

Abstract

The pyridine-borane (PyBH3) complex was analyzed by Raman vibrational spectroscopy and density functional theory to elucidate its structural and vibrational properties and to compare these with those for neat pyridine (Py). The borane–nitrogen (BN) bond length, the BN dative bond stretching frequency, and the effects of dative-bonded complex formation on Py are presented. Rather than having a single isolated stretching motion, the complex exhibits multiple BN dative bond stretches that are coupled to Py’s vibrations. These modes exhibit large shifts that are higher in energy relative to neat Py, similar to previous observations of Py/water mixtures. However, significantly higher charge transfer was observed in the dative-bonded complex when compared to the hydrogen-bonded complex with water. A linear relationship between charge transfer and shifts to higher frequencies of pyridine’s vibrational modes agrees well with earlier observations. The present work is of interest to those seeking a stronger relationship between charge-transfer events and concomitant changes in molecular properties.
Introduction
A covalent bond typically involves a sharing of electrons between atoms, i.e., both atoms donate a single electron to be shared. A coordinate covalent or dative bond, however, forms from one atom donating a pair of electrons to a Lewis acid.1−3 Boron’s three valence electrons allow for the formation of three covalent bonds, with an empty p orbital remaining. This empty p orbital serves as a Lewis acid site and readily dative bonds to nitrogenous Lewis bases.2,4 Dative bonds have been investigated both experimentally5−9 and computationally5−11 for the characterization of their vibrational frequencies, bond energies, and bond lengths. Vibrational frequencies vary because of substituent atoms, but the borane–nitrogen (BN) dative bond has a typical bond length of approximately 1.6 Å8,9 and an interaction energy of −43.91 kcal mol–1 at the estimated CCSD(T) complete basis set limit.11
The vibrational frequencies of the BN dative bond stretching motion and dative-bonded complexes as a whole are of interest because of their potential to offer evidence of the significant charge transfer in dative bond formation. Shifts in vibrational energy have been studied extensively as a probe of intermolecular interactions.12−19 In 2013,19 we offered evidence of a strong correlation among charge transfer, vibrational mode shifts to higher energy, and changes in bond length in charge-donating, nitrogen-containing heterocycles. This work showed a linear relationship between the total electronic charge donated in a molecular complex and a shift to higher frequency in vibrational modes, particularly the ring breathing mode. Here, we extend our investigations of the effects of electron transfer in nitrogen-containing heterocycles to include the effects of dative bonding in the pyridine-borane complex. By characterizing the vibrations in this complex through Raman spectroscopy and comparing the spectrum to hydrogen-bonded complexes showing known charge-transfer events with pyridine,16,17 a greater understanding of charge transfer in the dative bond, the correlation between charge transfer and changes in molecular properties, and the pyridine-borane complex can be obtained.
Since the development of its facile synthesis in 1954,20 the pyridine-borane complex (PyBH3) has been ubiquitous in molecular synthesis in roles such as a reducing agent,21−26 a key component in reductive amination,4,27−33 and a precursor to hydroborating alkenes at room temperature.34−36 To the best of our knowledge, neither the Raman vibrational spectrum nor the BN dative bond stretch of PyBH3 has been characterized. Borane–Lewis acid dative bonds have been studied extensively, as mentioned above, but pyridine (Py) has received little study as a Lewis base in dative bonds.5,7 Matthäus et al. in 20017 studied boron trichloride-pyridine fragments and complexes through their pioneering vibrational fragment mode analysis technique. By describing vibrational modes in a complex as percentages of their fragment mode, they show mode mixing between Py and BCl3 and large perturbations of the vibrational modes relative to their fragments. In 2003, Tao et al.5 studied Py bound to Si(100) through vibrational spectroscopy and density functional theory (DFT) calculations to show that Py molecules interact with Si(100) through Si:N dative bonding. Tao et al. in 20086 published a similar work studying pyrrolidine dative bonds to Si(111)-7 × 7. Their work shows shifts to higher energy for pyrrolidine’s ring breathing mode upon charge transfer and dative bond formation. The work by these groups lays a foundation for the study of the PyBH3 dative-bonded complex in the present work.
Here, in an effort to investigate the vibrations of the PyBH3 complex both in isolation and in comparison to Py and provide a greater correlation between charge transfer and vibrational mode shifts, we present a comprehensive Raman spectroscopic and DFT study. Raman spectra of Py and PyBH3 are acquired experimentally and compared to explore the effects of the addition of BH3 to Py’s vibrational modes. These experimental results are also compared to Raman spectra of Py/water mixtures of various mole fractions, similar to the work of Schlucker et al.,16 to compare the results of our work to a known charge-transfer event in Py. DFT calculations were performed to compare the structures of Py and PyBH3, their simulated Raman spectra, and charge-transfer events through the natural electron configuration (NEC) within natural bond orbital (NBO) analyses. These calculations will show changes in bond length and aid in the characterization of PyBH3’s Raman vibrational spectrum.
