

Abstract
The determination of intracellular drug concentrations can provide a better understanding of the drug function and efficacy. Ideally, this should be performed nondestructively, with no modification of either the drug or the target, and with the capability to detect low amounts of the molecule of interest, in many cases in the μM to nM range (pmol to fmol per million cells). Unfortunately, it is currently challenging to have an experimental technique that provides direct quantitative measurements of intracellular drug concentrations that simultaneously satisfies these requirements. Here, we show that magic-angle spinning dynamic nuclear polarization (MAS DNP) can be used to fulfill these requirements. We apply a quantitative 15N MAS DNP approach in combination with 15N labeling to quantify the intracellular amount of the drug [15N]CHIR-98014, an activator of the Wingless and Int-1 signaling pathway, determining intracellular drug amounts in the range of tens to hundreds of picomoles per million cells. This is, to our knowledge, the first time that MAS DNP has been used to successfully estimate intracellular drug amounts.
Introduction
In order to study the pharmacological activity of a drug, its amount at the intracellular effector site must be determined, which is often difficult to measure.1−3 Intracellular quantification should ideally be performed nondestructively, with no modification of either the drug or the target, and requires the capability to detect low amounts of the molecule of interest. Unfortunately, it is currently difficult to provide direct quantitative measurements of intracellular drug concentrations satisfying all these requirements simultaneously.4
Solution NMR is routinely used for the nondestructive, tag-free quantification of analytes even in complex mixtures.5−7 NMR can be used with success to identify components inside intact cells,8−12 however, in such applications, NMR is hindered by low sensitivity and is biased toward the analysis of molecules in a fast motional regime, which is not always the case in the intracellular environment, for example, in the case of molecules interacting with large proteins.13
Magic-angle spinning dynamic nuclear polarization (MAS DNP) can in principle be used to overcome these two drawbacks, providing both significantly increased sensitivity and the detection and characterization of immobilized molecules.14−16 MAS DNP has been shown to be relevant in pharmaceutical characterization problems,17 and over the last 3 years, MAS DNP of intact cells has attracted significant attention.18−21 MAS DNP enabled us to push the detection limit of NMR down to the ∼100 pmol range for 31P NMR spectra of phosphorothioester probe groups in DNA and microRNA strands.22 Indeed, MAS DNP has already been shown to allow the otherwise challenging detection of isotopically enriched proteins in cells19,23 and, recently, to be able to observe 31P NMR spectra from 16-mer siRNA fragments in cells20 or in lipid nanoparticles.24
However, to our knowledge, MAS DNP has not been used up to now for the detection of drug molecules or for the quantification of analytes in-cell. We recently demonstrated that it is possible to quantify analytes using DNP by adding an internal standard, analogously to approaches used in solution NMR, and using a multiple-contact cross-polarization (CP) acquisition scheme.25 Here, we apply the same approach to quantify the drug [15N]CHIR-98014 (Figure 1) in HEK293 cells. [15N]CHIR-98014 is an inhibitor of the glycogen synthase kinase-3 protein and acts as an activator of the Wingless and Int-1 (Wnt) signaling pathway.26 The selective detection of the drug through the signal of the NO2 group in 15N MAS DNP NMR spectra allows us to determine in-cell drug amounts between 20 and 490 pmol per million cells as a function of the incubation conditions. Ex situ liquid chromatography coupled with mass spectrometry (LC–MS) is used as an orthogonal quantification method.
Figure 1.
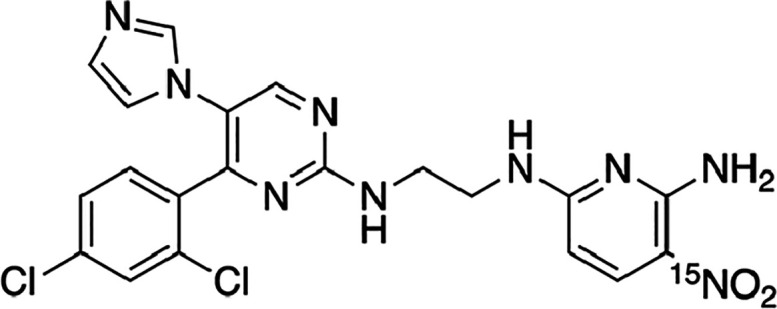
Structure and labeling of [15N]CHIR-98014.
Results and Discussion
In order to detect specific molecules in cells, the target molecule must be distinguished from the background. As a result, the most widespread approach to in-cell NMR is to study proteins that are both isotopically enriched (usually in 15N and 13C) and often present at concentrations that are higher than physiological.27−30 Such samples allow sophisticated multidimensional experiments to be performed to follow interactions,31,32 post-translational modifications,33,34 or complete structure determination.10,35 The in-cell studies of small molecules are by comparison much less widespread.12,36,37 A key limitation is the detection limit of solution-state NMR, which is combined with the need to distinguish the signals of the small molecule from the background at physiologically relevant concentrations, which will usually be much larger. To address these issues, and in order to maximize the limit of detection, here we use MAS DNP in combination with 15N labeling of the NO2 group to detect and quantify CHIR-98014 (Figure 1) in cells following incubation with the drug. The drug is selectively 15N labeled at the nitro group position to assure higher sensitivity and selectivity as it avoids any overlap with the cellular background. The 15N NO2 resonance is expected in a region of the spectrum which should otherwise be largely empty (i.e., there will be no overlap with natural abundance 15N signals from proteins, nucleic acids, or other metabolites present at significant concentrations). Nitro groups are otherwise not expected to be present at significant levels and should not be visible in the 15N NMR spectra at natural isotopic abundance (Figure S1).
Solution NMR and MAS DNP characterization of the pure drug show a single 15N resonance arising from the nitro group at 368 ppm (Figure S2). As can be seen in Figure S3, with a (1H)–15N CP experiment under MAS DNP conditions, a detection limit below 100 pmol can be achieved over 3 days of acquisition time. This represents an improvement of a factor of 4 with respect to standard 1H solution NMR at the same magnetic field, and of a factor of almost 200 compared to 15N solution NMR. Note also that these estimations were obtained for pure solutions. In the complex intracellular environment, the detection limits for solution NMR are expected to be higher due to the slower motional regime experienced by the drug and, in the case of 1H NMR, to the overlap of the signals with the cellular background.
