

Summary
This protocol summarizes the pipeline for analysis of tumor-derived cell-free DNA (cfDNA) from cerebrospinal fluid (CSF) using low-coverage whole-genome sequencing (lcWGS). This approach enables resolution of chromosomal and focal copy-number variations (CNVs) as oncologic signatures, particularly for patients with central nervous system tumors. Our strategy tolerates sub-nanogram cfDNA input and is thus optimized for CSF samples where cfDNA yields are typically low. Overall, the detection of tumor-specific signatures in CSF-derived cfDNA is a promising biomarker for personalization of brain-tumor therapy.
For complete details on the use and execution of this protocol, please refer to Liu et al. (2021).
Subject areas: Bioinformatics, Sequence analysis, Cancer, Health Sciences, Clinical Protocol, Genomics, Sequencing, Molecular Biology, Neuroscience
Graphical abstract
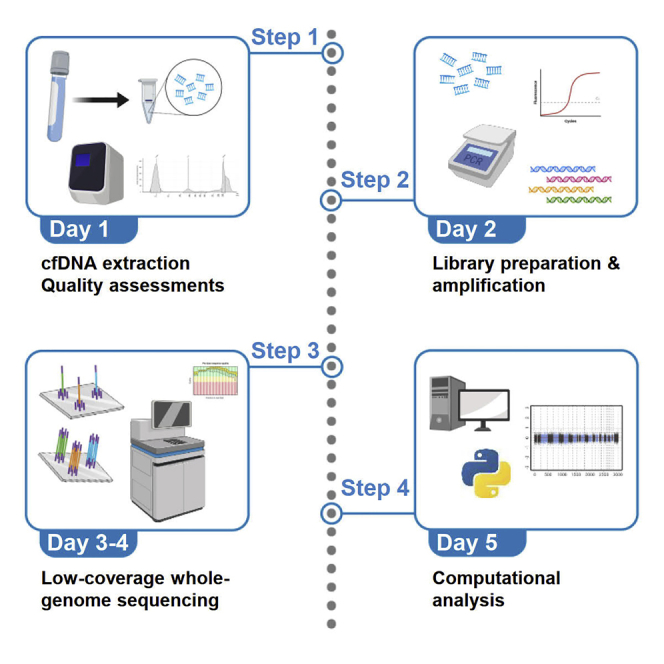
Highlights
-
•
Protocol for analyzing circulating tumor DNA in CSF of patients with brain tumors
-
•
Optimized designated workflow to promote successful lcWGS
-
•
Detection of somatic CNVs as a potential marker of minimal residual disease
This protocol summarizes the pipeline for analysis of tumor-derived cell-free DNA (cfDNA) from cerebrospinal fluid (CSF) using low-coverage whole-genome sequencing (lcWGS). This approach enables resolution of chromosomal and focal copy-number variations (CNVs) as oncologic signatures, particularly for patients with central nervous system tumors. Our strategy tolerates sub-nanogram cfDNA input and is thus optimized for CSF samples where cfDNA yields are typically low. Overall, the detection of tumor-specific signatures in CSF-derived cfDNA is a promising biomarker for personalization of brain-tumor therapy.
Before you begin
The protocol below describes the specific steps for isolating and processing human CSF-derived cfDNA. However, we have also used this protocol for cfDNA isolated from other sources (i.e., plasma). This protocol is optimized for low quantities of cfDNA in the sub-nanogram range.
Institutional permissions
Experiments outlined in the protocol were conducted according to the principles expressed in the Declaration of Helsinki. Ethical approval was obtained from the Institutional Review Board of St. Jude Children’s Research Hospital under protocol #Pro00008912. All patients provided written informed consent for sample and clinical data collection and subsequent analyses. Corresponding permissions from relevant regulatory bodies should be sought for before performing the described experiments.
CSF collection
Timing: 30 min
-
1.CSF is preferentially collected through lumbar puncture but can also be obtained by tapping of a CSF reservoir or intra-operatively. CSF collected during operation will inadvertently be contaminated with blood cells and CSF from lumbar puncture is also the standard source when cytologic examination is considered (Gajjar et al., 1999). Furthermore, only a limited subset of patients will have CSF reservoirs inserted to allow CSF sampling.