Methods
Py (99.8%) and PyBH3 (8 M BH3Py) were commercially acquired from Sigma-Aldrich and used without further purification. All experimental data was acquired at ambient temperature using a Horiba LabRAM HR Evolution Raman Spectroscopy system (Horiba Scientific, Kyoto, Japan) equipped with a 600 grooves/mm grating and a charge-coupled device (CCD) detector. A 532 nm Nd:YAG laser (Oxxius, Lannion, France) was used to excite the solutions of Py, PyBH3, and Py/water held in 1 cm cuvettes.
Calculations were carried out using the Gaussian 16 software suite.37 Geometry optimizations (using the Opt = Very Tight option) and Raman activities were carried out using the Minnesota 06 functional M06-2X38 with a correlation-consistent basis set augmented with diffuse functions (aug-cc-pVTZ).39 This method and basis set will be denoted as M06-2X/aVTZ for the remainder of this work. To calculate the harmonic vibrational frequencies, a pruned numerical integration grid composed of 99 radial shells and 590 angular points per shell was employed, and these frequencies were scaled by an empirically determined scaling factor of 0.970 to partially account for anharmonicity and intermolecular interactions in condensed phases. Spectra were simulated by summing Lorentzian profiles constructed from the calculated harmonic frequencies and normalized Raman optical activities.
Results and Discussion
Theoretical Predictions
Geometry optimizations and Raman vibrational frequency calculations of both Py and PyBH3 were performed to compare and accurately characterize the vibrations of interest in the experimental data. Optimized structures of the PyBH3 complex and Py are shown in Figure 1 with their associated bond lengths expressed in angstroms (Å) in Table 1. Cartesian coordinates for these molecules are available in the Supporting Information. The bond lengths here are reported to three decimal places to allow comparison; the precision is not intended to reflect the accuracy of the computed geometrical parameters. Upon formation of the PyBH3 complex, NC1 and C5N bonds elongate by 0.006 Å (0.45%), the C2C3 and C3C4 bonds elongate by 0.001 Å (0.07%), and the C1C2 and C4C5 bonds shorten by 0.007 Å (0.50%). Steric hindrance could be present in the complex between the three boro-hydrogens and the neighboring pyridyl-hydrogens. This is relieved by the elongation of the NC1 and C5N bonds and the shortening of all CH bonds. The C1H8 and C5H11 bonds closest to the BH3 group undergo shortenings greater in magnitude (0.004 Å, 0.37%) than those further removed (C2H9, C3H7, C4H10, 0.001 Å, 0.09%). We attribute the shortening of the C1C2 and C4C5 bonds to relieved ring strain in the stretched pyridine ring. The BN dative bond length is recorded as 1.615 Å and is in agreement with past works.8,9 The BH14, BH13, and BH15 bonds are slightly longer than what has been recorded experimentally for BH3 (1.190 Å)40 and more closely agree with BH bond lengths for the boron dihydride anion (BH2–) or the boron hydride cation (BH+), both with BH bond lengths of 1.205 Å.40 The antiplanar BH bond, BH13, is slightly longer than its neighboring BH bonds, alluding to a more delocalized, weaker bond here.
Figure 1.
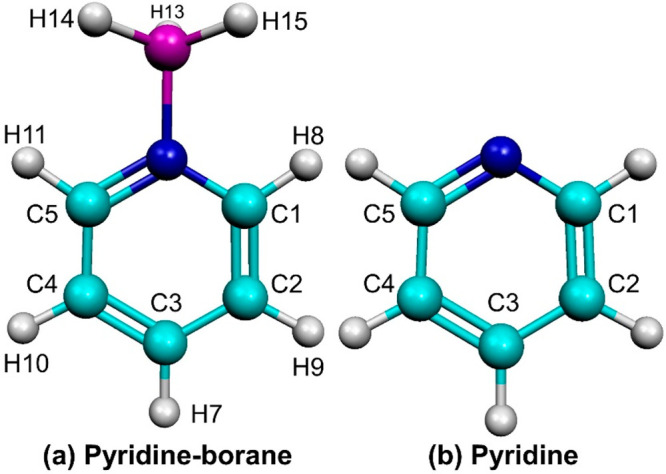
Optimized structures of PyBH3 and Py at the M06-2X/aVTZ level of theory.