Figure 2 shows the (1H)–15N multiple-contact CP DNP enhanced spectrum acquired from a sample of HEK cells (3 × 106 cells) incubated for 6 h with a 5 μM solution of [15N]CHIR-98014. The spectrum was acquired over ∼36 h at 10.5 kHz MAS and ∼100 K on a 9.4 T (400 MHz) spectrometer. Washing of the cells, as described in the Methods section, assures the removal of any residual incubation buffer. The cells are suspended in a DMSO-d6/D2O/H2O solution containing 14 mM AMUPol as a polarizing agent.38 An exactly known amount of 13C, 15N-labeled histidine (2.22 nmol) is added to the polarizing solution as a quantification standard.
Figure 2.
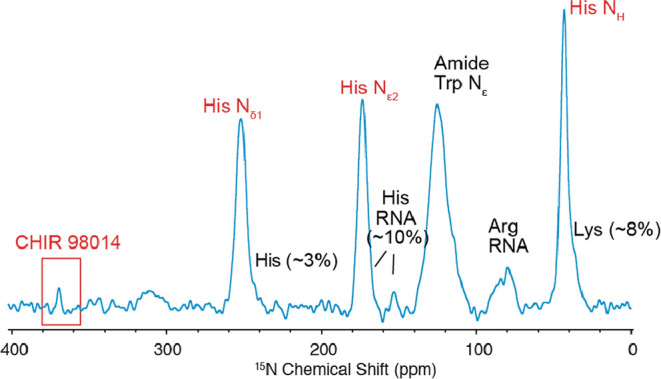
(1H)–15N multiple-contact CP41 (number of CP loops = 10, inter-CP delay = 2.5 s) DNP enhanced spectrum of HEK293 cells incubated with 5 μM [15N]CHIR-98014 for 6 h, then washed and resuspended in DMSO-d6/D2O/H2O 60:30:10v/v, AMUpol 14 mM and 15N, 13C l-histidine·HCl 343 μM. The spectrum was acquired at 10.5 kHz MAS and 100 K on a 9.4 T (400 MHz for 1H) spectrometer. The spectrum was acquired in 5120 scans, using a recycle delay of 0.1 s for a total acquisition time of 36 h. The number in parenthesis represents the total contribution of the cellular background to the signals of histidine.
In the spectrum, resonances arising from the cellular background are clearly observable between 100 and 120 ppm (assignable mostly to protein amide backbone resonances and tryptophan, asparagine, and glutamine side chains) and between 80 and 100 ppm (assignable mostly to arginine side chains and nucleic acids).39 Resonances from the histidine standard are observable at ∼40 ppm (amine group) and ∼170 and ∼250 ppm (aromatics). The presence of two distinct resonances for the aromatics indicates that the pH of the suspended solution is probably close to neutral.40 Histidine signals are partially overlapped with resonances from the cellular background, notably lysine side chains, nucleic acids, and naturally occurring histidine side chains. The contribution of these background resonances to the integral of the standard histidine signals was evaluated from a sample prepared without the addition of any standard, and is reported in brackets in Figure 2.
Finally, a clearly observable signal assignable to [15N]CHIR-98014 is seen at ∼370 ppm. The signal has a chemical shift almost identical to that observed in frozen solutions.
The quantification of the drug is performed from the integrals of the drug and of the standard, histidine. However, here, a complication arises from the fact that the DNP enhancement is expected to be nonuniform within the cells.19,42−44 To resolve this, ideally, one would measure the enhancement from the comparison of the 15N multiple-contact CP spectra acquired in the presence and absence of microwave irradiation. However, by the very nature of these experiments, where the signal from the drug is already barely detectable in the microwave-on spectrum, acquiring the microwave-off spectrum is not feasible. Therefore, we measured enhancements from the (1H)–13C CP spectra (Figure S4), where the enhancement measured for the solvent (ε = 244 ± 6) is almost 1 order of magnitude larger than the lowest enhancement measured for components of the cells (ε = 25 ± 4 for the aromatics, mostly nucleic acids).
Measuring the enhancement for the standard, histidine, is hampered by the background in the 13C experiments; however, a large fraction of the sample (see Methods) is occupied by the polarizing solution and one of the aromatic peaks of histidine in the (1H)–13C CP spectrum, which is expected also to have a lower (but still non-negligible) contribution from the cellular background, displays a 1H DNP enhancement of ∼210. This indicates that only a negligible fraction of the total histidine permeates the cells, and we can therefore assume that the histidine enhancement is the same as that of the solvent.42,43 For the drug, two limiting cases can be considered: either the drug is buried in the cells, and therefore its enhancement is as low as the lowest measured enhancement for the cells (ε = 25 ± 4), or it is adsorbed on the cell surface, which is essentially in the same phase as the polarizing solution and shares the same enhancement as the solvent (ε = 244 ± 6). In the first case, this yields an amount of [15N]CHIR-98014 per million cells, nCHIR, of 196 ± 112 pmol, while in the other case, nCHIR = 20 ± 9 pmol.
In order to validate the result, we performed an ex situ quantification of cell extracts using LC–MS.45−48 Briefly, the sample used for DNP MAS was recovered and resuspended in acetonitrile and subjected to two freeze and thaw cycles. The supernatant was then harvested, diluted in methanol, and analyzed using LC–MS. From an external calibration curve (Figure S5), we determined nCHIR = 81 ± 12 pmol, which falls within the range identified by MAS DNP, validating our in-cell detection and quantification approach.
Using the same strategy, it is possible to quantify different classes of metabolites in the cell whose signals, once quantified, can be used as internal standards in future experiments. As mentioned, the region in the 15N spectrum comprised between 100 and 120 ppm is largely assignable to protein amide resonances, and therefore represents a good candidate for the estimation of the total protein content in the cell. A large fraction of the proteins in the cell are buried in the cytoplasm, and therefore the lowest enhancement (25 ± 4) can be assumed in this case.