-
a.Initial CSF collection by lumbar puncture should be deferred to ∼14 days post-surgical resection.
-
b.Care should be taken to avoid traumatic lumbar puncture, with anesthesia given as necessary and procedure conducted by experienced providers.
-
c.2–3 mLs of CSF can be drawn, ensuring sufficient quantity of input cfDNA and mitigating concerns for removing excessive amount of CSF in young patients.
-
a.
-
2.
Sample should be collected in a standard sterile CSF collection tube and transferred immediately on ice for pre-processing.
CSF preprocessing
-
3.
Centrifuge CSF sample at 1,500 g for 10 min at 4°C.
-
4.
Transfer and aliquot supernatant to new 1.5 mL Eppendorf tubes leaving behind 50 μl with the cell pellet undisturbed.
-
5.
CSF supernatant can be used immediately for cfDNA extraction or stored at −80°C. Avoid unnecessary freeze-thaw cycles.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Cerebrospinal fluid samples (human; age = 3–21 years; male:female = 81:42) | Participating study centers | N/A |
| Tumor samples (human; age = 3–21 years; male:female = 81:42) | Participating study centers | N/A |
| Oligonucleotides | ||
| ALU115-F′ | Umetani et al. (2006) | CCTGAGGTCAGGAGTTCGAG |
| ALU115-R′ | Umetani et al. (2006) | CCCGAGTAGCTGGGATTACA |
| ALU247-F′ | Umetani et al. (2006) | GTGGCTCACGCCTGTAATC |
| ALU247-R′ | Umetani et al. (2006) | CAGGCTGGAGTGCAGTGG |
| Reagent R1 Primer 1 | Illumina | AATGATACGGCGACCACCGAGATC |
| Reagent R1 Primer 2 | Illumina | CAAGCAGAAGACGGCATACGA |
| Critical commercial assays | ||
| NucleoSnap cfDNA kit | MACHEREY-NAGEL | 740300.50 |
| Tapestation High Sensitivity D1000 ScreenTape and reagents | Agilent | 5067-5584 / 5067-5585 |
| Tapestation D5000 ScreenTape and reagents | Agilent | 5067-5588 / 5067-5589 |
| SYBR Green I | Sigma-Aldrich | S9430 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | A25780 |
| KAPA HiFi HotStart Ready Mix | Roche | KK2602 |
| Quant-iT PicoGreen ds DNA assay | Invitrogen | P11496 |
| 2S Hyb DNA Library Kit | Integrated DNA Technologies | 10009881 (previously Accel-NGS 2S Hyb DNA Library Kit, 23096, Swift Biosciences) |
| 2S MID Adapter S1-S4 | Integrated DNA Technologies | 10009907 (previously 2S Set S1-S4 MID Indexed Adapters, 279384, Swift Biosciences) |
| Deposited data | ||
| Raw sequencing data | This study | EGA: EGAS00001005592 |
| Code for data analysis | This study | Github: https://github.com/kylessmith/cfdna (https://doi.org/10.5281/zenodo.6335445) |
| Software and algorithms | ||
| R software (version 3.6.3) | R Project for Statistical Computing | https://www.r-project.org/ |
| Python (version > 3.6) | Python Software Foundation | https://www.python.org/ |
| Others | ||
| 4150/4200 TapeStation instruments | Agilent | G2992AA/G2991BA |
| NovaSeq 6000 | Illumina | 20013850 |
Step-by-step method details
cfDNA extraction
Here, cfDNA is isolated from CSF supernatant using the NucleoSnap cfDNA kit (Macherey-Nagel) according to the manufacturer’s instructions (Protocol Version November 2019: https://www.mn-net.com/media/pdf/56/35/fa/Instruction-NucleoSnap-cfDNA.pdf). Other cfDNA extraction kits used by the authors include the QIAamp Circulating Nucleic Acid Kit (Qiagen), QIAamp MinElute ccfDNA Mini Kit (Qiagen), and Maxwell RSC ccfDNA Plasma Kit (RSC). The NucleoSnap kit is chosen due to a combination of factors including flexibility of input volume, high cfDNA yield (Maass et al., 2021), and simplicity of protocol.
-
1.
