Figure 4). There are two types of mechanisms, the styrene nucleophilic attack occurring after N2 removal. N2 removal is the rate-limiting step and this step determines the preference of a given mechanism. In previous studies,17 it has been suggested that the interaction of boron yields a carbene form that facilitates N2 removal, and our calculation results also support this hypothesis. In addition, under B(C6F5)3 activation, the carbonyl O atom is more likely to provide an unshared electron pair and π–π stacking is possible between the two benzene rings in the transition state Z-TS3C. These factors would reduce the Gibbs free energy of the reaction, resulting in easier N2 removal and the formation of a more stable carbene intermediate. In contrast, for N and C atoms activated by B(C6F5)3, as there are fewer outer electrons, it is difficult to obtain a lone electron pair. Furthermore, steric hindrance in the transition state makes N2 removal difficult and increases the Gibbs free energy of the reaction.
Figure 4.
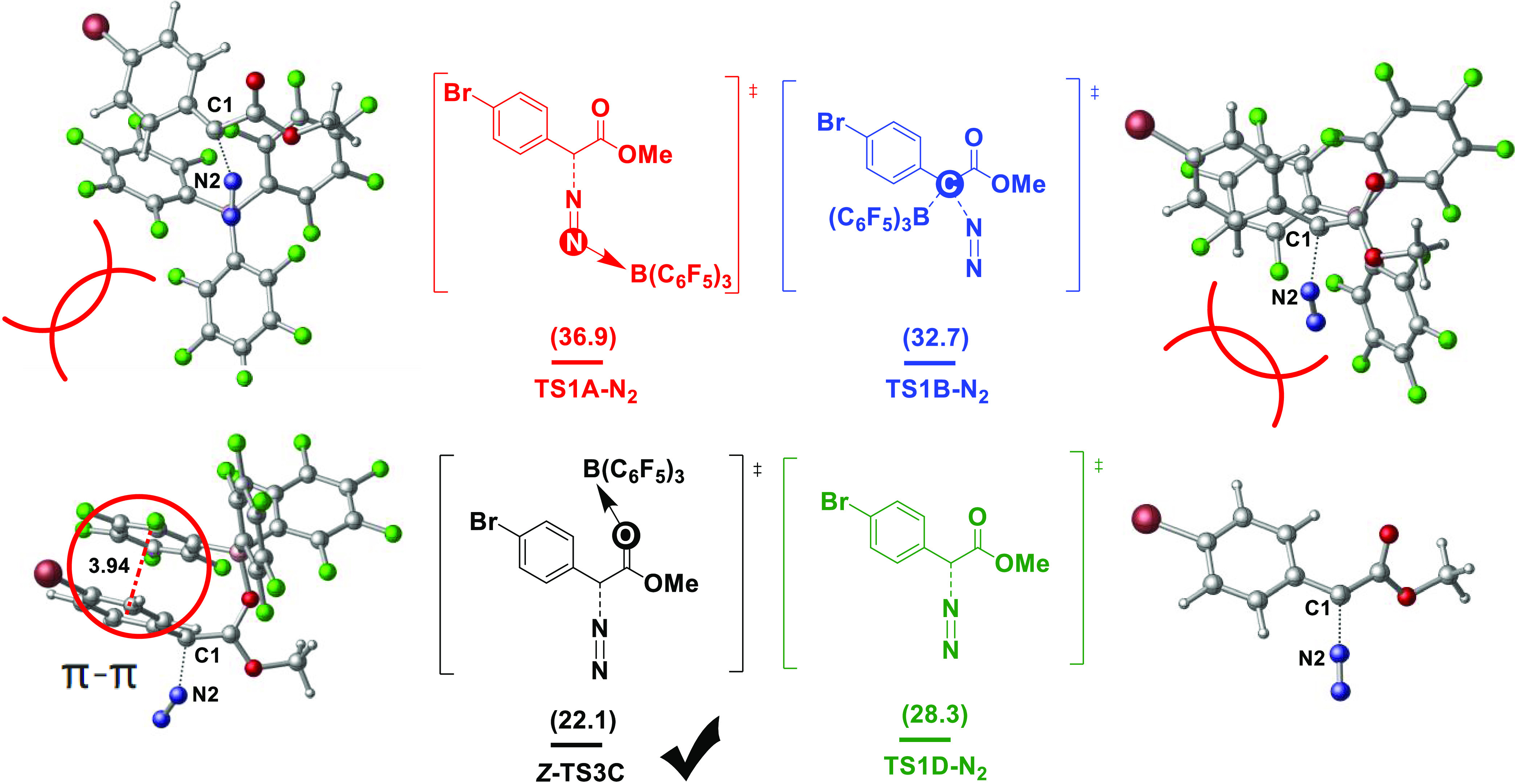
Comparison of key transition states for the four mechanisms. Free energies (kcal/mol) are relative to 1a.
