

Abstract

Leishmaniasis is a tropical disease caused by Leishmania parasites, which are transmitted through the bites of infected sandflies. We focused on the emergence of leishmaniasis in Thailand caused by a species (Leishmania orientalis). Treatment by chemotherapy is not effective against L. orientalis. Hence, we intended to solve this issue using a proteomics approach to investigate protein profiles and in silico analysis for the identification of antigenic proteins from L. orientalis, Leishmania martiniquensis, and Leishmania donovani. Using principal component analysis (PCA), protein profile comparisons indicated that different species of Leishmania are different at the protein level. Proteomics analysis identified 6099 proteins. Among these proteins, 1065 proteins were used for further analysis. There were 16 proteins that were promising candidates for therapeutic aspects as they were abundantly expressed and common to all species. In silico analysis of protein’s antigenicity revealed that eight proteins had the potential for the development of antigenic molecules. Protein profile information and these antigenic proteins may play key roles in the pathogeny of leishmaniasis and can be used as novel therapeutic targets against leishmaniasis in the future.
Introduction
Leishmaniasis is a tropical disease caused by Leishmania spp., an important zoonotic pathogen. Leishmaniasis infections have increased in over 100 countries including Thailand.1 The symptoms associated with the Leishmania parasite are dependent on Leishmania species, as well as parasite interactions with the host immune system.1Leishmania is a digenetic parasite that requires both invertebrate and vertebrate hosts to complete its life cycle. For the invertebrate hosts, sandflies are the main hosts that act as a vector to transmit the Leishmania to vertebrate hosts.2 In sandflies, Leishmania parasite would undergo many developmental steps in the hindgut and midgut until the infectious stage called metacyclic promastigote, which inhabits the salivary glands, is reached.3 Once vertebrate hosts, such as humans, are bitten by the sandflies, Leishmania parasites are transmitted to humans via the phagocytosis of neutrophils and macrophages.4 The symptomatic cases of Leishmania-infected humans show various kinds of clinical manifestations. Symptoms could range from cutaneous, mucocutaneous, to visceral, depending on Leishmania species. In the visceral form, this manifestation is life-threatening as the parasites could go through the bloodstream and target many internal organs such as the spleen, liver, and bone marrow.5 Since different Leishmania species seem to exhibit different mechanisms in the human cells, exploring the proteome of each species is required to reach a complete understanding of species-dependent mechanisms.
In visceral leishmaniasis treatment, most chemotherapeutic drugs have adverse side effects. Amphotericin B is the standard of care. Most of the patients obtaining this drug regimen experience adverse effects such as nephrotoxicity, hypokalemia, myocarditis, and even death.6 Amphotericin targets membrane-bound ergosterol and affects the formation of small pores, altering the osmotic integrity of the cell membranes.7,8 Furthermore, Miltefosine is effective against Leishmania parasites and can be used to treat visceral leishmaniasis and cutaneous leishmaniasis.9 However, this drug also causes gastrointestinal side effects in breast cancer patients.10 The mechanism of action of Miltefosine was through the inhibition of ATP-binding cassette (ABC) transporter P-glycoprotein.11 For our alternative therapeutic approach, we aim to discover new proteins or pathways that are different from known drugs to reduce the adverse effects. We believe targets such as kinases that are present in all Leishmania species may provide better therapeutic outcomes.
Proteomics approaches are used as a tool for addressing the functional aspects of proteins and their roles in living cells, specifically particular species of Leishmania, to fill the gaps in discovering promising drug targets. This tool has provided a high-throughput analysis of proteins on a large scale and has contributed to unraveling key protein-protein interactions, signaling cascades, characterizing the pathogens’ infection mechanisms.12 Among pathogenic species of Leishmania, the proteomics analysis of Leishmania mexicana was previously reported with a total number of 1764 proteins using a gel-free-based proteomics approach.13 Moreover, the protein information of Leishmania donovani was also addressed by proteogenomic analysis with a total number of 3999 proteins identified.14 Despite these data, the proteome of another pathogenic species, Leishmania orientalis, is still unavailable. Proteomics is increasingly being used for studying the mechanism of species-specific proteins, especially, those proteins involved in replication and transmission processes. For drug discovery, it is critical to understand the molecular systems of these pathogens to develop more effective therapeutic strategies. More than 20 species of pathogenic Leishmania are found worldwide.15 Those species are relevant to clinical forms of the disease and therapeutic drugs.16 However, the medications for leishmaniasis are limited in choices. Moreover, the mechanism of Leishmania replication and transmission is still lacking.
The prevention of leishmaniasis is an important priority. Vaccination is widely used as one of the most reliable preventive approaches. Vaccination relies on the protein antigen, which interacts specifically with a group of certain cell mediators such as T-cell or B-cell receptors. The prediction of protein antigenicity can be done using proteomics data.17 Identification of common Leishmania antigen proteins is important for practical purposes ranging from diagnostics to the development of vaccines for the prevention of leishmaniasis. Moreover, we need to establish the potential drug targets that interrupt the replication and transmission of the pathogen to promote drug discovery. Therefore, the objective of this work was to provide a public proteome database that would be useful for the establishment of novel protein antigenicity for therapeutic purposes. This information would introduce alternative protein targets for leishmaniasis, allowing the development of therapeutic strategies for the disease.