Avoid exogenous DNA contamination by working in a pre-PCR area with designated equipment, regularly decontaminating pipettes and surfaces with diluted bleach, and wearing clean gloves and lab coats at start of experiment.
-
2.
Thaw CSF supernatant (if necessary) at room temperature (15°C–25°C) and note volume for each sample.
-
3.
Prepare sample by centrifugation at 4,500 g for 10 min at room temperature, transfer supernatant to a new 50 mL conical tube.
-
4.
Add 15 μL Proteinase K per mL of sample, close lid and mix carefully by swirling tube without moistening lid, incubate at room temperature for 5 min.
-
5.
Add 1 mL of Buffer VL per mL of sample, mix by vortexing, incubate tube at 56°C for 5 min in bead or water bath.
-
6.
Add 1 mL of 96%–100% ethanol per mL of sample, mix by vortexing.
-
7.
Set up the NucleoSnap cfDNA columns (one per sample) by connecting them to a vacuum manifold.
-
8.
Prepare the column by adding 500 μL of Buffer CC to the column and applying vacuum (0.4–0.6 bar) until the solution has passed through the column. Perform this step 1–5 min before the sample lysate is to be added according to the next step.
-
9.
Decant the sample lysate into the column and apply vacuum (0.4–0.6 bar) until the lysate has passed through the column.
-
10.
Wash membrane by adding 1 mL of Buffer VW1 to column, apply vacuum (0.2–0.4 bar) until buffer has passed through the column.
-
11.
Wash membrane by adding 0.5 mL of Buffer WB to column, apply vacuum (0.2–0.4 bar) until buffer has passed through the column.
-
12.
Remove the column from the vacuum manifold and insert it into a 2 mL collection tube, break off the upper part of the column and discard it.
-
13.
Centrifuge the lower part of the column in the collection tube at >11,000 g for 3 min at room temperature, discard collection tube with flow through and insert column into a new 1.5 mL collection tube.
-
14.
Add 50 μL of Elution Buffer, incubate at room temperature for 3 min, and centrifuge at 11,000 g for 1 min.
Optional: When connecting NucleoSnap cfDNA columns to the vacuum manifold, the use of disposable NucleoVac Mini Adapter is recommended to avoid cross-contamination, and the addition of NucleoVac Valves or equivalent stopcocks can be used if samples with different volumes are expected.
Pause point: If not proceeding with quantification and fragment size determination, extracted cfDNA can be stored at 4°C for a short duration or at -20°C for longer term, freeze-thaw cycles should be avoided.
Quantification and fragment size determination
As part of quality control, extracted cfDNA is subjected to quantification and fragment size determination. In view of the expected low cfDNA concentration, a quantitative PCR (qPCR)-based assay targeting a repeat sequence (such as ALU or LINE-1) is adopted to maximize sensitivity (Rago et al., 2007), although fluorometric quantification such as using the Qubit fluorometer and dsDNA HS Assay Kit can be considered.
-
15.cfDNA is quantified by qPCR by comparison against standards of genomic DNA (gDNA) with known concentrations using primers targeting the ALU sequences (ALU115-F’: CCTGAGGTCAGGAGTTCGAG, ALU115-R’: CCCGAGTAGCTGGGATTACA) (Umetani et al., 2006).
-
a.Using gDNA at known concentration (e.g., Promega G1521), prepare serially diluted gDNA samples at (10 ng/μL, 1 ng/μL, 100 pg/μL, 10 pg/μL, 1 pg/μL) as qPCR standards.
-
b.Run qPCR according to the following setup with gDNA standards (1 pg/μL – 10 ng/μL) and samples in triplicates (1 μL of standard or extracted cfDNA per well) to obtain the mean concentration of samples relative to the standard curve.
-
a.
PCR reaction master mix
| Reagent | Working stocks | Volume (μL) |
|---|---|---|
| PowerUp SYBR Green Master Mix | 2× | 12.5 |
| Forward primer | 0.2 μM | 0.5 |
| Reverse primer | 0.2 μM | 0.5 |
| H2O | – | 10.5 |
| cfDNA | – | 1 |
| Total | – | 25 |
PCR cycling conditions
| Step | Temp | Duration | Cycles |
|---|---|---|---|
| Pre-activation | 95°C | 15 min | 1 |
| Denaturation | 94°C | 15 s | 35 |
| Annealing | 64°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 10 min | 1 |
-
16.Fragment size is determined using the Agilent 4150/4200 TapeStation instruments.