3.3. Origin of Diastereoselectivity
The origin of diastereoselectivity can be explained by the transition state of the most favorable mechanism. As shown in Figure 5, the π–π stacking interaction in Z-TS4C is obviously stronger than that in E-TS4C-iso (the distance between the two benzene ring centers is 3.51 Å vs 3.85 Å, and the included angle between the benzene ring planes is 40.1° vs 54.6°). In addition, In E-TS4C-iso, there is steric hindrance between the styrene aryl group and the large B(C6F5)3 catalyst. Furthermore, the analysis and comparison of LUMO frontier orbits of the two transition states can also prove this point of view. To rationalize the nonpair selectivity, we also compared Z-TS4C and E-TS4C-iso with an energy difference of 3.9 kcal/mol (7.2 kcal/mol vs 11.1 kcal/mol). This energy difference is consistent with the observed diastereoselectivity.17 As shown in Figure 6, we also the adopted distortion/interaction analysis. The distortion energies of Z-TS4C and E-TS4C-iso are 2.6 and 4.6 kcal/mol, respectively. The calculated interaction energies of Z-TS4C and E-TS4C-iso are −10.7 and −13.6 kcal/mol, respectively. It is pointed out that diastereoselectivity is the main cause of interaction and distortion energy. To stimulate the effect of interaction energy, Z-TS4C