Results
Protein Profiles of Leishmania Species
Our study presented the first proteomic characterization of the three Leishmania species. Proteomics analysis allowed us to assess the extent of the changes in protein abundances and the degree of variance in the proteome profiles in Leishmania from different ecological pandemics. To obtain an overview of the proteomic variability between the three strains, a principal component analysis (PCA) was undertaken (Figure 1).
Figure 1.

Two-dimensional principal component analysis (2D-PCA) of the protein abundance values of L. donovani, Leishmania martiniquensis, and L. orientalis. (A) PCA loadings of the first and second principal components (PC1 vs PC2), (B) PCA loadings of the first and third principal components (PC1 vs PC3). The explained variances are shown in brackets. The different groups discriminated by each component (PC1, PC2, and PC3) are highlighted using colored ellipses and dashed lines.
The use of 2D-PCA mainly allowed the effective discrimination of proteins extracted from different Leishmania species. The first principal component (PC1) revealed the highest variance of the data, followed by the second (PC2) and third principal components (PC3), with total variances of 60.7, 33.4, and 2.9%, respectively. PC1 and PC2 analyses revealed that the group of L. orientalis clustered in the bottom-left quadrant, while L. donovani and L. martiniquensis clustered in the bottom-right and top-left quadrant, respectively.
Biomarkers and Differentially Expressed Protein Analysis
Proteomics data were obtained using the Orbitrap mass spectrometry and resulted in 6099 proteins for all three species cultivated under the same conditions with four technical replicates for each species (12 samples in total). We performed postdata processing to obtain the high-certainty protein lists, which provided unbiased results for the downstream analysis. Mass deviation, digestion specificity, and false discovery rate (FDR) are the crucial factors that controlled the level of confidence in the data. The mass deviation analysis, which revealed that 97.76% of identified peptide groups had ≤2 ppm (−2 ≤ Δm ≤ 2), suggests that the major peptides achieve a good mass precision (Figure 2A). The narrow peptide mass deviation ensures the reduction in the number of false-positive peptides. Although a few miscleavage sites of tryptic peptides are a common phenomenon in the proteomics analysis, we observed that 91.57% of all identified peptides had no miscleavage site and 7.66% had a miscleavage site. We used FDR < 0.01 (1%), which defined the high confidence of identified proteins in this analysis. The qualified data were subjected to the Venn diagram and differential expression analysis.
Figure 2.

Postdata processing of the LC–MS/MS data set. (A) Delta mass is the deviation of the measured mass from the theoretical mass of the peptide shown in ppm. (B) Missed cleavage site distribution of identified peptides.
The high-confidence protein data revealed 1065 proteins with an estimated FDR close to 0 across all 12 LC runs (Table S1). We determined the unique and common proteins of all species as depicted in the Venn diagram (Figure 3 and Table S2).
Figure 3.
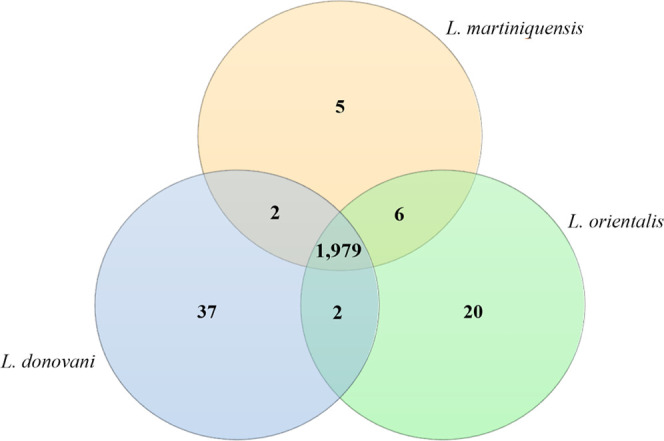
Venn diagram depicting the number of unique and common proteins identified in L. donovani, L. martiniquensis, and L. orientalis. The protein lists used to create this figure are provided in Table S2.
The Venn diagram in Figure 3 shows commonly and uniquely identified proteins in all three groups. This diagram reveals 1979 proteins commonly found among all species. The common proteins between L. martiniquensis and L. orientalis (six proteins) were higher than those between L. martiniquensis and L. donovani (two proteins). To classify the major function of the proteins exclusively detected in L. orientalis, a proteome data set was subjected to GO analysis according to biological function and molecular function as illustrated in Table 1.
