

Summary
Combating fungal pathogens poses metabolic challenges for neutrophils, key innate cells in anti-Candida albicans immunity, yet how host-pathogen interactions cause remodeling of neutrophil metabolism is unclear. We show that neutrophils mediate renal immunity to disseminated candidiasis by upregulating glucose uptake via selective expression of glucose transporter 1 (Glut1). Mechanistically, dectin-1-mediated recognition of β-glucan leads to activation of PKCδ, which triggers phosphorylation, localization and early glucose transport by a pool of pre-formed Glut1 in neutrophils. These events are followed by increased Glut1 gene transcription leading to more sustained Glut1 accumulation, also dependent on the β-glucan/dectin-1/CARD9-axis. Card9-deficient neutrophils show diminished glucose incorporation in candidiasis. Neutrophil-specific Glut1 ablated mice exhibit increased mortality in candidiasis, caused by compromised neutrophil phagocytosis, ROS and NET formation. In human neutrophils, β-glucan triggers metabolic remodeling and enhances candidacidal function. Our data show that the host-pathogen interface increases glycolytic activity in neutrophils by regulating Glut1 expression, localization and function.
Graphical Abstract
eTOC blurb
Neutrophils protect against fungal infections including candidiasis. Neutrophils kill Candida albicans in an energy demanding process. The mechanisms by which neutrophils meet their metabolic demand are unknown. Li and colleagues show how fungal sensing rewires metabolism in neutrophils via regulating glucose transporter 1 expression and function during renal host defense.
Introduction
Neutrophils are the most abundant, short lived, and terminally differentiated leukocytes in circulation (Mantovani et al., 2011; Mayadas et al., 2014). Neutrophils are the first to arrive at the site of infection where they execute rapid antimicrobial activities to eliminate and prevent pathogen dissemination to sterile sites (Burn et al., 2021). Encountering invading pathogens in diverse tissues exert metabolic challenges for these cells. Despite advances in understanding immunometabolism in T cells and macrophages (Geltink et al., 2018; Makowski et al., 2020; O’Neill et al., 2016; Pearce and Pearce, 2013), surprisingly little is known about the mechanisms by which neutrophils meet the high metabolic demand for effector functions in infectious settings. A comprehensive understanding of the antimicrobial metabolic shift could allow for selective targeting of metabolic pathways to improve neutrophil function without causing collateral tissue damage during host defense.
Unlike most cell types, mitochondria are not required for energy production in neutrophils in the presence of glucose, thus making this innate cell type metabolically unique (Fossati et al., 2003; Maianski et al., 2004; Reiss and Roos, 1978; Zucker-Franklin, 1968). Instead, neutrophils rely on glycolysis and pentose phosphate pathway (PPP) not only to fulfill their energy requirements, but also to support specialized effector functions including degranulation, phagocytosis, reactive oxygen species (ROS) production and neutrophil extracellular trap (NET) formation (Jeon et al., 2020; Kumar and Dikshit, 2019). Individuals with rare genetic deficiency of glucose-6-phopspahte dehydrogenase or glucose-6-phosphate transporter, key enzymes in glycolytic pathway, show impaired ATP and ROS generation (Ardati et al., 1997; Hiraiwa et al., 1999). Hence, neutrophils must tightly regulate glucose uptake, intracellular glucose cycling and glycolysis to execute rapid antimicrobial activities.
Most cells import glucose by facilitative diffusion across the cell membrane, a process mediated by the glucose transporter (Glut) family of membrane transport proteins. There are 14 Glut-family transporters in mammals (Glut 1–14) (Thorens and Mueckler, 2010). Of these, Glut5, Glut6 and Glut13 do not participate in glucose transport and Glut11 and Glut14 are not in the mouse genome (Douard and Ferraris, 2008; Maedera et al., 2019; Scheepers et al., 2005; Uldry et al., 2001; Wu and Freeze, 2002). The range of Glut transporters utilized by neutrophils has not yet been defined. Moreover, the mechanisms that regulate Glut expression in neutrophils are poorly understood. Unlike Glut4, which is insulin-sensitive, expression of Glut1 (encoded by SLC2A1) at the cell membrane is not regulated by blood glucose levels (Leto and Saltiel, 2012). Rather, Glut1 expression is controlled by transcription, translation and/or translocation of the protein from a pre-formed intracellular pool (Buller et al., 2008; Griffin et al., 2004; Hwang and Ismail-Beigi, 2006; Lee et al., 2015; Wang et al., 2019). Glut1 in CD4 T cells and macrophage are essential for activation, expansion and survival of these immune cells (Freemerman et al., 2014; Macintyre et al., 2014). However, our understanding of the role of neutrophil specific Glut1 expression and its regulation in host defense is far from complete.
Neutrophils are critical for controlling disseminated candidiasis, a bloodstream nosocomial infection caused by the dimorphic commensal fungus Candida albicans, that causes high mortality (30–40%) (Horn et al., 2009; Pappas et al., 2018; Pasqualotto and Denning, 2008; Zaoutis et al., 2005). Humans with neutropenia have a higher risk of disseminated candidiasis and mice depleted of neutrophils are susceptible to invasive infection (Desai and Lionakis, 2018; Fortun and Gioia, 2017; Lionakis and Netea, 2013; Uzun et al., 2001; Velasco and Bigni, 2008). Following disseminated candidiasis, neutrophils traffic to the infected organs such as kidney, brain, spleen, and liver, within 2 h (Lionakis et al., 2012). Neutrophils sense fungi by recognizing pathogen associated molecular patterns (PAMPs) via pathogen recognition receptors (PRRs), particularly C-type lectin receptors (CLRs) (Netea et al., 2006; Taylor et al., 2007). The fungal recognition via PRRs authorizes neutrophils to eliminate fungi by oxidative and non-oxidative mechanisms, and also deploy NET to neutralize pathogenic hyphal form of C. albicans (Brown, 2011; Urban et al., 2006). Additionally, the host-pathogen interaction is essential for the development of “trained” immunity in innate cells (Cheng et al., 2014). All these specialized antifungal functions require energy and neutrophils respond to the increasing metabolic demand by adapting to simpler metabolic pathways such as glycolysis (Weerasinghe and Traven, 2020). Indeed, recognition of fungal β-glucan by dectin-1 in monocytes leads to epigenetic changes and increased glycolysis in an Akt/mTOR/HIF1α-dependent manner, which are critical for the development of ‘trained’ immunity. While it is evident that host-pathogen interface is a critical determinant of antifungal functions, the mechanisms by which fungal sensing machinery in neutrophils rewire cell-intrinsic metabolic pathways to meet the increasing metabolic requirement in the kidney is poorly understood.
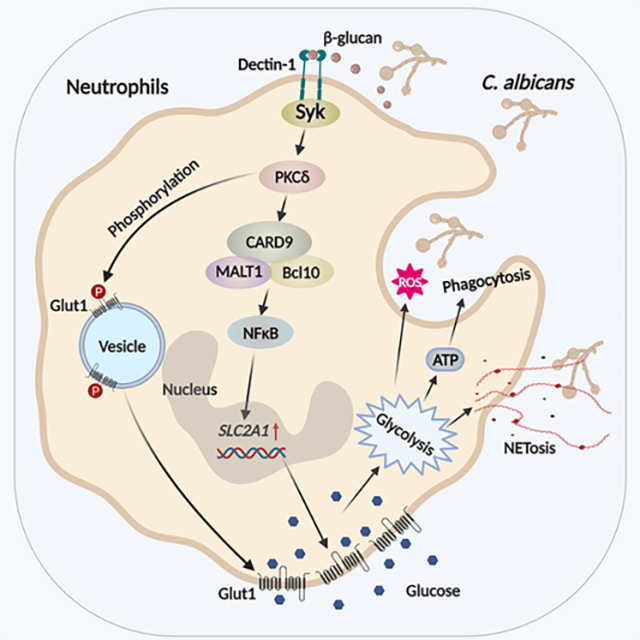
In this report, we demonstrate that renal immunity to disseminated candidiasis requires Glut1-dependent glucose uptake, which is controlled at multiple levels including Glut1 gene transcription, surface expression and glucose uptake. Recognition of fungal β-glucan via dectin-1 promotes PKCδ-mediated phosphorylation and surface localization of pre-formed Glut1, a process that is essential for early glucose uptake and antifungal activity of neutrophils in the kidney. This is later followed by increased transcription of Glut1 that allows for sustained neutrophil function, which is dependent on the β-glucan/dectin-1/CARD9-axis. Accordingly, mice with neutrophil-specific deletion of Glut1 showed increased susceptibility to DC. Glut1-mediated glucose uptake by neutrophils is indispensable for renal antifungal function including phagocytosis, ROS production and NET formation. Finally, β-glucan metabolically remodeled healthy donor neutrophils by upregulating Glut1 function, which is essential for fungicidal activity. Collectively, these data advance our fundamental understanding of how recognition of fungal pathogens triggers glycolytic activity by selectively regulating Glut1 expression and function in neutrophils during antifungal immunity.
Results
Glut1 transcription and phosphorylation are upregulated in neutrophils during disseminated candidiasis
The mechanisms by which neutrophils meet their high metabolic demand for rapid antimicrobial activities are poorly understood. We induced disseminated candidiasis by intra-venous (i.v.) infection with C. albicans (strain SC5314, 105 CFU) and found that glucose uptake is increased in neutrophils within the infected kidney compared to sham-infected mice at 24 h post-infection (p.i.), as measured by the enhanced incorporation of glucose analog 2-NBDG (Fig 1A, S1A). Moreover, in vitro 2-NBDG incorporation assay revealed increased glucose transport by murine bone marrow (BM)-derived neutrophils at 1.5 h post-C. albicans stimulation (first time point analyzed) (Fig S1B).
Fig 1: Neutrophils upregulate Glut1 transcription and translocation during fungal stimulation.
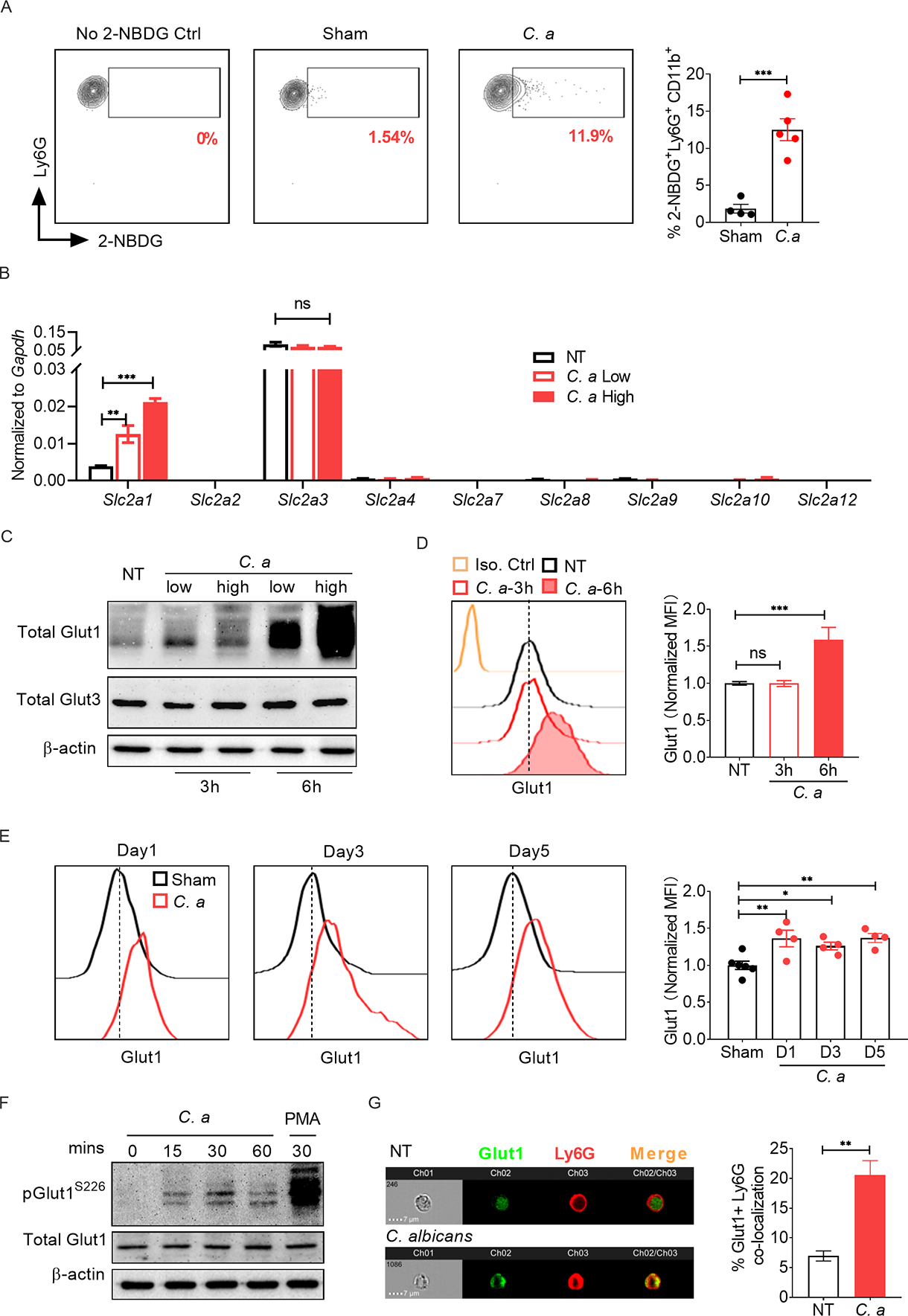
(A) WT mice (C57BL/6) were infected with 105 CFU of C. albicans (C. a) for 24 h (n=4–5). Glucose uptake by kidney infiltrating neutrophils was detected by injecting 2-NBDG followed by flow cytometry. (B) BM neutrophils from WT mice were stimulated with C. a [multiplicity of infection (MOI)=0.2 (low) or 1 (high)] for 3 h. Gene expression of different glucose transporters was measured by qPCR, normalized to Gapdh. Neutrophils were stimulated with C. a for 3 h or 6 h. Glut1 and Glut3 protein were evaluated by (C) western blot and (D) flow cytometry. (E) WT mice were infected with C. a (n=4–6). Glut1 expression in kidney-infiltrating neutrophils was measured by flow cytometry at indicated days p.i. (F) Neutrophils were stimulated with C. a (MOI=1) and phosphorylation of Glut1 (pGlut1S226) was assessed by western blot. Phorbol 12-myristate 13-acetate (PMA) (100 ng/ml) was used as a positive control. (G) Neutrophils were stimulated with C. a (MOI=1) for 1.5 h. Surface localization of Glut1 in neutrophils was visualized by ImageStream analysis. The surface translocation of Glut1 was expressed as percentage of surface expressed (Ly6G co-localized) Glut1 out of total Glut1 protein. Data pooled from 2 (A and E) and 3–4 independent experiments (B-D, F and G) and representative images and histogram plots are shown. Statistical analysis by Student’s T test (A, G), One-way ANOVA (D, E) and Two-way ANOVA (B). Data are represented as mean ± SEM (A, B, D, E, F). See also Fig S1.
