

Abstract
Tetrazines (Tz) have been applied as bioorthogonal agents for various biomedical applications, including pretargeted imaging approaches. In radioimmunoimaging, pretargeting increases the target-to-background ratio while simultaneously reducing the radiation burden. We have recently reported a strategy to directly 18F-label highly reactive tetrazines based on a 3-(3-fluorophenyl)-Tz core structure. Herein, we report a kinetic study on this versatile scaffold. A library of 40 different tetrazines was prepared, fully characterized, and investigated with an emphasis on second-order rate constants for the reaction with trans-cyclooctene (TCO). Our results reveal the effects of various substitution patterns and moreover demonstrate the importance of measuring reactivities in the solvent of interest, as click rates in different solvents do not necessarily correlate well. In particular, we report that tetrazines modified in the 2-position of the phenyl substituent show high intrinsic reactivity toward TCO, which is diminished in aqueous systems by unfavorable solvent effects. The obtained results enable the prediction of the bioorthogonal reactivity and thereby facilitate the development of the next generation of substituted aryltetrazines for in vivo applications.
Introduction
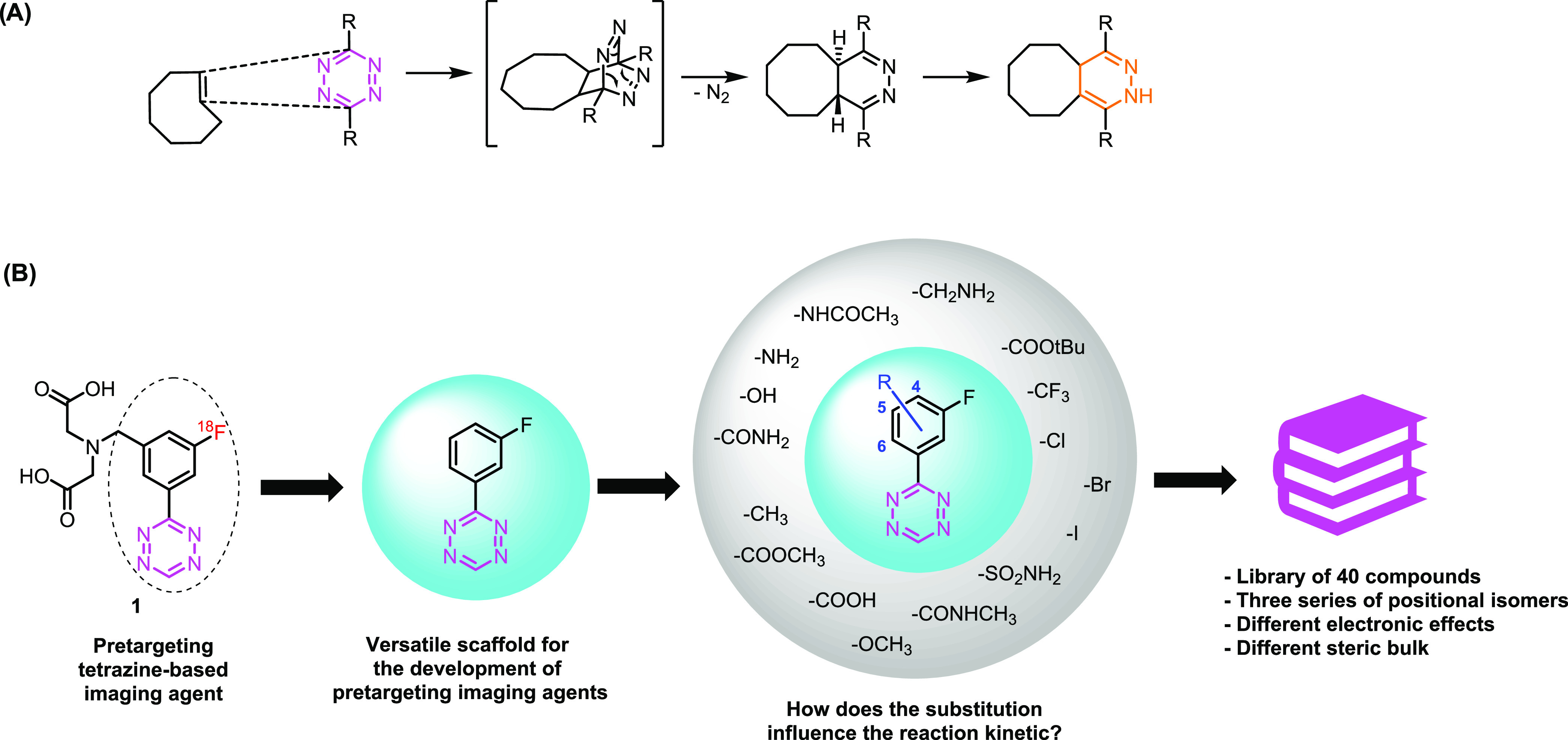
Bioorthogonal reactions are molecular transformations not present in nature.1 The term was introduced by Bertozzi and referred to a chemical reaction that does not interact or interfere with biological systems and functionalities.2 In recent years, bioorthogonal reactions have become increasingly popular, enabling a number of applications in various fields.1,3 The common features of these transformations include fast and efficient reaction at low concentrations in biological media at physiological pH, high selectivity and no cross-reactivity with functional groups present in biomolecules, and the formation of stable ligation products.1,3 Bioorthogonal reactions have attracted increasing interest in studying biological processes due to their unique properties.3,4 Amongst all bioorthogonal reactions reported so far, the [4 + 2] cycloaddition of 1,2,4,5-tetrazines (Tz) and trans-cyclooctenes (TCOs) stands out due to exceptional reaction kinetics (Figure 1).5,6 This inverse electron-demand Diels–Alder (IEDDA)-initiated bioorthogonal reaction was shown to be highly selective and compatible with biological systems, even enabling its application in vivo.6 In recent years, tetrazine ligations have been exploited in biomedical research, in particular for rapid bioconjugation, drug delivery, molecular imaging, drug-target identification, and radiochemistry.7 Tetrazine ligations have been extensively used in pretargeted positron emission tomography (PET) imaging in which high reaction rate constants are essential due to the very low concentrations of the radiolabeled compounds in vivo.7 Several preclinical examples exist demonstrating the potential of this approach.8−13
Figure 1.
(A) Tetrazine ligation mechanism. (B) Compound 1 and the different substituents employed for the structure–kinetics relationship on the 3-(3-fluorophenyl)-1,2,4,5-tetrazine core.
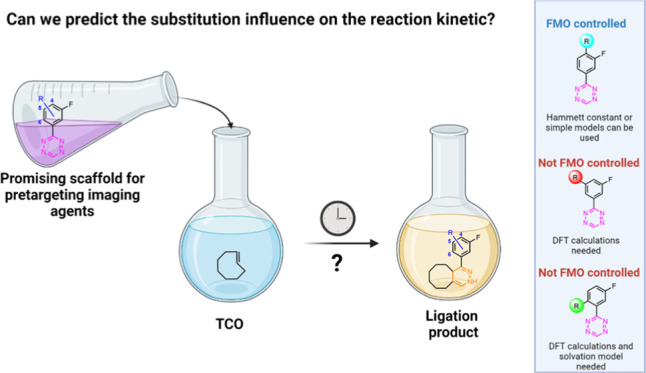
Since fluorine-18 (18F) possesses almost ideal physical characteristics for molecular imaging, an 18F-labeled Tz would be ideal for PET applications in a clinical setting.14−16 However, due to the intrinsic instability of the tetrazine core to basic conditions typically employed in direct 18F-fluorination approaches, no 18F-tetrazine was synthesized until a few years ago.17−22 Recently, we reported the first direct 18F-labeling of highly reactive tetrazines, applying copper-mediated oxidative [18F]fluorination.23 This method allowed us to develop a hydrophilic, fast-clearing, and highly reactive bioorthogonal click imaging agent (1, Figure 1B).23 3-(3-Fluorophenyl)-1,2,4,5-tetrazine, the scaffold of compound 1, could be further modified with different moieties to obtain pretargeting agents with peculiar physicochemical parameters as we have recently shown.23 To enable the rational design of tetrazines, it is, however, important to understand the relationship of the substitution pattern of Tz derivatives and their reactivity toward TCOs, which motivated this study (Figure 1). In general, it is well known that electron-withdrawing substituents increase, and electron-donating groups decrease the reactivity.24,25 Nevertheless, only a few systematic studies of the effect of various substituents of the phenyl ring of Tz derivatives on reaction kinetics have been reported so far, while most of them focus on the substituents on the Tz itself.6,26 Consequently, we prepared a library of 40 Tz derivatives with a set of 16 different phenyl substituents and evaluated the reaction kinetics of the ligation with TCO both in Dulbecco’s phosphate-buffered saline (DPBS) and acetonitrile (CH3CN). Based on the obtained data, we developed a method to predict the IEDDA reactivity of differently substituted Tzs.
Result and Discussion
Library Design
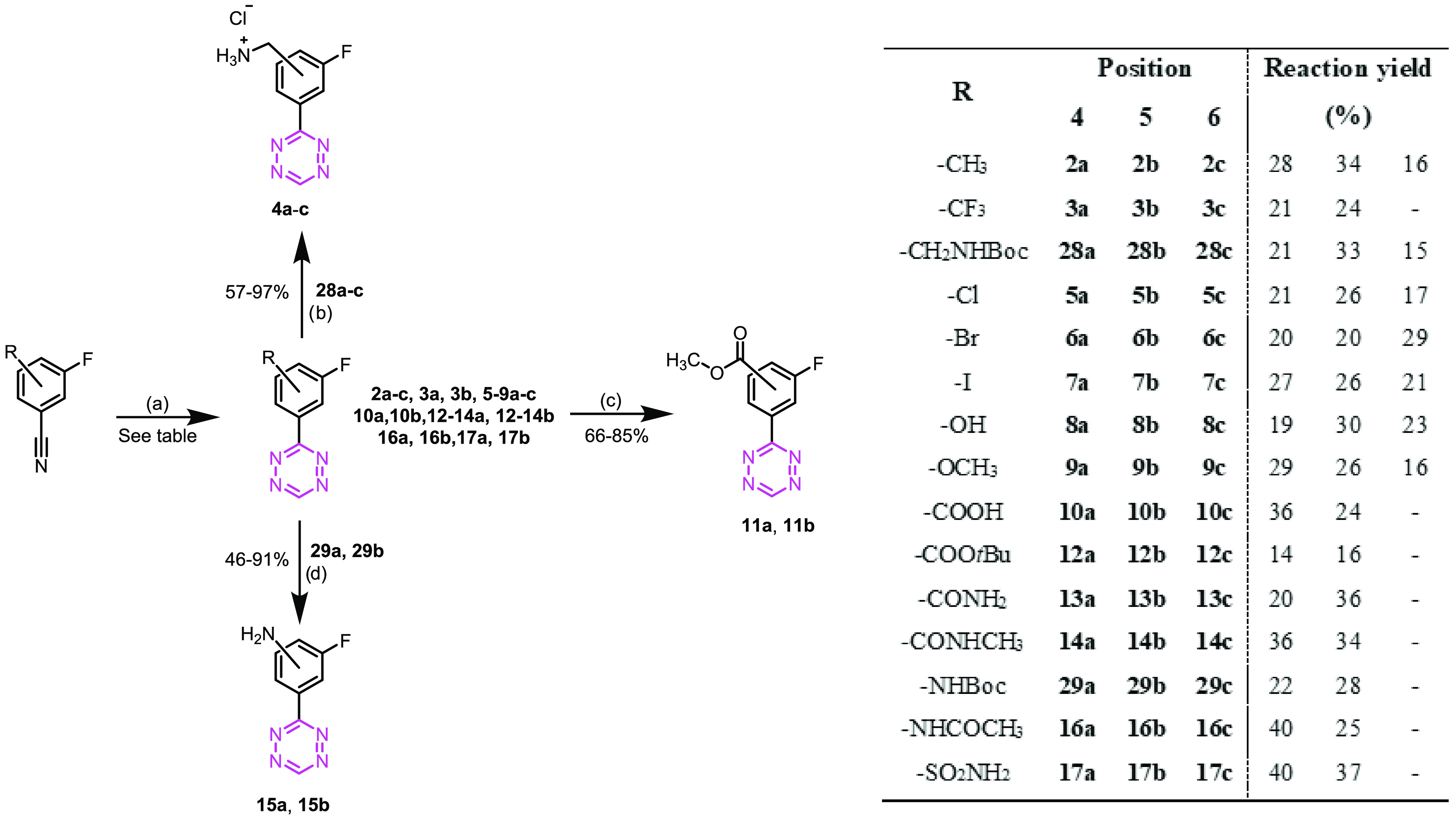
Sixteen different moieties were chosen to study the effect of different substituents on the reactivity of the 3-(3-fluorophenyl)-1,2,4,5-tetrazine core (Figure 1) covering a broad set of electron-withdrawing, electron-donating, and sterically demanding groups. In addition, 4-, 5-, and 6-modified isomers were included in the library to study the effects of the substitution pattern.
Synthesis
For the synthesis of tetrazines 2a–c, 3a–c, and 5–10a–c, the required nitriles were commercially available. Boc-protected nitriles for the synthesis of Tzs 4a–c were obtained by reacting the corresponding bromobenzyl compounds with the phthalimide potassium salt, followed by hydrazine deprotection.27 The primary amines were then reacted with di-tert-butyl dicarbonate to afford the protected compounds in good overall yields (Scheme 1).
Scheme 1. Synthesis of (Aminomethyl)benzonitrile Derivatives.
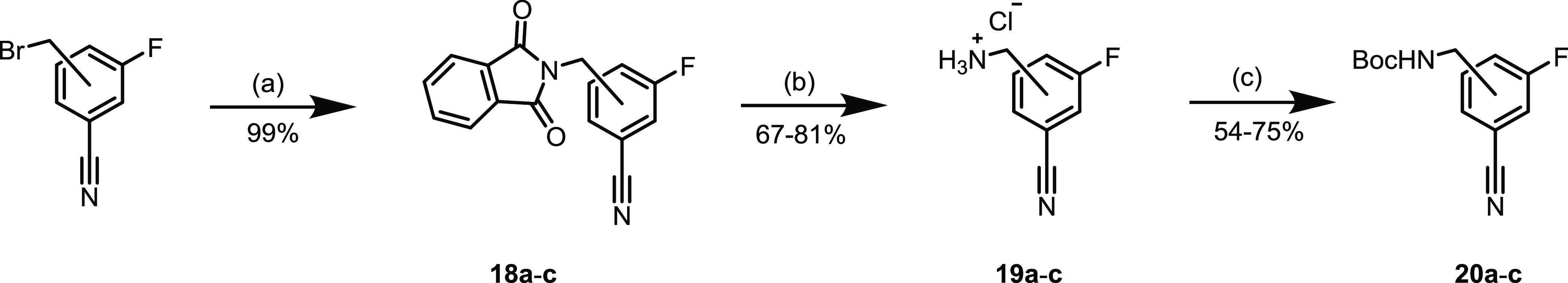
(a) Phthalimide potassium salt, N,N-dimethylformamide (DMF), 9 h, 130 °C; (b) (i) N2H4·H2O, EtOH, reflux, and 2 h; (ii) HCl, Et2O, 0 °C, and 1 h; and (c) Boc2O, Et3N, CH3CN, rt, and 12 h.
The tert-butyl ester nitriles for the synthesis of Tzs 12a–c were obtained from the corresponding carboxylic acids (Scheme 2A).28 Similarly, the primary amides for Tzs 13a–c and the secondary amides for Tzs 14a–c were obtained in yields ranging from 76 to 80% by CDI-mediated activation of the carboxyl group and reaction with ammonia and methylamine, respectively (Scheme 2A). Boc-protection and acylation of the corresponding aniline yielded the nitriles as starting materials for the synthesis of Tzs 15a–c and Tzs 16a–c (Scheme 2B). Finally, the sulfonamides Tzs 17a–c were prepared using a modified Sandmeyer procedure followed by a reaction with ammonia (Scheme 2B).29
Scheme 2. (A) Synthesis of tert-Butyl Ester and Amide Derivatives. (B) Synthesis of Boc-Protected Aniline, Acetamide, and Sulfonamide Derivatives.
