

Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPis) have become a mainstay of therapy in ovarian cancer and other malignancies, including BRCA-mutant breast, prostate, and pancreatic cancers. However, a growing number of patients develop resistance to PARPis, highlighting the need to further understand the mechanisms of PARPi resistance and develop effective treatment strategies. Targeting cell cycle checkpoint protein kinases, e.g., ATR, CHK1, and WEE1, which are upregulated in response to replication stress, represents one such therapeutic approach for PARPi-resistant cancers. Mechanistically, activated cell cycle checkpoints promote cell cycle arrest, replication fork stabilization, and DNA repair, demonstrating the interplay of DNA repair proteins with replication stress in the development of PARPi resistance. Inhibitors of these cell cycle checkpoints are under investigation in PARPi-resistant ovarian and other cancers. In this review, we discuss the cell cycle checkpoints and their roles beyond mere cell cycle regulation as part of the arsenal to overcome PARPi-resistant cancers. We also address the current status and recent advancements as well as limitations of cell cycle checkpoint inhibitors in clinical trials.
Keywords: ATR/CHK1/WEE1 pathway, cell cycle checkpoint, DNA damage response, ovarian cancer, PARP inhibitor resistance, replication stress
Graphical Abstract

1. Introduction
The past decade has witnessed the development and approval of numerous targeted cancer therapeutics. One such class of drugs, poly (ADP-ribose) polymerase (PARP) inhibitors (PARPis), has become a mainstay of therapy in ovarian cancer and other malignancies, including BRCA-mutant breast, prostate, and pancreatic cancers [1]. Broadly speaking, PARPis are nicotinamide adenine dinucleotide (NAD) mimetics that interfere with the catalytic activity of PARP1, preventing subsequent poly ADP-ribosylation necessary for DNA repair. PARPis also cause DNA damage by inducing toxic PARP-DNA complex. Double-stranded breaks (DSBs) accompanied by PARP trapping strongly blocks replication and induces S phase checkpoints, resulting in further DNA breaks and cytotoxicity, particularly in cells with underlying homologous recombination (HR) deficiency (HRD) [2]. The PARPi olaparib was first United States (U.S.) Food and Drug Administration (FDA)–approved in 2014 for germline breast cancer gene 1/2 (BRCA1/2)-mutated advanced ovarian cancer after three or more lines of chemotherapy, and rucaparib followed in 2016 for both germline as well as somatic BRCA-mutated advanced ovarian cancer after two or more lines of chemotherapy [3, 4]. Subsequently, PARPis, e.g., olaparib, niraparib, and rucaparib, have been approved in recurrent platinum-sensitive ovarian cancer, and olaparib and niraparib in the upfront maintenance setting, as maintenance therapy for those whose disease has not progressed on platinum-based chemotherapy [4]. PARPis have also shown clinical activity beyond ovarian cancer. Olaparib and talazoparib are U.S. FDA–approved for the treatment of locally advanced or metastatic breast cancer with germline BRCA mutations [5, 6]. In addition, PARPis demonstrate efficacy as upfront maintenance therapy in BRCA-mutated pancreatic cancer [7] and as monotherapy in metastatic, castration-resistant prostate cancer in patients with alterations in DNA damage repair genes (e.g., BRCA2 and ataxia-telangiectasia mutation [ATM]) [8, 9].
With the increased use of PARPis in the clinic, resistance has emerged as a pressing problem, creating unmet needs requiring a further understanding of resistance mechanisms and development of more effective treatment strategies. Thus far, known key mechanisms of resistance against PARPis include restoration of the HR repair pathway, through BRCA-dependent means (e.g., reversion mutations and epigenetic upregulation of BRCA1), as well as BRCA-independent means (e.g., loss of negative regulators of HR like p53-binding protein 1 (53BP1) or arrest deficient 2 like 2 [MAD2L2, also known as REV7], or reversion mutations of non-BRCA HR pathway genes like RAD51 paralog C [RAD51C] and partner and localizer of BRCA2 [PALB2]) [10]. PARPi resistance may also arise independently of HR, including via replication fork protection, upregulation of survival pathways, drug efflux, and other mechanisms (Table 1). Of note, these resistance mechanisms are not mutually exclusive, especially in the clinical setting. Individual patients may exhibit heterogeneous mechanisms of PARPi resistance simultaneously, suggesting that multiple molecular pathways act in parallel to confer clinical drug resistance [10]. Furthermore, PARPi resistance is generally associated with platinum resistance, defined clinically as disease relapse within six months of completion of platinum-based chemotherapy. Platinum derivatives induce DNA-intrastrand crosslinks resolved by nucleotide excision repair (NER) and share multiple overlapping resistance mechanisms with PARPis [11]. As such, elucidating PARPi resistance mechanisms that arise in both platinum-resistant and platinum-sensitive settings is crucial for better stratification of patients and further drug development.
Table 1.
PARP inhibitor resistance mechanisms.
| HR-dependent mechanisms | HR-independent mechanisms |
|---|---|
BRCA-dependent HR restoration
|
Replication fork stabilization
|
Abbreviations; HR, homologous recombination; PAR, poly (ADP ribose); PARG, poly (ADP ribose) glycohydrolase.
It appears that tumors resistant to PARPis frequently show signs of replication stress, or perturbations in DNA replication, ultimately resulting in genomic instability, which can be leveraged to overcome PARPi resistance [12, 13]. Replication stress is a relatively broad term, including any events blocking optimal DNA replication processes, and its precise definition evolves as our knowledge of cell biology expands. For the purposes of a common understanding, we herein define replication stress as the slowing or stalling of replication fork progression and/or DNA synthesis in part by DNA lesions, decreased pool of nucleotides, or transcription-replication collisions [14]. Elevated levels of replication stress activate checkpoint protein kinases including ataxia telangiectasia and Rad3-related serine/threonine kinase (ATR), checkpoint kinase 1 (CHK1), and WEE1 G2 checkpoint kinase (WEE1), triggering cell cycle arrest, replication fork stabilization, and DNA repair for optimal DNA replication [15]. Obstructions to replication fork progression cause fork stalling which leads to the accumulation of single-stranded DNA (ssDNA), and recruitment of phosphorylated replication protein A (pRPA). These actions subsequently activate ATR and its downstream effector, CHK1, as well as WEE1, with inhibition of cyclin-dependent kinases (CDKs) and cell division cycle 25A (CDC25A) and cell division cycle 25B (CDC25B), therefore halting the progression of the cell cycle to avoid mitotic catastrophe [16]. In addition, there is emerging evidence that beyond its role in regulation of the cell cycle, the ATR/CHK1/WEE1 pathway regulates HR repair and the process of DNA replication [16, 17], described below.
In this review, we discuss the rationale for targeting ATR, CHK1, and WEE1 both as monotherapy (Table 2) and in combination with PARPis or other drugs to address unmet clinical needs for PARPi-resistant tumors. We use ovarian cancer as an example and refer to other BRCA-mutant malignancies, e.g., breast cancer, where appropriate. We also report the current status and recent advancements as well as limitations of cell cycle checkpoint inhibitors in clinical trials.
Table 2.
ATR, CHK1, and WEE1 inhibitors in current clinical trials.
| ATR inhibitors | CHK1 inhibitors | WEE1 inhibitors |
|---|---|---|
|
|
|
2. Preclinical Studies
2.1. Functions of Cell Cycle Checkpoint Pathways: from Checkpoints to DNA Repair
ATR is a well-known regulator in the control of DNA repair apart from its roles in the cell cycle. Both ATR and CHK1 are involved in HR repair, which predominates in the S and G2 phases to repair DSBs (Figure 1). During HR, BRCA1 promotes end resection of DSBs to create 3′ ssDNA overhangs. The resected DNA ends are coated by hyperphosphorylated RPA, which further activates ATR. ATR then phosphorylates H2AX (γH2AX) to assist with the recruitment and accumulation of BRCA1. Thereafter, BRCA1 recruits the PALB2-BRCA2 complex to the sites of DSBs for RAD51-ssDNA filament formation, homology sequence searching, and strand invasion [18].
Figure 1. Role of ATR/CHK1 pathway in homologous recombination repair.
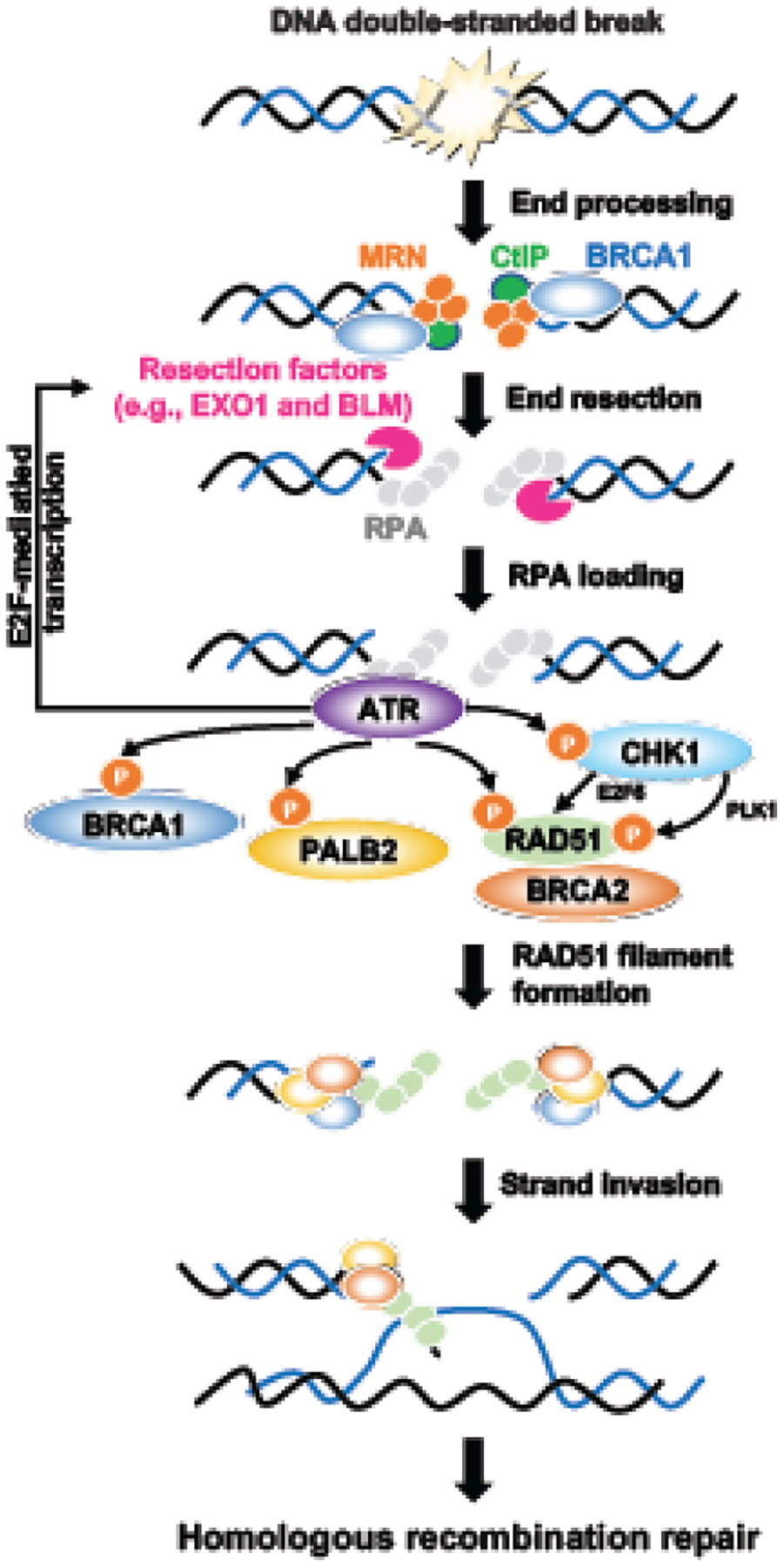
During homologous recombination repair, the double-stranded break is recognized by the MRE11-RAD50-NBS1 (MRN) complex, CtIP, and BRCA1, which resects DNA with EXO1 and BLM helicase to create 3’-overhang single-stranded DNAs. The single-stranded DNAs will be protected by RPA, which further activates the ATR/CHK1 pathway. ATR promotes HR repair by phosphorylating key HR proteins, including BRCA1, PALB2, and RAD51. ATR also maintains the pool of resection factors by promoting E2F-mediated transcription. CHK1 mainly assists HR repair by phosphorylating RAD51 via PLK1 or promoting E2F6-mediated RAD51 transcription. The BRCA1-PALB2-BRCA2 complex then mediates the replacement of RPA by RAD51. RAD51 filaments further invade the complementary DNA template, leading to branch migration, resolution, and faithful DNA repair.