To define how neutrophils incorporate glucose in disseminated candidiasis, we evaluated expression of multiple glucose transporters in BM neutrophils in the presence or absence of C. albicans (yeast and pseudo-hyphae) for 3 h under in vitro condition. Of 9 glucose transporters tested, only the Slc2a1 (encoding Glut1) and Slc2a3 (encoding Glut3) transcripts were expressed in neutrophils (Fig 1B). Slc2a3 was highly expressed at baseline and remained unchanged during C. albicans stimulation. In contrast, Slc2a1 mRNA was low at baseline but upregulated in neutrophils at 3 h post-C. albicans stimulation in a dose-dependent manner. Congruent with mRNA expression, there was increased Glut1 protein at 6 h but not 3 h post-fungal stimulation (Fig 1C, D). There was no alteration in Glut3 protein levels in C. albicans stimulated neutrophils at these time points. Consistent with these in vitro studies, kidney-infiltrating neutrophils showed increased Glut1 expression during disseminated candidiasis, assessed at 1, 3 and 5 days p.i. (Fig 1E). These data indicate that Glut1 is transcriptionally regulated in neutrophils in systemic fungal infection.
From these results, it was clear that neutrophils incorporate glucose as early as 1.5 h post-C. albicans stimulation, even though there was no evident increase in Glut1 transcript or protein over baseline at this time point. A previous study showed that Glut1 is stored in intracellular vesicles and PKC-dependent phosphorylation of Glut1 is required for surface localization and glucose transport (Lee et al., 2015). We hypothesized that neutrophil activation would lead to rapid phosphorylation and subsequent translocation of a pool of pre-formed Glut1 from cytoplasmic vesicles to the cell membrane, which will aid in early glucose uptake at 1.5 h post-stimulation. Indeed, there was rapid phosphorylation of Glut1 (pGlut1S226) within 30 minutes of C. albicans stimulation (Fig 1F), followed by Glut1 translocation to the cell membrane at 1.5 h post-stimulation, measured by ImageStream analysis (Fig 1G). Overall, these data indicate that C. albicans stimulation regulates Glut1 transcription, phosphorylation, and cell membrane translocation in neutrophils.
Fungal β-glucan regulates Glut1 transcription and phosphorylation in neutrophils
To define how host-pathogen interaction regulates Glut1 function in neutrophils, we assessed the role of fungal PRRs. Heat-killed (HK) C. albicans has exposed cell wall components such as β-glucans, which bind to the CLR dectin-1. Indeed, HK-C. albicans-stimulated neutrophils induced expression of Slc2a1 mRNA and phosphorylation of Glut1 at levels similar to live fungus at 3 h post-stimulation, suggesting that fungal non-protein and heat- resistant protein ligands control Glut1 function in neutrophils (Fig 2A, B). Accordingly, we activated neutrophils with purified fungal cell wall components including mannan, curdlan and zymosan. Curdlan and zymosan but not mannan increased Slc2a1 transcription and phosphorylation of Glut1 in neutrophils in a dose-dependent manner (Fig 2A, B). Since both curdlan and zymosan are β-glucans, we used curdlan for subsequent experiments. The curdlan-stimulated neutrophils demonstrated increased membrane translocation of Glut1 and in vitro glucose uptake (Fig 2C, D). In keeping with these results, BM neutrophils from Clec7a (Dectin-1)-deficient mice but not Myd88−/− animals exhibited diminished expression of Slc2a1 mRNA and Glut1 phosphorylation upon C. albicans or curdlan stimulation (Fig 2E, F and Fig S1C, D). These results indicate that dectin-1-dependent recognition of fungal β-glucan regulates Glut1 transcription and phosphorylation in neutrophils.
Fig 2: Fungal β-glucan and dectin-1 interaction controls Glut1 function in neutrophils.
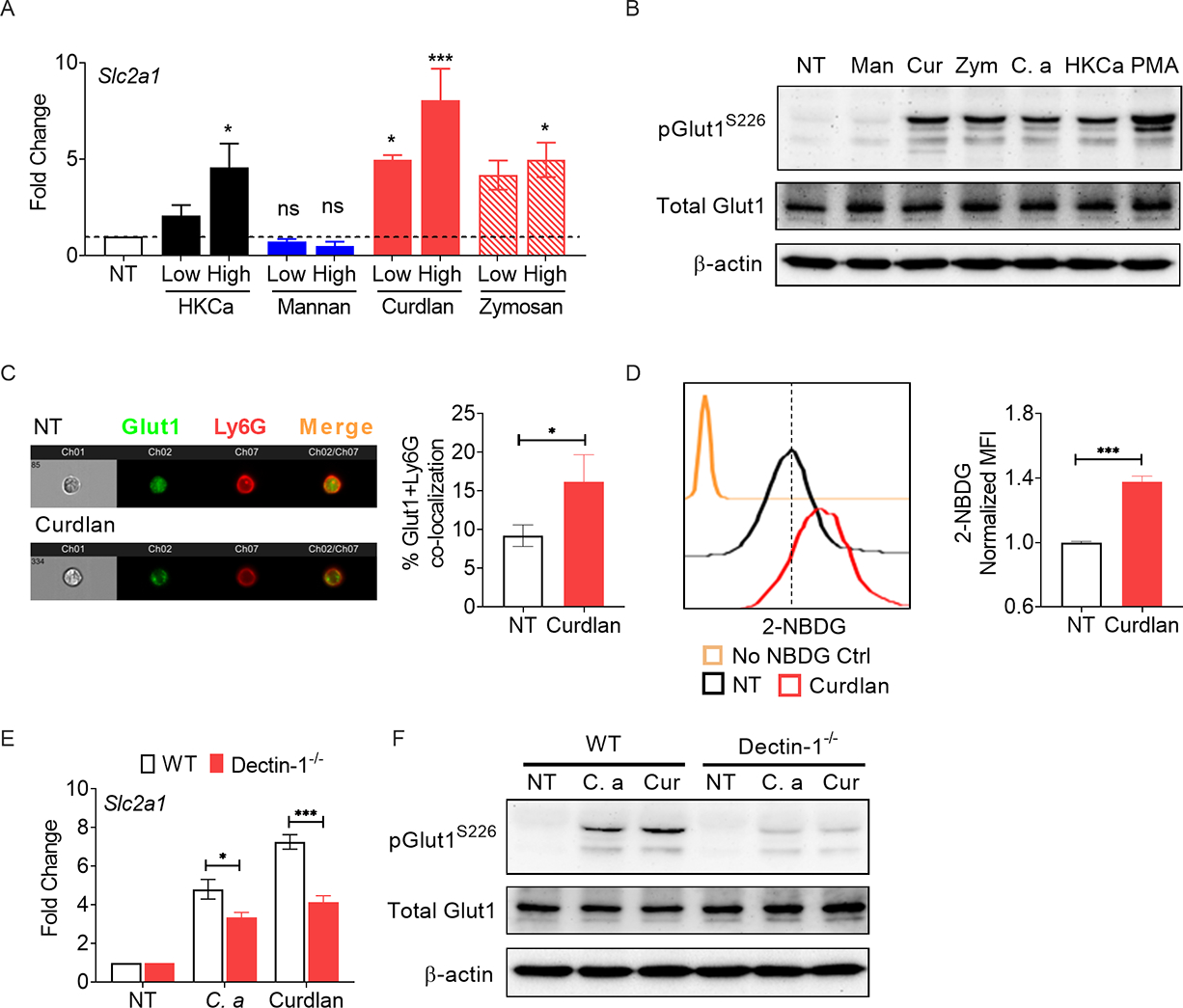
(A) BM neutrophils from WT mice were stimulated with heat-killed C. albicans (C. a) [HKCa, MOI=1 (low) or 5 (high)], mannan [Man: 10 (low) or 100 μg/ml (high)], curdlan [Cur: (10 (low) or 100 μg/ml (high)], or zymosan [Zym: (10 (low) or 100 μg/ml (high)] for 3 h. Gene expression of Slc2a1 was measured by qPCR. (B) Neutrophils were stimulated with C. a (MOI=1), HKCa (MOI=1), mannan (100 μg/ml), curdlan (100 μg/ml) or zymosan (100 μg/ml) for 30 min and Glut1 phosphorylation was assessed by western blot. Neutrophils were stimulated with curdlan (100 μg/ml) for 1.5 h. (C) Localization of Glut1 in neutrophils was visualized by ImageStream analysis and (D) glucose uptake (2-NBDG+ cells) was detected by flow cytometry. (E) Neutrophils from WT or Dectin-1−/− mice were stimulated with C. albicans (MOI=0.2) or curdlan (10 μg/ml) for 3 h. Gene expression of Slc2a1 was measured by qPCR. (F) Neutrophils from WT or Dectin-1−/− mice were stimulated with C. a (MOI=1) or curdlan (100 μg/ml). Phosphorylation of Glut1 was assessed by western blot at 30 min post-stimulation. Data pooled from at least 3 independent experiments (A-F) and representative images and histogram plots are shown. Statistical analysis by One-way ANOVA (A), Student’s T test (C, D) and Two-way ANOVA (E). Data are represented as mean ± SEM (A, C, D, E). See also Fig S1.
Activation of the dectin-1/Syk pathway controls Glut1 transcription and phosphorylation in neutrophils
The signaling pathways downstream of CLRs include Syk-dependent activation of PKCδ and CARD9, as well as a Syk-independent Raf-1 pathway (Geijtenbeek and Gringhuis, 2009). Both Syk-dependent and Syk-independent pathways activate NF-κB (Fig 3A). Selective blocking of Syk, PKCδ, CARD9 or NF-κB activity with pharmacological signaling inhibitors or gene knock-out mice impaired upregulation of Slc2a1 transcription in neutrophils in response to C. albicans or curdlan, assessed at 3 h (Fig 3B). The inhibitors showed no effect on neutrophil viability at the working concentrations (Fig S1E). In contrast, Raf-1 inhibition did not impact Slc2a1 expression (Fig S2A). These results demonstrate a specific role for the Syk/PKCδ/CARD9/NF-κB pathway in promoting Slc2a1 transcription following fungal recognition.
Fig 3: Glut1 regulation is mediated by dectin-1/PKCδ pathway in neutrophils.