Table 1. Bond Lengths (R, Å) Associated with Py and PyBH3 Structures Optimized at the M06-2X/aVTZ Level of Theory.
| molecule | R(N C1)a | R(C1 H8) | R(C1C2) | R(C2H9) | R(C2C3) | R(C3H7) | R(C3C4) | R(C4H10) | R(C4C5) | R(C5H11) | R(C5N) | R(NB) | R(BH14) | R(BH13) | R(BH15) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pyridine | 1.330 | 1.084 | 1.388 | 1.081 | 1.385 | 1.082 | 1.385 | 1.081 | 1.388 | 1.084 | 1.330 | X | X | X | X |
| pyridine-borane | 1.336 | 1.080 | 1.381 | 1.080 | 1.386 | 1.081 | 1.386 | 1.080 | 1.381 | 1.080 | 1.336 | 1.615 | 1.206 | 1.209 | 1.206 |
| %Δ | 0.45 | 0.37 | 0.50 | 0.09 | 0.07 | 0.09 | 0.07 | 0.09 | 0.50 | 0.37 | 0.45 |
To provide a demonstrative quantification of charge transfer in the formation of the dative bond, natural electron configurations (NEC) were calculated for Py and BH3 molecules each in isolation and in the PyBH3 complex. These results are demonstrated in Table 2. To note, we do not present these data as a true value of charge transfer in the complex but rather as a relative quantification at the M06-2X level of theory. Charge transfer has been quantified with a wide range of theoretical methods,41 and a variation in numeric value is expected. However, there should be no discrepancy between the methods in terms of the nature of the charge transferred, be it donation or receipt. The nitrogen atom experiences a large decrease in natural electron population as the complex forms. This is correlated with a decrease in the populations of carbons 1, 3, and 5 as well. Carbons 1 and 5 are closest to the dative bond and undergo a larger magnitude of charge transfer than carbon 3. Interestingly, carbons 2 and 4 are unperturbed within the precision of our calculations. Hydrogens 8 and 11, those proximal to the nitrogen atom, undergo a relatively large depletion of natural population as the dative-bond formation draws charge from the ring structure. Hydrogens 7, 9, and 10 are still seen to lose charge in the PyBH3 complex but to a smaller degree than do the other pyridyl hydrogens. The borohydrogens, hydrogen 13, 14, and 15, also are depleted of electronic population in the dative-bonded complex, with the highest depletion in antiplanar hydrogen 13. The depletion of charge in the Py molecule is coupled with the large increase in the natural population of the boron atom upon complex formation. This charge transfer is consistent with established BN dative-bond formation and should correlate with shifts in the Raman spectra of Py upon complex formation.
Table 2. Charge Transfer (me–) Associated with the Py Atoms after the Formation of the Dative Bond in PyBH3.
| atoma | Δq (me–) | |
|---|---|---|
| C | 1 | –50 |
| C | 2 | 0 |
| C | 3 | –30 |
| C | 4 | 0 |
| C | 5 | –50 |
| N | 6 | –50 |
| H | 7 | –10 |
| H | 8 | –40 |
| H | 9 | –10 |
| H | 10 | –10 |
| H | 11 | –40 |
| B | 12 | 530 |
| H | 13 | –80 |
| H | 14 | –60 |
| H | 15 | –60 |
Simulated Raman spectra of both Py (bottom, black) and PyBH3 (top, green) at the M06-2X/aVTZ level of theory are shown in Figure 2, with tabulated spectral shifts recorded in Table 3. Full simulated spectra are available in the Supporting Information. Predicted spectral shifts are demonstrated with red arrows. Mode assignments for Py are based upon work by Wilmshurst and Bernstein42 and were employed to assign the modes in PyBH3. In the simulated spectrum of PyBH3, the BN dative-bond stretch appears at two separate energies, both coupled to a pyridine vibration. The first appears at 695 cm–1 and is coupled to pyridine’s ν6a vibrational mode. This mode is predicted to occur in neat pyridine at 594 cm–1 and involves a symmetric stretching and contracting of the NC1–C5N and C2C3–C3C4 bonds. The +101 cm–1 shift seen upon formation of the PyBH3 complex is correlated with these changes in bond lengths and charge transfer, perturbing the vibrational frequency. The second BN dative-bond stretch is predicted to occur at 1083 cm–1, coupled to pyridine’s ν18a vibrational mode. Neat Py’s ν18a vibrational mode is predicted to occur at 1070 cm–1. This vibration features a shift to higher energy relative to neat Py, correlated with the stretching motions by the elongated NC1–C5N and C2C3–C3C4 bonds.
Figure 2.