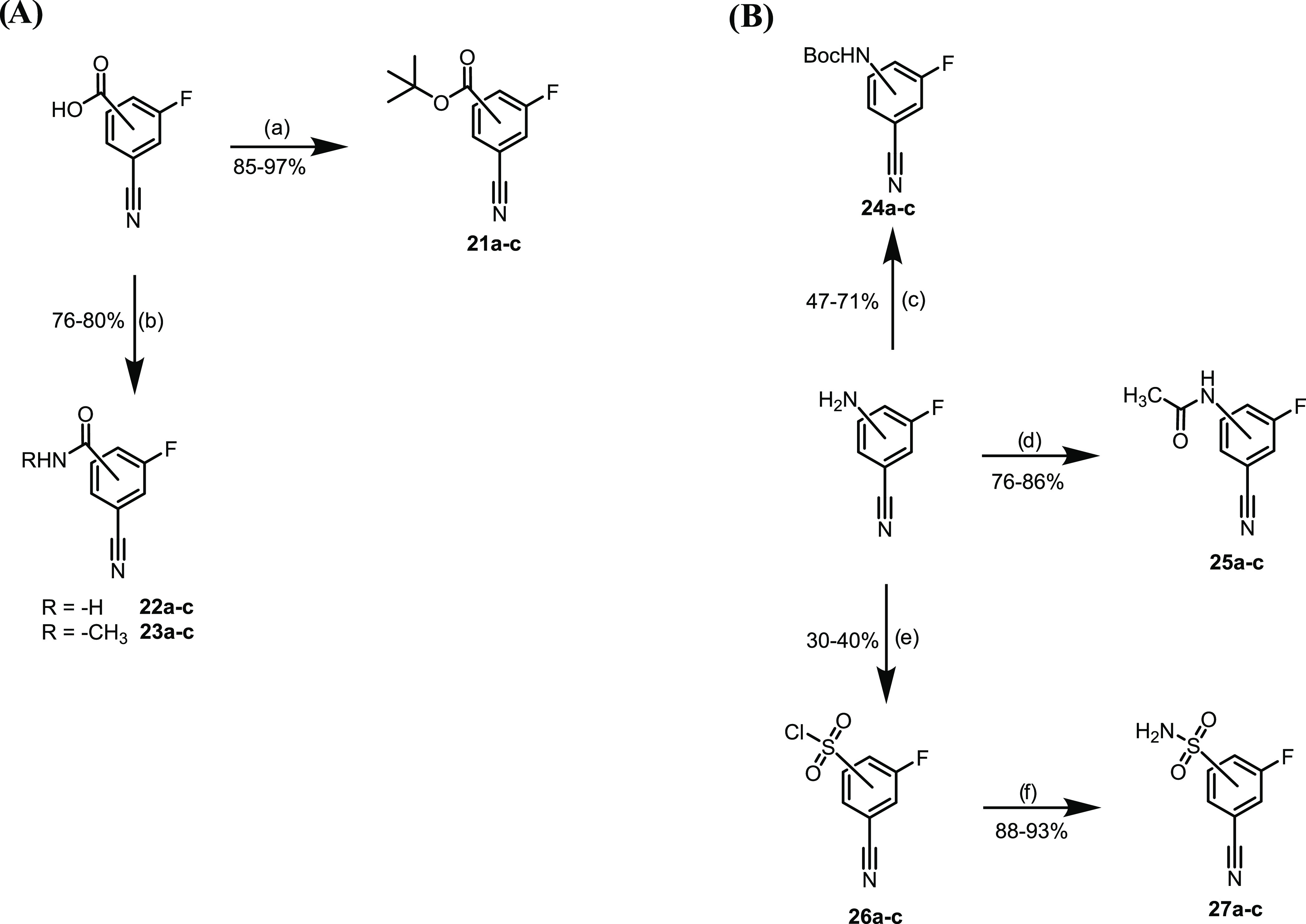
(a) tBuOH, 4-dimethylaminopyridine (DMAP), Boc2O, tetrahydrofuran (THF), rt, and 12 h; (b) NH3 (35% in H2O) or CH3NH2 (40% in H2O), CDI, CH3CN, rt, and 2 h; (c) Boc2O, Et3N, CH2Cl2, rt, and 12 h; (d) Ac2O, CH2Cl2, rt, and 24 h; (e) (i) HCl, NaNO2, 0 °C to rt, and 2 h; (ii) SO2, CuCl, AcOH, 0 °C to rt, and 1 h; and (f) NH3, MeOH, CH3CN, 0 °C to rt, and 2 h.
Tetrazines were synthesized using a metal-free synthetic approach reported by Qu et al. (Scheme 3).30 4- and 5-substituted derivatives were isolated in modest yields using this procedure. 6-Substituted derivatives were more difficult to prepare. Only 2c and 4c–9c could be obtained, suggesting that bulky groups at the 6-position might hinder the reaction or generate highly unstable tetrazines. Deprotection of Tzs 28a–c, 29a, and 29b resulted in Tzs 4a–c, 15a, and 15b in yields ranging from 46 to 97%. Methyl esters 11a and 11b were prepared by acid-catalyzed esterification of the corresponding carboxylic acids 10a and 10b, respectively, in 85 and 66% yields.
Scheme 3. Synthesis of 3-(3-Fluorophenyl)-1,2,4,5-tetrazine Derivatives.
(a) (i) NH2NH2·H2O, CH2Cl2, S8, EtOH, 50 °C, and 24 h; (ii) NaNO2, AcOH, 0 °C to rt, and 30 min; (b) HCl, dioxane, CH2Cl2, rt, and 2 h; (c) HCl, dioxane, MeOH, rt, and 3 h; and (d) trifluoroacetic acid (TFA), CH2Cl2, rt, and 2 h.
Reaction Kinetics
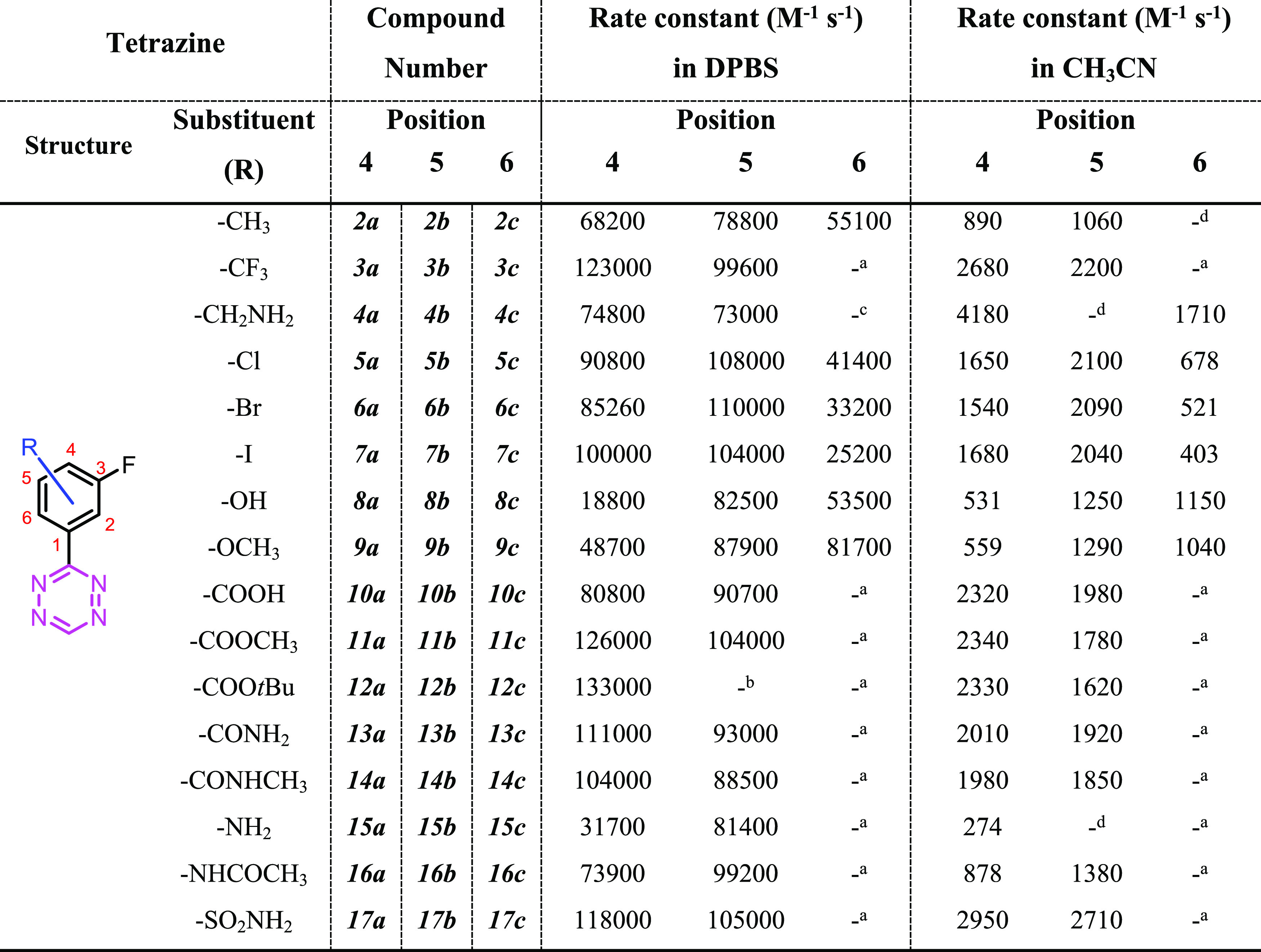
Reactivities of the tetrazines in the reaction with TCO were determined by pseudo-first-order measurements in CH3CN at 25 °C and in DPBS at 37 °C by stopped-flow spectrophotometry.31 Solutions of TCO in anhydrous CH3CN and axTCO-PEG4 in DPBS were employed for the analysis.31,32 The data obtained are reported in Table 1.
Table 1. Second-Order Rate Constants for the Reaction of Tetrazines with trans-Cyclooctenes Determined by Stopped-Flow Spectrophotometryd.
The corresponding Tz could not be obtained or isolated.
The compound is not soluble in DPBS.
The compound is decomposed in DPBS.
The compound is not soluble in CH3CN.
Reactivity Analysis
Measured rate constants in aqueous solution are considerably higher with accelerations of about two orders of magnitude compared to the aprotic and less polar acetonitrile. This effect has been observed before and is well known for Diels–Alder reactions between reactants that are able to form hydrogen bonds.33−35
Importantly, the rate constants measured in CH3CN show poor correlation to those obtained in DPBS (Figure 2). Several of the used tetrazines change their protonation state between these two solvents. Carboxylic acids (10a and 10b) are protonated in acetonitrile but deprotonated in DPBS at pH 7.4. Similarly, phenols can be deprotonated at this pH. We used the computational method by Shields and co-workers to estimate the pKa of 8a–c.36 Our calculations gave pKa values for 8b and 8c of 11.4 and 14.5, respectively, indicating minimal deprotonation at pH 7.4. The high pKa of 14.5 of 8c is caused by the intramolecular hydrogen bond that must be broken to deprotonate this phenol and the unfavored high energy anion that is formed. In the deprotonated 8c, a strong repulsion between the negatively charged oxygen and the tetrazine nitrogen causes the two aromatic rings to break the favorable conjugation and rotate. The ωB97X-D/def2-TZVPD-SMD (water) calculated dihedral angle between the two arenes is 0° in the protonated form and 46° in the anion. Tetrazine 8a is assumed to be partly deprotonated in buffered solution based on a calculated pKa of 7.8. This deprotonation in DPBS leads to electron donation (in contrast to the electron-withdrawing effect of the protonated carboxyl group in acetonitrile), thus resulting in a lower reactivity in aqueous solution. Compounds 4a–c were prepared as HCl salts. Hence, 4a was protonated in acetonitrile but mostly in its neutral state at pH 7.4. Eliminating tetrazines with different protonation states from the analysis results in a linear relationship between the second-order rate constants in acetonitrile and DPBS (R2 = 0.83). The possibility of different protonation states and the limited correlation highlights the importance of performing kinetic measurements in aqueous and pH-controlled solutions if data is needed to estimate the reactivity for bioorthogonal application in biological systems. Therefore, the following analyses are based on the values obtained in DPBS.
Figure 2.

Correlation between second-order rate constants measured in DPBS and CH3CN. Compounds are shown in purple (a-series), green (b-series), and black (c-series), with compounds depicted in red not included in the correlation due to different protonation states.
The measured second-order rate constants for the reaction of tetrazines with axTCO-PEG4 in the DPBS range from approximately 20,000 to 130,000 M–1 s–1 for 4-substituted derivatives, while in the case of 5-substituted compounds, the values range from 70,000 to 110,000 M–1 s–1. 6-Substituted tetrazines show lower rates in the range of 25,000 to 80,000 M–1 s–1. In most cases, the 5-substituted derivatives show higher reactivity than the respective 4-substituted isomer, with the 6-substituted compound being the least reactive one. A notable exception is a-series 8a–c, where 8a shows the lowest reactivity due to the deprotonated nature of this Tz. Most substituents influence the electronics of the π-system through resonance. These mesomeric effects are strongest in the ortho and para positions, which explains the stronger influence and higher variation in the a-series (substituent in the para-position to the tetrazine) compared to compounds b (substituents in the meta-position to the tetrazine). The reactivities of 4-substituted tetrazines correlate well with Hammett δM parameters (Figure 3A and Table S2), demonstrating a main frontier molecular orbital (FMO) control of reactivity.37 The rate constants measured for 5- and 6-substituted phenyltetrazines, however, did not correlate well with Hammett constants (Figure 3B,C). This model only considers the influence of one substituent in isolation, hence not considering the fluorination of the phenyl substituent.
Figure 3.

Correlation between measured second-order rate constants and Hammett parameters δ for (A) 4-substituted, (B) 5-substituted, and (C) 6-substituted phenyltetrazines. (D) Correlation between the steric parameter υ and rate constants for 6-substituted phenyltetrazines.11,37
In the case of 6-substituted derivatives, we have focused on potential steric effects, which need to be considered. Plotting the rate constants of these tetrazines against the υ steric parameter shows that sterically more demanding substituents lead to lower reaction rates (Figure 3D and Table S2).11
From these data, it is evident that simple models, such as the Hammett equation, are not able to correctly predict the reactivity of all of these tetrazines. Using density functional theory (DFT), we investigated the reactivity of 22 tetrazines (2–9a, 2–9b, 2c, and 5–9c), selected out of our library, with trans-cyclooctene in more detail. Gas-phase ωB97X-D/def2-TZVPD calculated reaction barriers show no correlation to the logarithm of measured rate constants (Figure 4A). While gas-phase calculations are usually not able to reproduce accurate barrier heights for reactions in the condensed phase, they often produce the correct trends and relative reactivities. In addition, they provide additional information about the reaction when compared to solution-phase calculations. In our case, for compounds with substituents in the 6-position, the calculated barriers are underestimated by several kcal/mol compared to 5- and 6-substituted derivatives. The reactivity of 8a is likely to be overestimated due to the use of the protonated species in these gas-phase calculations. Including water solvent effects through the implicit SMD model and using deprotonated 8a, however, leads to a good correlation and a predictive model for the reactivity of such tetrazines (Figure 4B).
Figure 4.

(A) Correlation between the gas-phase calculated ΔG‡ values and the natural logarithm of the experimental rate constant. (B) Correlation between SMD (water) calculated ΔG‡ values and the natural logarithm of the experimental rate constant.