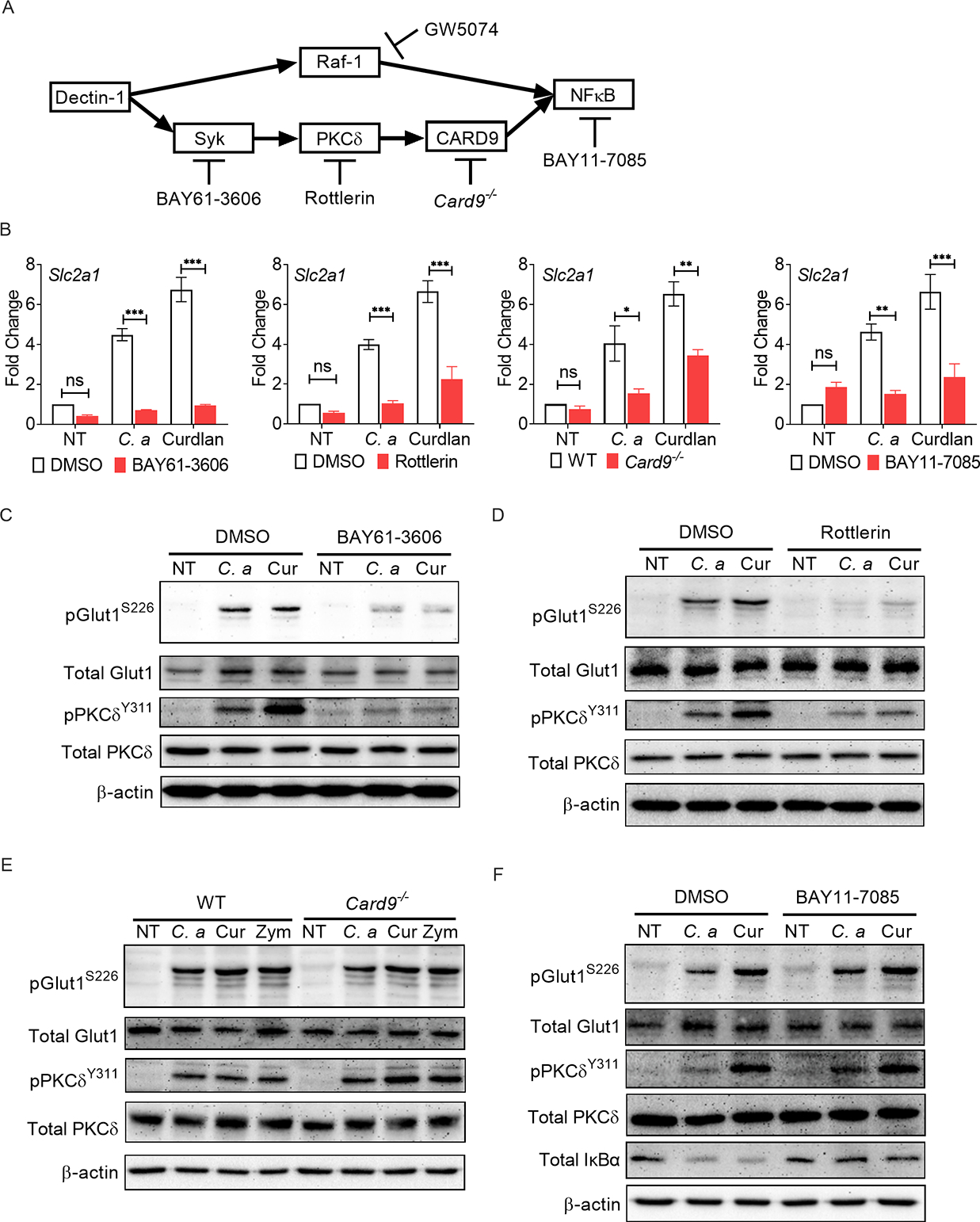
(A) Schematic diagram of downstream signaling pathways of dectin-1. The diagram also shows different inhibitors or gene knockout mice used to sequentially block the signaling intermediates in dectin-1 signaling pathway. (B) BM neutrophils from WT or Card9−/− mice were stimulated with C. albicans (C. a) (MOI=0.2) or curdlan (Cur) (10 μg/ml) for 3 h with or without indicated inhibitors. Gene expression of Slc2a1 was measured by qPCR. (C-F) BM neutrophils from WT or Card9−/− mice were stimulated with C. a (MOI=1) or curdlan (100 μg/ml) in the presence or absence of indicated inhibitors. Phosphorylation of Glut1 was assessed by western blot at 30 min post-stimulation. Data pooled from at least 3 independent experiments (B-F) and representative images are shown. Statistical analysis by Two-way ANOVA (B). Data are represented as mean ± SEM (B). See also Fig S2.
We also detected phosphorylation of Glut1 following selective blocking of Syk, PKCδ, CARD9 or NF-κB activity with pharmacological inhibitors or knock-out mice in neutrophils (Fig 3A). Neutrophils stimulated with C. albicans or curdlan showed increased phosphorylation of PKCδ and Glut1 relative to vehicle treated group at 30 min post-stimulation (Fig 3C). Pharmacological inhibition of Syk or PKCδ completely blocked Glut1 phosphorylation (Fig 3C, D). Inhibition of Raf-1 showed no impact on the phosphorylation of Glut1 (Fig S2B). Surprisingly, loss of CARD9 or treatment with an NF-κB inhibitor also had no effect on Glut1 phosphorylation (Fig 3E, F), indicating that PKCδ activation upstream of CARD9 is required and sufficient for Glut1 phosphorylation. Overall, these data highlight that activation of Syk and PKCδ downstream of dectin-1 activation is required for the phosphorylation of Glut1 in neutrophils but CARD9, NF-κB or Raf-1, are not.
Compromised glucose uptake by Card9−/− neutrophils in disseminated candidiasis
In order to test the role of dectin-1/Syk/CARD9 pathway in regulation of Glut1 function, we next assessed Glut1 translocation and glucose uptake in neutrophils following sequential inhibition of signaling molecules in Syk-dependent pathway, as in Fig 3A. In line with the phosphorylation data (Fig 3C, D), Syk or PKCδ inhibitors blocked the surface translocation of Glut1 at 1.5 h post-curdlan stimulation (Fig 4A, B, Fig S2C, D). Accordingly, glucose uptake by neutrophils was reduced in the presence of Syk or PKCδ inhibitors at 1.5 h post-stimulation (Fig 4E, F, Fig S2G, H). In contrast, CARD9 deficiency or NF-κB inhibition showed no impact on the cell membrane translocation of Glut1 (Fig 4C, D, Fig S2E, F). The glucose incorporation in Card9−/− neutrophils or NF-κB inhibition was diminished at 6 h but not at 1.5 h post-stimulation (Fig 4G, H, I, Fig S2I, J, K). These results indicate that Glut1 function and glucose uptake in early stage (1.5 h post-stimulation) is mediated by the rapid phosphorylation and membrane translocation of pre-formed and intracellularly stored Glut1 by activation of PKCδ. This is proceeded by a late stage of glucose uptake (6 h post-stimulation), which is regulated by increased Glut1 transcription and protein expression in a Syk/PKCδ/CARD9/NF-κB-axis-dependent manner.
Fig 4: Increased glucose uptake in fungal stimulated neutrophils is mediated by Syk-PKCδ/CARD9/NFκB-axis.
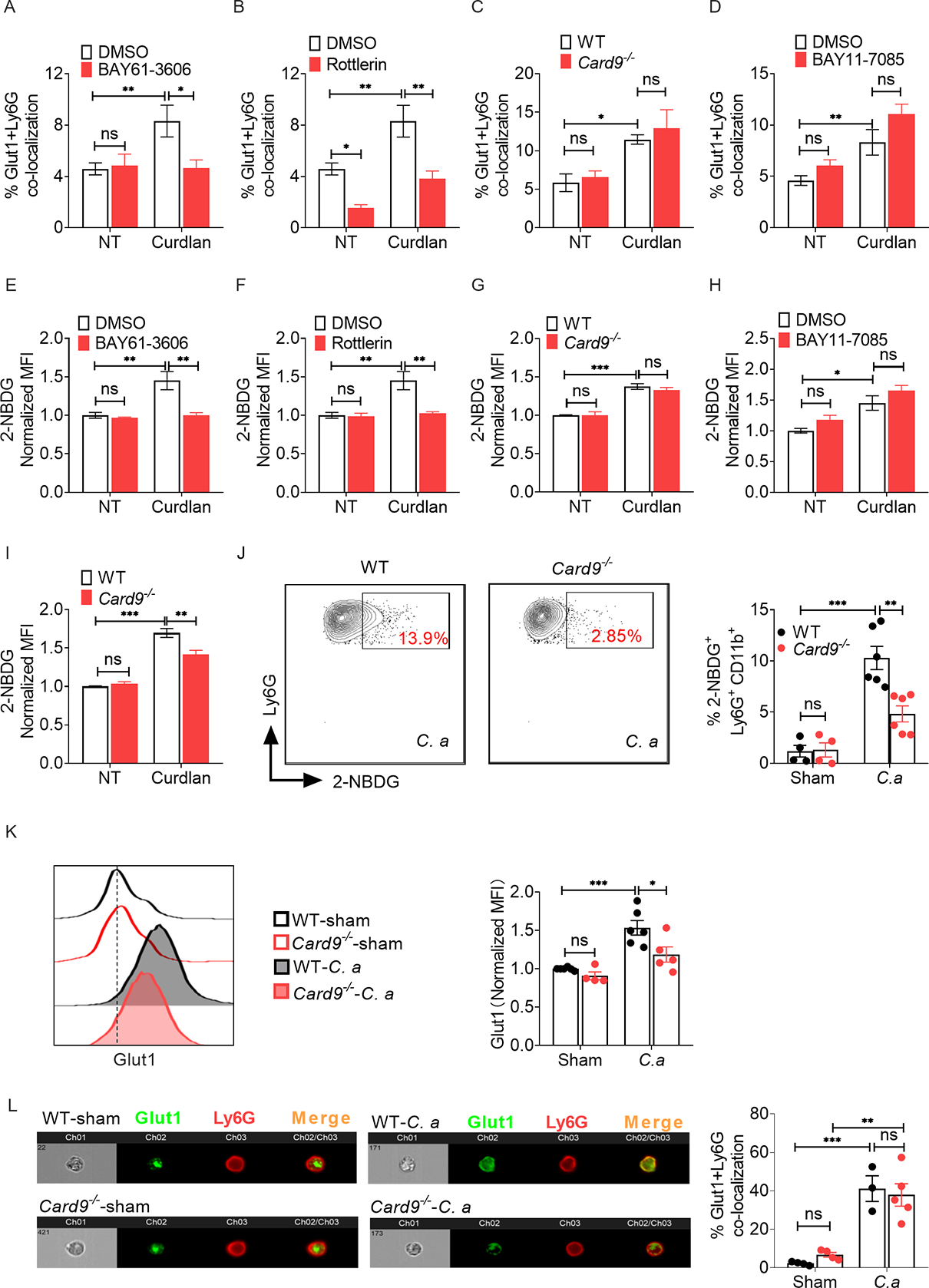
BM neutrophils from WT or Card9−/− mice were stimulated with curdlan (Cur) (100 μg/ml) for 1.5 h in the presence or absence of indicated inhibitors. (A-D) Glut1 localization in neutrophils was visualized by ImageStream and (E-H) glucose uptake (2-NBDG+ cells) by neutrophils was detected by flow cytometry. (I) Neutrophils from WT or Card9−/− mice were stimulated with curdlan (100 μg/ml) for 6 h, and glucose uptake by neutrophils was detected by flow cytometry. WT and Card9−/− mice were infected with C. albicans (C. a) for 24 h (n=4–6). (J) Glucose uptake and (K) Glut1 protein level in kidney infiltrating neutrophils were detected by flow cytometry. (L) Glut1 localization in neutrophils was visualized by ImageStream. Data pooled from at least 2–3 independent experiments (A-L) and representative images and flow cytometry plots are shown. Statistical analysis by Two-way ANOVA. Data are represented as mean ± SEM (A-L). See also Fig S2, S3.
To assess the contribution of these pathways to a physiological fungal infection setting, we measured glucose incorporation by Card9−/− neutrophils in kidney following disseminated candidiasis. WT and Card9-deficient mice showed comparable recruitment of neutrophils to the kidney at 24 h p.i., as described (Drummond et al., 2015) (Fig S3A). However, kidney-infiltrating neutrophils in Card9−/− mice showed reduced 2-NBDG+ cells compared to WT animals (Fig 4J, Fig S3B). Card9−/− neutrophils also exhibited diminished Glut1 protein expression at this time point (Fig 4K). However, the percentage of total Glut1 protein localized to the cell surface of kidney-infiltrating neutrophils was comparable between groups at 24 h p.i. (Fig 4L). These results indicate that Glut1 transcription but not phosphorylation or translocation in neutrophils is dependent on CARD9 during C. albicans infection and reveal functionally and kinetically distinct mechanisms by which glucose uptake is controlled in neutrophils during fungal encounter.