Most studies describe the roles of ATR and CHK1 after end resection [18]. However, recent studies provide additional insights into how the ATR/CHK1 pathway can act both before and after end resection to promote HR repair. During the initial stage of HR, CDK activity promotes DNA resection and activates the ATR/CHK1 pathway [19]. ATR maintains the pool of resection factors such as CtBP-interacting protein (CtIP) and bloom syndrome protein (BLM) helicase, likely through the activation of E2F-mediated transcription [20, 21]. As ATR/CHK1 activation is gradually increased, it inhibits CDK activation and drives HR to the next stage. In post-resection, ATR phosphorylates PALB2 at the S59 site, which is necessary for attaining optimal levels of PALB2 localization to DSBs and strengthening its interaction with BRCA1 [19]. ATR also directly increases BRCA1 or RAD51 phosphorylation, promoting their loading on DSBs in cells with intact HR. These data suggest that ATR controls HR both through the E2F-mediated transcription of DNA end resection and HR factors (e.g., BRCA1, CtIP, and BLM) and through the direct phosphorylation of key HR proteins (e.g., BRCA1, PALB2, and RAD51).
As a downstream target of ATR, CHK1 mainly assists HR through RAD51. CHK1 increases the E2F-mediated transcription of RAD51 by inhibiting the E2F6 repressor on its promoter [22]. Also, CHK1 can regulate the activation of RAD51 via polo like kinase 1 (PLK1). A recent study demonstrated that CHK1 and PARP1 coordinate PLK1 enzymatic activity to promote HR repair. More specifically, CHK1 phosphorylates PLK1 at S137, which directly phosphorylates RAD51 at S14. This process primes RAD51 for CHK1-mediated phosphorylation at T309, which is essential for full RAD51 activation [23]. In contrast to the broad-acting ATR and CHK1, WEE1 seems to have a rather conservative scale of effectors to arrest the cell cycle for cells to repair damaged DNA, rather than directly coordinating HR repair [24]. However, the WEE1 inhibitor (WEE1i, MK-1775) resulted in inactivation of both ATR and CHK1 in BRCA-wildtype (BRCAwt) U2OS and Panc1 cells treated with gemcitabine [25]. This finding suggests that WEE1 is required for tumor cells to sustain ATR/CHK1 activation, and the crosstalk among these kinases should be taken into consideration in further investigation of their roles in HR repair.
Of note, different treatment schemes of ATR and CHK1 inhibitors might affect the outcomes of mechanism studies. For instance, short-term treatment (<24 hours) with ATR inhibitor (ATRi, VE-821) decreases RAD51 phosphorylation and prevents its loading on DSBs [19–21], but has minor effects on end resection or RAD52 homolog DNA repair protein (RAD52) foci formation [20]. On the contrary, long-term treatment (5 or 8 days) with ATRi (VE-821) impairs end resection, RAD51 and RAD52 foci formation, and the pool of resection factors (BRCA1, CtIP, and BLM) by increasing the activation of E2F-mediated transcription [20]. Similarly, another study found that long-term, but not acute, inhibition of ATR (VE-821) or CHK1 (CHK1i, UCN-01) decreases the transcription of BRCA1, DNA topoisomerase II binding protein 1 (TOPBP1), and RAD51, leading to decreased HR-mediated repair [21]. These results may explain why earlier studies [26, 27] mainly saw the effects of ATRi or CHK1i on only RAD51-mediated HR repair but not end resection, as they mostly treated cells with these inhibitors within 3 days.
2.2. Another Function of Cell Cycle Checkpoint Pathways: a Master Regulator of Fork Protection during DNA Replication
The ATR/CHK1 pathway has extended roles in regulation of fork protection during DNA replication (Figure 2) besides its involvement in HR repair. ATR, CHK1, and WEE1 protect replication forks by phosphorylating and inhibiting distinct nucleases or helicases that target or degrade stalled replication forks during DNA replication [17, 28, 29]. Specifically, ATR phosphorylates exonuclease 1 (EXO1) at S714, which promotes ubiquitylation-dependent degradation of EXO1 and minimizes its processing activity on stalled forks [30, 31]. ATR also phosphorylates SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily A-like 1 (SMARCAL1) translocase to suppress DNA cleavage by synthetic lethal of unknown function 4 (SLX4)-dependent nucleases, thereby limiting replication progression and preventing subsequent fork degradation [32]. In addition, ATR phosphorylates minichromosome maintenance complex component 2–7 (MCM2–7) helicases to promote Fanconi anemia complementation group D2 (FANCD2) loading onto replication forks. FANCD2 then prevents replication fork collapse by either slowing down DNA polymerase activity or preventing meiotic recombination 11 (MRE11) double strand break repair nuclease from digesting exposed DNA [33].
Figure 2. Role of the ATR/CHK1/WEE1 pathway in replication fork protection.

In response to replication stress, the replication fork stalls at the site of damage. Stalled replication forks are unstable structures that can cause the fork to collapse and generate DNA double-stranded breaks. After fork stalling, single-stranded DNAs are coated by RPA, which activates ATR. ATR further phosphorylates the replication fork remodeler SMARCAL1 and RAD51 for replication fork reversal. The reversed forks are protected by several fork protectors from deleterious nuclease-mediated fork degradation that can destabilize stalled forks. During DNA replication, the ATR/CHK1/WEE1 pathway protects stalled replication forks by phosphorylating RAD51 and other fork protectors (e.g., BRCA2 and FANCD2) as well as inhibiting nucleases (e.g., EXO1, MUS81, and MRE11) that degrade stalled replication forks.
CHK1 also regulates nucleases involved in replication fork protection. CHK1 phosphorylates EXO1 at a different site (S746), which creates a docking site for binding to 14-3-3 proteins, thereby preventing the recruitment of EXO1 on the stalled replication forks [31]. CHK1 may also regulate the methyl methanesulfonate ultraviolet sensitive gene clone 81 (MUS81)/essential meiotic structure-specific endonuclease 1 (EME1) nuclease complex in an ATR-independent manner [34]. One study indicated that CHK1 depletion or CHK1i (UCN-01)-induced stalled replication forks are processed into DSBs by the MUS81/EME1 nuclease complex in yeast cells, whereas fork collapse is not dependent on the MUS81/EME1 nuclease complex when the same cells are ATR depleted or treated with ATRi (VE-822) [34]. This finding suggests that CHK1, but not ATR, may increase replication fork protection by inhibiting MUS81/EME1 activity. As a downstream kinase of CHK1, WEE1 also functions in both checkpoint activation and replication fork stability [35]. WEE1 regulates the MUS81/EME1 complex, either directly by phosphorylating and inhibiting it or indirectly through inhibiting CDK2 to block the activation of MUS81 [36]. Accordingly, WEE1 depletion or inhibition (MK-1775) slows replication fork progression and MUS81-dependent replication fork collapse [37]. Collectively, these examples highlight how ATR, CHK1, and WEE1 protect replication fork integrity via direct regulation of nucleases and helicases.
However, the mechanism studies described above warrant further evaluation in various contexts as these studies mainly used BRCAwt osteoblastoma U2OS cells or other cells [19–21], and the unique molecular characteristics of each cancer may modulate its response to cell cycle checkpoint inhibition differently. Nevertheless, most preclinical studies elucidating the roles of the ATR pathway in both HR repair and fork protection support blocking ATR/CHK1/WEE1 as one of the therapeutic strategies to overcome PARPi resistance (Figure 3).
Figure 3. Targeting the ATR/CHK1/WEE1 pathway overcomes PARP inhibitor resistance.