and E-TS4C-iso were also analyzed by noncovalent interactions. Therefore, for Z-TS4C, two strong π–π contacts, namely, the interaction between the aryl group of the diazo compound and the huge tri(pentafluorophenyl) group of B(C6F5)3 or the styrene aryl group, are the main factors determining the noncovalent interaction with different diastereoselective transition states. The noncovalent interactions of E-TS4C-iso only involve the π–π-superposition between aryl functional groups of diazo compounds and styrene aryl groups. This difference in noncovalent interactions may be the reason for the final diastereoselectivity.
Figure 5.
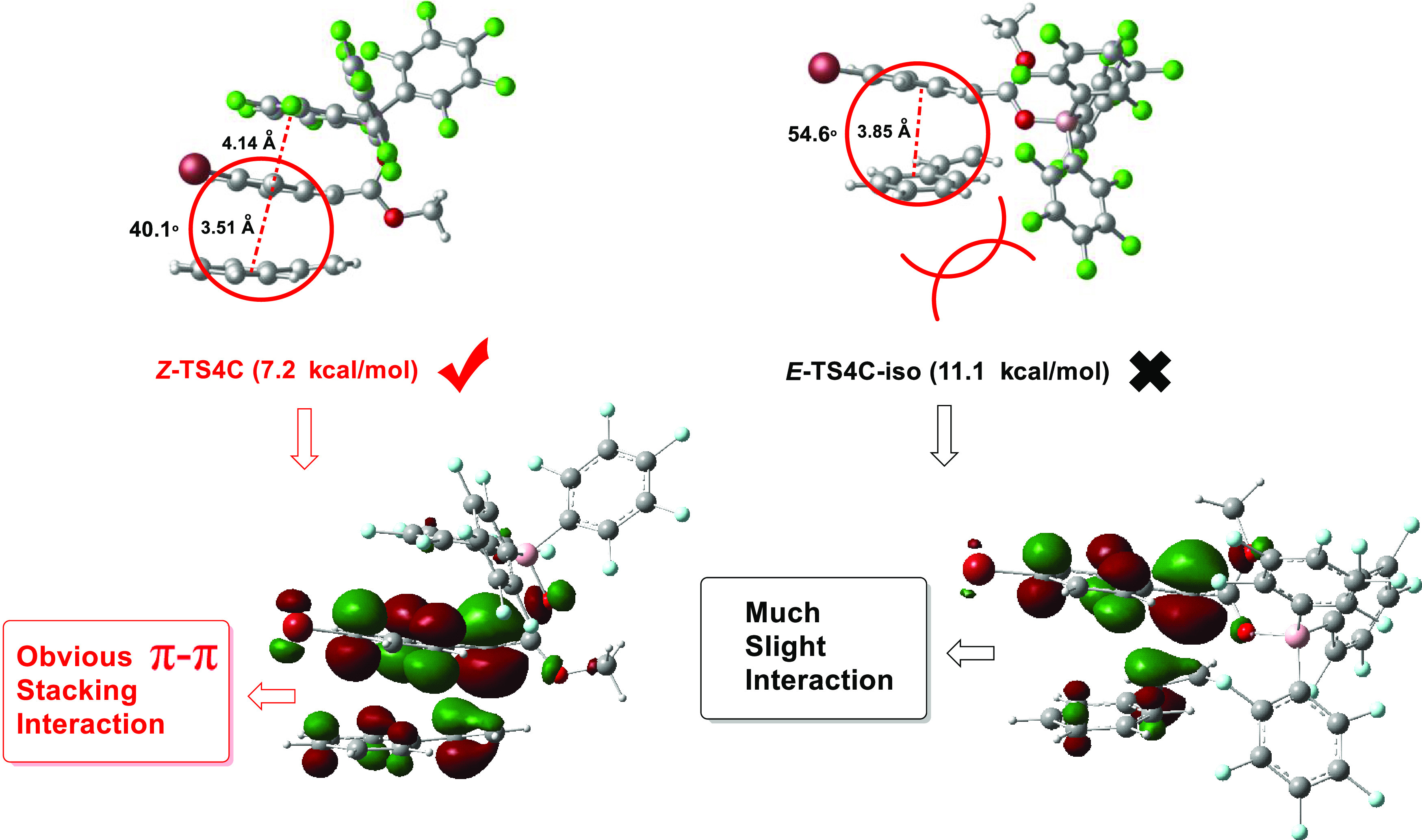
Key geometric parameters (in Å) and free energies (kcal/mol) are relative to 1a.
Figure 6.
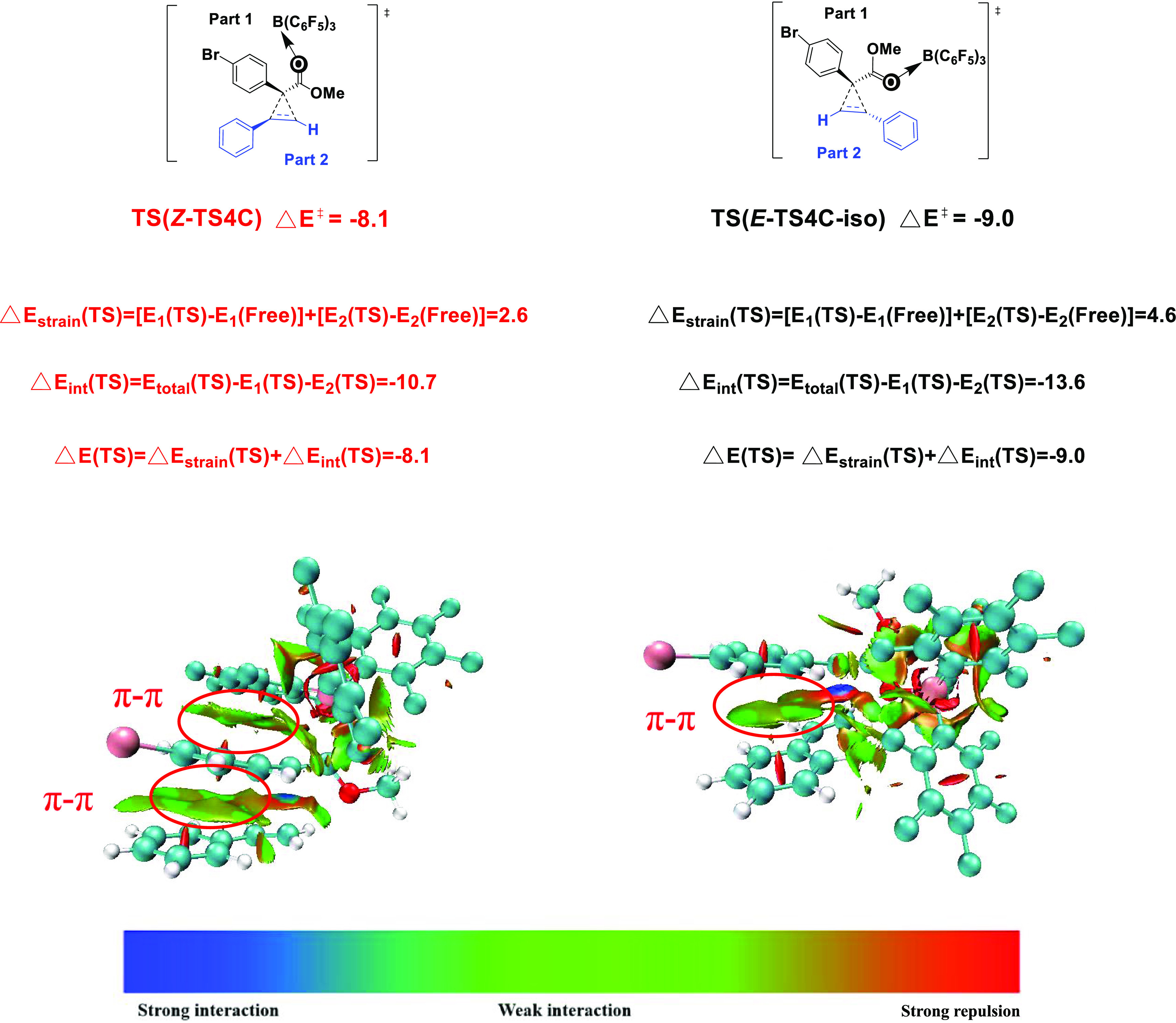
Distortion/interaction analysis and noncovalent interaction analysis for the [2+1] cycloaddition reaction. ΔE‡ is the activation energy, ΔEstrain is the distortion energy, and ΔEint is the interaction energy. The free energies are given in kcal/mol.
3.4. Substitution Effect of Diazoester and Styrene Cyclopropyl on Aromatic Rings
As shown in Figure 7, we demonstrated the substitution effect present at the aryl ring of both diazoester and styrene toward cyclopropanation. Experimental results showed that diazocarbonyl and styrene substrates without aryl substituents were ineffective, and the yield was less than 5%.17 According to the calculation results, it is suggested that the cleavage of the C–N bond and the removal of N2 are the key steps to determine the reaction rate. The strong p−π conjugation (carbene C and aryl substituent) can promote the formation of carbene because it stabilizes the key transition state and reduces the free activation energy. In contrast, diazo substrates Re1, Re2, and Re3 may affect the calculation results of carbene formation and combination due to the lack of the aryl substituent. The activation energies of C–N bond cleavage and N2 removal of substrates Re1, Re2, and Re3 are 35.2, 34.0, and 42.0 kcal/mol, respectively, indicating that they have high activation barriers, which makes it extremely difficult for C–N bond cleavage and N2 removal in the reaction process, which may be the main factor for a low product yield when Re1, Re2, and Re3 are used as substrates. This also indicates the substitution effect present at the aryl ring of diazoester toward cyclopropanation. Furthermore, Ariafard25 pointed out that the stronger π-donor the para substituent of the aryl ring, the more efficient the B(C6F5)3 catalyst.
Figure 7.