Glut1 deficiency inhibits glycolytic activity in neutrophils
To interrogate the role of neutrophil-specific Glut1 in disseminated candidiasis, we crossed MRP8-Cre-IRES/GFP and Slc2a1fl/fl mice to specifically delete Glut1 in this cell type (henceforth referred to as PMNΔGLUT1) (Lagasse and Weissman, 1994; Reber et al., 2017). In MRP8Cre mice, Cre recombinase gene, internal ribosomal entry site, and an enhanced green fluorescent protein gene were placed downstream of the human S100 calcium binding protein A8 (calgranulin A) (MRP8 or S100A8) promoter. GFP is conditionally expressed as a bicistronic transcript linked to MRP8 expression in the transgenic mice and reports MPR8Cre positive cells. PMNΔGLUT1 mice showed successful Glut1 deletion in neutrophils from blood, BM and spleen, as evidenced by reduced Glut1 protein expression (Fig 5A, B and Fig S4A). The lack of Glut1 did not impact Glut3 mRNA and protein expression in neutrophils at the baseline or after C. albicans stimulation (Fig 5B, Fig S4B). There was no detectable MRP8Cre activity in the kidney-resident macrophages and infiltrating monocytes/macrophages at the baseline (Fig S4C). Furthermore, PMNΔGLUT1 mice demonstrated normal neutrophil morphology and granulopoiesis, number of granulocyte-macrophage progenitors (GMPs) and neutrophils in the blood and spleen (Fig 5C, Fig S4D–F). The BM GMPs showed no MRP8Cre activity at the baseline and after infection (Fig S4G). These results indicate that Glut1 is not required for neutrophil development and survival.
Fig 5: Glut1 deficiency suppresses glycolysis in neutrophils during fungal stimulation.
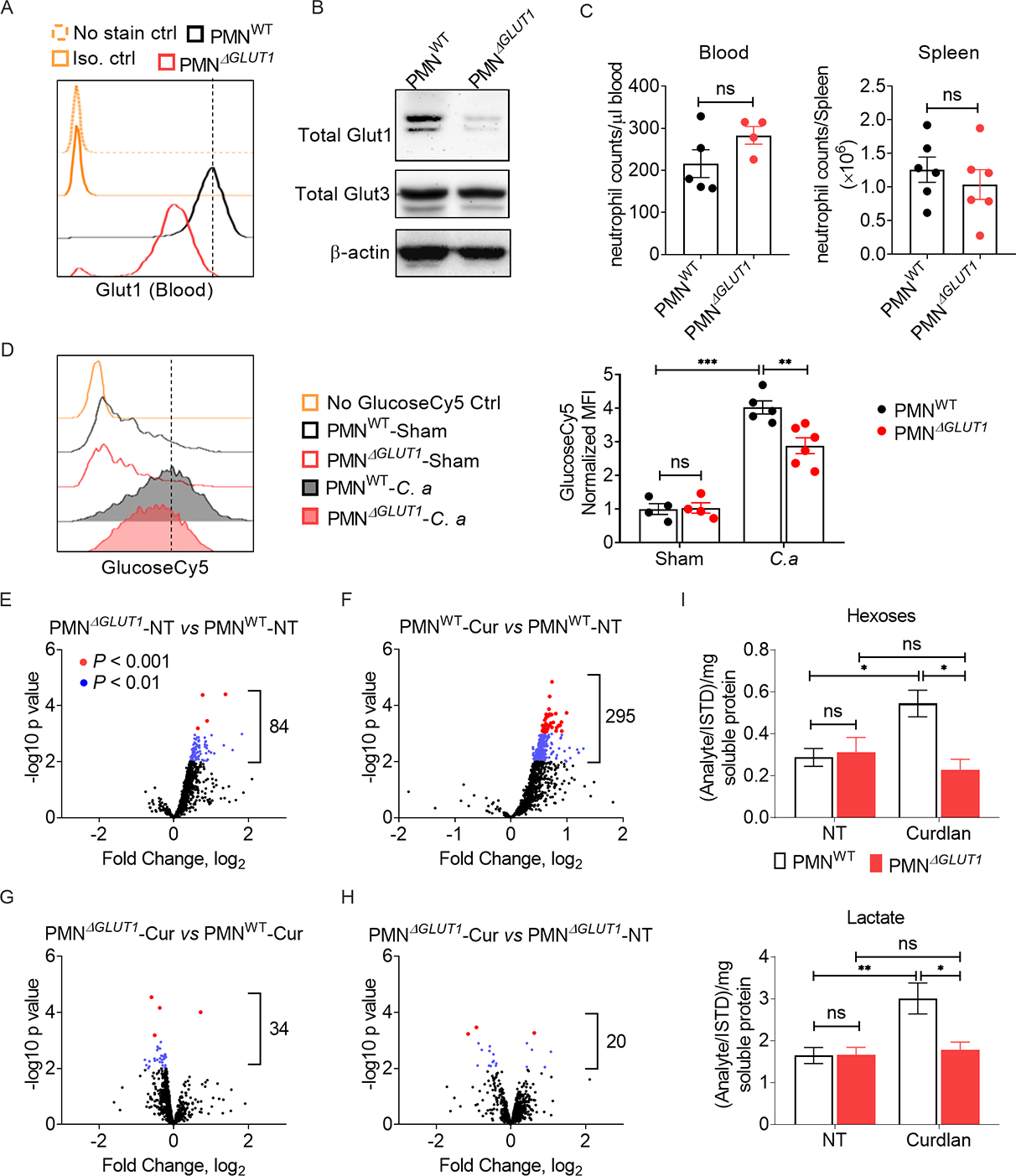
(A) Glut1 expression in mouse peripheral blood neutrophils was measured by flow cytometry. (B) Glut1 and Glut3 protein levels in BM neutrophils were measured by western blot. (C) The number of neutrophils in blood and spleen of 6–7 weeks old male mice was assessed by flow cytometry. (D) PMNWT and PMNΔGLUT1 mice were infected with C. albicans (C. a) for 24 h (n=4–6). Glucose uptake by kidney infiltrating neutrophils (GlucoseCy5+ cells) was detected by flow cytometry. (E-I) BM neutrophils from PMNWT or PMNΔGLUT1 mice were stimulated with curdlan (Cur) (10 μg/ml) for 3 h. Cell pellets were subjected to metabolomics by untargeted high-resolution LC-HRMS. Data pooled from at least 3 independent experiments (A-I) and representative images and flow cytometry plots are shown. Statistical analysis by Student’s T test (C) or Two-way ANOVA (D and I). Data are represented as mean ± SEM (C, D, I). See also Fig S4, S5.
We measured glucose uptake by kidney-infiltrating neutrophils from control (PMNWT) or PMNΔGLUT1 mice after systemic infection with C. albicans. The glucose analog 2-NBDG (FITC labeled) is incompatible with the GFP expressed in MRP8-Cre-IRES/GFP mice. So, we used another fluorescent glucose tracer, Cy5-linked 1-amino-glucose (GlucoseCy5), to monitor the in vivo glucose uptake by neutrophils (Watson et al., 2021). While glucose incorporation in kidney infiltrating neutrophils was comparable between the sham-infected groups, PMNΔGLUT1 neutrophils showed reduced glucose transport at 24 h p.i. (Fig 5D). These results indicate that while Glut1 is dispensable at the basal level, it is essential for glucose uptake in kidney-infiltrating neutrophils during disseminated candidiasis.
To further dissect the impact of Glut1 deletion on the energy metabolism, we performed metabolomics study on neutrophils from PMNΔGLUT1 and PMNWT mice. Since it is impossible to distinguish between fungal and mammalian metabolites, we used curdlan to stimulate the neutrophils. Metabolomics analysis revealed ~950 metabolites in neutrophils at 3 h post-curdlan stimulation, as assessed by untargeted high-resolution LC-HRMS. Curdlan-stimulated PMNWT neutrophils showed increased expression of 30% of the measured metabolites (295/950) compared to unstimulated control (Fig 5E, F, G, H). In contrast, very few metabolites (20/950) were changed in curdlan stimulated PMNΔGLUT1 neutrophils than control, suggesting impaired metabolic regulation in the absence of Glut1. The PMNΔGLUT1 neutrophils showed reduced expression of ~33 metabolites compared to PMNWT neutrophils after curdlan stimulation. Notably, the metabolites related to glycolysis including hexoses, lactate and hexose-6-phosphate were reduced in stimulated PMNΔGLUT1 neutrophils compared to PMNWT group (Fig 5I, Fig S5A). Interestingly, the effect of Glut1 deficiency on metabolites related to the TCA cycle and glutamate metabolism was minimal in curdlan-stimulated neutrophils (Fig S5B). When subjected to a glycolysis stress test, absence of Glut1 did not impact the basal glycolysis and glycolytic capacity of neutrophils (Fig S5C). However, glycolysis and glycolytic capacity was reduced in PMNΔGLUT1 neutrophils following curdlan stimulation. Overall, our data indicate that the absence of Glut1 compromised glucose uptake and glycolysis in neutrophils during fungal stimulation.
Loss of Glut1 in neutrophils increases susceptibility to disseminated candidiasis
We next assessed the susceptibility of PMNΔGLUT1 mice to invasive fungal infection. Mice were systemically infected with C. albicans and evaluated for survival over 14 days. PMNΔGLUT1 mice showed exacerbated susceptibility to systemic infection compared to PMNWT mice (Fig 6A). PMNΔGLUT1 mice succumbed to infection (~100% mortality) by day 7 p.i., a time point at which majority (~80%) of control mice were alive. Accordingly, PMNΔGLUT1 kidneys showed increased fungal burden and hyphal invasion into the renal parenchyma at day 3 p.i. (Fig 6B, C).
Fig 6: Deficiency of Glut1 in neutrophils exacerbates susceptibility of mice to disseminated candidiasis.
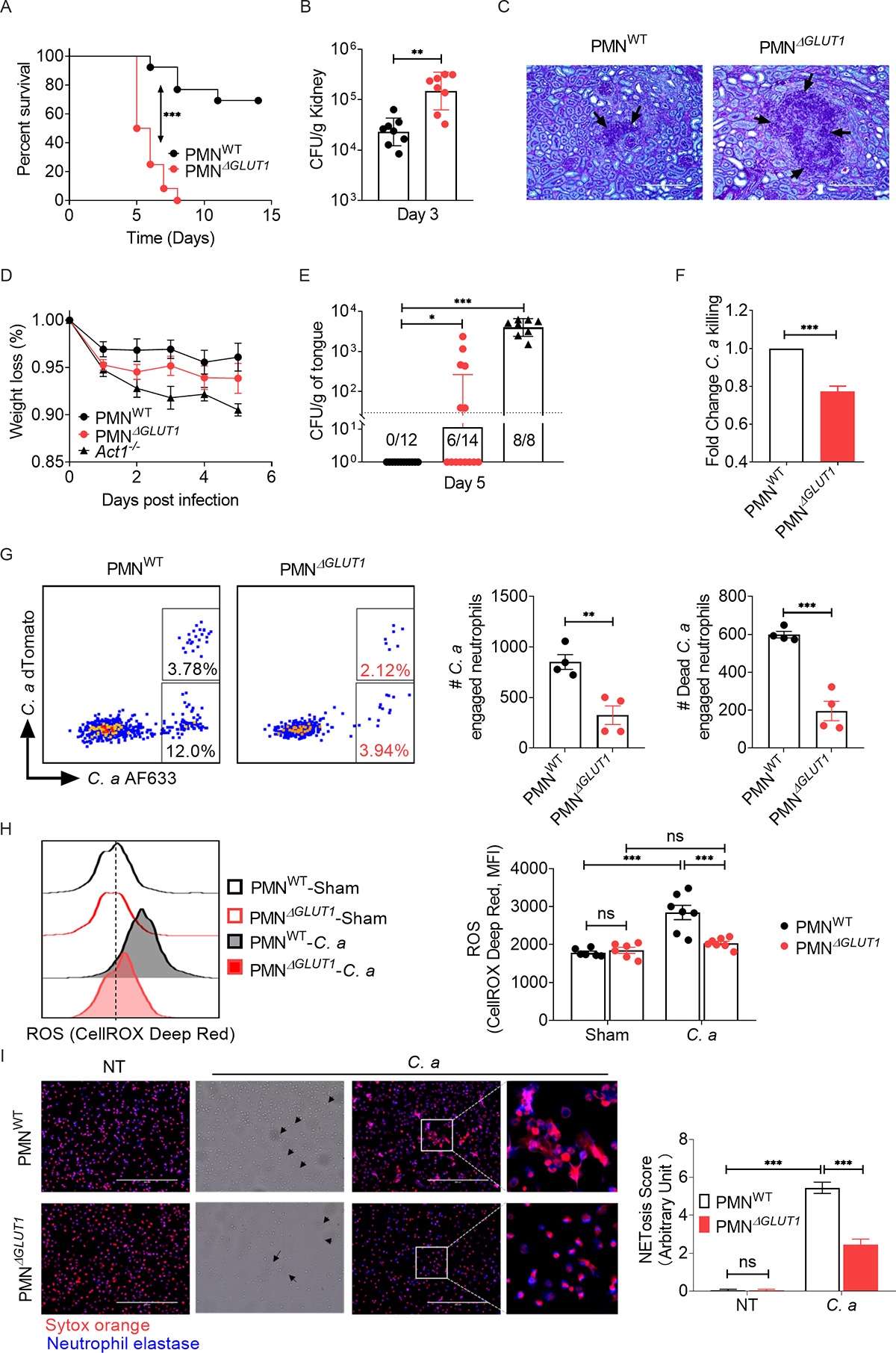
PMNWT and PMNΔGLUT1 mice were infected with C. albicans (C. a). (A) Mice were evaluated for survival over a period of 14 days (n=12). (B) Kidney fungal burden (n=8) and (C) renal histopathology (PAS staining) was evaluated at day 3 p.i. Black arrows indicate C. a hyphae in renal parenchyma. Magnification: 20×10. PMNWT and PMNΔGLUT1 mice were sublingually inoculated with a cotton ball saturated in C. a for 75 min (n=8–14). (D) Mice body weight were monitored daily and (E) fungal burden of the tongue was evaluated at day 5 p.i. (F) BM neutrophils from PMNWT or PMNΔGLUT1 mice were incubated with C. a (MOI=0.2) for 3 h. Candidacidal capacity was assessed by in vitro fungal killing assay. (G) PMNWT and PMNΔGLUT1 mice were infected with 5 × 106 CFU of Streptavidin-AF633+dTomato+ C. a. Mice were sacrificed 2 h p.i. and phagocytosis and intracellular killing by kidney-infiltrating neutrophils were measured by flow cytometry analysis. Live C. a engaged neutrophils emit two fluorescence signals (dTomato+AF633+) while dead C. a engaged neutrophils just emit only AF633 fluorescence signal (dTomato−AF633+). (H) Mice were infected with C. a for 24 h. ROS production in kidney infiltrating neutrophils were measured by CellROX® Deep Red reagent. (I) Neutrophils were incubated with C. a (MOI=0.2) for 3 h and NET formation was visualized by staining with Sytox Orange and anti-neutrophil elastase antibody. Black triangles indicate fungal hyphae. Magnification: 40×10. Frequency of NETosis score was blindly evaluated and expressed as arbitrary units. Data pooled from at least 2–3 independent experiments (A-I) and representative images and FACS plots are shown. Statistical analysis by log-rank test (A), Mann-Whitney test (B, E), Student’s T test (F, G) or Two-way ANOVA (H, I). Data are represented as geometric mean with geometric SD (B, E) or mean ± SEM (D, F, G, H, I). See also Fig S6, S7.