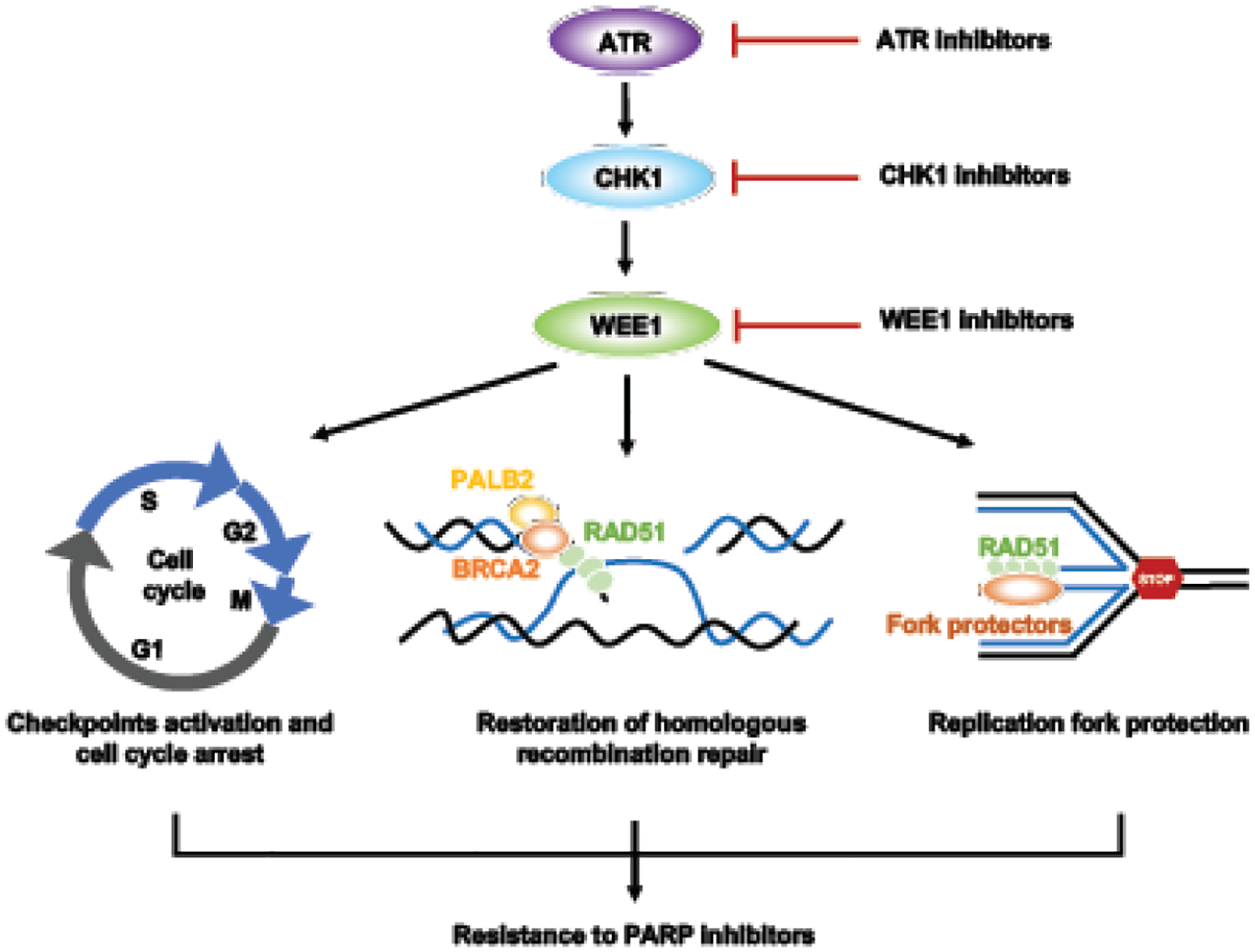
Monotherapy or combination therapies with ATR/CHK1/WEE1 inhibitors overcome PARP inhibitor resistance by disrupting both HR and replication fork protection, leading to accumulation of DNA damage throughout the cell cycle and increased cell death.
2.3. Cell Cycle Checkpoint Inhibition as a Way to Re-sensitize Tumors to PARPis
Upregulation of the ATR/CHK1 pathway has been found in PARPi-resistant BRCA-mutant HGSOC cell lines (BRCA1-mutant JHOS4 and BRCA2-mutant PEO1) [38]. Kim and colleagues also reported increased activity of the ATR/CHK1 pathway in various BRCA-mutant HGSOC cells with acquired PARPi resistance (platinum-sensitive, BRCA1-mutant JHOS4-PR and BRCA2-mutant PEO1-PR, as well as platinum-resistant, BRCA2-mutant PEO1-CR) [39]. It is worth noting that discordant resistance mechanisms between platinum drugs and PARPis have been described in unique subsets of tumors. For instance, HR restoration conferring PARPi resistance did not necessarily confer cross-resistance to platinum in BRCAwt or BRCA-deficient ovarian cancer cells with loss of NER (e.g., ERCC excision repair 4 [ERCC4]- and ERCC excision repair 6 [ERCC6]-mutant) [40]. A similar phenomenon is also found in BRCAwt clear cell ovarian cancer or BRCA1-deficient breast cancer with loss of REV7, which is involved in translesion DNA synthesis and blocks end resection to promote non-homologous end-joining DSB repair [41, 42]. In this regard, the status of platinum sensitivity or HR restoration may not always be an accurate predictor of PARPi sensitivity. Further investigation into ATR/CHK1 pathway activation in PARPi-resistant BRCA-deficient models with NER or REV7 alteration is necessary.
Therapeutic intervention of the ATR/CHK1 pathway can also apply to tumors with intact HR and fork protection. ATRis (VE-821 and VE-822) and CHK1i (prexasertib) attenuate RAD51-mediated HR repair and fork protection by disrupting recruitment of the PALB2-BRCA2 complex and RAD51 loading in BRCAwt glioblastoma cancer cells [43] and BRCAwt HGSOC models [27]. In addition, ATRi (VE-821) or CHK1i (prexasertib) monotherapy has demonstrated cytotoxicity in BRCAwt triple-negative breast cancer (TNBC, MDA-MB-231), BRCAwt (HT29), and BRCA2-mutant (HCC116) colon cancer cells that are Schlafen family member 11 (SLFN11)-deficient, which have intact replication fork protection, thus resistant to PARPis [44, 45]. These data further provide the preclinical evidence for utilizing cell cycle checkpoint inhibitors in tumors, independent of their HR or fork stabilization status.
As such, blockade of the ATR/CHK1/WEE1 axis has been proposed to sensitize cancer cells to PARPis in various contexts [17]. ATRi in combination with PARPi was synergistic in PARPi-sensitive BRCA2-depleted HeLa and mouse mammary tumor KB2P1.21 cells [46] or PARPi-resistant BRCA-deficient HGSOC (platinum-sensitive, BRCA1-mutant JHOS4-PR and BRCA2-mutant PEO1-PR, as well as platinum-resistant, BRCA2-mutant PEO1-CR) [39], and BRCA2-mutant prostate cancer cells (22Rv1) [47]. ATRi in combination with PARPi also showed greater cytotoxicity compared to either monotherapy in ATM-deficient prostate cancer [48] or SLFN11-negative TNBC (MDA-MB-231) and colon cancer (HT29 and HCC116) [44, 45] preclinical models. CHK1is (MK-8776, prexasertib) produced similar synergistic cytotoxic effects when combined with PARPi (olaparib) in both BRCA-mutant and BRCAwt HGSOC models [27, 49]. Likewise, inhibition of WEE1 also sensitizes cancer cells to PARPis in BRCAwt pancreatic cancer [50], Kirsten rat sarcoma 2 viral oncogene homolog (KRAS)-mutated non-small cell lung cancer (NSCLC) [51], and multiple BRCAwt HGSOC cell lines [52].
It has been hypothesized that targeting more than one of these kinases may be more effective than monotherapy in PARPi-resistant models, since ATR, CHK1, and WEE1 protect stalled replication forks via distinct mechanisms as previously mentioned. Accordingly, combined inhibition of WEE1 and ATR is effective in several BRCAwt TNBC models compared to either monotherapy [53]. The combination of WEE1i and ATRi has not yet been evaluated in the clinical setting, likely due to overlapping hematopoietic side effects. But given its potential in BRCAwt models which exhibit limited or moderate response to PARPi treatment, further exploration in future clinical trials may be warranted.
2.4. Cell Cycle Checkpoint Inhibitor Combinations with Other Drugs in the PARPi-Resistant Preclinical Setting
Targeting alternative DNA repair pathways or replication progress with ATR pathway inhibitors has been proposed as a therapeutic strategy in the PARPi-resistant setting. For instance, gemcitabine, a nucleoside analog that leads to decreased DNA replication, dNTP depletion, and fork stalling, has been reported to synergize with ATRis (AZD6738, M4344) or CHK1i (prexasertib) in BRCAwt tumor cells including HGSOC [54, 55]. Also, ATRis (VE-821, VX-970) sensitize BRCAwt colon (HT29 and COLO 205) and TNBC (MDA-MD-231) cells to topoisomerase I inhibitors by disabling DNA replication initiation and fork elongation responses [56]. However, most of the combination strategies with chemotherapy interrupt DNA replication and DNA repair pathways, with overlapping downstream targets, thus leading to the rapid emergence of resistance [24, 57]. Therefore, combination strategies beyond replication process with cell cycle checkpoint inhibitors to minimize overlapping side effects and to avoid similar resistance mechanisms are being explored.
One field warranting further investigation is the link between replication stress–related DNA damage with the immune response (Figure 4). Sato and colleagues reported that ATM, ATR, and CHK1 can drive programmed death ligand-1 (PD-L1) expression on U2OS cells with BRCA2 or X-Ray repair cross complementing 6 and 5 (XRCC6/XRCC5, encoding Ku 70/80 protein) depletion [58]. ATR/CHK1 pathway inhibition leads to increased cytosolic DNA fragments, thus activating the cyclic guanosine monophosphate-AMP synthase–stimulator of interferon genes (cGAS-STING) pathway and upregulating the transcription of type I interferon genes, which drive an innate immune response [59, 60]. Accordingly, ATR or CHK1 inhibition decreases the expression of PD-L1 by destabilizing PD-L1 in a proteasome-dependent manner to attenuate programmed cell death protein 1 (PD-1)/PD-L1 interaction and sensitizes cancer cells to T-cell–mediated killing [59, 61]. A recent study also showed that combining a low dose of gemcitabine (40 mg/kg, once per week) with CHK1i (SRA737) and anti-PD-L1 increases antitumorigenic CD8+ cytotoxic T-cells and upregulates the mRNA expressions of chemokines C-X-C motif chemokine 10 (CXCL10) and C-C motif chemokine ligand 5 (CCL5), which are key in CD4+ and CD8+ T-cell chemotaxis in small cell lung cancer animal models [62]. Similarly, dual inhibition of WEE1 (adavosertib) and ATM (AZD0156) reduced tumor growth and downregulated the expression of PD-L1, CKLF like MARVEL transmembrane domain containing 6 (CMTM6), cluster of differentiation 163 (CD163), and C-X-C chemokine receptor 2 (CXCR2) in BRCA2-mutated pancreatic cancer cell line models, which contribute to tumor immune evasion [63]. Therefore, immune checkpoint blockade might be a potential combination treatment for cell cycle checkpoint pathway inhibitor(s) to bypass PARPi resistance by improving the immunoreactive microenvironment.
Figure 4. DNA sensing and innate immune response in cancer cells.