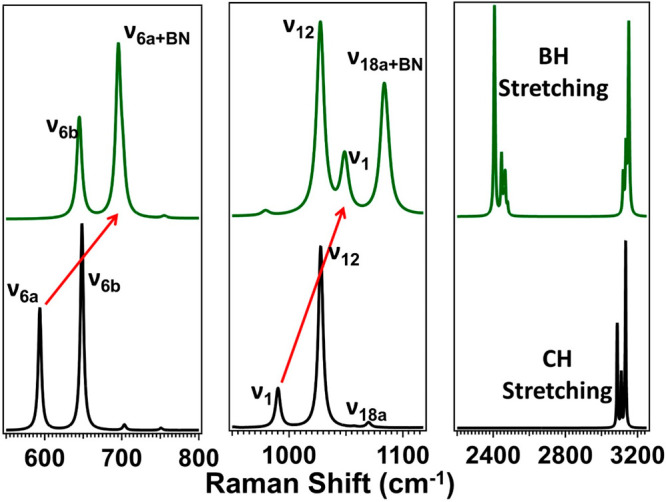
Simulated Raman spectra of Py (black, bottom) and PyBH3 (green, top) at the M06-2X/aVTZ level of theory. Red arrows denote large shifts in the Raman spectra.
Table 3. Spectral Shifts Associated with Py and PyBH3 Simulated Raman Spectra (Figure 2)a.
| mode | Py (cm–1) | PyBH3 (cm–1) | Δν (cm–1) |
|---|---|---|---|
| ν1 | 990 | 1049 | 59 |
| ν6a | 594 | 695 | 101 |
| ν6b | 648 | 645 | –3 |
| ν12 | 1028 | 1028 | 0 |
| ν18a | 1070 | 1083 | 13 |
The computed energies have been scaled by 0.97.
In Py’s simulated Raman spectrum, the ν18a vibrational mode is relatively weak compared to the Raman activities of the adjacent vibrations. In the PyBH3 simulated spectrum, ν18a has undergone a large increase in Raman activity relative to its adjacent modes, indicating an increase in proportion to this vibration.
The ν6b vibration is predicted to be relatively unperturbed, shifting from 648 cm–1 in Py to 645 cm–1 in PyBH3. This small shift to lower frequency is attributed to an overall lack of bond stretching and compression in this vibrational mode. A similar phenomenon is seen with the ν12 mode, predicted to occur at 1028 cm–1 in both Py and PyBH3. The ν12 vibrational mode is sometimes denoted as a triangle mode and is similar in motion to a ring breathing mode. This mode features a symmetric radial contraction and expansion of C1, C3, and C5. Neither vibrational mode mentioned involves any vibration of the nitrogen atom or the BN dative bond, which correlates with their small predicted shift. There is a small peak observed at 703 cm–1 in the simulated spectrum of isolated pyridine. This mode is an out-of-plane symmetric C–H wagging motion and is present at 690 cm–1 in the PyBH3 spectrum but is unresolved because of the breadth of the ν6a peak.
As mentioned above, the ring breathing mode (ν1) has long been a probe of interactions in the azabenzenes. This mode, not unlike the triangle mode, involves a symmetrical radial contraction and expansion of N, C2, and C4. Py’s ν1 mode is predicted to occur at 990 cm–1 and undergo a large shift higher in frequency to 1049 cm–1 in PyBH3. Similar to the vibrations discussed above, it involves a vibration of the nitrogen atom/BN dative bond and is significantly shifted to higher frequency. Theory also predicts a slight shift higher in frequency of the CH stretching modes (approximately 3100 cm–1) in PyBH3 relative to neat Py. This change in vibrational frequency is correlated with a shortening and stiffening of the CH bonds upon complex formation.
A 2013 study19 performed by our group examined shifts in vibrational frequency as a function of total electronic charge transferred by a nitrogen-containing heterocycle in complex with several Lewis acid solvents. These earlier results showed a linear relationship between charge transfer and spectral shift, which was strongest with the ν1 ring breathing mode and had an R2 value of 0.952 for >100 unique microsolvated structures. Interestingly, taking the total computed charge transfer into account (Table 2, 180 me–), the computationally predicted shift in ν1 in nitrogen-containing heterocycles using our previously reported results is about 60 cm–1, in excellent agreement with the computed prediction in Table 3. This result simultaneously strengthens the evidence for the charge-transfer event in the PyBH3 dative-bond formation and the correlation between charge transfer and shifts to higher vibrational frequency in nitrogen heterocyclic complexes.