We also subjected PMNΔGLUT1 mice to oropharyngeal candidiasis (OPC), a setting where neutrophils play an important role in fungal clearance (Huppler et al., 2014). Following oral infection with C. albicans, PMNΔGLUT1 mice lost more body weight than controls (Fig 6D). At day 5 p.i., a time point when WT mice are known to fully clear C. albicans from the oral mucosa, PMNΔGLUT1 mice exhibited elevated oral CFU compared to PMNWT mice (Fig 6E). Collectively, these data highlight a critical role for neutrophil-specific Glut1 expression in antifungal host defense.
Cellular mechanisms of Glut1-dependent immunity to C. albicans
To define the cellular mechanisms for susceptibility of PMNΔGLUT1 mice to disseminated candidiasis, we quantified inflammatory cells infiltration in the kidney at 24 h p.i. Surprisingly, PMNΔGLUT1 and PMNWT mice showed comparable numbers of neutrophils (liveCD45+Ly6G+CD11b+ cells), monocytes (liveCD45+Ly6G−CD11c+CD11bloF4/80lo cells) and macrophages (liveCD45+Ly6G−CD11c+CD11bhiF4/80hi cells) in the infected kidney (Fig S6A). Similar results were obtained when neutrophil infiltration in the kidney was evaluated at an earlier time point (2 h p.i) (Fig S6B). Moreover, PMNΔGLUT1 neutrophils showed no defect in migration in the presence of the neutrophil-attracting chemokine CXCL2, assessed in a transwell migration assay (Fig S6C). There were also no obvious changes in patterns of cell death in these mice, with similar numbers of early (AnnexinV+7-AAD−) and late (AnnexinV+7-AAD+) apoptotic neutrophils in the infected kidney between groups (Fig S6D). These results indicate that the defect in renal fungal clearance in PMNΔGLUT1 mice cannot be attributed to impaired migration or increased apoptosis of neutrophils.
Neutrophils kill C. albicans mainly by phagocytosis, ROS formation and NETosis (Desai and Lionakis, 2018; Lionakis and Netea, 2013; Netea MG, 2015; Urban et al., 2006). An in vitro C. albicans killing assay demonstrated that Glut1 deficiency led to impaired fungicidal activity of neutrophils at 3 h post-incubation (Fig 6F). To determine the fungicidal capacity of neutrophils lacking Glut1 in vivo, we infected PMNΔGLUT1 and PMNWT mice with a streptavidin AF633-dTomato+ C. albicans reporter strain that enables simultaneous fluorescent visualization of phagocytosis and fungal killing in the same cells: AF633+dTomato+ signal report intracellular live fungi and AF633+dTomato− signal denote killed fungal cells within the neutrophils. As shown, kidney-infiltrating neutrophils in PMNΔGLUT1 mice had compromised phagocytosis and intracellular fungal killing activity compared to PMNWT mice (Fig 6G, S7A, B).
Our data show that PMNΔGLUT1 neutrophils are defective in glycolysis, a pre-requisite for generating NADPH oxidase-mediated ROS production (Fig 5) (Mayadas et al., 2014). Hence, we assessed ROS generation by kidney-infiltrating neutrophils during candidiasis. As expected, PMNΔGLUT1 neutrophils produced diminished ROS compared to control neutrophils in the infected kidney, as measured by CellROX® Deep Red at 24 p.i. (Fig 6H). Since NET formation is partially dependent on ROS generation (DeSouza-Vieira et al., 2016), we evaluated NETosis by PMNΔGLUT1 neutrophils in the presence of fungi. To that end, neutrophils were incubated with C. albicans for 3 h and NET formation was visualized by co-staining with Sytox Orange and an anti-neutrophil elastase antibody. Indeed, PMNΔGLUT1 neutrophils demonstrated reduced NET formation around fungal hyphae than PMNWT neutrophils (Fig 6I). Overall, these results demonstrate that Glut1-mediated glucose uptake in neutrophils is critical for physiological fungicidal activity and control of systemic C. albicans infection.
Glut1 in human neutrophils is essential for fungicidal activity
There are a number of differences between mouse and human neutrophils, including the absolute number in the blood, composition of granules, expression of neutrophil-derived antimicrobial peptides and ability to kill C. albicans (Zschaler et al., 2014). To assess the role of Glut1 in human PMNs, we isolated neutrophils from peripheral blood from 10 healthy volunteers and activated the cells with C. albicans or curdlan. As in murine neutrophils, SLC2A1 mRNA was upregulated 3 h after C. albicans or curdlan stimulation (Fig 7A). We also observed increased phosphorylation of Glut1 within 30 minutes of fungal or curdlan stimulation (Fig 7B). Moreover, human neutrophils exhibited increased in vitro glucose uptake (measured by 2-NBDG incorporation) during C. albicans stimulation. A Glut1-specific inhibitor (WZB117) blocked both glucose uptake and fungicidal activity in neutrophils (Fig 7C, D). Collectively, these data show an essential role for Glut1 in ensuring the ‘metabolic fitness’ of human neutrophils that enable them to mediate immunity to C. albicans infections.
Fig 7: Glut1 is critical for fungicidal function of human neutrophils.
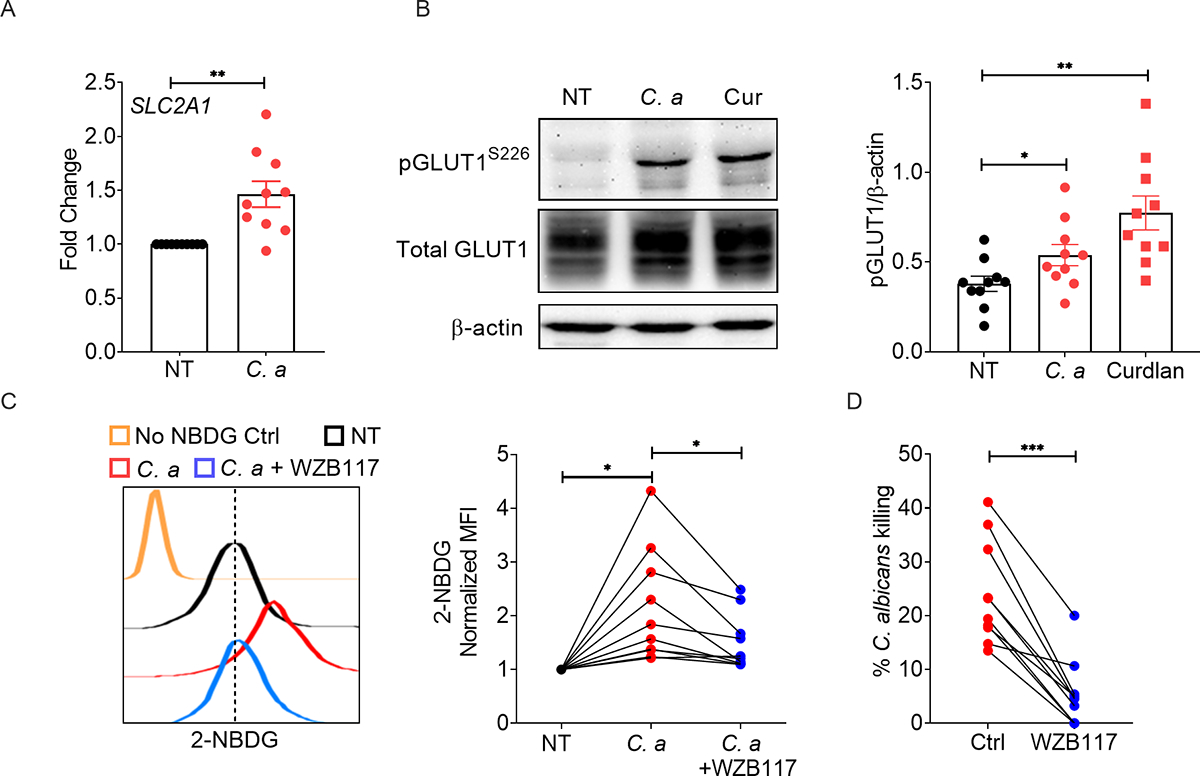
Healthy donor neutrophils (n=10) were stimulated with C. albicans (C. a) (MOI=0.2) for 3 h. (A) Gene expression of SLC2A1 was measured by qPCR. (B) Neutrophils isolated from healthy volunteers were stimulated with C. a (MOI=1) or curdlan (100 μg/ml) for 30 min. Glut1 phosphorylation was assessed by western blot. Representative image and ImageJ quantification of band intensities are shown. Healthy donor neutrophils were stimulated with C. a (MOI=1) in the presence or absence of Glut1 inhibitor (WZB117: 10 μM) for 1.5 h. (C) Glucose uptake by neutrophils was detected by flow cytometry, and (D) candidacidal activity of neutrophils was assessed by in vitro fungal killing assay. Statistical analysis by Student’s T test (A, D) and One-way ANOVA (B, C). Data are represented as mean ± SEM (A, B). See also Table S1.
Discussion
The regulation of cellular metabolic pathways is a critical determinant of immune function, a field that has gained renewed appreciation in recent years. To date, most studies of immunometabolism focus on how T cells and macrophages are impacted, with surprisingly little emphasis on understanding metabolic regulation of neutrophil function. Neutrophils are short lived, terminally differentiated cells and execute rapid antimicrobial function in diverse tissues and have unique metabolic requirements (Burn et al., 2021). How neutrophils dynamically regulate metabolism to fuel antifungal activity is poorly understood. We show that neutrophils upregulate Glut1 expression and glucose transport across the cell membrane in candidiasis. Recognition of fungal β-glucan via CLRs induced rapid phosphorylation, surface localization and transcription of Glut1 in a dectin-1 dependent manner. Mice with a neutrophil-specific deletion of Glut1 showed increased susceptibility to candidiasis. We also demonstrated that β-glucan-mediated upregulation of Glut1 is essential for antifungal functions in human neutrophils. Overall, these results reveal that fungal sensing ensures ‘metabolic fitness’ of neutrophils, which is required for host defense against candidiasis.
We demonstrate that Glut1 transcript and protein expression were upregulated in neutrophils in disseminated candidiasis. Although baseline Glut3 mRNA was expressed at a higher level than Glut1, Glut3 mostly remained unchanged during fungal stimulation, indicating that Glut1 and Glut3 are differentially regulated in neutrophils. The transcription factor RelA binds to the Glut1 promoter (Wang et al., 2019), thus confirming the role of NF-κB in the selective regulation of Glut1 in neutrophils. In contrast, Glut3 is upregulated in primary human monocytes following C. albicans stimulation (Wu et al., 2021). In a recently published report, mice with a neutrophil-specific deletion of Glut3 showed diminished number of neutrophils in the blood, indicating that Glut3 is indispensable for neutrophil development and survival (Ancey et al., 2021). Deletion of Glut1 partially inhibited neutrophil-mediated glucose transport, suggesting that Glut3 may contribute to increased glucose uptake during fungal stimulation. In-depth studies utilizing tamoxifen inducible Cre mice to ablate Glut3 are required to interrogate the role of Glut3 in host defense against candidiasis.