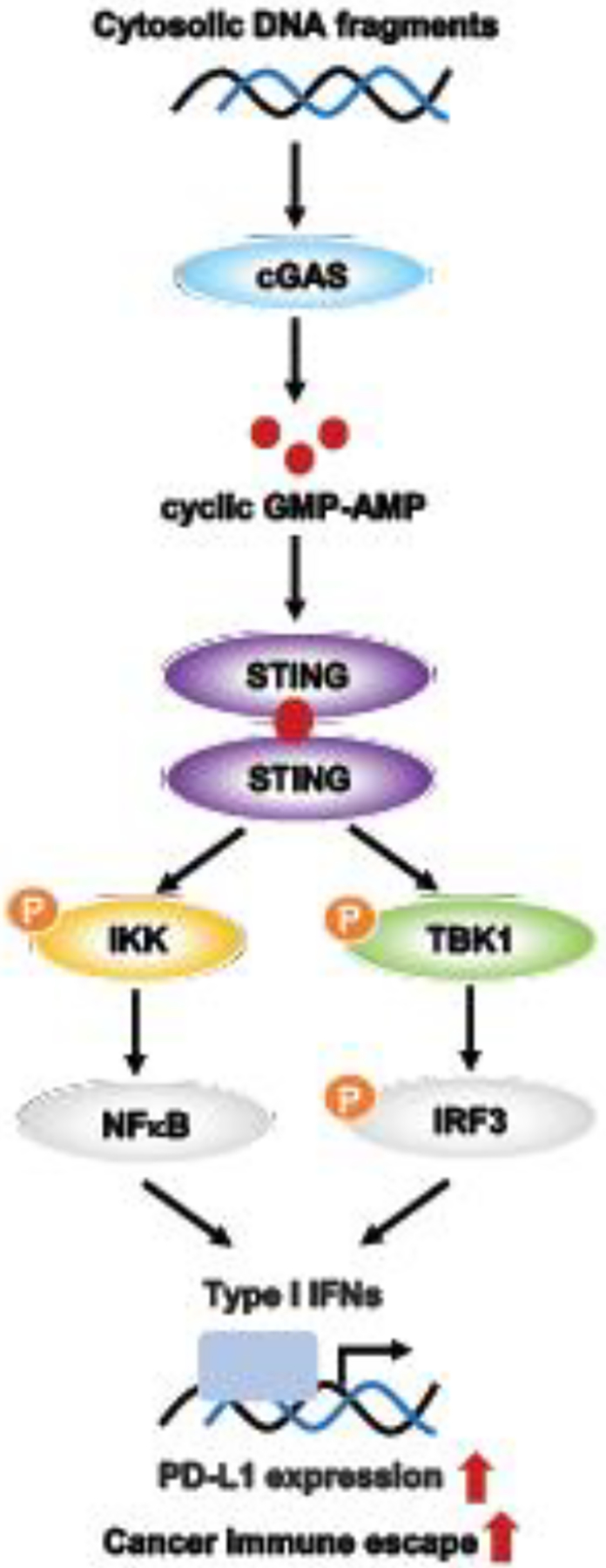
Radiotherapy or chemotherapy induces DNA damage and the formation of cytosolic DNA fragments. Cytosolic DNA fragment–induced cGAS activation leads to the endogenous generation of cyclic GMP-AMP, which binds to STING. STING then activates the TANK-binding kinase 1 (TBK1)/interferon regulatory factor 3 (IRF3) axis and NF-κB pathway, resulting in the transcription of type I interferons (IFNs) and other cytokines. These cytokines upregulate PD-L1 expression, which binds to PD-1 on the surface of activated T-cell or B-cells, contributing to cancer immune escape.
Another approach to circumventing PARPi resistance is to block cell survival pathway activation. Increased phosphoinositide 3-kinase (PI3K)/ mammalian target of rapamycin (mTOR) activation has been demonstrated in PARPi-resistant HGSOC cells and is thought to be one of the key mechanisms for survival in PARPi-resistant tumors [39]. As such, Huang et al. reported that inhibition of the PI3K/mTOR axis augments CHK1i-induced replication stress and cell death in both PARPi-sensitive and -resistant HGSOC preclinical models [64]. Similarly, CHK1is (PF477736, AZD7762, prexasertib) in combination with PI3K inhibitors (BYL719, GDC0941, GSK1059615) exerted synergistic anticancer effects in TP53-mutant/low EGFR oral cavity squamous cell carcinoma cell lines and patient-derived xenografts [65]. Notably, combined inhibition of the PI3K/mTOR axis (LY3023414, AZD8186, AZD2014) with CHK1 (prexasertib, SRA737, AZD7762) showed promising antitumor activity in HGSOC cell lines with PARPi resistance [64], suggesting this combination strategy may be used in the PARPi-resistant clinical setting.
In summary, this section provides an insight into the growing evidence supporting further development of cell cycle checkpoint inhibitors in PARPi-resistant cancer models. As with PARPi combinations, the key challenge is appropriate dosing and scheduling to achieve antitumor activity and limit toxicity. Although the preclinical data give some early indications of which tumor subtypes might benefit from treatment with ATR/CHK1 inhibition, such as SLFN11-deficiency in NSCLC, leukemia, TNBC, and colon cancers, much is still unknown regarding determinants of sensitivity to cell cycle checkpoint inhibitors.
3. Clinical Studies
3.1. Cell Cycle Checkpoint Inhibitor Monotherapy in Advanced/Recurrent Solid Tumors with PARPi Resistance
The promising preclinical studies of ATR/CHK1/WEE1 inhibitors in various PARPi-resistant cancers have prompted the initiation of early phase clinical trials (Table 3). However, the results from these clinical trials to date have been somewhat limited and include heterogenous patient populations. Also worth noting is the fact that unlike platinum sensitivity or resistance, which is a relatively reliable clinical predictor of response to PARPis [66], there is no universal consensus as to what constitutes PARPi clinical resistance. In general, documented progression of disease while on PARPi therapy is how PARPi resistance is defined in the clinic, although each trial may have slight variation or more specific criteria that must be considered when evaluating outcomes from different trials. Most cell cycle checkpoint inhibitors are combined with PARPis or other drugs, but there are a few monotherapy trials with PARPi-resistant ovarian cancer patients that are worth highlighting.
Table 3.
Clinical trials targeting cell cycle checkpoints as monotherapy and in combination with chemotherapy and other targeted agents.
| Trial | Phase | Eligible Patients | Drugs | Dose/Schedule | Findings of Primary/Secondary Endpoints | Notable Common Grade AEs (≥10%) |
|---|---|---|---|---|---|---|
| ATR Inhibitors | ||||||
| DUETTE NCT04239014 |
Randomized, double-blinded, placebo-controlled phase II | Platinum-sensitive, PARPi-resistant HGSOC | Ceralasertib + olaparib | Arm 1: Ceralasertib 160 mg PO QD on days 1–7; olaparib 300 mg PO BID q28-day cycle Arm 2: Olaparib 300 mg PO BID Arm 3: Placebo PO BID |
N/A Note: Withdrawn by the company sponsor in January 2021 based on the interim analysis of the VIOLETTE study NCT03330847 investigating the combination of ceralasertib and olaparib, where study closure was recommended due to insufficient activity. |
|
| NCT04655183 | Phase I/II | PARPi-resistant, germline BRCA1/2-mutated, HER2-advanced breast cancer | M4344 + niraparib | Niraparib PO QD; M4344 at the dose determined in part 1 of study | N/A Note: Withdrawn by the company sponsor. |
|
| NCT04149145 | Phase I | PARPi-resistant recurrent ovarian cancer Estimated enrollment: n = 40 |
M4344 + niraparib | 1st phase: 3+3 design of fixed dose niraparib PO QD (with 4-week lead-in); M4344 will be escalated from 100–200 mg PO QD q28-day cycle |
Not yet recruiting as of October 2021
Estimated study start date: December 2021 |
|
| NCT03704467 | Phase Ib | Platinum-sensitive, PARPi-resistant, recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer n = 3 |
Carboplatin + berzosertib (M6620) + avelumab |
M6620 90 mg/m2 IV on day 2; avelumab 1600 mg IV on day 1; carboplatin AUC 5 on day 1 q3-week cycle for a maximum of 6 cycles | Note: Originally designed as phase Ib/II trial, but sponsor did not conduct phase II after completing phase Ib and confirming safe combination dose. | |
| NCT04267939 | Phase I/Ib | Escalation cohort: All solid tumors (excluding prostate cancer) and positive for DDR deficiency Expansion cohort: Ovarian cancer, PARPi naïve, and with a platinum resistant/refractor y disease and DDR deficiency Expansion cohort: Ovarian cancer, PARPi-resistant, unselected for BRCA mutation status, platinum-status unspecified |
BAY189534 4 + niraparib | BAY189534 4 BID QD; niraparib PO QD q28-day cycle |
Recruiting
Estimated study completion date: December 5, 2025 |
|
| NCT04826341 | Phase I/II | Phase I: Advanced or recurrent solid tumors Phase II: Multi-cohort single arm study HRD cohort: Known HRD cancer and documented evidence of germline or somatic BRCA mutation or other HRD germline mutation, or tumor is HRD positive; PD while taking a PARPi or within 6 months of completing PARPi SCLC cohort: Recurrent SCLC after ≥ 1 prior platinum-based therapy Estimated enrollment: n = 70 |
Berzosertib (M6620) + sacituzumab govitecan | M6620 IV 20 mg/mL on days 2 and 9; sacituzumab govitecan IV on days 1 and 8 q21-day cycles |
Recruiting
Estimated study completion date: March 1, 2026 |
|
| CAPRI NCT03462342 | Single-arm, multi-cohort phase II | Cohort A: HGSOC with unknown or negative BRCA germline mutation status with no limit on prior number of regimens Cohort B: HGSOC with unknown or negative BRCA germline mutation status with no more than 3 prior cytotoxic therapies since the development of platinum-resistance Cohort C: Platinum-sensitive HGSOC with germline or somatic BRCA1/2 mutation, other HRD mutation, or positive HRD score on Myriad My Choice immediately following prior PARPi treatment (n = 13) |
Ceralasertib + olaparib | Ceralasertib 160 mg PO QD on days 1–7; olaparib 300 mg PO BID q28-day cycle | Cohort C: ORR: 46% (6 PR) Median duration of treatment: 8 mths (range 3–23) |
Cohort C: Grade 3 thrombocytopenia: 23% (n = 3); anemia: 16% (n = 2); neutropenia: 16% (n = 2) No grade 4 toxicities |
| OLAPCO NCT02576444 |
Single-arm phase II | BRCA-mutated, PARPi pretreated/resistant HGSOC n = 7 |
Ceralasertib + olaparib | Ceralasertib 160 mg PO QD on days 1–7; olaparib 300 mg PO BID q28-day cycle | 1 PR, 3 SD, 3 PD | Anemia, thrombocytopenia (rates not reported) |
| CHK1 Inhibitors | ||||||
| NCT03057145 | Phase I | Advanced solid tumors n = 29; 25 evaluable BRCA-mutant, PARPi-resistant expansion (n = 18) |
Prexasertib + olaparib | Olaparib PO on an intermittent schedule during each 28-day cycle; prexasertib IV on days 1 and 15 q28-day cycle | ORR: 22% (4/18 PR) 10/18 pts, (56%) remained on study for 4+ cycles |
Neutropenia: (86%); leukopenia (83%); anemia (72%); thrombocytopenia (66%) |
| NCT02203513 | Single-arm, multi-cohort phase II | Cohort 1: Germline or somatic BRCA-mutated HGSOC n = 22 (21 with prior PARPi); 18 evaluable |
Prexasertib | Prexasertib 105 mg/m2 IV q14 days q28-day cycle | ORR: 11% (1 CR, 1 PR) | Grade 3/4 AEs: neutropenia (82%); leukopenia (64%); thrombocytopenia (14%) |
| WEE1 Inhibitors | ||||||
| STAR NCT04197713 |
Single-arm phase I | Advanced solid tumors in a post-PARPi population Estimated enrollment: n = 54 |
Adavosertib + olaparib | Olaparib PO BID on days 1–5 and 15–19; adavosertib PO QD on days 8–12 and 22–26 q28 day cycle for 2 years |
Recruiting
Estimated study completion date: June 30, 2022 |
|
| NCT02482311 | Phase Ib |
BRCA1/2-mutant, PARPi-resistant advanced HGSOC cohort n = 30 |
Adavosertib | Adavosertib PO BID on days 1–3 and 8–10 q21-day cycle | ORR: 3% (n = 1 PR) DCR: 77% (n = 23) Median PFS: 3.9 mths |