Experimental Results
Mode Assignments
Characterization of the simulated Raman spectrum of PyBH3 allows for the assignment of vibrational modes in the experimental spectrum. The simulated Raman spectrum of PyBH3 at the M06-2X/aVTZ level of theory is compared to experimental data for PyBH3 in Figure 3. Full spectra are available in the Supporting Information. Table 4 contains the locations of the assigned modes of interest. A low activity peak is present at approximately 990 cm–1 experimentally, corresponding to a peak at 979 cm–1 in the simulated spectrum. Because of its wavenumber location, this peak could be assumed to be ν1; however, an analysis of the vibration reveals it to be a BN rocking motion. The ν1 mode is assigned to an ill-resolved shoulder of the ν12 peak at approximately 1032 cm–1 in the experimental data. Figure S3 compares an experimental Raman spectrum of PyBH3 to a less-resolved simulated spectrum. This simulated spectrum was obtained by increasing the Lorentzian fwhm of the vibrational modes. This comparison better shows the similarity between the simulated and experimental data for the ν1 mode in PyBH3 and offers strong evidence for its location as a shoulder of the ν12 mode. A peak is present at approximately 610 cm–1 in the experimental data that does not appear in the simulated data. Because this feature is present in neat pyridine (Figure 4), we believe that this mode may be a result of some dissociated Py in PyBH3. The BH stretching region (approximately 2400 cm–1) is less resolved, with a larger fwhm for each peak. Although this could stem from rovibrational populations within the sample, a more likely explanation is contributions from boron’s two isotopes. The breadth of this region can be replicated in simulated Raman spectra, not unlike what is demonstrated in Figure S3.
Figure 3.

Simulated Raman spectra of PyBH3 (top, green) at the M06-2X/aVTZ level of theory compared to experimental data for the same complex (bottom, blue).
Table 4. Predicted (M06-2X/aVTZ) and Experimental Locations of the Vibrational Modes of Interest in PyBH3a.
| mode | theory (cm–1) | experiment (cm–1) |
|---|---|---|
| ν1 | 1049 | 1032 |
| ν6a | 695 | 714 |
| ν6b | 645 | 651 |
| ν12 | 1028 | 1024 |
| ν18a | 1083 | 1093 |
The computed energies have been scaled by 0.97.
Figure 4.

Experimental Raman spectra of Py (bottom, black) and PyBH3 (top, blue). Red arrows denote large shifts in the Raman spectra.
Comparison to Neat Pyridine
The assignment of vibrational modes in PyBH3’s Raman spectrum allows for comparison to neat Py’s Raman spectrum in order to track shifts in Py’s peaks as a function of complex formation. Experimental Raman spectra for Py (bottom, black) and PyBH3 (top, blue) are compared in Figure 4. Full experimental spectra are available in the Supporting Information. The results for selected modes of interest are recorded in Table 4. In agreement with theory, both ν1 and ν6a shift higher in energy upon complex formation, shifting higher in energy than ν12 and ν6b, respectively. Vibrations ν6b and ν12 undergo small wavenumber shifts to lower energy, and ν18a undergoes a shift to higher energy and an increase in intensity relative to ν1 and ν12, also in agreement with theory. The CH stretching region shifts slightly higher in energy, as predicted.
Comparison to Pyridine/Water Clusters
Past studies12,19 have connected charge transfer in nitrogen-containing heterocycles to shifts to higher energy in vibrational modes, particularly ν1. When forming the dative bond, both of the pyridyl-nitrogen’s lone pairs are donated to the boron atom’s empty orbital. This charge transfer is large in magnitude and should result in a shift to a higher energy of ν1 that is larger in magnitude than that resulting from a smaller charge transfer. This effect is demonstrated by comparing the results obtained here to spectral shifts observed in Py–water hydrogen-bonded networks, similar to work performed by Schlucker et al.16 The experimental shifts of the above studied modes and the CH stretching region due to hydrogen bonding with increasing mole fractions of water are compared with those in the PyBH3 complex in Figure 5. Shifts for the characterized vibrational modes are recorded in Table 5. Full experimental spectra are available in the Supporting Information.
Figure 5.

Experimental Raman spectra of decreasing mole fractions of Py/water compared to the PyBH3 complex.
Table 5. Spectral Shifts Induced in Py by Hydrogen Bonding and PyBH3 Complex Formation.