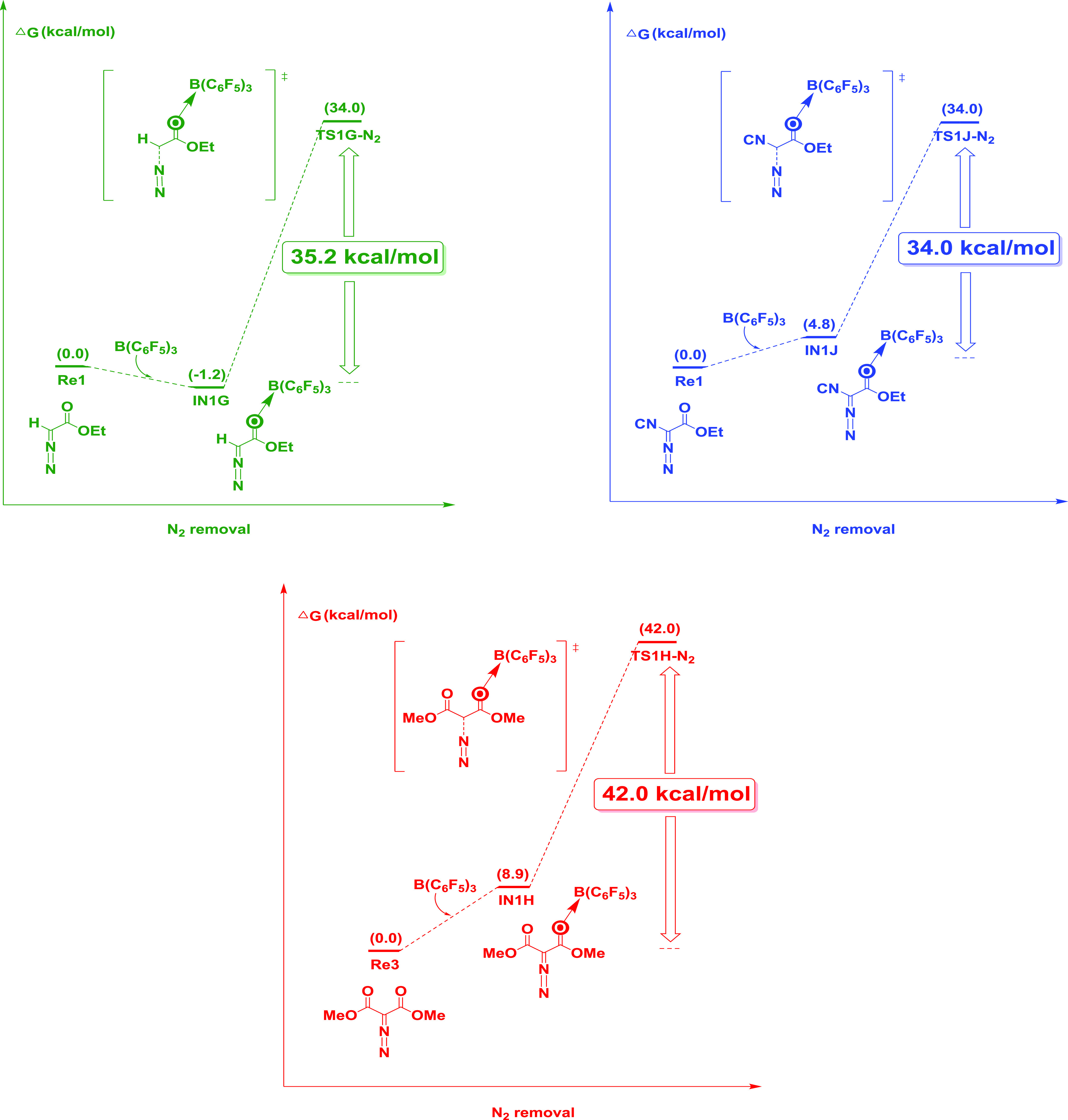
Energy profile calculated for the reactions with ethyl diazoacetate (Re1), ethyl diazocyanoacetate (Re2), or dimethyl diazomalonate (Re3). The free energies are given in kcal/mol.