Table 1. Description and Functions of the Proteins Uniquely Identified from the L. orientalis Groupa.
| accession | biological process | molecular function |
|---|---|---|
| A0A5B8Z0A1 | glycolytic process | magnesium ion binding; phosphopyruvate hydratase activity |
| A0A1E1ISJ7 | GTPase activity; GTP binding; translation elongation factor activity | |
| A0A1E1J0Y4 | translation | structural constituent of ribosome |
| A0A640KYQ2 | 2 iron, 2 sulfur cluster binding; metal ion binding; ubiquinol-cytochrome-c reductase activity | |
| A0A640KG80 | protein ubiquitination | ATP binding; ubiquitin-like modifier activating enzyme activity |
| A0A640KDZ6 | cytoplasmic translational elongation | structural constituent of ribosome |
| A0A088RW67 | arginyl-tRNA aminoacylation | arginine-tRNA ligase activity; ATP binding |
| A4HD14 | tricarboxylic acid cycle | electron transfer activity; iron–sulfur cluster binding |
| A0A088RR95 | carbohydrate metabolic process | aldose 1-epimerase activity; carbohydrate binding |
| A0A1E1IVQ5 | cellular glucose homeostasis; glycolytic process | ATP binding; glucose binding; hexokinase activity |
| A0A4D5YJB0 | cysteine-type peptidase activity | |
| A0A640KIC6 | serine-type endopeptidase inhibitor activity | |
| A0A640KH80 | translation | RNA binding; structural constituent of ribosome |
This information was obtained from UniProt.org.
All unique protein entries are not fully annotated according to the GO analysis, only 13 out of 20 proteins were classified. The classification of unique proteins according to their roles in biological and molecular systems allows us to analyze the relationships and interactions between them.
Bioinformatic Analysis of Proteomics Data
According to the Venn diagram, proteome profiling was established. The difference in protein expressions among these species was clustered hierarchically as represented in the heatmap in Figure 4.
Figure 4.

Heatmap with hierarchical clustering of differentially expressed proteins. Color key expression: green and red represent the lower and higher differential abundance, respectively.
The columns show the relative protein expression of L. donovani, L. martiniquensis, and L. orientalis. The rows present significantly expressed proteins with a maximum distance of 2.5. The heatmap depicts their relative abundance level by color-coding (lower and higher expression levels: green and red, respectively). Patterns of expression changes across all species were observed. There are 22 clusters of notably differentially expressed patterns. Cluster number 11 has the highest number of differentially expressed proteins. This cluster contains 201 differentially expressed proteins. This evidence suggests distinct species-specific expression profiles.
Common proteins from a proteome data set that corresponds to the disease can be screened for therapeutic protein targeting. Therefore, using a thorough proteomics study of all Leishmania species enables the identification of a specific protein candidate, which can be further developed into a novel protein marker for drug discovery. This proteomics analysis also find kinetoplastid membrane protein-11 (KMP-11) expressed in all species. The search for new effective protein antigens is required for the most comprehensive prevention of leishmaniasis. Hence, highly abundant proteins in the three species should be targeted. Our finding reveals 16 proteins that exhibit relative abundance >200 (normalized abundance). These 16 proteins are promising candidates for both preventive and therapeutic aspects, as they are commonly found in all species with high abundance levels. They may be useful for the development of treatment and vaccination strategies for all Leishmania species. The proteins with high relative abundance (16 proteins) were compared against the KEGG database as illustrated in Table 2.
Table 2. KEGG Pathways Identified for High Relative Abundance Proteins from the Proteomics Data Set.
| accession | KEGG pathway |
|---|---|
| A4I1P9 | tyrosine metabolism |
| E8NHI0 | ribosome |
| A4IAU0 | ribosome |
| E9BID4 | endocytosis; protein processing in endoplasmic reticulum; spliceosome |
| A0A640KJ19 | |
| A0A451EJM4 | |
| Ndpk | |
| E9ART6 | oxidative phosphorylation |
| A0A3S7WPD8 | |
| A4HEA3 | tyrosine metabolism |
| E9ARK0 | ribosome |
| E9AQ19 | ribosome |
| Q4QHR7 | RNA transport |
| Q4Q8G4 | ribosome |
| E9B1E0 | phagosome; oxidative phosphorylation; metabolic pathways |
| A0A088RKF1 |
The analysis revealed that these candidate proteins were associated with eight common cellular pathways. Among 16 proteins, only two (E9BID4 and E9B1E0) were found to be involved in more than one pathway.
Antigenicity of Candidate Protein Evaluation
In silico assessment of antigenicity revealed that eight proteins had the potential for the development of antigenic molecules. Although most of the proteins produced by a given parasite can be considered antigens, eight of them are potential antigens (score >0.5). One of the general characteristics of the potential antigens is the ease of accessibility to the host immune system; therefore, the immune signaling cascade can be triggered. A total of eight proteins were potentially antigenic and one of them was novel (Table 3).