We note that, in principle, the carbonyl region of the 13C spectrum would represent a better choice for the direct estimation of the protein enhancement; however, as mentioned above, this is hampered by overlap with the histidine CO signal. Nevertheless, we observed that in samples prepared without the addition of histidine, the enhancements are relatively uniform among the different cellular components, as shown also by Ghosh et al.(44)
Taking into account that the protein signals arise from natural abundance 15N (0.4%), the total amount of protein per million of cells is estimated to be 2.95 ± 0.98 μmol, which is in good agreement with the 3.22 μmol value in the literature for HEK cells.49 (See Table S2 for additional details).
Once a constant intracellular reference is identified (even if the absolute quantification is uncertain), the need to add an external quantification standard is removed. This is exemplified in Figure 3, where the (1H)–15N multiple-contact CP DNP enhanced spectrum for HEK293 cells (3 × 106) incubated for 6 h with a 10 μM solution of CHIR 98014 is shown in orange. The spectrum is overlapped with the same spectrum shown in Figure 2, and both spectra are normalized to the amide region, as we can assume that the total protein content in the cells does not significantly vary. Following the previous approach, and using this time the amide signal as an intermediate standard, it is possible to identify a range for nCHIR comprised between 54 ± 19 and 490 ± 280 pmol, a factor of ∼3 higher than the previous case, which is in substantial agreement with the factor of 2 difference in the starting incubating solutions.
Figure 3.
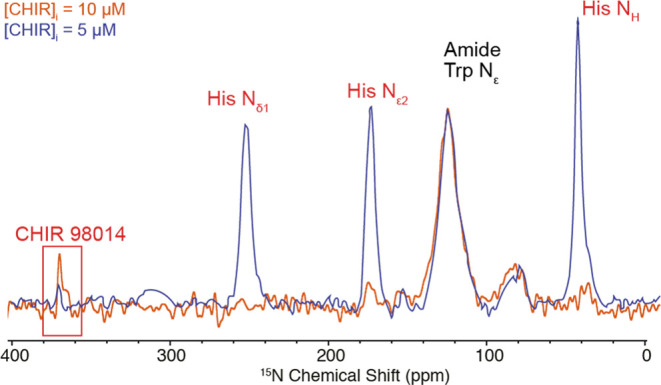
(1H)–15N multiple-contact CP MAS DNP enhanced spectrum of HEK293 cells incubated with [15N]CHIR-98014 for 6 h then washed and resuspended in DMSO-d6/D2O/H2O, AMUpol 14 mM 60:30:10v/v (orange) or DMSO-d6/D2O/H2O 60:30:10v/v, AMUpol 14 mM and 15N, 13C l-histidine·HCl 343 μM (blue). Blue: incubated with 5 μM [15N]CHIR-98014. Orange: incubated with 10 μM [15N]CHIR-98014. The spectra are normalized to the amide region. The spectra were acquired at 10.5 kHz MAS (blue) and 13 kHz MAS (blue) and 100 K on a 9.4 T (400 MHz) spectrometer with 5120 and 1536 scans for the blue and orange spectra, respectively.
Here, we used a protocol for sample preparation that has been widely used in the DNP community and also for biological samples.19,20 The protocol is detrimental to the viability of the cells.44,50 In the present study, however, we only require that the cells remain largely intact, without significant lysis, and we do not require the cells to be viable post-thawing. If cell viability post-thawing is required, this can be achieved for MAS DNP experiments using the recently introduced protocols18,44,50,51 that are otherwise perfectly compatible with the quantification approaches here.
Note that the spectrum of the polarizing solution (Figure S6) recovered ∼5 min post-thawing after the experiments does not show any signal from the drug itself after a comparable acquisition time, which excludes any significant release of the drug into the polarizing solution and shows only a barely detectable contribution from the cell background. From the spectrum in Figure S6, which was acquired over an experimental time comparable to the spectrum in Figure 3 and which displayed a comparable enhancement, we concluded that only a small fraction (∼10%) of the cells were lysed to the point that would significantly release the compound of interest. Moreover, a large (factor ∼10) difference in enhancement is observed between the cellular components and the polarizing solution. This is a strong indication that the cells are largely intact and, again, that there is negligible lysis of the cells during the ∼10 min period between first exposure to dimethyl sulfoxide (DMSO) and freezing (which applies also for the sample analyzed in Figure 2). This is also confirmed by a trypan blue assay performed on a parallel batch of HEK cells (Figure S7): cells exposed to 60% DMSO and frozen directly using the spectrometer lose their viability but overall preserve their shape without being extensively lysed.
We point out that in this system there is a fairly large variability in the drug uptake by the cells from one preparation to another, as is common (data not shown), and that this results in some cases in the drug concentrations, as confirmed by LC–MS, being below the MAS DNP detection limit.
In summary, using the MAS DNP approach in combination with a 15NO2 probe group and both an external and internal standard, we are able to detect and identify the drug amounts in the picomole range in HEK293 cells incubated with the drug. The drug amount is cross-validated by ex situ LC–MS. We show that the drug amount can be straightforwardly monitored as a function of, for example, the concentration in the incubating solution.
We note that the chemical shift observed for the 15NO2 group is very similar to that observed in the frozen solution. The spectrum also exhibits a narrow resonance. This suggests that either the bulk of the drug is being observed in the solution inside the cells, or that interactions with various other components, including the drug receptor, do not lead to significant changes in the 15NO2 chemical shift. It is also possible that some of the drug is found in environments with different 15N chemical shifts, but that these environments are below the detection limit here. To lead to a detectable signal in the experiments shown in Figures 2 and 3, we estimate the environments yielding a given chemical shift would need to have a concentration >1–10 μM in the rotor, depending on the localization within the cells.
We identify two main limitations to the widespread use of the approach discussed here that should be addressed in the future. The first is the detection limit, as discussed above. The second is the impossibility of directly measuring the DNP enhancement for the drug, which leads to some uncertainty in the quantification. This can be considerably reduced if the methodology presented here is complemented with information on the localization of the drug, which could allow a better estimation of its enhancements and, therefore, its quantification. We imagine that, for example, measurements of relayed DNP,42 chemical shift perturbations (even if it is not the case in the present work) or orthogonal bioassays could potentially provide information on the localization of the drug itself.
Conclusions
The determination of intracellular drug concentrations is fundamental in order to study their pharmacokinetics and pharmacodynamics. Although the plasma and blood concentrations of drugs often represent good surrogate measurements, this is not always the case, especially when the drugs are poorly permeable, metabolized in vivo, or actively transported.52
Here, we have introduced a new approach to quantifying drugs in cells using DNP MAS NMR. This does not require any modification of the drug itself, except for isotope labeling, which does not change the physico-chemical properties of the compound (hence the in-cell uptake propensity).