-
a.The High Sensitivity D1000 ScreenTape and Reagents allow sizing of DNA at the cell-free range and has a sensitivity down to 5 pg/μL (requires 2 μL of extracted cfDNA).
-
b.Alternatively, the Cell-free DNA ScreenTape and Reagents allow sizing of DNA within the cell-free range (150-200 bp) and at the same time determines the proportion of high-molecular weight gDNA contamination (sensitivity down to 20 pg/μL, requires 2 μL of extracted cfDNA).
-
a.
Note: Absence of a DNA peak within the cell-free range (150-200 bp) on a TapeStation trace does not preclude downstream processing of a sample as concentrations of tumor-derived cfDNA may be lower than the limit of detection for the TapeStation assay.
Library preparation
Library preparation for lcWGS using the 2S Hyb DNA Library Kit (Integrated DNA Technologies) and the 2S MID Adapter (S1-4, Integrated DNA Technologies). Other indexing options are also available from the manufacturer. Dephosphorylation, end repair, and ligation of adapters are performed according to protocol (Indexing by Ligation and Direct Sequencing workflow, Protocol PRT-037 Version 1 by Swift Biosciences, Document S1), with the following specifications and modifications.
-
17.
In our experience, starting material in the range of 50 pg-5 ng of cfDNA was compatible with library preparation (ideally determined by DNA concentration from qPCR in conjunction with proportion of cfDNA based on Cell-free DNA ScreenTape). Top off sample with low-EDTA TE to 40 μL for Repair I.
-
18.
After completion of Ligation II, proceed with pilot amplification and library amplification according to the next section.
Optional: Suggest to always include a negative control with 40 μL of low-EDTA TE as the pipeline can generate library even from negligible amounts of contaminating DNA.
Library amplification
Library amplification according to cfDNA quantity may not be effective due to the low cfDNA concentration and frequent inability to determine the proportion of cfDNA versus high-molecular weight gDNA. A pilot qPCR run is first performed to offer guidance on the PCR cycles required for actual library amplification.
-
19.
Library prepared after completion of Ligation II is used for a qPCR run as follows:
PCR reaction master mix
| Reagent | Working stocks | Volume (μL) |
|---|---|---|
| KAPA HiFi HotStart Ready Mix | 2× | 12.5 |
| Reagent R1 primer | 6 μM | 2.5 |
| SYBR Green I | 100× | 0.25 |
| H2O | – | 8.75 |
| Library | – | 1 |
| Total | – | 25 |
-
20.
Set up and run qPCR according to the following protocol (1 well per library):
PCR cycling conditions
| Step | Temp | Duration | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 45 s | 1 |
| Denaturation | 98°C | 15 s | 20 |
| Annealing | 60°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 1 min | 1 |
-
21.
CT value to be determined from the amplification curve for each library.
-
22.
Amplify library with number of PCR cycles being (CT value + 2), detailed as follows:
PCR reaction master mix
| Reagent | Working stocks | Volume (μL) |
|---|---|---|
| KAPA HiFi HotStart Ready Mix | 2× | 25 |
| Reagent R1 primer | 6 μM | 5 |
| H2O | – | 1 |
| Library | – | 19 |
| Total | – | 50 |
PCR cycling conditions
| Step | Temp | Duration | Cycles |
|---|---|---|---|
| Initial Denaturation | 98°C | 45 s | 1 |
| Denaturation | 98°C | 15 s | CT value + 2 |
| Annealing | 60°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 1 min | 1 |
| Hold | 4°C | Infinite | – |
-
23.
Clean up PCR reaction using SPRIselect beads at ratio of 0.85 (sample volume = 50 μL, bead volume = 42.5 μL). Elute in 20 μL of Tris HCl (pH 8.5) or water.
-
24.
Assess size profile and concentration of amplified and cleaned-up library using the D5000 ScreenTape and Reagents (Agilent).
-
25.
If needed, libraries with low concentration (<4 nM) can be further amplified for 2–4 more cycles.