Table 3. In Silico Evaluation of theIdentified Protein Antigenicity.
| accession | VaxiJen value | AntiGENpro value |
|---|---|---|
| A4I1P9 | 0.494 | 0.592 |
| E8NHI0a | 0.641 | 0.616 |
| A4IAU0a | 0.636 | 0.580 |
| E9BID4 | 0.488 | 0.923 |
| A0A640KJ19a | 0.516 | 0.571 |
| A0A451EJM4a | 0.506 | 0.774 |
| Ndpka | 0.658 | 0.718 |
| E9ART6 | 0.487 | 0.166 |
| A0A3S7WPD8a | 0.595 | 0.867 |
| A4HEA3a | 0.516 | 0.635 |
| E9ARK0 | 0.467 | 0.727 |
| E9AQ19 | 0.477 | 0.385 |
| Q4QHR7 | 0.444 | 0.784 |
| Q4Q8G4a | 0.613 | 0.872 |
| E9B1E0 | 0.598 | 0.116 |
| A0A088RKFa | 0.596 | 0.383 |
Highly antigenic proteins are shown in bold text (with scores >0.5 in both analyses).
Using ANTIGENpro and VaxiJen at >0.5 indicated that E8NHI0, A4IAU0, A0A640KJ19, A0A451EJM4, ndpk, A0A3S7WPD8, A4HEA3, and Q4Q8G4 were demonstrated to be antigenic. In addition, we also found that E8NHI0, A4IAU0, A0A640KJ19, and A0A451EJM4 were grouped in cluster 11, which is the largest cluster. Interestingly, A0A640KJ19 has not been reported elsewhere. These results indicate that the prediction model from both platforms revealed relevant characteristics of the most antigenic proteins.
Discussion
PCA suggests that proteome profile comparisons from different species of Leishmania are substantially different at the protein level. Although several polymerase chain reaction-based approaches have been employed for biotyping Leishmania species, these approaches are often limited by low sensitivity results. Modern mass spectrometry with data analysis has emerged to allow reliable, fast, and cost-effective identification of pathogenic organisms.18
In the peptide identification aspects, the characteristic of our tryptic peptides revealed high specificity and similarity to the known properties of the tryptic digestion approach.19 FDR in proteomics results is important to assess and maintain the quality of protein identification. The general approach to measure the FDR in the analysis is a target-decoy search that assigns a score to each peptide-spectrum match (PSM) based on the frequency of false discovery for peptide identification.20
The GO-based biological process showed that they were primarily involved in the glycolytic process and translation. In addition, GO-based molecular functions showed that they are involved in ATP binding. The analysis revealed that most of the proteins are involved in multimolecular functions. The biological functions of A0A1E1ISJ7, A0A640KYQ2, A0A4D5YJB0, and A0A640KIC6 were unavailable. ATP binding is a crucial molecular function in living organisms. According to Table 2, there were three proteins (A0A640KG80, A0A088RW67, A0A1E1IVQ5) that interacted selectively with ATP as universally important co-enzymes and enzyme regulators. Biologically speaking, several proteins are associated with the ATP binding for ATP transport of various molecules including therapeutic drugs across cell membranes.21 This finding is the first report of unique proteins that were only found in L. orientalis.
The proteomics analysis found kinetoplastid membrane protein-11 (KMP-11) expressed in all species. This protein was previously chosen as a potential vaccine therapy.14 However, we found that this protein was present in low abundance (<0.01-fold-abundance) in L. orientalis. The feasibility of a Leishmania vaccine using proteins such as 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase was identified as a potential proteins antigen in asymptomatic dog sera,22 many ribosomal proteins of Leishmania are immunologically activating molecules in mice such as ribosomal protein S3a.23 Heat shock protein70 of Leishmania could stimulate humoral immune responses in mice and humans.24 Putative V type ATPase subunit F gene was developed for a DNA vaccine against Leishmania tropica.25
KEGG pathway analysis is generally used as reference information for the integration and interpretation of high-throughput proteomics data sets.26 Currently, the KEGG database incorporates data from the complete genome sequences of Leishmania major Friedlin, infantum JPCM5, braziliensis MHOM/BR/75/M2904, mexicana MHOM/GT/2001/U1103, donovani, and panamensis MHOM/PA/94/PSC-1.26,27 The pathway analysis may be appropriated for identifying potentially useful protein markers against L. orientalis. Biological relevance among the protein markers and biological functions with the pathway analysis, which serves as a new basis for drug–target interaction predictions.28,29 These protein-pathway analyses suggest that the data would be informative for the development of new proteins as therapeutic targets. Moreover, other identified proteins also drew our attention because they were also crucial in molecular pathways. For instance, glycerol kinase, phosphoenolpyruvate carboxykinase, and pyruvate phosphate dikinase were proven to be essential proteins for parasitic survival.30 Specifically, we first found the high expression of pyruvate phosphate dikinase, Putative V type ATPase (subunit C), and HpcH-HpaI domain-containing protein in all species; hence, they could also be other feasible therapeutic targets. Because of various factors related to antigen abundance and immunodominance, not all possible antigens are recognized by natural immune responses. Various approaches have been proposed for antigen identification. To confirm the efficiency of these proteins for a therapeutic approach, a total of 16 proteins were analyzed using in silico analysis to test a predictor of protein antigenicity that can be utilized in many reliable preventive strategies.