Diarrhea (61%); nausea (50%); fatigue (43%) across safety population (n = 80) |
| EFFORT NCT03579316 |
Randomized phase II | Recurrent ovarian cancer in which progression has been documented following PARPi therapy n = 80 enrolled and randomized; 35 patients evaluable in each arm |
Adavosertib ± olaparib | Arm 1: Adavosertib PO QD on days 1–5 and 8–12 q21-day cycle (n = 39) Arm 2: Olaparib PO BID on days 1–21; adavosertib PO QD on days 1–3 and 8–10 q21-day cycle (n = 41) |
ORR (90% CI): Arm 1: 23% (12–38) Arm 2: 29% (16–44) CBR (90% CI): Arm 1: 63% (48–76) Arm 2: 89% (76–96) Median PFS (90% CI) Arm 1: 5.5 mths (3.9–6.9) Arm 2: 6.8 mths (4.3–8.3) |
Arm 1: 51% with grade 3/4 toxicities: neutropenia (13%); thrombocytopenia (10%); diarrhea (8%) Arm 2: 76% with grade 3/4 toxicities: thrombocytopenia (20%); neutropenia (15%); diarrhea (12%); fatigue (12%); anemia (10%) |
Abbreviations; AE, adverse event; AUC, area under curve; BID, twice daily; CBR, clinical benefit rate; CI, confidence interval; DCR, disease control rate; HRD, homologous recombination deficient; IV, intravenous; mths, months; NE, not evaluable, ORR, objective response rate; PARPi, poly (ADP-ribose) polymerase inhibitor; PD, progression of disease; PFS, progression-free survival; PO, by mouth; PR, partial response; pts, patients; QD, once daily; SCLC, small cell lung cancer.
Three ATR inhibitors, berzosertib (M6620), ceralasertib (AZD6738), and BAY1895344, have been evaluated as monotherapy in phase I trials, and have demonstrated safety but only limited activity outside of BRCA-mutated and ATM-deficient tumors [67–69]. In a phase I study of the ATRi BAY1895344, the most common all-grade treatment-emergent adverse events (AEs) were hematologic, i.e., anemia 81.8% (all grade 3), neutropenia 72.7% (grade 3/4, 54.5% n = 12]), and thrombocytopenia 45.5% (grade 3/4, 18.2% n = 4]) [69]. The common non-hematologic AEs included fatigue 68.2% (grade 2 requiring dose reduction, 4.5% n = 1]; grade 3, 9.1% n = 2]) and nausea 50.0% (grade 3, 9.1% n = 2]). Among all dose cohorts and schedules, four partial responses (PRs) were observed in 20 heavily pretreated evaluable patients, with a median duration of response of approximately 10.5 months among responders (advanced renal collective ductal carcinoma; metastatic appendiceal cancer; estrogen receptor/progesterone receptor–positive, HER2-negative, platinum-refractory breast cancer; and endometrial cancer) [69]. Fifty percent of patients, including all four responders, had ATM protein loss (renal collecting duct carcinoma) and/or deleterious ATM mutations (ATM_T2333fs* breast cancer, ATM_p.I2629fs* endometrial cancer, ATM_p.V1268fs* appendiceal cancer]), which indicates the limited utility of ATM deficiency as a predictive biomarker [69]. Given the lack of sufficient activity signal with ATRi monotherapy in unselected populations, efforts have instead been directed toward ATRi in combination with PARPis and chemotherapeutic agents.
For CHK1is, preliminary results of the phase II clinical trial of CHK1i prexasertib monotherapy in a BRCA-mutant HGSOC cohort have been reported (NCT02203513) [70]. The objective response rate (ORR) was 11% among all evaluable patients (n = 18) and only 6% among patients with prior PARPis (n = 17) [70]. The relatively low response rate (RR) is somewhat surprising given the 33% RR seen in the BRCAwt cohort from the same trial [71], suggesting that the levels of underlying replication stress might have been different between the two cohorts, and biomarker studies are ongoing. Other CHK1is, e.g., rabusertib, SRA737, AZD7762, and MK-8776, have been reported in phase I/II trials in advanced solid tumors, but not in PARPi-resistant HGSOC, showing modest antitumor activity with ORR ranging from 0–5% and dose-limiting toxicities including thrombocytopenia and gastrointestinal intolerability [24].
Bauer et al. reported a phase Ib trial of the WEE1i adavosertib in expansion cohorts in advanced tumor types with molecular biomarkers of interest, including germline BRCA1/2-mutated, PARPi-resistant HGSOC (n = 30) (NCT02482311) [72]. The activity was very modest, with only 1 out of 30 patients achieving a PR, for an ORR of 3%. Once again, the BRCAwt group (n = 16) had a higher ORR of 6%, although the difference here may be due to the small sample sizes [72]. Nonetheless, the RRs of cell cycle checkpoint inhibitor monotherapy in PARPi-resistant subgroups have largely been underwhelming, prompting drug combinations to achieve better efficacy [72].
3.2. Cell Cycle Checkpoint Inhibitors in Combination with PARPi in PARPi-Resistant Advanced/Recurrent Solid Tumors
Preclinical studies suggest that combination PARPi and cell cycle checkpoint inhibition may yield greater efficacy than either monotherapy in the PARPi-resistant setting [39]. This point has particular relevance in the clinical setting from a practical standpoint, as whether or not patients should be rechallenged with PARPis after progression is yet to be determined, although current ASCO PARPi guidelines do not recommend it outside of a clinical trial setting [4]. Thus, the potential for cell cycle checkpoint inhibitors to re-sensitize previously PARPi-resistant patients to PARPis is notable, as it could substantially broaden the scope of available treatment options for advanced cancer patients.
Two ongoing trials combining ATRi (ceralasertib) and PARPi (olaparib) are CAPRI (NCT03462342) [73] and OLAPCO (NCT02576444) [74]. CAPRI is a single-arm, multi-cohort phase II trial, where one of three cohorts includes platinum-sensitive HGSOC immediately following prior PARPi treatment using a 28-day cycle of olaparib 300 mg taken by mouth (PO) twice daily (BID) and ceralasertib 160 mg PO once daily (QD) on days 1–7. In this study, ATRi was given at a lower dose than the recommended phase II dose (RP2D) established as monotherapy (160 mg BID, 2 weeks on/2 weeks off [75]), while maintaining the standard dose of olaparib, given that both drugs can cause marrow toxicity. Eligibility criteria include a germline or somatic BRCA1/2 mutation, other HR repair gene mutation, or positive HRD score (>42 on Myriad My Choice), as well as clinical benefit from prior PARPi (defined as > 12 months on treatment for first line maintenance, > 6 months for ≥ second line maintenance, or treatment of recurrence with response by CA-125 or imaging), suggesting that the target population consists of those with acquired rather than de novo PARPi resistance in the platinum-sensitive disease setting. Preliminary results of the CAPRI study were recently reported [73]. Of the 13 patients enrolled, 6 had a PR (46% ORR), all with HRD, while no response was seen in HR-proficient patients. Importantly, the toxicity profile was manageable, with 4 patients (31%) experiencing grade 3 AEs of thrombocytopenia (n = 3), anemia (n = 2), and neutropenia (n = 2) [73].
The OLAPCO trial investigated the combination of olaparib and ceralasertib also in patients with PARPi-pre-treated/resistant (both acquired or de novo), platinum-resistant, BRCA-mutated HGSOC as well as in relapsed, refractory cancer patients with tumors harboring HR repair gene mutations [74]. The dosing regimen was the same as that used in the CAPRI trial [73]. For the HGSOC cohort, 7 patients had BRCA-mutated, PARPi-resistant, platinum-resistant disease: 1 had a PR (14% ORR) and 3 had SD > 4 months [74]. Meanwhile, a BRCA-mutant prostate and pancreatic cohort (n = 4) had a 0% ORR; whether these patients had prior PARPi or ATM mutation/loss is not reported, although knowing the PARPi resistance status is necessary in order to better characterize the differential response to ATRi/PARPi combination in various PARPi-resistant cancers [74].
Do et al. recently reported results from a phase I trial of the CHK1i prexasertib and PARPi olaparib combination in advanced solid tumors (NCT03057145), in which preliminary antitumor activity was seen in patients with platinum- and PARPi-resistant HGSOC [76]. The study followed a 3 + 3 dose escalation design with a 7-day lead-in of olaparib alone (cycle 0), followed by prexasertib administered intravenously on days 1 and 15 of a 28-day cycle in combination with olaparib. The RP2D was prexasertib 70 mg/m2 intravenously with olaparib at 100 mg PO BID [76]. Both of these doses are lower than the RP2D of each monotherapy due to overlapping marrow toxicity [76]. Four out of 18 patients with BRCA-mutant PARPi-resistant HGSOC (defined as progression on a prior PARPi after achieving some degree of clinical benefit, likely indicating acquired rather than de novo PARPi resistance), all platinum-resistant, achieved a confirmed PR ranging from 6–12 months (ORR 22%) [76]. It is unclear how much clinical activity was driven by CHK1i alone.