The discrepancy between calculations in the gas phase and in solution reveals two interesting effects. First, the intrinsic (gas phase) reactivity of 6-substituted derivatives is unexpectedly high compared to the respective 4- and 5-substituted isomers. One would expect that a bulky substituent in the ortho-position to the tetrazine leads to lower IEDDA reactivity due to increased steric demand, but the opposite seems to be the case. This high intrinsic reactivity is likely based on a distortion-lowering effect. Due to the substituent, the two aromatic systems are not coplanar in the reactant, which leads to a reduced energy penalty when forcing the tetrazine into transition state geometry. This effect was recently described for 2-pyridyl-substituted tetrazines, showing that intramolecular repulsion of the lone pairs of two nitrogen atoms leads to a similar effect.38 Second, a strong solvent effect is observed in which the inclusion of a solvent model lowers the barriers of 6-substituted tetrazines less than those of 4- and 5-substituted derivatives. In the rate-limiting step (Tz/TCO Diels–Alder cycloaddition), a polarized transition state is formed, which can be stabilized through the solvent. More polar solvents and hydrogen bonds stabilize the transition state better, which leads to a lowered barrier and the observed acceleration in water compared to aprotic, less polar solvents. In the case of 6-substituted tetrazines, the substituent partially blocks solvent access to the polarized part of the transition state structure, thus leading to less stabilization. While the average stabilization through the introduction of the SMD water model for 4- and 5-substituted derivatives is 3.7 kcal/mol (with a range of 3.4–4.2 kcal/mol), the average stabilization for 6-substituted tetrazines is only 1.8 kcal/mol (ranging from 0.9 to 3.4 kcal/mol). A larger size of the substituent also results in less solvent access, which leads to the observed correlation between the second-order rate constants and steric parameters. A more detailed investigation of these effects is ongoing and will be reported in due course.
For 4-substituted phenyltetrazines, the reactivity is purely FMO controlled, as already suspected due to the good correlation with Hammett constants. Since the reaction is an inverse electron-demand Diels–Alder cycloaddition, the relevant orbital of the tetrazine is the LUMO + 1.24 Plotting ωB97X-D/def2-TZVPD-SMD(water) LUMO + 1 energies against calculated barriers reveals an excellent correlation (Figure 5A). Calculations of orbital energies are therefore sufficient to estimate the reactivity of these tetrazines. For 5- and 6-substituted compounds, other effects come into play and FMO interactions are not indicative for the reactivity (Figure 5B,C). Therefore, an analysis of the reaction barrier must be conducted to correctly predict the reactivity.
Figure 5.

Correlation between LUMO + 1 energies and calculated ΔG‡ for (A) 4-substituted, (B) 5-substituted, and (C) 6-substituted derivatives.
Conclusions
Overall, our study sheds new light on the key parameters that affect the bioorthogonal reactivity of substituted fluorinated aryltetrazines. Kinetic investigations using a library of synthesized compounds revealed a substantial difference between the relative reactivities observed in CH3CN and in DPBS. This discrepancy is higher for ionic compounds and shows the importance of performing kinetic measurements in an aqueous solution. Furthermore, the substitution pattern on the phenyl moiety of the aryltetrazine scaffold plays a crucial role. In the case of 4-substituted phenyltetrazines, the reactivity is mainly FMO controlled and can be easily predicted based on Hammett δM parameters. In the case of 5- and 6-substituted phenyltetrazines, more detailed studies are required. In particular, the reactivities of 5-substituted analogues can be well explained by DFT studies in the gas phase, while for the 6-substituted isomers, the solvent needs to be taken into consideration. Our results indicate that the substituents at the 6-position prevent the stabilization effect of the solvent on the polarized transition state resulting in a lower reactivity. Considering the recent development of radiolabeled 18F-phenyltetrazines with improved bioorthogonal performance, our findings will aid the design of next-generation tetrazines for in vivo pretargeting.12,23,31
Experimental Section
Chemistry
All reagents and solvents were dried prior to use according to standard methods. Commercial reagents were used without further purification. Analytical thin-layer chromatography (TLC) was performed using silica gel 60 F254 (Merck) with detection by UV absorption and/or by charring following immersion in a 7% ethanolic solution of sulfuric acid or KMnO4 solution (1.5 g of KMnO4, 10 g K2CO3, and 1.25 mL 10% NaOH in 200 mL water). Purification of compounds was carried out by column chromatography on silica gel (40–60 μm, 60 Å) or employing a CombiFlash NextGen 300 + (Teledyne ISCO). 1H and 13C NMR spectra were recorded on Bruker (400 and 600 MHz instruments), using chloroform-d, methanol-d4, or DMSO-d6 as a deuterated solvent and with the residual solvent as the internal reference. For all NMR experiences, the deuterated solvent signal was used as the internal lock. Chemical shifts are reported in δ parts per million (ppm). Coupling constants (J values) are given in Hertz (Hz). Multiplicities of 1H NMR signals are reported as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; dt, doublet of triplets; t, triplet; q, quartet; m, multiplet; and br, broad signal. NMR spectra of all compounds are reprocessed in MestReNova software (version 12.0.22023) from original FID’s files. Mass spectra analysis was performed using an Agilent 6130A and an Agilent 1200 HPLC.
Synthesis
3-(3-Fluoro-4-methylphenyl)-1,2,4,5-tetrazine (2a)
The compound was obtained following the literature procedure.30 3-Fluoro-4-methylbenzonitrile (0.54 g, 4.00 mmol), CH2Cl2 (4.00 mmol, 0.256 mL), sulfur (0.256 g, 1.00 mmol), and ethanol (4.0 mL) were mixed together in a 20 mL microwave reaction tube. Hydrazine monohydrate (1.6 mL, 32.00 mmol) was added slowly with stirring afterward. The vessel was sealed, and the reaction mixture was heated to 50 °C for 24 h. Then, 3 mL of CH2Cl2 and sodium nitrite (2.76 g, 40.00 mmol) in 40 mL of H2O were added to the mixture. Excess acetic acid (14 mL) was then added slowly, during which the solution turned bright red in color. The reaction mixture was extracted with CH2Cl2 (3 × 30 mL). The organic phase was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (95/5 heptane/EtOAc) to yield 0.21 g (28%) of 2a as a red solid. Rf = 0.4 (heptane/EtAOc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.21 (s, 1H), 8.33 (dd, J = 8.0, 1.7 Hz, 1H), 8.27 (dd, J = 10.5, 1.7 Hz, 1H), 7.43 (t, J = 7.8 Hz, 1H), 2.41 (s, 3H); 13C NMR (101 MHz, Chloroform-d) δ 165.80, 161.82 (d, J = 246.1 Hz), 157.83, 132.50 (d, J = 8.3 Hz), 131.11 (d, J = 8.3 Hz), 130.94 (d, J = 17.4 Hz), 123.78 (d, J = 3.5 Hz), 114.69 (d, J = 25.1 Hz), 14.92 (d, J = 3.4 Hz); MS (ESI) m/z [M + H]+: 191.1.
3-(3-Fluoro-5-methylphenyl)-1,2,4,5-tetrazine (2b)
The compound was obtained from 3-fluoro-5-methylbenzonitrile (0.54 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.26 g (34%) of 2b as a red oil. Rf = 0.39 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.16 (s, 1H), 8.19 (d, J = 1.4 Hz, 1H), 8.05 (d, J = 9.4 Hz, 1H), 7.10 (d, J = 9.2 Hz, 1H), 2.43 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 165.83 (d, J = 3.4 Hz), 163.30 (d, J = 246.8 Hz), 157.95, 141.81 (d, J = 7.7 Hz), 133.31 (d, J = 8.9 Hz), 124.67 (d, J = 2.7 Hz), 120.79 (d, J = 21.2 Hz), 112.29 (d, J = 24.3 Hz), 21.45 (d, J = 1.8 Hz); MS (ESI) m/z [M + H]+: 191.0.
3-(3-Fluoro-5-methylphenyl)-1,2,4,5-tetrazine (2c)
The compound was obtained from 3-fluoro-6-methylbenzonitrile (0.54 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.12 g (16%) of 2c as a red solid. Rf = 0.37 (heptane/EtOAc 90/10); 1H NMR (600 MHz, chloroform-d) δ 10.23 (s, 1H), 7.72 (dd, J = 8.5, 3.2 Hz, 1H), 7.32–7.27 (m, 1H), 7.08 (dd, J = 9.1, 4.2 Hz, 1H), 3.90 (s, 3H); 13C NMR (151 MHz, chloroform-d) δ 167.77 (d, J = 2.2 Hz), 157.08 (d, J = 240.5 Hz), 156.99, 154.97 (d, J = 2.1 Hz), 122.91 (d, J = 7.8 Hz), 120.08 (d, J = 22.9 Hz), 118.48 (d, J = 25.3 Hz), 113.94 (d, J = 7.9 Hz), 56.96; MS (ESI) m/z [M + H]+: 191.1.
3-(3-Fluoro-4-(trifluoromethyl)phenyl)-1,2,4,5-tetrazine (3a)
The compound was obtained from 3-fluoro-4-trifluoromethylbenzonitrile (0.76 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.21 g (21%) of 3a as a red solid. Rf = 0.41 (80/20 heptane/EtOAc); 1H NMR (400 MHz, chloroform-d) δ 10.36 (s, 1H), 8.65–8.54 (m, 1H), 8.55–8.45 (m, 1H), 7.90 (t, J = 7.6 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 164.80 (d, J = 2.9 Hz), 160.26 (dd, J = 258.1, 2.1 Hz), 158.32, 137.35 (d, J = 8.3 Hz), 130.90–126.53 (m), 128.36 (dd, J = 4.4, 1.8 Hz), 123.75 (d, J = 4.1 Hz), 123.46, 123.12–121.35 (m), 120.76, 116.57 (d, J = 23.5 Hz); MS (ESI) m/z [M + H]+: 245.0.
3-(3-Fluoro-5-(trifluoromethyl)phenyl)-1,2,4,5-tetrazine (3b)
The compound was obtained from 3-fluoro-5-trifluoromethylbenzonitrile (0.76 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.23 g (24%) of 3b as a red solid. Rf = 0.39 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.25 (s, 1H), 8.65 (s, 1H), 8.44 (d, J = 9.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 164.72 (d, J = 3.0 Hz), 163.01 (d, J = 250.8 Hz), 158.33, 134.88 (d, J = 8.3 Hz), 133.95 (qd, J = 34.1, 7.9 Hz), 122.79 (dd, J = 272.9, 3.0 Hz), 121.41–120.21 (m), 118.41 (d, J = 24.2 Hz), 117.20 (dq, J = 24.6, 3.7 Hz).; MS (ESI) m/z [M + H]+: 245.0.
(2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine (4a)
4-((1,3-Dioxoisoindolin-2-yl)methyl)-3-fluorobenzonitrile (18a)
The compound was obtained following the literature procedure with minor modifications.27 2-(Bromomethyl)-3-fluorobenzonitrile (3.0 g, 14.01 mmol) was dissolved in DMF (20 mL). The phthalimide potassium salt (2.89 g, 15.41) was added, and the mixture was stirred for 9 h at 130 °C. After cooling to rt, the mixture was poured on ice. The solid obtained was filtered off. Ethyl acetate (100 mL) and water (100 mL) were added, and the organic layer was separated. The organic phase was washed with water (2 × 50 mL), dried over MgSO4, filtered, and evaporated to give a light brown solid. Crystallization from EtOAc afforded 3.30 g (84%) of the desired compound as a white solid. Rf = 0.24 (heptane/EtOAc 70/30); 1H NMR (400 MHz, chloroform-d) δ 7.92–7.86 (m, 2H), 7.81–7.74 (m, 2H), 7.52–7.33 (m, 3H), 4.98 (s, 2H); 13C NMR (101 MHz, chloroform-d) δ 167.55, 159.99 (d, J = 252.2 Hz), 134.38, 131.80, 130.99 (d, J = 4.3 Hz), 129.06 (d, J = 14.9 Hz), 128.29 (d, J = 4.0 Hz), 123.64, 119.34 (d, J = 24.9 Hz), 117.27 (d, J = 2.9 Hz), 113.24 (d, J = 9.5 Hz), 35.09 (d, J = 4.6 Hz); MS (ESI) m/z [M + H]+: 281.1.
(4-Cyano-2-fluorophenyl)methanaminium Chloride (19a)
The compound was obtained following the literature procedure with minor modifications.27 To a solution of 4-((1,3-dioxoisoindolin-2-yl)methyl)-3-fluorobenzonitrile (3.0 g, 10.70 mmol) in EtOH (5 mL) was added hydrazine hydrate (5 mL). The reaction mixture was then refluxed for 2 h, and a white precipitate was formed. The reaction mixture was diluted with NaOH solution (10%, 40 mL) and extracted with EtOAc (3 × 30 mL). The organic portion was dried over anhydrous Na2SO4, filtered, and the solvent was evaporated to dryness under reduced pressure. The crude was solubilized in Et2O, filtered, and treated with HCl in Et2O (2 mL, 2 M). The solid obtained was filtered and recrystallized from MeOH to give 1.51 g (76%) of the desired compound as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 3H), 7.95 (d, J = 9.9 Hz, 1H), 7.90–7.77 (m, 2H), 4.13 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.13 (d, J = 249.5 Hz), 132.78 (d, J = 3.9 Hz), 129.27 (d, J = 3.9 Hz), 127.73 (d, J = 14.8 Hz), 119.87 (d, J = 25.6 Hz), 117.84 (d, J = 3.0 Hz), 113.38 (d, J = 10.3 Hz), 35.87 (d, J = 4.4 Hz); MS (ESI) m/z [M + H]+: 151.0.
tert-Butyl 4-Cyano-2-fluorobenzylcarbamate (20a)
(4-Cyano-2-fluorophenyl)methanaminium chloride (1.5 g, 8.04 mmol) and triethylamine (2.35 mL, 16.87 mmol) were dissolved in anhydrous CH2Cl2 (40 mL) at 0 ° C. To this stirred solution was added di-tert-butyl dicarbonate (2.10 g, 9.64 mmol), and the reaction mixture was allowed to warm to room temperature and was stirred for 12 h. The reaction mixture was evaporated under reduced pressure, and the residue was redissolved in diethyl ether (50 mL), which was washed successively with 0.5 M aq. HCl (2 × 25 mL), saturated NaHCO3 (2 × 25 mL), and brine (25 mL). The organic layer was dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure to give a white solid. The residue was purified by flash column chromatography (heptane/EtOAc = 85/15) to afford 1.51 g (75%) of the desired compound as an orange solid. Rf = 0.24 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 7.48–7.30 (m, 2H), 7.22 (d, J = 9.4 Hz, 1H), 5.52 (t, J = 6.4 Hz, 1H), 4.29 (d, J = 6.4 Hz, 2H), 1.35 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 159.86 (d, J = 249.8 Hz), 155.96, 132.56 (d, J = 14.5 Hz), 130.10 (d, J = 5.0 Hz), 128.18 (d, J = 3.9 Hz), 118.69 (d, J = 25.0 Hz), 117.44 (d, J = 2.9 Hz), 112.10 (d, J = 9.5 Hz), 38.04, 28.21, MS (ESI) m/z [M + H]+: 251.1.
tert-Butyl 2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (28a)
The compound was obtained from tert-butyl 4-cyano-2-fluorobenzylcarbamate (0.76 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.39 g (21%) of the desired compound as a red solid. Rf = 0.25 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.25 (s, 1H), 8.41 (dd, J = 8.1, 1.6 Hz, 1H), 7.60 (t, J = 7.7 Hz, 1H), 5.11 (s, 1H), 4.49 (d, J = 6.3 Hz, 2H), 1.48 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 165.53 (d, J = 3.0 Hz), 161.21 (d, J = 247.6 Hz), 157.94, 155.82, 132.59 (d, J = 8.5 Hz), 131.64 (d, J = 15.0 Hz), 130.48, 124.13, 114.98 (d, J = 24.5 Hz), 79.97, 38.57, 28.36; MS (ESI) m/z [M + H]+: 306.1
(2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine
To a solution of tert-butyl 2-fluoro-4-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (0.300 g, 0.98 mmol) in CH2Cl2 (20 mL) was added a solution of HCl in dioxane (4.0 M, 10.0 mL). The mixture was stirred at room temperature for 2 h. The reaction mixture was then concentrated under reduced pressure to give 0.23 g (97%) of HCl salt of 4a as a pink solid. 1H NMR (400 MHz, methanol-d4) δ 10.34 (s, 1H), 8.43 (dd, J = 8.0, 1.6 Hz, 1H), 8.33 (dd, J = 10.9, 1.7 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 4.27 (s, 2H); 13C NMR (101 MHz, methanol-d4) δ 165.15, 161.44 (d, J = 248.4 Hz), 158.27, 135.65 (d, J = 8.5 Hz), 131.96 (d, J = 3.5 Hz), 124.80 (d, J = 15.3 Hz), 124.08 (d, J = 3.7 Hz), 114.72 (d, J = 24.5 Hz), 36.45 (d, J = 4.2 Hz); MS (ESI) m/z [M + H]+: 206.1.