Little is known about the immunogenicity, biological function, and molecular function of A0A640KJ19, except that it has a molecular weight of ∼30 kDa. The comparison of this protein sequence against other proteins using the BlastP revealed a high similarity in primary structure with A4HEA3 at a 97.4% similarity level (e-value = 5.2e–164). However, these two proteins exhibited different values in AntiGENpro analysis. The AntiGenpro algorithm used two-stage architecture based on multiple representations of the protein sequence and machine learning algorithms to estimate whole protein sequence antigenicity; therefore, different values suggested that the small difference in the sequence of A0A640KJ19 and A4HEA3 may affect the immunogenicity.
Using epitope prediction of A0A640KJ19 protein by SVMTriP,31 we found that the peptide at location 151–170 (VETAACIENLEEIMAVKGID; yellow ribbon structure) had the highest possibility to be an antigenic epitope (Figure 5).
Figure 5.
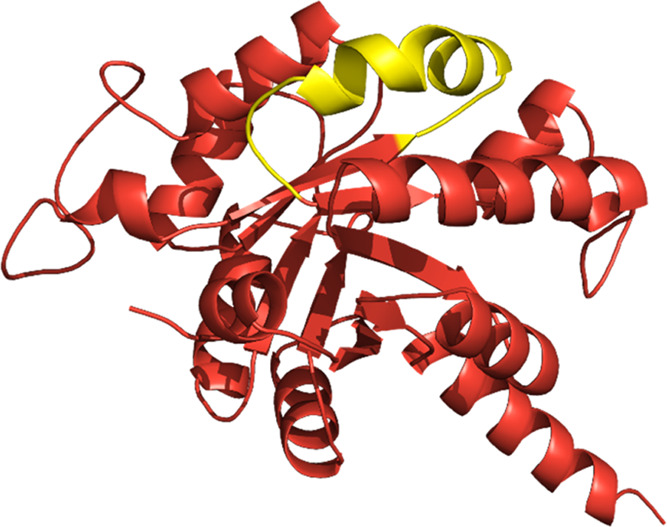
Antigenic epitope prediction of A0A640KJ19 protein. The highest antigenic region was shown in the yellow ribbon structure (location 151–170) which is 19 amino acid residue length.
The predicted antigenic epitope region is found in the prominently exposed regions of the protein surface. The bioinformatics coupled with the available protein database has shown several additional factors such as protein molecular mass, localization, and structural information that influence the determination of the therapeutic targets in addition to identifying the proteins of the pathogen as therapeutic targets. Based on the molecular weight of the immunogenic proteins, the proteins with smaller molecular mass were found very likely to be water-soluble, thus they are easily synthesized and purified.32 Characterization of these proteins, as well as investigation of their biological functions, are essential for establishing their practicability as novel therapeutic targets against leishmaniasis in the future.
In conclusion, the results reported here elucidated the protein profiles of L. orientalis in comparison with reference species including L. martiniquensis and L. donovani, as well as the benefit of integrating proteomics data with antigenic protein analysis for identifying proteins of the pathogen as therapeutic targets. Moreover, our findings suggest that the HpcH-HpaI domain-containing protein might be a novel immunogenic protein as a therapeutic target against leishmaniasis. However, there is no explicit understanding of the exact biological functions of this protein. Investigation of its functions is essential to establish its practicability for drug development in the future.
Materials and Methods
Parasite Culture
L. orientalis (MHOM/TH/2010/TR, zymodeme MON-324), L. martiniquensis (MHOM/MQ/92/MAR1, zymodeme MON-229) and L. donovani (MHOM/ET/67/HU3) promastigotes were cultured in Schneider’s insect medium (Sigma-Aldrich, France) supplemented with 20% heat-inactivated fetal bovine serum (Gibco BRL, France) with pH 6.7 at 26 °C. Promastigotes in the exponential phase of growth were conducted with the same conditions as previously studied.33 Cells were harvested by centrifugation at 3000g for 10 min at room temperature and washed three times in phosphate-buffered saline (PBS) buffer pH 7.4. Approximately 109 parasites were estimated by counting with a hemocytometer prior to extracting proteins.
Protein Extraction and Digestion for Gel-Free Proteomics
To investigate the protein expression profile of different Leishmania species, Leishmania cells were prepared as in previous studies.34,35 Briefly, the cells were lysed by lysis buffer solution (0.5% Triton X-100, 10 mM DTT, 10 mM NaCl in 50 mM HEPES-KOH pH 8.0) supplemented with a protease inhibitor cocktail. Proteins extracted were precipitated using ice-cold acetone and stored at −20 °C for 16 h. After precipitation, the protein pellet was reconstituted in 0.2% RapidGest SF (Waters, U.K.) in 10 mM ammonium bicarbonate. The protein concentrations were determined by the bicinchoninic acid assay kit (Pierce, New York, NY) using bovine serum albumin as the standard. The total protein amount of 20 μg was subjected to trypsin digestion. Reduction of the sulfhydryl bonds was done using 5 mM DTT in 10 mM ammonium bicarbonate at 72 °C for 1 h and alkylation of sulfhydryl at room temperature for 25 min in the dark. The solution was cleaned up by a desalting column (Zebra-spin). The flow-through solution was enzymatically digested by trypsin (Promega, Germany) at a ratio of 1:50 (enzyme/protein) and incubated at 37 °C for 3 h. The digested peptides were reconstituted in 0.1% formic acid and transferred to a TruView LC–MS vial (Waters, U.K.).