This is unlike fluorescent labeling, for example, the latter, despite being a highly sensitive technique, usually involves chemical tagging of the compound, which can modify its chemical properties and its transport, binding, or localization in the cell. Moreover, here, the quantification can be performed on intact cells, which can be complementary to MS. Indeed, MS is more sensitive than DNP NMR, but it usually requires lysing the cells in order to extract the compound of interest. Therefore, it is not an adequate technique whenever sample integrity is an issue or when spatial information is required. More importantly, here, it requires the ability to quantitatively fully extract the compound of interest, which can be an issue when it is poorly soluble or engaged in strong interactions with, for example, a binding partner. Furthermore, we note that the most recent developments in sample preparation protocols for MAS DNP NMR allow not just for cells to remain intact but also for cells to remain viable following thawing,44,50 which opens up the exciting possibility of following time courses of unmodified drugs using the NMR approach. The methodology introduced here is, of course, not intended to replace but rather to complement other quantification approaches currently used in biology.
We demonstrated the approach by measuring HEK293 cells incubated with [15N]CHIR-98104, an activator of the Wnt signaling pathway, where we were able to identify a concentration range for the drug between 196 ± 112 and 20 ± 9 pmol per million cells. The interval identified with MAS DNP corresponds to ex situ quantification from cell extracts with LC–MS, validating our in-cell detection and quantification approach.
With the same approach, we are able to determine the total amount of protein in the cells through the integration of the amide signal in the 15N spectrum, estimated at 2.95 ± 0.98 μmol per million cells, and we show how 15N protein signals can be used as an intermediate standard in the cells, avoiding the necessity to add an external standard.
Although the MAS DNP approach introduced here is a sensitive technique and considerably improves the sensitivity for NMR quantification, it still suffers from lower sensitivity compared to MS or fluorescence spectroscopy. The limit of detection determined for [15N]CHIR-98104 in the present work corresponds roughly to submicromolar intracellular concentrations. Although this is an extremely relevant range for drug activity, some targets of interest can have intracellular concentrations in the nanomolar or subnanomolar ranges. Moreover, in the case that no chemical shift distinction could be performed between the drug and the cellular background, the quantification of very diluted species can be difficult, even if with a careful blank subtraction approach this should be still potentially feasible.25 We note that the approach can also be used with 19F- or 31P-based probes, which have extensive precedent in the NMR literature, and have already been combined with MAS DNP experiments20,22 in the same way as for the 15NO2 probe used here. In particular, the limit of detection can potentially be pushed further down with the detection of the more sensitive 19F nucleus, especially given that it is present in many drugs and is almost absent in cells, obviating the problem of the cellular background. We expect that ongoing improvements in DNP instrumentation and the design of polarizing agents will further increase the sensitivity of the technique,53,54 and the applications of the more recently introduced preparation protocols44,50 will enhance the approach presented here. The present approach can also be affected if the drug is metabolized by the cells. As long as the metabolite signals do not overlap with the drug signal, the quantification of the drug itself should not be affected, but the overall sensitivity will be lowered by the fact that the integral associated with labeled 15N groups would be spread over the signals of all the products of the metabolization process, which, depending on the metabolic process, might also overlap with the cellular background. However, at least in principle, this could represent a powerful tool to study the pharmacokinetics of the drug.
The approach presented is a potent strategy for the quantification of drugs in cells and, more generally, of low concentrations of analytes in complex mixtures, which can complement other commonly used quantification techniques in biology and chemistry.
Methods
Cell Preparation
HEK293 cells were cultured in a T-75 cm2 culture flask with a medium composed of Dulbecco’s modified Eagle’s (DMEM, GIBCO, 12800-017), 10% fetal bovine serum (FBS) (Gibco Fetal Bovine Serum, Qualified, Cat. 26140095), 2 mmol/L glutamine, and penicillin–streptomycin 100 U/mL, and incubated at 37 °C in 5% CO2 and 95% saturated atmospheric humidity. The culture medium was changed every 2 days until the cells attained 80% confluence, then the cells were expanded. Briefly, the cells were rinsed with Ca2+/Mg2+ free Dulbecco’s phosphate-buffered saline (PBS). TrypLE Express Enzyme (1×), no phenol red, was added to detach the cell layer from the flask. The enzyme activity was stopped by adding culture medium containing serum to the flask. The cells were aspirated by gently pipetting, transferred to a tube, and centrifuged at 1000 rpm for 5. The cell pellet was resuspended with culture medium and dispensed into a T-75 cm2 culture flask at a density of 15·× 103 cells/cm2 and incubated at 37 °C and 5% CO2 for 24 h, followed by the replacement of the culture medium by prewarmed fresh medium containing 5 or 10 μM [15N]CHIR-98104 for an experimental period of 6 h. At the endpoint, the cells were detached from the culture flask using TrypLE, as described before. The cells were counted and checked for their viability before centrifugation. The cell pellet was resuspended in 1 mL of freezing medium containing 90% FBS + 10% DMSO to yield a cell suspension of 3·× 106 cells/mL. The cells were cryopreserved in an isopropanol chamber at −80 °C overnight. For long-term storage, the vials were transferred to liquid nitrogen and stored in the gas phase above the liquid nitrogen. All the steps were carried out under aseptic conditions.
[15N]CHIR-98104
[15N]CHIR-98104 was synthetized following an adaptation of a literature procedure.55 The detailed synthesis is reported in the Supporting Information.
Sample Preparation
Solution NMR of [15N]CHIR-98014
A bulk solution of CHIR98014 was prepared by dissolving the drug in DMSO-d6 to a final concentration of 1.06 mM and filtering. 500 μL of the solution were transferred to a 5 mm NMR tube and characterized by solution NMR.
MAS DNP NMR of [15N]CHIR-98014
A 1.06 mM solution in DMSO-d6 was diluted with D2O and H2O dissolving AMUpol38 in order to obtain a 10 mM AMUpol, DMSO-d6/D2O/H2O 60:30:10v/v solution. 20 μL of the solution were transferred into a 3.2 mm sapphire rotor, subsequently sealed with a silicon plug and closed with a zirconia cap, and transferred in a precooled DNP spectrometer.