4. Conclusions
In summary, we performed DFT calculations to clarify the catalytic mechanism and the origin of diastereoselectivity in the cyclopropanation of aryldiazodiacetate and styrene derivatives catalyzed by B(C6F5)3. Four possible reaction mechanisms were considered, namely, N-, C-, and O-bound boron activated by B(C6F5)3 and without B(C6F5)3. By comparing the calculated energy barriers for the different reaction mechanisms, the most plausible reaction mechanism was identified. In this pathway, B(C6F5)3 catalyzed O-bound boron to activate aryldiazodiacetate, followed by the removal of a N2 molecule, and finally the styrene nucleophilic attack produced [2+1] cyclopropane products. N2 removal is the rate-limiting step and this step determines the preference of a given mechanism. The calculated results are in agreement with experimental observations.17 This mechanism was also used to explain the origin of the observed diastereoselectivity. Steric hindrance between the styrene aryl group and the large catalyst, B(C6F5)3, as well as a favorable π–π stacking interaction between the benzene rings resulted in high diastereoselectivity and lowered the energy of the transition state (TS) in the corresponding reaction mechanism. The calculated results not only provide a more detailed explanation of the mechanism for the experimental study but also have certain references and guiding significance for other catalytic cyclopropanation reactions.
Acknowledgments
The authors thank the National Natural Science Foundation of China (NSFC, 21773312 and 21973113) and the Fundamental Research Funds for the Central Universities for the support given for this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00200.
Author Contributions
§ X.W. and P.L. contributed equally. All authors made contributions in writing the manuscript. All authors approve the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Kumar D.; de Visser S. P.; Sharma P. K.; Cohen S.; Shaik S. Radical clock substrates, their C–H hydroxylation mechanism by cytochrome P450, and other reactivity patterns: What Does Theory Reveal about the Clocks’ Behavior?. J. Am. Chem. Soc. 2004, 126, 1907–1920. 10.1021/ja039439s. [DOI] [PubMed] [Google Scholar]; b de Montellano P. R. O. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schneider T. F.; Kaschel J.; Werz D. B. A new golden age for donor-acceptor cyclopropanes. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. 10.1002/anie.201309886. [DOI] [PubMed] [Google Scholar]; b Cavitt M. A.; Phun L. H.; France S. Intramolecular donor-acceptor cyclopropane ring-opening cyclizations. Chem. Soc. Rev. 2014, 43, 804–818. 10.1039/C3CS60238A. [DOI] [PubMed] [Google Scholar]
- Talele T. T. The ‘cyclopropyl fragment’ is a versatile player that frequently appears in preclinical/clinical drug molecules. J. Med. Chem. 2016, 59, 8712–8756. 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- a Rubin M.; Rubina M.; Gevorgyan V. Transition metal chemistry of cyclopropenes and cyclopropanes. Chem. Rev. 2007, 107, 3117–3179. 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]; b Chen Y.; Fields K. B.; Zhang X. P. Bromoporphyrins as Versatile Synthons for Modular construction of chiral porphyrins: Cobalt-catalyzed highly enantioselective and diastereoselective cyclopropanation. J. Am. Chem. Soc. 2004, 126, 14718–14719. 10.1021/ja044889l. [DOI] [PubMed] [Google Scholar]; c Qin C.; Boyarskikh V.; Hansen J. H.; Hardcastle K. I.; Musaev D. G.; Davies H. M. L. D2-Symmetric Dirhodium catalysts derived from a 1,2,2,-triarylcyclopropanecarboxylate ligand: Design, Synthesis and application. J. Am. Chem. Soc. 2011, 133, 19198–19204. 10.1021/ja2074104. [DOI] [PubMed] [Google Scholar]