LC–MS/MS Setting for Proteomics Analysis
A total of 600 ng peptides were subjected to LC–MS/MS. The spectrum data were collected in a positive mode using an Orbitrap HF hybrid mass spectrometer combined with an EASY-nLC1000 nano-liquid chromatography (LC) system (Thermo Fisher Scientific, San Jose, CA) with a nano-C18 column. LC conditions were as follows: mobile phases A and B were used, with mobile phase A composed of 0.1% formic acid in water and mobile phase B comprising 95% acetonitrile with 0.1% formic acid. The samples were loaded directly onto the analytical column; C18 separation was a linear gradient separated for 100 min from 2 to 45% B from the nano-LC system at a constant flow rate of 300 nL·min–1. The analytical column was regenerated at 90% B for 10 min and re-equilibrated at 5% B for 25 min. The peptides were analyzed by applying the data-dependent TopN15 acquisition method, followed by a higher-energy collisional dissociation (HCD) at collision energy = 28. Full scan (MS) mass spectra were acquired from m/z 400 to 1600 with an AGC target set at 3 × 106 ions and a resolution of 120k. MS/MS scan was initiated when the ACG target reached 1 × 105 ions and a resolution was 15k. The raw mass spectra (.raw file) were processed by Proteome Discoverer 2.4 identified against the Uniprot protein database (organism: Leishmania; 100 302 sequences) retrieved on April 15, 2021.
For protein identification and quantification, the setting parameters were as follows: peptide tolerance, 20 ppm; fragment tolerance, 0.05 Da; minimum fragment ion matches per peptide = 3; digest enzyme = trypsin; cysteine carbamidomethylation as a fixed modification and methionine oxidation as a variable modification. The false discovery rate (FDR) of peptide and protein identification were both set to 0.01 (1%). The normalization of the relative protein abundance ratio was performed by total peptide amount for each of the LC runs (across all runs; n = 12) by the normalization algorithm (total intensity count) of the software. The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifiers PXD026304 and 10.6019/PXD026304.36
Data Analysis and Bioinformatic Analysis
To assemble a differential expressed protein list, multiple consensus workflows were used within the Proteome Discoverer software to compile the peptide-spectrum matches (PSMs) into peptide groups, protein database matches, and nonredundant protein groups using the principle of strict parsimony as defined by the software defaults. To investigate the heterogeneity of variation of the three strains of Leishmania, the principal component analysis (PCA) was used to visualize the differences among intra- and intergroup replicates. Venn diagrams grouping the identified proteins of different species were created by Microsoft word 365 to express the unique and common proteins. Protein expression values were transformed as the relative expression data by adding “1” to all expression values to avoid errors upon log transformation. The heatmap was obtained using MultiExperiment Viewer (MeV) for hierarchical clustering. A gene ontology (GO) enrichment analysis was manually performed using web-based software tools, and a Uniprot database (http://www.uniprot.org/uniprot) retrieved in April 30, 2021. For identification of the protein pathway, we used KEGG pathway analysis.27
Evaluation of the Antigenicity of Candidate Protein
To determine the efficiency of candidate target protein antigens and protein subunits, antigenicity was evaluated using bioinformatics software with an automatic cross-variance transformation into a vector of amino acid sequences by VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen).37 For the identification of pathogen-independent predictors, we used ANTIGENpro analysis with the primary sequence-based prediction algorithm (http://scratch.proteomics.ics.uci.edu/). Proteins with a score of >0.5 were considered as antigenic.
Statistical Analysis
All experiments were carried out with at least three independent replicates (n = 3), the results were reported as mean ± standard deviation (SD) and analyzed with GraphPad Prism software. One-way analysis of variance (one-way ANOVA) was performed using PD for proteomics software. The significance in differences was determined by Duncan’s multiple range test (p < 0.05).
Acknowledgments
The research project was supported by Kasetsart University Research and Development Institute: KURDI (FF(KU)6.64) and Center for Advanced Studies in Nanotechnology for Chemical, Food and Agricultural Industries, KU Institute for Advanced Studies, and Kasetsart University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c05792.