MAS DNP NMR of HEK Cells
Cryopreserved HEK cells previously incubated with [15N]CHIR-98014 5 μM for 6 h were thawed in 37 °C water bath for ∼2–3 min and pelleted at 200g for 5 min to remove the supernatant. Cells were resuspended in 100 μL of DMSO-d6/D2O/H2O 60:30:10v/v and pelleted again at 2000g for 5 min, and after removal of the supernatant, resuspended in 7.7 mg of a 14 mM AMUpol, 343 μM 13C,15N l-histidine·HCl, and a DMSO-d6/D2O/H2O 60:30:10v/v solution (total amount of histidine in the rotor was 2.22 nmol). The entire sample was transferred to a 3.2 mm sapphire rotor by centrifugation at 2000g for 1 min. The rotor was sealed with a silicon plug, closed with a zirconia cap, and transferred to a precooled DNP spectrometer. Cryopreserved cells previously incubated with [15N]CHIR-98014 10 μM for 6 h were prepared in the same way without the addition of 13C–15N l-histidine·HCl. After the DNP experiments, the sample was thawed and centrifuged in order to separate the supernatant from the cell pellet, and the supernatant (∼10 μL) was transferred to a 3.2 mm sapphire rotor, diluted with 5 μL of a DMSO/D2O/H2O 60:30:10v/v solution to ensure the rotor stability, and sealed with a silicon plug and closed with a zirconia cap. The time between removing the sample from the spectrometer (thawing) and separation from the pellet was ∼5 min. The rotor was transferred to a precooled DNP spectrometer, and DNP enhanced spectra (with measured 1H enhancements similar to that of the solvent in the frozen cell samples) were acquired to check for the presence of dissolved drugs and/or cellular components.
NMR Experiments
Solution NMR experiments were acquired on a 14.1 T (600 MHz) AVANCE III Bruker spectrometer equipped with a double resonance broad band 5 mm probe (Figure S3), and on a 9.4 T (400 MHz) equipped with a double resonance broad band 5 mm probe (Figure S2). DNP enhanced MAS NMR experiments were performed on a 400 MHz (9.4 T) AVANCE III HD Bruker NMR spectrometer, coupled with a 263 GHz gyrotron microwave source and equipped with a 3.2 mm triple resonance low-temperature probe. The magnetic field was adjusted using the internal sweep coil to match the maximum enhancement of AMUPOL. The MAS probe has been tuned to 1H, 13C, and 15N. Experiments were performed at ∼100 K and at 10.5 kHz MAS (CHIR98104 5 μM) or 13 kHz MAS (CHIR98104 10 μM). (1H)–15N multiple-contact CP experiments were acquired using the pulse sequence used in refs (41) and (56). The 1H rf field was ramped from 90 to 100% during the contact time. The recycle delay was set to 100 ms. Spectra were acquired with 10 CP loops and an inter-CP delay of 2.5 s = TB,on (CHIR98104 5 μM) or 5 CP loops and an inter-CP delay of 6 s = 2 TB,on (CHIR98104 10 μM). To check for the presence of dissolved drugs in the supernatant (Figure S6), a DNP enhanced (1H)–15N multiple-contact CP spectrum (with measured 1H enhancements similar to that of the solvent in the frozen cell samples) was acquired and compared to a DNP enhanced (1H)–15N CP multiple-contact CP spectrum of the cells acquired in the same amount of time. Furthermore, details are provided in Table S2.
LC–MS Experiments
After DNP experiments, the full sample from Figure 2 (20 μL total) was resuspended in 100 μL of acetonitrile and subjected to two cycles of freeze (−80 °C) and thaw (25 °C). 10 μL of the supernatant were mixed with 90 μL of methanol and 5 μL of DMSO and analyzed in the LC–MS. The calibration curve (Figure S5) was performed with the solutions of [15N]CHIR-98014 in methanol with the following concentrations: 500–250–100–50–25–10–1 nM.
Trypan Blue Assay
400 μL of a 3 ×·106 mL–1 suspension of HEK cells were divided into four identical aliquots and pelleted at 200g for 5 min. The first aliquot was resuspended in 15% DMSO, 75% D2O, and 10% H2O for 5 min. The second aliquot was resuspended in 15% DMSO, 75% D2O, and 10% H2O for 5 min, and the third in 60% DMSO, 30% D2O, and 10% H2O for 5 min. The second and third samples were then packed into 3.2 mm rotors as previously described, and frozen directly in the spectrometer, then thawed. All the three samples were then pelleted and resuspended in 500 μL of PBS pH = 7.2 buffer. The fourth aliquot was suspended in pure acetonitrile for 5 min. For the assay, all the four resulting suspensions were mixed 1:1 with a 0.4% trypan blue solution for 5 min before observation.
Quantification Procedure
The quantification for multiple-contact CP experiments is performed through the integrals of the 15N signals of interest using the following expression57
where the subscripts x and s refer to the analyte and the standard, respectively. n is the number of moles, I is the microwave-on NMR integral, ε is the DNP enhancement, and N is the number of nuclei associated with a given signal. P is a factor which takes into account the purity of the standard and the isotope abundance of 15N (natural abundance is assumed here to be 0.4%). For histidine signals, a correction factor taking into account the contribution to the signal from the cellular background is applied (as shown in Figure 2). In order to provide an estimation of the uncertainty, average integrals from four different processings of the same data set are used.
The error on nx, Δnx, is determined from the propagation of errors as it follows
where ΔIx and ΔIs represent the 95% confidence interval of the averaged integrals and Δεx and Δεs represent the uncertainty in the enhancements, respectively, determined according to Lelli et al.(58)
For LC–MS, the quantification is performed from a linear fitting of the calibration curve, after taking into account the dilution factors. The error is determined from the propagation of the errors of the intercept and the slope determined from the fitting.
Acknowledgments
We thank Dr. Daniel Ortiz (EPFL) for performing the LC–MS measurements, Irina Sinenko (EPFL) for acquiring cell pictures, and Prof. Olivier Ouari (Aix Marseille University) for providing the AMUpol radical. The cell graphic used in the TOC was designed by brgfx/Freepik.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c12442.