(3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine (4b)
5-((1,3-Dioxoisoindolin-2-yl)methyl)-3-fluorobenzonitrile (18b)
The compound was obtained from 3-(bromomethyl)-5-fluorobenzonitrile (1.25 g, 5.84 mmol) following the procedure reported above for 18a. Crystallization from EtOAc afforded 1.62 g (99%) of the desired compound as a white solid. Rf = 0.26 (heptane/EtOAc 70/30); 1H NMR (400 MHz, chloroform-d) δ 7.93–7.87 (m, 2H), 7.81–7.74 (m, 2H), 7.53 (d, J = 1.5 Hz, 1H), 7.41 (dt, J = 9.0, 2.0 Hz, 1H), 7.32–7.25 (m, 1H), 4.87 (s, 2H); 13C NMR (101 MHz, chloroform-d) δ 167.66, 162.27 (d, J = 251.4 Hz), 140.44 (d, J = 7.6 Hz), 134.43, 131.76, 128.05 (d, J = 3.5 Hz), 123.70, 120.67 (d, J = 21.8 Hz), 118.64 (d, J = 24.6 Hz), 117.19 (d, J = 3.3 Hz), 114.23 (d, J = 9.7 Hz), 40.41 (d, J = 1.9 Hz), MS (ESI) m/z [M + H]+: 281.0.
(3-Cyano-5-fluorophenyl)methanaminium Chloride (19b)
The compound was obtained from 5-((1,3-dioxoisoindolin-2-yl)methyl)-3-fluorobenzonitrile (1.6 g, 5.71 mmol) following the procedure reported above 19a. Recrystallization from MeOH/Et2O afforded 0.71 g (67%) of the desired compound as a yellow solid. 1H NMR (400 MHz, methanol-d4) δ 7.76 (t, J = 1.4 Hz, 1H), 7.73–7.62 (m, 2H), 4.26 (s, 2H); 13C NMR (101 MHz, methanol-d4) δ 163.76 (d, J = 249.8 Hz), 138.91 (d, J = 8.0 Hz), 130.12 (d, J = 3.8 Hz), 122.40 (d, J = 22.7 Hz), 120.79 (d, J = 25.2 Hz), 118.01, 115.74 (d, J = 10.1 Hz), 43.01 (d, J = 1.5 Hz), MS (ESI) m/z [M + H]+: 151.1.
tert-Butyl 3-Cyano-5-fluorobenzylcarbamate (20b)
The compound was obtained from (3-cyano-5-fluorophenyl)methanaminium chloride (0.71 g, 3.75 mmol) following the procedure reported above for 20a. Purification by flash chromatography (heptane/EtOAc = 85/15) afforded 0.51 g (54%) of the desired compound as an orange solid. Rf = 0.26 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 7.32 (s, 1H), 7.26–7.10 (m, 2H), 5.57 (t, J = 6.2 Hz, 1H), 4.26 (d, J = 6.2 Hz, 2H), 1.39 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 162.26 (d, J = 250.4 Hz), 156.01, 144.21 (d, J = 7.5 Hz), 126.53 (d, J = 3.2 Hz), 119.09 (d, J = 21.7 Hz), 117.56 (d, J = 27.6 Hz), 117.45, 113.65 (d, J = 9.7 Hz), 79.98, 43.34, 28.23, MS (ESI) m/z [M + H]+: 251.1.
tert-Butyl 3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (28b)
The compound was obtained from tert-butyl 3-cyano-5-fluorobenzylcarbamate (0.40 g, 1.60 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.16 g (33%) of the desired compound as a red solid. Rf = 0.26 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.26 (s, 1H), 8.35 (s, 1H), 8.20 (dt, J = 9.2, 2.0 Hz, 1H), 7.35–7.28 (m, 1H), 5.21 (t, J = 5.7 Hz, 1H), 4.45 (d, J = 5.7 Hz, 2H), 1.46 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 165.56 (d, J = 3.3 Hz), 163.46 (d, J = 248.1 Hz), 158.00, 143.45 (d, J = 5.8 Hz), 133.75 (d, J = 8.6 Hz), 122.49, 118.86 (d, J = 22.2 Hz), 113.91 (d, J = 24.3 Hz), 81.41, 80.04, 43.99, 28.35, MS (ESI) m/z [M + H]+: 306.1.
(3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine
To a solution of tert-butyl 3-fluoro-5-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (0.140 g, 0.46 mmol) in CH2Cl2 (10 mL) was added a solution of HCl in dioxane (4.0 M, 5.0 mL). The mixture was stirred at room temperature for 2 h. The reaction mixture was then concentrated under reduced pressure to 0.07 g (63%) of HCl salt of 4b as a pink solid. 1H NMR (400 MHz, methanol-d4) δ 10.33 (s, 1H), 8.48 (d, J = 1.6 Hz, 1H), 8.26 (dt, J = 9.4, 1.8 Hz, 1H), 7.52 (dt, J = 9.0, 1.9 Hz, 1H), 4.23 (s, 2H); 13C NMR (101 MHz, methanol-d4) δ 165.15 (d, J = 3.3 Hz), 163.32 (d, J = 247.4 Hz), 158.31, 137.02 (d, J = 8.0 Hz), 135.37 (d, J = 8.5 Hz), 124.09 (d, J = 3.1 Hz), 119.87 (d, J = 23.0 Hz), 114.92 (d, J = 24.3 Hz), 42.20; MS (ESI) m/z [M + H]+: 206.1.
(4-Fluoro-2-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine (4c)
2-((1,3-Dioxoisoindolin-2-yl)methyl)-5-fluorobenzonitrile (18c)
The compound was obtained from 2-(bromomethyl)-5-fluorobenzonitrile (2.0 g, 9.34 mmol) following the procedure reported above for 18a. Crystallization from EtOAc afforded 2.60 g (99%) of the desired compound as a white solid. Rf = 0.28 (heptane/EtOAc 70/30); 1H NMR (400 MHz, DMSO-d6) δ 8.01–7.76 (m, 5H), 7.60–7.42 (m, 2H), 4.93 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 172.71, 165.87 (d, J = 246.6 Hz), 141.61 (d, J = 3.4 Hz), 139.82, 136.79, 136.22 (d, J = 8.8 Hz), 128.53, 126.28 (d, J = 21.5 Hz), 125.05 (d, J = 26.3 Hz), 124.94, 121.23 (d, J = 2.8 Hz), 117.23 (d, J = 10.0 Hz), 44.14, MS (ESI) m/z [M + H]+: 281.1.
(2-Cyano-4-fluorophenyl)methanaminium Chloride (19c)
The compound was obtained from 2-((1,3-dioxoisoindolin-2-yl)methyl)-5-fluorobenzonitrile (2.6 g, 9.23 mmol) following the procedure reported above 19a. Recrystallization from MeOH/Et2O afforded 1.41 g (81%) of the desired compound as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 1H), 9.79 (s, 1H), 9.37 (s, 1H), 8.18 (dd, J = 8.6, 2.5 Hz, 1H), 7.82 (dd, J = 8.5, 4.8 Hz, 1H), 7.68 (td, J = 8.9, 2.5 Hz, 1H), 4.80 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 163.10 (d, J = 3.8 Hz), 162.20 (d, J = 244.3 Hz), 140.95, 130.40 (d, J = 10.2 Hz), 126.44 (d, J = 9.0 Hz), 121.63 (d, J = 23.5 Hz), 110.86, 51.40, MS (ESI) m/z [M + H]+: 151.1.
tert-Butyl 2-Cyano-4-fluorobenzylcarbamate (20c)
The compound was obtained from (2-cyano-4-fluorophenyl)methanaminium chloride (1.0 g, 5.36 mmol) following the procedure reported above for 20a. Purification by flash chromatography (heptane/EtOAc = 85/15) afforded 0.85 g (63%) of the desired compound as an orange solid. Rf = 0.29 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 7.65 (d, J = 8.1 Hz, 1H), 7.33 (dd, J = 8.4, 4.6 Hz, 1H), 7.22 (td, J = 8.6, 2.5 Hz, 1H), 4.71 (s, 2H), 1.58 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 162.89 (d, J = 246.9 Hz), 158.04, 152.55, 135.85, 133.99, 124.05 (d, J = 8.5 Hz), 119.59 (d, J = 23.8 Hz), 110.32 (d, J = 23.7 Hz), 83.19, 50.83, 28.24, MS (ESI) m/z [M + H]+: 251.1.
tert-Butyl 4-Fluoro-2-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (28c)
The compound was obtained from tert-butyl 2-cyano-4-fluorobenzylcarbamate (0.55 g, 2.20 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.100 g (15%) of the desired compound as a red solid. Rf = 0.29 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.30 (s, 1H), 8.15–7.89 (m, 1H), 7.70 (d, J = 7.0 Hz, 1H), 7.31 (td, J = 8.1, 2.9 Hz, 1H), 5.56 (s, 1H), 4.49 (d, J = 6.5 Hz, 2H), 1.41 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 167.76, 162.12 (d, J = 247.9 Hz), 157.11, 155.72, 135.75, 133.81, 132.47, 119.54 (d, J = 21.0 Hz), 117.91 (d, J = 23.8 Hz), 79.59, 42.61, 28.39, MS (ESI) m/z [M + H]+: 306.1.
(4-Fluoro-2-(1,2,4,5-tetrazin-3-yl)phenyl)methanamine
To a solution of tert-butyl 4-fluoro-2-(1,2,4,5-tetrazin-3-yl)benzylcarbamate (0.10 g, 0.33 mmol) in CH2Cl2 (10 mL) was added a solution of HCl in dioxane (4.0 M, 5.0 mL). The mixture was stirred at room temperature for 2 h. The reaction mixture was then concentrated under reduced pressure to 0.045 g (57%) of HCl salt of 4c as a pink solid. 1H NMR (400 MHz, methanol-d4) δ 10.42 (s, 1H), 8.15 (dd, J = 9.7, 2.8 Hz, 1H), 7.74 (dd, J = 8.6, 5.4 Hz, 1H), 7.44 (td, J = 8.2, 2.9 Hz, 1H), 4.35 (s, 2H); 13C NMR (101 MHz, methanol-d4) δ 166.62 (d, J = 2.7 Hz), 163.29 (d, J = 249.0 Hz), 157.59, 135.47 (d, J = 8.5 Hz), 134.74 (d, J = 8.4 Hz), 129.06 (d, J = 3.8 Hz), 119.25 (d, J = 21.6 Hz), 117.86 (d, J = 24.9 Hz), 41.22; MS (ESI) m/z [M + H]+: 206.0.
3-(4-Chloro-3-fluorophenyl)-1,2,4,5-tetrazine (5a)
The compound was obtained from 3-fluoro-4-chlorobenzonitrile (0.62 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.18 g (21%) of 5a as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.27 (s, 1H), 8.37 (dd, J = 9.3, 2.0 Hz, 1H), 8.31 (ddd, J = 8.4, 2.0, 0.7 Hz, 1H), 7.80 (dd, J = 8.4, 6.9 Hz, 1H); 13C NMR (101 MHz, Chloroform-d) δ 165.25 (d, J = 2.7 Hz), 159.76 (d, J = 249.0 Hz), 158.03, 134.75, 132.79 (d, J = 7.5 Hz), 124.79 (d, J = 3.7 Hz), 115.93 (d, J = 25.1 Hz), 115.17 (d, J = 21.2 Hz); MS (ESI) m/z [M + H]+: 211.0.
3-(5-Chloro-3-fluorophenyl)-1,2,4,5-tetrazine (5b)
The compound was obtained from 3-fluoro-5-chlorobenzonitrile (0.62 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.22 g (26%) of 5b as a red solid. Rf = 0.38 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.22 (s, 1H), 8.38 (t, J = 1.6 Hz, 1H), 8.17 (ddd, J = 9.0, 2.4, 1.4 Hz, 1H), 7.30 (dt, J = 8.0, 2.2 Hz, 1H); 13C NMR (101 MHz, Chloroform-d) δ 164.88 (d, J = 3.5 Hz), 163.15 (d, J = 251.2 Hz), 158.22, 136.59 (d, J = 10.2 Hz), 134.68 (d, J = 9.2 Hz), 124.27 (d, J = 3.3 Hz), 120.72 (d, J = 24.7 Hz), 113.64 (d, J = 24.2 Hz); MS (ESI) m/z [M + H]+: 211.0.
3-(6-Chloro-3-fluorophenyl)-1,2,4,5-tetrazine (5c)
The compound was obtained from 3-fluoro-5-chlorobenzonitrile (0.62 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.14 g (17%) of 5c as a red solid. Rf = 0.48 (heptane/EtOAc 80/20); 1H NMR (600 MHz, chloroform-d) δ 10.35 (s, 1H), 7.76 (dd, J = 8.4, 3.0 Hz, 1H), 7.62 (dd, J = 8.9, 4.8 Hz, 1H), 7.31 (ddd, J = 9.1, 7.5, 3.1 Hz, 1H); 13C NMR (151 MHz, chloroform-d) δ 167.66 (d, J = 2.3 Hz), 161.24 (d, J = 249.1 Hz), 157.29 (d, J = 2.9 Hz), 132.88 (d, J = 8.1 Hz), 132.74 (d, J = 8.0 Hz), 128.84 (d, J = 3.6 Hz), 119.94 (d, J = 22.5 Hz), 119.11 (d, J = 25.1 Hz); MS (ESI) m/z [M + H]+: 211.0.
3-(4-Bromo-3-fluorophenyl)-1,2,4,5-tetrazine (6a)
The compound was obtained from 3-fluoro-4-bromobenzonitrile (0.80 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.20 g (20%) of 6a as a red solid. Rf = 0.42 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.28 (s, 1H), 8.49–8.37 (m, 2H), 7.67 (dd, J = 8.6, 7.3 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 165.14 (d, J = 3.0 Hz), 158.72 (d, J = 250.5 Hz), 157.99, 131.98 (d, J = 7.3 Hz), 131.78, 126.67 (d, J = 17.7 Hz), 124.53 (d, J = 3.8 Hz), 116.17 (d, J = 23.7 Hz); MS (ESI) m/z [M + H]+: 254.9.
3-(5-Bromo-3-fluorophenyl)-1,2,4,5-tetrazine (6b)
The compound was obtained from 3-fluoro-5-bromobenzonitrile (0.80 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.20 g (20%) of 6b as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.21 (s, 1H), 8.52 (d, J = 1.6 Hz, 1H), 8.21 (ddd, J = 9.1, 2.5, 1.5 Hz, 1H), 7.45 (ddd, J = 7.8, 2.4, 1.8 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 164.74 (d, J = 3.3 Hz), 163.06 (d, J = 252.3 Hz), 158.21, 134.93 (d, J = 8.9 Hz), 127.16 (d, J = 3.3 Hz), 123.88 (d, J = 9.5 Hz), 123.59 (d, J = 24.5 Hz), 114.09 (d, J = 24.0 Hz); MS (ESI) m/z [M + H]+: 254.9.
3-(6-Bromo-3-fluorophenyl)-1,2,4,5-tetrazine (6c)
The compound was obtained from 3-fluoro-6-bromobenzonitrile (0.40 g, 2.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.15 g (29%) of 6c as a red solid. Rf = 0.45 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.34 (s, 1H), 7.78 (dd, J = 8.9, 5.1 Hz, 1H), 7.70 (dd, J = 8.5, 3.1 Hz, 1H), 7.22 (ddd, J = 8.9, 7.6, 3.1 Hz, 1H); 13C NMR (101 MHz, Chloroform-d) δ 168.38, 161.88 (d, J = 249.5 Hz), 157.31, 135.95 (d, J = 7.8 Hz), 134.91 (d, J = 8.0 Hz), 120.06 (d, J = 22.1 Hz), 119.34 (d, J = 24.9 Hz), 116.68 (d, J = 3.6 Hz); MS (ESI) m/z [M + H]+: 254.9.