| mode | Py | PyBH3 | Δν PyBH3 | X = 0.1 Py/water | Δν X = 0.1 Py/water |
|---|---|---|---|---|---|
| ν1 | 991 | 1032 | 41 | 1001 | 10 |
| ν6a | 603 | 714 | 111 | 615 | 12 |
| ν6b | 652 | 651 | –1 | 652 | 0 |
| ν12 | 1031 | 1024 | –7 | 1034 | 3 |
| ν18a | 1068 | 1093 | 25 | 1069 | 1 |
At the lowest mole fraction ratio of pyridine/water, the majority of the Py molecules are participating with surrounding water molecules through N···H–O hydrogen bonding, resulting in a shift in ν1 by +10 cm–1. When this shift is compared to the experimental PyBH3-induced shift of +41 cm–1, it can be predicted that a much larger amount of charge is transferred during dative-bond formation, in agreement with the results shown in Table 1. The ν6a vibrational mode serves as another probe of charge transfer in the hydrogen-bonded species, shifting by +12 cm–1; ν18a is also minutely affected by the introduction of water, compared to a large change in energy and activity in the PyBH3 complex. The coupling of BN stretching motions to ν1, ν6a, and ν18b in the PyBH3 complex is further linked to the large shifts in the Raman spectrum of Py. In the Py-water complexes, ν12 and ν6b are relatively unaffected, indicating a comparable degree of perturbation of energy to the PyBH3 complex. The CH stretching region as a whole also consistently shifts higher in energy upon charge transfer, with the highest shift occurring for the PyBH3 complex. The results here offer strong evidence of the significant amount of charge transferred during PyBH3 complex formation relative to that for hydrogen-bonded complexes. They also strengthen the conclusions stated previously correlating blue shifts with the charge transfer of vibrational modes.19
Conclusions
The Raman vibrational spectrum and charge-transfer properties of the pyridine-borane (PyBH3) complex are demonstrated in relation to neat pyridine by Raman vibrational spectroscopy and density functional theory (M06-2X/aug-cc-pVTZ method and basis set). This complex is found to have a length of 1.615 Å and two stretching frequencies coupled to pyridine’s vibrational modes at 714 and 1093 cm–1. A significant amount of electronic charge is transferred from the pyridine (Py) molecule upon dative-bonded complex formation. This charge transfer is correlated with a shift in the ring breathing mode of Py to higher vibrational frequency. This finding is in strong agreement with our past study which shows a linear relationship between charge transfer and spectral shifts to higher energy. Upon complex formation, there are also changes in bond length to relieve both steric hindrance and ring strain. Experimental shifts in vibrational modes for the PyBH3 complex were compared to shifts acquired for Py-water hydrogen-bonded complexes, which have been previously correlated to charge transfer. Larger vibrational frequency shifts were seen in the dative-bonded complex than in hydrogen-bonded complexes, indicating a higher degree of transferred electronic charge. The work employs DFT to demonstrate charge-transfer events in dative bond formation and is of interest to those attempting to characterize the physical properties of molecular complexes.
Glossary
Abbreviations
- aVTZ
aug-cc-pVTZ
- BN
borane–nitrogen
- DFT
density functional theory
- NBO
natural bond orbital
- NEC
natural electron configuration
- Py
pyridine
- PyBH3
pyridine-borane complex
- aVTZ
aug-cc-pVTZ;
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00636.
Full Raman spectra of all figures and Cartesian coordinates of optimized molecular structures (PDF)
Author Present Address
† Department of Chemistry, University of Georgia, Athens, Georgia 30602, United States
This article has been supported by the National Science Foundation (OIA-1757220 and CHE-1532079) and the Sally McDonnell Barksdale Honors College at the University of Mississippi. The computations in this article were performed using resources at the Mississippi Center for Supercomputing Resources (MCSR).
The authors declare no competing financial interest.
Supplementary Material
References
- Pupim C. F.; Catão A. J. L.; López-Castillo A. Boron–nitrogen dative bond. J. Mol. Model. 2018, 24 (10), 283. 10.1007/s00894-018-3810-9. [DOI] [PubMed] [Google Scholar]
- Frenking G. Peculiar boron startles again. Nature 2015, 522 (7556), 297–298. 10.1038/522297a. [DOI] [PubMed] [Google Scholar]