Note: Further amplification beyond 4 cycles is unlikely to yield libraries useful for sequencing.
Whole-genome sequencing
DNA libraries are then subjected to further quality assessment and sequencing to achieve 3× genome-wide coverage.
-
26.
Analyze for library size distribution using a 4200 TapeStation D5000 ScreenTape assay (Agilent).
-
27.
Quantify libraries using the Quant-iT PicoGreen ds DNA assay (ThermoFisher).
-
28.
Paired-end, 100 bp sequencing is then performed on a NovaSeq 6000 (Illumina), targeting approximately 50 million read pairs.
-
29.
After sequencing is complete, run bcl2fastq to translate and demultiplex BCL files into FASTQ files.
Computational analysis
Complete this section to detect focal and large-scale CNVs and confirm the presence of tumor-derived cfDNA in CSF based on lcWGS.
-
30.Process lcWGS reads (recommended QC thresholds are indicated):
-
a.Run FastQC on FASTQ files.
-
i.Check that the average sequence quality is >25.
-
ii.Check that there is not an over-representation of k-mers that would be indicative of adapter sequences.
-
iii.Check GC content is within the expected normal distribution.
-
i.
-
b.Run ddupk on FASTQ files to remove adaptors.
-
c.Run BWA-mem to map FASTQs to human reference genome (hg19).
-
i.Check >80% of reads mapped.
-
i.
-
d.Run Picard de-duplication to remove predicted duplicates.
-
i.The number of duplicates can vary and will be larger for lower input samples.
-
ii.Optionally, if unique molecular identifiers (UMIs) were included then fgbio (https://github.com/fulcrumgenomics/fgbio) can be used to collapse UMI families and remove duplicates.
-
i.
-
a.
-
31.Calculate tumor purity and CNVs.
-
a.cfdna python package: see quantification and statistical analysis.
-
a.
Expected outcomes
Expected cfDNA profile
We expect the isolated cfDNA to have a fragment size predominantly peaking between ∼160-180 bp (mononucleosomal) based on TapeStation assessment. Successive peaks at decreasing intensities might be seen corresponding to dinucleosomal (∼340 bp) and trinucleosomal (∼510 bp) fragments. High-molecular weight gDNA contamination might be evident but does not preclude successful downstream processing, as PCR for library preparation preferentially amplifies short DNA fragments and shorter library fragments also cluster more efficiently on flow cells (Figure 1).
Figure 1.

Electropherogram (High Sensitivity D1000 ScreenTape) of extracted cfDNA indicating an average fragment size of 163 bp
High-molecular weight gDNA is present distorting appearance of the upper marker.
cfDNA concentration
The quantity of cfDNA as defined by qPCR from CSF is expected to be lower than from plasma samples. In our experience with medulloblastoma patients who have CSF sampled at median of 2 weeks after resection of the primary lesion, the median concentration of cfDNA was 0.5 ng/mL. In cases where low quantities of cfDNA are isolated, the cfDNA peak may not be evident on TapeStation analysis.
NGS library
Success of the NGS libraries will first be apparent after TapeStation. The predominant library fragment peak should be centered at ∼300 bp (Figure 2). Successive peaks corresponding to libraries generated from dinucleosomal/polynucleosomal cfDNA fragments might be present.
Figure 2.

Electropherogram (D5000 ScreenTape) of amplified libraries indicating the main library size to be averaged at 310 bp
Additional peaks are present due to di-/polynucleosomal cfDNA fragments being present as input material (may not be apparent on original cfDNA trace.
Computational analysis
DNA fragment distributions should be recapitulated by the computational pipeline (see quantification and statistical analysis). Focal and chromosomal CNVs from tumor-derived cfDNA should be evident in the generated genome-wide copy number plots for cfDNA samples with tumor fraction >5%–10%.
Quantification and statistical analysis
cfDNA lcWGS copy number analysis:
The pipeline for determining the presence of CNVs was written in the Python programming language and is distributed as a package on the Python Package Index. The memory requirements for this analysis will increase as the sequencing depth increases. With about 50 million reads the analysis pipeline requires 4 GB of memory and can be run on a single CPU in about 10 min. If only CNV detection and tumor purity prediction is requested, this can be reduced to 2 GB of memory in about 3 min.