Similarly, the ongoing EFFORT phase II clinical trial evaluating the WEE1i adavosertib with and without olaparib in PARPi-resistant ovarian cancer (NCT03579316), in which the majority had platinum-resistant disease (64%), has shown modest activity [77]. Eighty patients were randomized to adavosertib only (300 mg PO QD on 5 days/week for 2 week-on and 1 week-off of a 21-day cycle; n = 39; 35 evaluable) or adavosertib plus olaparib (adavosertib 150 mg PO BID on 3 days/week and olaparib 200 mg PO BID of a 21-day cycle; n = 41; 35 evaluable) [77]. Forty-eight percent of patients had germline or somatic BRCA mutations. The combination arm trended toward better clinical activity relative to the monotherapy, although there was no statistical significance: 23% ORR (90% CI 12–38) and median PFS of 5.5 months (90% CI 3.9–6.9) in the adavosertib arm vs. 29% ORR (90% CI 16–44) and median PFS of 6.8 months (90% CI 4.3–8.3) in the combination arm [77]. Grade 3/4 toxicities occurred more frequently in the combination arm (76% vs. 51%), and the most common AEs were neutropenia (15% combination vs. 13% monotherapy), thrombocytopenia (20% vs. 10%), and diarrhea (12% vs. 8%) in both arms. Of note, approximately half of patients (20) in the adavosertib arm required dose reduction, whereas in the combination arm, 29 (71%) required dose reduction, and 4 (10%) discontinued due to toxicity [77]. Not surprisingly, the combination treatment elicited more serious dose-limiting toxicities than monotherapy despite using lower than the RP2D dose, suggesting that even attenuated doses and different dosing schedules may not necessarily mitigate adverse effects of the combination.
The preclinical activity of cell cycle checkpoint inhibitors in combination with PARPis does not match the largely mixed results in humans, underscoring the need for predictive biomarkers of sensitivity and resistance to combination therapy. To date, there are no validated biomarkers of replication stress. Some ongoing trials now include replication stress endpoints to better predict drug response, and a genomic or proteomic biomarker approach has been widely accepted. For instance, tumors with cyclin E1 (CCNE1) overexpression or amplification, MYC proto-oncogene, BHLH transcription factor (MYC) amplification, as well as retinoblastoma protein 1 (RB1) loss, or pRPA have been hypothesized as particularly susceptible to ATR/CHK1 pathway inhibition [27, 71, 78, 79]. However, their use as biomarkers of sensitivity to ATR/CHK1/WEE1 inhibitors has been limited in the clinical setting and warrants further validation in prospective clinical trials.
3.3. Cell Cycle Checkpoint Inhibitors in Combination with Other Drugs in the PARPi-Resistant Clinical Setting
Most combination trials of cell cycle checkpoint inhibitors involve PARPis, although there have been a few trials assessing ATRi in combination with other drugs in PARPi-resistant cancers. A first-in-human trial of the ATRi berzosertib was conducted to determine the safety, RP2D, pharmacokinetics, and antitumor activity of berzosertib monotherapy and combined with carboplatin in patients with advanced solid tumors [68]. While this study was not specifically limited to the PARPi-resistant setting, it is notable that in the combined berzosertib and carboplatin arm of the study, one patient with advanced germline BRCA1-mutant HGSOC achieved a PR lasting six months, despite being platinum-refractory and acquired PARPi-resistant. The combination of berzosertib and carboplatin is now being compared to carboplatin–docetaxel in men with pre-treated metastatic castrate-resistant prostate cancer without prospectively defined mutations (NCT03517969) [80].
In another phase I study of berzosertib in combination with gemcitabine (NCT02157792) [81], of the 4/48 patients with PR (8.3%), one with heavily pretreated advanced estrogen receptor/progesterone receptor–positive and HER2-negative breast cancer with BRCA2 mutation, who previously progressed on platinum chemotherapy and olaparib, achieved a durable PR (about 10 months). While the trial was not specifically conducted in the PARPi-resistant setting, these results again suggest that berzosertib may have a role in overcoming platinum and/or PARPi resistance, and further investigation in this subgroup is warranted for their molecular and clinical characteristics.
A phase I/II trial of berzosertib and sacituzumab govitecan in HRD, PARPi-resistant cancer (NCT04826341) just opened in 2021. Sacituzumab govitecan is an antibody-drug conjugate (ADC) comprising topoisomerase-I inhibiting camptothecin, SN-38, linked to a humanized antibody targeting trophoblastic cell-surface antigen 2 (Trop-2). Although no preclinical data on the ADC combination have been reported yet, the data from this clinical trial are eagerly awaited.
Regarding immunotherapy combinations, which have shown some promise preclinically, there is a dearth of published data on cell cycle checkpoint and immune checkpoint inhibitor combinations. A phase Ib/II clinical trial combining carboplatin, berzosertib, and anti–PD-L1 avelumab (NCT03704467) was unfortunately withdrawn by the sponsor [82]. There are two early-phase (phase I/II) clinical trials combining ATR inhibition and immune checkpoint blockade (berzosertib and avelumab; BAY1895344 and pembrolizumab) in patients with advanced malignancies, including prostate cancer (NCT04266912 and NCT04095273, respectively), and data from these trials are forthcoming. These trials are not being conducted specifically in PARPi-resistant populations; however, there is a high likelihood that a number of patients in both trials will have prior PARPi exposure given the expanding use of PARPis in ovarian, breast, pancreatic, and prostate cancers.
Though there are currently no ongoing trials of CHK1i in combination with non-PARPi drugs, Hong et al. recently published the results from a phase Ib study of the CHK1i, prexasertib, in combination with the PI3K/mTOR inhibitor, samotolisib, in three sub-arms: patients with advanced/metastatic solid tumors (dose escalation phase), patients with tumors containing phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutations, and patients with TNBC (median of 3 prior treatment regimens, range 1–9) [83]. The combination was associated with substantial toxicity. The most frequently reported treatment-related AEs in the overall safety population were leukopenia/neutropenia (94.3%; grade ≥3, 92.5%), thrombocytopenia (62.3%; grade ≥3, 47.2%), and nausea (52.8%; grade ≥3, 1.9%). An RP2D was not established because even at the reduced dose of samotolisib (from 200 mg BID to 150 mg BID) and standard monotherapy RP2D of prexasertib 105 mg/m2, there were many grade 3/4 AEs as noted above, suggesting that this combination may need an intermittent dosing schedule with more aggressive supportive care [83]. Among the 53 evaluable patients, 8 PRs were observed, for an overall ORR of 15.1%. The ORR differed substantially across sub-arms, with an ORR of 15.4% in the dose-escalation phase, 0% in the solid tumor expansion arm, 13.3% in the PIK3CA mutation arm, and 25% in the TNBC arm, both with and without PIK3CA mutation [83]. In addition to this study, other CHK1i trials have been completed in the past in combination with chemotherapeutic agents such as gemcitabine in advanced solid tumors and lymphomas (e.g., NCT00779584 [84], NCT01564251 [85]). Of note, most of the CHK1is that have been combined with chemotherapy in clinical trials (e.g., AZD7762, PF-00477736, GDC-0575, MK-8776, Sch 900776) are no longer under active clinical investigation, likely a result of their de-prioritization due to dose-limiting toxicities with only a modest clinical response [86].
4. Conclusion
The growing prevalence of PARPi resistance across various malignancies is an unmet medical need. Targeting the ATR/CHK1/WEE1 pathway is a rational approach to address this pressing matter. Preclinical studies of cell cycle checkpoint inhibitor monotherapy as well as in combination with PARPis show promise, whereas clinical trials have had somewhat more middling results. A biologically relevant consensus on what constitutes PARPi resistance in the clinic, as well as a unifying definition of replication stress, would aid greatly in standardizing results from clinical trials and facilitate more direct comparisons across different settings and development of biomarkers to identify subgroups of PARPi-resistant patients who may have benefit from such treatments. Overlapping toxicities between PARPis and cell cycle checkpoint inhibitors pose a potential challenge, although carefully designed treatment schedules and sequences are being tested and may help to circumvent a substantial increase or worsening of AEs. In addition, most clinical trials now incorporate pre- and on-treatment tissue biopsies and cell-free DNA to perform biomarker analyses, which will ultimately aid in furthering our knowledge of subsets of patients. As PARPi resistance continues to increase, tailoring clinical trials to PARPi-resistant patients and including PARPi-resistant subgroups within larger trials will be critically important in understanding the therapeutic combinations that best serve this unique but growing population.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute (J.M.L., #ZIA BC011525), Center for Cancer Research, NCI, USA. This research was made possible through the National Institutes of Health (NIH) Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation (DDCF Grant #2014194), the American Association for Dental Research, the Colgate-Palmolive Company, Genentech, Elsevier and other private donors (N.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
The authors declare no potential conflicts of interest.
CRediT authorship contribution statement
Nitasha Gupta: Writing – original draft, Tzu-Ting Huang: Writing – original draft, Visualization, Sachi Horibata: Writing – review & editing, Jung-Min Lee: Supervision, Funding acquisition, Writing – review & editing.