- a Ebner C.; Carreira E. M. Cyclopropanation strategies in recent total synthesis. Chem. Rev. 2017, 117, 11651–11679. 10.1021/acs.chemrev.6b00798. [DOI] [PubMed] [Google Scholar]; b Chen D. Y.-K.; Pouwer R. H.; Richard J.-A. Recent advances in the total synthesis of cyclopropane-containing natural products. Chem. Soc. Rev. 2012, 41, 4631–4642. 10.1039/c2cs35067j. [DOI] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Chang W.; Liu H. Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 2012, 112, 1681–1709. 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Díaz-Requejo M. M.; Perez P. J. Copper, silver and gold-based cactalyts for carbene addition or insertion reactions. J. Organomet. Chem. 2005, 690, 5441–5450. 10.1016/j.jorganchem.2005.07.092. [DOI] [Google Scholar]; b Chanthamath S.; Iwasa S. Enantioselective cyclo-propanation of a wide variety of olefins catalyzed by Ru(II)-phenox complexes. Acc. Chem. Res. 2016, 49, 2080–2090. 10.1021/acs.accounts.6b00070. [DOI] [PubMed] [Google Scholar]
- a Pellicciari R.; Natalini B.; Sadeghpour B. M.; Marinozzi M.; Snyder J. P.; Williamson B. L.; Kuethe J. T.; Padwa A. The reaction of α-diazo-β-hydroxy esters with boron trifluoride etherate: generation and rearrangement of destabilized vinyl cations. A detailed experimental and theoretical study. J. Am. Chem. Soc. 1996, 118, 1–12. 10.1021/ja950971s. [DOI] [Google Scholar]; b Draghici C.; Brewer M. Lewis acid promoted carbon-carbon bond cleavage of γ-silyloxy-α-hydroxy-β-diazoesters. J. Am. Chem. Soc. 2008, 130, 3766–3767. 10.1021/ja801004d. [DOI] [PubMed] [Google Scholar]; c Cleary S.; Hensinger M.; Brewer M. Remote C–H insertion of vinyl cations leading to cyclo-pentenones. Chem. Sci. 2017, 8, 6810–6814. 10.1039/C7SC02768K. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hensinger M. J.; Dodge N. J.; Brewer M. Substituted S-alkylidene cyclopentenones via the intramolecular reaction of vinyl cations with alkenes. Org. Lett. 2020, 22, 497–500. 10.1021/acs.orglett.9b04255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu L.; Azzouzi-Zriba K.; Bonnet-Deplon D.; Crousse B. Nonmetal catalyzed insertion reactions of diazocarbonyls to acid derivatives in fluorinated alcohols. Org. Lett. 2011, 13, 692–695. 10.1021/ol102923h. [DOI] [PubMed] [Google Scholar]
- a Légaré M.; Courtemanche M. A.; Rochette E.; Fontaine F. G. Metal-Free Catalytic C–H Bond Activation and Borylation of Heteroarenes. Science 2015, 349, 513–516. 10.1126/science.aab3591. [DOI] [PubMed] [Google Scholar]; b Chernichenko K.; Lindqvist M.; Kotai B.; Nieger M.; Sorochkina K.; Papai I.; Repo T. Metal-Free sp2-C–H Borylation as a Common Reactivity Pattern of Frustrated 2-Aminophenylboranes. J. Am. Chem. Soc. 2016, 138, 4860–4868. 10.1021/jacs.6b00819. [DOI] [PubMed] [Google Scholar]; c Bähr S.; Oestreich M. Electrophilic Aromatic Substitution with Silicon Electrophiles: Catalytic FriedelCrafts C–H Silylation. Angew. Chem., Int. Ed. 2017, 56, 52–59. 10.1002/anie.201608470. [DOI] [PubMed] [Google Scholar]; d Ma Y.; Wang B.; Zhang L.; Hou Z. Boron-Catalyzed Aromatic C–H Bond Silylation with Hydrosilanes. J. Am. Chem. Soc. 2016, 138, 3663–3666. 10.1021/jacs.6b01349. [DOI] [PubMed] [Google Scholar]
- a So S. S.; Mattson A. E. Urea activation of U-nitrodiazoesters: an organocatalytic approach to N–H insertion reactions. J. Am. Chem. Soc. 2012, 134, 8798–8801. 10.1021/ja3031054. [DOI] [PubMed] [Google Scholar]; b Auvil T. J.; So S. S.; Mattson A. E. Double arylation of nitrodiazoesters via a transient N–H insertion organocascade. Angew. Chem., Int. Ed. 2013, 52, 11317–11320. 10.1002/anie.201304921. [DOI] [PubMed] [Google Scholar]; c Fisher T. J.; Mattson A. E. Synthesis of S-peroxyesters via organocatalyzed O–H insertion of hydroperoxides and aryl diazoesters. Org. Lett. 2014, 16, 5316–5319. 10.1021/ol502494h. [DOI] [PubMed] [Google Scholar]; d Zheng H.; Dong K.; Wherritt D.; Arman H.; Doyle M. P. Brønsted acid catalyzed Friedel-Crafts-type coupling and dedini-trogenation reactions of vinyl diazo compounds. Angew. Chem., Int. Ed. 2020, 59, 13613–13617. 10.1002/anie.202004328. [DOI] [PubMed] [Google Scholar]