Additional experimental details, additional tables, additional figures, and link to all the raw NMR data (PDF)
Author Contributions
The manuscript was written through the contributions of all the authors. All the authors have given approval to the final version of the manuscript.
This work was funded by the Swiss National Centre of Competence in Research Chemical Biology and AstraZeneca.
The authors declare no competing financial interest.
Supplementary Material
References
- Dollery C. T. Intracellular Drug Concentrations. Clin. Pharmacol. Ther. 2013, 93, 263–266. 10.1038/clpt.2012.240. [DOI] [PubMed] [Google Scholar]
- Rizk M.; Zou L.; Savic R.; Dooley K. Importance of Drug Pharmacokinetics at the Site of Action. Clin. Transl. Sci. 2017, 10, 133–142. 10.1111/cts.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuespée R.; Theile D.; Fresnais M.; Burhenne J.; Weiss J.; Haefeli W. E. Approaching sites of action of drugs in clinical pharmacology: New analytical options and their challenges. Br. J. Clin. Pharmacol. 2021, 87, 858–874. 10.1111/bcp.14543. [DOI] [PubMed] [Google Scholar]
- Ryadnov M. G. Where is the drug gone? – Measuring intracellular delivery and localization. Methods 2014, 68, 281–282. 10.1016/j.ymeth.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Wishart D. S. Quantitative metabolomics using NMR. TrAC, Trends Anal. Chem. 2008, 27, 228–237. 10.1016/j.trac.2007.12.001. [DOI] [Google Scholar]
- Bharti S. K.; Roy R. Quantitative 1H NMR spectroscopy. TrAC, Trends Anal. Chem. 2012, 35, 5–26. 10.1016/j.trac.2012.02.007. [DOI] [Google Scholar]
- Vignoli A.; Ghini V.; Meoni G.; Licari C.; Takis P. G.; Tenori L.; Turano P.; Luchinat C. High-Throughput Metabolomics by 1D NMR. Angew. Chem., Int. Ed. 2019, 58, 968–994. 10.1002/anie.201804736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand K.; Reverdatto S.; Burz D. S.; Zitomer R.; Shekhtman A. Structure of Proteins in Eukaryotic Compartments. J. Am. Chem. Soc. 2012, 134, 12798–12806. 10.1021/ja304809s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedberg D. I.; Selenko P. Live Cell NMR. Annu. Rev. Biophys. 2014, 43, 171–192. 10.1146/annurev-biophys-051013-023136. [DOI] [PubMed] [Google Scholar]
- Theillet F.-X.; Binolfi A.; Bekei B.; Martorana A.; Rose H. M.; Stuiver M.; Verzini S.; Lorenz D.; van Rossum M.; Goldfarb D.; Selenko P. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- Luchinat E.; Banci L. In-Cell NMR in Human Cells: Direct Protein Expression Allows Structural Studies of Protein Folding and Maturation. Acc. Chem. Res. 2018, 51, 1550–1557. 10.1021/acs.accounts.8b00147. [DOI] [PubMed] [Google Scholar]
- Siegal G.; Selenko P. Cells, drugs and NMR. J. Magn. Reson. 2019, 306, 202–212. 10.1016/j.jmr.2019.07.018. [DOI] [PubMed] [Google Scholar]
- Kyne C.; Ruhle B.; Gautier V. W.; Crowley P. B. Specific ion effects on macromolecular interactions in Escherichia coli extracts. Protein Sci. 2015, 24, 310–318. 10.1002/pro.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Q. Z.; Daviso E.; Can T. V.; Markhasin E.; Jawla S. K.; Swager T. M.; Temkin R. J.; Herzfeld J.; Griffin R. G. High Frequency Dynamic Nuclear Polarization. Acc. Chem. Res. 2013, 46, 1933–1941. 10.1021/ar300348n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossini A. J.; Zagdoun A.; Lelli M.; Lesage A.; Copéret C.; Emsley L. Dynamic Nuclear Polarization Surface Enhanced NMR Spectroscopy. Acc. Chem. Res. 2013, 46, 1942–1951. 10.1021/ar300322x. [DOI] [PubMed] [Google Scholar]
- Reif B.; Ashbrook S. E.; Emsley L.; Hong M. Solid-state NMR spectroscopy. Nat. Rev. Methods Primers 2021, 1, 2. 10.1038/s43586-020-00002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Xu W.; Su Y. Solid-state NMR spectroscopy in pharmaceutical sciences. TrAC, Trends Anal. Chem. 2021, 135, 116152. 10.1016/j.trac.2020.116152. [DOI] [Google Scholar]
- Albert B. J.; Gao C.; Sesti E. L.; Saliba E. P.; Alaniva N.; Scott F. J.; Sigurdsson S. T.; Barnes A. B. Dynamic Nuclear Polarization Nuclear Magnetic Resonance in Human Cells Using Fluorescent Polarizing Agents. Biochemistry 2018, 57, 4741–4746. 10.1021/acs.biochem.8b00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan S.; Scherpe S.; Lucini Paioni A.; van der Zwan J.; Folkers G. E.; Ovaa H.; Baldus M. DNP-Supported Solid-State NMR Spectroscopy of Proteins Inside Mammalian Cells. Angew. Chem., Int. Ed. 2019, 58, 12969–12973. 10.1002/anie.201903246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlagnitweit J.; Friebe Sandoz S.; Jaworski A.; Guzzetti I.; Aussenac F.; Carbajo R. J.; Chiarparin E.; Pell A. J.; Petzold K. Observing an Antisense Drug Complex in Intact Human Cells by in-Cell NMR Spectroscopy. ChemBioChem 2019, 20, 2474–2478. 10.1002/cbic.201900297. [DOI] [PubMed] [Google Scholar]