The recognition of fungal PAMPs via CLRs is crucial for innate immunity and clearance of fungal pathogens (Netea et al., 2006; Taylor et al., 2007). Here, we show that fungal β-glucan recognition via dectin-1 is also required for metabolic reprogramming of neutrophils, which is essential for the antifungal activities. Intriguingly, the lack of dectin-1 partially inhibited the upregulation of Glut1, indicating the contribution of additional PRRs. Accordingly, selective inhibitors of Syk and PKCδ completely abrogated Glut1 upregulation in neutrophils. We observed that mannan was unable to induce Glut1 expression, which may be due to low dectin-2 expression on mouse neutrophils (Taylor et al., 2014). The impact of fungal PAMPs on alternative metabolic pathways including fatty acid oxidation and glutaminolysis, known to regulate neutrophil function during limited glucose availability in tissues, warrants careful investigation (Kumar and Dikshit, 2019).
While most antifungal activities were diminished in the absence of Glut1, chemotaxis was unaffected in neutrophils. This observation is in line with our prior study showing that uremic neutrophils with compromised Glut1-mediated glucose uptake exhibited normal migration during disseminated candidiasis (Jawale et al., 2020). The neutrophils respond to chemotactic signals instantly and make extensive cytoskeleton reorganization, which is an energy dependent process. However, there are several possibilities that can explain this result. Numerous reports suggested that mitochondria and TCA cycle regulate chemotaxis in neutrophils (Bao et al., 2015; Chen et al., 2006; Zhou et al., 2018). Accordingly, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone treatment, a potent uncoupler of mitochondrial oxidative phosphorylation, suppressed neutrophil migration (Fossati et al., 2003). Alternatively, energy derived from break down of stored glycogen by glycogenolysis may compensate for the loss of glucose uptake during chemotaxis.
We show a dynamic and kinetically distinct regulation of Glut1-mediated glucose uptake in neutrophils during candidiasis. Following fungal recognition via CLRs, the early stage enables rapid glucose transport and is mediated by PKCδ-driven phosphorylation of pre-formed Glut1 in cytoplasmic vesicle. These early events are followed by increased Glut1 gene transcription and Glut1 accumulation to replenish Glut1 level inside the cell and sustain antifungal activities by neutrophils, dependent on the β-glucan/dectin-1/CARD9-axis. The physiological significance of early glucose uptake is underscored by the fact that PKCδ, but not CARD9, promotes neutrophil ROS production and C. albicans killing within 3 h (Li et al., 2016). Interestingly, dectin-1/Akt/mTOR/HIF-1α and the noncanonical Raf-1 pathways have been implicated in increased glucose consumption and a shift towards glycolysis dependent metabolic pathway in ‘trained’ human monocytes with trained immunity (Cheng et al., 2014). Although Raf-1 pathway is dispensable for Glut1-mediated glucose uptake in neutrophils, we showed that mTORC1 inhibition suppressed glucose incorporation in neutrophils in fungal infection (Jawale et al., 2020). Moreover, only Syk inhibition but not PKCδ or NK-κB inhibitors showed a greater reduction in Slc2a1 mRNA expression than Card9−/− neutrophils, suggesting a Syk dependent but Card9 independent signaling mechanisms in Slc2a1 gene regulation in neutrophils. Although the small molecule inhibitors used in the present study showed no effect on neutrophil viability, potential off-target effects of these inhibitors on Glut1 regulation cannot be completely ruled out.
The neutrophils are exposed to a variety of tissue micro-environments during infection, which poses metabolic stress on these innate cells. The neutrophils respond to these changing environments by enhancing glucose uptake to execute antimicrobial activities (Weerasinghe and Traven, 2020). Moreover, recent studies have indicated that neutrophils exhibit preferences for utilizing alternative metabolic pathways depending on the availability of nutrients in tissues (Jeon et al., 2020; Kumar and Dikshit, 2019). Hence, we tested the ability of PMNΔGLUT1 animals to control fungal infection in OPC, where neutrophils play an important role in fungal clearance in the oral cavity. In contrast to kidney fungal burden in DC, PMNΔGLUT1 mice exhibited moderate impairment in fungal clearance in the oral cavity during OPC, thus phenocopying Card9−/− mice (Bishu et al., 2014). In this context, it is important to note that MRP8 promoter was originally described to drive Cre-mediated deletion in 80–90% of neutrophils, with minor leakage (<20%) into some monocyte/macrophage populations (Abram et al., 2014).
A comprehensive understanding of the metabolic regulator(s) in neutrophils could be helpful in unraveling the fundamental mechanisms of antifungal immunity, with broader implications for other infections and autoinflammatory diseases. Additionally, this knowledge may allow timely and selective targeting of metabolic pathways to augment antifungal effector mechanisms of neutrophils without causing bystander damage to host tissues.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Partha S. Biswas (psb13@pitt.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The study did not create large data sets that required deposition in a public data base. Microscopy data reported in this paper will be shared by the lead contact upon request.
The study did not create original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental models and subject details
Mice
C57BL/6 (WT) mice were purchased from Taconic Biosciences Inc. MRP8-Cre+, Card9−/−, Clec7a−/− and Myd88−/− mice were purchased from the Jackson laboratory. Slc2a1fl/fl mice are under Material Transfer Agreement (MTA) from University of Colorado, USA. To generate mice with conditional deletion of Glut1 in neutrophils, we crossed the Slc2a1fl/fl mice with MRP8-Cre-IRES/GFP mice. All mice were housed in specific pathogen free conditions under the supervision of Division of Laboratory Animal Resources, University of Pittsburgh. All animal experiments were conducted following the NIH guidelines under protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Mouse model of disseminated candidiasis and oropharyngeal candidiasis (OPC)
C. albicans (Strain SC5314) was grown in yeast extract-peptone-dextrose (YPD) broth at 30°C for 16 to 20 hours in a shaker incubator. Eight to ten weeks old male mice were infected with 105 CFU of C. albicans per mouse via the lateral tail vein and daily monitored for signs of illness, weight loss, and survival. Mice showing >20% weight loss or signs of severe pain or distress were sacrificed for humane purpose. For fungal burden evaluation, mice were sacrificed at day 3 p.i. and kidneys were weighed and homogenized using a GentleMACS. Serial dilutions of homogenates were plated on YPD agar, and fungal burden was represented as CFU per gram of kidney. Formalin-fixed tissue sections were stained with Periodic Acid-Schiff (PAS) stain for visualization of C. albicans.
OPC was induced as previously described (Solis and Filler, 2012). Briefly, mice were sublingually inoculated with a cotton ball saturated in C. albicans (Strain SC5314) for 75 min under anesthesia. Then the mice were daily monitored for signs of illness and weight loss. On day 5 p.i., mice tongue was weighed and homogenized. Fungal burdens were determined by plating serial dilutions of homogenates on YPD agar, and fungal burden was represented as CFU per gram of tongue.
Human subjects
All human subjects’ studies were approved by the University of Pittsburgh Institutional Review Board. The biological samples were obtained after providing written informed consent. The healthy donor blood samples (n=10) were obtained as part of a study entitled “Banking of Biological Samples and Collection of Clinical Data for Connective Tissue Disease” at the Division of Rheumatology and Clinical Immunology, University of Pittsburgh. All demographic information including race, gender and age of the study subjects are provided as Supplementary Table 1.
Neutrophil Culture
Isolated mouse or human neutrophils were used immediately for in vitro experiments. Neutrophils were cultured in RPMI 1640 Complete Medium at 37°C with 5% CO2. The cells were seeded in 24-well flat bottom plates with a concentration of 1.5 × 106/well and stimulated for 30 min to 6 hours depending on the experiment.
Method details
Mouse and human neutrophil isolation
Mouse femur and tibia BM-derived neutrophils were isolated using Neutrophil Isolation Kit, following manufacturer instructions. Neutrophil purity (>90%) was checked by FACS (liveLy6G+CD11b+). Human peripheral blood neutrophils were isolated from healthy donors using MACSxpress Human Neutrophil Isolation Kit, following manufacturer instructions, and checked for purity (>95%) by FACS.
Glucose uptake measurement
BM neutrophils were seeded in 96-well flat bottom plates and incubated at 37°C for 30 min. Cells were stimulated with C. albicans (MOI=1) or curdlan (100 μg/ml) for 1.5 hours or 6 hours. The cells were washed with PBS and 100 μl 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG, 100 μM in PBS) was added. Cells were incubated at 37 °C for another 30 min. The glucose uptake by neutrophils was detected by FACS.
For glucose uptake measurement in kidney-infiltrating neutrophils, mice were infected with 105 CFU of C. albicans via the lateral tail vein. After 24 hours, mice were injected i.v. with 200 μL 2-NBDG (2.5 mM in PBS) or glucoseCy5 (37 μM in PBS). Thirty min later, kidney infiltrating neutrophils were analyzed for glucose uptake by flow cytometry (FACS).
Glut1 expression
BM neutrophils were seeded in 24-well flat bottom plates and incubated at 37°C for 30 min. Cells were stimulated with C. albicans (MOI=1) or curdlan (100 μg/ml) for 3 hours or 6 hours. For FACS analysis, the cells were fixed and anti-Glut1 antibody or Rabbit IgG isotype control antibody were used for intracellular staining. For Western blot detection of Glut1, neutrophils were lysed and total protein was stained for Glut1 using anti-Glut1 antibody.
For Glut1 protein level measurement in kidney-infiltrating neutrophils, mice were systemically infected with 105 CFU of C. albicans. After 24 hours, kidney cell suspensions were prepared and Glut1 protein expression in neutrophils was measured by FACS.
Imagestream analysis
BM neutrophils were seeded in 24-well flat bottom plates and incubated at 37°C for 30 min. Cells were stimulated with C. albicans (MOI=1) or curdlan (100 μg/ml) for 1.5 hours. For Imagestream analysis, the cells were stained with anti-Ly6G antibody followed by intracellular staining with anti-Glut1 antibody. The co-localization of Ly6G and Glut1 fluorescence was detected by Imagestream analysis and expressed as percentage of surface expressed (Ly6G co-lacalized) Glut1 out of total Glut1 protein.
For Glut1 localization in kidney-infiltrating neutrophils, mice were infected with 105 CFU of C. albicans. After 24 hours, kidney cell suspensions were subjected to Imagestream analysis to detect Glut1 localization in neutrophils.
Inhibitors for in vitro studies
Inhibitors used for the in vitro experiments include 2 μM BAY61–3606 (Syk inhibitor), 10–50 μM Rottlerin (PKCδ inhibitor), 1 and 10 μM GW5074 (Raf-1 inhibitor), 1 μM BAY11–7085 (NFκB inhibitor), and 10 μM WZB117 (Glut1 inhibitor). All the inhibitors were incubated with neutrophils 30 min before stimulation. The stock solution of BAY61–3606 (10 mM), Rottlerin (20 mM), GW5074 (10 mM), BAY11–7085 (10 mM) and WZB117 (10 mM) was made by dissolving each inhibitor in DMSO. The stock solutions were further diluted using media to make working concentrations.
Flow cytometry
Single cell suspensions from tissues were prepared as previously described (Jawale et al., 2020). Briefly, spleens were subjected to mechanical dissociation followed by red blood cell lysis. For kidneys, mice were perfused with PBS containing 10 mM EDTA before the harvest. Kidneys were digested with collagenase IV for 20 min and homogenates were passed through 70 μm cell strainers.
Staining of mouse cells was performed with surface antibodies (CD45, CD11b, Ly6G, Ly6C, F4/80, CD34, CD3, CD19, B220 and C-kit) and intracellular antibody (Glut1). Cells were fixed and permeabilized before intracellular staining. Dead cells were excluded using the Live/Dead Ghost Dye Violet 510 dye. To quantify the number of cells, cell counting beads were added before the run. The samples were acquired on BD LSR Fortessa cytometer and analyzed with FlowJo software.
In vivo neutrophil phagocytosis and Candida killing assay
In vivo neutrophil phagocytosis and Candida killing assay was performed as previously described (Jawale et al., 2020). Briefly, dTomato+ C. albicans were incubated with 0.5 mg/mL Biotin-XX, sulfo-SE for 2 h and then 0.02 mg/mL streptavidin conjugated to Alexa Fluor 633 for another 30 min. Mice were infected with 5 × 106 CFU of AF633+tdTomato+ C. albicans via lateral tail vein. Mice were sacrificed at 2 h p.i., and phagocytosis/intracellular killing by kidney-infiltrating neutrophils were measured by FACS. The streptavidin-AF633+dTomato+ C. albicans emit two fluorescence signals: dTomato+AF633+ is emitted by live C. albicans, while dead C. albicans emits only AF633 fluorescence signal (dTomato−AF633+).
In vitro C. albicans killing assay
BM neutrophils (5 × 104/well) were seeded in a flat bottom 96-well plate. After 30 min, 104 CFU of C. albicans were added to the wells. The number of live C. albicans was assessed by plating serial dilutions on YPD agar at 3 h post-incubation. The percent killing was expressed as [1 − (CFU of C. albicans in the presence of neutrophils/CFU of C. albicans cells in the absence of neutrophils)] × 100%. Fold change C. albicans killing was expressed as percent killing in experimental group/percent killing in control group.