Supplementary proteomics data including Table S1: the protein list of high-confidence data and Table S2: accession number of proteins from the Venn diagram comparing proteins identified from three Leishmania species (PDF)
Author Contributions
◆ S.K. and Y.Y. contributed equally to this work and are considered co-first authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Alvar J.; Velez I. D.; Bern C.; Herrero M.; Desjeux P.; Cano J.; Jannin J.; den Boer M. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 2012, 7, e35671 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killick-Kendrick R. The biology and control of phlebotomine sand flies. Clin. Dermatol. 1999, 17, 279–289. 10.1016/S0738-081X(99)00046-2. [DOI] [PubMed] [Google Scholar]
- Gossage S. M.; Rogers M. E.; Bates P. A. Two separate growth phases during the development of Leishmania in sand flies: implications for understanding the life cycle. Int. J. Parasitol. 2003, 33, 1027–1034. 10.1016/S0020-7519(03)00142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach R.; Kiilu G.; Hendricks L.; Oster C.; Leeuwenburg J. Cutaneous leishmaniasis in Kenya: transmission of Leishmania major to man by the bite of a naturally infected Phlebotomus duboscqi. Trans. R. Soc. Trop. Med. Hyg. 1984, 78, 747–751. 10.1016/0035-9203(84)90006-3. [DOI] [PubMed] [Google Scholar]
- McCall L. I.; Zhang W. W.; Matlashewski G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathog. 2013, 9, e1003053 10.1371/journal.ppat.1003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laniado-Laborín R.; Cabrales-Vargas M. N. Amphotericin B: side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. 10.1016/j.riam.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Mbongo N.; Loiseau Philippe M.; Billion Marie A.; Robert-Gero M. Mechanism of Amphotericin B Resistance inLeishmania donovani Promastigotes. Antimicrob. Agents Chemother. 1998, 42, 352–357. 10.1128/AAC.42.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourshafie M.; Morand S.; Virion A.; Rakotomanga M.; Dupuy C.; Loiseau P. M. Cloning of S-adenosyl-L-methionine:C-24-Delta-sterol-methyltransferase (ERG6) from Leishmania donovani and characterization of mRNAs in wild-type and amphotericin B-Resistant promastigotes. Antimicrob. Agents Chemother. 2004, 48, 2409–2414. 10.1128/AAC.48.7.2409-2414.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlo T. P.; Balasegaram M.; Beijnen J. H.; de Vries P. J. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. 10.1093/jac/dks275. [DOI] [PubMed] [Google Scholar]
- Sindermann H.; Engel J. Development of miltefosine as an oral treatment for leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, S17–S20. 10.1016/j.trstmh.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Menez C.; Buyse M.; Chacun H.; Farinotti R.; Barratt G. Modulation of intestinal barrier properties by miltefosine. Biochem. Pharmacol. 2006, 71, 486–496. 10.1016/j.bcp.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Gavin A. C.; Bosche M.; Krause R.; Grandi P.; Marzioch M.; Bauer A.; Schultz J.; Rick J. M.; Michon A. M.; Cruciat C. M.; Remor M.; Hofert C.; Schelder M.; Brajenovic M.; Ruffner H.; Merino A.; Klein K.; Hudak M.; Dickson D.; Rudi T.; Gnau V.; Bauch A.; Bastuck S.; Huhse B.; Leutwein C.; Heurtier M. A.; Copley R. R.; Edelmann A.; Querfurth E.; Rybin V.; Drewes G.; Raida M.; Bouwmeester T.; Bork P.; Seraphin B.; Kuster B.; Neubauer G.; Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Paape D.; Barrios-Llerena M. E.; Le Bihan T.; Mackay L.; Aebischer T. Gel free analysis of the proteome of intracellular Leishmania mexicana. Mol. Biochem. Parasitol. 2010, 169, 108–114. 10.1016/j.molbiopara.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Nirujogi R. S.; Pawar H.; Renuse S.; Kumar P.; Chavan S.; Sathe G.; Sharma J.; Khobragade S.; Pande J.; Modak B.; Prasad T. S.; Harsha H. C.; Patole M. S.; Pandey A. Moving from unsequenced to sequenced genome: reanalysis of the proteome of Leishmania donovani. J. Proteomics 2014, 97, 48–61. 10.1016/j.jprot.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhoundi M.; Kuhls K.; Cannet A.; Votypka J.; Marty P.; Delaunay P.; Sereno D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349 10.1371/journal.pntd.0004349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouttaki T.; Morales-Yuste M.; Merino-Espinosa G.; Chiheb S.; Fellah H.; Martin-Sanchez J.; Riyad M. Molecular diagnosis of cutaneous leishmaniasis and identification of the causative Leishmania species in Morocco by using three PCR-based assays. Parasit. Vectors 2014, 7, 420 10.1186/1756-3305-7-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Zeller M.; Kayala M. A.; Vigil A.; Randall A.; Felgner P. L.; Baldi P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasch P.; Schneider A.; Blumenscheit C.; Doellinger J. Identification of Microorganisms by Liquid Chromatography-Mass Spectrometry (LC-MS 1) and in Silico Peptide Mass Libraries. Mol. Cell. Proteomics 2020, 19, 2125–2139. 