- Nandi A.; Kozuch S. History and Future of Dative Bonds. Chemistry – A European Journal 2020, 26 (4), 759–772. 10.1002/chem.201903736. [DOI] [PubMed] [Google Scholar]
- Molander G. A.; Cooper D. J. Functionalization of organotrifluoroborates: Reductive amination. J. Org. Chem. 2008, 73 (10), 3885–3891. 10.1021/jo800383e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao F.; Qiao M. H.; Wang Z. H.; Xu G. Q. Dative and Di−σ Binding States of Pyridine on Si(100) and Their Thermal Stability. J. Phys. Chem. B 2003, 107 (26), 6384–6390. 10.1021/jp030259e. [DOI] [Google Scholar]
- Tao F.; Cai Y.; Ning Y.; Xu G.-Q.; Bernasek S. L. Transfer of Electron Density and Formation of Dative Bonds in Chemisorption of Pyrrolidine on Si(111)-7 × 7. J. Phys. Chem. C 2008, 112 (39), 15474–15482. 10.1021/jp8056866. [DOI] [Google Scholar]
- Matthäus C.; Wheeler R. A. Fragment mode analysis and its application to the vibrational normal modes of boron trichloride–ammonia and boron trichloride–pyridine complexes. Spectrochim. Acta, Part A 2001, 57 (3), 521–534. 10.1016/S1386-1425(00)00400-5. [DOI] [PubMed] [Google Scholar]
- Dreux K. M.; McNamara L. E.; Kelly J. T.; Wright A. M.; Hammer N. I.; Tschumper G. S. Probing Dative and Dihydrogen Bonding in Ammonia Borane with Electronic Structure Computations and Raman under Nitrogen Spectroscopy. J. Phys. Chem. A 2017, 121 (31), 5884–5893. 10.1021/acs.jpca.7b03509. [DOI] [PubMed] [Google Scholar]
- Reinemann D. N.; Wright A. M.; Wolfe J. D.; Tschumper G. S.; Hammer N. I. Vibrational Spectroscopy of N-Methyliminodiacetic Acid (MIDA)-Protected Boronate Ester: Examination of the B–N Dative Bond. J. Phys. Chem. A 2011, 115 (24), 6426–6431. 10.1021/jp112016j. [DOI] [PubMed] [Google Scholar]
- De Silva C. C.; Holme T. A. Computational studies of dative bond containing heterocyclic ring structures. Comput. Theor. Chem. 2013, 1019, 78–84. 10.1016/j.comptc.2013.06.022. [DOI] [Google Scholar]
- Karthikeyan S.; Sedlak R.; Hobza P. on the Nature of Stabilization in Weak, Medium, and Strong Charge-Transfer Complexes: CCSD(T)/CBS and SAPT Calculations. J. Phys. Chem. A 2011, 115 (34), 9422–9428. 10.1021/jp1112476. [DOI] [PubMed] [Google Scholar]
- Howard A. A.; Tschumper G. S.; Hammer N. I. Effects of hydrogen bonding on vibrational normal modes of pyrimidine. J. Phys. Chem. A 2010, 114 (25), 6803–6810. 10.1021/jp101267w. [DOI] [PubMed] [Google Scholar]
- Howard J. C.; Hammer N. I.; Tschumper G. S. Structures, energetics and vibrational frequency shifts of hydrated pyrimidine. ChemPhysChem 2011, 12 (17), 3262–3273. 10.1002/cphc.201100457. [DOI] [PubMed] [Google Scholar]
- Cabaco M. I.; Besnard M.; Yarwood J. Raman spectroscopic studies of vibrational relaxation and chemical exchange broadening in hydrogen bonded systems. I. Investigation of relaxation processes in binary mixtures of pyridine and ethanol. Mol. Phys. 1992, 75 (1), 139–155. 10.1080/00268979200100111. [DOI] [Google Scholar]
- Schlücker S.; Koster J.; Singh R. K.; Asthana B. P. Hydrogen-bonding between pyrimidine and water: A vibrational spectroscopic analysis. J. Phys. Chem. A 2007, 111 (24), 5185–5191. 10.1021/jp0702292. [DOI] [PubMed] [Google Scholar]
- Schlücker S.; Singh R. K.; Asthana B. P.; Popp J.; Kiefer W. Hydrogen-bonded pyridine-water complexes studied by density functional theory and raman spectroscopy. J. Phys. Chem. A 2001, 105 (43), 9983–9989. 10.1021/jp0122272. [DOI] [Google Scholar]
- Takahashi H.; Mamola K.; Plyler E. K. Effects of hydrogen bond formation on vibrations of pyridine, pyrazine, pyrimidine, and pyridazine. J. Mol. Spectrosc. 1966, 21 (1–4), 217–230. 10.1016/0022-2852(66)90139-1. [DOI] [Google Scholar]
- Zoidis E.; Yarwood J.; Danten Y.; Besnard M. Spectroscopic studies of vibrational relaxation and chemical exchange broadening in hydrogen-bonded systems. III. Equilibrium processes in the pyridine/water system. Mol. Phys. 1995, 85 (2), 373–383. 10.1080/00268979500101161. [DOI] [Google Scholar]
- Wright A. M.; Howard A. A.; Howard J. C.; Tschumper G. S.; Hammer N. I. Charge Transfer and Blue Shifting of Vibrational Frequencies in a Hydrogen Bond Acceptor. J. Phys. Chem. A 2013, 117 (26), 5435–5446. 10.1021/jp401642b. [DOI] [PubMed] [Google Scholar]