NETosis assay
BM neutrophils were incubated with C. albicans for 3 h. The cells were fixed (4% PFA) and permeabilized and NET formation was measured by staining with Sytox Orange and an anti-neutrophil elastase polyclonal antibody. The secondary antibody used was goat anti-rabbit Cy5. The staining was visualized using an EVOS FL Auto microscope (Life Technologies).
Giemsa Staining of blood smear
Peripheral blood smears of PMNWT and PMNΔGLUT1 mice were stained with Giemsa stain and microscopically evaluated for the morphology of neutrophils. The staining was visualized using an EVOS FL Auto microscope.
Transwell migration assay
BM neutrophils were subjected to transwell migration assay (inserts with 3 μm pore size) in the presence or absence of recombinant murine CXCL2 (100 ng/ml). Ninety min post-incubation, the numbers of cells in the lower and upper chambers were quantified by flow cytometry analysis and expressed as Chemotactic Index (Number of cells in the lower chamber/Number of cells in the upper chamber).
Granulopoiesis measurement
BM cells from femur and tibia were stained with anti-Ly6G, anti-CD34, anti-CD3, anti-CD19, anti-B220 and anti-C-kit antibodies and analyzed by flow cytometry analysis.
Apoptosis detection
Mice were infected with 105 CFU of C. albicans via the lateral tail vein. After 24 hours, kidney cell suspensions were prepared and early and late apoptotic neutrophils were detected using Annexin V Apoptosis Detection Kit, according to manufacturer instructions, followed by flow cytometry analysis.
ROS measurement
Mice were infected with 105 CFU of C. albicans via the lateral tail vein. After 24 hours, kidney cell suspensions were prepared and resuspended with RPMI complete medium. CellROX® Deep Red reagent was added to the cell suspensions and incubated at 37°C for 30 min. The ROS production was measured by flow cytometry analysis.
Western blot analysis
Neutrophils were lysed by 1×NP-40 lysis buffer supplemented with protease inhibitor cocktail. Lysates were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. After incubation with primary and secondary antibodies, protein bands were detected using an enhanced chemiluminescence detection system and developed with a FluorChem E imager (ProteinSimple).
RNA extraction and qPCR
RNA was extracted from tissues or neutrophils using RNeasy kits. Complementary DNA was synthesized by SuperScript III First Strand Kits. Quantitative real-time PCR (qPCR) was performed with the PerfeCTa SYBR Green FastMix and analyzed on an ABI 7300 real-time instrument. Primers were obtained from QuantiTect Primer Assays. The expression of each gene was normalized to that of Gapdh.
Seahorse metabolic assay
BM neutrophils from PMNWT and PMNΔGLUT1 mice were isolated and plated on Cell-Tak coated Seahorse culture plates (5 × 105 cells/well) in DMEM without glucose. After stimulated with/without curdlan (Cur) (10 μg/ml) at 37°C for 3 h, the cells were analyzed using a Seahorse XFe96 Analyzer (Agilent). EACR (Basal extracellular acidification rate) was detected in the presence of glucose (25mM), oligomycin (2 mM), 2-deoxyglucose (100 mM) to obtain maximal and control EACR values.
Untargeted high-resolution LC-HRMS
Neutrophils from PMNWT and PMN△GLUT1 mice were either stimulated with curdlan (10 μg/ml) for 3 h (8 replications per condition) or left unstimulated. Metabolic quenching and polar metabolite pool extraction from neutrophils was performed by adding 500μL 80% methanol/ 0.1% formic acid spiked with deuterated (D3)-creatinine and (D3)-alanine, (D4)-taurine and (D3)-lactate as an internal standard for a final concentration of 10μM. After 5 min of vertexing, the supernatant was cleared of protein by centrifugation at 16,000×g. 2μL of cleared supernatant was subjected to online LC-MS analysis.
Analyses were performed by untargeted LC-HRMS. Briefly, samples were injected via a Thermo Vanquish UHPLC and separated over a reversed phase Thermo HyperCarb porous graphite column (2.1×100mm, 3μm particle size) maintained at 55°C. For the 20 min LC gradient, the mobile phase consisted of the following: solvent A (water / 0.1% FA) and solvent B (ACN / 0.1% FA). The gradient was the following: 0–1min 1% B, increase to 15%B over 5 minutes, continue increasing to 98%B over 5 min, hold at 98%B for 5 min, re-equilibrate at 1%B for 5 min. The Thermo IDX tribrid mass spectrometer was operated in both positive and ion mode, scanning in ddMS2 mode (2 μ scans) from 70 to 800 m/z at 120,000 resolutions with an AGC target of 2×105 for full scan, 2×104 for ms2 scans using HCD fragmentation at stepped 15, 35, 50 collision energies. Source ionization setting was 3.0 and 2.4kV spray voltage, respectively for positive and negative mode. Source gas parameters were 35 sheath gas, 12 auxiliary gas at 320°C, and 8 sweep gas. Calibration was performed prior to analysis using the PierceTM FlexMix Ion Calibration Solutions. Integrated peak areas were then extracted manually using Quan Browser (Xcalibur ver. 2.7) and normalized to the peak areas of the spiked internal standards to control for both sample and instrument variation. Untargeted differential comparisons were performed using Compound Discoverer 3.0 to generate a ranked list of significant compounds with tentative identifications from BioCyc, KEGG, and internal compound databases.
Quantification and statistical analysis
All data are expressed as means ± SEM. Statistical analyses were performed using the analysis of variance (ANOVA), log-rank, Mann-Whitney, or Student’s t test through GraphPad Prism 8 program. *P < 0.05; **P < 0.01; ***P < 0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PerCP-Cy™5.5 Rat Anti-Mouse CD45 (Clone 30-F11 (RUO)) | BD Biosciences | Cat # 561869; RRID: AB_10895563 |
| Anti-phospho GLUT-1 Antibody (Ser226) | Millipore | Cat # ABN991 |
| APC anti-mouse CD45 Antibody (Clone 30-F11) | Biolegend | Cat # 103112; RRID: AB_312977 |
| CD45 Monoclonal Antibody, eFluor 450, eBioscience™ (Clone 30-F11) | Invitrogen | Cat # 48-0451-82; RRID: AB_1518806 |
| Alexa Fluor® 700 anti-mouse/human CD11b Antibody (Clone M1/70) | Biolegend | Cat # 101222; RRID:AB_493705 |
| Ly-6G Monoclonal Antibody, FITC, eBioscience™ (Clone 1A8-Ly6g) | Invitrogen | Cat# 11-9668-82; RRID: AB_2572532 |
| Ly-6G Monoclonal Antibody (1A8-Ly6g) | Invitrogen | Cat# 48-9668-82; RRID:AB_2637124 |
| Ly-6G Monoclonal Antibody, PE, eBioscience™ (Clone 1A8-Ly6g) | Invitrogen | Cat# 12-9668-82; RRID: AB_2572720 |
| Ly-6C Monoclonal Antibody, PerCP-Cyanine5.5, eBioscience™ (Clone HK1.4) | Invitrogen | Cat# 45-5932-80; RRID: AB_2723342 |
| Brilliant Violet 605™ anti-mouse F4/80 antibody, BioLegend (Clone BM8) | Biolegend | Cat# 123133; RRID:AB_2562305 |
| F4/80 Monoclonal Antibody, APC, eBioscience™ (Clone BM8) | Invitrogen | Cat# 17-4801-82; RRID: AB_469452 |
| Rabbit IgG Isotype Control (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | Cat# 4340S; RRID:AB_561545 |
| Rabbit (DA1E) mAb IgG XP® Isotype Control | Cell Signaling Technology | Cat# 3900; RRID:AB_1550038 |
| Alexa Fluor 647-AffiniPure Goat Anti-Rabbit IgG (H+L) antibody, | Jackson Immuno | Cat# 111-605-003; RRID: AB_2338072 |
| PE Rat Anti-Mouse c-KIT (Clone 2B8) | BD Biosciences | Cat# 553355; RRID: AB_394806 |
| Alexa Fluor® 700 Rat Anti-Mouse CD34 (Clone RAM34) | BD Biosciences | Cat# 560518; RRID: AB_1727471 |
| PerCP anti-mouse/human CD45R/B220 Antibody (Clone RA3-6B2) | Biolegend | Cat# 103233; RRID: AB_893355 |
| PerCP/Cy5.5 anti-mouse CD19 Antibody (Clone 1D3/CD19) | Biolegend | Cat# 152405; RRID: AB_2629814 |
| PerCP Hamster Anti-Mouse CD3e (Clone 145-2C11) | BD Biosciences | Cat# 553067; RRID: AB_394599 |
| Pacific Blue™ anti-mouse Lineage Cocktail | Biolegend | Cat# 133310; RRID:AB_11150779 |
| CD16/CD32 Monoclonal Antibody (93), PerCP-Cyanine5.5, | eBioscience™ | Cat # 45-0161-80; RRID:AB_996661 |
| APC anti-mouse Ly-6A/E (Sca-1) Antibody | Biolegend | Cat # 108111; RRID:AB_313348 |
| APC anti-mouse CX3CR1 Antibody | Biolegend | Cat # 149007; RRID:AB_2564491 |
| Anti-Glucose Transporter GLUT1-FITC antibody | Abcam | Cat # ab195359; RRID:AB_2714026 |
| Phospho-PKCdelta (Tyr311) Antibody | Cell Signaling Technology | Cat # 2055; RRID:AB_330876 |
| PKCδ (D10E2) Rabbit mAb | Cell Signaling Technology | Cat # 9616; RRID:AB_10949973 |
| IκB-α Antibody (C-21): sc-371 | Santa Cruz | Cat # SC-371; RRID:AB_2235952 |
| Anti-GLUT-1 Antibody, CT | Millipore | Cat # 07-1401; RRID:AB_1587074 |
| Anti-Glucose Transporter GLUT1 antibody [EPR3915] | Abcam | Cat# ab115730; RRID:AB_10903230 |
| Anti-Glucose Transporter GLUT3 antibody [EPR10508(N)1 - N-terminal | Abcam | Cat# ab191071; RRID: AB_2736916 |
| Anti-beta Actin antibody [AC-15] (HRP) | Abcam | Cat# ab49900; AB_867494 |
| Anti-Neutrophil Elastase antibody | Abcam | Cat# ab21595; RRID:AB_446409 |
| Goat anti-rabbit IgG (HPL) HRP conjugated | Invitrogen | Cat# G21234; RRID:AB_2536530 |
| Bacterial and virus strains | ||
| Biological Samples | ||
| Healthy human peripheral blood samples | Division of Rheumatology and Clinical Immunology, University of Pittsburgh. | See Table S1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| WZB117 | Sigma | Cat# SML0621; CAS: 1223397-11-2 |
| mannan | sigma | Cat# M7504; CAS: 903688-8 |
| curdlan | sigma | Cat# C7821; CAS: 54724-00-4 |
| Zymosan | sigma | Cat# Z4250; CAS: 58856-93-2 |
| Rottlerin | Tocris | Cat# 1610; CAS: 82-08-6 |
| Bay-61-3606 | sigma-Calbiochem | Cat# 574714; CAS: 732938-37-8 |
| Bay 11-7085 | sigma-Calbiochem | Cat# B5681; CAS: 196309-76-9 |
| GW5074 | Sigma | Cat# G6416; CAS: 220904-83-6 |
| CellROX® Deep Red reagent | Invitrogen | Cat#C10422 |
| SYTOX™ Orange Nucleic Acid Stain | Sigma | Cat#S11368 |
| Phorbol-12-myristate-13-acetate | Sigma-Calbiochem | Cat# 524400; CAS: 16561-29-8 |
| Recombinant Murine MIP-2 (CXCL2) | Peprotech | Cat# 250-15 |
| Ghost Dye™ Violet 510 | TONBO biosciences | Cat# 13-0870-T100 |
| CountBright™ Absolute Counting Beads | Invitrogen | Cat# C36950 |
| GlucoseCy5 | Dr. Greg M. Delgoffe, University of Pittsburgh | Watson et al., 2021 |
| 2-NBDG | Cayman Chemical Company | Cat#11046; CAS: 186689-07-6 |
| Biotin-XX, sulfo-SE | Invitrogen | Cat# B6352 |
| Collagenase IV | ThermoFisher Scientific | Cat# 17104019 |
| Gibco™ ACK Lysing Buffer | ThermoFisher Scientific | Cat# A1049201 |
| Corning™ EDTA, 0.5M, pH8.0 | Fisher Scientific | Cat# MT-46034CI |
| DNAsel | MP Biomedicals | Cat# 190062 |
| Streptavidin, Alexa Fluor 633 conjugate | Life Technologies Corporation | Cat# S21375 |
| Giemsa Stain (Powder/Certified Biological Stain) | Fisher Chemical™ | Cat# G146-5 |
| Protease inhibitor cocktail | Roche | Cat# 11697498001 |
| Critical Commercial Assays | ||
| PE Annexin V Apoptosis Detection Kit I | BD Pharmingen | Cat# 559763 |
| Neutrophil Isolation kit mouse | Miltenyi Biotec | Cat# 130-097-658 |
| SuperScript™ III First-Strand Synthesis kit | Invitrogen | Cat# 18080051 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| MACSxpress® Neutrophil Isolation kit, Human | Miltenyi Biotec | Cat# 130-104-434 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6NTac | Taconic Biosciences | Model# B6-M |
| Mouse: Card9−/−: B6.129-Card9tm1Xlin/J | The Jackson Laboratory | Cat# 028652 |
| Mouse: Dectin-1−/−: B6.129S6-Clec7atm1Gdb/J | The Jackson Laboratory | Cat# 012337 |
| Mouse: MRP8-Cre+: B6.Cg-Tg(S100A8-cre,-EGFP)1Ilw/J | The Jackson Laboratory | Cat # 021614 |
| Mouse: Myd88−/−: B6.129P2(SJL)-Myd88tm1.1Defr/J | The Jackson Laboratory | Cat# 009088 |
| Mouse: Slc2a1fl/fl | University of Colorado; MTA | N/A |
| Mouse: Act1−/− | Dr. Sarah L. Gaffen University of Pittsburgh | N/A |
| C. albicans: SC5314 | Dr. Sarah Gaffen, University of Pittsburgh | ATCC: MYA-2876 |
| dTomato+ C. albicans reporter strain | Dr. Michail Lionakis, NIH | N/A |
| Oligonucleotides | ||
| Primers | QIAGEN | See Table S2 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Image J | National Institute of Health | https://imagej.nih.gov/ij/ |
| FlowJo v10 | Tree Star | https://www.flowjo.com/ |
| Alpha View SA v3.4.0 | ProteinSimple | https://www.proteinsimple.com/ |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/ |
| IDEAS applications 6.1 | Luminex | https://www.luminexcorp.com/flow-cytometry-and-imaging/ |
| EndNote ×9 | EndNote | https://endnote.com/ |
| Compound Discoverer 3.0 | Thermo Fisher | https://assets.thermofisher.com/ |
| Other | ||
Highlights.