3-(4-Iodo-3-fluorophenyl)-1,2,4,5-tetrazine (7a)
The compound was obtained from 3-fluoro-4-iodobenzonitrile (0.99 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 Heptane/EtOAc) afforded 0.33 g (27%) of 7a as a red solid. Rf = 0.39 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.19 (s, 1H), 8.19 (dd, J = 8.7, 2.0 Hz, 1H), 8.07 (dd, J = 8.3, 1.9 Hz, 1H), 7.92 (dd, J = 8.3, 6.2 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 164.29 (d, J = 2.9 Hz), 161.40 (d, J = 246.8 Hz), 157.04, 139.61 (d, J = 1.9 Hz), 132.83 (d, J = 7.6 Hz), 123.98 (d, J = 3.6 Hz), 113.81 (d, J = 26.8 Hz), 87.16 (d, J = 25.7 Hz); MS (ESI) m/z [M + H]+: 302.9.
3-(5-Iodo-3-fluorophenyl)-1,2,4,5-tetrazine (7b)
The compound was obtained from 3-fluoro-5-iodobenzonitrile (0.99 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.31 g (26%) of 7b as a red solid. Rf = 0.39 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.21 (s, 1H), 8.72 (d, J = 1.4 Hz, 1H), 8.23 (dt, J = 9.1, 1.9 Hz, 1H), 7.65 (dt, J = 7.6, 1.9 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 164.51 (d, J = 3.2 Hz), 162.68 (d, J = 253.2 Hz), 158.18, 134.97 (d, J = 8.5 Hz), 133.04 (d, J = 3.3 Hz), 129.29 (d, J = 23.7 Hz), 114.77 (d, J = 24.1 Hz), 94.36 (d, J = 8.0 Hz); MS (ESI) m/z [M + H]+: 302.9.
3-(6-Iodo-3-fluorophenyl)-1,2,4,5-tetrazine (7c)
The compound was obtained from 3-fluoro-6-iodobenzonitrile (0.99 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.25 g (21%) of 7c as a red solid. Rf = 0.37 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.26 (s, 1H), 7.99 (dd, J = 8.8, 5.3 Hz, 1H), 7.64 (dd, J = 8.8, 3.0 Hz, 1H), 7.00 (ddd, J = 8.7, 7.8, 3.0 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 169.14 (d, J = 2.5 Hz), 162.94 (d, J = 250.3 Hz), 157.34, 142.62 (d, J = 7.5 Hz), 138.35 (d, J = 7.7 Hz), 120.15 (d, J = 21.6 Hz), 119.01 (d, J = 24.5 Hz), 88.53 (d, J = 3.7 Hz); MS (ESI) m/z [M + H]+: 302.9.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenol (8a)
The compound was obtained from 3-fluoro-4-hydroxybenzonitrile (0.55 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.15 g (19%) of 8a as a red solid. Rf = 0.26 (heptane/EtOAc 80/20); 1H NMR (400 MHz, methanol-d4) δ 10.24 (s, 1H), 8.40–8.12 (m, 2H), 7.14 (t, J = 8.7 Hz, 1H); 13C NMR (101 MHz, methanol-d4) δ 165.50, 157.39, 151.85 (d, J = 241.9 Hz), 149.87 (d, J = 12.9 Hz), 124.85 (d, J = 3.1 Hz), 123.47 (d, J = 6.7 Hz), 118.09 (d, J = 3.1 Hz), 115.20 (d, J = 21.1 Hz); MS (ESI) m/z [M + H]+: 193.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenol (8b)
The compound was obtained from 3-fluoro-5-hydroxybenzonitrile (0.55 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (80/20 Heptane/EtOAc) afforded 0.23 g (30%) of 8b as a red solid. Rf = 0.28 (heptane/EtOAc 80/20); 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 10.50 (s, 1H), 7.79 (t, J = 1.8 Hz, 1H), 7.67 (dt, J = 9.7, 1.9 Hz, 1H), 6.91 (dd, J = 10.6, 2.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.12 (d, J = 3.9 Hz), 163.96 (d, J = 243.1 Hz), 160.36 (d, J = 12.2 Hz), 158.77, 134.88 (d, J = 10.9 Hz), 111.30 (d, J = 2.4 Hz), 107.31 (d, J = 23.8 Hz), 105.34 (d, J = 24.4 Hz); MS (ESI) m/z [M + H]+: 193.0.
4-Fluoro-2-(1,2,4,5-tetrazin-3-yl)phenol (8c)
The compound was obtained from 3-fluoro-6-hydroxybenzonitrile (0.55 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.18 g (23%) of 8c as a red solid. Rf = 0.37 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.89 (s, 1H), 10.28 (s, 1H), 8.33 (dd, J = 9.3, 3.2 Hz, 1H), 7.28 (ddd, J = 9.2, 7.4, 3.2 Hz, 1H), 7.10 (dd, J = 9.2, 4.6 Hz, 1H); 13C NMR (101 MHz, chloroform-d) δ 166.76 (d, J = 3.1 Hz), 157.06, 156.72 (d, J = 1.8 Hz), 156.30 (d, J = 238.9 Hz), 123.33 (d, J = 23.9 Hz), 120.32 (d, J = 7.7 Hz), 114.02 (d, J = 8.3 Hz), 113.87 (d, J = 25.6 Hz); MS (ESI) m/z [M + H]+: 193.0.
3-(3-Fluoro-4-methoxyphenyl)-1,2,4,5-tetrazine (9a)
The compound was obtained from 3-fluoro-4-methoxybenzonitrile (0.60 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.24 g (29%) of 9a as a red solid. Rf = 0.39 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.18 (s, 1H), 8.45 (ddd, J = 8.6, 2.1, 1.3 Hz, 1H), 8.38 (dd, J = 12.1, 2.2 Hz, 1H), 7.18 (t, J = 8.5 Hz, 1H), 4.04 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 165.53, 157.51, 152.72 (d, J = 247.7 Hz), 152.06 (d, J = 10.7 Hz), 125.29 (d, J = 3.6 Hz), 124.35 (d, J = 7.2 Hz), 115.80 (d, J = 20.8 Hz), 113.52 (d, J = 2.2 Hz), 56.36; MS (ESI) m/z [M + H]+: 207.0.
3-(3-Fluoro-5-methoxyphenyl)-1,2,4,5-tetrazine (9b)
The compound was obtained from 3-fluoro-5-methoxybenzonitrile (0.60 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.21 g (26%) of 9b as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.24 (s, 1H), 7.98 (d, J = 2.0 Hz, 1H), 7.94 (ddd, J = 9.1, 2.4, 1.4 Hz, 1H), 6.90 (dd, J = 10.1, 2.4 Hz, 1H), 3.93 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 165.63 (d, J = 3.9 Hz), 164.07 (d, J = 246.5 Hz), 161.75 (d, J = 11.4 Hz), 158.02, 133.96 (d, J = 10.7 Hz), 108.96 (d, J = 2.8 Hz), 107.73 (d, J = 24.7 Hz), 106.98 (d, J = 24.9 Hz), 55.96; MS (ESI) m/z [M + H]+: 207.0.
3-(3-Fluoro-6-methoxyphenyl)-1,2,4,5-tetrazine (9c)
The compound was obtained from 3-fluoro-6-methoxybenzonitrile (0.60 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.13 g (16%) of 9c as a red solid. Rf = 0.18 (heptane/EtOAc 90/10); 1H NMR (600 MHz, chloroform-d) δ 10.23 (s, 1H), 8.26 (s, 1H), 8.12 (d, J = 9.4 Hz, 1H), 7.17 (d, J = 9.1 Hz, 1H), 2.50 (s, 3H); 13C NMR (151 MHz, chloroform-d) δ 165.99 (d, J = 3.4 Hz), 164.27 (d, J = 247.1 Hz), 158.11, 141.97 (d, J = 7.8 Hz), 133.45 (d, J = 8.9 Hz), 124.81, 120.95 (d, J = 21.2 Hz), 112.44 (d, J = 24.2 Hz), 21.60; MS (ESI) m/z [M + H]+: 207.0.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzoic Acid (10a)
The compound was obtained from 4-cyano-2-fluorobenzoic acid (0.66 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.32 g (36%) of 10a as a red solid. Rf = 0.31 (heptane/EtOAc 30/70); 1H NMR (600 MHz, methanol-d4) δ 10.44 (s, 1H), 8.50 (dd, J = 8.1, 1.6 Hz, 1H), 8.40 (dd, J = 11.5, 1.7 Hz, 1H), 8.20 (t, J = 7.7 Hz, 1H); 13C NMR (151 MHz, methanol-d4) δ 165.11 (d, J = 3.3 Hz), 165.04 (d, J = 2.7 Hz), 162.10 (d, J = 259.0 Hz), 158.24 (d, J = 10.2 Hz), 137.97 (d, J = 8.8 Hz), 132.85 (d, J = 1.2 Hz), 123.10 (d, J = 4.0 Hz), 122.84 (d, J = 10.7 Hz), 115.83 (d, J = 25.7 Hz); MS (ESI) m/z [M – H]−: 219.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzoic Acid (10b)
The compound was obtained from 5-cyano-3-fluorobenzoic acid (0.66 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.21 g (24%) of 10b as a red solid. Rf = 0.33 (heptane/EtOAc 30/70); 1H NMR (400 MHz, methanol-d4) δ 10.32 (s, 1H), 8.95 (t, J = 1.5 Hz, 1H), 8.42 (ddd, J = 9.2, 2.6, 1.5 Hz, 1H), 7.90 (ddd, J = 8.8, 2.7, 1.4 Hz, 1H); 13C NMR (101 MHz, methanol-d4) δ 166.04, 165.10, 163.08 (d, J = 247.1 Hz), 158.29, 134.91 (d, J = 8.2 Hz), 134.49 (d, J = 7.3 Hz), 124.63 (d, J = 3.1 Hz), 119.87 (d, J = 23.2 Hz), 118.29 (d, J = 24.7 Hz); MS (ESI) m/z [M – H]−: 219.0.
Methyl 2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzoate (11a)
Compound 10a (0.20 g, 0.90 mmol) was solubilized in MeOH (30 mL), and then a solution of HCl in dioxane (4 M, 2.0 mL) was added. The reaction mixture was stirred for 3 h, and then the solvent was removed under reduced pressure. The compound was purified by flash chromatography (90/10 Heptane/EtoAc) to give 0.14 g (66%) of 11a as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.32 (s, 1H), 8.50 (dd, J = 8.3, 1.6 Hz, 1H), 8.44 (dd, J = 11.3, 1.6 Hz, 1H), 8.18 (dd, J = 8.2, 7.2 Hz, 1H), 4.01 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 165.07 (d, J = 2.8 Hz), 164.10 (d, J = 3.8 Hz), 162.14 (d, J = 261.3 Hz), 158.15, 137.32 (d, J = 8.9 Hz), 133.17 (d, J = 1.3 Hz), 123.47 (d, J = 4.1 Hz), 122.51, 116.72 (d, J = 25.6 Hz), 52.73; MS (ESI) m/z [M + H]+: 235.0.
Methyl 3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzoate (11b)
Compound 10b (0.20 g, 0.90 mmol) was solubilized in MeOH (30 mL) and then a solution of HCl in dioxane (4 M, 2.0 mL) was added. The reaction mixture was stirred for 3 h, and then the solvent was removed under reduced pressure. The compound was purified by flash chromatography (90/10 Heptane/EtoAc) to give 0.18 g (85%) of 11b as a red solid. Rf = 0.43 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.31 (s, 1H), 9.13 (t, J = 1.5 Hz, 1H), 8.54 (ddd, J = 9.0, 2.6, 1.5 Hz, 1H), 8.02 (ddd, J = 8.5, 2.6, 1.4 Hz, 1H), 4.00 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 165.17 (d, J = 3.2 Hz), 165.04 (d, J = 3.1 Hz), 163.11 (d, J = 249.1 Hz), 158.22, 134.11 (d, J = 8.1 Hz), 133.72 (d, J = 7.5 Hz), 125.12 (d, J = 3.1 Hz), 121.01 (d, J = 23.3 Hz), 119.27 (d, J = 24.2 Hz), 52.78; MS (ESI) m/z [M + H]+: 235.0.
tert-Butyl 2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzoate (12a)
tert-Butyl 4-Cyano-2-fluorobenzoate (21a)
The compound was obtained following the literature procedure.28 4-Cyano-2-fluorobenzoic acid (1.09 g, 6.54 mmol) was dissolved in t-BuOH (9 mL) and THF (3 mL). Boc anhydride (2.90 g, 13.27 mmol) was added, followed by DMAP (0.24 g, 1.99 mmol). The mixture was stirred at rt under N2 for 12 h. The solvents were removed. The residue was dissolved in EtOAc (30 mL) and washed with saturated aqueous NaHCO3 (2 × 30 mL) and brine (2 × 30 mL). It was dried over anhydrous MgSO4 and concentrated under reduced pressure to give 1.25 g (85%) of the desired compound as a white solid. Rf = 0.30 (heptane/EtOAc 80/20); 1H NMR (600 MHz, chloroform-d) δ 7.98 (dd, J = 8.0, 7.1 Hz, 1H), 7.50 (dd, J = 8.1, 1.5 Hz, 1H), 7.43 (dd, J = 9.7, 1.5 Hz, 1H), 1.62 (s, 9H); 13C NMR (151 MHz, chloroform-d) δ 161.93 (d, J = 3.8 Hz), 161.02 (d, J = 262.6 Hz), 132.88 (d, J = 1.9 Hz), 127.55 (d, J = 4.6 Hz), 125.14 (d, J = 10.5 Hz), 120.76 (d, J = 26.3 Hz), 116.85 (d, J = 9.5 Hz), 116.79 (d, J = 2.7 Hz), 83.36, 28.09; MS (ESI) m/z [M + H]+: 222.1.
tert-Butyl 2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzoate
The compound was obtained from tert-butyl 4-cyano-2-fluorobenzoate (1.19 g, 5.38 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.21 g (14%) of 12a as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.29 (s, 1H), 8.46 (dd, J = 8.2, 1.6 Hz, 1H), 8.39 (dd, J = 11.3, 1.6 Hz, 1H), 8.08 (dd, J = 8.1, 7.2 Hz, 1H), 1.64 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 165.19 (d, J = 2.7 Hz), 162.75 (d, J = 3.7 Hz), 162.04 (d, J = 260.2 Hz), 158.10, 136.64 (d, J = 8.8 Hz), 132.93, 124.65 (d, J = 10.5 Hz), 123.34 (d, J = 4.0 Hz), 116.65 (d, J = 25.8 Hz), 82.84, 28.19; MS (ESI) m/z [M + H]+: 291.1.
tert-Butyl 3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzoate (12b)
tert-Butyl 5-Cyano-3-fluorobenzoate (21b)
The compound was obtained following the literature procedure.28 5-Cyano-3-fluorobenzoic acid (1.09 g, 6.54 mmol) was dissolved in t-BuOH (9 mL) and THF (3 mL). Boc anhydride (2.90 g, 13.27 mmol) was added, followed by DMAP (0.24 g, 1.99 mmol). The mixture was stirred at rt under N2 for 12 h. The solvents were removed. The residue was dissolved in EtOAc (30 mL) and washed with saturated aqueous NaHCO3 (2 × 30 mL) and brine (2 × 30 mL). It was dried over anhydrous MgSO4 and concentrated under reduced pressure to give 1.42 g (97%) of the desired compound as a white solid. Rf = 0.32 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 8.05 (dt, J = 4.2, 1.4 Hz, 1H), 7.93–7.85 (m, 1H), 7.54–7.45 (m, 1H), 1.59 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 162.40 (d, J = 3.0 Hz), 162.07 (d, J = 251.6 Hz), 135.79 (d, J = 7.3 Hz), 129.08 (d, J = 3.5 Hz), 122.49 (d, J = 25.1 Hz), 121.18 (d, J = 22.8 Hz), 116.86 (d, J = 3.0 Hz), 114.01 (d, J = 9.1 Hz), 83.06, 28.01; MS (ESI) m/z [M + H]+: 222.1.