- Taylor M. D.; Grant L. R.; Sands C. A. A Convenient Preparation of Pyridine-Borane. J. Am. Chem. Soc. 1955, 77 (6), 1506–1507. 10.1021/ja01611a031. [DOI] [Google Scholar]
- Ambrogelly A.; Cutler C.; Paporello B. Screening of reducing agents for the pegylation of recombinant human IL-10. Protein Journal 2013, 32 (5), 337–342. 10.1007/s10930-013-9491-4. [DOI] [PubMed] [Google Scholar]
- Gurram S.; Srivastava G.; Badve V.; Nandre V.; Gundu S.; Doshi P. Pyridine Borane as Alternative Reducing Agent to Sodium Cyanoborohydride for the PEGylation of L-asparaginase. Appl. Biochem. Biotechnol. 2022, 194, 827. 10.1007/s12010-021-03657-y. [DOI] [PubMed] [Google Scholar]
- Kawase M.; Kikugawa Y. Chemistry of amine-boranes. Part 5. Reduction of oximes, O-acyl-oximes, and O-alkyl-oximes with pyridine-borane in acid. J. Chem. Soc., Perkin Trans. 1 1979, 643–645. 10.1039/P19790000643. [DOI] [Google Scholar]
- Kikugawa Y.; Saito K.; Yamada S. I. Reduction of heterocycles with pyridine: Borane in acetic acid. Synthesis (Germany) 1978, 1978 (6), 447–448. 10.1055/s-1978-24776. [DOI] [Google Scholar]
- Matsuoka M.; Hayashi T. Chemical Nickel plating using pyridine borane as a reducing agent. Plat. Surf. Finish. 1981, 68 (7), 66–69. [Google Scholar]
- Wong W. S. D.; Osuga D. T.; Feeney R. E. Pyridine borane as a reducing agent for proteins. Anal. Biochem. 1984, 139 (1), 58–67. 10.1016/0003-2697(84)90388-9. [DOI] [PubMed] [Google Scholar]
- Abdel-Magid A. F.; Carson K. G.; Harris B. D.; Maryanoff C. A.; Shah R. D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 1996, 61 (11), 3849–3862. 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- Bomann M. D.; Guch I. C.; DiMare M. A Mild, Pyridine-Borane-Based Reductive Animation Protocol. J. Org. Chem. 1995, 60 (18), 5995–5996. 10.1021/jo00123a049. [DOI] [Google Scholar]
- Khanna I. K.; Weier R. M.; Lentz K. T.; Swenton L.; Lankin D. C. Facile, Regioselective Syntheses of N-Alkylated 2, 3-Diaminopyridines and Imidazo[4, 5-b]pyridines. J. Org. Chem. 1995, 60 (4), 960–965. 10.1021/jo00109a029. [DOI] [Google Scholar]
- Krusemark C. J.; Ferguson J. T.; Wenger C. D.; Kelleher N. L.; Belshaw P. J. Global amine and acid functional group modification of proteins. Anal. Chem. 2008, 80 (3), 713–720. 10.1021/ac7019317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moormann A. E. Reductive amination of piperidines with aldehydes using borane-pyridine. Synth. Commun. 1993, 23 (6), 789–795. 10.1080/00397919308009840. [DOI] [Google Scholar]
- Pelter A.; Rosser R. M.; Mills S. Reductive aminations of ketones and aldehydes using borane-pyridine. J. Chem. Soc., Perkin Trans. 1 1984, 717–720. 10.1039/P19840000717. [DOI] [Google Scholar]
- Stults N. L.; Asta L. M.; Lee Y. C. Immobilization of proteins on oxidized crosslinked sepharose preparations by reductive amination. Anal. Biochem. 1989, 180 (1), 114–119. 10.1016/0003-2697(89)90097-3. [DOI] [PubMed] [Google Scholar]
- Brown H. C.; Vara Prasad J. V. N.; Zee S. H. Hydroboration. 75. Directive Effects in the Hydroboration of Vinyl and Propenyl Heterocycles with Representative Hydroborating Agents. J. Org. Chem. 1986, 51 (4), 439–445. 10.1021/jo00354a004. [DOI] [Google Scholar]
- Clay J. M.; Karatjas A. G.; Vedejs E. Hydroboration with pyridine borane at room temperature. J. Am. Chem. Soc. 2008, 130 (32), 10828. 10.1021/ja8046602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay J. M.; Vedejs E. Hydroboration with pyridine borane at room temperature. J. Am. Chem. Soc. 2005, 127 (16), 5766–5767. 10.1021/ja043743j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J., et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2016
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120 (1), 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90 (2), 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Kuchitsu K.Inorganic molecules. In Structure of Free Polyatomic Molecules: Basic Data; Kuchitsu K., Ed.; Springer: Berlin, 1998; pp 25–78. [Google Scholar]
- Weinhold F.; Landis C. R.; Glendening E. D. What is NBO analysis and how is it useful?. Int. Rev. Phys. Chem. 2016, 35 (3), 399–440. 10.1080/0144235x.2016.1192262. [DOI] [Google Scholar]
- Wilmshurst J. K.; Bernstein H. J. the Vibrational Spectra of Pyridine, Pyridine-4-d, Pyridine-2,6-d2, and Pyridine-3,5-d2. Can. J. Chem. 1957, 35 (10), 1183–1194. 10.1139/v57-159. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.