- So S. S.; Oottikkal S.; Badjic J.; Hadad C. M.; Mattson A. E. Urea-catalyzed N–H insertion-arylation reactions of nitrodiazoesters. J. Org. Chem. 2014, 79, 4832–4842. 10.1021/jo500698q. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Li Y.; Shi J.; Ma B.; Liu L.; Zhang J. (C6F5)3B catalyzed chemoselective and ortho-selective substitution of phenols with α-aryl α-diazoesters. Angew. Chem., Int. Ed. 2016, 55, 14807–14811. 10.1002/anie.201608937. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Zhang X.-F.; Li M.; Li C.; Liu J.-Q.; Jiang Y.-Y.; Ji X.; Liu L.; Wu Y.-C. Mechanistic insights into the chemo- and regioselective B(C6F5)3 catalyzed C–H functionalization of phenols with diazoesters. J. Org. Chem. 2019, 84, 14508–14519. 10.1021/acs.joc.9b02035. [DOI] [PubMed] [Google Scholar]
- Tang C.; Liang Q.; Jupp A. R.; Johnstone T. C.; Neu R. C.; Song D.; Grimme S.; Stephan D. W. 1,1-Hydroboration and a boron adduct of diphenyldiazomethane: A Potential prelude to FLP-N2 chemistry. Angew. Chem. 2017, 129, 16815–16819. 10.1002/ange.201710337. [DOI] [PubMed] [Google Scholar]
- Dasgupta A.; Bambaahmadi R.; Slater B.; Yates B. F.; Ariafard A.; Melen R. L. Borane-catalyzed stereoselective C–H insertion, cyclopropanation, and ring-opening reactions. Chem 2020, 6, 2364–2381. 10.1016/j.chempr.2020.06.035. [DOI] [Google Scholar]
- Mancinelli J. P.; Wilkerson-Hill S. M. Tris(pentafluorophenyl)borane-Catalyzed Cyclopropanation of Styrenes with Aryldiazoacetates. ACS Catal. 2020, 10, 11171–11176. 10.1021/acscatal.0c03218. [DOI] [Google Scholar]
- a He Z.; Kajdlik A.; Yudin A. K. F-Borylcarbonyl compounds: from transient intermediates to robust building blocks. Dalton Trans. 2014, 43, 11434–11451. 10.1039/C4DT00817K. [DOI] [PubMed] [Google Scholar]; b Rao S.; Kapanaiah R.; Prabhu K. R. Boron catalyzed C–C functionalization of allylic alcohols. Adv. Synth. Catal. 2019, 361, 1301–1306. 10.1002/adsc.201801389. [DOI] [Google Scholar]
- Cao T.; Gao C.; Kirillov A. M.; Fang R.; Yang L. DFT quest for mechanism and stereoselectivity in B (C6F5) 3-catalyzed cyclopropanation of alkenes with aryldiazoacetates. Mol. Catal. 2021, 516, 111980 10.1016/j.mcat.2021.111980. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2013.
- a Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; c Stephens P. J.; Devlin F.; Chabalowski C.; Frisch M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. A 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- a Fukui K. Formulation of the reaction coordinate. J. Phys. Chem. B 1970, 74, 4161–4163. 10.1021/j100717a029. [DOI] [Google Scholar]; b Fukui K. The path of chemical reactions - the IRC approach. Acc. Chem. Res. 1981, 14, 363. 10.1021/ar00072a001. [DOI] [Google Scholar]
- Legault C. Y.CYLview, 1.0 b; Université de Sherbrooke: Canada. http://www.cylview.org, 2009.
- Manzetti S.; Lu T. The geometry and electronic structure of aristolochic acid: possible implications for a frozen resonance. J. Phys. Org. Chem. 2013, 26, 473–483. 10.1002/poc.3111. [DOI] [Google Scholar]
- Babaahmadi R.; Dasgupta A.; Hyland C. J. T.; Yates B. F.; Melen R. L.; Ariafard A. Understanding the Influence of Donor-Acceptor Diazo Compounds on the Catalyst Efficiency of B (C6F5)3 Towards Carbene Formation. Chem. – Eur. J. 2022, 28, e202104376 10.1002/chem.202104376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.