tert-Butyl 3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzoate
The compound was obtained from tert-butyl 5-cyano-3-fluorobenzoate (1.22 g, 5.51 mmol) following the procedure employed for 2a. Purification by flash chromatography (95/5 heptane/EtOAc) afforded 0.25 g (16%) of 12b as a red solid. Rf = 0.37 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 10.32 (s, 1H), 9.04 (t, J = 1.5 Hz, 1H), 8.50 (ddd, J = 9.0, 2.7, 1.6 Hz, 1H), 7.97 (ddd, J = 8.6, 2.7, 1.4 Hz, 1H), 1.66 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 165.31 (d, J = 3.1 Hz), 163.58 (d, J = 2.9 Hz), 163.05 (d, J = 248.6 Hz), 158.15, 135.70 (d, J = 7.3 Hz), 133.84 (d, J = 8.0 Hz), 124.92 (d, J = 3.1 Hz), 120.87 (d, J = 23.1 Hz), 118.74 (d, J = 24.4 Hz), 82.51, 28.14; MS (ESI) m/z [M + H]+: 291.1.
tert-Butyl 2-Cyano-5-fluorobenzoate (21c)
The compound was obtained following the literature procedure.28 2-Cyano-4-fluorobenzoic acid (1.09 g, 6.54 mmol) was dissolved in t-BuOH (9 mL) and THF (3 mL). Boc anhydride (2.90 g, 13.27 mmol) was added, followed by DMAP (0.24 g, 1.99 mmol). The mixture was stirred at rt under N2 for 12 h. The solvents were removed. The residue was dissolved in EtOAc (30 mL) and washed with saturated aqueous NaHCO3 (2 × 30 mL) and brine (2 × 30 mL). It was dried over anhydrous MgSO4 and concentrated under reduced pressure to give 1.10 g (75%) of the desired compound as a white solid. Rf = 0.354 (heptane/EtOAc 80/20); 1H NMR (400 MHz, chloroform-d) δ 8.04 (dd, J = 8.8, 5.5 Hz, 1H), 7.37 (dd, J = 8.1, 2.7 Hz, 1H), 7.28 (ddd, J = 8.8, 7.7, 2.6 Hz, 1H), 1.55 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 163.95 (d, J = 256.4 Hz), 162.13, 130.53 (d, J = 3.6 Hz), 121.50 (d, J = 25.1 Hz), 119.78 (d, J = 21.2 Hz), 116.46 (d, J = 2.6 Hz), 114.76 (d, J = 9.9 Hz), 83.90; MS (ESI) m/z [M + H]+: 222.1.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzamide (13a)
4-Cyano-2-fluorobenzamide (22a)
To a solution of 4-cyano-2-fluorobenzoic acid (0.99 g, 6.0 mmol) in acetonitrile (20 mL) was added 1,1′-carbonyldiimidazole (1.46 g, 9.0 mmol). The mixture was stirred at room temperature for 45 min before the addition of aqueous ammonium hydroxide solution (35%, 20 mL). The reaction mixture was stirred for 45 min, and ice-cold water (20 mL) was added. The precipitate was collected by filtration and dried to give 0.78 g (79%) of the desired compound as a white solid. Rf = 0.28 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 8.13–7.92 (m, 2H), 7.86 (s, 0H), 7.84–7.68 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.56, 158.89 (d, J = 251.4 Hz), 131.56 (d, J = 4.0 Hz), 129.60 (d, J = 15.7 Hz), 129.16 (d, J = 4.0 Hz), 120.79 (d, J = 26.7 Hz), 117.68 (d, J = 2.8 Hz), 114.58 (d, J = 10.0 Hz); MS (ESI) m/z [M + H]+: 165.0.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzamide
The compound was obtained from 4-cyano-2-fluorobenzamide (0.78 g, 4.75 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.21 g (20%) of 13a as a red solid. Rf = 0.32 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.39 (dd, J = 8.0, 1.6 Hz, 1H), 8.30 (dd, J = 11.1, 1.6 Hz, 1H), 7.97 (s, 1H), 7.93 (t, J = 7.7 Hz, 1H), 7.83 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.07, 164.84 (d, J = 2.9 Hz), 159.91 (d, J = 250.1 Hz), 158.83, 136.15 (d, J = 8.5 Hz), 131.81 (d, J = 3.4 Hz), 128.25 (d, J = 15.2 Hz), 124.16 (d, J = 3.4 Hz), 115.57 (d, J = 25.4 Hz); MS (ESI) m/z [M + H]+: 220.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzamide (13b)
5-Cyano-3-fluorobenzamide (22b)
The compound was obtained from 5-cyano-3-fluorobenzoic acid (0.99 g, 6.0 mmol) as reported above for 22a to give 0.77 g (78%) of the desired compound as a white solid. Rf = 0.30 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 8.21 (s, 1H), 8.16 (t, J = 1.5 Hz, 1H), 8.05 (ddd, J = 8.4, 2.6, 1.3 Hz, 1H), 8.01 (ddd, J = 9.6, 2.5, 1.4 Hz, 1H), 7.78 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 165.12 (d, J = 2.4 Hz), 162.04 (d, J = 247.5 Hz), 138.39 (d, J = 7.3 Hz), 128.11 (d, J = 3.1 Hz), 122.37 (d, J = 25.7 Hz), 120.16 (d, J = 22.9 Hz), 117.69 (d, J = 3.1 Hz), 113.52 (d, J = 9.9 Hz); MS (ESI) m/z [M + H]+: 165.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzamide
The compound was obtained from 5-cyano-3-fluorobenzamide (0.75 g, 4.57 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.36 g (36%) of 13b as a red solid. Rf = 0.31 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 8.88 (s, 1H), 8.48–8.20 (m, 2H), 8.16–7.92 (m, 1H), 7.71 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 166.08 (d, J = 2.3 Hz), 164.94 (d, J = 3.3 Hz), 162.85 (d, J = 245.6 Hz), 158.89, 138.32 (d, J = 6.9 Hz), 134.93 (d, J = 8.2 Hz), 123.56 (d, J = 2.9 Hz), 118.85 (d, J = 23.0 Hz), 117.31 (d, J = 24.1 Hz); MS (ESI) m/z [M + H]+: 220.0.
2-Cyano-4-fluorobenzamide (22c)
To a solution of 2-cyano-4-fluorobenzoic acid (0.99 g, 6.0 mmol) in acetonitrile (20 mL) was added 1,1′-carbonyldiimidazole (1.46 g, 9.0 mmol). The mixture was stirred at room temperature for 45 min before the addition of aqueous ammonium hydroxide solution (35%, 20 mL). The reaction mixture was stirred for 45 min, and ice-cold water (20 mL) was added. The precipitate was collected by filtration and dried to give 0.71 g (71%) of the desired compound as a white solid. Rf = 0.18 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 9.67 (s, 1H), 8.79–7.87 (m, 2H), 7.50 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 172.27 (d, J = 20.8 Hz), 165.44 (d, J = 249.8 Hz), 163.41 (d, J = 9.7 Hz), 136.63, 129.38, 125.29 (d, J = 9.8 Hz), 119.66 (d, J = 23.8 Hz), 110.01 (d, J = 25.5 Hz); MS (ESI) m/z [M + H]+: 165.0.
2-Fluoro-N-methyl-4-(1,2,4,5-tetrazin-3-yl)benzamide (14a)
4-Cyano-2-fluoro-N-methylbenzamide (23a)
The compound was obtained from 4-cyano-2-fluorobenzoic acid (0.99 g, 6.0 mmol) and aqueous methylamine solution (40%, 20 ml) as reported above for 22a to give 0.86 g (80%) of the desired compound as a white solid. Rf = 0.29 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 8.50 (d, J = 6.3 Hz, 1H), 8.02–7.92 (m, 1H), 7.76 (d, J = 5.9 Hz, 2H), 2.79 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.25, 158.82 (d, J = 251.2 Hz), 131.56 (d, J = 3.8 Hz), 129.54 (d, J = 15.6 Hz), 129.23 (d, J = 3.9 Hz), 120.78 (d, J = 26.6 Hz), 117.67 (d, J = 2.9 Hz), 114.55 (d, J = 10.1 Hz), 26.69; MS (ESI) m/z [M + H]+: 179.0.
2-Fluoro-N-methyl-4-(1,2,4,5-tetrazin-3-yl)benzamide
The compound was obtained from 4-cyano-2-fluoro-N-methylbenzamide (0.77 g, 4.32 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.36 g (36%) of 14a as a red solid. Rf = 0.36 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 10.68 (s, 1H), 8.50 (d, J = 5.3 Hz, 1H), 8.40 (dd, J = 8.1, 1.6 Hz, 1H), 8.30 (dd, J = 11.1, 1.6 Hz, 1H), 7.91 (t, J = 7.7 Hz, 1H), 2.83 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.83 (d, J = 2.9 Hz), 163.79, 159.81 (d, J = 249.8 Hz), 158.82, 136.08 (d, J = 8.5 Hz), 131.76 (d, J = 3.5 Hz), 128.25 (d, J = 15.4 Hz), 124.23 (d, J = 3.3 Hz), 115.57 (d, J = 25.4 Hz), 26.75; MS (ESI) m/z [M + H]+: 234.0.
3-Fluoro-N-methyl-5-(1,2,4,5-tetrazin-3-yl)benzamide (14b)
5-Cyano-3-fluoro-N-methylbenzamide (23b)
The compound was obtained from 5-cyano-3-fluorobenzoic acid (0.99 g, 6.0 mmol) and aqueous methylamine solution (40%, 20 mL) as reported above for 22a to 0.81 g (76%) of the desired compound as a white solid. Rf = 0.32 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 5.8 Hz, 1H), 8.11 (t, J = 1.5 Hz, 1H), 8.05 (ddd, J = 8.1, 2.7, 1.4 Hz, 1H), 7.97 (ddd, J = 9.5, 2.7, 1.5 Hz, 1H), 2.81 (d, J = 4.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.96 (d, J = 2.5 Hz), 162.02 (d, J = 247.5 Hz), 138.49 (d, J = 7.4 Hz), 127.76 (d, J = 3.3 Hz), 122.17 (d, J = 25.6 Hz), 119.85 (d, J = 23.1 Hz), 117.66 (d, J = 3.4 Hz), 113.55 (d, J = 10.0 Hz), 26.83; MS (ESI) m/z [M + H]+: 179.0.
3-Fluoro-N-methyl-5-(1,2,4,5-tetrazin-3-yl)benzamide
The compound was obtained from 5-cyano-3-fluoro-N-methylbenzamide (0.62 g, 3.48 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.28 g (34%) of 14b as a red solid. Rf = 0.31 (heptane/EtOAc 30/70); 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H), 8.87 (s, 2H), 8.39 (dd, J = 9.3, 1.9 Hz, 1H), 7.99 (dt, J = 9.5, 2.0 Hz, 1H), 2.85 (d, J = 4.5 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.88 (d, J = 11.1 Hz), 162.89 (d, J = 245.8 Hz), 158.92, 138.41 (d, J = 7.5 Hz), 135.00 (d, J = 8.2 Hz), 134.22–131.41 (m), 123.08, 118.56 (d, J = 22.8 Hz), 117.16 (d, J = 24.0 Hz), 26.89; MS (ESI) m/z [M + H]+: 234.1.
2-Cyano-4-fluoro-N-methylbenzamide (23c)
The compound was obtained from 2-cyano-4-fluorobenzoic acid (0.99 g, 6.0 mmol) and aqueous methylamine solution (40%, 20 mL) as reported above for 22a to give 0.74 g (69%) of the desired compound as a white solid. Rf = 0.24 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 0H), 9.45 (s, 0H), 8.03 (dd, J = 8.4, 2.3 Hz, 0H), 7.82 (dd, J = 8.3, 4.8 Hz, 0H), 7.72–7.54 (m, 0H), 7.72–7.16 (m, 0H), 3.12 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 166.97, 165.59 (d, J = 250.2 Hz), 158.55, 134.52 (d, J = 10.3 Hz), 127.39 (d, J = 2.5 Hz), 125.39 (d, J = 9.7 Hz), 119.64 (d, J = 23.9 Hz), 110.51 (d, J = 25.8 Hz), 24.92; MS (ESI) m/z [M + H]+: 179.0.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)aniline (15a)
tert-Butyl (4-Cyano-2-fluorophenyl)carbamate (24a)
4-Amino-3-fluorobenzonitrile (1.0 g, 7.34 mmol) was heated at reflux with Boc2O (4.81 g, 22.04 mmol) and DMAP (0.09 g, 0.73 mmol) in THF (50 mL) overnight. The reaction mixture was evaporated to dryness in vacuo, and the residue was dissolved in dichloromethane (50 mL). TFA (1.6 mL) was then added. The mixture was stirred at room temperature for 3 h. The mixture was made basic using concentrated aqueous ammonia and then extracted with water (3 × 30 mL). The organic portion was dried over anhydrous Na2SO4, and the solvents were evaporated to dryness under reduced pressure. The crude was purified by flash chromatography (90/10 heptane/EtOAc) to give 1.21 g (70%) of the selected compound as a white solid. Rf = 0.31 (90/10 heptane/EtOAc); 1H NMR (400 MHz, DMSO-d6) δ 9.54 (s, 1H), 7.99 (t, J = 8.3 Hz, 1H), 7.82 (dd, J = 10.9, 1.9 Hz, 1H), 7.67–7.56 (m, 1H), 1.48 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 152.87, 152.36 (d, J = 247.9 Hz), 132.59 (d, J = 11.0 Hz), 129.70 (d, J = 3.5 Hz), 122.96 (d, J = 2.7 Hz), 119.82 (d, J = 23.3 Hz), 118.43 (d, J = 2.6 Hz), 105.64 (d, J = 9.3 Hz), 80.91, 28.38; MS (ESI) m/z [M + H]+: 237.1.
tert-Butyl (2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)carbamate (29a)
The compound was obtained from tert-butyl (4-cyano-2-fluorophenyl)carbamate (1.15 g, 4.87 mmol) following the procedure employed for 2a. Purification by flash chromatography (90/10 heptane/EtOAc) afforded 0.31 g (22%) of the desired compound as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (600 MHz, chloroform-d) δ 10.19 (s, 1H), 8.45 (d, J = 1.8 Hz, 2H), 8.35 (dd, J = 12.1, 1.8 Hz, 1H), 7.02 (d, J = 3.9 Hz, 1H), 1.58 (s, 9H); 13C NMR (151 MHz, chloroform-d) δ 165.44 (d, J = 3.3 Hz), 157.57 (d, J = 2.8 Hz), 151.84 (d, J = 243.2 Hz), 151.80, 131.86 (d, J = 9.9 Hz), 125.74 (d, J = 7.9 Hz), 125.20 (d, J = 2.9 Hz), 119.67, 114.38 (d, J = 22.0 Hz), 81.96, 28.23; MS (ESI) m/z [M + H]+: 292.1.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)aniline
To a solution of tert-butyl (2-fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)carbamate (0.20 g, 0.69 mmol) in CH2Cl2 (4 mL) was added TFA (4 mL). The reaction mixture was stirred at room temperature for 1 h. The solvent was then evaporated under reduced pressure to give 0.14 g of crude. Purification by flash chromatography (70/30 heptane/EtOAc) afforded 0.12 g (91%) of 15a as a red solid. Rf = 0.33 (70/30 heptane/EtOAc); 1H NMR (400 MHz, chloroform-d) δ 10.10 (s, 1H), 8.78–8.05 (m, 2H), 6.94 (t, J = 8.6 Hz, 1H), 4.17 (s, 2H); 13C NMR (101 MHz, chloroform-d) δ 165.77 (d, J = 3.1 Hz), 157.12, 151.28 (d, J = 239.9 Hz), 139.71 (d, J = 12.9 Hz), 125.55 (d, J = 2.9 Hz), 121.33 (d, J = 7.3 Hz), 116.39 (d, J = 4.0 Hz), 115.12 (d, J = 20.9 Hz); MS (ESI) m/z [M + H]+: 192.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)aniline (15b)
tert-Butyl (5-Cyano-3-fluorophenyl)carbamate (24b)
The compound was obtained from 3-amino-5-fluorobenzonitrile (1.0 g, 7.34 mmol) as described above for 24a. The crude was purified by flash chromatography (90/10 heptane/EtOAc) to give 1.23 g (71%) of the desired compound as a white solid. Rf = 0.32 (90/10 heptane/EtOAc); 1H NMR (400 MHz, chloroform-d) δ 7.37–7.29 (m, 2H), 7.19 (dt, J = 8.9, 2.2 Hz, 1H), 1.46 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 162.00 (d, J = 251.2 Hz), 150.67, 141.84 (d, J = 10.5 Hz), 128.07 (d, J = 3.6 Hz), 120.90 (d, J = 22.6 Hz), 118.12 (d, J = 24.6 Hz), 116.85 (d, J = 3.5 Hz), 113.74 (d, J = 10.8 Hz), 84.12, 27.86; MS (ESI) m/z [M + H]+: 237.1.