10.1074/mcp.TIR120.002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y. Q.; Gilar M.; Lee P. J.; Bouvier E. S.; Gebler J. C. Enzyme-friendly, mass spectrometry-compatible surfactant for in-solution enzymatic digestion of proteins. Anal. Chem. 2003, 75, 6023–6028. 10.1021/ac0346196. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Xin L.; Shan B.; Chen W.; Xie M.; Yuen D.; Zhang W.; Zhang Z.; Lajoie G. A.; Ma B. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteomics 2012, 11, M111.010587 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C. F. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- Agallou M.; Athanasiou E.; Samiotaki M.; Panayotou G.; Karagouni E. Identification of Immunoreactive Leishmania infantum Protein Antigens to Asymptomatic Dog Sera through Combined Immunoproteomics and Bioinformatics Analysis. PLoS One 2016, 11, e0149894 10.1371/journal.pone.0149894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro-Da-Silva A.; Borges M. C.; Guilvard E.; Ouaissi A. Dual role of the Leishmania major ribosomal protein S3a homologue in regulation of T- and B-cell activation. Infect. Immun. 2001, 69, 6588–6596. 10.1128/IAI.69.11.6588-6596.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati S.; Gholami E.; Hassani N.; Ghaemimanesh F.; Taslimi Y.; Taheri T.; Soong L. Leishmania major heat shock protein 70 (HSP70) is not protective in murine models of cutaneous leishmaniasis and stimulates strong humoral responses in cutaneous and visceral leishmaniasis patients. Vaccine 2007, 25, 4159–4169. 10.1016/j.vaccine.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Orabi A.; Maarouf M.; Alammori M. Evaluation of Immune Response and Protection Induced by V-ATPase Subunit F as DNA Vaccine Against Leishmania tropica (LCED Syrian 01) After Detection and Sequencing. Avicenna J. Med. Biotechnol. 2020, 12, 9–16. [PMC free article] [PubMed] [Google Scholar]
- Arakawa K.; Kono N.; Yamada Y.; Mori H.; Tomita M. KEGG-based pathway visualization tool for complex omics data. In Silico Biol. 2005, 5, 419–423. [PubMed] [Google Scholar]
- Aoki-Kinoshita K. F.; Kanehisa M. Gene annotation and pathway mapping in KEGG. Methods Mol. Biol. 2007, 396, 71–91. 10.1007/978-1-59745-515-2_6. [DOI] [PubMed] [Google Scholar]
- Chen L.; Chu C.; Lu J.; Kong X.; Huang T.; Cai Y. D. Gene Ontology and KEGG Pathway Enrichment Analysis of a Drug Target-Based Classification System. PLoS One 2015, 10, e0126492 10.1371/journal.pone.0126492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B.; Shi C.; Jiang H. X.; Qin S. Y. Identification of novel therapeutic target genes and pathway in pancreatic cancer by integrative analysis. Medicine 2017, 96, e8261 10.1097/MD.0000000000008261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Contreras D.; Hamilton N. Gluconeogenesis in Leishmania mexicana: contribution of glycerol kinase, phosphoenolpyruvate carboxykinase, and pyruvate phosphate dikinase. J. Biol. Chem. 2014, 289, 32989–33000. 10.1074/jbc.M114.569434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B.; Zheng D.; Liang S.; Zhang C. SVMTriP: A Method to Predict B-Cell Linear Antigenic Epitopes. Methods Mol. Biol. 2020, 2131, 299–307. 10.1007/978-1-0716-0389-5_17. [DOI] [PubMed] [Google Scholar]
- Duffield M.; Cooper I.; McAlister E.; Bayliss M.; Ford D.; Oyston P. Predicting conserved essential genes in bacteria: in silico identification of putative drug targets. Mol. BioSyst. 2010, 6, 2482–2489. 10.1039/c0mb00001a. [DOI] [PubMed] [Google Scholar]
- Siripattanapipong S.; Boontanom P.; Leelayoova S.; Mungthin M.; Tan-Ariya P. In vitro growth characteristics and morphological differentiation of Leishmania martiniquensis promastigotes in different culture media. Acta Trop. 2019, 197, 105039 10.1016/j.actatropica.2019.05.030. [DOI] [PubMed] [Google Scholar]
- Krobthong S.; Choowongkomon K.; Suphakun P.; Kuaprasert B.; Samutrtai P.; Yingchutrakul Y. The anti-oxidative effect of Lingzhi protein hydrolysates on lipopolysaccharide-stimulated A549 cells. Food Biosci. 2021, 41, 101093 10.1016/j.fbio.2021.101093. [DOI] [Google Scholar]
- Krobthong S.; Yingchutrakul Y.; Visessanguan W.; Mahatnirunkul T.; Samutrtai P.; Chaichana C.; Papan P.; Choowongkomon K. Study of the Lipolysis Effect of Nanoliposome-Encapsulated Ganoderma lucidum Protein Hydrolysates on Adipocyte Cells Using Proteomics Approach. Foods 2021, 10, 2157 10.3390/foods10092157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Cote R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; O’Kelly G.; Schoenegger A.; Ovelleiro D.; Perez-Riverol Y.; Reisinger F.; Rios D.; Wang R.; Hermjakob H. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2012, 41, D1063–D1069. 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I. A.; Flower D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.