Dependencies:
>pip=20.1.1
The software can be downloaded on the Python Package Index or directly from GitHub at the kylessmith/cfdna repository (https://doi.org/10.5281/zenodo.6335445).
>$ pip install cfdna
>$ python -m cfdna summarize --bam sample.bam --prefix cfdna_lcwgs --nfrags 25000000 --segs
-
•
sample.bam = deduplicated mapped read file
-
•
cfdna_lcwgs = prefix to be added to output file name
-
•
nfrags∗ = number of fragments to process (this number will depend on your sequencing coverage)
-
•
segs∗ = output a segmentation file
-
•
∗ = optional parameter
This command will run the entire analysis pipeline and output a summary plot of the results, a segmentation file for querying called CNVs, and a cache of the analysis in the format of an h5df file. In addition to CNVs, the fragment size distribution and average TSS window protection scores are plotted and can be used as quality control metrics (Figure 3). There should be a general depletion at the TSS as quantified in the nucleosome free score (NFR) also provided.
Figure 3.

Exemplary output from the cfdna package
Panels inclusive of (A) quality-control metrics, (B) predicted fragment size distribution (corresponding to fragment size of input cfDNA), (C) plot of average TSS window protection score, and (D) genome-wide CNV plot with predicted segmental gains labeled in red and losses in blue.
Each copy-number segment is color coded by whether they are predicted to be an amplification (red), deletion (blue), or neutral (gray). Resulting h5df files containing the measurements used to create this plot can be merged into a multi-sample cache file in order to conduct cohort level analyses.
>$ python -m cfdna merge --inputs cfdna_outputs/∗.h5 --output cfdna_multisample.h5
These .h5 files can be read using the h5py package in python or the h5df library in R.
Limitations
The cfdna computational pipeline will yield variable predictions of tumor purity if the actual tumor purity is below 5%. In theory, deeper sequencing could improve the limit of detection although this is also dependent on library complexity. Additionally, tumor purity estimations are predicated on the assumption that there are CNVs in the tumor and are therefore not compatible with copy-number neutral samples. We do not expect mutation calling to be reliable based on the data generated from lcWGS due to the inadequate sequencing depth for such purpose.
Troubleshooting
Problem 1
Clogging of column during cfDNA extraction (step 9).
Potential solution
The sample may contain residual cells or debris. Make sure to use only samples that have been centrifuged in order to remove cells and debris.
Problem 2
No cfDNA peak detected using the TapeStation assay (step 16).
Potential solution
Review TapeStation assay protocols and ensure that the corresponding TapeStation reagents are used for the High Sensitivity ScreenTapes. cfDNA at low concentrations might not be detected during fragment analysis but does not preclude the sample from successful library preparation. Consider including cfDNA samples (or sheared gDNA samples) with known concentration as a positive control during TapeStation analysis.
Problem 3
High DNA concentration by qPCR but cfDNA peak still absent on TapeStation (steps 15–16).
Potential solution
Review TapeStation trace for high-molecular weight gDNA contamination. qPCR with the ALU115 primers cannot distinguish between cfDNA fragments and gDNA. Consider using up to 40 μL of eluted cfDNA as input for library preparation in cases without cfDNA peak on TapeStation, despite apparently adequate concentration by qPCR. The inclusion of ALU247 primers (ALU247-F’: GTGGCTCACGCCTGTAATC, ALU247-R’: CAGGCTGGAGTGCAGTGG) in qPCR will be able to selectively quantify high-molecular weight gDNA for more precise delineation of cfDNA concentration (Hussein et al., 2019).
Problem 4
Presence of small-sized DNA fragments in post-amplification (step 24) library TapeStation traces (Figure 4).
Figure 4.

Electropherogram (D5000 ScreenTape) of amplified library from low cfDNA input indicating presence of adapter dimers (133 bp) in addition to NGS library peaks (315 bp and 514 bp)
Potential solution
This is likely a result of adapter dimers carried over in libraries prepared from low quantities of cfDNA input. A repeat SPRI bead clean-up using 1:1 bead to library volume ratio may be carried out.