Neutrophils upregulate Glut1 and glucose uptake in disseminated candidiasis
The β-glucan/dectin-1-axis transcriptionally and post-translationally modulates Glut1
Glut1 deficiency in neutrophils exacerbates susceptibility to disseminated candidiasis
Glut1 is indispensable for antifungal activity in mice and human neutrophils
Acknowledgments
This work is supported in part by NIH grants AI142354, DK104680 and AI145242 to P.S.B.; AI45242, DE022550 and AI147383 to S.L.G.; HL135476, HL15423 and AI153549 to G.J.T.; NIHS10OD023402 to S.G.W. and Division of Intramural Research (DIR) of the NIAID, NIH to M.S.L. We thank Unified Flow Core, Department of Immunology; Center for Metabolism and Mitochondrial Medicine, Vascular Medicine Institute; and Metabolomics and Lipidomics Core, University of Pittsburgh for flow cytometry, Seahorse bioanalyzer assay and metabolomics study, respectively.
Footnotes
Declaration of interests
The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancey PB, Contat C, Boivin G, Sabatino S, Pascual J, Zangger N, Perentes JY, Peters S, Abel ED, Kirsch DG, et al. (2021). GLUT1 Expression in Tumor-Associated Neutrophils Promotes Lung Cancer Growth and Resistance to Radiotherapy. Cancer Res 81, 2345–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardati KO, Bajakian KM, and Tabbara KS (1997). Effect of glucose-6-phosphate dehydrogenase deficiency on neutrophil function. Acta Haematol 97, 211–215. [DOI] [PubMed] [Google Scholar]
- Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, and Junger WG (2015). mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol 210, 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishu S, Hernandez-Santos N, Simpson-Abelson MR, Huppler AR, Conti HR, Ghilardi N, Mamo AJ, and Gaffen SL (2014). The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun 82, 1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD (2011). Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol 29, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL, and Brosius FC 3rd (2008). A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol 295, C836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn GL, Foti A, Marsman G, Patel DF, and Zychlinsky A (2021). The Neutrophil. Immunity 54, 1377–1391. [DOI] [PubMed] [Google Scholar]
- Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, and Junger WG (2006). ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314, 1792–1795. [DOI] [PubMed] [Google Scholar]
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. (2014). mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai JV, and Lionakis MS (2018). The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep 5, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSouza-Vieira T, Guimaraes-Costa A, Rochael NC, Lira MN, Nascimento MT, Lima-Gomez PS, Mariante RM, Persechini PM, and Saraiva EM (2016). Neutrophil extracellular traps release induced by Leishmania: role of PI3Kgamma, ERK, PI3Ksigma, PKC, and [Ca2+]. J Leukoc Biol 100, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douard V, and Ferraris RP (2008). Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab 295, E227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, Fink DL, Hsu AP, Zhai B, Karauzum H, et al. (2015). CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog 11, e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, and Gioia F (2017). Invasive candidiasis in the neutropenic patient. Rev Esp Quimioter 30 Suppl 1, 22–25. [PubMed] [Google Scholar]
- Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, and Edwards SW (2003). The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol 170, 1964–1972. [DOI] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, and Makowski L (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem 289, 7884–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijtenbeek TB, and Gringhuis SI (2009). Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol 9, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geltink RIK, Kyle RL, and Pearce EL (2018). Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin ME, Hamilton BJ, Roy KM, Du M, Willson AM, Keenan BJ, Wang XW, and Nichols RC (2004). Post-transcriptional regulation of glucose transporter-1 by an AU-rich element in the 3’UTR and by hnRNP A2. Biochem Biophys Res Commun 318, 977–982. [DOI] [PubMed] [Google Scholar]
- Hiraiwa H, Pan CJ, Lin B, Moses SW, and Chou JY (1999). Inactivation of the glucose 6-phosphate transporter causes glycogen storage disease type 1b. J Biol Chem 274, 5532–5536. [DOI] [PubMed] [Google Scholar]
- Horn DL, Neofytos D, Anaissie EJ, Fishman JA, Steinbach WJ, Olyaei AJ, Marr KA, Pfaller MA, Chang CH, and Webster KM (2009). Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis 48, 1695–1703. [DOI] [PubMed] [Google Scholar]
- Huppler AR, Conti HR, Hernandez-Santos N, Darville T, Biswas PS, and Gaffen SL (2014). Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J Immunol 192, 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DY, and Ismail-Beigi F (2006). Control of Glut1 promoter activity under basal conditions and in response to hyperosmolarity: role of Sp1. Am J Physiol Cell Physiol 290, C337–344. [DOI] [PubMed] [Google Scholar]
- Jawale CV, Ramani K, Li DD, Coleman BM, Oberoi RS, Kupul S, Lin L, Desai JV, Delgoffe GM, Lionakis MS, et al. (2020). Restoring glucose uptake rescues neutrophil dysfunction and protects against systemic fungal infection in mouse models of kidney disease. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JH, Hong CW, Kim EY, and Lee JM (2020). Current Understanding on the Metabolism of Neutrophils. Immune Netw 20, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, and Dikshit M (2019). Metabolic Insight of Neutrophils in Health and Disease. Front Immunol 10, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagasse E, and Weissman IL (1994). bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med 179, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EE, Ma J, Sacharidou A, Mi W, Salato VK, Nguyen N, Jiang Y, Pascual JM, North PE, Shaul PW, et al. (2015). A Protein Kinase C Phosphorylation Motif in GLUT1 Affects Glucose Transport and is Mutated in GLUT1 Deficiency Syndrome. Mol Cell 58, 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto D, and Saltiel AR (2012). Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol 13, 383–396. [DOI] [PubMed] [Google Scholar]
- Li X, Cullere X, Nishi H, Saggu G, Durand E, Mansour MK, Tam JM, Song XY, Lin X, Vyas JM, and Mayadas T (2016). PKC-delta activation in neutrophils promotes fungal clearance. J Leukoc Biol 100, 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionakis MS, Fischer BG, Lim JK, Swamydas M, Wan W, Richard Lee CC, Cohen JI, Scheinberg P, Gao JL, and Murphy PM (2012). Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog 8, e1002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionakis MS, and Netea MG (2013). Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathog 9, e1003079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedera S, Mizuno T, Ishiguro H, Ito T, Soga T, and Kusuhara H (2019). GLUT6 is a lysosomal transporter that is regulated by inflammatory stimuli and modulates glycolysis in macrophages. FEBS Lett 593, 195–208. [DOI] [PubMed] [Google Scholar]
- Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, and Kuijpers TW (2004). Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ 11, 143–153. [DOI] [PubMed] [Google Scholar]
- Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: From basic mechanisms to translation. Immunol Rev 295, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Cassatella MA, Costantini C, and Jaillon S (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11, 519–531. [DOI] [PubMed] [Google Scholar]
- Mayadas TN, Cullere X, and Lowell CA (2014). The multifaceted functions of neutrophils. Annu Rev Pathol 9, 181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, et al. (2006). Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest 116, 1642–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, van der Meer JL JWM, Kullberg BJ, van de Veerdonk FL. (2015). Immune defence against Candida fungal infections. Nature Reviews Immunology 15, 630–642 [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat Rev Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, and Kullberg BJ (2018). Invasive candidiasis. Nat Rev Dis Primers 4, 18026. [DOI] [PubMed] [Google Scholar]
- Pasqualotto AC, and Denning DW (2008). New and emerging treatments for fungal infections. J Antimicrob Chemother 61 Suppl 1, i19–30. [DOI] [PubMed] [Google Scholar]
- Pearce EL, and Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber LL, Gillis CM, Starkl P, Jonsson F, Sibilano R, Marichal T, Gaudenzio N, Berard M, Rogalla S, Contag CH, et al. (2017). Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med 214, 1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss M, and Roos D (1978). Differences in oxygen metabolism of phagocytosing monocytes and neutrophils. J Clin Invest 61, 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheepers A, Schmidt S, Manolescu A, Cheeseman CI, Bell A, Zahn C, Joost HG, and Schurmann A (2005). Characterization of the human SLC2A11 (GLUT11) gene: alternative promoter usage, function, expression, and subcellular distribution of three isoforms, and lack of mouse orthologue. Mol Membr Biol 22, 339–351. [DOI] [PubMed] [Google Scholar]
- Solis NV, and Filler SG (2012). Mouse model of oropharyngeal candidiasis. Nat Protoc 7, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Roy S, Leal SM Jr., Sun Y, Howell SJ, Cobb BA, Li X, and Pearlman E (2014). Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat Immunol 15, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, and Brown GD (2007). Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, and Mueckler M (2010). Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab 298, E141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uldry M, Ibberson M, Horisberger JD, Chatton JY, Riederer BM, and Thorens B (2001). Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J 20, 4467–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban CF, Reichard U, Brinkmann V, and Zychlinsky A (2006). Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8, 668–676. [DOI] [PubMed] [Google Scholar]
- Uzun O, Ascioglu S, Anaissie EJ, and Rex JH (2001). Risk factors and predictors of outcome in patients with cancer and breakthrough candidemia. Clin Infect Dis 32, 1713–1717. [DOI] [PubMed] [Google Scholar]
- Velasco E, and Bigni R (2008). A prospective cohort study evaluating the prognostic impact of clinical characteristics and comorbid conditions of hospitalized adult and pediatric cancer patients with candidemia. Eur J Clin Microbiol Infect Dis 27, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu R, Qu X, Yu H, Chu H, Zhang Y, Zhu W, Wu X, Gao H, Tao B, et al. (2019). alpha-Ketoglutarate-Activated NF-kappaB Signaling Promotes Compensatory Glucose Uptake and Brain Tumor Development. Mol Cell 76, 148–162 e147. [DOI] [PubMed] [Google Scholar]
- Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, Menk AV, Rittenhouse NL, DePeaux K, Whetstone RD, et al. (2021). Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerasinghe H, and Traven A (2020). Immunometabolism in fungal infections: the need to eat to compete. Curr Opin Microbiol 58, 32–40. [DOI] [PubMed] [Google Scholar]
- Wu X, and Freeze HH (2002). GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics 80, 553–557. [DOI] [PubMed] [Google Scholar]
- Wu X, Zhang G, Yang WH, Cui JT, Zhang L, Xiao M, and Xu YC (2021). GLUT3 as an Intersection of Glycerophospholipid Metabolism and the Innate Immune Response to Candida albicans. Front Cell Infect Microbiol 11, 648988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaoutis TE, Argon J, Chu J, Berlin JA, Walsh TJ, and Feudtner C (2005). The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis 41, 1232–1239. [DOI] [PubMed] [Google Scholar]
- Zhou W, Cao L, Jeffries J, Zhu X, Staiger CJ, and Deng Q (2018). Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis Model Mech 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschaler J, Schlorke D, and Arnhold J (2014). Differences in innate immune response between man and mouse. Crit Rev Immunol 34, 433–454. [PubMed] [Google Scholar]
- Zucker-Franklin D (1968). Electron microscopic studies of human granulocytes: structural variations related to function. Semin Hematol 5, 109–133. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not create large data sets that required deposition in a public data base. Microscopy data reported in this paper will be shared by the lead contact upon request.
The study did not create original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.