tert-Butyl (3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)carbamate (29b)
The compound was obtained from tert-butyl (5-cyano-3-fluorophenyl)carbamate (0.94 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (90/10 heptane/EtOAc) afforded 0.33 g (28%) of the desired compound as a red solid. Rf = 0.36 (heptane/EtOAc 80/20); 1H NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 9.95 (s, 1H), 8.54 (t, J = 1.7 Hz, 1H), 7.82 (ddd, J = 9.4, 2.5, 1.4 Hz, 1H), 7.64 (dt, J = 11.2, 2.3 Hz, 1H), 1.52 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 165.12 (d, J = 3.8 Hz), 163.18 (d, J = 241.7 Hz), 158.81, 153.08, 142.96 (d, J = 11.6 Hz), 134.64 (d, J = 10.1 Hz), 113.57 (d, J = 2.6 Hz), 108.96 (d, J = 26.6 Hz), 107.88 (d, J = 24.3 Hz), 80.49, 28.50; MS (ESI) m/z [M + H]+: 292.1.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)aniline
To a solution of tert-butyl (3-fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)carbamate (0.15 g, 0.51 mmol) in CH2Cl2 (4 mL) was added TFA (4 mL). The reaction mixture was stirred at room temperature for 1 h. The solvent was then evaporated under reduced pressure to give 0.11 g of crude. Purification by flash chromatography (70/30 heptane/EtOAc) afforded 0.45 g (46%) of 15b as a red solid. Rf = 0.31 (70/30 heptane/EtOAc); 1H NMR (400 MHz, methanol-d4) δ 10.31 (s, 1H), 7.72 (t, J = 1.7 Hz, 1H), 7.48 (dt, J = 9.6, 1.9 Hz, 1H), 6.66 (dt, J = 10.9, 2.3 Hz, 1H); 13C NMR (101 MHz, methanol-d4) δ 165.87 (d, J = 3.8 Hz), 164.41 (d, J = 241.6 Hz), 157.91, 151.23 (d, J = 11.5 Hz), 134.13 (d, J = 10.8 Hz), 109.48 (d, J = 2.0 Hz), 104.62 (d, J = 25.0 Hz), 102.28 (d, J = 25.2 Hz); MS (ESI) m/z [M + H]+: 192.0.
tert-Butyl (2-Cyano-4-fluorophenyl)carbamate (24c)
The compound was obtained from 2-amino-5-fluorobenzonitrile (1.0 g, 7.34 mmol) as described above for 24a. The crude was purified by flash chromatography (90/10 heptane/EtOAc) to give 0.81 g (47%) of the desired compound as a white solid. Rf = 0.41 (90/10 heptane/EtOAc); 1H NMR (400 MHz, chloroform-d) δ 8.12 (dd, J = 9.4, 4.8 Hz, 1H), 7.26–7.11 (m, 2H), 6.90 (s, 1H), 1.46 (s, 9H); 13C NMR (101 MHz, chloroform-d) δ 157.15 (d, J = 245.5 Hz), 151.94, 137.95 (d, J = 2.9 Hz), 121.67 (d, J = 22.1 Hz), 121.54 (d, J = 7.8 Hz), 118.26 (d, J = 25.5 Hz), 115.37 (d, J = 2.7 Hz), 101.79 (d, J = 9.1 Hz), 82.09, 28.16; MS (ESI) m/z [M + H]+: 237.1.
N-(2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)acetamide (16a)
N-(4-Cyano-2-fluorophenyl)acetamide (25a)
To a solution of 4-amino-3-fluorobenzonitrile (0.82 g, 6.00 mmol) in CH2Cl2 (30.0 mL) was added acetic anhydride (0.80 mL, 8.40 mmol). The mixture was stirred at room temperature for 12 h. The suspension was filtered, and the solvent was removed under reduced pressure. Purification by flash chromatography (70/30 heptane/EtOAc) afforded 0.90 g (85%) of the desired compound as a white solid. Rf = 0.5 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 8.28 (t, J = 8.2 Hz, 1H), 7.88 (dd, J = 11.1, 1.9 Hz, 1H), 7.65 (dt, J = 8.5, 1.3 Hz, 1H), 2.15 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.94, 152.05 (d, J = 247.2 Hz), 132.10 (d, J = 11.2 Hz), 129.82 (d, J = 3.6 Hz), 123.31 (d, J = 2.9 Hz), 119.77 (d, J = 23.4 Hz), 118.36 (d, J = 2.7 Hz), 106.18 (d, J = 9.4 Hz), 24.31; MS (ESI) m/z [M + H]+: 179.0.
N-(2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)phenyl)acetamide
The compound was obtained from tert-butyl N-(4-cyano-2-fluorophenyl)acetamide (0.71 g, 4.00 mmol) following the procedure employed for 2a. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.37 g (40%) of 16a as a red solid. Rf = 0.25 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 10.09 (s, 1H), 8.45–8.21 (m, 3H), 2.18 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.79, 164.91 (d, J = 3.0 Hz), 158.44, 153.21 (d, J = 246.0 Hz), 131.38 (d, J = 11.3 Hz), 128.01 (d, J = 7.9 Hz), 124.72 (d, J = 3.3 Hz), 123.67, 114.75 (d, J = 22.1 Hz), 24.32; MS (ESI) m/z [M + H]+: 234.1.
N-(3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)acetamide (16b)
N-(3-Cyano-5-fluorophenyl)acetamide (25b)
The compound was obtained from 3-amino-5-fluorobenzonitrile (0.82 g, 6.00 mmol) as described above for 25a. The crude was purified by flash chromatography (70/30 heptane/EtOAc) to give 0.92 g (86%) of the desired compound as a white solid. Rf = 0.31 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.86–7.70 (m, 2H), 7.57–7.37 (m, 1H), 2.09 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.69, 162.24 (d, J = 244.3 Hz), 142.35 (d, J = 11.8 Hz), 118.65, 118.09 (d, J = 3.6 Hz), 113.70 (d, J = 25.5 Hz), 113.25 (d, J = 12.1 Hz), 110.95 (d, J = 26.2 Hz), 24.52; MS (ESI) m/z [M + H]+: 179.0.
N-(3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)phenyl)acetamide
The compound was obtained from tert-butyl N-(3-cyano-5-fluorophenyl)acetamide (0.58 g, 3.25 mmol) following the procedure employed for 2a. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.19 g (25%) of 16b as a red solid. Rf = 0.24 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 10.48 (s, 1H), 8.52 (t, J = 1.7 Hz, 1H), 7.98–7.81 (m, 2H), 2.12 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.54, 165.03 (d, J = 3.8 Hz), 163.08 (d, J = 242.2 Hz), 158.83, 142.45 (d, J = 11.5 Hz), 134.61 (d, J = 10.1 Hz), 114.40 (d, J = 2.6 Hz), 109.87 (d, J = 26.6 Hz), 108.74 (d, J = 24.4 Hz), 24.58; MS (ESI) m/z [M + H]+: 234.1.
N-(2-Cyano-4-fluorophenyl)acetamide (25c)
The compound was obtained from 2-amino-5-fluorobenzonitrile (0.82 g, 6.00 mmol) as described above for N-(4-cyano-2-fluorophenyl)acetamide. The crude was purified by flash chromatography (70/30 heptane/EtOAc) to give 0.81 g (76%) of the desired compound as a white solid. Rf = 0.41 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.16 (s, 1H), 7.81 (dd, J = 8.4, 1.8 Hz, 1H), 7.58 (dd, J = 6.7, 1.7 Hz, 2H), 2.09 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 169.22, 158.86 (d, J = 244.3 Hz), 137.55 (d, J = 3.0 Hz), 128.41 (d, J = 8.7 Hz), 121.74 (d, J = 22.4 Hz), 120.01 (d, J = 26.0 Hz), 116.21 (d, J = 2.7 Hz), 109.34, 23.49; MS (ESI) m/z [M + H]+: 179.0.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzenesulfonamide (17a)
4-Cyano-2-fluorobenzene-1-sulfonyl Chloride (26a)
The compound was synthesized as described in the literature.29 A portion of glacial acetic acid (30 mL) was saturated for 30 min with gaseous sulfur dioxide. To the resulting solution, cooled on an ice bath, was added under stirring an aqueous solution of CuCl2 (1.5 g in 10 mL) (suspension A). 4-amino-3-fluorobenzonitrile (5 g, 36.75 mmol) was dissolved in a mixture of glacial acetic acid (30 mL) and concentrated HCl (15 mL). To the resulting solution, cooled in an ice–salt bath (−5 °C), was added dropwise under stirring an aqueous solution of NaNO2 (3.37 g in 10 mL, 48.88 mmol). At the end of the addition, the resulting solution was slowly mixed with suspension A. After being stirred for 15 min, the suspension was poured onto ice. The resulting precipitate was collected by filtration and washed with water to give 3.09 g (40%) of the desired compound as a white solid. 1H NMR (400 MHz, chloroform-d) δ 8.15 (dd, J = 8.2, 6.8 Hz, 1H), 7.90–7.22 (m, 1H); 13C NMR (101 MHz, chloroform-d) δ 158.25 (d, J = 266.7 Hz), 135.53 (d, J = 13.0 Hz), 130.36, 128.61 (d, J = 4.7 Hz), 121.95 (d, J = 23.9 Hz), 120.92 (d, J = 9.4 Hz), 115.43 (d, J = 2.6 Hz).
4-Cyano-2-fluorobenzenesulfonamide (27a)
To a solution of 4-cyano-2-fluorobenzene-1-sulfonyl chloride (0.60 g, 2.73 mmol) in CH3CN at 0 °C was added dropwise a solution of NH3 in MeOH (5.00 mL, 7 M). The reaction mixture was stirred at rt for 2 h, and then the solvent was removed under reduced pressure. The residue was solubilized in EtOAc (30 mL) and washed with water (2 × 30 mL). The organic phase was dried over Na2SO3, filtered, and concentrated under reduced pressure to give 0.51 g (93%) of the desired compound as a white solid. Rf: 0.38 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 8.12 (dd, J = 10.0, 1.5 Hz, 1H), 8.03–7.92 (m, 3H), 7.89 (dd, J = 8.1, 1.5 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 158.01 (d, J = 255.5 Hz), 136.37 (d, J = 14.7 Hz), 129.94, 129.64 (d, J = 4.3 Hz), 121.76 (d, J = 25.5 Hz), 117.21 (d, J = 2.6 Hz), 116.77 (d, J = 9.8 Hz); MS (ESI) m/z [M + H]+: 201.0.
2-Fluoro-4-(1,2,4,5-tetrazin-3-yl)benzenesulfonamide
The compound was obtained from 4-cyano-2-fluorobenzenesulfonamide (0.51 g, 2.54 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.26 g (40%) of 17a as a red solid. Rf = 0.43 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.50 (dd, J = 8.2, 1.6 Hz, 1H), 8.43 (dd, J = 10.9, 1.6 Hz, 1H), 8.11 (t, J = 7.8 Hz, 1H), 7.92 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.56, 158.89, 158.86 (d, J = 254.2 Hz), 138.15 (d, J = 8.1 Hz), 135.35 (d, J = 14.9 Hz), 130.14, 124.44 (d, J = 4.0 Hz), 116.42 (d, J = 24.2 Hz); MS (ESI) m/z [M + H]+: 256.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzenesulfonamide (17b)
3-Cyano-5-fluorobenzene-1-sulfonyl Chloride (26b)
The compound was obtained from 3-amino-5-fluorobenzonitrile (5 g, 36.75 mmol) as described above for 26a to give 2.98 (38%) of the desired compound as a white solid. 1H NMR (400 MHz, chloroform-d) δ 8.17 (s, 1H), 8.02 (dt, J = 7.0, 2.1 Hz, 1H), 7.77 (ddd, J = 7.4, 2.5, 1.3 Hz, 2H); 13C NMR (101 MHz, chloroform-d) δ 162.01 (d, J = 259.6 Hz), 146.83 (d, J = 7.4 Hz), 126.44 (d, J = 4.2 Hz), 125.68 (d, J = 24.4 Hz), 119.00 (d, J = 25.1 Hz), 116.14 (d, J = 8.9 Hz), 115.13 (d, J = 2.7 Hz).
3-Cyano-5-fluorobenzenesulfonamide (27b)
The compound was obtained from 3-cyano-5-fluorobenzene-1-sulfonyl chloride (0.60 g, 2.73 mmol) as described above for 27a to give 0.50 g (91%) of the desired compound as a white solid. Rf = 0.31 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 8.17 (dt, J = 8.5, 1.9 Hz, 1H), 8.11 (d, J = 1.4 Hz, 1H), 8.01 (dt, J = 8.2, 2.0 Hz, 1H), 7.85 (s, 2H), 13C NMR (101 MHz, DMSO-d6) δ 161.79 (d, J = 251.2 Hz), 147.98 (d, J = 7.0 Hz), 126.25 (d, J = 3.5 Hz), 123.40 (d, J = 25.6 Hz), 118.48 (d, J = 24.2 Hz), 117.03 (d, J = 3.0 Hz), 114.31 (d, J = 10.0 Hz); MS (ESI) m/z [M + H]+: 201.0.
3-Fluoro-5-(1,2,4,5-tetrazin-3-yl)benzenesulfonamide
The compound was obtained from 3-cyano-5-fluorobenzenesulfonamide (0.49 g, 2.45 mmol) following the procedure employed for 2a. Purification by flash chromatography (98/2 CH2Cl2/MeOH) afforded 0.21 g (37%) of 17b as a red solid. Rf = 0.25 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 8.81 (t, J = 1.5 Hz, 1H), 8.49 (ddd, J = 9.3, 2.5, 1.4 Hz, 1H), 7.95 (ddd, J = 8.1, 2.5, 1.6 Hz, 1H), 7.76 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 164.47 (d, J = 2.9 Hz), 162.61 (d, J = 249.5 Hz), 159.00, 147.99 (d, J = 6.8 Hz), 135.87 (d, J = 8.2 Hz), 121.44 (d, J = 3.0 Hz), 118.06 (d, J = 24.2 Hz), 117.10 (d, J = 24.4 Hz); MS (ESI) m/z [M + H]+: 256.0.
2-Cyano-4-fluorobenzenesulfonamide
2-Cyano-4-fluorobenzene-1-sulfonyl Chloride (26c)
The compound was obtained from 2-amino-5-fluorobenzonitrile (5 g, 36.75 mmol) as described above for 26a to give 2.41 (30%) of the desired compound as a white solid. 1H NMR (400 MHz, chloroform-d) δ 8.15 (dd, J = 8.2, 6.8 Hz, 1H), 7.78–7.69 (m, 2H); 13C NMR (101 MHz, chloroform-d) δ 158.25 (d, J = 266.7 Hz), 135.53 (d, J = 13.0 Hz), 130.36, 128.61 (d, J = 4.7 Hz), 121.95 (d, J = 23.9 Hz), 120.92 (d, J = 9.4 Hz), 115.43 (d, J = 2.6 Hz).
2-Cyano-4-fluorobenzenesulfonamide (27c)
The compound was obtained from 2-cyano-4-fluorobenzene-1-sulfonyl chloride (0.60 g, 2.73 mmol) as described above for 27a to give 0.48 g (88%) of the desired compound as a white solid. Rf = 0.35 (heptane/EtOAc 40/60); 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.90 (s, 1H), 8.15–7.96 (m, 2H), 7.69 (td, J = 8.6, 2.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.96 (d, J = 250.9 Hz), 159.95, 138.90, 131.53 (d, J = 9.7 Hz), 123.95 (d, J = 9.9 Hz), 121.13 (d, J = 24.1 Hz), 111.22 (d, J = 26.0 Hz), MS (ESI) m/z [M + H]+: 201.0.