Problem 5
Low number of mapped reads (step 31) despite adequate raw read depth.
Potential solution
This could be a result of adapters being sequenced and/or significant PCR duplicates. Adapters can be removed prior to sequencing (see problem 4) while higher quantities of input cfDNA will be required to increase library complexity and thus reduce the proportion of duplicated reads.
Resource availability
Lead contact
Paul A. Northcott: paul.northcott@stjude.org.
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This study was principally supported by the National Cancer Institute (P.A.N.; R21CA256386). Additional funding was provided by the American Lebanese Syrian Associated Charities, National Cancer Institute Cancer Center Grant (P30CA021765), St. Jude Comprehensive Cancer Center Developmental Funds (P.A.N.), and Conquer Cancer – #cureMEdullo powered by Carson Leslie Foundation Young Investigator Award (A.P.Y.L.). A.P.Y.L. was supported by the Li Shu Pui Medical Foundation Training Grant and the Lin Kin Pang-HKU Foundation Scholarship. P.A.N. is a Pew-Stewart Scholar for Cancer Research (Margaret and Alexander Stewart Trust), Sontag Foundation Distinguished Scientist, and recipient of the Robert J. Arceci Innovation Award (St. Baldrick’s Foundation). We thank K. Maass for providing critical feedback on the manuscript.
Author contributions
Conceptualization and methodology, all authors; bioinformatic pipeline, K.S.S.; data analysis, A.P.Y.L., K.S.S., and R.K.; writing – original draft, A.P.Y.L. and K.S.S.; writing – review & editing, all authors; funding and supervision, P.A.N.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101292.
Supplemental information
Data and code availability
The code generated during this study is available through Github at https://github.com/kylessmith/cfdna (https://doi.org/10.5281/zenodo.6335445). Sequencing data used in the manuscript have been deposited to the European Genome-Phenome Archive (EGA: EGAS00001005592) and are publicly available as of the date of publication.
References
- Gajjar A., Fouladi M., Walter A.W., Thompson S.J., Reardon D.A., Merchant T.E., Jenkins J.J., Liu A., Boyett J.M., Kun L.E., Heideman R.L. Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors. J. Clin. Oncol. 1999;17:1825. doi: 10.1200/JCO.1999.17.6.1825. [DOI] [PubMed] [Google Scholar]
- Hussein N.A., Mohamed S.N., Ahmed M.A. Plasma ALU-247, ALU-115, and cfDNA integrity as diagnostic and prognostic biomarkers for breast cancer. Appl. Biochem. Biotechnol. 2019;187:1028–1045. doi: 10.1007/s12010-018-2858-4. [DOI] [PubMed] [Google Scholar]
- Liu A.P., Smith K.S., Kumar R., Paul L., Bihannic L., Lin T., Maass K.K., Pajtler K.W., Chintagumpala M., Su J.M. Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell. 2021;39:1519–1530. doi: 10.1016/j.ccell.2021.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass K.K., Schad P.S., Finster A.M., Puranachot P., Rosing F., Wedig T., Schwarz N., Stumpf N., Pfister S.M., Pajtler K.W. From sampling to sequencing: a liquid biopsy pre-analytic workflow to maximize multi-layer genomic information from a single tube. Cancers. 2021;13:3002. doi: 10.3390/cancers13123002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rago C., Huso D.L., Diehl F., Karim B., Liu G., Papadopoulos N., Samuels Y., Velculescu V.E., Vogelstein B., Kinzler K.W., Diaz L.A., Jr. Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007;67:9364–9370. doi: 10.1158/0008-5472.CAN-07-0605. [DOI] [PubMed] [Google Scholar]
- Umetani N., Giuliano A.E., Hiramatsu S.H., Amersi F., Nakagawa T., Martino S., Hoon D.S. Prediction of breast tumor progression by integrity of free circulating DNA in serum. J. Clin. Oncol. 2006;24:4270–4276. doi: 10.1200/JCO.2006.05.9493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code generated during this study is available through Github at https://github.com/kylessmith/cfdna (https://doi.org/10.5281/zenodo.6335445). Sequencing data used in the manuscript have been deposited to the European Genome-Phenome Archive (EGA: EGAS00001005592) and are publicly available as of the date of publication.