References
- [1].Mateo J, Lord CJ, Serra V, Tutt A, Balmaña J, Castroviejo-Bermejo M, Cruz C, Oaknin A, Kaye SB, de Bono JS, A decade of clinical development of PARP inhibitors in perspective, Ann. Oncol 30 (2019) 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y, Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors, Cancer Res. 72 (2012) 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, Lisyanskaya A, Floquet A, Leary A, Sonke GS, Gourley C, Banerjee S, Oza A, Gonzalez-Martin A, Aghajanian C, Bradley W, Mathews C, Liu J, Lowe ES, Bloomfield R, DiSilvestro P, Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer, N. Engl. J. Med 379 (2018) 2495–2505. [DOI] [PubMed] [Google Scholar]
- [4].Tew WP, Lacchetti C, Ellis A, Maxian K, Banerjee S, Bookman M, Jones MB, Lee JM, Lheureux S, Liu JF, Moore KN, Muller C, Rodriguez P, Walsh C, Westin SN, Kohn EC, PARP Inhibitors in the Management of Ovarian Cancer: ASCO Guideline, J. Clin. Oncol 38 (2020) 3468–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roche H, Im YH, Quek RGW, Markova D, Tudor IC, Hannah AL, Eiermann W, Blum JL, Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation, N. Engl. J. Med 379 (2018) 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, Masuda N, Delaloge S, Li W, Armstrong A, Wu W, Goessl C, Runswick S, Domchek SM, OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer, Ann. Oncol 30 (2019) 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, Reinacher-Schick A, Tortora G, Algul H, O’Reilly EM, McGuinness D, Cui KY, Schlienger K, Locker GY, Kindler HL, Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer, N. Engl. J. Med 381 (2019) 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, Gillman A, Figueiredo I, Paulding C, Seed G, Jain S, Ralph C, Protheroe A, Hussain S, Jones R, Elliott T, McGovern U, Bianchini D, Goodall J, Zafeiriou Z, Williamson CT, Ferraldeschi R, Riisnaes R, Ebbs B, Fowler G, Roda D, Yuan W, Wu YM, Cao X, Brough R, Pemberton H, A’Hern R, Swain A, Kunju LP, Eeles R, Attard G, Lord CJ, Ashworth A, Rubin MA, Knudsen KE, Feng FY, Chinnaiyan AM, Hall E, de Bono JS, DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer, N. Engl. J. Med 373 (2015) 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Messina C, Cattrini C, Soldato D, Vallome G, Caffo O, Castro E, Olmos D, Boccardo F, Zanardi E, BRCA Mutations in Prostate Cancer: Prognostic and Predictive Implications, J. Oncol 2020 (2020) 4986365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J, PARP inhibitor resistance: the underlying mechanisms and clinical implications, Mol. Cancer 19 (2020) 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McMullen M, Karakasis K, Madariaga A, Oza AM, Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer, Cancers 12 (2020) 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gaillard H, Garcia-Muse T, Aguilera A, Replication stress and cancer, Nat. Rev. Cancer 15 (2015) 276–289. [DOI] [PubMed] [Google Scholar]
- [13].McDermott N, Buechelmaier ES, Powell SN, Capitalizing on Cancer Replication Stress by Preventing PAR Chain Turnover: A New Type of Synthetic Lethality, Cancer Cell 35 (2019) 706. [DOI] [PubMed] [Google Scholar]
- [14].Zeman MK, Cimprich KA, Causes and consequences of replication stress, Nat. Cell Biol 16 (2014) 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gralewska P, Gajek A, Marczak A, Rogalska A, Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer, J. Hematol. Oncol 13 (2020) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cleary JM, Aguirre AJ, Shapiro GI, D’Andrea AD, Biomarker-Guided Development of DNA Repair Inhibitors, Mol. Cell 78 (2020) 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Haynes B, Murai J, Lee JM, Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition, Cancer Treat. Rev 71 (2018) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Prakash R, Zhang Y, Feng W, Jasin M, Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins, Cold Spring Harb. Perspect. Biol 7 (2015) a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, Masson JY, Zou L, Coupling of Homologous Recombination and the Checkpoint by ATR, Mol. Cell 65 (2017) 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dibitetto D, Sims JR, Ascencao CFR, Feng K, Kim D, Oberly S, Freire R, Smolka MB, Intrinsic ATR signaling shapes DNA end resection and suppresses toxic DNA-PKcs signaling, NAR Cancer 2 (2020) zcaa006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim D, Liu Y, Oberly S, Freire R, Smolka MB, ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells, Nucleic Acids Res. 46 (2018) 8311–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bertoli C, Klier S, McGowan C, Wittenberg C, de Bruin RA, Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription, Curr. Biol 23 (2013) 1629–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Peng B, Shi R, Bian J, Li Y, Wang P, Wang H, Liao J, Zhu WG, Xu X, PARP1 and CHK1 coordinate PLK1 enzymatic activity during the DNA damage response to promote homologous recombination-mediated repair, Nucleic Acids Res. 49 (2021) 7554–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gorecki L, Andrs M, Korabecny J, Clinical Candidates Targeting the ATR–CHK1–WEE1 Axis in Cancer, Cancers 13 (2021) 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saini P, Li Y, Dobbelstein M, Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress, Oncotarget 6 (2015) 13072–13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Krajewska M, Fehrmann RS, Schoonen PM, Labib S, de Vries EG, Franke L, van Vugt MA, ATR inhibition preferentially targets homologous recombination-deficient tumor cells, Oncogene 34 (2015) 3474–3481. [DOI] [PubMed] [Google Scholar]
- [27].Parmar K, Kochupurakkal BS, Lazaro J-B, Wang ZC, Palakurthi S, Kirschmeier PT, Yang C, Sambel LA, Färkkilä A, Reznichenko E, Reavis HD, Dunn CE, Zou L, Do KT, Konstantinopoulos PA, Matulonis UA, Liu JF, D’Andrea AD, Shapiro GI, The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition, Clin. Cancer Res 25 (2019) 6127–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Simoneau A, Zou L, An extending ATR-CHK1 circuitry: the replication stress response and beyond, Curr. Opin. Genet. Dev 71 (2021) 92–98. [DOI] [PubMed] [Google Scholar]
- [29].Yazinski SA, Zou L, Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway, Annu. Rev. Genet 50 (2016) 155–173. [DOI] [PubMed] [Google Scholar]
- [30].Tomimatsu N, Mukherjee B, Harris JL, Boffo FL, Hardebeck MC, Potts PR, Khanna KK, Burma S, DNA-damage-induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair, J. Biol. Chem 292 (2017) 10779–10790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li S, Lavagnino Z, Lemacon D, Kong L, Ustione A, Ng X, Zhang Y, Wang Y, Zheng B, Piwnica-Worms H, Vindigni A, Piston DW, You Z, Ca(2+)-Stimulated AMPK-Dependent Phosphorylation of Exo1 Protects Stressed Replication Forks from Aberrant Resection, Mol. Cell 74 (2019) 1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, Cortez D, ATR phosphorylates SMARCAL1 to prevent replication fork collapse, Genes Dev. 27 (2013) 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A, FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling, Mol. Cell 51 (2013) 678–690. [DOI] [PubMed] [Google Scholar]
- [34].Techer H, Koundrioukoff S, Carignon S, Wilhelm T, Millot GA, Lopez BS, Brison O, Debatisse M, Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage, Cell Rep. 14 (2016) 1114–1127. [DOI] [PubMed] [Google Scholar]
- [35].Ghelli Luserna di Rora A Cerchione C, Martinelli G, Simonetti G A WEE1 family business: regulation of mitosis, cancer progression, and therapeutic target, J. Hematol. Oncol 13 (2020) 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Martin Y, Dominguez-Kelly R, Freire R, Novel insights into maintaining genomic integrity: Wee1 regulating Mus81/Eme1, Cell Div. 6 (2011) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, Sorensen CS, Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption, Mol. Cell Biol 32 (2012) 4226–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, Morgan M, Herlyn M, Brown E, Simpkins F, Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models, Clin. Cancer Res 23 (2017) 3097–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, Medvedev S, Kinose Y, Devins K, Verma P, Ly K, Wang Y, Greenberg RA, Schwartz L, Johnson N, Scharpf RB, Mills GB, Zhang R, Velculescu VE, Brown EJ, Simpkins F, Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models, Nat. Commun 11 (2020) 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ceccaldi R, O’Connor KW, Mouw KW, Li AY, Matulonis UA, D’Andrea AD, Konstantinopoulos PA, A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance, Cancer Res. 75 (2015) 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, Jaspers JE, Watanabe K, Pieterse M, Kersbergen A, Sol W, Celie PHN, Schouten PC, van den Broek B, Salman A, Nieuwland M, de Rink I, de Ronde J, Jalink K, Boulton SJ, Chen J, van Gent DC, Bartek J, Jonkers J, Borst P, Rottenberg S, REV7 counteracts DNA double-strand break resection and affects PARP inhibition, Nature 521 (2015) 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Niimi K, Murakumo Y, Watanabe N, Kato T, Mii S, Enomoto A, Asai M, Asai N, Yamamoto E, Kajiyama H, Shibata K, Kikkawa F, Takahashi M, Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells, Cancer Sci. 105 (2014) 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, Lees J, Zou L, Martuza RL, Wakimoto H, Rabkin SD, Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma, Nat. Commun 10 (2019) 2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, Pommier Y, Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition, Oncotarget 7 (2016) 76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, Al Abo M, Rajapakse VN, Nakamura E, Jenkins LMM, Aladjem MI, Pommier Y, SLFN11 Blocks Stressed Replication Forks Independently of ATR, Mol. Cell 69 (2018) 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schoonen PM, Kok YP, Wierenga E, Bakker B, Foijer F, Spierings DCJ, van Vugt M, Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells, Mol. Oncol 13 (2019) 2422–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wengner AM, Siemeister G, Lucking U, Lefranc J, Wortmann L, Lienau P, Bader B, Bomer U, Moosmayer D, Eberspacher U, Golfier S, Schatz CA, Baumgart SJ, Haendler B, Lejeune P, Schlicker A, von Nussbaum F, Brands M, Ziegelbauer K, Mumberg D, The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models, Mol. Cancer Ther 19 (2020) 26–38. [DOI] [PubMed] [Google Scholar]
- [48].Neeb A, Herranz N, Arce-Gallego S, Miranda S, Buroni L, Yuan W, Athie A, Casals T, Carmichael J, Rodrigues DN, Gurel B, Rescigno P, Rekowski J, Welti J, Riisnaes R, Gil V, Ning J, Wagner V, Casanova-Salas I, Cordoba S, Castro N, Fenor de la Maza MD, Seed G, Chandran K, Ferreira A, Figueiredo I, Bertan C, Bianchini D, Aversa C, Paschalis A, Gonzalez M, Morales-Barrera R, Suarez C, Carles J, Swain A, Sharp A, Gil J, Serra V, Lord C, Carreira S, Mateo J, de Bono JS, Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors, Eur. Urol 79 (2021) 200–211. [DOI] [PubMed] [Google Scholar]