- Overall S. A.; Price L. E.; Albert B. J.; Gao C.; Alaniva N.; Judge P. T.; Sesti E. L.; Wender P. A.; Kyei G. B.; Barnes A. B. In Situ Detection of Endogenous HIV Activation by Dynamic Nuclear Polarization NMR and Flow Cytometry. Int. J. Mol. Sci. 2020, 21, 4649. 10.3390/ijms21134649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder B. J.; Berk C.; Liao W.-C.; Rossini A. J.; Schwarzwälder M.; Pradere U.; Hall J.; Lesage A.; Copéret C.; Emsley L. One- and Two-Dimensional High-Resolution NMR from Flat Surfaces. ACS Cent. Sci. 2019, 5, 515–523. 10.1021/acscentsci.8b00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Caporini M. A.; Im S.-C.; Waskell L.; Ramamoorthy A. Cellular solid-state NMR investigation of a membrane protein using dynamic nuclear polarization. Biochim. Biophys. Acta, Biomembr 2015, 1848, 342–349. 10.1016/j.bbamem.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viger-Gravel J.; Schantz A.; Pinon A. C.; Rossini A. J.; Schantz S.; Emsley L. Structure of Lipid Nanoparticles Containing siRNA or mRNA by Dynamic Nuclear Polarization-Enhanced NMR Spectroscopy. J. Phys. Chem. B 2018, 122, 2073–2081. 10.1021/acs.jpcb.7b10795. [DOI] [PubMed] [Google Scholar]
- Bertarello A.; Berruyer P.; Skantze U.; Sardana S.; Sardana M.; Elmore C. S.; Schade M.; Chiarparin E.; Schantz S.; Emsley L. Quantification of magic angle spinning dynamic nuclear polarization NMR spectra. J. Magn. Reson. 2021, 329, 107030. 10.1016/j.jmr.2021.107030. [DOI] [PubMed] [Google Scholar]
- Ring D. B.; Johnson K. W.; Henriksen E. J.; Nuss J. M.; Goff D.; Kinnick T. R.; Ma S. T.; Reeder J. W.; Samuels I.; Slabiak T.; Wagman A. S.; Hammond M.-E. W.; Harrison S. D. Selective Glycogen Synthase Kinase 3 Inhibitors Potentiate Insulin Activation of Glucose Transport and Utilization In Vitro and In Vivo. Diabetes 2003, 52, 588–595. 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- Reckel S.; Hänsel R.; Löhr F.; Dötsch V. In-cell NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 91–101. 10.1016/j.pnmrs.2007.02.002. [DOI] [Google Scholar]
- Selenko P.; Wagner G. Looking into live cells with in-cell NMR spectroscopy. J. Struct. Biol. 2007, 158, 244–253. 10.1016/j.jsb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Inomata K.; Ohno A.; Tochio H.; Isogai S.; Tenno T.; Nakase I.; Takeuchi T.; Futaki S.; Ito Y.; Hiroaki H.; Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- Barbieri L.; Luchinat E.; Banci L. Characterization of proteins by in-cell NMR spectroscopy in cultured mammalian cells. Nat. Protoc. 2016, 11, 1101–1111. 10.1038/nprot.2016.061. [DOI] [PubMed] [Google Scholar]
- Burz D. S.; Dutta K.; Cowburn D.; Shekhtman A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR). Nat. Methods 2006, 3, 91–93. 10.1038/nmeth851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P.; Serber Z.; Gadea B.; Ruderman J.; Wagner G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 11904–11909. 10.1073/pnas.0604667103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selenko P.; Frueh D. P.; Elsaesser S. J.; Haas W.; Gygi S. P.; Wagner G. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat. Struct. Mol. Biol. 2008, 15, 321–329. 10.1038/nsmb.1395. [DOI] [PubMed] [Google Scholar]
- Amata I.; Maffei M.; Igea A.; Gay M.; Vilaseca M.; Nebreda A. R.; Pons M. Multi-phosphorylation of the Intrinsically Disordered Unique Domain of c-Src Studied by In-Cell and Real-Time NMR Spectroscopy. ChemBioChem 2013, 14, 1820–1827. 10.1002/cbic.201300139. [DOI] [PubMed] [Google Scholar]
- Sakakibara D.; Sasaki A.; Ikeya T.; Hamatsu J.; Hanashima T.; Mishima M.; Yoshimasu M.; Hayashi N.; Mikawa T.; Wälchli M.; Smith B. O.; Shirakawa M.; Güntert P.; Ito Y. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 2009, 458, 102–105. 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- Xie J.; Thapa R.; Reverdatto S.; Burz D. S.; Shekhtman A. Screening of Small Molecule Interactor Library by Using In-Cell NMR Spectroscopy (SMILI-NMR). J. Med. Chem. 2009, 52, 3516–3522. 10.1021/jm9000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvier G.; Simenel C.; Jang J.; Kalia N. P.; Choi I.; Nilges M.; Pethe K.; Izadi-Pruneyre N. Target Engagement and Binding Mode of an Antituberculosis Drug to Its Bacterial Target Deciphered in Whole Living Cells by NMR. Biochemistry 2019, 58, 526–533. 10.1021/acs.biochem.8b00975. [DOI] [PubMed] [Google Scholar]
- Sauvée C.; Rosay M.; Casano G.; Aussenac F.; Weber R. T.; Ouari O.; Tordo P. Highly Efficient, Water-Soluble Polarizing Agents for Dynamic Nuclear Polarization at High Frequency. Angew. Chem., Int. Ed. 2013, 52, 10858–10861. 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- Nygaard R.; Romaniuk J. A. H.; Rice D. M.; Cegelski L. Whole Ribosome NMR: Dipolar Couplings and Contributions to Whole Cells. J. Phys. Chem. B 2017, 121, 9331–9335. 10.1021/acs.jpcb.7b06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila J. A. Limiting Values of the 15N Chemical Shift of the Imidazole Ring of Histidine at High pH. J. Phys. Chem. B 2012, 116, 6665–6669. 10.1021/jp211196r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R. L.; Schmidt-Rohr K. Quantitative solid-state 13C NMR with signal enhancement by multiple cross polarization. J. Magn. Reson. 2014, 239, 44–49. 10.1016/j.jmr.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Pinon A. C.; Schlagnitweit J.; Berruyer P.; Rossini A. J.; Lelli M.; Socie E.; Tang M.; Pham T.; Lesage A.; Schantz S.; Emsley L. Measuring Nano- to Microstructures from Relayed Dynamic Nuclear Polarization NMR. J. Phys. Chem. C 2017, 121, 15993–16005. 10.1021/acs.jpcc.7b04438. [DOI] [Google Scholar]