Stopped-Flow Kinetic measurements
Stopped-flow measurements were performed using an SX20-LED stopped-flow spectrophotometer (Applied Photophysics) equipped with a 535 nm LED (optical pathlength 10 mm, full width half-maximum 34 nm) to monitor the characteristic tetrazine visible light absorbance (520–540 nm). Solutions of TCO in anhydrous CH3CN and axTCO-PEG4 in DPBS were prepared at an approximate concentration above 2 mM.32 The exact concentration was determined by absorbance titration with 3,6-dimethyltetrazine.39 These initial stock solutions were diluted before stopped-flow analysis to reach a final TCO concentration of 2 mM. Stock solutions of tetrazines were prepared in DMSO at a concentration of 10 mM. Serial dilution into CH3CN or DPBS (10 mM) was used to prepare solutions for stopped-flow analysis at a Tz concentration of 100 μM and thus well below the buffer capacity to avoid any influence of acidic/basic Tz on the pH of the buffer. The reagent syringes were loaded with solutions of the Tz and TCO or axTCO-PEG4, and the instrument was primed. Subsequent data were collected in triplicate to sextuplicate for each tetrazine. Reactions were conducted at 25 °C (CH3CN) or 37 °C (DPBS) and recorded automatically at the time of acquisition. Data sets were analyzed by fitting an exponential decay using Prism 6 (Graphpad) to calculate the observed pseudo-first-order rate constants that were converted into second-order rate constants by dividing through the concentration of excess TCO compound.
DFT Calculations
Density functional theory calculations were performed in Gaussian 16 Revision A.03. The ωB97X-D functional was used in combination with the def2-TZVPD basis set.40 The basis set definition was obtained from the basis set exchange.41 Solvent effects were included using the SMD model. Conformer searches of all stationary points (starting materials and transition states) were conducted using CREST, and all obtained conformers were reoptimized using ωB97X-D/def2-TZVPD with or without solvation.42 Only the lowest energy conformers were used for further analysis. Stationary points were confirmed by having zero (minima) or exactly one (transition states) imaginary frequency. Free energies were corrected using the Truhlar quasiharmonic approximation with a cutoff of 100 cm–1 in GoodVibes.43
pKa calculations were conducted using the method introduced by Shields and co-workers.36 Scheme “S7” was used for the determination of solvation energies. Structures were optimized using HF/6–31+G(d) with the CPCM solvent models, and free energies were calculated at the HF/6–31G(d) level of theory, with or without CPCM solvation to determine the solvation energy. CBS-QB3 was used to obtain accurate free energies and corrected using the HF-calculated solvation energies. Equation 8 in the manuscript by Shields and co-workers was used to estimate the absolute pKa in water.36
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.2c00042.
Author Contributions
∥ U.M.B. and R.G.-V. contributed equally to this work. The organic synthesis was performed by U.M.B. and R.G.-V. Stopped-flow measurements were obtained by B.H. and A.L. Reactivity analysis were performed by D.S. The study was conceptionally designed by U.M.B., H.M., and M.M.H. This manuscript was written by U.M.B. and M.M.H. with contributions from all authors. All authors approved the final version of the manuscript.
This project has received funding from the European Unionʼs EU Framework Program for Research and Innovation Horizon 2020, under grant agreement no. 668532 and from the European Unionʼs Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 813528. H.M. and M.M.H. have received funding from the European Unionʼs EU Framework Program for Research and Innovation Horizon 2020 (grant agreement no. 670261). The Lundbeck Foundation, the Novo Nordisk Foundation, the Innovation Fund Denmark, and the Research Council for Independent Research (grant agreement no. 8022-00187B) are further acknowledged. Quantum chemical calculations were performed on the Vienna Scientific Cluster (Austria). The authors also acknowledge Xiaosong Xue (Nankai University) and Aneta Turlik (University of California, Los Angeles) for fruitful discussions.
The authors declare no competing financial interest.
Supplementary Material
References
- Devaraj N. K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang H. C.; Yu C.; Kato D. L.; Bertozzi C. R. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 14846. 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk M. L. W. J.; Agramunt J.; Bonger K. M. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr. Opin. Chem. Biol. 2021, 60, 79–88. 10.1016/j.cbpa.2020.09.002. [DOI] [PubMed] [Google Scholar]
- Nguyen S. S.; Prescher J. A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat. Rev. Chem. 2020, 4, 476–489. 10.1038/s41570-020-0205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Stéen E. J. L.; Edem P. E.; Nørregaard K.; Jørgensen J. T.; Shalgunov V.; Kjaer A.; Herth M. M. Pretargeting in nuclear imaging and radionuclide therapy: Improving efficacy of theranostics and nanomedicines. Biomaterials 2018, 179, 209–245. 10.1016/j.biomaterials.2018.06.021. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Renart Verkerk P.; van den Bosch S. M.; Vulders R. C. M.; Verel I.; Lub J.; Robillard M. S. In Vivo Chemistry for Pretargeted Tumor Imaging in Live Mice. Angew. Chem., Int. Ed. 2010, 49, 3375–3378. 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- van Onzen A. H. A. M.; Versteegen R. M.; Hoeben F. J. M.; Filot I. A. W.; Rossin R.; Zhu T.; Wu J.; Hudson P. J.; Janssen H. M.; ten Hoeve W.; Robillard M. S. Bioorthogonal Tetrazine Carbamate Cleavage by Highly Reactive trans-Cyclooctene. J. Am. Chem. Soc. 2020, 142, 10955–10963. 10.1021/jacs.0c00531. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Brand C.; Abdel-Atti D.; Carnazza K. E.; Cook B. E.; Carlin S.; Reiner T.; Lewis J. S. Optimization of a Pretargeted Strategy for the PET Imaging of Colorectal Carcinoma via the Modulation of Radioligand Pharmacokinetics. Mol. Pharmaceutics 2015, 12, 3575–3587. 10.1021/acs.molpharmaceut.5b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charton M.Volume and Bulk Parameters; Austel V.; Balaban A. T.; Bonchev D.; Charton M.; Fujita T.; Iwamura H.; Mekenyan O.; Motoc I., Eds.; Steric Effects in Drug Design, Berlin, Heidelberg, 1983; Springer Berlin Heidelberg: Berlin, Heidelberg, 1983; pp 107–118. [Google Scholar]
- Stéen E. J. L.; Jørgensen J. T.; Denk C.; Battisti U. M.; Nørregaard K.; Edem P. E.; Bratteby K.; Shalgunov V.; Wilkovitsch M.; Svatunek D.; Poulie C. B. M.; Hvass L.; Simón M.; Wanek T.; Rossin R.; Robillard M.; Kristensen J. L.; Mikula H.; Kjaer A.; Herth M. M. Lipophilicity and Click Reactivity Determine the Performance of Bioorthogonal Tetrazine Tools in Pretargeted In Vivo Chemistry. ACS Pharmacol. Transl. Sci. 2021, 4, 824–833. 10.1021/acsptsci.1c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Kwon K.; Jana S.; Kim S.; Avila-Crump S.; Tae G.; Mehl R. A.; Kwon I. Temporal Control of Efficient In Vivo Bioconjugation Using a Genetically Encoded Tetrazine-Mediated Inverse-Electron-Demand Diels–Alder Reaction. Bioconjugate Chem. 2020, 31, 2456–2464. 10.1021/acs.bioconjchem.0c00497. [DOI] [PubMed] [Google Scholar]
- Li Z.; Cai H.; Hassink M.; Blackman M. L.; Brown R. C. D.; Conti P. S.; Fox J. M. Tetrazine-trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem. Commun. 2010, 46, 8043–8045. 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šečkutė J.; Devaraj N. K. Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol. 2013, 17, 761–767. 10.1016/j.cbpa.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. C.; Richter S.; Wuest M.; Way J. D.; Wuest F. Synthesis and evaluation of an 18F-labelled norbornene derivative for copper-free click chemistry reactions. Org. Biomol. Chem. 2013, 11, 3817–3825. 10.1039/c3ob40548f. [DOI] [PubMed] [Google Scholar]
- Meyer J.-P.; Houghton J. L.; Kozlowski P.; Abdel-Atti D.; Reiner T.; Pillarsetty N. V. K.; Scholz W. W.; Zeglis B. M.; Lewis J. S. 18F-based pretargeted PET imaging based on bioorthogonal Diels–Alder click chemistry. Bioconjugate Chem. 2016, 27, 298–301. 10.1021/acs.bioconjchem.5b00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Li S.; Wängler C.; Wängler B.; Lennox R. B.; Schirrmacher R. Synthesis of 3-chloro-6-((4-(di-tert-butyl [18 F] fluorosilyl)-benzyl) oxy)-1, 2, 4, 5-tetrazine ([18 F] SiFA-OTz) for rapid tetrazine-based 18 F-radiolabeling. Chem. Commun. 2015, 51, 12415–12418. 10.1039/C5CC03623B. [DOI] [PubMed] [Google Scholar]
- Denk C.; Svatunek D.; Filip T.; Wanek T.; Lumpi D.; Fröhlich J.; Kuntner C.; Mikula H. Development of a 18F-Labeled Tetrazine with Favorable Pharmacokinetics for Bioorthogonal PET Imaging. Angew. Chem., Int. Ed. 2014, 53, 9655–9659. 10.1002/anie.201404277. [DOI] [PubMed] [Google Scholar]
- Keinänen O.; Li X.-G.; Chenna N. K.; Lumen D.; Ott J.; Molthoff C. F. M.; Sarparanta M.; Helariutta K.; Vuorinen T.; Windhorst A. D. A new highly reactive and low lipophilicity fluorine-18 labeled tetrazine derivative for pretargeted PET imaging. ACS Med. Chem. Lett. 2016, 7, 62–66. 10.1021/acsmedchemlett.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Cai H.; Hassink M.; Blackman M. L.; Brown R. C. D.; Conti P. S.; Fox J. M. Tetrazine–trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem. Commun. 2010, 46, 8043–8045. 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K.; Thurber G. M.; Keliher E. J.; Marinelli B.; Weissleder R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 4762–4767. 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Vázquez R.; Battisti U. M.; Jørgensen J. T.; Shalgunov V.; Hvass L.; Stares D. L.; Petersen I. N.; Crestey F.; Löffler A.; Svatunek D.; Kristensen J. L.; Mikula H.; Kjaer A.; Herth M. M. Direct Cu-mediated aromatic 18F-labeling of highly reactive tetrazines for pretargeted bioorthogonal PET imaging. Chem. Sci. 2021, 12, 11668–11675. 10.1039/D1SC02789A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Liang Y.; Houk K. N. Theoretical Elucidation of the Origins of Substituent and Strain Effects on the Rates of Diels–Alder Reactions of 1,2,4,5-Tetrazines. J. Am. Chem. Soc. 2014, 136, 11483–11493. 10.1021/ja505569a. [DOI] [PubMed] [Google Scholar]
- Kronister S.; Svatunek D.; Denk C.; Mikula H. Acylation-Mediated ‘Kinetic Turn-On’ of 3-Amino-1,2,4,5-tetrazines. Synlett 2018, 29, 1297–1302. 10.1055/s-0036-1591764. [DOI] [Google Scholar]
- Mboyi C. D.; Vivier D.; Daher A.; Fleurat-Lessard P.; Cattey H.; Devillers C. H.; Bernhard C.; Denat F.; Roger J.; Hierso J.-C. Bridge-Clamp Bis(tetrazine)s with [N]8 π-Stacking Interactions and Azido-s-Aryl Tetrazines: Two Classes of Doubly Clickable Tetrazines. Angew. Chem., Int. Ed. 2020, 59, 1149–1154. 10.1002/anie.201911947. [DOI] [PubMed] [Google Scholar]
- Le T. G.; Kundu A.; Ghoshal A.; Nguyen N. H.; Preston S.; Jiao Y.; Ruan B.; Xue L.; Huang F.; Keiser J.; Hofmann A.; Chang B. C. H.; Garcia-Bustos J.; Jabbar A.; Wells T. N. C.; Palmer M. J.; Gasser R. B.; Baell J. B. Optimization of Novel 1-Methyl-1H-Pyrazole-5-carboxamides Leads to High Potency Larval Development Inhibitors of the Barber’s Pole Worm. J. Med. Chem. 2018, 61, 10875–10894. 10.1021/acs.jmedchem.8b01544. [DOI] [PubMed] [Google Scholar]
- Stumpf A.; Cheng Z. K.; Beaudry D.; Angelaud R.; Gosselin F. Improved Synthesis of the Nav1.7 Inhibitor GDC-0276 via a Highly Regioselective SNAr Reaction. Org. Process Res. Dev. 2019, 23, 1829–1840. 10.1021/acs.oprd.9b00082. [DOI] [Google Scholar]
- Drapier T.; Geubelle P.; Bouckaert C.; Nielsen L.; Laulumaa S.; Goffin E.; Dilly S.; Francotte P.; Hanson J.; Pochet L.; Kastrup J. S.; Pirotte B. Enhancing Action of Positive Allosteric Modulators through the Design of Dimeric Compounds. J. Med. Chem. 2018, 61, 5279–5291. 10.1021/acs.jmedchem.8b00250. [DOI] [PubMed] [Google Scholar]
- Qu Y.; Sauvage F.-X.; Clavier G.; Miomandre F.; Audebert P. Metal-Free Synthetic Approach to 3-Monosubstituted Unsymmetrical 1,2,4,5-Tetrazines Useful for Bioorthogonal Reactions. Angew. Chem., Int. Ed. 2018, 57, 12057–12061. 10.1002/anie.201804878. [DOI] [PubMed] [Google Scholar]
- Battisti U. M.; Bratteby K.; Jørgensen J. T.; Hvass L.; Shalgunov V.; Mikula H.; Kjær A.; Herth M. M. Development of the First Aliphatic 18F-Labeled Tetrazine Suitable for Pretargeted PET Imaging—Expanding the Bioorthogonal Tool Box. J. Med. Chem. 2021, 64, 15297–15312. 10.1021/acs.jmedchem.1c01326. [DOI] [PubMed] [Google Scholar]
- Svatunek D.; Denk C.; Rosecker V.; Sohr B.; Hametner C.; Allmaier G.; Fröhlich J.; Mikula H. Efficient low-cost preparation of trans-cyclooctenes using a simplified flow setup for photoisomerization. Monatsh. Chem. 2016, 147, 579–585. 10.1007/s00706-016-1668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk C.; Svatunek D.; Filip T.; Wanek T.; Lumpi D.; Fröhlich J.; Kuntner C.; Mikula H. Development of a (18) F-labeled tetrazine with favorable pharmacokinetics for bioorthogonal PET imaging. Angew. Chem., Int. Ed. 2014, 53, 9655–9659. 10.1002/anie.201404277. [DOI] [PubMed] [Google Scholar]
- Otto S.; Engberts J. B. F. N. Diels_Alder reactions in water. Pure Appl. Chem. 2000, 72, 1365–1372. 10.1351/pac200072071365. [DOI] [Google Scholar]
- Yang Z.; Doubleday C.; Houk K. N. QM/MM Protocol for Direct Molecular Dynamics of Chemical Reactions in Solution: The Water-Accelerated Diels–Alder Reaction. J. Chem. Theory Comput. 2015, 11, 5606–5612. 10.1021/acs.jctc.5b01029. [DOI] [PubMed] [Google Scholar]
- Liptak M. D.; Gross K. C.; Seybold P. G.; Feldgus S.; Shields G. C. Absolute pKa Determinations for Substituted Phenols. J. Am. Chem. Soc. 2002, 124, 6421–6427. 10.1021/ja012474j. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Svatunek D.; Wilkovitsch M.; Hartmann L.; Houk K.; Mikula H. Uncovering the key role of distortion in tetrazine ligations guides the design of bioorthogonal tools with high reactivity and superior stability. ChemRxiv 2021, 10.26434/chemrxiv-2021-vnwzk. [DOI] [Google Scholar]
- Versteegen R. M.; Rossin R.; ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem., Int. Ed. 2013, 52, 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Pracht P.; Bohle F.; Grimme S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. 10.1039/C9CP06869D. [DOI] [PubMed] [Google Scholar]
- bobbypaton/GoodVibes: GoodVibes v3.0.0, v3.0.0; Zenodo, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.