- [49].Brill E, Yokoyama T, Nair J, Yu M, Ahn YR, Lee JM, Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer, Oncotarget 8 (2017) 111026–111040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, Marsh KB, Davis MA, Zhao L, Maybaum J, Lawrence TS, Morgan MA, Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer, Clin. Cancer Res 20 (2014) 5085–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parsels LA, Karnak D, Parsels JD, Zhang Q, Velez-Padilla J, Reichert ZR, Wahl DR, Maybaum J, O’Connor MJ, Lawrence TS, Morgan MA, PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors, Mol. Cancer Res 16 (2018) 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, Jeong KJ, Ding Z, Liang J, Wang SW, Dai H, Lee S, Sahni N, Mercado-Uribe I, Kim TB, Chen K, Lin SY, Peng G, Westin SN, Liu J, O’Connor MJ, Yap TA, Mills GB, Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy, Cancer Cell 35 (2019) 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jin J, Fang H, Yang F, Ji W, Guan N, Sun Z, Shi Y, Zhou G, Guan X, Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer, Neoplasia 20 (2018) 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, Rajapakse VN, Elloumi F, Takahashi N, Schultz CW, Thomas A, Zenke FT, Pommier Y, Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents, Mol. Cancer Ther 20 (2021) 1431–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Nair J, Huang TT, Murai J, Haynes B, Steeg PS, Pommier Y, Lee JM, Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer, Oncogene 39 (2020) 5520–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Josse R, Martin SE, Guha R, Ormanoglu P, Pfister TD, Reaper PM, Barnes CS, Jones J, Charlton P, Pollard JR, Morris J, Doroshow JH, Pommier Y, ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses, Cancer Res. 74 (2014) 6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bradbury A, Hall S, Curtin N, Drew Y, Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations?, Pharmacol. Ther 207 (2020) 107450. [DOI] [PubMed] [Google Scholar]
- [58].Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S, Yoshimoto Y, Held KD, Suzuki Y, Kono K, Miyagawa K, Nakano T, Shibata A, DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells, Nat. Commun 8 (2017) 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mouw KW, Konstantinopoulos PA, From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation, Br. J. Cancer 118 (2018) 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ngoi NYL, Pham MM, Tan DSP, Yap TA, Targeting the replication stress response through synthetic lethal strategies in cancer medicine, Trends Cancer 7 (2021) 930–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, Chan LC, Yang Y, Hsu JL, Lai YJ, Hung MC, Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing, Am. J. Cancer Res 8 (2018) 1307–1316. [PMC free article] [PubMed] [Google Scholar]
- [62].Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, Gay CM, Stewart CA, Fan Y, Shen L, Hansen RJ, Strouse B, Hedrick MP, Hassig CA, Heymach JV, Wang J, Byers LA, Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC, J. Thorac. Oncol 14 (2019) 2152–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jin MH, Nam AR, Park JE, Bang JH, Bang YJ, Oh DY, Therapeutic Co-targeting of WEE1 and ATM Downregulates PD-L1 Expression in Pancreatic Cancer, Cancer Res. Treat 52 (2020) 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Huang TT, Brill E, Nair JR, Zhang X, Wilson KM, Chen L, Thomas CJ, Lee JM, Targeting the PI3K/mTOR Pathway Augments CHK1 Inhibitor-Induced Replication Stress and Antitumor Activity in High-Grade Serous Ovarian Cancer, Cancer Res. 80 (2020) 5380–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yang CY, Liu CR, Chang IY, OuYang CN, Hsieh CH, Huang YL, Wang CI, Jan FW, Wang WL, Tsai TL, Liu H, Tseng CP, Chang YS, Wu CC, Chang KP, Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts, Cancers 12 (2020) 1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, Chang DK, Garsed DW, Jonkers J, Ledermann JA, Nik-Zainal S, Ray-Coquard I, Shah SP, Matias-Guiu X, Swisher EM, Yates LR, ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer, Ann. Oncol 31 (2020) 1606–1622. [DOI] [PubMed] [Google Scholar]
- [67].Dillon MT, Boylan Z, Smith D, Guevara J, Mohammed K, Peckitt C, Saunders M, Banerji U, Clack G, Smith SA, Spicer JF, Forster MD, Harrington KJ, PATRIOT: A phase I study to assess the tolerability, safety and biological effects of a specific ataxia telangiectasia and Rad3-related (ATR) inhibitor (AZD6738) as a single agent and in combination with palliative radiation therapy in patients with solid tumours, Clin. Transl. Radiat. Oncol 12 (2018) 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, Tunariu N, Perez-Lopez R, Rodrigues DN, Riisnaes R, Figueiredo I, Carreira S, Hare B, McDermott K, Khalique S, Williamson CT, Natrajan R, Pettitt SJ, Lord CJ, Banerji U, Pollard J, Lopez J, de Bono JS, Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors, J. Clin. Oncol 38 (2020) 3195–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yap TA, Tan DSP, Terbuch A, Caldwell R, Guo C, Goh BC, Heong V, Haris NRM, Bashir S, Drew Y, Hong DS, Meric-Bernstam F, Wilkinson G, Hreiki J, Wengner AM, Bladt F, Schlicker A, Ludwig M, Zhou Y, Liu L, Bordia S, Plummer R, Lagkadinou E, de Bono JS, First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors, Cancer Discov. 11 (2021) 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lampert EJ, An D, McCoy A, Kohn EC, Annunziata CM, Trewhitt K, Zimmer ADS, Lipkowitz S, Lee J, Prexasertib, a cell cycle checkpoint kinase 1 inhibitor, in BRCA mutant recurrent high-grade serous ovarian cancer (HGSOC): A proof-of-concept single arm phase II study, J. Clin. Oncol 38 (2020) 6038–6038. [Google Scholar]
- [71].Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, Bagheri MH, Botesteanu DA, Steinberg SM, Minasian L, Ekwede I, Kohn EC, Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study, Lancet Oncol. 19 (2018) 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bauer TM, Moore K, Rader JS, Simpkins F, Mita A, Beck JT, Hart L, Chu Q, Oza A, Tinker AV, So K, Imedio ER, Kumar S, Mugundu GM, Jenkins S, Chmielecki J, Jones S, Spigel DR, Fu S, Abstract CT012: Open-label, multicenter, Phase Ib study to assess safety, tolerability and efficacy of adavosertib monotherapy in patients with advanced solid tumors: Expansion cohorts, Cancer Res. 79 (2019) CT012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wethington SL, Shah PD, Martin LP, Tanyi JL, Latif NA, Morgan MA, Torigian DA, Pagan C, Rodriguez D, Domchek SM, Drapkin R, Shih I-M, Smith S, Dean E, Armstrong DK, Gaillard S, Simpkins F, Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer, J. Clin. Oncol 39 (2021) 5516. [Google Scholar]
- [74].Eder JP, Sohal D, Mahdi H, Do K, Keedy V, Hafez N, Doroshow D, Avedissian M, Mortimer P, Glover C, LoRusso P, Juergensmeier JM, Shapiro GI, Abstract A080: Olaparib and the ATR inhibitor AZD6738 in relapsed, refractory cancer patients with homologous recombination (HR) repair mutations – OLAPCO, Mol. Cancer Ther 18 (2019) A080. [Google Scholar]
- [75].Dillon M, Guevara J, Mohammed K, Smith S, Dean E, McLellan L, Boylan Z, Spicer J, Forster M, Harrington K, 450PDA phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B), Ann. Oncol 30 (2019) v159–v193. [Google Scholar]
- [76].Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A, Quinn N, Gannon C, Vuong L, Parmar K, Lazaro J-B, D’Andrea AD, Shapiro GI, Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors, Clin. Cancer Res 27 (2021) 4710–4716. [DOI] [PubMed] [Google Scholar]
- [77].Westin SN, Coleman RL, Fellman BM, Yuan Y, Sood AK, Soliman PT, Wright AA, Horowitz NS, Campos SM, Konstantinopoulos PA, Levenback CF, Gershenson DM, Lu KH, Bayer V, Tukdi S, Rabbit A, Ottesen L, Godin R, Mills GB, Liu JF, EFFORT: EFFicacy Of adavosertib in parp ResisTance: A randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer, J. Clin. Oncol 39 (2021) 5505. [Google Scholar]
- [78].Guerrero Llobet S, van der Vegt B, Jongeneel E, Bense RD, Zwager MC, Schröder CP, Everts M, Fehrmann RSN, de Bock GH, van Vugt MATM, Cyclin E expression is associated with high levels of replication stress in triple-negative breast cancer, NPJ Breast Cancer 6 (2020) 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shapiro GI, Wesolowski R, Devoe C, Lord S, Pollard J, Hendriks BS, Falk M, Diaz-Padilla I, Plummer R, Yap TA, Phase 1 study of the ATR inhibitor berzosertib in combination with cisplatin in patients with advanced solid tumours, Br. J. Cancer 125 (2021) 520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choudhury AD, Xie W, Parikh M, Lee D, Kessler ER, Einstein DJ, Kochupurakkal B, Mouw KW, Allen EMV, Doyle LA, D’Andrea AD, Taplin M-E, Shapiro G, A phase II study of M6620 in combination with carboplatin compared with docetaxel in combination with carboplatin in metastatic castration-resistant prostate cancer, J. Clin. Oncol 38 (2020) TPS5597. [Google Scholar]
- [81].Plummer ER, Dean EJ, Evans TRJ, Greystoke A, Herbschleb K, Ranson M, Brown J, Zhang Y, Karan S, Pollard J, Penney MS, Asmal M, Fields SZ, Middleton MR, Phase I trial of first-in-class ATR inhibitor VX-970 in combination with gemcitabine (Gem) in advanced solid tumors (NCT02157792), J. Clin. Oncol 34 (2016) 2513. [Google Scholar]
- [82].Banerjee S, Vergotte I, Colombo N, Barve M, Grisham R, Mehr KT, Falk M, Beier F, Hennessy M, Schroeder A, Birrer M, 1060TiP - Randomized, phase Ib/II study of M6620 + avelumab + carboplatin vs standard care (sc) in patients (pts) with platinum-sensitive poly (ADP-ribose) polymerase inhibitor-(PARPi)-resistant ovarian cancer, Ann. Oncol 30 (2019) v431–v432. [Google Scholar]
- [83].Hong DS, Moore KN, Bendell JC, Karp DD, Wang JS, Ulahannan SV, Jones S, Wu W, Donoho GP, Ding Y, Capen A, Wang X, Bence Lin A, Patel MR, Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/mTOR Inhibitor, Clin. Cancer Res 27 (2021) 1864–1874. [DOI] [PubMed] [Google Scholar]
- [84].Daud AI, Ashworth MT, Strosberg J, Goldman JW, Mendelson D, Springett G, Venook AP, Loechner S, Rosen LS, Shanahan F, Parry D, Shumway S, Grabowsky JA, Freshwater T, Sorge C, Kang SP, Isaacs R, Munster PN, Phase I Dose-Escalation Trial of Checkpoint Kinase 1 Inhibitor MK-8776 As Monotherapy and in Combination With Gemcitabine in Patients With Advanced Solid Tumors, J. Clin. Oncol 33 (2015) 1060–1066. [DOI] [PubMed] [Google Scholar]
- [85].Italiano A, Infante JR, Shapiro GI, Moore KN, LoRusso PM, Hamilton E, Cousin S, Toulmonde M, Postel-Vinay S, Tolaney S, Blackwood EM, Mahrus S, Peale FV, Lu X, Moein A, Epler J, DuPree K, Tagen M, Murray ER, Schutzman JL, Lauchle JO, Hollebecque A, Soria JC, Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors, Ann. Oncol 29 (2018) 1304–1311. [DOI] [PubMed] [Google Scholar]
- [86].Dent P, Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer, Expert Opin. Investig. Drugs 28 (2019) 1095–1100. [DOI] [PubMed] [Google Scholar]