- Prisco N. A.; Pinon A. C.; Emsley L.; Chmelka B. F. Scaling analyses for hyperpolarization transfer across a spin-diffusion barrier and into bulk solid media. Phys. Chem. Chem. Phys. 2021, 23, 1006–1020. 10.1039/d0cp03195j. [DOI] [PubMed] [Google Scholar]
- Ghosh R.; Xiao Y.; Kragelj J.; Frederick K. K. In-Cell Sensitivity-Enhanced NMR of Intact Viable Mammalian Cells. J. Am. Chem. Soc. 2021, 143, 18454–18466. 10.1021/jacs.1c06680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a Novel B-Cell Lymphoma 6 (BCL6) PROTAC To Provide Insight into Small Molecule Targeting of BCL6. ACS Chem. Biol. 2018, 13, 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- Linnane E.; Davey P.; Zhang P.; Puri S.; Edbrooke M.; Chiarparin E.; Revenko A. S.; Macleod A. R.; Norman J. C.; Ross S. J. Differential uptake, kinetics and mechanisms of intracellular trafficking of next-generation antisense oligonucleotides across human cancer cell lines. Nucleic Acids Res. 2019, 47, 4375–4392. 10.1093/nar/gkz214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adihou H.; Gopalakrishnan R.; Förster T.; Guéret S. M.; Gasper R.; Geschwindner S.; Carrillo García C.; Karatas H.; Pobbati A. V.; Vazquez-Chantada M.; Davey P.; Wassvik C. M.; Pang J. K. S.; Soh B. S.; Hong W.; Chiarparin E.; Schade D.; Plowright A. T.; Valeur E.; Lemurell M.; Grossmann T. N.; Waldmann H. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun. 2020, 11, 5425. 10.1038/s41467-020-19224-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt M.; Bellavita R.; Gerber A.; Efrém N. L.; Ramshorst T.; Pearce N. M.; Davey P. R. J.; Everard I.; Vazquez-Chantada M.; Chiarparin E.; Grieco P.; Hennig S.; Grossmann T. N. Bicyclic β-Sheet Mimetics that Target the Transcriptional Coactivator β-Catenin and Inhibit Wnt Signaling. Angew. Chem., Int. Ed. 2021, 60, 13937–13944. 10.1002/anie.202102082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietmair S.; Hodson M. P.; Quek L.-E.; Timmins N. E.; Gray P.; Nielsen L. K. A Multi-Omics Analysis of Recombinant Protein Production in Hek293 Cells. PLoS One 2012, 7, e43394 10.1371/journal.pone.0043394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall S. A.; Barnes A. B. Biomolecular Perturbations in In-Cell Dynamic Nuclear Polarization Experiments. Front. Mol. Biosci. 2021, 8, 743829. 10.3389/fmolb.2021.743829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Ghosh R.; Frederick K. K. In-Cell NMR of Intact Mammalian Cells Preserved with the Cryoprotectants DMSO and Glycerol Have Similar DNP Performance. Front. Mol. Biosci. 2022, 8, 789478. 10.3389/fmolb.2021.789478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X.; Korzekwa K.; Elsby R.; Fenner K.; Galetin A.; Lai Y.; Matsson P.; Moss A.; Nagar S.; Rosania G. R.; Bai J. P. F.; Polli J. W.; Sugiyama Y.; Brouwer K. L. R.; Consortium I. T. Intracellular Drug Concentrations and Transporters: Measurement, Modeling, and Implications for the Liver. Clin. Pharmacol. Ther. 2013, 94, 126–141. 10.1038/clpt.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanato G.; Casano G.; Kubicki D. J.; Rao Y.; Esteban Hofer L.; Menzildjian G.; Karoui H.; Siri D.; Cordova M.; Yulikov M.; Jeschke G.; Lelli M.; Lesage A.; Ouari O.; Emsley L. Open and Closed Radicals: Local Geometry around Unpaired Electrons Governs Magic-Angle Spinning Dynamic Nuclear Polarization Performance. J. Am. Chem. Soc. 2020, 142, 16587–16599. 10.1021/jacs.0c04911. [DOI] [PubMed] [Google Scholar]
- Berruyer P.; Björgvinsdóttir S.; Bertarello A.; Stevanato G.; Rao Y.; Karthikeyan G.; Casano G.; Ouari O.; Lelli M.; Reiter C.; Engelke F.; Emsley L. Dynamic Nuclear Polarization Enhancement of 200 at 21.15 T Enabled by 65 kHz Magic Angle Spinning. J. Phys. Chem. Lett. 2020, 11, 8386–8391. 10.1021/acs.jpclett.0c02493. [DOI] [PubMed] [Google Scholar]
- Wagman A. S.; Boyce R. S.; Brown S. P.; Fang E.; Goff D.; Jansen J. M.; Le V. P.; Levine B. H.; Ng S. C.; Ni Z.-J.; Nuss J. M.; Pfister K. B.; Ramurthy S.; Renhowe P. A.; Ring D. B.; Shu W.; Subramanian S.; Zhou X. A.; Shafer C. M.; Harrison S. D.; Johnson K. W.; Bussiere D. E. Synthesis, Binding Mode, and Antihyperglycemic Activity of Potent and Selective (5-Imidazole-2-yl-4-phenylpyrimidin-2-yl)[2-(2-pyridylamino)ethyl]amine Inhibitors of Glycogen Synthase Kinase 3. J. Med. Chem. 2017, 60, 8482–8514. 10.1021/acs.jmedchem.7b00922. [DOI] [PubMed] [Google Scholar]
- Björgvinsdóttir S.; Walder B. J.; Matthey N.; Emsley L. Maximizing nuclear hyperpolarization in pulse cooling under MAS. J. Magn. Reson. 2019, 300, 142–148. 10.1016/j.jmr.2019.01.011. [DOI] [PubMed] [Google Scholar]
- Malz F.; Jancke H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. 10.1016/j.jpba.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Lelli M.; Chaudhari S. R.; Gajan D.; Casano G.; Rossini A. J.; Ouari O.; Tordo P.; Lesage A.; Emsley L. Solid-State Dynamic Nuclear Polarization at 9.4 and 18.8 T from 100 K to Room Temperature. J. Am. Chem. Soc. 2015, 137, 14558–14561. 10.1021/jacs.5b08423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.